 Stefanie A. G. Black
Stefanie A. G. Black Peter K. Stys2,3
Peter K. Stys2,3 Gerald W. Zamponi
Gerald W. Zamponi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 28 August 2014
Sec. Cell Death and Survival
Volume 2 - 2014 | https://doi.org/10.3389/fcell.2014.00045
This article is part of the Research TopicPromiscuous functions of the prion protein gene familyView all 13 articles
Although it is well established that misfolding of the cellular prion protein (PrPC) into the β-sheet-rich, aggregated scrapie conformation (PrPSc) causes a variety of transmissible spongiform encephalopathies (TSEs), the physiological roles of PrPC are still incompletely understood. There is accumulating evidence describing the roles of PrPC in neurodegeneration and neuroinflammation. Recently, we identified a functional regulation of NMDA receptors by PrPC that involves formation of a physical protein complex between these proteins. Excessive NMDA receptor activity during conditions such as ischemia mediates enhanced Ca2+ entry into cells and contributes to excitotoxic neuronal death. In addition, NMDA receptors and/or PrPC play critical roles in neuroinflammation and glial cell toxicity. Inhibition of NMDA receptor activity protects against PrPSc-induced neuronal death. Moreover, in mice lacking PrPC, infarct size is increased after focal cerebral ischemia, and absence of PrPC increases susceptibility of neurons to NMDA receptor-dependent death. Recently, PrPC was found to be a receptor for oligomeric beta-amyloid (Aβ) peptides, suggesting a role for PrPC in Alzheimer's disease (AD). Our recent findings suggest that Aβ peptides enhance NMDA receptor current by perturbing the normal copper- and PrPC-dependent regulation of these receptors. Here, we review evidence highlighting a role for PrPC in preventing NMDA receptor-mediated excitotoxicity and inflammation. There is a need for more detailed molecular characterization of PrPC-mediated regulation of NMDA receptors, such as determining which NMDA receptor subunits mediate pathogenic effects upon loss of PrPC-mediated regulation and identifying PrPC binding site(s) on the receptor. This knowledge will allow development of novel therapeutic interventions for not only TSEs, but also for AD and other neurodegenerative disorders involving dysfunction of PrPC.
Misfolding of the cellular prion protein (PrPC) into the β-sheet-rich, aggregate-prone scrapie conformation (PrPSc) is well known to result in several progressive and often fatal diseases termed prionopathies. This group of disorders, also known as transmissible spongiform encephalopathies (TSEs), includes scrapie, bovine spongiform encephalopathy (BSE, or mad cow disease), and the human diseases Creutzfeld–Jakob disease, Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, and Kuru (Aguzzi et al., 2008). PrPSc aggregation and formation of amyloid-like plaques disrupt neuronal physiology, and most TSEs involve eventual loss of neurons (Aguzzi et al., 2008). Although much work has been done investigating the deleterious effects of misfolded/aggregated prion proteins, the physiological roles of PrPC remain incompletely understood.
Mature PrPC is a glycoprotein that is anchored to the extracellular leaflet of the plasma membrane through a carboxyl (C)-terminal glycosylphosphatidylinositol (GPI) anchor. Initially, PrPC is synthesized as a precursor polypeptide (254 amino acids for mouse PrPC) containing an amino (N)-terminal signal sequence that directs PrPC to the endoplasmic reticulum and is post-translationally cleaved, and a C-terminal GPI anchor sequence that is removed and replaced with a GPI anchor (Figure 1; Biasini et al., 2012). The N-terminal portion of PrPC is intrinsically unstructured, contains the octarepeat region, and has multiple copper binding sites both within and outside of the octarepeat region (Figure 1; Klewpatinond et al., 2008; Stanyon et al., 2014). The PrPC N-terminus also contains sites for binding of oligomeric β-amyloid (Aβ) peptides (Figure 1; Lauren et al., 2009; Chen et al., 2010). In contrast to the N-terminus, the C-terminus of PrPC is globular in structure, with three α-helical and two short β-strand regions, and is the location of two asparagine residues that undergo glycosylation (Figure 1; Biasini et al., 2012). In addition, PrPC can undergo physiological cleavage of different types: α-cleavage in the vicinity of residue 109 (mouse sequence) generates N1 and C1 fragments; β-cleavage in the vicinity of residue 90 (mouse sequence) produces N2 and C2 fragments; and cleavage at the GPI anchor releases full-length PrPC into the extracellular environment (Altmeppen et al., 2013; McDonald et al., 2014). However, recent data from McDonald and co-workers using recombinant mouse PrPC reveal that cleavage of PrPC is quite complex, generating a number of different N1, C1, N2, and C2 fragments (McDonald et al., 2014), although the biological functions of these cleavage products remain to be determined.

Figure 1. Schematic representation of PrPC. Numbering of amino acid residues corresponds to the mouse PrPC sequence. Shown are the locations of histidine (H) residues that bind copper ions (residues 60, 68, 76, 84, 95, 110), the amino (N)-terminal signal peptide (SP; residues 1–22), β-amyloid oligomer (Aβ) binding regions (residues 23–27, 95–110), the octapeptide repeat region (OR; residues 51–90), the central hydrophobic domain (HD; residues 111–130), α-helical regions (α; residues 143–152, 171–191, 199–221), β-sheet regions (β; residues 127–129, 166–168), sites of asparagine (N)-linked glycosylation (residues 180, 196), and the carboxyl (C)-terminal glycosylphosphatidylinositol (GPI) signal peptide (GSP; residues 231–254).
PrPC interacts with and signals through many different cell surface proteins, including the α7 nicotinic acetylcholine receptor (Beraldo et al., 2010), metabotropic glutamate receptors mGluR1 (Beraldo et al., 2011) and mGluR5 (Beraldo et al., 2011; Um et al., 2013), kainate receptor GluR6/7 (Carulla et al., 2011), and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor subunits GluA1 (Watt et al., 2012) and GluA2 (Kleene et al., 2007; Watt et al., 2012). Furthermore, Aβ oligomers can bind to PrPC and signal through mGluR5 to activate Fyn kinase (Um et al., 2013), and an Aβ-PrPC-mGluR5 pathway is also involved in facilitation of long-term depression (LTD) (Hu et al., 2014). The diversity of signaling mediated by PrPC highlights the need for a better understanding of how the interaction of PrPC with its binding partners is regulated in both physiological and pathological conditions. Work from our laboratory revealed that PrPC also interacts with N-methyl-D-aspartate (NMDA) receptors; PrPC-deficient mice display increased NMDA receptor-dependent neuronal excitability and are more susceptible to NMDA-induced excitotoxicity (Khosravani et al., 2008). Here, we briefly review properties of the NMDA receptor and focus on evidence supporting a role for PrPC in preventing NMDA receptor-mediated excitotoxicity, a process that may contribute to the pathogenesis of a variety of disorders including ischemic stroke, Alzheimer's disease (AD), Huntington's disease, and epilepsy (Lai et al., 2014; Parsons and Raymond, 2014). Similar mechanisms may be involved in PrPSc-induced toxicity (Müller et al., 1993; Riemer et al., 2008; Resenberger et al., 2011).
Glutamate is the major excitatory transmitter in the mammalian central nervous system (CNS), and it exerts its actions by binding to a variety of different receptor proteins. Glutamate receptors (GluRs) are divided into two families, ionotropic and metabotropic. Metabotropic GluRs are G protein-coupled receptors, while ionotropic GluRs are ion channels. There are three types of ionotropic GluRs, namely AMPA, kainate, and NMDA receptors (Mayer, 2005).
NMDA receptors are cation channels that mediate entry of Na+ and Ca2+ ions and are activated by the co-agonists glutamate (or NMDA) and glycine or D-serine. Activation of NMDA receptors thus contributes to the excitatory postsynaptic potential (EPSP), as well as both long-term potentiation (LTP) and LTD (Bartlett and Wang, 2013). In addition to co-agonist binding, activation of NMDA receptors requires membrane depolarization to remove the Mg2+ ion block of the channel pore that occurs at resting membrane potentials. NMDA receptors are heterotetrameric channels formed by the assembly of two obligatory GluN1 and two GluN2/GluN3 subunits. GluN1 subunits contain the binding site for glycine, while glutamate binds to GluN2 subunits. To date, there are seven known NMDA receptor subunits: GluN1, GluN2A-GluN2D, and GluN3A-GluN3B, with alternative splicing of GluN1 and GluN3 subunits generating further diversity (Paoletti et al., 2013). GluN3 subunits also bind glycine, and thus NMDA receptors containing only GluN1 and GluN3 subunits give rise to “glycine-only” receptors that cannot be activated by glutamate or NMDA (Chatterton et al., 2002; Pina-Crespo et al., 2010). Identity of the GluN2 subunits that combine with GluN1 subunits to form the tetrameric channel dictates NMDA receptor properties, such as activation and deactivation kinetics, ion conductance, and affinity for glutamate (Cull-Candy and Leszkiewicz, 2004). Pharmacological characterization using ligands for specific GluN2 subunits allows differentiation of receptor subtypes (Wyllie et al., 2013).
NMDA receptor localization can be synaptic, perisynaptic, extrasynaptic, or even presynaptic, with receptor activity at each location coupling to specific cellular events (Corlew et al., 2008; Hardingham, 2009; Hardingham and Bading, 2010; Paoletti et al., 2013). In general, activation of synaptic NMDA receptors activates pro-survival signaling, while extrasynaptic NMDA receptor activity mediates pro-death signaling (Hardingham, 2009; Hardingham and Bading, 2010). Thus, while NMDA receptors mediate key physiological functions such as learning and memory under normal conditions, they also play a role in glutamate excitotoxicity, which can occur in response to ischemia (Lau and Tymianski, 2010) and is involved in many neurodegenerative conditions, including AD (Parsons and Raymond, 2014). In these abnormal situations, excessive glutamate can spill over from synaptic to extrasynaptic sites, thus activating not only the desired synaptically-localized NMDA receptors, but also those receptors located extrasynaptically. Because NMDA receptors are highly permeable to Ca2+ ions, this overstimulation leads to enhanced Ca2+ influx that ultimately can be fatal to cells (Lau and Tymianski, 2010). NMDA receptors are found not only in neurons, but also in oligodendrocytes and myelin, and ischemia can cause NMDA receptor-dependent damage to both myelin and oligodendrocyte processes (Karadottir et al., 2005; Salter and Fern, 2005; Micu et al., 2006). Hence, tight regulation of NMDA receptor activity is of extreme importance in maintaining physiological signaling while preventing excitotoxicity. NMDA receptor activity is regulated by a number of different mechanisms, one of which involves an intrinsic desensitization mechanism that results in termination of channel activity even in the prolonged presence of glutamate, a process that is potently regulated by the receptor co-agonist glycine (Mayer et al., 1989). Recently, we demonstrated that the absence of PrPC enhances NMDA receptor glycine affinity, leading to persistent NMDA receptor activity upon prolonged agonist application (You et al., 2012), which likely underlies the greater susceptibility of PrPC-null neurons to NMDA receptor-mediated damage and dysfunction (Rangel et al., 2007; Khosravani et al., 2008; Gadotti and Zamponi, 2011; Gadotti et al., 2012; You et al., 2012; Fleisch et al., 2013).
In support of a role for PrPC in preventing NMDA receptor hyperactivity, data from our and other laboratories reveal that NMDA receptor activity is enhanced when PrPC is absent. Our work using mouse hippocampal slices showed that the enhanced neuronal excitability in PrPC-null neurons can be reduced by the NMDA receptor blocker aminophosphonovaleric acid (APV), and that NMDA receptor-mediated miniature excitatory postsynaptic currents (mEPSCs) recorded from cultured hippocampal neurons from PrPC-null mice had significantly larger amplitudes and longer decay times than those recorded from wild-type neurons (Khosravani et al., 2008). In this study we also showed that NMDA application resulted in whole-cell currents with a prolonged time course of deactivation in cultured PrPC-null hippocampal neurons that could be restored to wild-type levels by ectopic expression of mouse PrPC and mimicked by reducing PrPC expression in cultured wild-type hippocampal neurons. Co-immunoprecipitation experiments demonstrated that PrPC and GluN2D subunits were found in the same protein complex, and immunofluorescent staining of hippocampal neurons showed co-localization of these two proteins. Indeed, the properties of the NMDA-evoked currents we observed in PrPC-null neurons in this study appeared to resemble those of heterologously-expressed GluN2D-containing NMDA receptors (Cull-Candy and Leszkiewicz, 2004). Furthermore, siRNA knockdown of GluN2D expression dramatically accelerated NMDA receptor current deactivation kinetics and reduced current amplitude. Taken together, these observations point to an upregulation of GluN2D-containing NMDA receptor activity in the absence of PrPC. However, it is also possible that activity of NMDA receptors containing other subunits is altered by the lack of PrPC, producing GluN2D-like deactivation kinetics. Finally, in support of a role for PrPC in protecting against NMDA receptor-mediated toxicity, we observed significantly more cell death in response to NMDA treatment of PrPC-null hippocampal cultures than in wild-type cultures, and that focal injection of NMDA into the hippocampus resulted in a lesion of significantly greater size in PrPC-null mice than in wild-type mice (Khosravani et al., 2008). This increase in NMDA receptor activity not only gives rise to an increased susceptibility of neurons to cell death, but also affects other physiological processes that are linked to NMDA receptor function. For example, NMDA receptors are important for the transmission of pain signals at the spinal level. Consequently, in models of inflammatory and neuropathic pain, we found enhanced nociceptive responses in PrPC-null mice that could be restored to control levels by MK-801 (Gadotti and Zamponi, 2011). It is also known that NMDA receptors play a role in depressive-like behavior at the level of the hippocampus. Consistent with this notion, we found that PrPC-null mice displayed increased depressive-like behavior that could be abrogated by NMDA receptor blockers (Gadotti et al., 2012).
It is well established that PrPC is a copper-binding protein, with up to six copper ions binding to histidine residues in the N-terminal region both within (mouse PrPC residues 60, 68, 76, and 84) and outside of (mouse PrPC residues 95 and 110) the octarepeat region (Figure 1; Klewpatinond et al., 2008; Quintanar et al., 2013). In addition, copper may bind to histidine residues 176 and 186 (mouse sequence) in the C-terminus of PrPC (Quintanar et al., 2013), and recent evidence shows that copper can bind to the N-terminal amino group of PrPC (Stanyon et al., 2014). Occupancy of these copper sites is known to alter PrPC structure (Qin et al., 2000; Thakur et al., 2011; Younan et al., 2011; Quintanar et al., 2013), and thus likely its interactions with other protein partners. The ability to examine NMDA receptor activity as an indirect readout of the interaction of the receptors with PrPC has provided interesting insights into the roles of the copper binding sites for PrPC function. Under normal circumstances, NMDA receptors undergo desensitization in the prolonged presence of glutamate or NMDA, and this process is modulated by the co-agonist glycine (Mayer et al., 1989). In cultured mouse hippocampal neurons lacking PrPC, the desensitization kinetics appear to be slowed such that a non-desensitizing current is observed (You et al., 2012). In wild-type neurons, copper chelation using bathocuproine sulfonate (BCS) or cuprizone induced a persistent NMDA current similar to that seen in PrPC-null neurons (You et al., 2012). Acute treatment with phosphatidylinositol-specific phospholipase C (PI-PLC), which enzymatically removes proteins such as PrPC that are linked to the extracellular leaflet of the plasma membrane via GPI anchors, also induced a persistent NMDA-dependent current in rat hippocampal neurons that was similar to both the current seen with copper chelation and to that recorded from PrPC-null mouse hippocampal neurons (You et al., 2012). Moreover, PI-PLC treatment did not alter the steady-state NMDA current in PrPC-null neurons, indicating that the enzymatic cleavage did not measurably affect other potential regulatory proteins (You et al., 2012). Experiments investigating the effect of glycine concentration on steady-state NMDA current revealed that at a given glycine concentration, a higher level of steady-state current was seen in neurons lacking PrPC than in wild-type neurons; a similar effect was seen upon copper chelation (You et al., 2012). In rat hippocampal cultures, copper chelation induced cell death that could be prevented by NMDA receptor inhibition or re-addition of excess copper (You et al., 2012). Taken together, these results indicate that copper ions modulate NMDA receptors by virtue of their interactions with PrPC. Furthermore, these findings suggests that copper ions are key regulators of NMDA receptor function, thus adding another layer of metal ion regulation of these receptors in addition to the well described role of zinc ions.
Further support for PrPC-mediated regulation of NMDA receptor activity comes from a study using zebrafish with a targeted mutation of the prp2 gene encoding the zebrafish prion protein PrP2, which has characteristics similar to mammalian PrPC (Fleisch et al., 2013). Consistent with observations in mammalian brain, these mutants displayed increased convulsant-induced seizure activity compared to wild-type animals. Analysis of mEPSCs recorded in vivo from intact zebrafish hindbrain neurons showed that the frequency of NMDA receptor-mediated mEPSCs was reduced in prp2−/− fish compared to controls, and although NMDA receptor current amplitudes were the same in both groups, NMDA receptor currents had longer decay times in prp2−/− zebrafish. These recent observations agree with the earlier findings of Walz and colleagues showing that mice lacking PrPC were more sensitive to convulsant-induced seizures, and significantly more PrPC-null mice died than wild-type mice (Walz et al., 1999). However, a recent study found elevated thresholds for convulsant-induced epileptiform activity in hippocampal slices from PrPC-null mice compared to wild-type controls (Ratte et al., 2011). The reason for this disparity in the literature remains to be determined.
The presence of misfolded prion proteins could induce NMDA receptor hyperfunction by preventing normal, PrPC-mediated control of NMDA receptor activity. Indeed, there are several examples of augmented NMDA receptor function in the presence of mutant or scrapie forms of the prion protein. It has been demonstrated that NMDA receptor antagonists memantine and MK-801 prevented PrPSc-induced toxicity in cultured rat cortical neurons (Müller et al., 1993). Memantine also delayed death of scrapie-infected mice (Riemer et al., 2008) and blocked the PrPSc-induced increase in apoptosis of PrPC-expressing SH-SY5Y neuroblastoma cells (Resenberger et al., 2011). Recently, Biasini and co-workers revealed a role for aberrant NMDA receptor activity in the toxic effects of a prion protein lacking residues 105–125 in the central region, termed ΔCR PrP (Biasini et al., 2013). In this study, organotypic cerebellar slice cultures from ΔCR PrP-expressing mice were more susceptible to glutamate-, kainate-, and NMDA-induced cell death compared to those from PrP+/− controls, an effect that was rescued by overexpression of wild-type PrPC. Notably, NMDA had a more marked effect on cell death compared to the other treatments. These authors also observed that ΔCR PrP expression induces spontaneous inward currents, which may contribute to enhanced NMDA receptor activity via depolarization-induced relief of the Mg2+ ion block of these receptors. Importantly, the toxic NMDA receptor-dependent effects of ΔCR PrP in this study were prevented by overexpression of wild-type PrPC, again supporting a neuroprotective role of PrPC via prevention of NMDA receptor-mediated toxicity. In another recent study, the toxicity of the misfolded form of a prion protein fragment comprised of residues 90–231 was found to be mediated in part by NMDA receptor-dependent excitotoxicity (Thellung et al., 2013). In this study, treatment of cerebellar granule neuron cultures for 1–3 days with the misfolded prion protein 90–231 fragment augmented both intracellular Ca2+ levels and apoptosis compared to that measured in control cells, effects that were nearly completely abolished by NMDA receptor blockade.
Altogether, the data obtained from studies using PrPC-null animals suggest that there is aberrant NMDA receptor activity in the absence of normal PrPC function, which is further supported by the fact that exogenous expression of PrPC can prevent the NMDA receptor-dependent toxic effects of misfolded or mutant prion proteins. This raises the possibility of restoring PrPC-mediated regulation of NMDA receptors in pathologies where PrPC function is perturbed as a therapeutic intervention to control NMDA receptor-mediated toxicity. This could in theory be accomplished by mimicking the interaction of PrPC with NMDA receptors. To achieve this, more details regarding the molecular determinants governing the interaction of PrPC and NMDA receptor proteins are required. Additionally, if loss of physiological PrPC function results in augmented activity of certain NMDA receptor subtypes, such as those containing GluN2D subunits, a thorough molecular characterization of the PrPC-NMDA receptor interaction will allow development of targeted pharmacotherapies to reduce hyperfunction of only those receptors affected whose activity is aberrantly upregulated. This approach could prevent undesired effects of blocking physiological NMDA receptor signaling.
Given the neuroprotective effects of PrPC, it may not be surprising that absence of PrPC can exacerbate the neuronal damage that ensues following an ischemic insult. Several studies have demonstrated increased ischemic brain damage in PrPC-null mice (McLennan et al., 2004; Sakurai-Yamashita et al., 2005; Spudich et al., 2005; Weise et al., 2006; Mitteregger et al., 2007; Steele et al., 2009). However, ischemia can induce an upregulation of PrPC mRNA (McLennan et al., 2004; Mitsios et al., 2007; Mitteregger et al., 2007) and protein (McLennan et al., 2004; Weise et al., 2004; Shyu et al., 2005; Adle-Biassette et al., 2006; Mitsios et al., 2007; Mitteregger et al., 2007), presumably as a compensatory protective mechanism. The idea of compensatory upregulation of PrPC expression protecting against cell death in ischemia is supported by the findings that adenovirus-mediated overexpression of PrPC reduced ischemia-induced infarct volume in rats (Shyu et al., 2005) and PrPC-overexpressing mice showed smaller infarct volumes in response to ischemia than wild-type controls (Weise et al., 2008). However, another showed no difference in infarct sizes between wild-type and PrPC-overexpressing mice exposed to ischemia (Spudich et al., 2005). Of note, these studies used different PrPC-overexpressing mouse models and times of ischemia, which could account for the observed differences. Currently, the role of NMDA receptors in the enhanced ischemic cell death seen in the absence of PrPC is unknown. However, because NMDA receptor hyperfunction contributes to cell death in ischemia (Lau and Tymianski, 2010; Lai et al., 2014), and since NMDA receptor desensitization is reduced in the absence of PrPC (You et al., 2012), enhanced NMDA receptor-mediated Ca2+ influx may very well contribute to the increased ischemia-induced cell death that is observed in PrPC-null mice.
Increases in extracellular glutamate concentration, such as those seen in ischemic conditions, cause activation of both synaptic and extrasynaptic receptors (Hardingham and Bading, 2010; Parsons and Raymond, 2014). Extrasynaptic NMDA receptor activity can induce apoptotic signaling by promoting nuclear import of FOXO transcription factor proteins (Hardingham and Bading, 2010). FOXO3a has recently been identified as a negative regulator of the gene encoding PrPC, prnp (Liu et al., 2013), and in this study it was shown that insulin-like growth factor-1 (IGF-1)-induced PI3K/Akt activity promoted prnp expression by preventing nuclear import of FOXO proteins, thus inhibiting their negative effect on prnp expression. Therefore, it is possible that conditions where extrasynaptic NMDA receptor activity is upregulated may lead to a down-regulation of PrPC protein expression, which may initiate a positive feedback loop of further dysregulation of NMDA receptor activity and thus toxicity. Hence, even if there is an initial protective upregulation of PrPC, this may not be able to be sustained due to persistent activation of extrasynaptic NMDA receptors in the presence of prolonged elevation of glutamate levels.
Another important observation concerns increased shedding of PrPC in response to neurotoxic conditions (Wang et al., 2012). PrPC shedding results from cleavage of PrPC near the C-terminal GPI anchor, releasing full-length, soluble PrPC into the extracellular milieu (Altmeppen et al., 2013; McDonald et al., 2014). Wang and colleagues showed that upon treatment of cultured rat cortical neurons with NMDA, soluble PrPC was released into the culture medium, while total PrPC protein levels remained unchanged (Wang et al., 2012). As noted earlier, our data demonstrated that treatment with PI-PLC, which cleaves PrPC from its GPI anchor thus releasing it from the cell surface, resulted in persistent NMDA receptor-mediated current in cultured rat hippocampal neurons that was similar to those seen in PrPC-null hippocampal neurons (You et al., 2012). If loss of cell surface anchored-PrPC results in aberrant NMDA receptor activity, one could envision a scenario where a positive feedback loop contributes to further shedding of PrPC and enhanced NMDA receptor currents, which would be of detriment to cells. However, it has been suggested that PrPC shedding could serve a protective role, especially in preventing Aβ-mediated toxicity (discussed in Altmeppen et al., 2013; Beland et al., 2014). A recent study by Beland and colleagues showed that shed PrPC co-immunoprecipitated with Aβ (Beland et al., 2014), suggesting that extracellular Aβ can be sequestered by shed PrPC to prevent Aβ-induced cellular damage. As discussed below, we found that Aβ1-42 induced a PrPC-dependent increase in steady-state NMDA receptor current (You et al., 2012). Shedding of PrPC may thus aid in preventing Aβ 1-42-induced dysregulation of NMDA receptor function. Hence, PrPC shedding may ultimately be protective in situations where PrPC function is compromised, for example by binding of Aβ peptides. On the other hand, shedding of PrPC may promote aberrant NMDA receptor activity by altering desensitization kinetics that could contribute to cell death in conditions such as ischemia where glutamate concentrations are elevated.
There is also evidence that ischemia induces cleavage of PrPC into its N- and C-terminal fragments, and that these cleavage products can be neuroprotective. Mitteregger and coworkers also observed that ischemia caused increased cleavage of PrPC, as detected by elevated levels of PrPC C1 fragment on immunoblots of mouse brain homogenates (Mitteregger et al., 2007). In another study, oxygen glucose deprivation increased the secretion of the PrPC N1 fragment from cultured retinal ganglion cells (Guillot-Sestier et al., 2009). Moreover, application of recombinant PrPC N1 (PrPC residues 23–110) was able to prevent ischemia-induced death of retinal ganglion cells (Guillot-Sestier et al., 2009). The details of how PrPC N1 protects against ischemic cell death were not fully explored, but the mechanism did involve a reduction of ischemia-induced caspase-3 activation and p53 expression (Guillot-Sestier et al., 2009). An interesting possibility is that the PrPC N1 fragment, which contains the octarepeat region that binds copper, binds to NMDA receptors, thus providing copper-dependent regulation of receptor desensitization. Of note, inhibition of NMDA receptor activity has been shown to attenuate death of retinal ganglion cells following ischemia-reperfusion injury (Li et al., 2014).
Further support of a role for PrPC in protecting against ischemic cell death comes from the work of Mitteregger and coworkers where PrPC-null mice and mice expressing a prion protein lacking amino acid residues 32–93 had infarcts of similar volumes (Mitteregger et al., 2007), suggesting that this region of PrPC provides protection against ischemic injury. One reason for increased ischemic injury in mice expressing this form of prion protein could be that cleavage of this mutant would not produce the normal PrPC N1 fragment. However, this deletion mutant lacks the copper-binding octarepeat region of PrPC. As mentioned previously, we found a copper-dependent regulation of NMDA receptors that is mediated in large part through PrPC, as demonstrated by no further augmentation of NMDA receptor currents by copper chelation in the absence of PrPC (You et al., 2012). Moreover, exogenous copper can protect neurons from NMDA receptor-mediated death (Schlief et al., 2006), whereas copper chelation induced cell death that could be prevented by NMDA receptor inhibition (You et al., 2012). Recently, data from in vitro experiments have revealed that copper enhances one type of α-cleavage, termed α2-cleavage, of mouse PrPC (McDonald et al., 2014). Copper induced α2-cleavage such that the resultant N-terminal PrPC fragments terminate at alanine residues 116 or 117, in contrast to α1-cleavage observed in the absence of copper, which produces PrPC N1 ending at lysine residue 109 (McDonald et al., 2014). These observations suggest that copper levels may regulate PrPC processing by influencing the amount and identity of the N1 fragment produced, although the biological outcomes of this copper-dependent differential PrPC cleavage are not yet known. Of further interest is the finding that the prion proteins carrying pathogenic mutations have decreased α2-cleavage upon addition of copper (McDonald et al., 2014), suggesting aberrant copper-dependent PrPC processing could be detrimental to the health of cells. These findings reveal the possibility of copper-dependent processing of PrPC as another mechanism contributing to regulation of NMDA receptor activity by copper. Thus, copper may alter the activity of NMDA receptors in multiple ways: PrPC-independent control of receptor activity, as evidenced by restoration of NMDA receptor desensitization in PrPC-null neurons by addition of excess copper ions (You et al., 2012); by mediating interaction of PrPC and NMDA receptors, as shown by a decrease in the amount of GluN1 subunit that co-precipitated with PrPC upon copper chelation (You et al., 2012); and by influencing PrPC cleavage to generate PrPC N1 fragments, which could bind to NMDA receptors to modulate their function. Although evidence exists for the former two mechanisms, the latter remains to be investigated.
As mentioned previously, the activity of myelinic NMDA receptors is increased by ischemic conditions, resulting in damage to myelin (Micu et al., 2006). Thus, a loss of PrPC-mediated regulation of NMDA current may not only be neurotoxic, but may also contribute to demyelination that has been observed in PrPC-null mice and in mice expressing mutant prion proteins (Radovanovic et al., 2005; Baumann et al., 2007; Bremer et al., 2010).
PrPC has been shown to be a receptor for oligomeric Aβ peptides (Lauren et al., 2009; Chen et al., 2010). Since these discoveries, many studies have investigated the role of PrPC in the pathological effects of Aβ (reviewed in Lauren, 2014). Overall, these findings suggest a role for PrPC in protecting from the pathological effects of Aβ peptides that occur in AD, although the requirement of PrPC for Aβ-induced toxicity has been controversial, with some investigators finding that PrPC was not required for the pathological effects of Aβ (Balducci et al., 2010; Calella et al., 2010; Kessels et al., 2010; Cisse et al., 2011). Recent findings by Nicoll and coworkers may help to reconcile these discrepancies. Their work showed that Aβ protofibrils, and not monomers or fibrils, bind with highest affinity to full-length PrPC and the PrPC N1 fragment (Nicoll et al., 2013). Furthermore, protofibril-rich Aβ preparations blocked LTP in a PrPC-dependent manner, whereas LTP inhibition by fibrillar Aβ was independent of PrPC. These observations underscore the need for careful characterization of Aβ preparations in order to determine the nature of the Aβ species used for experiments, which allow better interpretation of data resulting from these experiments.
Notably, many studies have demonstrated that Aβ can alter NMDA receptor function (reviewed in Rush and Buisson, 2014). Recently, we demonstrated that PrPC is required for Aβ-induced alterations in NMDA receptor kinetics (You et al., 2012). Application of oligomerized Aβ1-42, which is known to be a high-affinity copper chelator, to hippocampal neurons gave rise to a persistent (i.e., non-desensitizing) NMDA current, similar to what was observed in the presence of the copper chelator BCS or in the absence of PrPC. Of note, the effects of Aβ1-42 and copper chelation on NMDA current were not additive, suggesting that they acted via the same mechanism. Finally, in neurons isolated from 5XFAD mice (a mouse model of AD), NMDA current displayed a qualitatively similar persistent component. Altogether, these data then suggest that Aβ-mediated perturbation of NMDA receptor activity depends on both PrPC and copper ions. Furthermore, the Aβ1-42-induced upregulation of NMDA receptor activity may in part be responsible for the Aβ1-42-mediated neurodegeneration observed in AD. If this were the case, then why is neurodegeneration observed in 5XFAD mice, but relatively absent in PrPC-null mice even though they both display similar non-desensitizing NMDA receptor currents? Slowed receptor desensitization can only manifest itself pathologically under conditions where there is a prolonged excess of glutamate. It has been shown that Aβ1-42 prevents glutamate re-uptake into neurons and glial cells (Danysz and Parsons, 2012), and induces release of glutamate from astrocytes (Talantova et al., 2013), which then would be expected to lead to just such an excess of glutamate. Then, together with the slowed desensitization kinetics of the receptor, this excess glutamate may damage neuronal structures such as spines. In PrPC-null mice, this accumulation of glutamate would not be expected to occur, thus rendering the slowed desensitization kinetics physiologically inert (Figure 2). Nonetheless, as we discussed earlier, there are other pathophysiological events such as chemically-induced seizures or ischemia that are known to lead to an accumulation of glutamate that then causes increased brain damage in PrPC-null mice (Walz et al., 1999; McLennan et al., 2004; Sakurai-Yamashita et al., 2005; Spudich et al., 2005; Weise et al., 2006; Mitteregger et al., 2007; Steele et al., 2009).
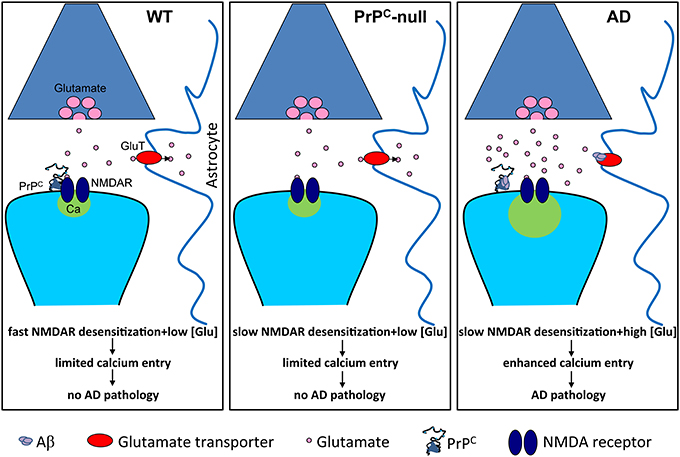
Figure 2. Slowed NMDA receptor desensitization is pathological only in conditions of prolonged excess of glutamate. Under normal conditions (WT), glutamate homeostasis is unperturbed and prevents glutamate accumulation, and NMDA receptors (NMDAR) undergo fast desensitization, thus limiting calcium entry. In the absence of PrPC (PrPC-null), NMDA receptor desensitization is slowed but glutamate homeostasis remains unperturbed, thus limited calcium entry occurs. However, in Alzheimer's disease (AD), Aβ oligomers bind to PrPC and cause slowed NMDA receptor desensitization. This, in combination with Aβ-induced elevation of glutamate levels, for example by inhibiting glutamate re-uptake by astrocytic glutamate transporters, leads to enhanced calcium entry and pathology.
Work by Um and colleagues provides further evidence for the requirement of PrPC on the effects of Aβ on NMDA receptor function (Um et al., 2012). In their study, they found that phosphorylation of the GluN2B subunit of the NMDA receptor at tyrosine residue 1472 was transiently increased by Aβ oligomers in Fyn-overexpressing neuroblastoma cells and in cortical cultures; longer treatment with Aβ oligomers (1–3 h) resulted in decreased phospho-GluN2B levels, which correlated with an increase in the level of STEP protein phosphatase. Moreover, the Aβ oligomer-induced increase in GluN2B phosphorylation was absent in PrPC- and Fyn-null cultures. Indeed, they found that Aβ oligomers activated Fyn kinase in a PrPC-dependent manner. Furthermore, brain extract from AD patients induced Fyn activation in cultured mouse cortical neurons in a PrPC-dependent manner, whereas extract from healthy controls did not activate Fyn. The initial GluN2B phosphorylation was paralleled by an increase in surface levels of GluN2B, and with longer Aβ oligomer treatment a reduction in GluN2B surface expression was observed; again, both PrPC and Fyn were required for these Aβ oligomer-mediated effects on GluN2B surface expression. In cultures from wild-type mice, pre-treatment for 15 min with Aβ oligomers significantly enhanced the NMDA-induced increase in intracellular Ca2+ levels, whereas a 60 min Aβ oligomer pre-treatment significantly reduced the NMDA-induced Ca2+ signal; in PrPC-null cultures, Aβ oligomers had no effect on the Ca2+ signal induced by NMDA. Thus, Aβ oligomers, acting via PrPC, induce a transient increase then a decrease in NMDA receptor-mediated Ca2+ influx into neurons. LDH release from cortical cultures was increased upon 90 min Aβ oligomer treatment and no further toxicity was observed with a 72 h treatment. This toxicity was NMDA receptor-dependent, as it was reduced by NMDA receptor inhibition with both APV and ifenprodil, the latter being a specific GluN2B antagonist. Thus, brief exposure to Aβ oligomers causes toxicity, likely by Ca2+ influx mediated by the transient increase in surface localization of GluN2B-containing NMDA receptors. In this study, it was also shown that Aβ oligomers induced spine loss in a PrPC- and Fyn-dependent manner, and that the seizures and death due to status epilepticus in a mouse model of AD were prevented by genetic deletion of PrPC. Taken together, these data suggest that Aβ oligomers signal through PrPC to induce aberrant Fyn activation, leading to first an increase then later a decrease in activity of GluN2B-containing receptors, which is ultimately deleterious to neurons. Altogether, the recent data indicate that Aβ oligomers, by interacting with PrPC, can alter both NMDA receptor kinetics (You et al., 2012) and the signaling pathways that modulate NMDA receptor function (Um et al., 2012).
A study by Resenberger and colleagues showed that Aβ- and PrPSc-induced cell death of PrPC-expressing SH-SY5Y neuroblastoma cells were not additive, suggesting that these species act via the same pathway (Resenberger et al., 2011). Furthermore, pre-treatment with the NMDA receptor blocker memantine prevented apoptosis of PrPC-expressing SH-SY5Y neuroblastoma cells induced by PrPSc, Aβ, and a synthetic peptide designed to be rich in β-sheet structure. This suggests that several types of β-sheet-containing proteins/peptides share a common mechanism of toxicity, aberrant NMDA receptor activity. Thus, PrPC could contribute to toxicity in disorders other than TSEs and AD, possibly by mediating aberrant NMDA receptor activity in the presence of β-sheet-rich peptides.
Of further interest is the finding that the toxicity induced by β-sheet-rich conformers was dependent upon the N-terminal region of PrPC and was prevented by a soluble N-terminal domain of PrPC (Resenberger et al., 2011). Similar protective effects of the PrPC N1 fragment against Aβ-induced toxicity have been observed in other studies (Beland et al., 2012, 2014; Guillot-Sestier et al., 2012; Nieznanski et al., 2012; Fluharty et al., 2013). This raises the possibility that increased PrPC cleavage to produce the N1 fragment is neuroprotective. Interestingly, cleavage of PrPC was increased in a mouse model of AD (Ostapchenko et al., 2013) and in post-mortem human AD brain (Beland et al., 2014). Furthermore, the recent findings from Beland and coworkers provide strong evidence for a protective role of the soluble N1 fragment that is produced by α-cleavage of PrPC. In this study, the binding of PrPC N1 and Aβ induced a conformational change that produced amorphous aggregates, the amount of PrPC N1 in guanidine hydrochloride extracts from insoluble amyloid deposits from AD brain was increased compared to non-demented controls, and there was a significant correlation between the amount of α-cleavage, which generates the N1 fragment, and duration of AD (Beland et al., 2014). Altogether, these observations indicate that cleavage of PrPC may reduce Aβ-mediated toxicity via N1-mediated neutralization of Aβ peptides, and N-terminal fragments may be useful as a therapy to prevent pathological effects of Aβ or other β-sheet-rich peptides, including alterations in NMDA receptor activity.
Recent evidence has emerged that glutamatergic mechanisms operate not only in the nervous system, but also in a wide variety of non-neuronal cells (reviews: Gill and Pulido, 2001; Skerry and Genever, 2001; Hinoi et al., 2004), including in the immune system (reviews: Boldyrev et al., 2005, 2012). GluRs, both ionotropic and metabotropic, are highly expressed in various immune cells, such as T cells, B cells, neutrophils, macrophages and dendritic cells, which are thought to subserve communication between the immune and nervous systems (review: Ganor and Levite, 2012). Interestingly, different GluRs, or different levels of certain GluRs, are expressed in resting and activated T cells (for review see Ganor and Levite, 2012). In particular, NMDA receptors expressed on T cells are involved in a wide variety of T cell functions, such as regulation of cytokine secretion (Kahlfuβ et al., 2014), proliferation (Kahlfuβ et al., 2014), apoptosis (Affaticati et al., 2011), and induction of reactive oxygen species (Tuneva et al., 2003).
As described above, PrPC interacts with NMDA receptors on neurons and modulates NMDA receptor-dependent neuronal excitability and excitotoxicity (Khosravani et al., 2008). Although the precise functions of PrPC in immune cells remain unclear, PrPC expression is detected in the lymphoid system (Bendheim et al., 1992; Ford et al., 2002). Human T lymphocytes constitutively express PrPC and its surface expression is influenced by the activation state of the cells (Cashman et al., 1990; Mabbott et al., 1997). In addition, PrPC is upregulated on mouse T cells and dendritic cells after activation, and the lack of PrPC increases T cell proliferation and causes T cell over-activation in mouse (Tsutsui et al., 2008). Therefore, PrPC may play a role as a regulator of NMDA receptors in the immune system as we observed in the nervous system (Khosravani et al., 2008; You et al., 2012), with a common thread being “hyperfunction” when PrPC is absent and cannot restrain normal physiological mechanisms.
The CNS is conventionally recognized as being “immunologically privileged” (Bailey et al., 2006) and is anatomically separated from the peripheral immune system by the presence of the blood-brain barrier (BBB) (Xiao and Link, 1998; Weller et al., 2009). Despite the presence of the BBB, which limits the entry of cells and pathogens to the brain, lymphocytes can traffic into the CNS to surveil the local environment (Xiao and Link, 1998; Kivisakk et al., 2003). It is increasingly recognized that the CNS is capable of shaping the immune response (for review see Xiao and Link, 1998); it is now clear that neuroinflammation is a well-established hallmark of a number of neurodegenerative diseases, including AD, and is frequently detrimental to neurological function (Akiyama et al., 2000; Rubio-Perez and Morillas-Ruiz, 2012; Solito and Sastre, 2012; Cappellano et al., 2013; Enciu and Popescu, 2013; Lynch, 2014). In the AD brain, the inflammatory response is thought to be a secondary response caused by an initial brain insult, which is provided by damaged neurons, neurites, insoluble Aβ aggregates, and neurofibrillary tangles as stimuli (Akiyama et al., 2000). However, recent evidence suggests that inflammatory mediators may stimulate amyloid precursor protein processing, and therefore, play a role as a driving force to establish a deleterious cycle to AD progression (Heneka et al., 2010). As described above, PrPC is a receptor for oligomeric Aβ peptides (Lauren et al., 2009; Chen et al., 2010; Lauren, 2014), and Aβ oligomers, acting via PrPC, modulate NMDA receptor-mediated Ca2+ influx into neurons. Perhaps, even soluble forms of small oligomers, may directly stimulate PrPC and modulate NMDA receptor-mediated Ca2+ influx into immune cells to trigger signaling pathways to secrete cytokines, reactive oxygen species, and/or regulate T cell polarization at the early stage of AD. Gaining more insight into the role of PrPC-NMDA receptor interactions in neuroinflammation may reveal novel approaches to visualize early AD pathogenesis and diagnosis.
It is becoming clear that loss of PrPC regulation of NMDA receptors can result in toxicity in a variety of pathological conditions, and that Aβ can cause aberrant activation of NMDA receptors in a PrPC-dependent manner. The possibility of restoring the NMDA receptor-PrPC interaction as a way to protect against excitotoxicity will need to be tested experimentally, and in order to accomplish this, elucidation of the molecular details of the interaction between PrPC and NMDA receptors is needed. In light of the recent findings regarding cleavage of PrPC and how this is altered in disease, there is a need for investigation of how physiological processing of PrPC impacts NMDA receptor activity in both health and disease. Overall, a better understanding of the molecular mechanisms that determine how PrPC regulates NMDA receptor function will open up new therapeutic avenues in situations where the regulation of NMDA receptors by PrPC is perturbed, such as in AD and prion diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Stefanie A. G. Black is supported by a Postdoctoral Fellowship from the Multiple Sclerosis Society of Canada (MSSOC). Peter K. Stys is supported by an Alberta Innovates-Health Solutions (AI-HS) Scientist Award; work in the lab of Peter K. Stys is supported by the Canadian Institutes of Health Research (CIHR), MSSOC, the Alberta Prion Research Institute (APRI), Canada Research Chairs. Gerald W. Zamponi is a Canada Research Chair and an AI-HS Scientist; work in the Gerald W. Zamponi lab is supported by funds from APRI and CIHR. Work in the lab of Shigeki Tsutsui is supported by APRI/Alzheimer Society of Alberta and Northwest Territories (ASANT).
Adle-Biassette, H., Verney, C., Peoc'h, K., Dauge, M. C., Razavi, F., Choudat, L., et al. (2006). Immunohistochemical expression of prion protein (PrPC) in the human forebrain during development. J. Neuropathol. Exp. Neurol. 65, 698–706. doi: 10.1097/01.jnen.0000228137.10531.72
Affaticati, P., Mignen, O., Jambou, F., Potier, M., Klingel-Schmitt, I., Degrouard, J., et al. (2011). Sustained calcium signalling and caspase-3 activation involve NMDA receptors in thymocytes in contact with dendritic cells. Cell Death Differ. 18, 99–108. doi: 10.1038/cdd.2010.79
Aguzzi, A., Baumann, F., and Bremer, J. (2008). The prion's elusive reason for being. Annu. Rev. Neurosci. 31, 439–477. doi: 10.1146/annurev.neuro.31.060407.125620
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G., et al. (2000). Inflammation and Alzheimer's disease. Neurobiol. Aging 21, 383–421. doi: 10.1016/S0197-4580(00)00124-X
Altmeppen, H. C., Prox, J., Puig, B., Dohler, F., Falker, C., Krasemann, S., et al. (2013). Roles of endoproteolytic alpha-cleavage and shedding of the prion protein in neurodegeneration. FEBS J. 280, 4338–4347. doi: 10.1111/febs.12196
Bailey, S., Carpentier, P., McMahon, E., Begolka, W., and Miller, S. (2006). Innate and adaptive immune responses of the central nervous system. Crit. Rev. Immunol. 26, 149–188. doi: 10.1615/CritRevImmunol.v26.i2.40
Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., et al. (2010). Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 2295–2300. doi: 10.1073/pnas.0911829107
Bartlett, T. E., and Wang, Y. T. (2013). The intersections of NMDAR-dependent synaptic plasticity and cell survival. Neuropharmacology 74, 59–68. doi: 10.1016/j.neuropharm.2013.01.012
Baumann, F., Tolnay, M., Brabeck, C., Pahnke, J., Kloz, U., Niemann, H. H., et al. (2007). Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 26, 538–547. doi: 10.1038/sj.emboj.7601510
Beland, M., Bedard, M., Tremblay, G., Lavigne, P., and Roucou, X. (2014). Abeta induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol. Aging 35, 1537–1548. doi: 10.1016/j.neurobiolaging.2014.02.001
Beland, M., Motard, J., Barbarin, A., and Roucou, X. (2012). PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J. Neurosci. 32, 13255–13263. doi: 10.1523/JNEUROSCI.2236-12.2012
Bendheim, P., Brown, H., Rudelli, R., Scala, L., Goller, N., Wen, G., et al. (1992). Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 42, 149–156. doi: 10.1212/WNL.42.1.149
Beraldo, F. H., Arantes, C. P., Santos, T. G., Machado, C. F., Roffe, M., Hajj, G. N., et al. (2011). Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin γ1 chain. FASEB J. 25, 265–279. doi: 10.1096/fj.10-161653
Beraldo, F. H., Arantes, C. P., Santos, T. G., Queiroz, N. G., Young, K., Rylett, R. J., et al. (2010). Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 285, 36542–36550. doi: 10.1074/jbc.M110.157263
Biasini, E., Turnbaugh, J. A., Unterberger, U., and Harris, D. A. (2012). Prion protein at the crossroads of physiology and disease. Trends Neurosci. 35, 92–103. doi: 10.1016/j.tins.2011.10.002
Biasini, E., Unterberger, U., Solomon, I. H., Massignan, T., Senatore, A., Bian, H., et al. (2013). A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J. Neurosci. 33, 2408–2418. doi: 10.1523/JNEUROSCI.3406-12.2013
Boldyrev, A., Bryushkova, E., and Vladychenskaya, E. (2012). NMDA receptors in immune competent cells. Biochemistry (Mosc.) 77, 128–134. doi: 10.1134/S0006297912020022
Boldyrev, A., Carpenter, D., and Johnson, P. (2005). Emerging evidence for a similar role of glutamate receptors in the nervous and immune systems. J. Neurochem. 95, 913–918. doi: 10.1111/j.1471-4159.2005.03456.x
Bremer, J., Baumann, F., Tiberi, C., Wessig, C., Fischer, H., Schwarz, P., et al. (2010). Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, 310–318. doi: 10.1038/nn.2483
Calella, A. M., Farinelli, M., Nuvolone, M., Mirante, O., Moos, R., Falsig, J., et al. (2010). Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314. doi: 10.1002/emmm.201000082
Cappellano, G., Carecchio, M., Fleetwood, T., Magistrelli, L., Cantello, R., Dianzani, U., et al. (2013). Immunity and inflammation in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2, 89–107.
Carulla, P., Bribián, A., Rangel, A., Gavín, R., Ferrer, I., Caelles, C., et al. (2011). Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell 22, 3041–3054. doi: 10.1091/mbc.E11-04-0321
Cashman, N., Loertscher, R., Nalbantoglu, J., Shaw, I., Kascsak, R., Bolton, D., et al. (1990). Cellular isoform of the scrapie agent protein participates in lymphocyte activation. Cell 61, 185–192. doi: 10.1016/0092-8674(90)90225-4
Chatterton, J. E., Awobuluyi, M., Premkumar, L. S., Takahashi, H., Talantova, M., Shin, Y., et al. (2002). Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415, 793–798. doi: 10.1038/nature715
Chen, S., Yadav, S. P., and Surewicz, W. K. (2010). Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role of N-terminal residues. J. Biol. Chem. 285, 26377–26383. doi: 10.1074/jbc.M110.145516
Cisse, M., Sanchez, P. E., Kim, D. H., Ho, K., Yu, G. Q., and Mucke, L. (2011). Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J. Neurosci. 31, 10427–10431. doi: 10.1523/JNEUROSCI.1459-11.2011
Corlew, R., Brasier, D. J., Feldman, D. E., and Philpot, B. D. (2008). Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist 14, 609–625. doi: 10.1177/1073858408322675
Cull-Candy, S. G., and Leszkiewicz, D. N. (2004). Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004:re16. doi: 10.1126/stke.2552004re16
Danysz, W., and Parsons, C. G. (2012). Alzheimer's disease, beta-amyloid, glutamate, NMDA receptors and memantine–searching for the connections. Br. J. Pharmacol. 167, 324–352. doi: 10.1111/j.1476-5381.2012.02057.x
Enciu, A., and Popescu, B. (2013). Is there a causal link between inflammation and dementia? Biomed. Res. Int. 2013:316495. doi: 10.1155/2013/316495
Fleisch, V. C., Leighton, P. L., Wang, H., Pillay, L. M., Ritzel, R. G., Bhinder, G., et al. (2013). Targeted mutation of the gene encoding prion protein in zebrafish reveals a conserved role in neuron excitability. Neurobiol. Dis. 55, 11–25. doi: 10.1016/j.nbd.2013.03.007
Fluharty, B. R., Biasini, E., Stravalaci, M., Sclip, A., Diomede, L., Balducci, C., et al. (2013). An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 288, 7857–7866. doi: 10.1074/jbc.M112.423954
Ford, M., Burton, L., Morris, R., and Hall, S. (2002). Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience 113, 177–192. doi: 10.1016/S0306-4522(02)00155-0
Gadotti, V. M., Bonfield, S. P., and Zamponi, G. W. (2012). Depressive-like behaviour of mice lacking cellular prion protein. Behav. Brain. Res. 227, 319–323. doi: 10.1016/j.bbr.2011.03.012
Gadotti, V. M., and Zamponi, G. W. (2011). Cellular prion protein protects from inflammatory and neuropathic pain. Mol. Pain 7:59. doi: 10.1186/1744-8069-7-59
Ganor, Y., and Levite, M. (2012). “Glutamate in the immune system: glutamate receptors in immune cells, potent effects, endogenous production and involvement in disease,” in Nerve-Driven Immunity, ed M. Levite (Wien: Springer-Verlag/Wien), 121–161. doi: 10.1007/978-3-7091-0888-8
Gill, S., and Pulido, O. (2001). Glutamate receptors in peripheral tissues: current knowledge, future research, and implications for toxicology. Toxicol. Pathol. 29, 208–223. doi: 10.1080/019262301317052486
Guillot-Sestier, M. V., Sunyach, C., Druon, C., Scarzello, S., and Checler, F. (2009). The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 284, 35973–35986. doi: 10.1074/jbc.M109.051086
Guillot-Sestier, M. V., Sunyach, C., Ferreira, S. T., Marzolo, M. P., Bauer, C., Thevenet, A., et al. (2012). alpha-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J. Biol. Chem. 287, 5021–5032. doi: 10.1074/jbc.M111.323626
Hardingham, G. E. (2009). Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem. Soc. Trans. 37, 1147–1160. doi: 10.1042/BST0371147
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Heneka, M., O'banion, M., Terwel, D., and Kummer, M. (2010). Neuroinflammatory processes in Alzheimer's disease. J. Neural Transm. 117, 919–947. doi: 10.1007/s00702-010-0438-z
Hinoi, E., Takarada, T., Ueshima, T., Tsuchihashi, Y., and Yoneda, Y. (2004). Glutamate signaling in peripheral tissues. Eur. J. Biochem. 271, 1–13. doi: 10.1046/j.1432-1033.2003.03907.x
Hu, N. W., Nicoll, A. J., Zhang, D., Mably, A. J., O'Malley, T., Purro, S. A., et al. (2014). mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat. Commun. 5, 3374. doi: 10.1038/ncomms4374
Kahlfuβ, S., Simma, N., Mankiewicz, J., Bose, T., Lowinus, T., Klein-Hessling, S., et al. (2014). Immunosuppression by N-methyl-D-aspartate receptor antagonists is mediated through inhibition of Kv1.3 and KCa3.1 channels in T cells. Mol. Cell. Biol. 34, 820–831. doi: 10.1128/MCB.01273-13
Karadottir, R., Cavelier, P., Bergersen, L. H., and Attwell, D. (2005). NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438, 1162–1166. doi: 10.1038/nature04302
Kessels, H. W., Nguyen, L. N., Nabavi, S., and Malinow, R. (2010). The prion protein as a receptor for amyloid-beta. Nature 466, E3–E4. discussion: E4–E5. doi: 10.1038/nature09217
Khosravani, H., Zhang, Y., Tsutsui, S., Hameed, S., Altier, C., Hamid, J., et al. (2008). Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 181, 551–565. doi: 10.1083/jcb.200711002
Kivisakk, P., Mahad, D., Callahan, M., Trebst, C., Tucky, B., Wei, T., et al. (2003). Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. U.S.A. 100, 8389–8394. doi: 10.1073/pnas.1433000100
Kleene, R., Loers, G., Langer, J., Frobert, Y., Buck, F., and Schachner, M. (2007). Prion protein regulates glutamate-dependent lactate transport of astrocytes. J. Neurosci. 27, 12331–12340. doi: 10.1523/JNEUROSCI.1358-07.2007
Klewpatinond, M., Davies, P., Bowen, S., Brown, D. R., and Viles, J. H. (2008). Deconvoluting the Cu2+ binding modes of full-length prion protein. J. Biol. Chem. 283, 1870–1881. doi: 10.1074/jbc.M708472200
Lai, T. W., Zhang, S., and Wang, Y. T. (2014). Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog. Neurobiol. 115, 157–188. doi: 10.1016/j.pneurobio.2013.11.006
Lau, A., and Tymianski, M. (2010). Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 460, 525–542. doi: 10.1007/s00424-010-0809-1
Lauren, J. (2014). Cellular prion protein as a therapeutic target in Alzheimer's disease. J. Alzheimers Dis. 38, 227–244. doi: 10.3233/JAD-130950
Lauren, J., Gimbel, D., Nygaard, H., Gilbert, J., and Strittmatter, S. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Li, J. B., Lu, Z. G., Xu, L., Wang, Q., Zhang, Z. H., and Fang, J. H. (2014). Neuroprotective effects of bis(7)-tacrine in a rat model of pressure-induced retinal ischemia. Cell Biochem. Biophys. 68, 275–282. doi: 10.1007/s12013-013-9707-4
Liu, T., Yi, W., Feng, B., Zhou, Z., and Xiao, G. (2013). IGF-1-induced enhancement of PRNP expression depends on the negative regulation of transcription factor FOXO3a. PLoS ONE 8:e71896. doi: 10.1371/journal.pone.0071896
Lynch, M. (2014). The impact of neuroimmune changes on development of amyloid pathology; relevance to Alzheimer's disease. Immunology 141, 292–301. doi: 10.1111/imm.12156
Mabbott, N., Brown, K., Manson, J., and Bruce, M. (1997). T-lymphocyte activation and the cellular form of the prion protein. Immunology 92, 161–165. doi: 10.1046/j.1365-2567.1997.00331.x
Mayer, M. L. (2005). Glutamate receptor ion channels. Curr. Opin. Neurobiol. 15, 282–288. doi: 10.1016/j.conb.2005.05.004
Mayer, M. L., Vyklicky, L. Jr., and Clements, J. (1989). Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature 338, 425–427. doi: 10.1038/338425a0
McDonald, A. J., Dibble, J. P., Evans, E. G., and Millhauser, G. L. (2014). A new paradigm for enzymatic control of alpha-cleavage and beta-cleavage of the prion protein. J. Biol. Chem. 289, 803–813. doi: 10.1074/jbc.M113.502351
McLennan, N. F., Brennan, P. M., McNeill, A., Davies, I., Fotheringham, A., Rennison, K. A., et al. (2004). Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 165, 227–235. doi: 10.1016/S0002-9440(10)63291-9
Micu, I., Jiang, Q., Coderre, E., Ridsdale, A., Zhang, L., Woulfe, J., et al. (2006). NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439, 988–992. doi: 10.1038/nature04474
Mitsios, N., Saka, M., Krupinski, J., Pennucci, R., Sanfeliu, C., Miguel Turu, M., et al. (2007). Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J. Neurosci. Res. 85, 602–611. doi: 10.1002/jnr.21142
Mitteregger, G., Vosko, M., Krebs, B., Xiang, W., Kohlmannsperger, V., Nolting, S., et al. (2007). The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 17, 174–183. doi: 10.1111/j.1750-3639.2007.00061.x
Müller, W. E., Ushijima, H., Schroder, H. C., Forrest, J. M., Schatton, W. F., Rytik, P. G., et al. (1993). Cytoprotective effect of NMDA receptor antagonists on prion protein (PrionSc)-induced toxicity in rat cortical cell cultures. Eur. J. Pharmacol. 246, 261–267. doi: 10.1016/0922-4106(93)90040-G
Nicoll, A. J., Panico, S., Freir, D. B., Wright, D., Terry, C., Risse, E., et al. (2013). Amyloid-beta nanotubes are associated with prion protein-dependent synaptotoxicity. Nat. Commun. 4, 2416. doi: 10.1038/ncomms3416
Nieznanski, K., Choi, J. K., Chen, S., Surewicz, K., and Surewicz, W. K. (2012). Soluble prion protein inhibits amyloid-beta (Abeta) fibrillization and toxicity. J. Biol. Chem. 287, 33104–33108. doi: 10.1074/jbc.C112.400614
Ostapchenko, V. G., Beraldo, F. H., Guimaraes, A. L., Mishra, S., Guzman, M., Fan, J., et al. (2013). Increased prion protein processing and expression of metabotropic glutamate receptor 1 in a mouse model of Alzheimer's disease. J. Neurochem. 127, 415–425. doi: 10.1111/jnc.12296
Paoletti, P., Bellone, C., and Zhou, Q. (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. doi: 10.1038/nrn3504
Parsons, M. P., and Raymond, L. A. (2014). Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82, 279–293. doi: 10.1016/j.neuron.2014.03.030
Pina-Crespo, J. C., Talantova, M., Micu, I., States, B., Chen, H. S., Tu, S., et al. (2010). Excitatory glycine responses of CNS myelin mediated by NR1/NR3 “NMDA” receptor subunits. J. Neurosci. 30, 11501–11505. doi: 10.1523/JNEUROSCI.1593-10.2010
Qin, K., Yang, D. S., Yang, Y., Chishti, M. A., Meng, L. J., Kretzschmar, H. A., et al. (2000). Copper(II)-induced conformational changes and protease resistance in recombinant and cellular PrP. Effect of protein age and deamidation. J. Biol. Chem. 275, 19121–19131. doi: 10.1074/jbc.275.25.19121
Quintanar, L., Rivillas-Acevedo, L., Grande-Aztatzi, R., Gómez-Castro, C. Z., Arcos-López, T., and Vela, A. (2013). Copper coordination to the prion protein: insights from theoretical studies. Coordin. Chem. Rev. 257, 429–444. doi: 10.1016/j.ccr.2012.06.026
Radovanovic, I., Braun, N., Giger, O. T., Mertz, K., Miele, G., Prinz, M., et al. (2005). Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J. Neurosci. 25, 4879–4888. doi: 10.1523/JNEUROSCI.0328-05.2005
Rangel, A., Burgaya, F., Gavin, R., Soriano, E., Aguzzi, A., and Del Rio, J. A. (2007). Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: role of AMPA/kainate receptors. J. Neurosci. Res. 85, 2741–2755. doi: 10.1002/jnr.21215
Ratte, S., Vreugdenhil, M., Boult, J. K., Patel, A., Asante, E. A., Collinge, J., et al. (2011). Threshold for epileptiform activity is elevated in prion knockout mice. Neuroscience 179, 56–61. doi: 10.1016/j.neuroscience.2011.01.053
Resenberger, U. K., Harmeier, A., Woerner, A. C., Goodman, J. L., Muller, V., Krishnan, R., et al. (2011). The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 30, 2057–2070. doi: 10.1038/emboj.2011.86
Riemer, C., Burwinkel, M., Schwarz, A., Gultner, S., Mok, S. W., Heise, I., et al. (2008). Evaluation of drugs for treatment of prion infections of the central nervous system. J. Gen. Virol. 89, 594–597. doi: 10.1099/vir.0.83281-0
Rubio-Perez, J., and Morillas-Ruiz, J. (2012). A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal 2012:756357. doi: 10.1100/2012/756357
Rush, T., and Buisson, A. (2014). Reciprocal disruption of neuronal signaling and Abeta production mediated by extrasynaptic NMDA receptors: a downward spiral. Cell Tissue Res. 356, 279–286. doi: 10.1007/s00441-013-1789-1
Sakurai-Yamashita, Y., Sakaguchi, S., Yoshikawa, D., Okimura, N., Masuda, Y., Katamine, S., et al. (2005). Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience 136, 281–287. doi: 10.1016/j.neuroscience.2005.06.095
Salter, M. G., and Fern, R. (2005). NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 438, 1167–1171. doi: 10.1038/nature04301
Schlief, M. L., West, T., Craig, A. M., Holtzman, D. M., and Gitlin, J. D. (2006). Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc. Natl. Acad. Sci. U.S.A. 103, 14919–14924. doi: 10.1073/pnas.0605390103
Shyu, W. C., Lin, S. Z., Chiang, M. F., Ding, D. C., Li, K. W., Chen, S. F., et al. (2005). Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 25, 8967–8977. doi: 10.1523/JNEUROSCI.1115-05.2005
Skerry, T., and Genever, P. (2001). Glutamate signalling in non-neuronal tissues. Trends Pharmacol. Sci. 22, 174–181. doi: 10.1016/S0165-6147(00)01642-4
Solito, E., and Sastre, M. (2012). Microglia function in Alzheimer's disease. Front. Pharmacol. 3:14. doi: 10.3389/fphar.2012.00014
Spudich, A., Frigg, R., Kilic, E., Kilic, U., Oesch, B., Raeber, A., et al. (2005). Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 20, 442–449. doi: 10.1016/j.nbd.2005.04.002
Stanyon, H. F., Patel, K., Begum, N., and Viles, J. H. (2014). Copper(II) sequentially loads onto the N-terminal amino group of the cellular prion protein before the individual octarepeats. Biochemistry 53, 3934–3939. doi: 10.1021/bi500643b
Steele, A. D., Zhou, Z., Jackson, W. S., Zhu, C., Auluck, P., Moskowitz, M. A., et al. (2009). Context dependent neuroprotective properties of prion protein (PrP). Prion 3, 240–249. doi: 10.4161/pri.3.4.10135
Talantova, M., Sanz-Blasco, S., Zhang, X., Xia, P., Akhtar, M. W., Okamoto, S., et al. (2013). Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. U.S.A. 110, E2518–E2527. doi: 10.1073/pnas.1306832110
Thakur, A. K., Srivastava, A. K., Srinivas, V., Chary, K. V., and Rao, C. M. (2011). Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J. Biol. Chem. 286, 38533–38545. doi: 10.1074/jbc.M111.265645
Thellung, S., Gatta, E., Pellistri, F., Corsaro, A., Villa, V., Vassalli, M., et al. (2013). Excitotoxicity through NMDA receptors mediates cerebellar granule neuron apoptosis induced by prion protein 90-231 fragment. Neurotox. Res. 23, 301–314. doi: 10.1007/s12640-012-9340-9
Tsutsui, S., Hahn, J., Johnson, T., Ali, Z., and Jirik, F. (2008). Absence of the cellular prion protein exacerbates and prolongs neuroinflammation in experimental autoimmune encephalomyelitis. Am. J. Pathol. 173, 1029–1041. doi: 10.2353/ajpath.2008.071062
Tuneva, E., Bychkova, O., and Boldyrev, A. (2003). Effect of NMDA on production of reactive oxygen species by human lymphocytes. Bull. Exp. Biol. Med. 136, 159–161. doi: 10.1023/A:1026366907084
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
Walz, R., Amaral, O. B., Rockenbach, I. C., Roesler, R., Izquierdo, I., Cavalheiro, E. A., et al. (1999). Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia 40, 1679–1682. doi: 10.1111/j.1528-1157.1999.tb01583.x
Wang, K. K., Zoltewicz, J. S., Chiu, A., Zhang, Z., and Rubenstein, R. (2012). Release of full-length PrP(C) from cultured neurons following neurotoxic challenges. Front. Neurol. 3:147. doi: 10.3389/fneur.2012.00147
Watt, N. T., Taylor, D. R., Kerrigan, T. L., Griffiths, H. H., Rushworth, J. V., Whitehouse, I. J., et al. (2012). Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 3, 1134. doi: 10.1038/ncomms2135
Weise, J., Crome, O., Sandau, R., Schulz-Schaeffer, W., Bahr, M., and Zerr, I. (2004). Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 372, 146–150. doi: 10.1016/j.neulet.2004.09.030
Weise, J., Doeppner, T. R., Muller, T., Wrede, A., Schulz-Schaeffer, W., Zerr, I., et al. (2008). Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor. Neurol. Neurosci. 26, 57–64.
Weise, J., Sandau, R., Schwarting, S., Crome, O., Wrede, A., Schulz-Schaeffer, W., et al. (2006). Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 37, 1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4
Weller, R., Preston, S., Subash, M., and Carare, R. (2009). Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res. Ther. 1:6. doi: 10.1186/alzrt6
Wyllie, D. J., Livesey, M. R., and Hardingham, G. E. (2013). Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 74, 4–17. doi: 10.1016/j.neuropharm.2013.01.016
Xiao, B., and Link, H. (1998). Immune regulation within the central nervous system. J. Neurol. Sci. 157, 1–12. doi: 10.1016/S0022-510X(98)00049-5
You, H., Tsutsui, S., Hameed, S., Kannanayakal, T., Chen, L., Xia, P., et al. (2012). Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 1737–1742. doi: 10.1073/pnas.1110789109
Keywords: NMDA receptor, cellular prion protein, excitotoxicity, neuroinflammation, ischemia, beta-amyloid, Alzheimer's disease
Citation: Black SAG, Stys PK, Zamponi GW and Tsutsui S (2014) Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity. Front. Cell Dev. Biol. 2:45. doi: 10.3389/fcell.2014.00045
Received: 30 June 2014; Paper pending published: 21 July 2014;
Accepted: 09 August 2014; Published online: 28 August 2014.
Edited by:
Sophie Mouillet-Richard, INSERM Unit 1124, FranceCopyright © 2014 Black, Stys, Zamponi and Tsutsui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerald W. Zamponi, Department of Physiology and Pharmacology, Hotchkiss Brain Institute, University of Calgary, 3330 Hospital Drive NW, Calgary, AB T2N 4N1, Canada e-mail:emFtcG9uaUB1Y2FsZ2FyeS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.