 Xin Guo1†*
Xin Guo1†* Juan Zheng2,3†
Juan Zheng2,3† Shixiu Zhang1
Shixiu Zhang1 Xiaofan Jiang1
Xiaofan Jiang1 Ting Chen2,3Jiayu Yu2,3Shu'e Wang1Xiaomin Ma1
Ting Chen2,3Jiayu Yu2,3Shu'e Wang1Xiaomin Ma1 Chaodong Wu4*
Chaodong Wu4*- 1Department of Nutrition and Food Hygiene, School of Public Health, Cheeloo College of Medicine, Shandong University, Jinan, China
- 2Department of Endocrinology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Hubei Provincial Clinical Research Center for Diabetes and Metabolic Disorders, Wuhan, China
- 4Department of Nutrition, Texas A&M University, College Station, TX, United States
Unhealthy diets and lifestyle result in various metabolic conditions including metabolic syndrome and non-alcoholic fatty liver disease (NAFLD). Much evidence indicates that disruption of circadian rhythms contributes to the development and progression of excessive hepatic fat deposition and inflammation, as well as liver fibrosis, a key characteristic of non-steatohepatitis (NASH) or the advanced form of NAFLD. In this review, we emphasize the importance of nutrition as a critical factor in the regulation of circadian clock in the liver. We also focus on the roles of the rhythms of nutrient intake and the composition of diets in the regulation of circadian clocks in the context of controlling hepatic glucose and fat metabolism. We then summarize the effects of unhealthy nutrition and circadian dysregulation on the development of hepatic steatosis and inflammation. A better understanding of how the interplay among nutrition, circadian rhythms, and dysregulated metabolism result in hepatic steatosis and inflammation can help develop improved preventive and/or therapeutic strategies for managing NAFLD.
Introduction
With the ongoing pandemic of obesity, 25% of the population worldwide, including children, adolescents and adults, are suffering from non-alcoholic fatty liver disease (NAFLD) (1). NAFLD is characterized by excess accumulation of triglycerides in the hepatocytes (hepatic steatosis) due to both increased inflow of free fatty acids and de novo lipogenesis. When the liver exhibits overt inflammatory damage and fibrosis, NAFLD progresses to its advanced stage, nonalcoholic steatohepatitis (NASH). The latter has increased risk to progress to liver cirrhosis and hepatocellular carcinoma. Metabolic abnormalities related to unhealthy nutrition, such as central obesity, insulin resistance, dyslipidemia and hypertension, are closely related to NAFLD (2). Although the etiology and progression of NAFLD remain to be elucidated, more and more studies have shown that the circadian clocks play a key role in the regulation of key aspects of the pathogenesis of NAFLD (3, 4).
Circadian rhythms are widely present in animals and plants. In mammals, circadian rhythms function to coordinate a diverse panel of physiological processes that are influenced by environmental rhythmic signals such as food and light. In addition, circadian dysfunction is associated with sleep disorders, elevated incidence of cancer, and metabolic abnormalities. Located in the suprachiasmatic nucleus (SCN) of the hypothalamus, the master clock plays an important role in governing biological rhythms, coordinating the peripheral clocks in peripheral tissues such as the liver, muscle, adipose tissue and gastrointestinal tract. The master clock receives light and generates timing signals to govern peripheral clocks, shaping whole body’s circadian rhythms. Also, the master clock drives circadian rhythms of behavior such as rest and activity cycles while peripheral clocks play more important roles in physiological regulation of each peripheral tissue (5, 6).
Recent studies have indicated that the misalignment of the central clock and the peripheral clock results in dysregulated metabolism of glucose and fat. Indeed, abnormal rhythmic cycles are related to metabolic diseases such as obesity, diabetes and NAFLD (7). Of note, the liver is considered a clock organ because many genes related to metabolism in the liver exhibit diurnal rhythmicity and are regulated by the circadian clock.
Description of the Molecular Clock
A large number of physiological events follow circadian rhythmicity. At the cellular level, circadian rhythms are generated by an auto regulatory transcriptional and translational feedback loop (TTFL) consisting of the clock genes Period (PER) 1, PER2, Cryptochrome (CRY) 1, CRY2, brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1), circadian locomotor output cycles kaput (CLOCK) and their protein products (8–12). In the core loop of the mammalian clocks, the CLOCK: BMAL1 heterodimer binds to enhancer elements (E-boxes) to initiate the transcription of PER, CRY and other clock-controlled genes (CCGs). PER: CRY dimers accumulate in the nucleus over the course of the day to inhibit the activity of the CLOCK: BMAL1 heterodimer, thereby suppressing their own transcription. PER and CRY also can be broken down by β-transducing repeat-containing protein (β–TrCP) and F-box/LRR-repeat protein 3 (FBXL3) respectively to reset the cycle. In addition, the CLOCK: BMAL1 heterodimer also regulates the transcription of REV-ERBs, which are nuclear receptors and compete with retinoic acid-related orphan receptors (RORs) to inhibit the transcription of BMAL1 and delay the transcription of CRY1 (5, 13, 14).
Coordination at the cellular level is necessary for tissue-specific oscillations that regulate circadian physiology (15–17) and the alignment of these clocks between tissues is essential for the maintenance of metabolic homeostasis. The possibility of misalignment arises from the differential responsiveness of tissues to the environmental cues that synchronize the clock (zeitgebers). Although light is the dominant environmental rhythmic cues that resents the master clock of the SCN, many other tissues are sensitive to cues derived from nutritional rhythms (18).
The liver is involved in the decomposition of three major macronutrients. About one tenth of the liver transcriptome are expressed rhythmically (19). In the liver of wild-type (WT) animals, the rhythm of a CLOCK: BMAL1 is coordinated during the day, accompanied with histone acetylation and RNA polymerase II accumulation, which triggers the peak of transcription in the early phase of night (16, 20). While the translation of inhibitory CRY and PER proteins increases, the levels of CLOCK and BMAL1 decrease and CLOCK:BMAL1-driven transcription is inhibited (20). This forms a core feedback inhibitory loop of circadian clock. REV-ERBα, also functioning to adjust CLOCK: BMAL1, reaches its peak expression at ZT8-10 in the liver, and returns to the lowest levels at ZT20-22. This constitutes another negative feedback loop (14, 21). However, it should be noted that only a very small number (less than 10%) of rhythmic genes are common to the liver, kidney and heart whereas most of rhythmic genes are tissue-specific (22, 23). Upon reconstituting circadian expression of BMAL1 only in the liver of BMAL1-/- mice, the liver recovered about 10% of total hepatic rhythmic transcripts and 20% of oscillatory metabolites, including oscillation for glycogen and NAD+ salvage metabolism (16).
Energy metabolism and nutrient absorption are integrated with the peripheral clock of the liver (24). The amplitude of BMAL1, a critical driver of metabolic homeostasis under physiological conditions, is decreased in light and dark periods in response to high-fat feeding, resulting in the dysregulation of oscillation rhythms (25, 26). Global disruption of BMAL1 in mice exhibited increased body weight and fat content and decreased insulin secretion. When disrupting BMAL1 only in the liver, mice did not show differences in body weight, fat content and serum insulin levels compared to wild type mice. However, the rhythmic expression of clock-regulated metabolic genes such as glucose transporter 2 (GLUT2), glucokinase, liver pyruvate kinase, phosphoenolpyruvate carboxykinase 1 (PEPCK1), carnitine palmitoyltransferase 1 (CPT1), etc. are abolished. In addition, liver-specific disruption of BMAL1 increased glucose clearance and induced hypoglycemia (27).
Metabolism in the liver is closely synchronized with the clock, making the pace of demand and supply consistent. For example, upon food consumption after late-night fasting during sleep, circadian clock provides rhythmic and baseline regulation and repetitive activities, resulting in rapid consumption of glucose in the morning to meet energy demand (4). Furthermore, the liver acts as a transit point for toxins. For example, amino acids (exogenous substances) are converted to ammonia (transformation) and then urea (water soluble metabolites) in the liver, which is a well-known process of detoxification. The first step of transformation involves hepatic nuclear receptors which are controlled by rhythmic expression, such as REV-ERB and ROR subfamilies (28). Core clock and clock-controlled genes regulate hepatic glucose metabolism. For example, in the liver, glucagon stimulates gluconeogenesis by activating cAMP/cAMP-response element binding protein (CREB) signaling during fasting. PER inhibits glucagon-stimulated cAMP production to decrease gluconeogenesis via interacting with G protein–coupled receptors (29). Peroxisome proliferator-activated receptor (PPAR), which is a family of nuclear receptor proteins serving as the transcription factors to regulate gene expression related to lipid metabolism, is controlled by the circadian clock. Hepatic PPARα and PPARγ are activated during day time and are inhibited during night time, while hepatic PPARδ is induced during night time (28). REV-ERBα modulates the oscillation of sterol regulatory element-binding protein (SREBP) activity, which is involved in cholesterol and lipid metabolism. In addition, REV-ERBα also is involved in the circadian transcription of cholesterol-7a-hydroxylase (CYP7A1), which is the key enzyme in bile acid synthesis (30).
Predictably, any factor that alters circadian rhythms can cause hepatic metabolic dysregulation, in the course of time, leading to metabolic diseases. In the liver, circadian mRNA and protein expression of clock genes such as PER1, PER2, BMAL1 and CLOCK and circadian-related metabolic regulators, such as AMPK, lipogenic enzymes, and gluconeogenic proteins are changed in response to HFD feeding, leading to obesity and insulin resistance (29, 31). Based on this, it is conceivable that circadian rhythms control various metabolic processes in the liver. When the phases of metabolic genes are misaligned, diseases and disorders often occur.
Nutrient Control of the Circadian Clock
Nutrient supply and circadian rhythms are intimately linked. Feeding a high fat diet (HFD) disrupts circadian rhythms and causes an unexpectedly large-scale genesis of de novo oscillating transcripts, resulting in reorganization of the coordinated oscillations between transcripts and metabolites. The mechanisms underlying this reprogramming involve both the impairment of CLOCK: BMAL1 chromatin recruitment and a pronounced cyclic activation of surrogate pathways through the transcriptional regulator peroxisome proliferator-activated receptor γ (PPARγ) (32). An in vivo study revealed that it takes only 3 days for an HFD to change the circadian clock in the liver (20). Moreover, HFD feeding for 4 weeks significantly altered the rhythms of fatty acid synthesis rate-limiting enzyme hepatic acetyl-CoA carboxylase (ACACA), REV-ERBα and histone regulator HDAC3 (33). Studies have shown that insulin fluctuations caused by nutrient supply can “reset” the biological clock of the liver (34). In insulin-deficient mice, this change was not observed (35) but injection of insulin reset the rhythmicity of genes. This process involved phosphoinositide 3-kinase (PIK3) and mitogen-activated protein kinase (MAPK) pathways (36).
Conversely, food restriction can almost reset some peripheral clocks entirely (37–39). During fasting, increased phosphorylation of adenosine monophosphate (AMP)-activated protein kinase (AMPK) destroyed the abundance of CRY and targeted CRYI for its subsequent degradation, thus preventing CRY from inhibiting CLOCK: BMAL 1 target genes (40). The duration period of fasting also affects the peripheral biological clocks. Compared with short-term fasting, long-term fast is shown to cause a stronger stimulatory effect on the peripheral biological clock system (41, 42). Temporal feeding restriction under light-dark or dark-dark conditions is shown to change the phase of circadian gene expression in peripheral cell types by up to 12 h, while leaving the phase of rhythmic gene expression in the SCN unaffected. As such, changes in nutrient supply can uncouple peripheral oscillators and the central pacemaker. The persistence of circadian clock gene oscillation in both normal chow and HFD validates the notion that circadian oscillation within the core clock genes is highly resistant to perturbation, whereas clock output genes are more sensitive to food as a zeitgeber (39).
Compared with direct circadian control, nutrient rhythmic supply in the form of feeding/fating rhythms is an indirect but vital contributor of rhythmic hepatic transcripts (43). In general, the liver oscillator is controlled by signals from the SCN, and these signals are amplified by normal feeding rhythms (39). When feeding rhythm changes, central and peripheral rhythmic signals clash, leading to metabolic changes. Studies have shown that in rodents disordered feeding rhythms led to weight gain (44, 45). In human subjects, changes in meal schedules cause the central circadian rhythm to be out of sync with environmental signals, resulting in the circadian dysregulation (46). The molecular basis for this metabolic disruption likely is attributable to the dissociation of metabolic gene rhythms caused by inconsistency between the central clock and nutrient rhythms. Feeding a high-fat food in limited time does not change the overall caloric intake as a whole, but improved diurnal. This timed feeding in mice was shown to improve diurnal rhythms in metabolic regulators and the circadian oscillator, thereby improving insulin sensitivity and reducing adiposity in adipose tissue and liver, compared to mice fed with an HFD ad libitum (45). While exploring how nutrients couple clock regulation, two studies have demonstrated that the cellular pathways of NAD+ metabolism (47) and the concentrations of adenosine monophosphate (48) appear to link the availability of nutrients and rhythmic regulation. This led to the creation of the concept of chrononutrition (24), which aims to use timed nutrient supply as effective strategy to regulate circadian clocks in the liver (49, 50).
In addition to food intake rhythm, the nutrient composition of the food could also disrupt the normal outputs of body clock signals. For example, saturated fatty acids have been shown to alter the expression of clock genes in cell lines (51–53). Certain studies also have shown that in cell cultures, palmitate, the most common type of saturated fatty acid found in obese animals, disrupted protein–protein interaction between CLOCK and BMAL1 in a dose- and time-dependent manner. This inhibitory effect of palmitate was reversed by Sirtuin 1 (SIRT1) activator (52). In addition, docosahexaenoic acid (DHA), a polyunsaturated fatty acid (PUFA), functioned to alleviate the effect of palmitate on decreasing BMAL1 in cells; although DHA per se altered the circadian rhythm of BMAL1, suggesting a protective role (54, 55). Notably, the effect of saturated fat on the body clock has never been tested in vivo. A recent study suggested non-obesogenic doses of palmitate appeared to change circadian rhythms. In addition, non-obesogenic doses of oleate, a monounsaturated fatty acid (MUFA), also functioned to cause circadian dysregulation. Overall, saturated fatty acids are more harmful (56), although unsaturated fatty acids also alter circadian rhythms.
Additional to fats, amino acids also regulate liver clocks. For instance, the peak of liver PER2 is altered in response to an intravenous injection of mixture of 18 amino acids (57). Another recent study showed that feeding mice a diet containing only protein and/or amino acids changed liver circadian rhythms but not increased insulin levels, which is likely because pure protein diets and cysteine stimulated glucagon secretion and/or increased the production of insulin-like growth factor (IGF-1) (58). Collectively, the existing evidence indicates that altering food composition and the timing of nutrient supply appears to be a feasible approach for managing metabolic diseases. Of note, the effectiveness of nutritional regulation of the circadian clocks may vary depending on age and sex (15).
In short, nutrients regulate hepatic circadian rhythm through directly or indirectly modulating the expression of hepatic circadian-related genes. The investigation of hepatic circadian regulation by nutrition is expected to provide new knowledge concerning the mechanisms of liver metabolism and metabolic dysregulation-related liver diseases.
Circadian Dysregulation in the Pathogenesis of NAFLD
The pathogenesis of NAFLD and NASH were illustrated largely by a “two hit hypothesis”. The “first hit” is characterized by fat accumulation in liver, leading to hepatic steatosis and liver inflammatory damage. The “second hit” is referred to the concept that the damaged liver is susceptible to proinflammatory cytokines, adipokines and oxidative stress, resulting in steatohepatitis and liver fibrosis (59). Much evidence has shown that circadian rhythmic dysregulation is related to the occurrence and the progression of NAFLD/NASH (60, 61). Therefore, circadian rhythm-related hepatic lipid metabolism and inflammatory response has attracted much attention with hope to better manage NAFLD.
Circadian Dysregulation Promotes Hepatic Steatosis
In terms of the pathogenesis of NAFLD, the primary stage of NAFLD onset is excessive accumulation of triglycerides (TG) (59, 62). Fats depose in the liver with the following routes: 1) free fatty acids from adipose tissue lipolysis are transported to the liver. In most cases, insulin resistance results in an increase in lipolysis in the adipose tissue, leading to increased free fatty acids mobilization to the bloodstream and increased influx of free fatty acids to hepatocytes (63); 2) dietary fats digested and absorbed into lymph vessels by forming a lipoprotein called chylomicron. Some free fatty acids from chylomicron goes to deposit in adipose tissue, while other fatty acids are still in chylomicron remnants which are delivered to liver (64); 3) de novo lipogenesis is increased, especially by increasing dietary simple sugar (65); 4) fatty acid oxidation is decreased (66); 5) fatty acids, which derived from diets, de novo lipogenesis, and lipolysis of fats in adipose tissue are bound to glycerol to produce TG (67).
Nutritional signal (glucose) and hormonal signal (insulin) up-regulate carbohydrate responsive element-binding protein (ChREBP) and Sterol Regulatory Element-binding Protein-1c (SREBP-1c) via liver X receptors (LXRs) to increase the expression of genes for lipogenic enzymes such as acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), and stearoyl-CoA desaturase (SCD-1), thereby promoting hepatic lipid accumulation in the liver in response to feeding. Of note, the results from the studies involving multi-stable isotope labeling methods prove that fat formation is the key point in the process of hepatic steatosis (68, 69). It is now widely accepted that elevated TG formation in liver causes simple steatosis, and when liver lipotoxicity is overt, it has promoted the progression of simple steatosis to NASH. While the fatty acids used to synthesize TG are derived from diets, de novo lipogenesis, and lipolysis of fats in adipose tissue (66).
Excessive accumulation of fats is the key characteristic of NAFLD. Circadian disorders are involved in metabolic disruption, especially liver metabolic pathways (Table 1), which contribute to the development of NAFLD (3, 64, 76). According to the findings from a lipidomic analysis (3), TG is a significant component of oscillating lipids in the liver. Under normal circumstances, TG accumulate and disappear quantitatively every day. X-box binding protein 1 (XBP1) regulated the hepatic 12-h cistrome and was found to adjust TG transport and the levels of VLDL-TG (77). Disruption of XBP1 promoted NAFLD development, likely through altering the temporal 12-h transcription of lecithin–cholesterol acyltransferase, lysophosphatidylcholine acyltransferase 3, and stearoyl-CoA desaturase 1, and through impairing phosphatidylcholine and lysophosphatidylcholines cycle, as well as fatty acid monounsaturation (73). Differentiated embryo-chondrocyte expressed gene 1 (DEC1), which is a regulator of the circadian clock, inhibited the expression of SREBP-1c to reduce hepatic lipogenesis and ameliorated fatty liver phenotype in NAFLD mouse models (70). In rat primary hepatocytes, circadian transcriptional regulators such as DEC1 and Kruppel-like factor-10 (KLF-10) formed a feedback loop and were involved in the regulation of hepatic lipogenesis via ChREBP (71, 72). Moreover, both cytochrome P450 and 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) enzymes are involved in the synthesis and decomposition of fats as rate limiting enzymes with circadian rhythms (78). For example, cholesterol 7-alpha-monooxygenase (CYP7A1), which is a cytochrome P450 enzyme in cholesterol metabolism, was largely upregulated in patients with NAFLD (79). CYP7A1 was also reported to control circadian clock in the liver. Inhibition of HMG-CoA reductase increased the expression of CYP7A1 and altered clock gene expression such as BMAL1, PER2, and PER3 in the liver (74).

Table 1 The relationship between selective circadian related genes and pathogenic pathways in NAFLD.
High-fat food intake and liver insulin resistance affect NAFLD via disruption of circadian clock (80, 81). If high-fat foods are ingested randomly, metabolic regulators will be disturbed, accompanied by weakened CREB oscillation, reduced AMPK activity, inhibited biological clock components (REV-ERBα, PER2), and elevated FASN expression. This resulted in increased long-chain free fatty acids in the liver (14, 45), at the levels of the synthesis, extension and/or desaturation of fatty acids.
Several genetic variations of clock genes are related to hepatic steatosis. Mice with circadian-related genes mutations underwent metabolic dysregulation and revealed more severe hepatic steatosis than did WT mice under conventional and high-fat food feeding conditions (82). Depletion of nuclear receptor REV-ERBα and REV-ERBβ in the liver disrupted clock genes and output genes such as CLOCK, BMAL1, CRY1, PER2, POR, PPARα, and SCO2. Moreover, liver-specific depletion of REV-ERBα and REV-ERBβ led to increased plasma glucose and TG levels and decreased FFAs levels (14). In addition, liver-specific depletion of both histone deacetylase 3 (HDAC3) and REV-ERBα in mice caused increases in hepatic TG levels (21). Finally, another study also showed that long-term deficiency of REV-ERBα activity led to moderate hepatic steatosis (76). Although the amount of HDAC3 is constant, its genome recruitment in the liver corresponds to the expression pattern of circadian rhythm REV-ERBα (21). The REV-ERBα binding site coincides with most HDAC3 binding sites at ZT10. Disruption of steroid receptor coactivator-2 (SRC-2), which regulates clock genes, resulted in hepatic steatosis. Furthermore, when circadian rhythm was chronically disrupted in SRC-2-/- mice, more severe hepatic steatosis phenotype was generated (75). In summary, the synthesis and decomposition of fat and TG are altered in patterns to promote fat accumulation in the liver when the circadian rhythm is dysregulated.
Circadian Dysregulation Enhances Liver Inflammation
Although excessive accumulation of TG in hepatocytes is characteristic of NAFLD, steatosis alone is not necessarily pathogenic, because as in the early stages, NAFLD is reversible after weight loss and exercise. Oxidative stress, endoplasmic reticulum stress and the release of pro-inflammatory cytokines (such as tumor necrosis factor α, TNFα) are the main consequences of hepatic lipid overload, and are key factors in the progression of NAFLD to NASH. Free fatty acids and cholesterol, especially when accumulated in mitochondria, lead to increase TNFα and reactive oxygen species (ROS) production and play an early “inflammatory” role in promoting NASH. The increase in TNFα and ROS production due to excessive fat accumulation is known as lipotoxicity, which in turn causes inflammation, apoptosis, and consequently, the progression to hepatic fibrosis (62, 83). This process involves two alterations due to mitochondrial dysfunction including impared β-oxidation and endoplasmic reticulum stress, thereby resulting in lipid peroxidation. The resultant increase in ROS and the destruction of antioxidants’ activities are responsible for the initiation or exacerbation of inflammation (84). In the pathogenesis of NASH, hepatic resident cells (such as Kupffer cells and hepatic stellate cells) and cells recruited by injury (such as monocytes and macrophages) all release pro-inflammatory signals and participate in apoptosis or necrotic death of hepatocytes (85, 86). Infiltration of immune cells and the proinflammatory activation of immune cells are the two significant features of inflammation in NASH (87). The activation of nuclear factor kappa light chain enhancer of activated B cells (NF-κB) in hepatocytes, which is a nuclear transcription factor to regulate the expressions of inflammatory cytokines such as TNFα, interleukin 6 (IL-6), and interleukin 1β (IL-1β), leads to the recruitment and activation of Kupffer cells to mediate inflammation in NASH (88).
Much evidence suggests that circadian dysregulation is involved in the pathogenesis of inflammation in NASH (89). Recent studies have shown that inflammatory cytokines such as TNFα and IL-6 can resynchronize the circadian clock through activating NF-κB, which inhibits transcription of clock repressors (90, 91). In macrophages, BMAL1 increases the response of nuclear factor erythroid 2-related factor 2 (NRF2) to lipopolysaccharides (LPS) challenge, inhibiting the production of IL-1β (92). On the other hand. interleukin 10 (IL-10), an anti-inflammatory cytokine, is regulated by REV-ERBs throughout the circadian day (93). In a human study, HFD feeding altered cortisol rhythms, and brought about changes in diurnal oscillations of clock genes, inflammatory genes, and fat metabolic genes in monocytes (94). In addition, monocytes exhibit diurnal oscillations in the expression of clock genes (95).
Disruption of circadian rhythm (via using a chronic light-dark cycle shift paradigm) exacerbated the severity of HFD-induced inflammation, which was characterized by increases in the proinflammatory activation status and the expressions of inflammatory cytokines in bone marrow-derived macrophages (96). Myeloid-specific circadian disruption was reported to be sufficient to induce inflammatory responses. In myeloid-specific BMAL1 knockout mice, loss of BMAL1 led to disruption of rhythmic oscillations of clock genes such as ARNTL and nuclear receptor subfamily 1 group D member 1 (NR1D1), increased serum inflammatory cytokines such as IL-1β, IL-6, interferon gamma (INFγ), and monocyte chemoattractant protein-1 (MCP-1), and exacerbated metabolic dysregulation including increased adiposity and insulin resistance (95). Furthermore, myeloid cell-specific PER1/2 disruption via bone morrow transplantation worsened HFD-induced liver inflammation, which was accompanied with increased severity of hepatic steatosis and insulin resistance (97).
It suggested that the circadian disruption in macrophage is sufficient to bring about in vivo changes in hepatic steatosis and inflammation. SIRT1, an NAD+-dependent protein deacetylase, participates the maintenance of interaction between CLOCK and BMAL1 (52). Disruption of SIRT1 only in macrophage exacerbated HFD-induced hepatic steatosis via activating SREBP-1, activated hepatic fibrogenesis via stimulating collagen secretion, and elevated hepatic inflammation through activating NF-κB pathway and increasing hepatic macrophage infiltration (98).
Collectively, circadian rhythm dysregulation resulted from high-fat diet appears to trigger chain reactions, which lead to activation of pro-inflammatory pathways in macrophages, excessive accumulation of fats in hepatocytes and liver inflammation (Figure 1). This underlies the pathogenesis of hepatic steatosis and inflammation in NAFLD.
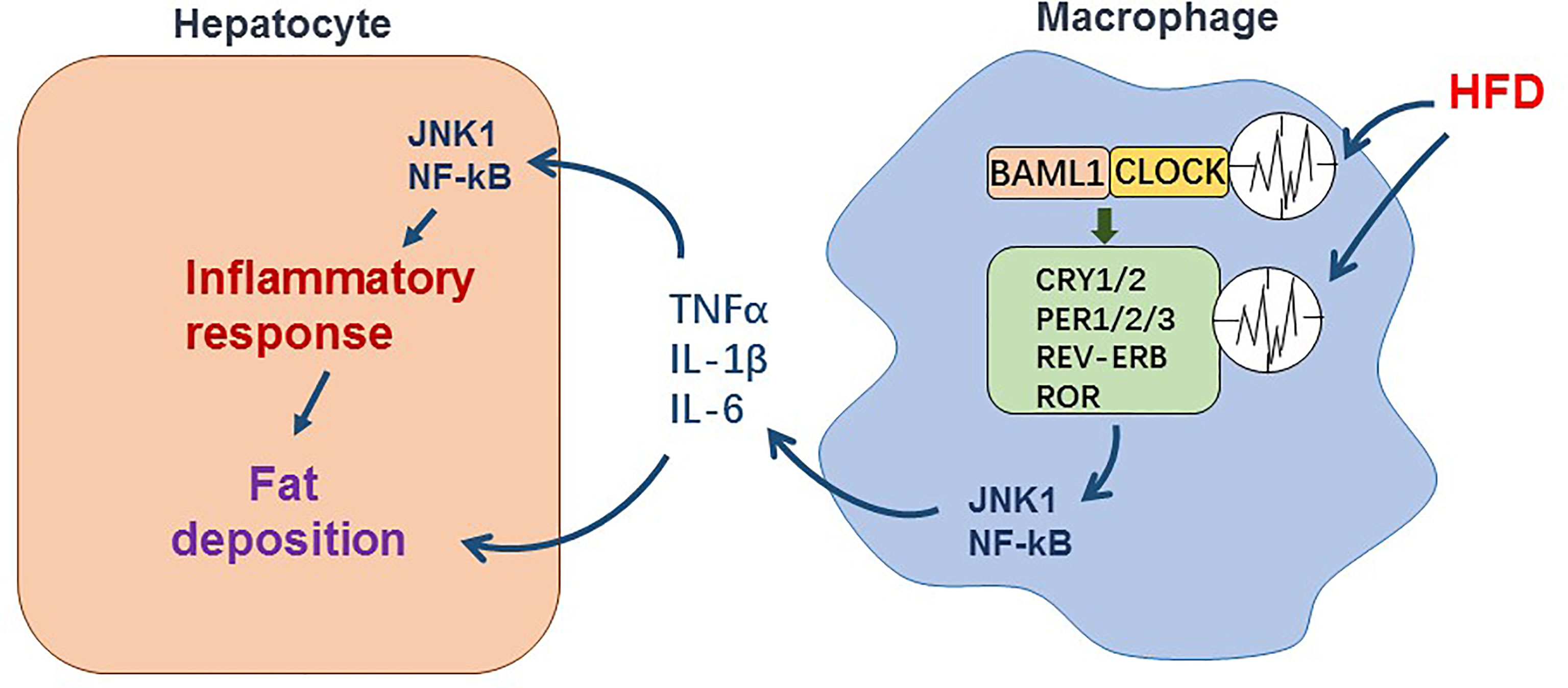
Figure 1 The effect of circadian dysregulation in macrophage induced by HFD on liver inflammation in NAFLD. Macrophage itself exhibits diurnal oscillations in expression of clock genes. During NAFLD, disruption of diurnal oscillations of clock genes induced by HFD promotes pro-inflammatory response in macrophage. The inflammatory cytokines such as TNFα, IL-1β, and IL-6 released by macrophage enhance inflammatory response and fat deposition in hepatocytes.
Distal Circadian Disruption Promotes NAFLD
Since adipose tissue and gut microbiota contribute significantly to NAFLD/NASH aspects including hepatic steatosis and inflammation, a “multiple hit hypothesis” has recently been proposed to better elucidate the pathogenesis of NAFLD and NASH. Adipose tissue is an endocrine organ which releases cytokines to influence other organs and regulate the systemic metabolism. A significant body of literature has validated that NAFLD is closely associated with the quantity, location and the types of fats in adipose tissue (99, 100). Moreover, visceral white fats appear be more critical in terms of promoting the development of NAFLD, although overall increased fat content is a risk factor for NAFLD (101, 102). During obesity, nutrition stress, e.g., nutrient overload or unhealthy nutrition, and environmental factors promote inflammation via activating macrophages and increasing macrophage infiltration into adipose tissue, leading to adipocyte dysfunction. The dysfunctional adipocytes reveal increased lipolysis and release a large amount of free fatty acids to distal tissues such as the liver. This in turn activates proinflammatory responses in hepatocytes and liver immune cells (103). Unhealthy diets and environmental factors also disturb gut microbiota, leading to increased gut permeability and release of gut microbiota metabolites and toxins such as LPS. These changes work collectively to increase fat synthesis and accumulation, as well as liver lipotoxicity. Mechanistically, lipotoxicity causes mitochondrial dysfunction and increases ROS production and ER stress, leading to hepatic inflammation and activation of liver fibrogenic program (104).
Adipokines, such as adiponectin, leptin, and resistin, are polypeptides produced by adipocytes, and are shown to contribute to NAFLD pathogenesis (105). In particular, adiponectin and leptin are capable of decrease liver fat accumulation and insulin resistance. Also, adiponectin is shown to reduce hepatic fibrosis and inflammation by blocking the activation of NF-κB and inhibiting the release of pro-inflammatory cytokines such as TNFα and IL-6 (106). In contrast, resistin have the opposite effects in the liver (107, 108). Leptin decreases fat accumulation in the liver and generally functions to improve insulin sensitivity, but promotes insulin resistance during leptin resistance states (108). During NAFLD, leptin increases liver inflammation and fibrosis through stimulating the production of transforming growth factor-β1 (TGF-β1) and activating hepatic stellate cells (109, 110). Adipokines, such as leptin and adiponectin oscillate within 24 hours to ensure the rational use of energy (111). Adiponectin was shown to regulate proinflammatory responses because its effect on increasing liver fat oxidation via inactivating ACC and activating AMPK. The latter likely involves increased expression of PPARα gene, which is controlled by circadian (28, 112). Adiponectin also is shown to reduce the activity of FASN, another enzyme involved in fatty acid synthesis (113). Environmental factor-mediated circadian rhythm disruption also amplified the pro-inflammatory responses of adipose tissue macrophages (114), leading to adipocyte dysfunction. The latter released a large amount of pro-inflammatory factors TNF α and IL-6 to inhibit adiponectin production (115–117). The diurnal oscillation of leptin secretion from adipose tissue mediated by C/EBPα was regulated by BMAL1/CLOCK. Disruption of circadian, for example, deletion of PER1/PER2 or under jet-lag, caused leptin resistance (118).
In addition, regulating the expression of genes in adipose tissue is shown to influence fat accumulation and inflammation in the liver. For instance, adipocyte-selective overexpression of PFKFB3/iPFK2, which activates 6-phosphofructo-1-kinase (6PFK1) to enhance glycolysis, decreased HFD-induced liver proinflammatory response and improved insulin signaling; although leading to increased hepatic steatosis (119–121). Also, disruption of interferon (α and β) receptor 1 (IFNAR1) only in adipose tissue exacerbated HFD-induced hepatic steatosis and systemic metabolic dysregulation (122).
The synthesis and storage of fats in adipose tissue reveal circadian rhythms (123). Deletion of adipose tissue BMAL1 disrupted circadian clocks in adipocytes and exacerbated HFD-induced obesity through increasing food intake during daytime and reducing energy expenditure. In addition, adipose tissue-specific BMAL1-disrupted mice exhibited decreases in a number of key enzymes involved in polyunsaturated fatty acid biosynthesis, affecting the energy metabolism homeostasis of the body (124). Moreover, CLOCK/BMAL1 was involved in fat formation and stimulation of lipid uptake by adipocytes via PPARγ activation (28, 125). PPARγ regulates the expression of REV-ERBα in adipocytes (126). Loss-of-PPARγ function in fat not only resulted in increases in FFAs and TG levels in plasma, but also elevated liver glucose production and insulin resistance (127, 128).
Given what mentioned above and described in Figure 2, NAFLD is a metabolic disease of the liver. Circadian rhythm disorder accelerates the accumulation of excessive TG and promotes inflammation and abdominal obesity. Therefore, obese people with high visceral adipose tissue content reveal significantly increased incidences of NAFLD.
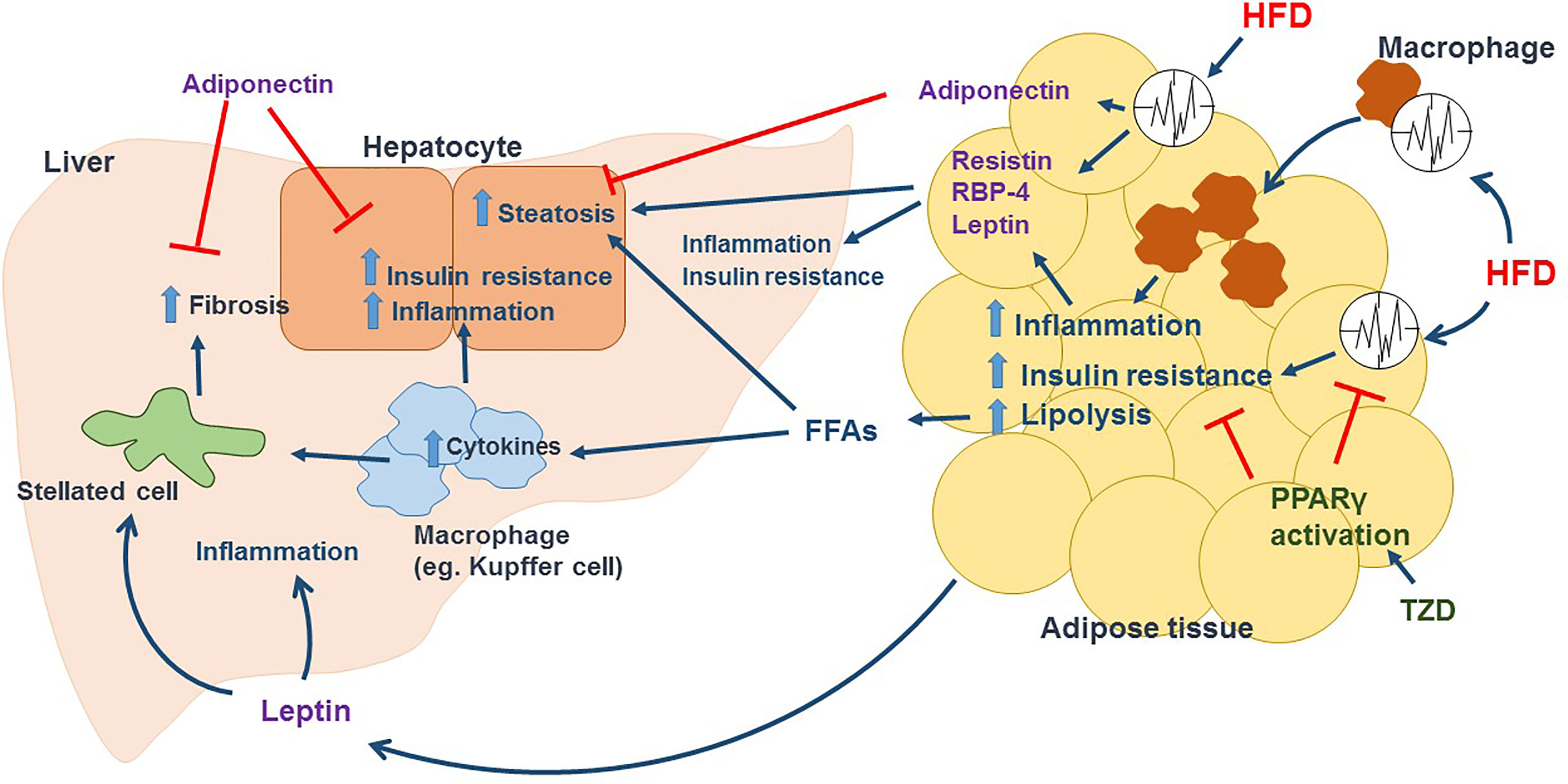
Figure 2 HFD induced circadian dysregulation in adipose tissue promotes the development of NAFLD. In adipose tissue, HFD disrupts circadian rhythm and promotes macrophage activation and infiltration, leading to inflammation and insulin resistance in adipocytes and cause adipocyte dysfunction. Lipolysis is enhanced in dysfunctional adipocytes, releasing large amount of FFAs in circulation, which increase fat accumulation in hepatocytes and activate immune cells such as Kupffer cells in liver. Activated immune cells release tons of inflammatory cytokines including TNFα and IL-6 to promote hepatic inflammation and insulin resistance. In addition, recruited immune cells and increased inflammation activate hepatic stellated cells, which cause collagen deposition and fibrosis. HFD also change the secretion of adipokines mediated by circadian clock. Dysfunctional adipocytes release Resistin and RBP-4, increasing steatosis, inflammation and insulin resistance in liver. HFD induces a leptin resistance condition, that leptin promotes inflammation, activates stellated cells and increase fibrosis in liver. Adiponectin, which can decrease hepatic steatosis, inflammation, insulin resistance, and fibrosis, is reduced in dysfunctional adipocytes induced by HFD. TZD such as rosiglitazone and pioglitazone activate PPARγ, restoring adipose tissue circadian rhythm and lowering inflammation and insulin resistance in adipose tissue, and further decreasing FFAs in circulation and the aspects of NAFLD/NASH.
Management of NAFLD/NASH Involving Circadian
To manage NAFLD, it is of a particular importance to control the prevalence of obesity, considering that obesity significantly promotes hepatic fat accumulation as a key characteristic of NAFLD. During the progression from NAFLD to NASH, inflammation plays a key role, and reducing inflammation can prevent liver damage and fibrosis. Lifestyle interventions and medications, which reduce weight and fat content, insulin resistance, and/or inflammation, are considered as effective ways to treat NAFLD. Although many medications were proved to be safe for NAFLD treatment, lifestyle interventions including dietary intervention and exercise are still the first choices. As such, patients are recommended to manage routine life according to the recommendations of healthy diet, loss of body weight, and exercise.
Caloric restriction, with or without exercise, was reported to reduce hepatic fat accumulation, inflammation and fibrosis, and to improve liver function through decreasing the serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in NAFLD (129, 130). However, BMAL1 knockout mice failed to increase life span under caloric restriction (131). In addition, peripheral circadian clock genes, timeless (TIM) and PER, were required for caloric restriction-mediated increase in lifespan and improvement in metabolic changes such as increased fat turnover and decreased TG synthesis in drosophila (132). Without caloric restriction, limiting feeding to certain times of the day that align with activity patterns is reported to reduce hepatic steatosis (45). Feeding an HFD within 8 hours per day (time-restricted feeding, TRF) is shown to protect against HFD-induced hepatic steatosis and liver damage. TRF also reduced HFD-induced elevation of hepatic gluconeogenesis by increasing CRY expression and suppressing CREB expression, leading to down-regulation of the expression of gluconeogenic genes such as pyruvate carboxylase and glucose-6-phosphatase (45). Compared to mice fed an HFD only during the 12 h dark phase, mice fed an HFD only during the 12 h light phase gain more body weight (44). This indicates the importance of timed feeding to maintaining weight or losing weight.
Mediterranean diet, which contains up to 40% of the calories from fat (mainly unsaturated fats) and 40% of the calories from carbohydrate (usually 50-60% in a low-fat diet), was reported to provide beneficial effects on NAFLD including reduction of liver fat and inflammation, as well as improvement of insulin resistance (133, 134). CLOCK gene polymorphisms were found to predict the loss of body weight of obese patients after consumption of Mediterranean diet (135). N-3 polyunsaturated fatty acids (PUFAs) were shown to reduce hepatic lipid synthesis and insulin resistance via regulating the expression of PPARα and SREBP-1, which were expressed with diurnally rhythms according to circadian clock (14, 136). Moreover, n-3 PUFAs also improved insulin signal pathway via modulating membrane fluidity and decreased proinflammatory responses via reducing the levels of TNF-α and IL-6 in NAFLD (133). Monounsaturated fatty acids (MUFAs) were known to suppress hepatic inflammation and ROS production, increase insulin sensitivity, and provide protective effects against NAFLD, although MUFAs might increase hepatic steatosis (137, 138). High carbohydrate diet and refined carbohydrates such as sucrose and fructose were found to promote the development of NAFLD through stimulating hepatic de novo lipogenesis and, thus, increase lipid accumulation in the liver (134). BMAL1 participated in hepatic de novo lipogenesis via the insulin-mTORC2-AKT signaling pathway as this was suggested by the findings that BMAL1 knockout mice exhibited decreased de novo lipogenesis in the liver and that de novo lipogenesis was recovered when restored AKT activity by insulin (139).
Sedentary lifestyle is a risk factor of the development of obesity and NAFLD. Exercise was found to improve insulin sensitivity in adipose tissue and decrease the flow of fatty acids to the liver, thereby reducing hepatic fat accumulation and insulin resistance. The effects of exercise on improving metabolic regulation were mediated by activation of AMPK, which inhibited hepatic lipogenesis through SREBP-1 to suppress the expression of lipogenic genes (such as ACC1 and FASN) and elevated hepatic fatty acid β-oxidation by reducing malonyl CoA and stimulating the expression of carnitine palmitoyltransferase 1 (CPT1) (140). A circadian phosphoproteome study in mouse liver indicated that 25% of phosphopeptides including AKT, AMPK and mTOR oscillated and signaling pathways for metabolism were regulated rhythmically by phosphorylation (141). Choosing the right time of the day to exercise was shown to obtain optimal beneficial effects due to that hypoxia-inducible factor 1a (HIF1a), a transcription factor responsible for regulating glycolysis under low oxygen condition, was activated in a time-dependent manner upon exercise (142). In addition to exercise, sleep-awake cycle plays an important role in maintaining metabolic homeostasis as indicated by the finding that late bedtime was positively associated with the incidence of NAFLD in human (143).
There are several pharmaco-therapeutic options for NAFLD/NASH. Thiazolidinediones (TZDs), which induce PPARγ activation, are considered an effective medication to increase insulin sensitivity, reduce adipose tissue inflammation and improve biopsy parameters of steatohepatitis (144). For example, pioglitazone was reported to reduce liver fibrosis and adipose tissue insulin resistance in NASH patients with and without type 2 diabetes, but more effective in patients with type 2 diabetes (145). Rosiglitazone, a ligand of PPARγ, recovered HFD-induced the changes of hepatic BMAL1 function, which increased the recruitment and activity of BMAL1 target genes, such as DBP, CHRONO, and FABP2, and increased liver insulin sensitivity as well (146). Glucagon-like peptide 1 (GLP-1), which is a peptide hormone secreted by intestine L cell after food intake, stimulates insulin release and inhibits glucagon release, thereby improving glucose homeostasis. The secretion of GLP-1 was diurnally oscillated and paralleled with the expression of BMAL1, which was suppressed by palmitate (147). GLP-1 receptor (GLP-1R) agonism was found to reduce body weight and hepatic steatosis, as well as increased hepatic fatty acid oxidation and insulin sensitivity in NAFLD/NASH (148). Dipeptidyl peptidase-4 (DPP-4) inhibitors suppressed DPP-4 enzyme, thereby preventing inactivation of GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) to stimulate insulin secretion. DPP-4 inhibitors also were reported to decrease body mass index, liver triglyceride levels, serum aminotransferase levels, and the progression of NAFLD (149). Blocking sodium-glucose cotransporter 2 (SGLT2) inhibited glucose absorption. Also, SGLT2 inhibitors reduced body weight, blood pressure, and insulin resistance, which is considered a potential medication for NAFLD/NASH. Metformin, a first-line anti-diabetic drug, was shown to provide beneficial effects in patients with NAFLD/NASH including reduction of serum lipids and glucose levels. However, the improvement of liver histology and function by metformin was not significant (150). The anti-diabetic effect of metformin also was considered to be mediated by gut microbiota, which was supported by the study that transferring fecal samples from metformin-treated mice to germ-free mice improved glucose tolerance (151). Gut microbiota has its own diurnal compositional and functional oscillations. Nutrition and environmental factors such as HFD and disruption of feeding time are shown to impair microbiota diurnal rhythmicity and cause microbiota dysbiosis, leading to metabolic dysregulation (152). Obeticholic acid (OCA), which is a synthetic bile acid derivative and farnesoid X receptor agonist, was recently found to reduce hepatic inflammation and improve the histological features of NASH (153). This was attributable to alteration of gut microbiota composition by increased population of Blautia and decreased levels of taurine-bound bile acid induced by an HFD after OCA treatment (154). Also, patients with NAFLD are in a need to consider the time and intervals of taking certain drugs, given that liver detoxification is affected by biological rhythms.
Conclusion and Future Directions
Nutrition and lifestyle are known to affect metabolism, thereby profoundly influencing human health, quality of life and longevity. Circadian clock plays an important role in maintaining metabolic homeostasis. Dysregulation of circadian rhythms induced by high-fat and/or high-sugar diets and unhealthy eating patterns such as eating throughout the day and late-night eating are considered pathogenic factors for NAFLD/NASH. As summarized in this review, we provide the current state of knowledge for NAFLD pathogenesis from a circadian perspective. However, the pathogenesis of NAFLD/NASH is multifactorial and complex, which makes it difficult to develop an effective therapy for treating NAFLD/NASH. Dietary and lifestyle interventions, which promote weight loss, are still considered effective to improve NAFLD/NASH aspects.
Author Contributions
XG and JZ wrote the manuscript. SZ, XJ, TC, and JY participated in revision. SW, XM, and CW organized the manuscript. XG and CW came up with the concept. All authors contributed to the article and approved the submitted version.
Funding
The development of this review was supported by The Fundamental Research Funds of Shandong University (Grant No. 2017TB0028), Young Scholars Program of Shandong University (Grant No. 2018WLJH33), National Natural Science Foundation of China (Grant No. 81803224) and National Natural Science Foundation of China (Grant No. 81770772).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Younossi Z, Tacke F, Arrese M, Sharma BC, Mostafa I, Bugianesi E, et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology (2019) 69(6):2672–82. doi: 10.1002/hep.30251
2. Younossi ZM. Non-Alcoholic Fatty Liver Disease - A Global Public Health Perspective. J Hepatol (2019) 70(3):531–44. doi: 10.1016/j.jhep.2018.10.033
3. Adamovich Y, Rousso-Noori L, Zwighaft Z, Neufeld-Cohen A, Golik M, Kraut-Cohen J, et al. Circadian Clocks and Feeding Time Regulate the Oscillations and Levels of Hepatic Triglycerides. Cell Metab (2014) 19(2):319–30. doi: 10.1016/j.cmet.2013.12.016
4. Reinke H, Asher G. Circadian Clock Control of Liver Metabolic Functions. Gastroenterology (2016) 150(3):574–80. doi: 10.1053/j.gastro.2015.11.043
5. Partch CL, Green CB, Takahashi JS. Molecular Architecture of the Mammalian Circadian Clock. Trends Cell Biol (2014) 24(2):90–9. doi: 10.1016/j.tcb.2013.07.002
6. Dibner C, Schibler U, Albrecht U. The Mammalian Circadian Timing System: Organization and Coordination of Central and Peripheral Clocks. Annu Rev Physiol (2010) 72(1):517–49. doi: 10.1146/annurev-physiol-021909-135821
7. Schroeder AM, Colwell CS. How to Fix a Broken Clock. Trends Pharmacol Sci (2013) 34(11):605–19. doi: 10.1016/j.tips.2013.09.002
8. Feng D, Lazar MA. Clocks, Metabolism, and the Epigenome. Mol Cell (2012) 47(2):158–67. doi: 10.1016/j.molcel.2012.06.026
9. Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, et al. Transcriptional Architecture and Chromatin Landscape of the Core Circadian Clock in Mammals. Science (2012) 338(6105):349–54. doi: 10.1126/science.1226339
10. Morf J, Rey G, Schneider K, Stratmann M, Fujita J, Naef F, et al. Cold-Inducible RNA-Binding Protein Modulates Circadian Gene Expression Posttranscriptionally. Science (2012) 338(6105):379–83. doi: 10.1126/science.1217726
11. Masri S, Patel VR, Eckel-Mahan KL, Peleg S, Forne I, Ladurner AG, et al. Circadian Acetylome Reveals Regulation of Mitochondrial Metabolic Pathways. Proc Natl Acad Sci USA (2013) 110(9):3339–44. doi: 10.1073/pnas.1217632110
12. Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, Naef F, et al. Genome-Wide and Phase-Specific DNA-Binding Rhythms of BMAL1 Control Circadian Output Functions in Mouse Liver. PloS Biol (2011) 9(2). doi: 10.1371/journal.pbio.1000595
13. Rosensweig C, Green CB. Periodicity, Repression, and the Molecular Architecture of the Mammalian Circadian Clock. Eur J Neurosci (2020) 51(1):139–65. doi: 10.1111/ejn.14254
14. Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, et al. Regulation of Circadian Behaviour and Metabolism by REV-ERB-Alpha and REV-ERB-Beta. Nature (2012) 485(7396):123–7. doi: 10.1038/nature11048
15. Srour B, Plancoulaine S, Andreeva VA, Fassier P, Julia C, Galan P, et al. Circadian Nutritional Behaviours and Cancer Risk: New Insights From the NutriNet-Sante Prospective Cohort Study. Int J Cancer (2018) 143(10):2369–79. doi: 10.1002/ijc.31584
16. Koronowski KB, Kinouchi K, Welz P-S, Smith JG, Zinna VM, Shi J, et al. Defining the Independence of the Liver Circadian Clock. Cell (2019) 177(6):1448–62.e14. doi: 10.1016/j.cell.2019.04.025
17. Pilorz V, Helfrich-Foerster C, Oster H. The Role of the Circadian Clock System in Physiology. Pflugers Archiv-European J Physiol (2018) 470(2):227–39. doi: 10.1007/s00424-017-2103-y
18. Zhao M, Tuo H, Wang S, Zhao L. The Effects of Dietary Nutrition on Sleep and Sleep Disorders. Mediators Inflamm (2020) 2020. doi: 10.1155/2020/3142874
19. Hunter AL, Ray DW. Circadian Clock Regulation of Hepatic Energy Metabolism Regulatory Circuits. Biol (Basel) (2019) 8:79. doi: 10.3390/biology8040079
20. Venkatesh S, Workman JL. Histone Exchange, Chromatin Structure and the Regulation of Transcription. Nat Rev Mol Cell Biol (2015) 16(3):178–89. doi: 10.1038/nrm3941
21. Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A Circadian Rhythm Orchestrated by Histone Deacetylase 3 Controls Hepatic Lipid Metabolism. Science (2011) 331(6022):1315–9. doi: 10.1126/science.1198125
22. Beytebiere JR, Trott AJ, Greenwell BJ, Osborne CA, Vitet H, Spence J, et al. Tissue-Specific BMAL1 Cistromes Reveal That Rhythmic Transcription Is Associated With Rhythmic Enhancer–Enhancer Interactions. Genes Dev (2019) 33(5-6):294–309. doi: 10.1101/gad.322198.118
23. Zhang Y, Fang B, Emmett MJ, Damle M, Sun Z, Feng D, et al. Discrete Functions of Nuclear Receptor Rev-Erbα Couple Metabolism to the Clock. Science (2015) 348(6242):1488–92. doi: 10.1126/science.aab3021
24. Tahara Y, Shibata S. Circadian Rhythms of Liver Physiology and Disease: Experimental and Clinical Evidence. Nat Rev Gastroenterol Hepatol (2016) 13(4):217–26. doi: 10.1038/nrgastro.2016.8
25. Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, et al. High-Fat Diet Disrupts Behavioral and Molecular Circadian Rhythms in Mice. Cell Metab (2007) 6(5):414–21. doi: 10.1016/j.cmet.2007.09.006
26. Honma K, Hikosaka M, Mochizuki K, Goda T. Loss of Circadian Rhythm of Circulating Insulin Concentration Induced by High-Fat Diet Intake Is Associated With Disrupted Rhythmic Expression of Circadian Clock Genes in the Liver. Metabolism (2016) 65(4):482–91. doi: 10.1016/j.metabol.2015.12.003
27. Lamia KA, Storch KF, Weitz CJ. Physiological Significance of a Peripheral Tissue Circadian Clock. Proc Natl Acad Sci USA (2008) 105(39):15172–7. doi: 10.1073/pnas.0806717105
28. Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, et al. Nuclear Receptor Expression Links the Circadian Clock to Metabolism. Cell (2006) 126(4):801–10. doi: 10.1016/j.cell.2006.06.050
29. Zhang EE, Liu Y, Dentin R, Pongsawakul PY, Liu AC, Hirota T, et al. Cryptochrome Mediates Circadian Regulation of cAMP Signaling and Hepatic Gluconeogenesis. Nat Med (2010) 16(10):1152–6. doi: 10.1038/nm.2214
30. Le Martelot G, Claudel T, Gatfield D, Schaad O, Kornmann B, Lo Sasso G, et al. REV-ERB Alpha Participates in Circadian SREBP Signaling and Bile Acid Homeostasis. PloS Biol (2009) 7(9):12. doi: 10.1371/journal.pbio.1000181
31. Barnea M, Madar Z, Froy O. High-Fat Diet Delays and Fasting Advances the Circadian Expression of Adiponectin Signaling Components in Mouse Liver. Endocrinology (2009) 150(1):161–8. doi: 10.1210/en.2008-0944
32. Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, et al. Reprogramming of the Circadian Clock by Nutritional Challenge. Cell (2013) 155(7):1464–78. doi: 10.1016/j.cell.2013.11.034
33. Christie S, Vincent AD, Li H, Frisby CL, Kentish SJ, O'Rielly R, et al. A Rotating Light Cycle Promotes Weight Gain and Hepatic Lipid Storage in Mice. Am J Physiol Gastrointest Liver Physiol (2018) 315(6):G932–g942. doi: 10.1152/ajpgi.00020.2018
34. Bae S-A, Androulakis IP. Mathematical Modeling Informs the Impact of Changes in Circadian Rhythms and Meal Patterns on Insulin Secretion. Am J Physiol-Regul Integr Comp Physiol (2019) 317(1):R98–R107. doi: 10.1152/ajpregu.00230.2018
35. Yamajuku D, Inagaki T, Haruma T, Okubo S, Kataoka Y, Kobayashi S, et al. Real-Time Monitoring in Three-Dimensional Hepatocytes Reveals That Insulin Acts as a Synchronizer for Liver Clock. Sci Rep (2012) 2:439–9. doi: 10.1038/srep00439
36. Sato M, Murakami M, Node K, Matsumura R, Akashi M. The Role of the Endocrine System in Feeding-Induced Tissue-Specific Circadian Entrainment. Cell Rep (2014) 8(2):393–401. doi: 10.1016/j.celrep.2014.06.015
37. Reznick J, Preston E, Wilks DL, Beale SM, Turner N, Cooney GJ, et al. Altered Feeding Differentially Regulates Circadian Rhythms and Energy Metabolism in Liver and Muscle of Rats. Biochim. Et Biophys. Acta-Mol Basis Dis (2013) 1832(1):228–38. doi: 10.1016/j.bbadis.2012.08.010
38. She Y, Sun J, Hou P, Fang P, Zhang Z. Time-Restricted Feeding Attenuates Gluconeogenic Activity Through Inhibition of PGC-1 Alpha Expression and Activity. Physiol Behav (2021) 231. doi: 10.1016/j.physbeh.2021.113313
39. Patke A, Young MW, Axelrod S. Molecular Mechanisms and Physiological Importance of Circadian Rhythms. Nat Rev Mol Cell Biol (2020) 21(2):67–84. doi: 10.1038/s41580-019-0179-2
40. Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, et al. AMPK Regulates the Circadian Clock by Cryptochrome Phosphorylation and Degradation. Science (2009) 326(5951):437–40. doi: 10.1126/science.1172156
41. Hirao A, Nagahama H, Tsuboi T, Hirao M, Tahara Y, Shibata S, et al. Combination of Starvation Interval and Food Volume Determines the Phase of Liver Circadian Rhythm in Per2::Luc Knock-in Mice Under Two Meals Per Day Feeding. Am J Physiol Gastrointest Liver Physiol (2010) 299(5):G1045–53. doi: 10.1152/ajpgi.00330.2010
42. Chaix A, Zarrinpar A, Miu P, Panda S. Time-Restricted Feeding is a Preventative and Therapeutic Intervention Against Diverse Nutritional Challenges. Cell Metab (2014) 20(6):991–1005. doi: 10.1016/j.cmet.2014.11.001
43. Weger BD, Gobet C, David FPA, Atger F, Martin E, Phillips NE, et al. Systematic Analysis of Differential Rhythmic Liver Gene Expression Mediated by the Circadian Clock and Feeding Rhythms. Proc Natl Acad Sci USA (2021) 118(3). doi: 10.1073/pnas.2015803118
44. Arble DM, Bass J, Laposky AD, Vitaterna MH, Turek FW. Circadian Timing of Food Intake Contributes to Weight Gain. Obes (Silver Spring) (2009) 17(11):2100–2. doi: 10.1038/oby.2009.264
45. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-Restricted Feeding Without Reducing Caloric Intake Prevents Metabolic Diseases in Mice Fed a High-Fat Diet. Cell Metab (2012) 15(6):848–60. doi: 10.1016/j.cmet.2012.04.019
46. Manoogian ENC, Panda S. Circadian Rhythms, Time-Restricted Feeding, and Healthy Aging. Ageing Res Rev (2017) 39:59–67. doi: 10.1016/j.arr.2016.12.006
47. Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide Mononucleotide, a Key NAD(+) Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab (2011) 14(4):528–36. doi: 10.1016/j.cmet.2011.08.014
48. Hardie DG. Sensing of Energy and Nutrients by AMP-Activated Protein Kinase. Am J Clin Nutr (2011) 93(4):891S–6. doi: 10.3945/ajcn.110.001925
49. Toledo M, Batista-Gonzalez A, Merheb E, Aoun ML, Tarabra E, Feng D, et al. Autophagy Regulates the Liver Clock and Glucose Metabolism by Degrading Cry1. Cell Metab (2018) 28(2):268. doi: 10.1016/j.cmet.2018.05.023
50. Oosterman JE, Kalsbeek A, la Fleur SE, Belsham DD. Impact of Nutrients on Circadian Rhythmicity. Am J Physiol-Regul Integr Comp Physiol (2015) 308(5):R337–50. doi: 10.1152/ajpregu.00322.2014
51. Fick LJ, Fick GH, Belsham DD. Palmitate Alters the Rhythmic Expression of Molecular Clock Genes and Orexigenic Neuropeptide Y mRNA Levels Within Immortalized, Hypothalamic Neurons. Biochem Biophys Res Commun (2011) 413(3):414–9. doi: 10.1016/j.bbrc.2011.08.103
52. Tong X, Zhang D, Arthurs B, Li P, Durudogan L, Gupta N, et al. Palmitate Inhibits SIRT1-Dependent BMAL1/CLOCK Interaction and Disrupts Circadian Gene Oscillations in Hepatocytes. PloS One (2015) 10(6). doi: 10.1371/journal.pone.0130047
53. Gil-Lozano M, Wu WK, Martchenko A, Brubaker PL. High-Fat Diet and Palmitate Alter the Rhythmic Secretion of Glucagon-Like Peptide-1 by the Rodent L-Cell. Endocrinology (2016) 157(2):586–99. doi: 10.1210/en.2015-1732
54. Greco JA, Oosterman JE, Belsham DD. Differential Effects of Omega-3 Fatty Acid Docosahexaenoic Acid and Palmitate on the Circadian Transcriptional Profile of Clock Genes in Immortalized Hypothalamic Neurons. Am J Physiol-Regul Integr Comp Physiol (2014) 307(8):R1049–60. doi: 10.1152/ajpregu.00100.2014
55. Kim S-M, Neuendorff N, Chapkin RS, Earnest DJ. Role of Inflammatory Signaling in the Differential Effects of Saturated and Poly-Unsaturated Fatty Acids on Peripheral Circadian Clocks. Ebiomedicine (2016) 7:100–11. doi: 10.1016/j.ebiom.2016.03.037
56. Tal Y, Chapnik N, Froy O. Non-Obesogenic Doses of Fatty Acids Modulate the Functionality of the Circadian Clock in the Liver. Cell Mol Life Sci (2019) 76(9):1795–806. doi: 10.1007/s00018-019-03023-6
57. Iwanaga H, Yano M, Miki H, Okada K, Azama T, Takiguchi S, et al. Per2 Gene Expressions in the Suprachiasmatic Nucleus and Liver Differentially Respond to Nutrition Factors in Rats. JPEN J Parenter Enteral Nutr (2005) 29(3):157–61. doi: 10.1177/0148607105029003157
58. Ikeda Y, Kamagata M, Hirao M, Yasuda S, Iwami S, Sasaki H, et al. Glucagon and/or IGF-1 Production Regulates Resetting of the Liver Circadian Clock in Response to a Protein or Amino Acid-Only Diet. Ebiomedicine (2018) 28:210–24. doi: 10.1016/j.ebiom.2018.01.012
59. Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of Non-Alcoholic Fatty Liver Disease. Qjm (2010) 103(2):71–83. doi: 10.1093/qjmed/hcp158
60. Asher G, Schibler U. Crosstalk Between Components of Circadian and Metabolic Cycles in Mammals. Cell Metab (2011) 13(2):125–37. doi: 10.1016/j.cmet.2011.01.006
61. Green CB, Takahashi JS, Bass J. The Meter of Metabolism. Cell (2008) 134(5):728–42. doi: 10.1016/j.cell.2008.08.022
62. Ferramosca A, Zara V. Modulation of Hepatic Steatosis by Dietary Fatty Acids. World J Gastroenterol (2014) 20(7):1746–55. doi: 10.3748/wjg.v20.i7.1746
63. Parry SA, Hodson L. Influence of Dietary Macronutrients on Liver Fat Accumulation and Metabolism. J Invest Med (2017) 65(8):1102–15. doi: 10.1136/jim-2017-000524
64. Rui L. Energy Metabolism in the Liver. Compr Physiol (2014) 4(1):177–97. doi: 10.1002/cphy.c130024
65. Schwarz J-M, Clearfield M, Mulligan K. Conversion of Sugar to Fat: Is Hepatic De Novo Lipogenesis Leading to Metabolic Syndrome and Associated Chronic Diseases? J Am Osteopathic Assoc (2017) 117(8):520–7. doi: 10.7556/jaoa.2017.102
66. Schleicher J, Tokarski C, Marbach E, Matz-Soja M, Zellmer S, Gebhardt R, et al. Zonation of Hepatic Fatty Acid Metabolism - The Diversity of Its Regulation and the Benefit of Modeling. Biochim Et Biophys Acta-Mol Cell Biol Lipids (2015) 1851(5):641–56. doi: 10.1016/j.bbalip.2015.02.004
67. Marchisello S, Di Pino A, Scicali R, Urbano F, Piro S, Purrello F, et al. Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. Int J Mol Sci (2019) 20(8):1948. doi: 10.3390/ijms20081948
68. Viscarra J, Sul HS. Epigenetic Regulation of Hepatic Lipogenesis: Role in Hepatosteatosis and Diabetes. Diabetes (2020) 69(4):525–31. doi: 10.2337/dbi18-0032
69. Oosterveer MH, Schoonjans K. Hepatic Glucose Sensing and Integrative Pathways in the Liver. Cell Mol Life Sci (2014) 71(8):1453–67. doi: 10.1007/s00018-013-1505-z
70. Shen L, Cui A, Xue Y, Cui Y, Dong X, Gao Y, et al. Hepatic Differentiated Embryo-Chondrocyte-Expressed Gene 1 (Dec1) Inhibits Sterol Regulatory Element-Binding Protein-1c (Srebp-1c) Expression and Alleviates Fatty Liver Phenotype. J Biol Chem (2014) 289(34):23332–42. doi: 10.1074/jbc.M113.526343
71. Iizuka K, Horikawa Y. Regulation of Lipogenesis via BHLHB2/DEC1 and ChREBP Feedback Looping. Biochem Biophys Res Commun (2008) 374(1):95–100. doi: 10.1016/j.bbrc.2008.06.101
72. Iizuka K, Takeda J, Horikawa Y. Kruppel-Like Factor-10 Is Directly Regulated by Carbohydrate Response Element-Binding Protein in Rat Primary Hepatocytes. Biochem Biophys Res Commun (2011) 412(4):638–43. doi: 10.1016/j.bbrc.2011.08.016
73. Meng H, Gonzales NM, Lonard DM, Putluri N, Zhu B, Dacso CC, et al. XBP1 Links the 12-Hour Clock to NAFLD and Regulation of Membrane Fluidity and Lipid Homeostasis. Nat Commun (2020) 11(1). doi: 10.1038/s41467-020-20028-z
74. Li W-K, Li H, Lu Y-F, Li Y-Y, Fu ZD, Liu J. Atorvastatin Alters the Expression of Genes Related to Bile Acid Metabolism and Circadian Clock in Livers of Mice. Peerj (2017) 5. doi: 10.7717/peerj.3348
75. Fleet T, Stashi E, Zhu B, Rajapakshe K, Marcelo KL, Kettner NM, et al. Genetic and Environmental Models of Circadian Disruption Link SRC-2 Function to Hepatic Pathology. J Biol Rhythms (2016) 31(5):443–60. doi: 10.1177/0748730416657921
76. Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, et al. Rev-Erbalpha and Rev-Erbbeta Coordinately Protect the Circadian Clock and Normal Metabolic Function. Genes Dev (2012) 26(7):657–67. doi: 10.1101/gad.186858.112
77. Heath RB, Karpe F, Milne RW, Burdge GC, Wootton SA, Frayn KN. Selective Partitioning of Dietary Fatty Acids Into the VLDL TG Pool in the Early Postprandial Period. J Lipid Res (2003) 44(11):2065–72. doi: 10.1194/jlr.M300167-JLR200
78. Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, et al. Coordinated Transcription of Key Pathways in the Mouse by the Circadian Clock. Cell (2002) 109(3):307–20. doi: 10.1016/S0092-8674(02)00722-5
79. Chiang JYL, Ferrell JM. Bile Acid Biology, Pathophysiology, and Therapeutics. Clin Liver Dis (2020) 15(3):91–4. doi: 10.1002/cld.861
80. Coomans CP, van den Berg SA, Lucassen EA, Houben T, Pronk AC, van der Spek RD, et al. The Suprachiasmatic Nucleus Controls Circadian Energy Metabolism and Hepatic Insulin Sensitivity. Diabetes (2013) 62(4):1102–8. doi: 10.2337/db12-0507
81. Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH. Circadian Disruption Leads to Insulin Resistance and Obesity. Curr Biol (2013) 23(5):372–81. doi: 10.1016/j.cub.2013.01.048
82. Mazzoccoli G, Vinciguerra M, Oben J, Tarquini R, De Cosmo S. Non-Alcoholic Fatty Liver Disease: The Role of Nuclear Receptors and Circadian Rhythmicity. Liver Int (2014) 34(8):1133–52. doi: 10.1111/liv.12534
83. Engin A. Non-Alcoholic Fatty Liver Disease. Adv Exp Med Biol (2017) 960:443–67. doi: 10.1007/978-3-319-48382-5_19
84. Arroyave-Ospina JC, Wu Z, Geng Y, Moshage H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants (2021) 10(2). doi: 10.3390/antiox10020174
85. Wang T, Ma C. The Hepatic Macrophage Pool in NASH. Cell Mol Immunol (2021) 18(8):2059–60. doi: 10.1038/s41423-021-00690-z
86. Anthony BJ, Ramm GA, McManus DP. Role of Resident Liver Cells in the Pathogenesis of Schistosomiasis. Trends Parasitol (2012) 28(12):572–9. doi: 10.1016/j.pt.2012.09.005
87. Cha J-Y, Kim D-H, Chun K-H. The Role of Hepatic Macrophages in Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Lab Anim Res (2018) 34(4):133–9. doi: 10.5625/lar.2018.34.4.133
88. Luo X, Li H, Ma L, Zhou J, Guo X, Woo S-L, et al. Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology (2018) 155(6):1971. doi: 10.1053/j.gastro.2018.09.010
89. Summa KC, Voigt RM, Forsyth CB, Shaikh M, Cavanaugh K, Tang Y, et al. Disruption of the Circadian Clock in Mice Increases Intestinal Permeability and Promotes Alcohol-Induced Hepatic Pathology and Inflammation. PloS One (2013) 8(6):e67102–2. doi: 10.1371/journal.pone.0067102
90. Kim S-M, Neuendorff N, Earnest DJ. Role of Proinflammatory Cytokines in Feedback Modulation of Circadian Clock Gene Rhythms by Saturated Fatty Acids. Sci Rep (2019) 9:8909. doi: 10.1038/s41598-019-45322-9
91. Gachon F, Yeung J, Naef F. Cross-Regulatory Circuits Linking Inflammation, High-Fat Diet, and the Circadian Clock. Genes Dev (2018) 32(21-22):1359–60. doi: 10.1101/gad.320911.118
92. Early JO, Menon D, Wyse CA, Cervantes-Silva MP, Zaslona Z, Carroll RG, et al. Circadian Clock Protein BMAL1 Regulates IL-1 Beta in Macrophages via NRF2. Proc Natl Acad Sci USA (2018) 115(36):E8460–8. doi: 10.1073/pnas.1800431115
93. Abo SMC, Layton AT. Modeling the Circadian Regulation of the Immune System: Sexually Dimorphic Effects of Shift Work. PloS Comput Biol (2021) 17(3):e1008514. doi: 10.1371/journal.pcbi.1008514
94. Pivovarova O, Jurchott K, Rudovich N, Hornemann S, Ye L, Mockel S, et al. Changes of Dietary Fat and Carbohydrate Content Alter Central and Peripheral Clock in Humans. J Clin Endocrinol Metab (2015) 100(6):2291–302. doi: 10.1210/jc.2014-3868
95. Nguyen KD, Fentress SJ, Qiu YF, Yun KR, Cox JS, Chawla A. Circadian Gene Bmal1 Regulates Diurnal Oscillations of Ly6C(hi) Inflammatory Monocytes. Science (2013) 341(6153):1483–8. doi: 10.1126/science.1240636
96. Kim SM, Neuendorff N, Alaniz RC, Sun YX, Chapkin RS, Earnest DJ. Shift Work Cycle-Induced Alterations of Circadian Rhythms Potentiate the Effects of High-Fat Diet on Inflammation and Metabolism. FASEB J (2018) 32(6):3085–95. doi: 10.1096/fj.201700784R
97. Xu H, Li H, Woo S-L, Kim S-M, Shende VR, Neuendorff N, et al. Myeloid Cell-Specific Disruption of Period1 and Period2 Exacerbates Diet-Induced Inflammation and Insulin Resistance. J Biol Chem (2014) 289(23):16374–88. doi: 10.1074/jbc.M113.539601
98. Kim KE, Kim H, Heo RW, Shin HJ, Yi CO, Lee DH, et al. Myeloid-Specific SIRT1 Deletion Aggravates Hepatic Inflammation and Steatosis in High-Fat Diet-Fed Mice. Korean J Physiol Pharmacol (2015) 19(5):451–U75. doi: 10.4196/kjpp.2015.19.5.451
99. Ruhl CE, Everhart JE. Trunk Fat Is Associated With Increased Serum Levels of Alanine Aminotransferase in the United States. Gastroenterology (2010) 138(4):1346–56.e13563. doi: 10.1053/j.gastro.2009.12.053
100. Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ, Nash CRN. The Association of Genetic Variability in Patatin-Like Phospholipase Domain-Containing Protein 3 (PNPLA3) With Histological Severity of Nonalcoholic Fatty Liver Disease. Hepatol (Baltimore Md) (2010) 52(3):894–903. doi: 10.1002/hep.23759
101. Loomba R, Sanyal AJ. The Global NAFLD Epidemic. Nat Rev Gastroenterol Hepatol (2013) 10(11):686–90. doi: 10.1038/nrgastro.2013.171
102. Younossi ZM, Henry L. Economic and Quality-Of-Life Implications of Non-Alcoholic Fatty Liver Disease. PharmacoEconomics (2015) 33(12):1245–53. doi: 10.1007/s40273-015-0316-5
103. Seki E, Schwabe RF. Hepatic Inflammation and Fibrosis: Functional Links and Key Pathways. Hepatology (2015) 61(3):1066–79. doi: 10.1002/hep.27332
104. Buzzetti E, Pinzani M, Tsochatzis EA. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism-Clin Exp (2016) 65(8):1038–48. doi: 10.1016/j.metabol.2015.12.012
105. Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in in Nonalcoholic Fatty Liver Disease. Metabolism-Clin Exp (2016) 65(8):1062–79. doi: 10.1016/j.metabol.2015.11.006
106. Bertolani C, Marra F. Role of Adipocytokines in Hepatic Fibrosis. Curr Pharm Design (2010) 16(17):1929–40. doi: 10.2174/138161210791208857
107. Nobili V, Carpino G, Alisi A, Franchitto A, Alpini G, De Vito R, et al. Hepatic Progenitor Cells Activation, Fibrosis, and Adipokines Production in Pediatric Nonalcoholic Fatty Liver Disease. Hepatology (2012) 56(6):2142–53. doi: 10.1002/hep.25742
108. Polyzos SA, Mantzoros CS. Leptin in Health and Disease: Facts and Expectations at Its Twentieth Anniversary. Metabolism-Clin Exp (2015) 64(1):5–12. doi: 10.1016/j.metabol.2014.10.017
109. Dattaroy D, Pourhoseini S, Das S, Alhasson F, Seth RK, Nagarkatti M, et al. Micro-RNA 21 Inhibition of SMAD7 Enhances Fibrogenesis via Leptin-Mediated NADPH Oxidase in Experimental and Human Nonalcoholic Steatohepatitis. Am J Physiol-Gastrointest Liver Physiol (2015) 308(4):G298–312. doi: 10.1152/ajpgi.00346.2014
110. Wang JH, Leclercq I, Brymora JM, Xu N, Ramezani-Moghadam M, London RM, et al. Kupffer Cells Mediate Leptin-Induced Liver Fibrosis. Gastroenterology (2009) 137(2):713–23. doi: 10.1053/j.gastro.2009.04.011
111. Fujii S-I, Shimizu K. Exploiting Antitumor Immunotherapeutic Novel Strategies by Deciphering the Cross Talk Between Invariant NKT Cells and Dendritic Cells. Front Immunol (2017) 8:886. doi: 10.3389/fimmu.2017.00886
112. Li H, Kim U-H, Yoon J-H, Ji H-S, Park H-M, Park H-Y, et al. Suppression of Hyperglycemia and Hepatic Steatosis by Black-Soybean-Leaf Extract via Enhanced Adiponectin-Receptor Signaling and AMPK Activation. J Agric Food Chem (2019) 67(1):90–101. doi: 10.1021/acs.jafc.8b04527
113. Moschen AR, Wieser V, Tilg H. Adiponectin: Key Player in the Adipose Tissue-Liver Crosstalk. Curr Med Chem (2012) 19(32):5467–73. doi: 10.2174/092986712803833254
114. Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, et al. Dysregulation of Inflammatory Responses by Chronic Circadian Disruption. J Immunol (2010) 185(10):5796–805. doi: 10.4049/jimmunol.1001026
115. Wang T, He C. Pro-Inflammatory Cytokines: The Link Between Obesity and Osteoarthritis. Cytokine Growth Factor Rev (2018) 44:38–50. doi: 10.1016/j.cytogfr.2018.10.002
116. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL Jr, Ferrante AW, et al. Obesity Is Associated With Macrophage Accumulation in Adipose Tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
117. Milić S, Lulić D, Štimac D. Non-Alcoholic Fatty Liver Disease and Obesity: Biochemical, Metabolic and Clinical Presentations. World J Gastroenterol (2014) 20(28):9330–7. doi: 10.3748/wjg.v20.i28.9330
118. Kettner NM, Mayo SA, Hua J, Lee C, Moore DD, Fu L, et al. Circadian Dysfunction Induces Leptin Resistance in Mice. Cell Metab (2015) 22(3):448–59. doi: 10.1016/j.cmet.2015.06.005
119. Huo YQ, Guo X, Li HG, Xu H, Halim V, Zhang WY, et al. Targeted Overexpression of Inducible 6-Phosphofructo-2-Kinase in Adipose Tissue Increases Fat Deposition But Protects Against Diet-Induced Insulin Resistance and Inflammatory Responses. J Biol Chem (2012) 287(25):21492–500. doi: 10.1074/jbc.M112.370379
120. Guo X, Xu KF, Zhang JF, Li HG, Zhang WY, Wang HA, et al. Involvement of Inducible 6-Phosphofructo-2-Kinase in the Anti-Diabetic Effect of Peroxisome Proliferator-Activated Receptor Gamma Activation in Mice. J Biol Chem (2010) 285(31):23711–20. doi: 10.1074/jbc.M110.123174
121. Huo YQ, Guo X, Li HG, Wang H, Zhang WY, Wang Y, et al. Disruption of Inducible 6-Phosphofructo-2-Kinase Ameliorates Diet-Induced Adiposity But Exacerbates Systemic Insulin Resistance and Adipose Tissue Inflammatory Response. J Biol Chem (2010) 285(6):3713–21. doi: 10.1074/jbc.M109.058446
122. Wieser V, Adolph TE, Grander C, Grabherr F, Enrich B, Moser P, et al. Adipose Type I Interferon Signalling Protects Against Metabolic Dysfunction. Gut (2018) 67(1):157–65. doi: 10.1136/gutjnl-2016-313155
123. Froy O, Garaulet M. The Circadian Clock in White and Brown Adipose Tissue: Mechanistic, Endocrine, and Clinical Aspects. Endocrine Rev (2018) 39(3):261–73. doi: 10.1210/er.2017-00193
124. Paschos GK, Ibrahim S, Song W-L, Kunieda T, Grant G, Reyes TM, et al. Obesity in Mice With Adipocyte-Specific Deletion of Clock Component Arntl. Nat Med (2012) 18(12):1768–77. doi: 10.1038/nm.2979
125. Lehrke M, Lazar MA. The Many Faces of PPARgamma. Cell (2005) 123(6):993–9. doi: 10.1016/j.cell.2005.11.026
126. Fontaine C, Dubois G, Duguay Y, Helledie T, Vu-Dac N, Gervois P, et al. The Orphan Nuclear Receptor Rev-Erbalpha Is a Peroxisome Proliferator-Activated Receptor (PPAR) Gamma Target Gene and Promotes PPARgamma-Induced Adipocyte Differentiation. J Biol Chem (2003) 278(39):37672–80. doi: 10.1074/jbc.M304664200
127. Gooley JJ, Chua EC-P. Diurnal Regulation of Lipid Metabolism and Applications of Circadian Lipidomics. J Genet Genomics (2014) 41(5):231–50. doi: 10.1016/j.jgg.2014.04.001
128. He W, Barak Y, Hevener S, Olson P, Liao D, Le J, et al. Adipose-Specific Peroxisome Proliferator-Activated Receptor Gamma Knockout Causes Insulin Resistance in Fat and Liver But Not in Muscle. Proc Natl Acad Sci USA (2003) 100(26):15712–7. doi: 10.1073/pnas.2536828100
129. Larson-Meyer DE, Newcomer BR, Heilbronn LK, Volaufova J, Smith SR, Alfonso AJ, et al. Effect of 6-Month Calorie Restriction and Exercise on Serum and Liver Lipids and Markers of Liver Function. Obesity (2008) 16(6):1355–62. doi: 10.1038/oby.2008.201
130. Kim KE, Jung Y, Min S, Nam M, Heo RW, Jeon BT, et al. Caloric Restriction of Db/Db Mice Reverts Hepatic Steatosis and Body Weight With Divergent Hepatic Metabolism. Sci Rep (2016) 6:14. doi: 10.1038/srep30111
131. Patel SA, Chaudhari A, Gupta R, Velingkaar N, Kondratov RV. Circadian Clocks Govern Calorie Restriction-Mediated Life Span Extension Through BMAL1-And IGF-1-Dependent Mechanisms. FASEB J (2016) 30(4):1634–42. doi: 10.1096/fj.15-282475
132. Katewa SD, Akagi K, Bose N, Rakshit K, Camarella T, Zheng XZ, et al. Peripheral Circadian Clocks Mediate Dietary Restriction-Dependent Changes in Lifespan and Fat Metabolism in Drosophila. Cell Metab (2016) 23(1):143–54. doi: 10.1016/j.cmet.2015.10.014
133. Godos J, Federico A, Dallio M, Scazzina F. Mediterranean Diet and Nonalcoholic Fatty Liver Disease: Molecular Mechanisms of Protection. Int J Food Sci Nutr (2017) 68(1):18–27. doi: 10.1080/09637486.2016.1214239
134. Zelber-Sagi S, Salomone F, Mlynarsky L. The Mediterranean Dietary Pattern as the Diet of Choice for Non-Alcoholic Fatty Liver Disease: Evidence and Plausible Mechanisms. Liver Int (2017) 37(7):936–49. doi: 10.1111/liv.13435
135. Garaulet M, Corbalan MD, Madrid JA, Morales E, Baraza JC, Lee YC, et al. CLOCK Gene Is Implicated in Weight Reduction in Obese Patients Participating in a Dietary Programme Based on the Mediterranean Diet. Int J Obes (2010) 34(3):516–23. doi: 10.1038/ijo.2009.255
136. Hernandez-Rodas MC, Valenzuela R, Echeverria F, Angel Rincon-Cervera M, Espinosa A, Illesca P, et al. Supplementation With Docosahexaenoic Acid and Extra Virgin Olive Oil Prevents Liver Steatosis Induced by a High-Fat Diet in Mice Through PPAR-Alpha and Nrf2 Upregulation With Concomitant SREBP-1c and NF-kB Downregulation. Mol Nutr Food Res (2017) 61(12). doi: 10.1002/mnfr.201700479
137. Guo X, Li HG, Xu H, Halim V, Zhang WY, Wang H, et al. Palmitoleate Induces Hepatic Steatosis But Suppresses Liver Inflammatory Response in Mice. PloS One (2012) 7(6):9. doi: 10.1371/journal.pone.0039286
138. Assy N, Nassar F, Nasser G, Grosovski M. Olive Oil Consumption and Non-Alcoholic Fatty Liver Disease. World J Gastroenterol (2009) 15(15):1809–15. doi: 10.3748/wjg.15.1809
139. Zhang DQ, Tong X, Arthurs B, Guha A, Rui LY, Kamath A, et al. Liver Clock Protein BMAL1 Promotes De Novo Lipogenesis Through Insulin-Mtorc2-AKT Signaling. J Biol Chem (2014) 289(37):25925–35. doi: 10.1074/jbc.M114.567628
140. Berzigotti A, Saran U, Dufour JF. Physical Activity and Liver Diseases. Hepatology (2016) 63(3):1026–40. doi: 10.1002/hep.28132
141. Robles MS, Humphrey SJ, Mann M. Phosphorylation Is a Central Mechanism for Circadian Control of Metabolism and Physiology. Cell Metab (2017) 25(1):118–27. doi: 10.1016/j.cmet.2016.10.004
142. Sato S, Basse AL, Schonke M, Chen SW, Samad M, Altintas A, et al. Time of Exercise Specifies the Impact on Muscle Metabolic Pathways and Systemic Energy Homeostasis. Cell Metab (2019) 30(1):92. doi: 10.1016/j.cmet.2019.03.013
143. Wang HL, Gu YQ, Zheng LX, Liu L, Meng G, Wu HM, et al. Association Between Bedtime and the Prevalence of Newly Diagnosed Non-Alcoholic Fatty Liver Disease in Adults. Liver Int (2018) 38(12):2277–86. doi: 10.1111/liv.13896
144. Tilg H, Moschen AR, Roden M. NAFLD and Diabetes Mellitus. Nat Rev Gastroenterol Hepatol (2017) 14(1):32–42. doi: 10.1038/nrgastro.2016.147
145. Bril F, Kalavalapalli S, Clark VC, Lomonaco R, Soldevila-Pico C, Liu IC, et al. Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin Gastroenterol Hepatol (2018) 16(4):558. doi: 10.1016/j.cgh.2017.12.001
146. Ribas-Latre A, Fekry B, Kwok C, Baumgartner C, Shivshankar S, Sun K, et al. Rosiglitazone Reverses High Fat Diet-Induced Changes in BMAL1 Function in Muscle, Fat, and Liver Tissue in Mice. Int J Obes (2019) 43(3):567–80. doi: 10.1038/s41366-018-0090-5
147. Martchenko A, Oh RH, Wheeler SE, Gurges P, Chalmers JA, Brubaker PL. Suppression of Circadian Secretion of Glucagon-Like Peptide-1 by the Saturated Fatty Acid, Palmitate. Acta Physiol (2018) 222(4):12. doi: 10.1111/apha.13007
148. Svegliati-Baroni G, Khan S, Khan C, Agostinelli L, De Minicis S, Candelaresi C, et al. Glucagon-Like Peptide-1 Receptor Activation Stimulates Hepatic Lipid Oxidation and Restores Hepatic Signalling Alteration Induced Bya High-Fat Diet in Nonalcoholic Steatohepatitis. Liver Int (2011) 31(9):1285–97. doi: 10.1111/j.1478-3231.2011.02462.x
149. Khan RS, Bril F, Cusi K, Newsome PN. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology (2019) 70(2):711–24. doi: 10.1002/hep.30429
150. Cholankeril R, Patel V, Perumpail BJ, Yoo ER, Iqbal U, Sallam S, et al. Anti-Diabetic Medications for the Pharmacologic Management of NAFLD. Diseases (2018) 6(4). doi: 10.3390/diseases6040093
151. Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Manneras-Holm L, et al. Metformin Alters the Gut Microbiome of Individuals With Treatment-Naive Type 2 Diabetes, Contributing to the Therapeutic Effects of the Drug. Nat Med (2017) 23(7):850–+. doi: 10.1038/nm.4345
152. Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, et al. Transkingdom Control of Microbiota Diurnal Oscillations Promotes Metabolic Homeostasis. Cell (2014) 159(3):514–29. doi: 10.1016/j.cell.2014.09.048
153. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet (2015) 385(9972):956–65. doi: 10.1016/S0140-6736(14)61933-4
Keywords: circadian, nutrition, metabolic diseases, NAFLD, hepatic steatosis, inflammation
Citation: Guo X, Zheng J, Zhang S, Jiang X, Chen T, Yu J, Wang S', Ma X and Wu C (2021) Advances in Unhealthy Nutrition and Circadian Dysregulation in Pathophysiology of NAFLD. Front. Clin. Diabetes Healthc. 2:691828. doi: 10.3389/fcdhc.2021.691828
Received: 07 April 2021; Accepted: 27 September 2021;
Published: 12 October 2021.
Edited by:
Elisabet Børsheim, University of Arkansas for Medical Sciences, United StatesReviewed by:
Soo Jin Yang, Seoul Women’s University, South KoreaEleni Beli, Queen’s University Belfast, United Kingdom
Copyright © 2021 Guo, Zheng, Zhang, Jiang, Chen, Yu, Wang, Ma and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Guo, eGd1b0BzZHUuZWR1LmNu; Chaodong Wu, Y2hhb2Rvbmcud3VAYWduZXQudGFtdS5lZHU=
†These authors have contributed equally to this work