
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bacteriol. , 21 August 2023
Sec. Molecular Bacteriology and Microbiome
Volume 2 - 2023 | https://doi.org/10.3389/fbrio.2023.1254305
This article is part of the Research Topic Editors' Showcase: Molecular Bacteriology and Microbiome View all 7 articles
 Edward A. P. Provencher1,2†
Edward A. P. Provencher1,2† Molly R. Ehrig1†
Molly R. Ehrig1† Andrew G. Cecere1,2Shyan C. Cousins1Michael A. Maybin1
Andrew G. Cecere1,2Shyan C. Cousins1Michael A. Maybin1 Timothy C. Meredith1
Timothy C. Meredith1 Tim I. Miyashiro1,2*
Tim I. Miyashiro1,2*Many animals form symbioses with environmental bacteria that provide biological functions beneficial to their hosts. The mechanisms that affect the acquisition of bacterial symbionts remain poorly understood but are important to identify to develop new ways to improve animal health. Vibrio fischeri is a Gram-negative bacterium that forms a mutualistic symbiosis with the Hawaiian bobtail squid Euprymna scolopes. From within a light organ, these V. fischeri populations engage in quorum sensing to produce bioluminescence for the host to eliminate its shadow. In our attempts to investigate how quorum sensing contributes to the evolution of V. fischeri, we unexpectedly isolated a strain that produced large structures resembling biofilms along glass surfaces that readily stain with crystal violet. Biofilm formation by this strain is independent of sypG, which encodes the primary activator of the symbiotic polysaccharide (syp) locus, suggesting a novel biofilm pathway. Squid colonization assays revealed that the isolate exhibited a colonization defect, which suggests that its biofilm phenotype inhibits establishment of symbiosis. Whole-genome sequencing and subsequent culture assays suggest that this biofilm phenotype is due to a single point mutation that confers an I125F substitution in the putative glycosyltransferase VF_0133. Expression of the wild-type copy of VF_0133 in trans eliminates the biofilm-like phenotypes in culture and restores the ability of the strain to establish symbiosis. Investigation of lipopolysaccharide (LPS) structure by silver stain suggests significant modifications to the oligosaccharide core and O-antigen in this strain. Taken together, these findings add knowledge to the role of LPS in V. fischeri physiology and light organ colonization, which provides important insight into how bacterial symbionts are acquired from the environment.
Bacterial symbionts, which are bacteria that form close and long-term associations with animal hosts, are major factors when considering overall host physiology (Ganesan et al., 2022). Many bacterial symbionts provide biological functions that contribute to basic host processes, including digestion and immune responses, and hosts are often at higher risk of developing disease if they lack such symbionts. Because bacterial symbionts are often acquired by the host from environmental reservoirs, the mechanisms that promote their colonization are critical for the symbiosis to form. In general, the molecular underpinnings of these mechanisms are unclear, but filling these knowledge gaps raises the possibility of improving animal health by developing new ways to promote the formation of symbioses with specific beneficial bacteria.
Vibrio fischeri (aka Aliivibrio fischeri) is a Gram-negative bacterium that can establish symbiosis with various fish and squid, including the Hawaiian bobtail squid Euprymna scolopes (Stabb and Visick, 2013). The symbiosis takes place within a symbiotic organ called the light organ that is located on the ventral side of the squid underneath its mantle. Populations of V. fischeri occupy epithelium-lined crypt spaces and produce bioluminescence that the host uses for counterillumination, a behavior by which the squid eliminates its shadow within the water column and avoids detection from below (Jones and Nishiguchi, 2004). The ability to cultivate and genetically manipulate V. fischeri (Visick et al., 2018; Christensen and Visick, 2020) as well as the capability to maintain a cohort of E. scolopes squid in mariculture facilities (Cecere and Miyashiro, 2022; Cecere et al., 2023) have led to this system becoming a powerful model for investigating the molecular details underlying the formation of host-bacterial symbioses.
The symbiosis is initially established when hatchlings are exposed to seawater containing V. fischeri cells and proceeds according to a series of discrete stages that have been the subject of multiple reviews, e.g., (Nyholm and Mcfall-Ngai, 2021). One stage that occurs early during symbiosis establishment involves the accumulation of environmental V. fischeri cells near superficial ciliated epithelial fields along the surface of the nascent light organ (Nyholm et al., 2000). Within these so-called aggregates, V. fischeri cells alter their transcriptional profile to produce symbiotic polysaccharide (Syp) (Yip et al., 2005), which in turn primes them for entering the host (Surrett et al., 2023). Following host entry, V. fischeri cells colonize crypt spaces, where they proliferate on host-derived nutrients (Graf and Ruby, 1998; Schwartzman et al., 2015; Wasilko et al., 2019). The resulting populations engage in LuxI/LuxR-based quorum sensing to promote the production of bioluminescence (Miyashiro and Ruby, 2012), thereby signifying completion of the initial stages of symbiosis establishment.
The strain ES114 has emerged as a preeminent model for investigating the regulation of Syp production by V. fischeri, which involves transcriptional activation of an 18-gene syp locus that encodes many of the factors that promote aggregation in vivo (Shibata et al., 2012). Under routine growth conditions, the syp locus is transcriptionally silent, and ES114 forms smooth colonies on solid surfaces and uniformly turbid cultures in liquid medium. However, ES114 can be induced to form Syp-dependent biofilms by stimulating specific regulatory pathways either through genetic modification or altering growth conditions (Yip et al., 2006; Tischler et al., 2018; Dial et al., 2023), which has led to molecular insight regarding the regulatory mechanisms underlying the aggregation stage of symbiosis establishment.
A recent study suggested that each population of V. fischeri within the light organ can detect the autoinducer produced by neighboring populations (Yount et al., 2022), which in turn can alter the extent to which each of them produces bioluminescence. The ability of autoinducer to impact V. fischeri physiology implies that the level of autoinducer within an environment, regardless of its source, can affect cellular fitness, thereby serving as a potential driver of adaptation. To test this possibility, we initiated a study to determine how V. fischeri strain ES114 adapts to 3-oxo-C6 HSL, and those results will be described elsewhere. Here, we report the phenotypes of a strain that was isolated while serially passaging ES114 in medium supplemented with N-3-oxohexanoyl-homoserine lactone (3-oxo-C6 HSL), which is the quorum-sensing molecule synthesized by synthase LuxI (Schaefer et al., 1996). We found that the isolated strain readily forms biofilms in culture, and this process depends on a disruption in lipopolysaccharide (LPS) biosynthesis rather than in Syp production. Genetic analysis suggests that an allele of VF_0133, which encodes a putative glycosyltransferase, results in a truncated LPS structure, which can be rescued through the introduction of the wild-type copy of VF_0133. We find that this strain exhibits a severe colonization defect with E. scolopes hatchlings, which suggests the truncated LPS structure is incompatible with V. fischeri establishing symbiosis. Taken together, the results reported here add knowledge of how LPS contributes to the general physiology of V. fischeri, which is significant for the bacterial symbiont to colonize its host.
Strains and plasmids used in this study are found in Table 1. Vibrio fischeri strains were grown under aerobic conditions at 28°C in LBS medium [1% (w/v) tryptone, 0.5% (w/v) yeast extract, 2% (w/v) NaCl, 50 mM Tris–HCl (pH 7.5)] or SWTO medium [0.5% (w/v) tryptone, 0.3% (w/v) yeast extract, 1% NaCl (w/v), 0.3% glycerol, 70% Instant Ocean (v/v)]. Solid medium contained 1.5% (w/v) agar. To maintain plasmids, chloramphenicol was used at a final concentration of 2.5 μg mL-1.

Table 1 Strains and plasmids used in this study.
Escherichia coli strains were grown under aerobic conditions at 37°C in LB medium [1% (w/v) tryptone, 0.5% (w/v) yeast extract, 1% (w/v) NaCl].
In general, starter cultures used to initiate the assays described below were generated by inoculating 3 mL LBS with an isolated colony of the indicated strain and incubating the culture at 28°C with shaking at 200 rpm overnight. However, for MRE010, which displays a severe growth defect in liquid LBS, starter cultures were prepared using SWTO medium instead of LBS.
Colonies were imaged using a Rebel T5 Camera (Canon) mounted on a SZX16 dissecting microscope (Olympus) equipped with an SDF PLFL 0.3× objective. Using ImageJ v. 1.52a software (NIH), images were converted from RAW format to TIFF with the DCRaw macro with the following settings: use_temporary_directory, white_balance = [Camera white balance], do_not_automatically_brighten, output_colorspace = [sRGB], read_as = [8-bit], interpolation = [High-speed, low-quality bilinear], and half_size.
MRE010 was isolated during the propagation of ES114 associated with another study that will be reported elsewhere. Briefly, an LBS starter culture of ES114 was washed twice by centrifugation at 9,000 x g for 2 min and resuspension in SWTO. A 20-μL sample of the cell suspension was diluted into 2 mL SWTO supplemented with 1 μM 3-oxo-C6 HSL and grown at 28°C shaking at 200 rpm. The culture was passaged every 12 h by diluting 20 μL into 2 mL SWTO, with each dilution corresponding to log2(100) = 6.6 generations. After every 8th passage (~53 generations), a frozen stock containing 16% (v/v) glycerol was generated and stored at -80°C. A small portion (<5 μL) of the frozen stock corresponding to the 32nd passage (~210 generations) was plated onto LBS agar, which was incubated at 28°C. One of the resulting colonies was randomly selected and labeled as MRE010.
To generate the ΔflrC mutant SSC014, pLosTfoX-dependent natural transformation was used to introduce into ES114 a deletion allele lacking codons encoding residues 8–471 of VF_1854. This region was replaced with a cassette containing ermR flanked by FRT sites constructed using the primers listed in Table 2 according to a protocol previously described (Visick et al., 2018). The ermR cassette was excised via FLP recombinase to yield SSC014, and the resulting scar was validated by sequencing the junction region.

Table 2 Primers used in this study.
Strains EAP010 and MRE011 were generated by allelic exchange according to a protocol previously described (Miyashiro et al., 2010). For EAP010, sypG in MRE010 was replaced with a sypG deletion allele (ΔsypG) that lacks codons encoding residues 49-478 (Surrett et al., 2023). For MRE011, the large chromosomal deletion associated with MRE010 [Δ(6,288 bp)] was amplified from MRE010 genomic DNA using the primers listed in Table 2 and cloned via XhoI/SpeI into pEVS79 to yield pMRE001. Plasmid pMRE001 was used to introduce Δ(6,288 bp) into ES114 to yield MRE011.
Plasmid pMRE003 was constructed by amplifying VF_0133 from ES114 genomic DNA using PfuUltra HF polymerase (Agilent) and the primers listed in Table 2 and subcloning the product via KpnI/SalI into pTM214 to yield pMRE003.
For each sample, a culture was inoculated with an isolated colony and incubated at 28°C with shaking. Following overnight incubation, each culture was supplemented with 0.3 mL 1% (w/v) crystal violet solution and incubated at 28°C with shaking. After 30 min, the aqueous solution was removed by aspiration, and tubes were rinsed 10 times with at least 3 mL DI water. To solubilize the crystal violet retained by the tube, 5 mL 200-proof ethanol was added and incubated at room temperature, with 10-sec vortex every 15 min. After 1 h, the OD600 of a 1.0-mL volume of the solution was measured using a BioPhotometer (Eppendorf).
For each strain, a cell suspension was generated by normalizing a starter culture to OD600 = 1.0, and a 30-μL volume was used to inoculate 3 mL of the corresponding media. Cultures were grown to OD600 = 1.0 and then diluted into 50 mL filter-sterilized seawater (FSSW) at 2× desired inoculum level. To initiate the assay, this cell suspension was combined with 50 mL FSSW containing hatchling E. scolopes. After 3.5 h, animals were washed twice in 100 mL FSSW and then transferred into individual vials containing 4 mL FSSW. After ~18 h, the bioluminescence of animals was determined using a GloMax 20/20 luminometer (Promega). Animals were then anesthetized in 3% ethanol/FSSW for 15 min and then euthanized by freezing at -80°C. To determine the abundance of V. fischeri, squid were thawed on ice and homogenized in 0.70-mL 70% (v/v) FSSW, which was serially diluted and plated onto solid medium. After 24 h of incubation at 28°C, CFU were enumerated, and the resulting CFU counts were used to calculate the bacterial abundance in each squid.
Soft-agar motility plates consisted of 0.5% (w/v) tryptone, 0.3% (w/v) yeast extract, 0.25% (w/v) agar, and 70% (v/v) Instant Ocean. A 30-μL volume of starter culture was used to inoculate 3 mL SWTO, which was grown at 28°C. After 1.5 h, cultures were normalized to OD600 = 0.20. To initiate the assay, a 5-μL volume was injected into the agar, and plates were stored at 28°C until assessed for motility ring diameter. Images were taken in jpeg format using the camera associated with a Samsung Galaxy S20 cellular phone.
Genomic DNA was isolated from 0.50-mL of starter cultures of ES114 and MRE010 using the MasterPure Complete DNA and RNA Purification kit (Epicentre) according to the manufacturer instructions. For MRE010 and ES114, DNA libraries were prepared and sequenced as paired end reads at a depth of 150 Mbps using the Illumina platform (SeqCenter, LLC), and the sequencing files were analyzed by the breseq pipeline (Deatherage and Barrick, 2014), with the consensus mode run using default settings and the reference genome for ES114 (BioProject PRJNA12986). The data for this project are available at the Sequence Read Archive (SRA) of NCBI under BioProject accession PRJNA999647 with BioSample accession numbers SAMN36745556 (MRE010) and SAMN36745557 (ES114).
Whole cell lysates were prepared in the manner of Hitchcock and Brown (Hitchcock and Brown, 1983) with a few modifications. Briefly, a 100-µL sample of an overnight culture in rich medium was pelleted by spinning at 9000 × g for 2 mins. Supernatant was discarded and pellet was resuspended in 50 µL of lysing/loading buffer (0.5 mM EDTA, 2% (w/v) SDS, 2% (v/v) 2-mercaptoethanol, 0.1% (w/v) bromophenol blue, 10% (v/v) glycerol, 50 mM Tris (pH 7.5)). Lysates were then incubated at 95 °C for 10 minutes, cooled, and proteinase K (PK) was added to a final concentration of 1 µg/µL. To degrade protein, lysates with PK were incubated at 60 °C for 60 minutes. Lysates were then either immediately run on SDS-PAGE gels or frozen for later use.
Gels were prepared as described by Lesse et al. (Lesse et al., 1990) with slight modification. Briefly, two acrylamide and bisacrylamide stock solutions of 30% T, 6% C and 30% T, 2.6% C were prepared where T represents the total percentage of acrylamide and C represents the percentage of the cross-linker (bis) to the total concentration of acrylamide. The resolving gels were prepared via the addition of 30% T, 6% C stock acrylamide solution (5.44 mL), gel buffer (3 M Tris-HCl, 0.3% SDS pH 8.5)(3.3 mL), glycerol (1.3 g), and water (160 µL). Gels were polymerized with the addition of ammonium persulfate (100 µL 10% APS) and TEMED (10 µL). Stacking gels were layered on top of the polymerized resolving gels and were prepared by the addition of 30% T, 2.6% C stock acrylamide solution (0.666 mL), gel buffer (3 M Tris-HCl, 0.3% SDS pH 8.5) (2 mL), and water (4 mL) before initializing polymerization via the addition of 10% APS (66 µL) and TEMED (6.6 µL). The final lower resolving gel consisted of 16.5% T, 6% C and was overlayed with a 3% T, 2.6% C upper stacking gel.
Whole cell lysates, prepared as described above, were heated at 95 °C for 5 minutes before loading onto gels. Electrophoresis was carried out in a BioRad® Mini-PROTEAN Tetra Vertical Electrophoresis Cell with a Tris-Tricine run buffer (0.1 M Tris, 0.1 M Tricine, 0.1% SDS) and run at a constant 50 V until the dye front reached the resolving gel, after which the voltage was increased to 120 V and the gel run until ~1 hour after the dye front ran of the gel.
Following electrophoresis, the gel was washed with water, then submerged in a fix solution of 40% ethanol and 5% acetic acid with 50 rpm shaking. Following overnight incubation, the gel was rinsed with water, then submerged in a solution of 40% ethanol, 5% acetic acid, and 0.7% periodic acid for 5 mins to oxidize. The gel was then subjected to four 30-min washes with water. The gel was then stained with ammonical silver as described by Hitchcock and Brown (Hitchcock and Brown, 1983). The gel was submerged in freshly-prepared stain solution (0.67% silver nitrate, 0.1 M ammonium hydroxide, and 18 mM sodium hydroxide) for 10 mins. The stain was discarded, and the gel was subjected to four 10-min washes in water. Finally, the gel was soaked in developer solution (0.26 mM citric acid and 0.019% formaldehyde, pre-warmed to 37 °C) until bands were visible, typically 5-15 minutes, before quenching by shaking in 1% acetic acid solution. Images were taken using a GelDoc Imager (Bio-Rad).
To detect the presence of MRE010-like phenotypes within passaged populations, each frozen stock of the propagated population was accessed three times by serially diluting 10-μL samples in SWTO and plating dilutions onto SWTO agar. The resulting colonies were counted and scored for the small, rough appearance of MRE010 colonies, which were used to calculate the frequency of MRE010-like CFU. For each sample, the corresponding limit of detection was determined as 1 CFU/total CFU, and the 95th percentile of the limit of detection for each frozen stock was determined using the geometric mean and geometric standard deviation of the three replicates.
Approximately 250 μL of each frozen stock of passaged culture was subjected to DNA extraction as described above. Each PCR was conducted in a 50-μL volume using 75 ng genomic DNA, 0.5 μM of each indicated primer, 0.3 mM dNTPs, and PFU polymerase. PCR conditions included a 3-min start at 95°C, 35 cycles [each cycle: (95°C for 30 sec, 56°C for 30 sec, 75°C for 1 min)], and 75°C for 5 min. Samples were assessed by gel electrophoresis and band sizes were determined using a 100-bp ladder (NEB) sample run in parallel.
Starter cultures were normalized to OD600 = 1.0 using SWTO as a diluent. Strains were combined, and 20 μL of the resulting cell suspension was added to 125-mL baffled Erlenmeyer flasks containing 20 mL SWTO + 1 µM 3-oxo-C6-HSL. To assess relative abundance, 100-µL volumes were sampled from each flask, serially diluted, and plated onto SWTO agar for CFU. After at least 48 h of incubation at 28 °C, colonies were enumerated and then scored for the wrinkled phenotype of MRE010. Every 12 h, 20 µL of each culture was diluted into 20 mL fresh medium.
While passaging V. fischeri strain ES114 in SWTO medium supplemented with 1 μM 3-oxo-C6 HSL, we isolated a strain (designated MRE010) that exhibits phenotypes remarkably distinct from the parental strain (Figure 1). On SWTO agar, MRE010 forms small colonies that appear rough and dry relative to the smooth colonies of ES114 (Figure 1A). In liquid SWTO, MRE010 forms thick structures at the air-liquid interface (Figure 1B). In V. fischeri, these phenotypes are linked to the formation of biofilms, and because biofilm formation is important for the ability of V. fischeri to establish symbiosis with E. scolopes, we further investigated these phenotypes of MRE010.
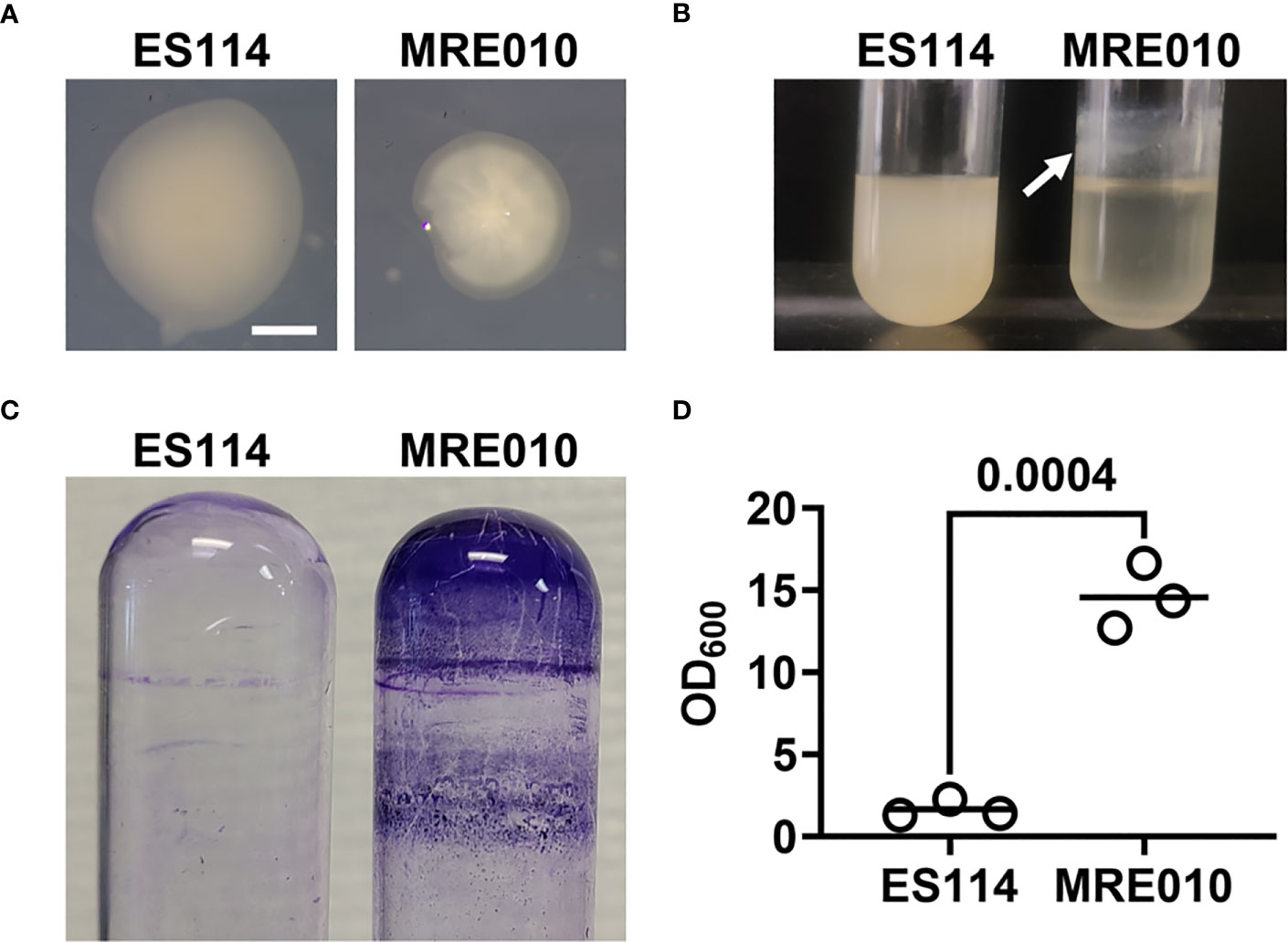
Figure 1 Biofilm properties of MRE010. (A) Colony morphology of indicated strains on SWTO agar after 48 h incubation at 28°C. (B) Liquid culture growth of indicated strains after 18 h incubation at 28°C. Arrow indicates ring at air-liquid interface. (C) Crystal violet staining of representative cultures grown as shown in (B). (D) Quantification of crystal violet staining for biofilm assay. Each point represents an individual culture (N = 3), and bar indicates group mean. P-value from two-tailed unpaired t-test.
To determine whether the structures MRE010 forms at the air-liquid interface are biofilms, we first used a biofilm assay based on staining spent culture tubes with crystal violet. Tubes that contained MRE010 cultures retained 15-fold more crystal violet than WT cells, which suggests growth of MRE010 results in more biomass adhered to the glass surface (Figures 1C, D). Taken together, these results suggest that MRE010 is an ES114-derived strain that can readily form biofilms.
Prior to entering the squid host, V. fischeri cells express Syp, which promotes their aggregation along the mucus-lined surface of the light organ (Yip et al., 2005). In culture, the production of Syp leads to increased biofilm formation, which includes the ability to form wrinkled colonies on solid medium and thick structures at air-liquid interface in liquid medium (Yip et al., 2005). In addition, ES114 cells engineered to overproduce Syp exhibits increased colonization of the squid host (Yip et al., 2006). To determine whether MRE010 also displays an increased capacity to colonize E. scolopes, we conducted squid colonization assays with this strain.
A 3.5-h exposure to a seawater inoculum of ES114 containing approximately 1,700 CFU/mL will result in half of hatchling population becoming bioluminescent by 24 h post-inoculation (p.i.) (Donnelly et al., 2023). However, we found that hatchlings exposed to inoculums of MRE010 exceeding 105 CFU/mL failed to emit bioluminescence (Figure 2). Examination of homogenized tissue for CFU revealed that the squid that did contain MRE010 had bacterial levels at least 10-fold lower than squid colonized with ES114 (Figure 2). These data suggest that MRE010 shows low symbiotic capacity despite possessing the trait of increased biofilm formation.
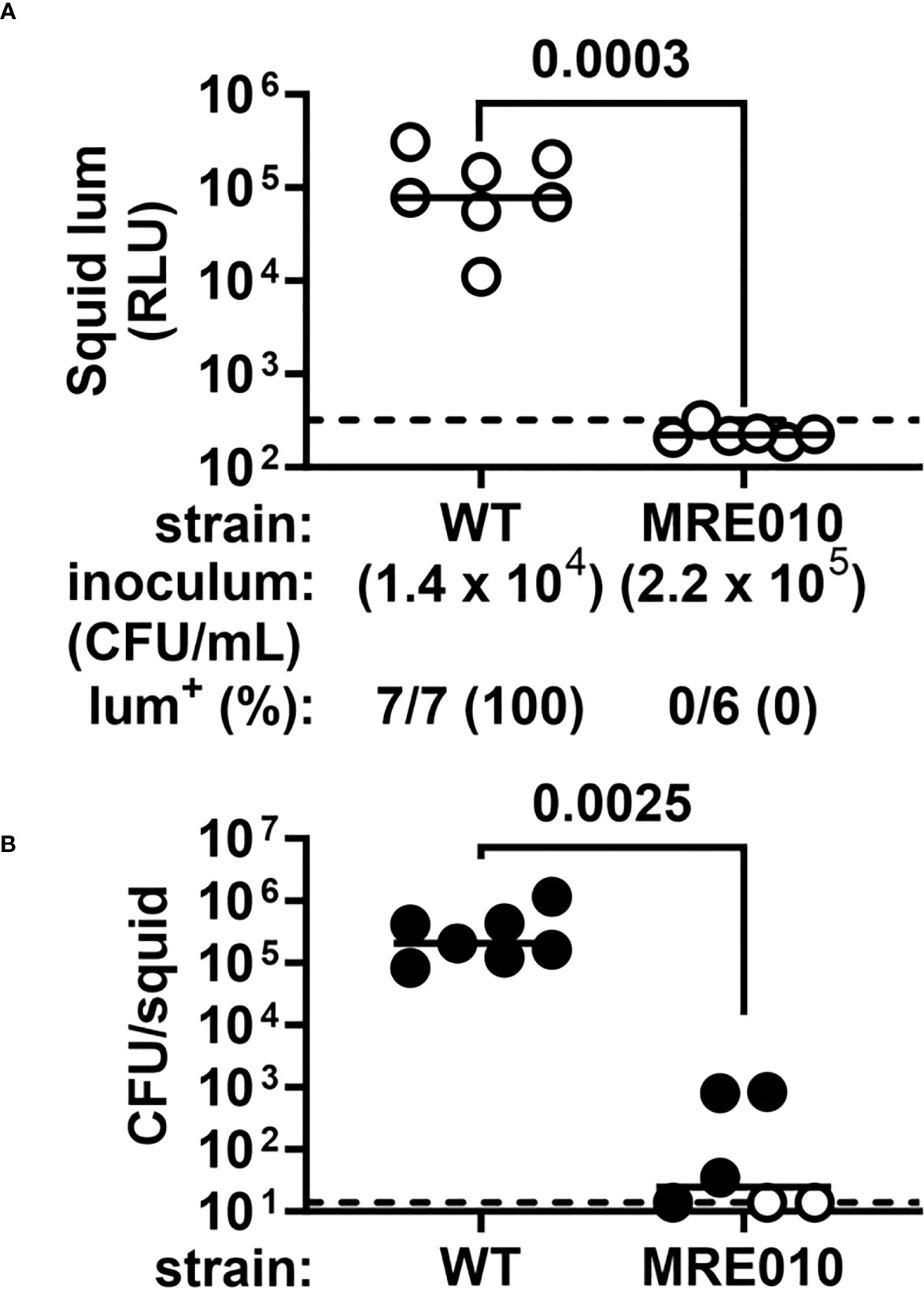
Figure 2 Squid colonization properties of MRE010. (A) Luminescence of squid at 24 h p.i. following a 3.5-h exposure to the indicated inoculum. Each point represents an individual animal, and each bar indicates group median. Dotted line indicates bioluminescence cutoff used to score whether a squid is bioluminescent (lum+). P-value from two-tailed two-proportion Z-test. (B) Abundance of indicated strain in squid shown in panel (A). Each point represents an individual animal, with closed (open) symbols indicating presence (absence) of CFU. Dotted line indicates limit of detection (14 CFU). P-value from two-tailed Mann-Whitney test of CFU+ squid.
Because MRE010 forms biofilms but exhibits a colonization defect, we next asked whether the biofilms produced by MRE010 are associated with Syp. The syp locus features 18 genes encoding factors involved in polysaccharide production, including several regulators that control expression of the syp locus. One regulatory factor is the bacterial enhancer binding protein SypG, which activates four σ54-dependent promoters located within the locus and is necessary for Syp-dependent biofilms (Ray et al., 2013). However, an MRE010 ΔsypG strain produces biofilms that are indistinguishable from MRE010 (Figure 3), which suggests the biofilms produced by MRE010 are independent of Syp.
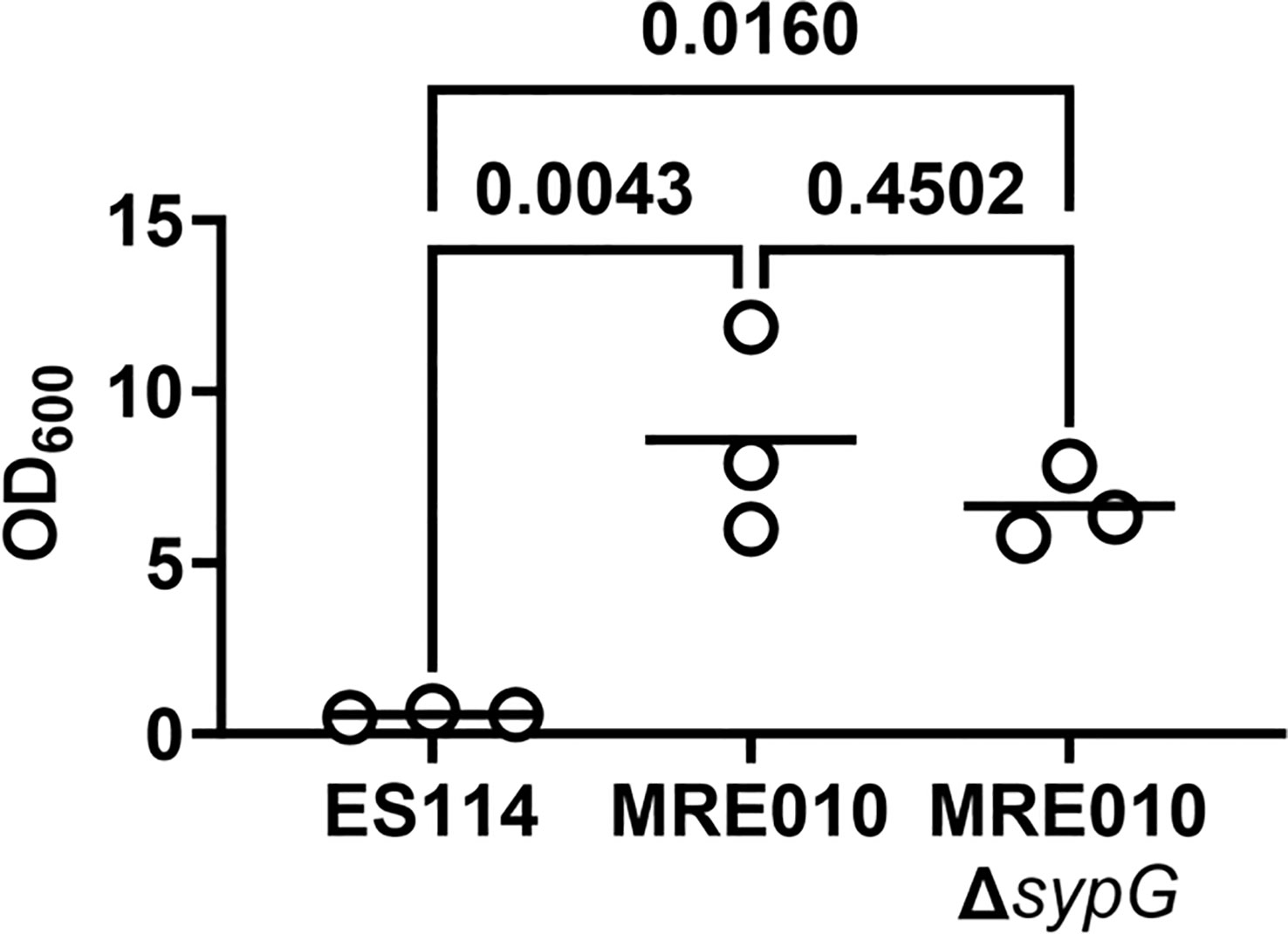
Figure 3 Biofilm properties of MRE010 are independent of Syp. Quantification of crystal violet staining for biofilm assay of indicated strains. Each point represents an individual culture (N = 3), and bar indicates group mean. One-way ANOVA test suggests group means are statistically different (F = 15.47, p-value = 0.0043), with indicated p-values from a Tukey’s post hoc test adjusted for multiple comparisons.
In addition to Syp-dependent biofilm formation, the ability for V. fischeri to establish symbiosis depends on cellular motility, which is important for cells to enter the light organ through pores, traverse the corresponding ducts, and access the designated colonization sites known as crypt spaces (Graf et al., 1994; Essock-Burns et al., 2020). Previous work has shown that amotile mutants exhibit a severe colonization defect, typified by low bacterial abundance (Brennan et al., 2013). The low symbiotic capacity of MRE010 prompted us to next ask whether MRE010 can swim like ES114. Using soft-agar motility assays, we found that in contrast to ES114, MRE010 forms a ring that is comparable to the amotile ΔflrC strain (Figure 4), which suggests that MRE010 has a severe motility defect. Taken together, the inability of MRE010 to swim provides a possible mechanism underlying its colonization defect.
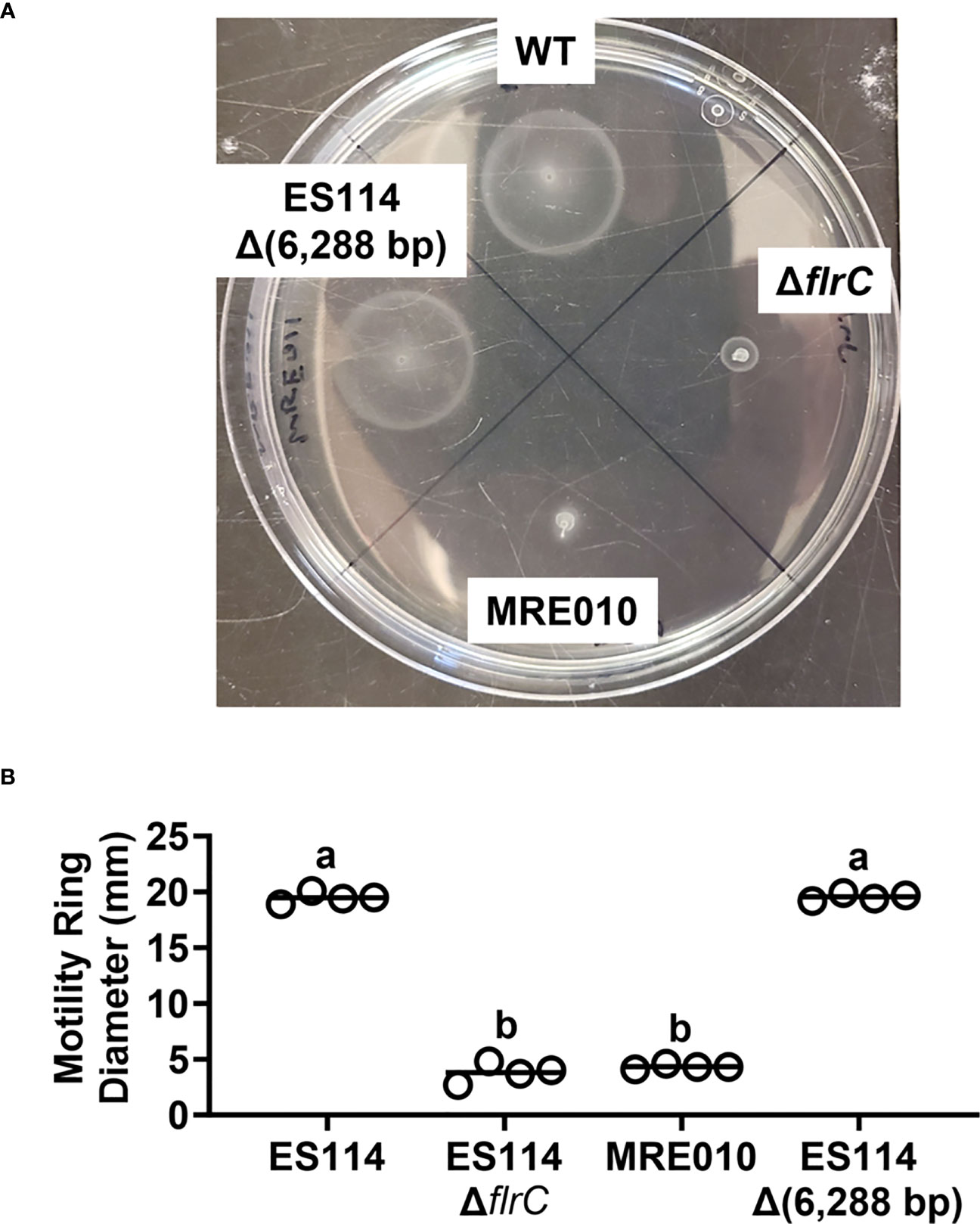
Figure 4 Motility properties of MRE010. (A) Representative soft-agar motility plate with indicated strains after 5.5 h of incubation at 24°C. (B) Motility ring diameter of indicated strains 5.5 h p.i. Each point represents an individual biological replicate (N = 4). One-way ANOVA test suggests group means are statistically different (F3,12 = 1122, p-value < 0.0001). Letters show different statistical groups from Tukey’s post hoc test (same letters: p ≥ 0.05, a/b: p < 0.0001, with p-values adjusted for multiple comparisons).
To investigate the mechanisms underlying the various phenotypes of MRE010, we sequenced its genome and compared the reads to that of ES114 sequenced in parallel. Overall, only six loci featured mutations relative to the parental strain (Table 3): a large 6,288-bp deletion (Δ6,288) linked to the lux operon, a nonsynonymous mutation in VF_0133, a synonymous point mutation in chiP, and three mutations located in intergenic regions.

Table 3 MRE010-specific mutations identified by breseq.
The large deletion was of interest because the corresponding region contains luxI and luxR, as well as luxC and part of luxD. Indeed, cultures of MRE010 showed no bioluminescence, including when the medium was supplemented with 3-oxo-C6 HSL (Figure S1), which suggests that MRE010 is a dark strain and likely explains why juveniles that become colonized with MRE010 do not produce bioluminescence.
To determine whether other phenotypes of MRE010 described above depend on the large deletion, we introduced the Δ6,288 allele into ES114 and assessed the resulting mutant (MRE011) for both biofilm formation and motility. However, MRE011 fails to produce biofilms (Figure S2) and displays wild-type motility (Figure 4), which suggests that the corresponding traits of MRE010 are independent of this large deletion.
To identify the genetic factor(s) of MRE010 that result in its biofilm phenotypes, we considered mutations aside from the large deletion (Table 3). Out of the remaining five mutations identified in MRE010, the point mutation within VF_0133 piqued our interest because VF_0133 is predicted to encode a glycosyltransferase and the corresponding mutation conferred a nonsynonymous point mutation (I125F). Initial attempts with BLAST to identify proteins with primary structures similar to VF_0133 revealed only uncharacterized homologs primarily in other Vibrionaceae species. However, analysis by HHpred (Zimmermann et al., 2018) identified structural orthologs of VF_0133 in the GT-4 family that feature a Rossmann-like fold (GT-B clan) (Liu and Mushegian, 2003). Indeed, the predicted structure of VF_0133 exhibits features associated with other GT-B glycosyltransferases, e.g., BshA of Staphylococcus aureus (PDB: 6D9T)(Royer and Cook, 2019), including a β/α/β-Rossmann fold as well as putative residues that comprise the active site (G13, K209, Q260, E280, and E288), despite low overall sequence identity (Figures S3A, B).
VF_0133 is located downstream of VF_0134 and VF_0135 (Figure 5A), which are predicted to function in lipopolysaccharide (LPS) biosynthesis through the synthesis of a heptose-containing sugar and its incorporation into the inner core, respectively. To test whether LPS structure is affected in MRE010, we examined whole-cell extracts by SDS-PAGE separation and silver staining (Figure 5B). As controls for specific LPS structures, we included E. coli strains that are derived from BL21(DE3), which produces a rough LPS chemotype that lacks the O-antigen. TXM331 and TXM333 both have deletions of eptA and arnA, which results in decreased variability in LPS structure due to the inability to modify lipid A with phosphoethanolamine and 4-deoxy-L-aminoarabinose, respectively. TXM333 also contains a deletion of lpcA, which encodes D-sedoheptulose 7-phosphate isomerase. Consequently, TXM333 produces Re LPS, which lacks all sugars on the saccharide core except 3-deoxy-α-D-manno-oct-2-ulosonic acid (Kdo). Wild-type V. fischeri samples feature two major bands that are consistent in size with an unmodified core or the addition of a single O-antigen repeating unit, which has been previously characterized for V. fischeri ES114 LPS (Post et al., 2012). MRE010 samples lack the high molecular weight band and have a smaller LPS core that migrates in line with deep rough Escherichia coli LPS core mutants. This result suggests that the VF_0133 variant prevents the addition of O-antigen, most likely due to the loss of the attachment point for an outer core sugar. Because this band typically corresponds to the oligosaccharide core and O-antigen, this result suggests that MRE010 is unable to produce full-length LPS.
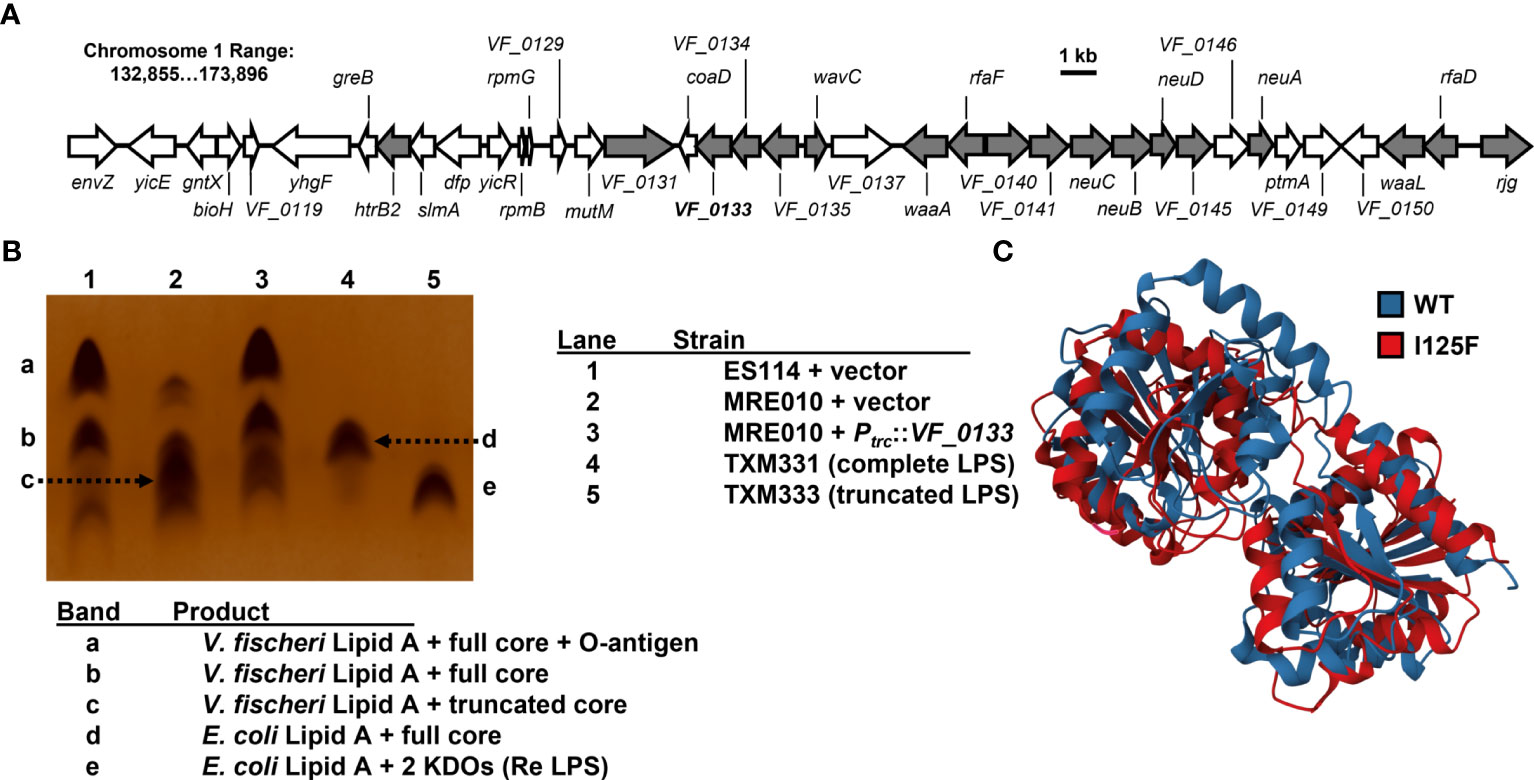
Figure 5 Impact of VF_0133 on LPS structure. (A) Genetic locus of VF_0133 on chromosome 1 with genes predicted to encode factors that promote LPS biosynthesis shown in grey. (B) Silver stain of whole-cell lysates for indicated strains. Lanes 4-5 correspond to E. coli size standards with complete (TXM331) and truncated (Re LPS, lacking heptose) LPS chemotypes. (C) Overlay of predicted structures of WT (blue) and I125F variant (red) of VF_0133.
C-I-TASSER analysis (Zheng et al., 2021) of VF_0133 suggests that I125 interacts with several other residues through noncovalent bonds to support formation of a particular tertiary structure (Figure 5C), and the I125F substitution greatly alters the predicted structure. Consequently, we hypothesized that the VF_0133I125F allele of MRE010 encodes a nonfunctional glycosyltransferase. To test this hypothesis, we initially attempted to introduce a deletion allele of VF_0133 into ES114 using standard genetic approaches but were unable to recover a ΔVF_0133 strain. Consistent with this observation, a transposon mutant library initially containing mutants with insertions within VF_0133 quickly became attenuated after propagating the library in culture (Brooks et al., 2014). Therefore, as an alternative way to test the VF_0133I125F allele, we expressed the wild-type copy of VF_0133 in MRE010 and examined its physiology using the various assays described above.
Colonies of MRE010 harboring the wild-type copy of VF_0133 exhibit smooth morphology comparable to those of ES114 (Figure 6A). In addition, the wild-type copy of VF_0133 in MRE010 abrogated the biofilm structures that MRE010 forms at the air-liquid interface of cultures (Figure 6B). Taken together, these data indicate that the VF_0133(I125F) allele is recessive to the wild-type allele and suggests that the biofilms produced by MRE010 arise due to its inability to properly synthesize LPS.
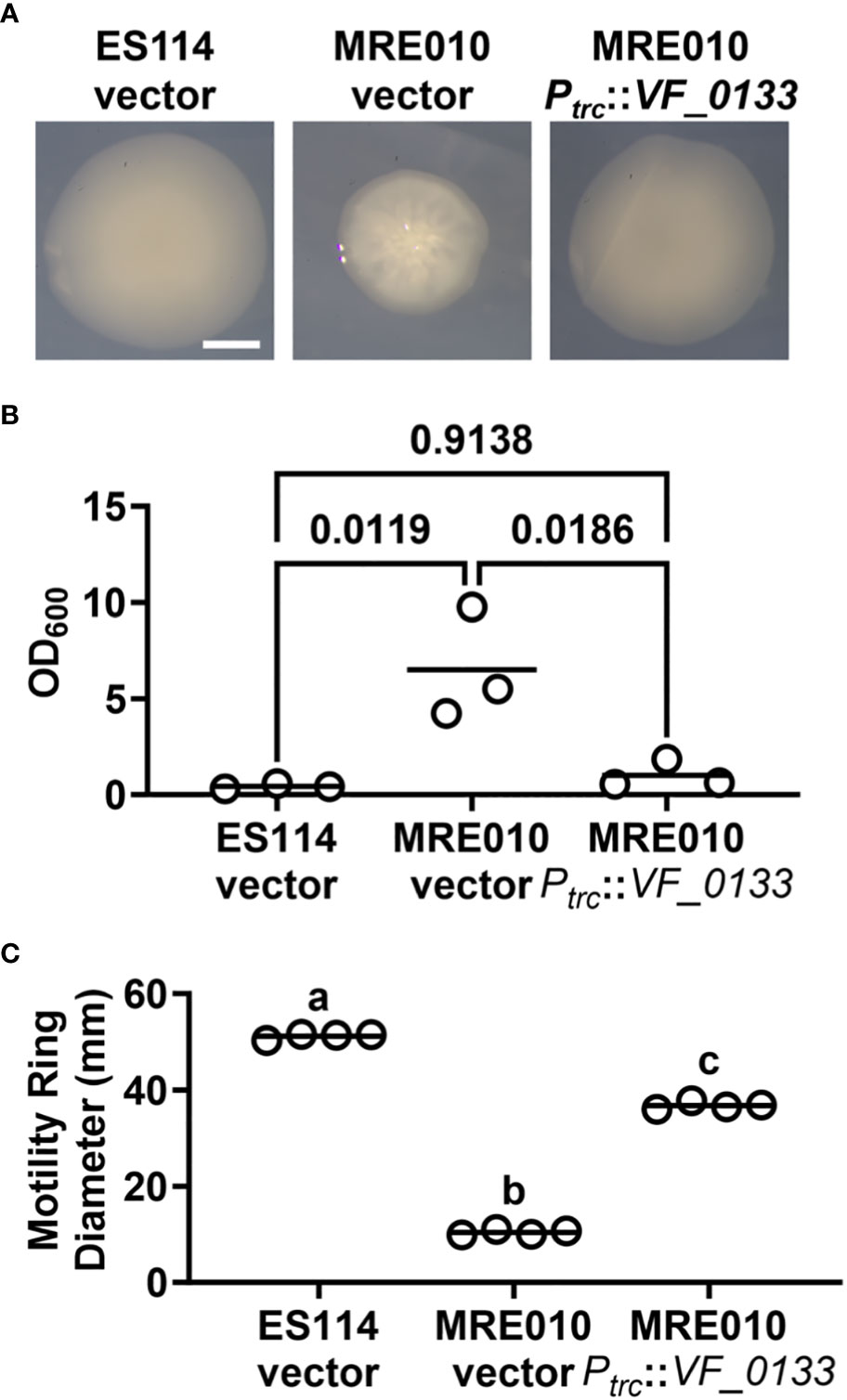
Figure 6 Dependence of MRE010 physiology on VF_0133. (A) Colony morphology of indicated strains on SWTO agar after 48 h incubation at 28°C. (B) Crystal violet staining of cultures for indicated strains. Each point represents an individual culture (N = 3), and each bar represents the group mean. One-way ANOVA test suggests group means are statistically different (F2,6 = 11.34, p = 0.0092). Indicated p-values are from Tukey’s post hoc test and adjusted for multiple comparisons. (C) Motility ring diameter of indicated strains at 6 h p.i. Each point represents an individual biological replicate (N = 4), and each bar represents the group mean. One-way ANOVA test suggests group means are statistically different (F2,9 = 4,166, p-value < 0.0001). Letters show different statistical groups from Tukey’s post hoc test (different groups: p < 0.0001, with p-values adjusted for multiple comparisons).
We also examined whether the motility defect of MRE010 is also linked to VF_0133. Expression of the wild-type copy of VF_0133 in MRE010 resulted in the formation of a motility ring only slightly smaller than ES114 (Figure 6C), which suggests that while most of the motility defect of MRE010 is also due to improper LPS synthesis other factors in MRE010 likely contribute as well. Finally, we assessed whether the colonization defect of MRE010 depends on VF_0133. Using an inoculum consisting of MRE010 cells harboring the wild-type copy of VF_0133 resulted in animals failing to produce bioluminescence but containing bacteria (Figure 7), which suggests that the structure of LPS altered by VF_0133(I125F) impaired the ability of V. fischeri to colonize the light organ.
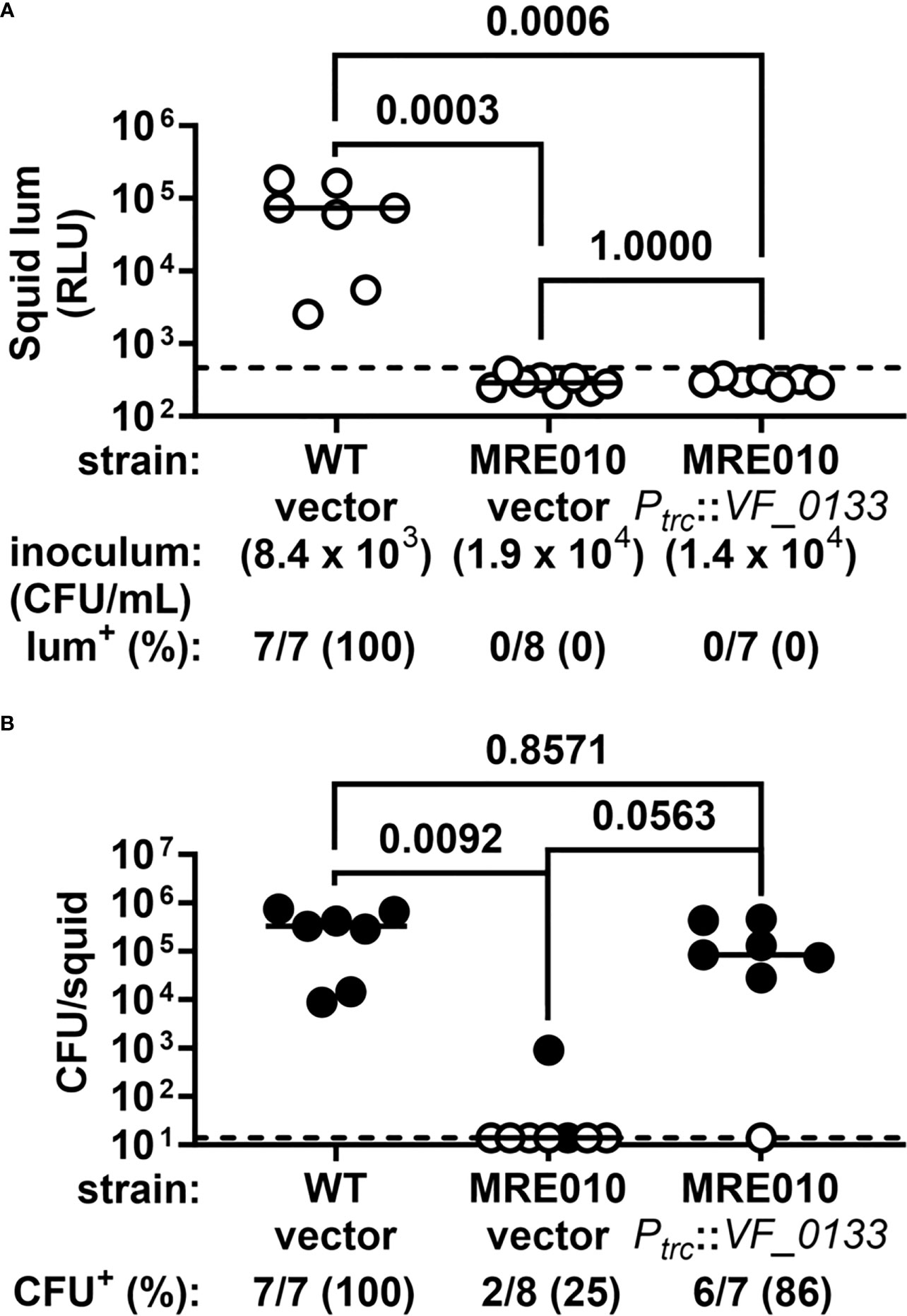
Figure 7 VF_0133 promotes colonization by V. fischeri. (A) Luminescence of squid at 24 h p.i. following a 3.5-h exposure to the indicated inoculum. Each point represents an individual animal, and each bar indicates group median. Dotted line indicates bioluminescence cutoff used to score whether a squid is bioluminescent (lum+). P-values from two-tailed two-proportion Z-test and adjusted for multiple comparisons (Bonferroni). (B) Abundance of indicated strain in squid shown in (A). Each point represents an individual animal, with closed (open) symbols indicating presence (absence) of CFU. Dotted line indicates limit of detection (14 CFU). P-values from two-tailed two-proportion Z-test and adjusted for multiple comparisons (Bonferroni).
As described above, MRE010 arose during the propagation of ES114. To determine when the biofilm-related allele appeared, we assessed the stocks of culture samples that were generated at earlier stages of propagation for the presence of MRE010-like phenotypes. Examination of the resulting CFU demonstrated that the MRE010-like CFU emerged between Generations 106 and 158, and then this allele swept the population by Generation 211 (Figure 8A). We next determined when the 6,288-bp deletion appeared in the population by using primers that anneal to either side of the junction formed by the large deletion. Endpoint PCR suggests that the 6,288-bp deletion appeared between Generations 53 and 106. Taken together, these data suggest that the large deletion is ancestral to the mutation within VF_0133 (Figure 8B).
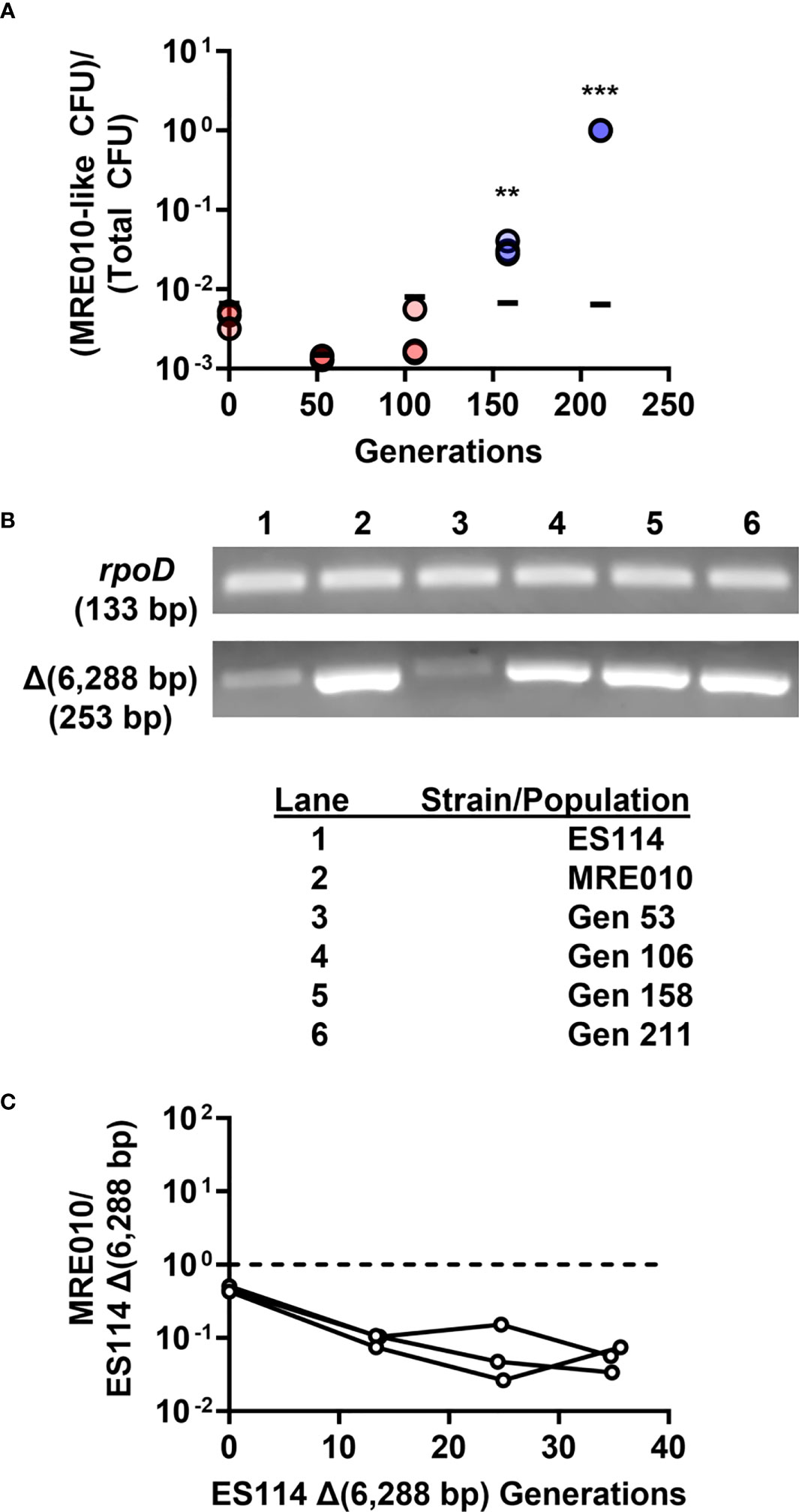
Figure 8 MRE010 exhibits lower fitness than its ancestor. (A) Frequency of CFU exhibiting MRE010-like colony morphology (small, rough colonies) in propagated culture. Each point represents an independent replicate of each stock (N = 3), with red (blue) symbols indicating lower (higher) frequency than the limit of detection. Bars represent the 95th percentile of the limit of detection established by replicates of each stock. Two-tailed t-test was performed for each group relative to limit of detection with p-values adjusted with Bonferroni correction for multiple tests (** = p < 0.01, *** = p < 0.001). (B) Endpoint PCR designed to detect junction generated by 6,288-bp deletion. (C) Relative abundance of MRE010 to MRE011 [ES114 Δ(6,288 bp)] in SWTO + 1 μM 3-oxo-C6 HSL. Each line represents independent culture (N = 3).
Because the MRE010-like phenotype had swept the population by Generation 211 (Figure 8A), we also examined the fitness of MRE010 to better understand how it emerged within the propagated culture. To assess fitness, we co-cultured MRE010 with the ES114-derived strain containing the 6,288-bp deletion, which our results described above suggest is similar to the ancestor of MRE010. However, the relative abundance of MRE010 decreased over time (Figure 8C), which suggests that the fitness of MRE010 is lower than that of the putative ancestor strain under these conditions and does not explain how MRE010 emerged in the original population unresolved.
In this study, we isolated mutant MRE010 following serial passaging of ES114 in the presence of 3-oxo-C6 HSL. Supplementing media with 3-oxo-C6 HSL induces bioluminescence production, which is the symbiotic trait of V. fischeri when housed in the light organ of E. scolopes (Miyashiro and Ruby, 2012). Because of the considerable energetic cost of bioluminescence production (Bose et al., 2008), the emergence of a dark strain while propagating ES114 in 3-oxo-C6 HSL was not unexpected, although the mechanisms by which bioluminescence production and its regulation evolve remain poorly understood. While only a limited number of mutations accumulated within MRE010 (Table 3), their impact on V. fischeri physiology was significant. The large 6,288-bp deletion that included part of the lux locus abrogated bioluminescence production (Figure S1), likely leading to an increase in fitness in the presence of autoinducer previously reported for dark strains (Bose et al., 2008). The large deletion resulted in MRE010 lacking luxI and luxR, which prevents MRE010 from synthesizing 3-oxo-C6 HSL and responding to the autoinducer by expressing the lux operon. MRE010 retains the luxAB genes that encode luciferase, but it likely cannot express the enzyme due to the large deletion having also eliminated the promoter region of the lux operon. These observations support the idea that the loss of lux genes represents one trajectory that permits V. fischeri to adapt to the circumstances of sustained autoinduction. We anticipate other trajectories are possible but their discovery requires further investigation.
Perhaps the most striking phenotype of MRE010 is its propensity to produce biofilms in culture, which we have linked to an allele of VF_0133. VF_0133 appears to play an important role in LPS biosynthesis, with its gene linkage to putative heptosyltransferase (VF_0134) and heptose-modifying glucosyltransferase (VF_0135) enzymes. A previous analysis of the LPS composition in V. fischeri revealed that L-glycero-D mannoheptose is one of the major sugars within the core polysaccharide (Post et al., 2012). Consequently, VF_0133 gene synteny raises the possibility that VF_0133 contributes to glyco-modification of the core, but future work is necessary to elucidate its enzymatic role.
LPS plays a significant role as a signaling molecule as the symbiosis between E. scolopes and V. fischeri initially forms. Colonization of the light organ by V. fischeri induces morphological changes within the symbiotic organ, with its LPS and a disaccharide-tetrapeptide monomer of peptidoglycan synergistically triggering cellular apoptosis within the superficial ciliated epithelium (Koropatnick et al., 2004). In addition, such immunogenic LPS is thought to be released during flagellar rotation (Brennan et al., 2014), which further promotes symbiont-dependent development of the light organ. Previous studies have demonstrated that factors that affect the acylation pattern of lipid A structure (htrB1 and msbB) or attach O-antigen (waaL) are important for symbiosis, as corresponding mutants show attenuated abundance in hatchlings (Adin et al., 2008; Post et al., 2012). Given that VF_0133 likely contributes to synthesis of LPS core, the large colonization defect associated with MRE010 provides some preliminary evidence of its involvement in establishing symbiosis. However, caution is warranted as the pleiotropy of this strain, e.g., its motility defect, likely also contributes to its lower capacity in colonizing the light organ. Thus, additional work with clean mutants of VF_0133 and other factors predicted to affect LPS core synthesis is necessary to clarify their roles in establishing the symbiosis.
How MRE010 became the dominant strain in the population remains an enigma. One possibility may be due to the medium used while propagating the culture. SWTO is an unbuffered medium, and previous work found that growth of ES114 in a related medium (SWT) leads to acidification that subsequently kills off wild-type cells (Studer et al., 2008). Furthermore, it is unclear whether mutations in luxI or other components of the lux operon predispose populations to accumulate secondary mutations, such as that in VF_0133. Future work will explore this possibility to better understand the evolutionary trajectory taken by the population that yielded MRE010.
In summary, this study reports on the traits, and their underlying genetic causes, for a strain of V. fischeri that was isolated while propagating a wild-type population. While the strain could produce a novel biofilm, its altered physiology greatly interfered with the ability of V. fischeri to establish symbiosis. Thus, this report serves as the foundation for a new direction to examine the mechanisms that promote the acquisition of bacterial symbionts by animal hosts.
The data presented in the study are deposited in the Sequence Read Archive (SRA) of NCBI, BioProject accession number PRJNA999647.
The animal study was approved by Penn State University IACUC (protocol # PROTO202101789). The study was conducted in accordance with the local legislation and institutional requirements.
EP: Conceptualization, Data curation, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. ME: Conceptualization, Data curation, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. AC: Investigation, Methodology, Writing – review & editing, Investigation, Methodology, Writing – review & editing. SC: Methodology, Writing – review & editing. MM: Data curation, Investigation, Methodology, Writing – review & editing. TCM: Methodology, Supervision, Writing – review & editing. TIM: Conceptualization, Formal Analysis, Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing.
This work was supported by the National Institute of General Medical Sciences Grant R01 GM129133 (to TIM) and by the National Institute of Allergy and Infectious Diseases Grant R21 AI138152 (to TCM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author TIM declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbrio.2023.1254305/full#supplementary-material
Adin D. M., Phillips N. J., Gibson B. W., Apicella M. A., Ruby E. G., Mcfall-Ngai M. J., et al. (2008). Characterization of htrB and msbB mutants of the light organ symbiont Vibrio fischeri. Appl. Environ. Microbiol. 74 (3), 633–644. doi: 10.1128/AEM.02138-07
Bose J. L., Rosenberg C. S., Stabb E. V. (2008). Effects of luxCDABEG induction in Vibrio fischeri: enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch. Microbiol. 190 (2), 169–183. doi: 10.1007/s00203-008-0387-1
Brennan C. A., Hunt J. R., Kremer N., Krasity B. C., Apicella M. A., Mcfall-Ngai M. J., et al. (2014). A model symbiosis reveals a role for sheathed-flagellum rotation in the release of immunogenic lipopolysaccharide. Elife 3, e01579. doi: 10.7554/eLife.01579
Brennan C. A., Mandel M. J., Gyllborg M. C., Thomasgard K. A., Ruby E. G. (2013). Genetic determinants of swimming motility in the squid light-organ symbiont Vibrio fischeri. Microbiologyopen 2 (4), 576–594. doi: 10.1002/mbo3.96
Brooks J. F. 2nd, Gyllborg M. C., Cronin D. C., Quillin S. J., Mallama C. A., Foxall R., et al. (2014). Global discovery of colonization determinants in the squid symbiont Vibrio fischeri. Proc. Natl. Acad. Sci. U.S.A. 111 (48), 17284–17289. doi: 10.1073/pnas.1415957111
Cecere A. G., Cook R. A., Miyashiro T. I. (2023). A case study assessing the impact of mating frequency on the reproductive performance of the Hawaiian bobtail squid Euprymna scolopes. Lab. Anim. Res. 39 (1), 17. doi: 10.1186/s42826-023-00168-1
Cecere A. G., Miyashiro T. I. (2022). Impact of transit time on the reproductive capacity of Euprymna scolopes as a laboratory animal. Lab. Anim. Res. 38 (1), 25. doi: 10.1186/s42826-022-00135-2
Christensen D. G., Visick K. L. (2020). Vibrio fischeri: laboratory cultivation, storage, and common phenotypic assays. Curr. Protoc. Microbiol. 57 (1), e103. doi: 10.1002/cpmc.103
Deatherage D. E., Barrick J. E. (2014). Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 1151, 165–188. doi: 10.1007/978-1-4939-0554-6_12
Dial C. N., Fung B. L., Visick K. L. (2023). Genetic Analysis Reveals a Requirement for the Hybrid Sensor Kinase RscS in para-Aminobenzoic Acid/Calcium-Induced Biofilm Formation by Vibrio fischeri. J. Bacteriol. 205 (7), e0007523. doi: 10.1128/jb.00075-23
Donnelly A. R., Giacobe E. J., Cook R. A., Francis G. M., Buddle G. K., Beaubrun C. L., et al. (2023). Quantification of the Capacity of Vibrio fischeri to Establish Symbiosis with Euprymna scolopes. PloS One 18 (7), e0287519. doi: 10.1371/journal.pone.0287519
Essock-Burns T., Bongrand C., Goldman W. E., Ruby E. G., Mcfall-Ngai M. J. (2020). Interactions of symbiotic partners drive the development of a complex biogeography in the squid-vibrio symbiosis. mBio 11 (3), e00853-20. doi: 10.1128/mBio.00853-20
Ganesan R., Wierz J. C., Kaltenpoth M., Flórez L. V. (2022). How it all begins: bacterial factors mediating the colonization of invertebrate hosts by beneficial symbionts. Microbiol. Mol. Biol. Rev. 86 (4), e0012621. doi: 10.1128/mmbr.00126-21
Graf J., Dunlap P. V., Ruby E. G. (1994). Effect of transposon-induced motility mutations on colonization of the host light organ by Vibrio fischeri. J. Bacteriol. 176 (22), 6986–6991. doi: 10.1128/jb.176.22.6986-6991.1994
Graf J., Ruby E. G. (1998). Host-derived amino acids support the proliferation of symbiotic bacteria. Proc. Natl. Acad. Sci. U.S.A. 95 (4), 1818–1822. doi: 10.1073/pnas.95.4.1818
Hitchcock P. J., Brown T. M. (1983). Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 154 (1), 269–277. doi: 10.1128/jb.154.1.269-277.1983
Jones B. W., Nishiguchi M. K. (2004). Counterillumination in the Hawaiian bobtail squid, Euprymna scolopes Berry (Mollusca:Cephalopoda). Mar. Biol. 144, 1151–1155. doi: 10.1007/s00227-003-1285-3
Komazin G., Maybin M., Woodard R. W., Scior T., Schwudke D., Schombel U., et al. (2019). Substrate structure-activity relationship reveals a limited lipopolysaccharide chemotype range for intestinal alkaline phosphatase. J. Biol. Chem. 294 (50), 19405–19423. doi: 10.1074/jbc.RA119.010836
Koropatnick T. A., Engle J. T., Apicella M. A., Stabb E. V., Goldman W. E., Mcfall-Ngai M. J. (2004). Microbial factor-mediated development in a host-bacterial mutualism. Science 306 (5699), 1186–1188. doi: 10.1126/science.1102218
Lesse A. J., Campagnari A. A., Bittner W. E., Apicella M. A. (1990). Increased resolution of lipopolysaccharides and lipooligosaccharides utilizing tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis. J. Immunol. Methods 126 (1), 109–117. doi: 10.1016/0022-1759(90)90018-q
Liu J., Mushegian A. (2003). Three monophyletic superfamilies account for the majority of the known glycosyltransferases. Protein Sci. 12 (7), 1418–1431. doi: 10.1110/ps.0302103
Mandel M. J., Stabb E. V., Ruby E. G. (2008). Comparative genomics-based investigation of resequencing targets in Vibrio fischeri: focus on point miscalls and artefactual expansions. BMC Genomics 9, 138. doi: 10.1186/1471-2164-9-138
Miyashiro T., Klein W., Oehlert D., Cao X., Schwartzman J., Ruby E. G. (2011). The N-acetyl-D-glucosamine repressor NagC of Vibrio fischeri facilitates colonization of Euprymna scolopes. Mol. Microbiol. 82 (4), 894–903. doi: 10.1111/j.1365-2958.2011.07858.x
Miyashiro T., Ruby E. G. (2012). Shedding light on bioluminescence regulation in Vibrio fischeri. Mol. Microbiol. 84 (5), 795–806. doi: 10.1111/j.1365-2958.2012.08065.x
Miyashiro T., Wollenberg M. S., Cao X., Oehlert D., Ruby E. G. (2010). A single qrr gene is necessary and sufficient for LuxO-mediated regulation in Vibrio fischeri. Mol. Microbiol. 77 (6), 1556–1567. doi: 10.1111/j.1365-2958.2010.07309.x
Nyholm S. V., Mcfall-Ngai M. J. (2021). A lasting symbiosis: how the Hawaiian bobtail squid finds and keeps its bioluminescent bacterial partner. Nat. Rev. Microbiol. 19 (10), 666–679. doi: 10.1038/s41579-021-00567-y
Nyholm S. V., Stabb E. V., Ruby E. G., Mcfall-Ngai M. J. (2000). Establishment of an animal-bacterial association: recruiting symbiotic vibrios from the environment. Proc. Natl. Acad. Sci. U.S.A. 97 (18), 10231–10235. doi: 10.1073/pnas.97.18.10231
Post D. M., Yu L., Krasity B. C., Choudhury B., Mandel M. J., Brennan C. A., et al. (2012). O-antigen and core carbohydrate of Vibrio fischeri lipopolysaccharide: composition and analysis of their role in Euprymna scolopes light organ colonization. J. Biol. Chem. 287 (11), 8515–8530. doi: 10.1074/jbc.M111.324012
Ray V. A., Eddy J. L., Hussa E. A., Misale M., Visick K. L. (2013). The syp enhancer sequence plays a key role in transcriptional activation by the sigma54-dependent response regulator SypG and in biofilm formation and host colonization by Vibrio fischeri. J. Bacteriol. 195 (23), 5402–5412. doi: 10.1128/JB.00689-13
Royer C. J., Cook P. D. (2019). A structural and functional analysis of the glycosyltransferase BshA from Staphylococcus aureus: Insights into the reaction mechanism and regulation of bacillithiol production. Protein Sci. 28 (6), 1083–1094. doi: 10.1002/pro.3617
Schaefer A. L., Val D. L., Hanzelka B. L., Cronan J. E. Jr., Greenberg E. P. (1996). Generation of cell-to-cell signals in quorum sensing: acyl homoserine lactone synthase activity of a purified Vibrio fischeri LuxI protein. Proc. Natl. Acad. Sci. U.S.A. 93 (18), 9505–9509. doi: 10.1073/pnas.93.18.9505
Schwartzman J. A., Koch E., Heath-Heckman E. A., Zhou L., Kremer N., Mcfall-Ngai M. J., et al. (2015). The chemistry of negotiation: rhythmic, glycan-driven acidification in a symbiotic conversation. Proc. Natl. Acad. Sci. U.S.A. 112 (2), 566–571. doi: 10.1073/pnas.1418580112
Shibata S., Yip E. S., Quirke K. P., Ondrey J. M., Visick K. L. (2012). Roles of the structural symbiosis polysaccharide (syp) genes in host colonization, biofilm formation, and polysaccharide biosynthesis in Vibrio fischeri. J. Bacteriol. 194 (24), 6736–6747. doi: 10.1128/JB.00707-12
Stabb E. V., Ruby E. G. (2002). RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol. 358, 413–426. doi: 10.1016/S0076-6879(02)58106-4
Stabb E. V., Visick K. L. (2013). “Vibrio fischeri: A Bioluminescent Light-Organ Symbiont of the Bobtail Squid Euprymna scolopes,” in The Prokaryotes - Prokaryotic Biology and Symbiotic Associations. Eds. Rosenberg E., Delong E. F., Stackebrand E., Lory S., Thompson F. (Berlin Heidelberg: Springer-Verlag).
Studer S. V., Mandel M. J., Ruby E. G. (2008). AinS quorum sensing regulates the Vibrio fischeri acetate switch. J. Bacteriol. 190 (17), 5915–5923. doi: 10.1128/JB.00148-08
Surrett E. D., Guckes K. R., Cousins S., Ruskoski T. B., Cecere A. G., Ludvik D. A., et al. (2023). Two enhancer binding proteins activate sigma(54)-dependent transcription of a quorum regulatory RNA in a bacterial symbiont. Elife 12, e78544. doi: 10.7554/eLife.78544
Tischler A. H., Lie L., Thompson C. M., Visick K. L. (2018). Discovery of calcium as a biofilm-promoting signal for vibrio fischeri reveals new phenotypes and underlying regulatory complexity. J. Bacteriol. 200 (15), e00016-18. doi: 10.1128/JB.00016-18
Visick K. L., Hodge-Hanson K. M., Tischler A. H., Bennett A. K., Mastrodomenico V. (2018). Tools for rapid genetic engineering of Vibrio fischeri. Appl. Environ. Microbiol. 84 (14), e00850-18. doi: 10.1128/AEM.00850-18
Wasilko N. P., Larios-Valencia J., Steingard C. H., Nunez B. M., Verma S. C., Miyashiro T. (2019). Sulfur availability for Vibrio fischeri growth during symbiosis establishment depends on biogeography within the squid light organ. Mol. Microbiol. 111 (3), 621–636. doi: 10.1111/mmi.14177
Yip E. S., Geszvain K., Deloney-Marino C. R., Visick K. L. (2006). The symbiosis regulator rscS controls the syp gene locus, biofilm formation and symbiotic aggregation by Vibrio fischeri. Mol. Microbiol. 62 (6), 1586–1600. doi: 10.1111/j.1365-2958.2006.05475.x
Yip E. S., Grublesky B. T., Hussa E. A., Visick K. L. (2005). A novel, conserved cluster of genes promotes symbiotic colonization and sigma-dependent biofilm formation by Vibrio fischeri. Mol. Microbiol. 57 (5), 1485–1498. doi: 10.1111/j.1365-2958.2005.04784.x
Yount T. A., Murtha A. N., Cecere A. G., Miyashiro T. I. (2023). Quorum Sensing Facilitates Interpopulation Signaling by Vibrio fischeri within the Light Organ of Euprymna scolopes. Isr. J. Chem. 63 (5–6), e202200061. doi: 10.1002/ijch.202200061
Zheng W., Zhang C., Li Y., Pearce R., Bell E. W., Zhang Y. (2021). Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 1 (3), 100014. doi: 10.1016/j.crmeth.2021.100014
Keywords: symbiosis, biofilm, Vibrio fischeri, LPS biosynthesis, quorum sensing, Euprymna scolopes
Citation: Provencher EAP, Ehrig MR, Cecere AG, Cousins SC, Maybin MA, Meredith TC and Miyashiro TI (2023) Inhibition of biofilm formation by a lipopolysaccharide-associated glycosyltransferase in the bacterial symbiont Vibrio fischeri. Front. Bacteriol. 2:1254305. doi: 10.3389/fbrio.2023.1254305
Received: 06 July 2023; Accepted: 02 August 2023;
Published: 21 August 2023.
Edited by:
Yanjiao Zhou, UCONN Health, United StatesReviewed by:
Eunhye Goo, Seoul National University, Republic of KoreaCopyright © 2023 Provencher, Ehrig, Cecere, Cousins, Maybin, Meredith and Miyashiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim I. Miyashiro, dGltMTRAcHN1LmVkdQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.