
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bacteriol., 22 September 2023
Sec. Pathogenesis, Vaccines, and Immunity of Bacterial Infections
Volume 2 - 2023 | https://doi.org/10.3389/fbrio.2023.1240698
This article is part of the Research TopicEditors' Showcase: Pathogenesis, Vaccines, and Immunity of Bacterial InfectionsView all 8 articles
 Sergei S. Biryukov1Christopher P. Klimko1Jennifer L. Dankmeyer1Ronald G. Toothman1Jennifer L. Shoe1Melissa Hunter1Nathaniel O. Rill1Yuli Talyansky1
Sergei S. Biryukov1Christopher P. Klimko1Jennifer L. Dankmeyer1Ronald G. Toothman1Jennifer L. Shoe1Melissa Hunter1Nathaniel O. Rill1Yuli Talyansky1 Michael L. Davies1Ju Qiu2David P. Fetterer2
Michael L. Davies1Ju Qiu2David P. Fetterer2 Joel A. Bozue1
Joel A. Bozue1 Susan L. Welkos1
Susan L. Welkos1 Christopher K. Cote1*
Christopher K. Cote1*Introduction: Plague is an ancient disease caused by Yersinia pestis, a widely disseminated Tier 1 pathogen that poses significant public health and biothreat risks. The rapid course and high mortality of pneumonic plague limit the efficacy of antibiotic treatment and mandate the need for an effective, licensed, and readily available vaccine. New candidate vaccines are being developed; however, their efficacy in nonhuman primates, optimal vaccination schedule and immune response, duration of protection, and breadth of coverage against various virulent strains are inadequately understood. In the current work, we explored homologous and heterologous vaccination schemes using the sensitive BALB/c mouse models of bubonic and pneumonic plague challenged with Y. pestis strain C12. This strain, a derivative of the wild-type strain CO92, lacks the anti-phagocytic F1 capsule yet remains highly virulent. Protection against such nonencapsulated strains has been particularly elusive.
Methods: We tested the efficacy of live attenuated vaccine (LAV) derivatives of Y. pestis CO92 or C12 with a deletion of a type 3 secretion-associated gene (ΔyscN) or the pgm pigmentation locus, and they were cured of the pPst (PCP1) plasmid (CO92 pgm− pPst−). The LAVs were evaluated alone or accompanied by a dose of a protein subunit vaccine (rF1V or rV).
Results: The most protective and immunogenic vaccination scheme, as tested under a variety of conditions in bubonic and pneumonic plague models, was heterologous vaccination with a LAV and the recombinant rF1V or rV protein subunit vaccine. Furthermore, in the heterologous scheme, different LAVs and subunit vaccines could be substituted, affording flexibility in vaccine component selection. We also evaluated a novel intervention strategy consisting of vaccination and post-exposure antibiotic treatment. The layering of vaccination with the LAVs and post-exposure treatment with streptomycin was synergistic, extending the time after the Y. pestis C12 challenge when treatment remained effective and affording a sparing of antibiotics.
Conclusion: The current work defined effective and flexible vaccination and treatment interventions that successfully prevented lethal infection with virulent, nonencapsulated Y. pestis.
The etiologic agent of plague, Yersinia pestis, is a globally distributed pathogen that is both a considerable public health risk and a potential biothreat agent. Plague is known as a fearsome menace of antiquity, as illustrated by the massive Black Death pandemic of the 14th century, with a fatality rate of more than 30%; yet, it remains an important current zoonotic hazard (Bertherat, 2016; Baril et al., 2019). In many countries, such as China, Madagascar, the United States of America, the Democratic Republic of the Congo, and elsewhere in South America and Africa, the foci of Y. pestis persist, causing periodic outbreaks of disease (Baril et al., 2019; Bevins et al., 2021). This situation is highlighted by the unusually large outbreak of bubonic and pneumonic plague in 2017 in Madagascar, where the majority of cases were the highly lethal pneumonic form (Randremanana et al., 2019). Furthermore, Y. pestis is also categorized as a Tier 1 select agent by the US Department of Health and Human Services (Nelson et al., 2021), largely due to its potential to be aersolized and produce and acute (in some cases rapidly fatal) disease. Attempts to harness Y. pestis as a bioweapon have long been described, from the early use of plague corpses in the Middle Ages to the experimental dissemination of plague in China by the infamous Japanese Unit 731 during World War II and possibly beyond, as documented in detail (Cowdrey, 1984; Williams and Wallace, 1989; Alibek, 1999; Worsham et al., 2018). Yersinia pestis is susceptible to antibiotics, but due to the rapid course, nonspecific symptomology, and high mortality rate of pneumonic plague, antibiotic treatment is often ineffective. Additionally, antibiotic-resistant strains have been identified previously and in the relatively recent outbreaks in Madagascar (Guiyoule et al., 2001; Hinnebusch et al., 2002; Cabanel et al., 2018). Thus, plague vaccines are greatly needed, yet none are licensed or widely available.
The first plague vaccine prototype was developed by Haffkine in 1897 and consisted of killed whole cells of Y. pestis (Meyer, 1970; Titball and Williamson, 2004; Wang et al., 2013; Sun, 2016). Such killed vaccines proved to be inefficient, providing only a short interval of protection against bubonic plague (Titball and Williamson, 2004; Wang et al., 2013). Live attenuated derivatives of Y. pestis were subsequently developed due to their potential ability to protect against both bubonic and pneumonic plague, to present multiple antigens to the immune system, and to induce both humoral and cellular immune responses, since both arms of the immune response appear to be critical in anti-plague vaccine strategies (Elvin and Williamson, 2004; Smiley, 2008; Feodorova and Motin, 2012; Wang et al., 2013; Sun, 2016; Verma and Tuteja, 2016). Despite their reactogenicity and short-lived efficacy, often requiring yearly vaccinations, some of these live attenuated vaccines (LAVs) have been employed as human vaccines, limited primarily to Russia and China (Stepanov et al., 1999; Feodorova and Motin, 2012).
The only vaccine for plague previously licensed in the United States was the immunogenic plague vaccine USP, commonly known as the Cutter vaccine. It contained a formalin-killed suspension of virulent plague bacilli. It is no longer available but had been administered routinely to military personnel posted in Vietnam and certain other individuals, such as field personnel working in plague endemic areas and laboratory personnel working with Y. pestis (Meyer, 1970). Although it was effective in reducing the occurrence of bubonic disease, as evidenced by the low incidence of plague in U.S. personnel situated in Vietnam, laboratory findings suggested that this vaccine might not protect against pneumonic plague (Sun, 2016; Verma and Tuteja, 2016). Also, the major protective antigen in these vaccines was fraction 1 (F1), a surface capsule composed of the Caf1 protein encoded by the caf1 operon on plasmid pFra (pMT1). Such vaccines do not readily protect against genetically engineered or naturally occurring F1-negative strains, which can be highly virulent despite the absence of a capsule (Welkos et al., 1995; Worsham et al., 1995; Andrews et al., 1999). A more recent human plague vaccine candidate is rF1V, a protein fusion of the strongly immunogenic F1 and LcrV (referred to as V), the low calcium response virulence protein. V is an essential anti-host virulence factor that is required for type 3 secretion system (T3SS)-mediated translocation of the toxic Yersinia outer protein effectors (Yops) into host cells. V also stimulates the production of immunosuppressive cytokines (Heath et al., 1998; Pettersson et al., 1999; Mueller et al., 2005). The rF1V vaccine was shown to be efficacious in mice and some, but not all, species of nonhuman primates (Pitt, 2004; Powell et al., 2005; Mizel et al., 2009; Quenee et al., 2011; Adamovicz and Worsham, 2012; Williamson and Oyston, 2013; Verma and Tuteja, 2016). Furthermore, the protection afforded by rF1V against F1-negative strains of Y. pestis relies solely on the V antigen component. Since there is evidence for V heterogeneity within Yersinia species (Roggenkamp et al., 1997; Anisimov et al., 2010; Miller et al., 2012; Daniel et al., 2019), the potential exists that naturally occurring or engineered strains harboring altered V antigens could overcome rF1V-induced immunity (Verma and Tuteja, 2016).
Thus, a more reliably effective plague vaccine is needed. Recent candidates have involved various platforms, e.g., attenuated and recombinant Yersinia strains, live recombinant viral and bacterial vectors constructed in adenovirus, Salmonella, Yersinia pseudotuberculosis, or other microbes, defined antigen subunit vaccines, outer membrane vesicles, DNA, mRNA, and nanoparticle constructs, as reviewed in detail elsewhere (Feodorova and Motin, 2012; Wang et al., 2013; Sun, 2016; Verma and Tuteja, 2016; Rosario-Acevedo et al., 2021; Rosenzweig et al., 2021; Aftalion et al., 2023; Kon et al., 2023).
While a number of vaccine candidates have induced significant protection against defined wild-type strains of virulent Y. pestis in animal models, their optimal vaccination schedule and immune response, duration of protection, and breadth of coverage, as well as efficacy in nonhuman primates and human vaccinees, are inadequately understood. The vaccine efforts in our laboratory are focused on the modification and optimization of our candidate LAVs and defined protein subunit vaccines (Swietnicki et al., 2011; Bozue et al., 2012). The LAV candidates evaluated in this study are highly attenuated due to a deletion in the yscN gene or of the pigmentation locus (pgm) accompanied by the curing of the pPst plasmid. The yscN gene encodes for an ATPase which provides energy to the T3SS and is necessary for Yop effector translocation. We have previously demonstrated that this mutant strain does not secrete LcrV into culture supernatant in vitro, but a small amount of LcrV can be found associated with whole-cell extracts using immunoassays (Bozue et al., 2012). The pgm mutation is a 102-kb deletion of the entire pgm locus that results in iron acquisition and storage deficiency (Perry et al., 1990; Pendrak and Perry, 1991; Fetherston et al., 1992). The pPst plasmid encodes the plasminogen activator enzyme that activates host fibrinolysis and permits bacterial systemic dissemination (Ferber and Brubaker, 1979; Sodeinde and Goguen, 1988; McDonough and Falkow, 1989).
Here, we explored homologous and heterologous vaccination schemes utilizing both the ΔyscN or pgm− pPst− mutants as standalone or paired with the rV or rF1V subunit vaccines in the sensitive BALB/c mouse model of protection against aerosol challenge with an F1 capsule-negative Y. pestis strain. Optimal protection against challenge with nonencapsulated Y. pestis was achieved by heterologous vaccination with a live strain and a protein subunit vaccine. In addition, we initiated studies for a novel intervention strategy layering vaccination and post-exposure antibiotic treatment. Disease outcome improved and a delayed initiation of antibiotics was still protective when vaccination was integrated with post-exposure antibiotic treatment. The layering of medical countermeasures will expand the treatment options available for the plague and potentially allow for vaccine and/or antibiotic dose-sparing.
The wild-type and mutant derivatives of Y. pestis and their construction and characteristics were described previously (Cote et al., 2021) and in the current study. These strains included the virulent F1-negative (nonencapsulated) derivative of CO92, which is C12 (Worsham et al., 1995). The recombinant strains had a mutation in the virulence-associated gene yscN or had a deletion of the entire pgm locus and were cured of the pPst (PCP1) plasmid encoding plasminogen activator (CO92 pgm− pPst−) (Welkos et al., 1997; Welkos et al., 2002; Swietnicki et al., 2011; Bozue et al., 2012; Bozue et al., 2014). Yersinia pestis strains were grown on 5% sheep blood agar plates (SBAP) or tryptose blood agar (TBA) base slants for approximately 48 h at 30°C. Liquid cultures of Y. pestis C12 used for mouse challenge studies were prepared in heart infusion broth (HIB) medium supplemented with 0.2% xylose (HIBX), and the live attenuated vaccine suspensions were prepared with broth cultures incubated in HIBX supplemented with 2.5 mM CaCl2, as described below (Cote et al., 2021). A solution of 10 mM potassium phosphate, pH 7.3–7.4 (KPhos), was used to prepare and dilute bacterial inocula. Challenge doses were determined by serial dilutions in KPhos buffer and plating on SBAP. Bacteriological media were obtained from Thermo Fisher-Remel (Rockville, MD, USA).
The rF1V vaccine was described previously (Heath et al., 1998; Amemiya et al., 2009; Biryukov et al., 2021). Toll-like receptor 9 (TLR9) oligonucleotide (ODN) CpG ODN 2006 (CpG2006) was purchased from InvivoGen (San Diego, CA, USA) and reconstituted in accordance with the manufacturer’s recommendations. The purified recombinant V (rV) protein was obtained from BEI Resources (Manassas, VA, USA). Alhydrogel was sourced from InvivoGen. Streptomycin for injection was obtained from X-Gen Pharmaceuticals, Inc. (Horseheads, NY, USA).
The day prior to vaccination, flasks were inoculated with a suspension of colonies from a freshly inoculated SBAP and incubated for 24 h at 28°C–30°C with shaking at 200 rpm. The next day, the cultures were adjusted to an OD600 of 0.1 in fresh medium and incubated to the OD600 determined to produce the target CFU concentration, which was 107 colony forming units (CFU) in doses of 0.2 mL (Cote et al., 2021). To confirm the actual delivered dose of bacteria, the final suspensions were diluted and plated for viable counts. All plates were incubated at 28°C–30°C for approximately 48 h before counting.
The doses of the rF1V vaccine contained 2 µg of rF1V in the presence or absence of 5 µg of CpG2006 (CpG) and included Alhydrogel (250 µg) in a total volume of 0.1 mL (Biryukov et al., 2021). In the studies indicated, 2 µg of rV with or without 5 µg of CpG was administered in a total volume of 0.1 mL.
The animal research was conducted under an animal use protocol approved by the USAMRIID Institutional Animal Care and Use Committee (IACUC) in compliance with the Animal Welfare Act, PHS Policy, and other Federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is accredited by AAALAC International and adheres to the principles stated in the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). Female BALB/c mice were obtained from Charles River (Frederick, MD, USA) and were 7–10 weeks of age at the time of vaccination; all groups contained 10 mice, with exceptions specified. Mice vaccinated with a single dose of vaccine were injected subcutaneously and exposed 4 weeks later by the aerosol or subcutaneous (SC) route to a lethal dose of Y. pestis C12. As indicated, mice vaccinated twice were administered the second dose 20–28 days after the initial vaccine dose. Sera and spleens were collected from a cohort of mice to assess immune responses to the vaccines. Mice were challenged 27–29 days post final vaccination.
Mice were exposed to aerosolized (pneumonic) or SC (bubonic) challenge doses of virulent Y. pestis C12. For the bubonic challenge, bacteria were harvested from TBA slants. Mice exposed by the SC route were inoculated with 0.2 mL volumes of the suspension in KPhos (Bozue et al., 2012; Bozue et al., 2014). The bacteria used for aerosol studies were prepared by using colonies from freshly inoculated TBA slants which were suspended in HIBX to an initial OD620 of approximately 0.01 and incubated for approximately 24 h at 28°C–30°C. For aerosol exposures, the cultures were harvested by centrifugation and suspended in HIB medium (no xylose) to the concentration yielding the number of LD50 doses indicated in the figures. Exposure to aerosolized bacteria was accomplished as previously described (Bozue et al., 2012; Bozue et al., 2014; Trevino et al., 2018). Briefly, mice were transferred to wire mesh cages and were placed in a whole-body aerosol chamber within a class 3 biological safety cabinet located inside a BSL-3 laboratory. Mice were exposed to aerosols of Y. pestis strain C12 created by a three-jet Collison nebulizer. Samples were collected from the all-glass impinger vessel and analyzed by performing CFU calculations to determine the inhaled dose of Y. pestis.
A solution of streptomycin was prepared in water for injection (Corning, Manassas, VA, USA), and a dose of 20 or 40 mg/kg was administered to mice by the intraperitoneal route every 6 h for 5 days. Mice were injected with the antibiotic beginning 48 h or 60 h after exposure to aerosolized Y. pestis C12.
The tissues collected from necropsied mice included lung, spleen, and blood. They were weighed and homogenized with disposable PRECISION™ homogenizers (Covidien, Dublin, Republic of Ireland), and the CFU of the homogenate was determined on SBA plates. Undiluted homogenate and 10-fold dilutions in KPhos were plated in duplicate to determine sterility. The limit of detection (LOD) was approximately 100 CFU/mL blood or 5 CFU/organ. After CFU determinations, samples were radiation-inactivated, sterility-checked, and stored at −80°C for immunological analyses.
Immunoglobulin (Ig) IgG antibody responses to the vaccines were determined by semiquantitative endpoint ELISA using sera from vaccinated BALB/c mice, as previously described (Biryukov et al., 2021; Cote et al., 2021). The sera were collected as terminal blood collections from axillary vessels and titrated against several capture antigens: the rF1V recombinant fusion protein, the rV protein, and the γ-radiation-inactivated whole cells of Y. pestis strains that were temperature-switched (TS) CO92 or TS C12. The strains had been grown at 30°C for 21 h followed by a temperature switch to 37°C and incubation for an additional 3 h to upregulate the presentation of potential antigens. The cultures were pelleted by centrifugation and then inactivated with γ-radiation; temperature-shifted antigens were designated TS. The rF1V fusion protein vaccine construct and the rV protein were diluted in 0.1 M of carbonate buffer, pH 9.5, to a concentration of 2 μg/mL, while inactivated Y. pestis TS CO92 or TS C12 whole cells were plated at a concentration of 10 μg/mL on 96-well Immulon 2HB plates (Thermo Fisher, Grand Island, NY, USA). Plates were stored at 4°C overnight, then washed and blocked, and samples were processed as previously described (Biryukov et al., 2021). Two-fold dilutions of the serum were made in triplicate, and the results are reported as the geometric mean (GM) and geometric standard error (GSE) of the reciprocal of the highest dilution giving a mean OD of at least 0.1 ± 1 SD at 450 nm with a reference filter (570 nm). Samples with an antibody titer of <50 were considered negative.
Four weeks following vaccination, purified splenocytes were prepared based on a previously published protocol (Amemiya et al., 2006; Cote et al., 2021). Briefly, spleens were excised from mice (n = 5 mice per group), weighed, and disaggregated in RPMI 1640 medium (Thermo Fisher, Grand Island, NY, USA). Red blood cells in the spleen homogenate were lysed with Ammonium-Chloride-Potassium (ACK) Lysing Buffer (BioWhittaker, Walkersville, MD, USA) after the extract was diluted with RPMI 1640 medium and cells were pelleted by centrifugation at 335×g for 10 min. Splenocytes were then resuspended in CTL-Medium (Cellular Technology Limited, Cleveland, OH, USA) supplemented with 1% l-glutamine and the cells were counted. Purified splenocytes were restimulated for ~48 h in the presence of rF1V (25 μg/mL) or irradiated Y. pestis TS C12 (5 μg/mL) in complete medium containing 10% heat-inactivated fetal calf serum (Thermo Fisher), 1 mM of sodium pyruvate, 0.1 mM of nonessential amino acids, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 50 μM of 2-mercaptoethanol. A solution of phorbol 12-myristate 13-acetate (PMA; 100 ng/mL) and ionomycin (0.5 μg/mL) was used as the positive control stimulant and resulted in uniformly strong signals (data not shown). Following restimulation, the plates were centrifuged at 1,200×g for 10 min at RT, and the cytokine expression in the supernatants was evaluated by Luminex Mag Pix 36-plex mouse panel as per manufacturer’s directions (Thermo Fisher Scientific, Grand Island, NY, USA).
In addition, 3 days post-challenge, the spleen and lung homogenates were prepared by removing and homogenizing the whole organs in KPhos using disposable PRECISION™ homogenizers (Covidien, Dublin, Republic of Ireland). Samples were irradiated with approximately 21 kGy of γ-radiation and confirmed sterile by testing 10% of the sample before use. Aliquots of the homogenates were saved for cytokine/chemokine determination and stored at −70°C. Cytokine and chemokine levels were assayed in spleen and lung homogenates, as previously described (Trevino et al., 2018). The homogenate samples were thawed and centrifuged at 10,000×g for 10 min, and the supernatant was then examined for cytokine expression by Luminex Mag Pix 36-plex mouse panel (Thermo Fisher Scientific, Grand Island, NY, USA) as per the manufacturer’s directions. The levels (pg/mL) were measured, and heat maps were generated to report the fold increase [vaccine group (pg/mL)/PBS control group (pg/mL)] in cytokine or chemokine levels.
ELISpot assays were performed to measure interferon-gamma (IFN-γ) expression. Purified splenocytes, 4 weeks after vaccination, were seeded in the presence of rF1V, TS C12, medium alone, or PMA/ionomycin. Briefly, 96-well plates were coated overnight at 4°C with 80 µL/well of capture anti-mouse IFN-γ monoclonal antibody. The plates were washed one time with PBS. Recombinant F1V (25 μg/mL) or TS C12 (5 μg/mL) was resuspended in CTL-Medium with 1% l-glutamine and 100 µl was added to each well. The plates were incubated at 37°C, 9% CO2 for 15 min. Splenocytes were resuspended in CTL-Medium with 1% l-glutamine and seeded at 8 × 105 cells per well for rF1V and 8 × 104 cells per well for TS C12 stimulations. The plates were incubated for 24 h at 37°C and 9% CO2, splenocytes were removed, and the plates were washed twice with PBS alone and then twice with PBS and 0.05% Tween. Eighty microliters per well of biotinylated detection anti-mouse IFN-γ monoclonal antibody was added. After 2 h of incubation at room temperature, the plates were washed three times with PBS and 0.05% Tween. Eighty microliters of Strep-AP antibody solution was added to the wells, and the plates were incubated for 30 min at room temperature. Development reagents were added and incubated for 15 min at room temperature according to the manufacturer’s recommendations. The colorimetric reaction was stopped by washing the plates three times with distilled water and air drying overnight. Spots were scanned and analyzed using an automated ELISpot reader (CTL-ImmunoSpot S6 Analyzer, CTL, Germany). The splenocyte response was assessed as spot-forming cells (SFCs), adjusted to 106 cells per well, which was automatically calculated by the ImmunoSpot® software for each stimulation condition and the medium-only control.
Statistical analyses were performed using SAS version 9.4, except as indicated. Survival curves of the vaccinated and control mice were estimated with the Kaplan–Meier method using GraphPad Prism 9.0 and were compared statistically using the log-rank test. Significant differences in survival rates on days 7 and 21 after the virulent challenge were determined using the Fisher exact test. The time-to-mortality (TTM) values were expressed as the median and interquartile range and compared with the log-rank test. The runs of aerosol-challenged mice were analyzed separately or combined, as indicated. The viable counts of Y. pestis from organ and blood samples were compared using the Wilcoxon rank sum test for pairwise comparisons test. The CFU was log-transformed and the mean results were summarized as GM and geometric standard deviation (GSD) values, using the LOD/SQRT(2) to replace the values with no recoverable CFU.
For the ELISA antibody titers, determined as described above, cytokine concentrations determined in Luminex assays, and ELISpot assays, pairwise treatment groups were compared by the Wilcoxon rank sum test. The median, Q1, Q3, GM, and GSE were reported.
The potential synergistic effects of vaccine and antibiotic on protection were analyzed using the Bliss synergy (Demidenko and Miller, 2019). Synergy score values indicated the ratio of median TTM and were based on a log-logistic or log-normal parametric survival model.
We demonstrated previously the efficacy of live attenuated vaccine strains of Y. pestis for the protection of mice against bubonic and pneumonic plague after exposure to the virulent encapsulated Y. pestis CO92 strain (Cote et al., 2021). We describe here that the same strategies could not similarly protect BALB/c mice against plague in animals exposed to Y. pestis strain C12, a nonencapsulated derivative of CO92. As demonstrated in Figure 1A, protection was afforded to mice vaccinated with a single dose of Y. pestis mutant CO92 ΔyscN or C12 ΔyscN or a combination of both ΔyscN vaccine strains (Combo LAV), against a SC challenge with approximately 214 LD50s of Y. pestis C12 (40%, 60%, or 70%, respectively); these survival rates were greater or nearly greater than the mean rate of the control mice, with p = 0.087, p = 0.011, and p = 0.003, respectively. Nevertheless, the vaccine-mediated protection was less robust than that reported previously for mice challenged with the wild-type Y. pestis CO92 strain, against which some of the live vaccines protected completely (Cote et al., 2021). Not surprisingly, these single-dose strategies offered no protection to mice after exposure to aerosolized Y. pestis C12 with only slight, statistically insignificant differences in mean TTM (Figure 1B).

Figure 1 Survival curves of groups of vaccinated BALB/c mice challenged with Yersinia pestis strain C12. The mice were vaccinated with a single dose of CO92 ΔyscN (1.3 × 107 CFU) or C12 ΔyscN (1.7 × 107 CFU) or both (Combo LAV), 1.54 × 107 CFU total; the control group received KPhos buffer alone. All groups contained 10 mice. Twenty-eight days later, the animals were exposed to 1.93 × 103 CFU (214 LD50s) by the SC route (A) or to 1.71 × 106 CFU (15 LD50s) by the aerosol route (B) and monitored for 21 days after challenge.
As demonstrated in Figure 2, two doses of the LAV (prime–boost) given on days 0 and 23 yielded only a modest increase in protection against the C12 challenge compared with the single-dose cohorts (Figure 1). In the bubonic model, the protection afforded after a booster vaccination by CO92 ΔyscN and C12 ΔyscN was increased by 20% (to 60% and 80%, respectively), and the extent of protection by the Combo LAV vaccine was unchanged (70%, Figure 2A); the mean survival rates were statistically greater than the mean survival rate of the control mice (p = 0.057 to p = 0.006). The two-dose homologous vaccination schemes with our live attenuated ΔyscN strains did not improve survival after exposure to aerosolized Y. pestis C12 (Figure 2B), but it slightly extended the TTMs on days 7 and 21 after challenge (p = 0.022 to p < 0.0001) as compared with the controls.
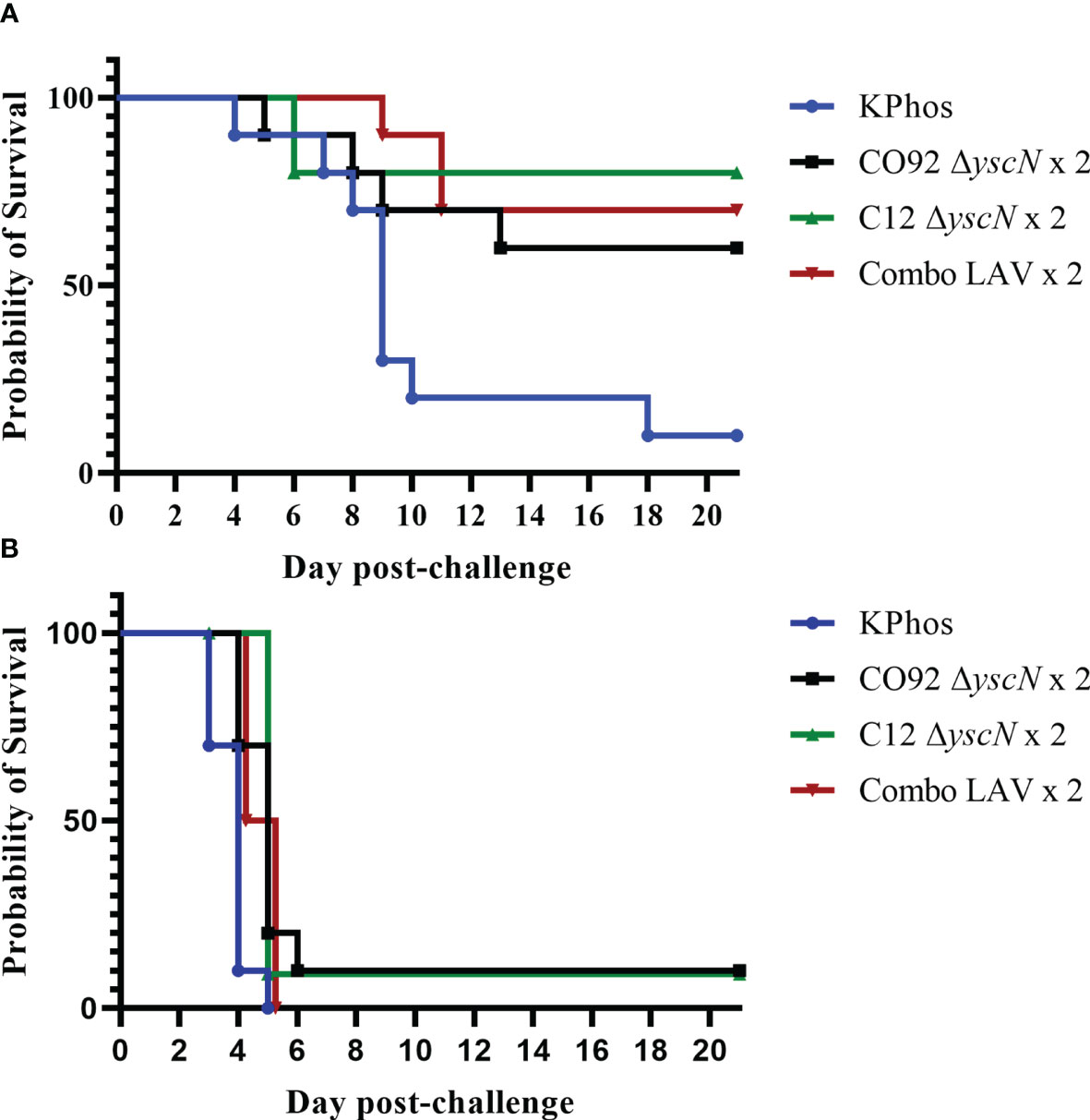
Figure 2 Survival curves of BALB/c mice vaccinated twice with the homologous LAV (days 0 and 23) and challenged 28 days later with Yersinia pestis strain C12 by the SC route (A) or by the aerosol route (B). The doses of C12 were 2.38 × 103 CFU (264 LD50s) or 2.5 × 105 CFU (3 LD50s), respectively. The mice were monitored for 21 days after challenge.
The LAV strains are clearly immunogenic as determined by humoral and cell-mediated immune responses that protect against Y. pestis CO92 (Cote et al., 2021), and these immune responses protect mice against bubonic, but not pneumonic, plague caused by Y. pestis C12. Encapsulated LAV candidate strains produce a robust anti-F1 humoral immune response, but due to possible immune interference, the anti-F1 responses may diminish the induction of a more diverse immunity against other protective antigens (Cote et al., 2021). To enhance the availability of other protective antigens but still retain the F1-mediated protection, approximately equal parts of CO92 ΔyscN and C12 ΔyscN vaccine mix (Combo LAV) were selected for further evaluation in heterologous vaccine strategies to improve the protection against Y. pestis C12 infection.
In these experiments, we primed mice with a dose of the Combo LAV. The mice were then boosted (28 days later) with either the same LAV given in the first injection or with a subunit vaccine: rF1V fusion protein or rV protein (groups 2–4, 7, 8) (Figure 3). We also tested the impact of further adjuvanting the recombinant protein boost with the addition of oligonucleotide CpG2006. Mice which received any of the four heterologous vaccines (groups 3, 4, 7, 8) were highly protected against bubonic disease when challenged 4 weeks after the boost vaccination, as illustrated in Figure 3A. Vaccination with the Combo LAV followed by rF1V + CpG, or rV +/− CpG, resulted in 100% survival (groups 4, 7, 8), p ≤ 0.001 vs. KPhos control; and 90% survival was observed in groups given either the Combo LAV first and then rF1V without CpG (group 3) or with two doses of the Combo LAV vaccine (group 2), p = 0.0001. Two groups receiving KPhos and CpG alone plus the Combo LAV, as either the prime or boost doses, resulted in 80% survival after the SC challenge (groups 6 and 9), p = 0.0007 vs. the control, respectively. Thus, in the bubonic model, the heterologous vaccine combinations were generally more protective against Y. pestis C12 than the homologous vaccine schemes.
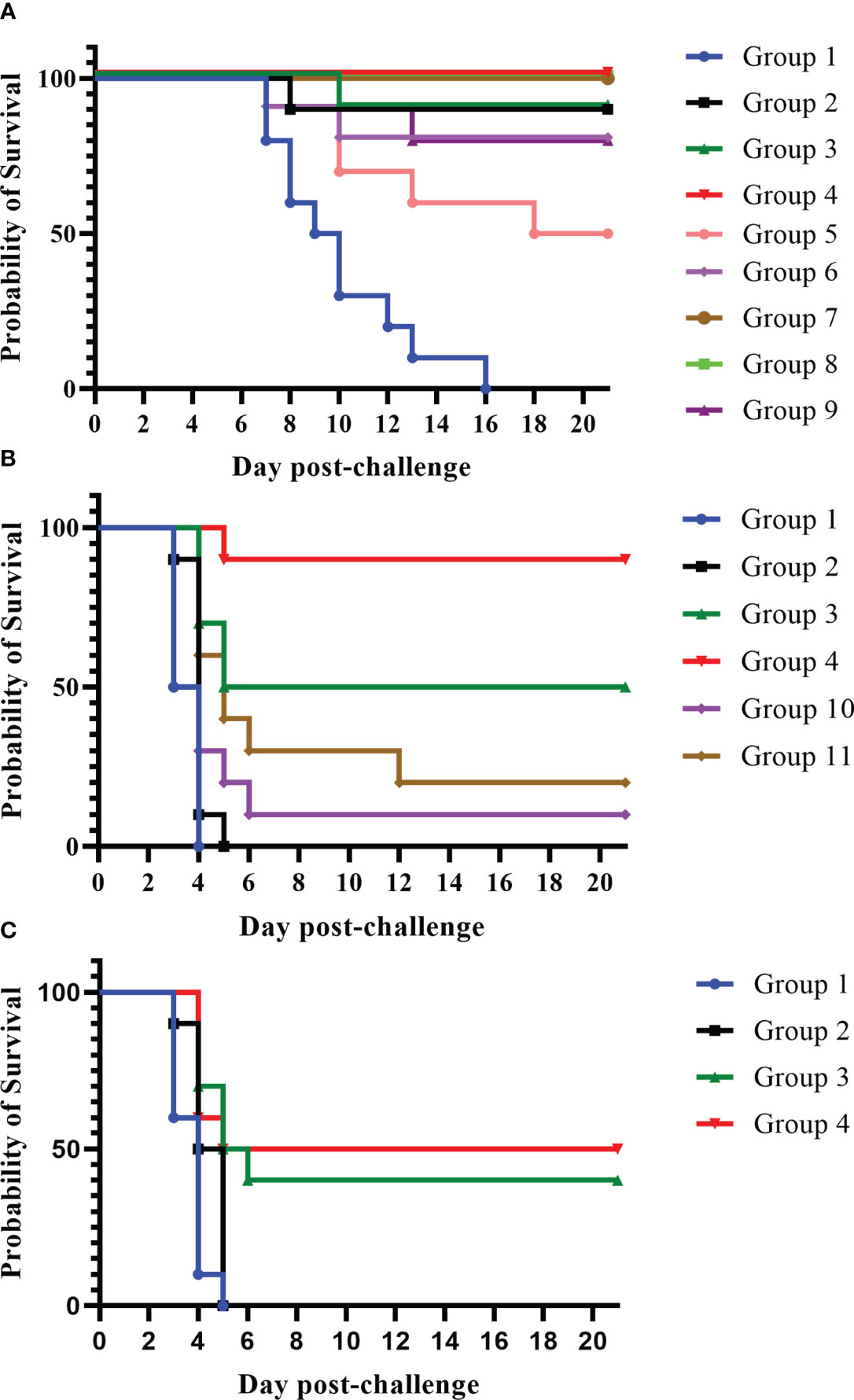
Figure 3 A comparison of protection elicited by homologous and heterologous vaccines against Yersinia pestis strain C12 challenge. The vaccine doses contained approximately 1 × 107 CFU total Combo LAV (ranging from 0.76 to 0.88 × 107 CFU) and/or 2 µg of protein subunit vaccine: rF1V or rV. Two vaccinations were given to the mice (n = 10/group). Vaccinated mice received two doses 28 days apart and were exposed to C12 by the SC route (3.62 × 103 CFU C12, 402 LD50s) 27 days later (A). Vaccinated mice received two doses 21 days apart and were then exposed to 7.93 × 105 CFU of aerosolized C12 (10 LD50s) 28 days later (B). Other groups of mice were vaccinated with two doses 21 days apart and then exposed to a larger challenge (1.65 × 106 CFU, 22 LD50s) of aerosolized Y. pestis C12 29 days later (C). The mice were monitored daily for 21 days after challenge. The vaccine regimens provided to the groups are described in Table 1 embedded into manuscript section 3.2.

Table 1 Vaccination regimens used in the experiment detailed in Figure 3.
Furthermore, heterologous approaches significantly improved the clinical outcome in mice exposed to aerosolized Y. pestis C12 (Figure 3B). The best survival against aerosolized Y. pestis C12 challenge was observed in mice vaccinated with the heterologous combinations of Combo LAV (prime) followed by a boost of rF1V with CpG (Figure 3B). The survival rates of these mice (90%) at both early (day 7) and final time points post-challenge (group 4) were significantly better than those of four of the other five groups (p-values ranged from p = 0.0001 to p = 0.048). Although 50% of group 3 (Combo LAV and rF1V) survived, this survival rate was not significantly different from that of group 4 (p = 0.141). However, the TTM of the latter was longer than that of mice given the Combo LAV and rF1V without CpG (p = 0.048). All five vaccinated groups survived slightly but significantly longer than did the KPhos controls (p ≤ 0.041), with mean TTMs for non-survivors of 3.5 days (controls) and 4.0 to 5.4 days for the vaccinated groups. Thus, the immunostimulant CpG enhanced the protection afforded by the heterologous Combo LAV and rF1V vaccine.
To further support these findings, mice were vaccinated again with the homologous or heterologous prime/boost scheme but were challenged with a mean inhaled dose of Y. pestis C12 approximately two-fold greater than the dose in the study described in Figure 3B, i.e., 1.65 × 106 CFU (22 LD50s). As shown in Figure 3C, the heterologous vaccines outperformed two doses of the Combo LAV. Whereas the latter provided no protection, the groups vaccinated with the LAV followed by rF1V, with or without CpG, were partially protected. The survival rate of the group receiving the heterologous vaccine with CpG was significantly greater than that of the control and Combo LAV ×2 groups on day 7 and day 21 after challenge (p = 0.0325); however, these survival rate comparisons to the control and Combo LAV ×2 groups were not statistically different for the mice receiving the heterologous vaccine without CpG. The TTMs of the two heterologous vaccination groups were greater than those of the KPhos control and LAV ×2 groups on both day 7 and day 21 post-challenge (p ≤ 0.0423). Overall, vaccination with heterologous combinations of LAVs and subunit protein vaccines was more strongly protective than homologous vaccine strategies of LAVs, especially against aerosol challenge (Figures 3B, C). Importantly, vaccination with the recombinant protein alone was shown to be less effective against similar challenge doses of C12 compared with mice challenged with Y. pestis CO92 (Biryukov et al., 2021).
Tissue samples and blood were collected from cohorts of mice in the study described in Figure 3B to assess bacterial load and antibody titers on day 3 after the aerosol challenge, and the results are shown in Tables 2, 3. Although there were large variations in organ and blood bacterial loads between mice in some groups, statistical analyses revealed significant vaccine-related differences. For the group vaccinated twice with the Combo LAV, there were no significant reductions in bacterial burden compared with the KPhos control mice in the lung, spleen, and blood. In contrast, mice vaccinated with the heterologous Combo LAV/rF1V + CpG regimen had a reduced burden in the lungs (GM of 274 CFU/g), and the bacterial load was significantly reduced compared with the other five groups (p = 0.026 to 0.0022). In addition, a GM of 123 CFU/g was isolated from the spleen samples of mice vaccinated with Combo LAV/rF1V + CpG, with four of six mice having no detectable CFU, and blood samples yielded a GM of 237 CFU/mL (Table 2); no bacteria were recovered from the blood of five out of six mice vaccinated with Combo LAV/rF1V + CpG.
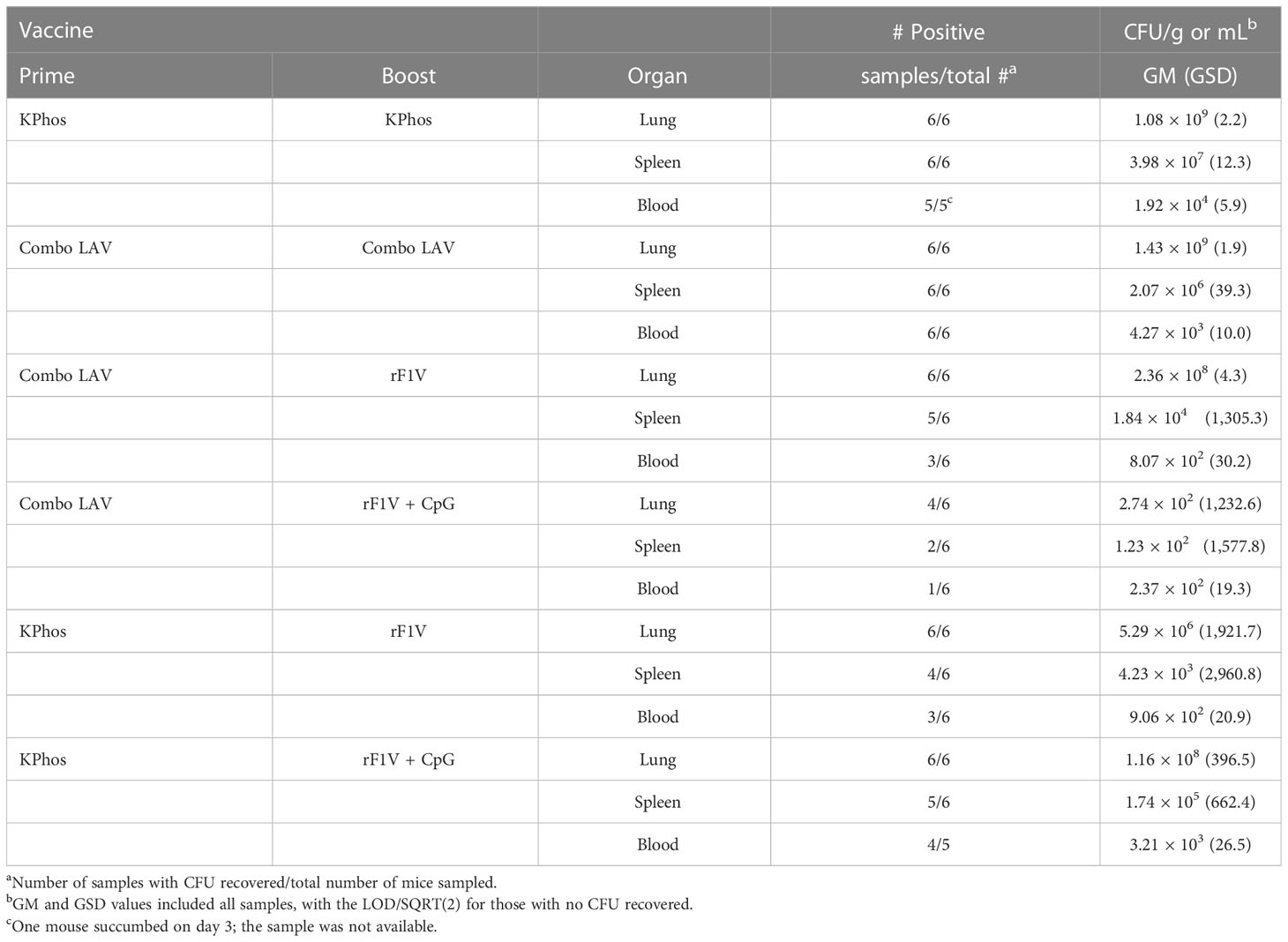
Table 2 The bacterial burden of mice collected three days after challenge with Yersinia pestis C12.
For the Combo LAV/rF1V-vaccinated mice, the lung CFUs were only significantly less than those recovered from the Combo LAV ×2-vaccinated mice (p = 0.041); the spleen and blood burdens were reduced, but there were no differences compared with the other five groups. Overall, the three vaccine groups, namely, Combo LAV/rF1V, KPhos/rF1V, and KPhos/rF1V + CpG, had generally similar bacterial loads. The GMs of the lung, spleen, and blood CFU of mice with positive cultures were not statistically different in comparison between these three vaccine groups. However, compared with the Combo LAV ×2 burdens, two of these vaccine groups had significant reductions of one to two logs in lung CFU (p = 0.026 for the KPhos/rF1V group and p = 0.041 for the Combo LAV/rF1V-vaccinated mice), at least one log lower spleen CFU counts, and no detectable CFU in half of the blood samples from the KPhos/rF1V and Combo LAV/rF1V-vaccinated mice. Thus, the bacterial burdens of these three groups were generally intermediate between those mice with the smallest bacterial loads (Combo LAV/rF1V + CpG) and the KPhos controls.
Antibody titers to four antigens were assessed by ELISA on sera from blood collected 3 days after challenge. The antigens included rF1V, rV, irradiated TS CO92 whole cells, and irradiated TS C12 whole cells, and the results are shown in Table 3. The most elevated antibody titers overall were those to the rF1V fusion protein or the rV antigen which reiterates the strong immunogenicity of the F1 and V antigens in BALB/c mice. All five vaccines induced anti-rF1V and rV titers that were significantly greater than those of the KPhos control mice, with p = 0.0159 for all groups (Table 3, Supplementary Table 1 describes statistical analyses performed). Mice vaccinated with Combo LAV/rF1V + CpG generated the highest anti-rV titers that were two- and three-fold higher than Combo LAV/rF1V and KPhos/rF1V + CpG, although these differences did not reach significance. Vaccination with a priming dose of the Combo LAV and a booster dose of rF1V induced the highest titers to TS CO92, whereas two doses of the Combo LAV vaccine induced the highest titers to TS C12 (p = 0.017). Likely, the antibody responses to TS CO92 whole-cell antigen were largely directed against the F1 capsule, and adding rF1V to the Combo LAV elicited higher titer to the rF1V and TS CO92 whole-cell antigens. The addition of CpG to Combo + rF1V in group 4 greatly increased titers to rF1V compared with the mean titers of group 1 (p = 0.0095) and group 10 (p = 0.0022). However, the addition of CpG was associated with repressed titers when TS CO92 whole cells were used as the capture antigen and did not significantly increase the titers to TS C12 cells. Specifically, the anti-TS CO92 levels in group 4 mice were less than those in group 3 (p = 0.0043), though not in group 2 (p = 0.643). The vaccines containing only a single dose of rF1V elicited the lowest titers overall, albeit these were still elevated against the rF1V antigen. Overall, the antibody levels against the TS C12 antigen were much less than those to the rF1V and TS CO92 antigens. This finding might suggest either that surface antigens other than the immune-dominant F1 protein do not elicit strong humoral responses or that the C12 ΔyscN LAV was cleared in the mice before reaching immunostimulatory levels due to the lack of an anti-phagocytic capsule.
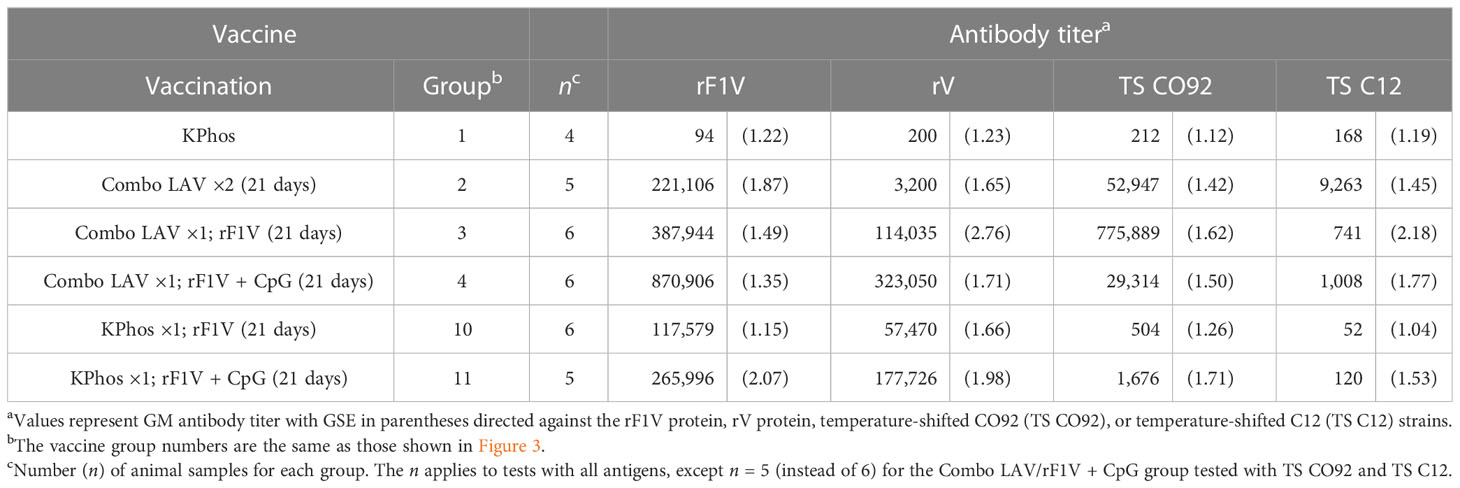
Table 3 Total IgG antibody response against rF1V, Yersinia pestis CO92, or Y. pestis C12.
Lungs and spleens were obtained from animals collected on day 3 post-challenge to analyze the cytokine responses. The lung sample data revealed that three vaccine groups had levels of cytokines that were repressed or not significantly different overall relative to the KPhos group levels (heat map in Table 4, Supplementary Table 2). The Combo LAV/rF1V + CpG and KPhos/rF1V groups had responses that were relatively most repressed. The KPhos/rF1V + CpG group cytokines also exhibited reduced expression, whereas the Combo LAV/rF1V group responses were not appreciably different from those of the KPhos group. For example, in the Combo LAV/rF1V + CpG group, the levels of three cytokines were approximately 100-fold lower than those of the controls (IL-6, MIP-2α, G-CSF), while seven additional cytokines were approximately 10- to 30-fold lower [Gro-α (CXCL1), LIF, IL-1β, GM-CSF, MIP-1α, IL-1α, and IFN-γ]. In contrast, the cytokine levels were generally significantly elevated in the Combo LAV ×2-vaccinated mice. The levels of IL-3, RANTES (CCL5), IL-4, and IFN-γ were from 5.3- to 7.1-fold greater than KPhos controls. These data were inversely associated in part with the vaccine-associated protection against the C12 challenge in that the Combo LAV/rF1V + CpG vaccine was the most protective and Combo/LAV ×2 the least protective (Figures 3B, C). The results of the analysis of spleen samples were similar overall to those of the lung samples, but the degree of differences in the levels between the vaccine groups was less (Table 5, Supplementary Table 3). In contrast to the lung results, IFN-γ, MCP-1, IL-1α, IP-10, and other cytokine levels were repressed in all vaccine groups, and IL-23 (a Th17 cytokine) was uniquely elevated in the Combo LAV/rF1V mice (but not in the Combo LAV/rF1V + CpG group). In aggregate, the spleen data do not appear to be as informative as the lung data in comparing cellular immune responses to vaccination of these female BALB/c mice, but they suggest that early post-challenge decreases in bacterial lung burdens of vaccine-protected mice might result in less dissemination to the spleen.
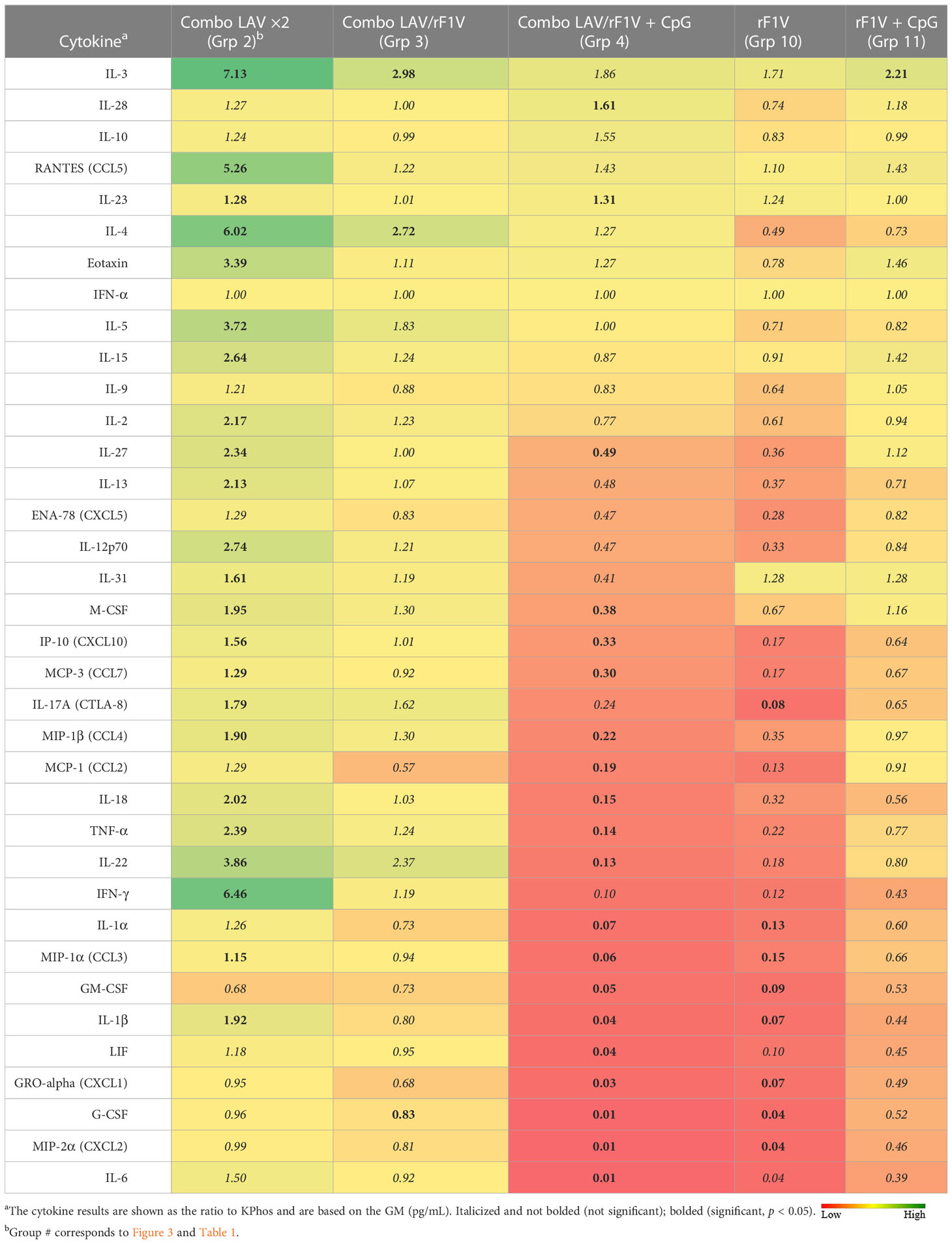
Table 4 Fold change in cytokine levels of lung homogenates from vaccinated groups relative to the KPhos group.
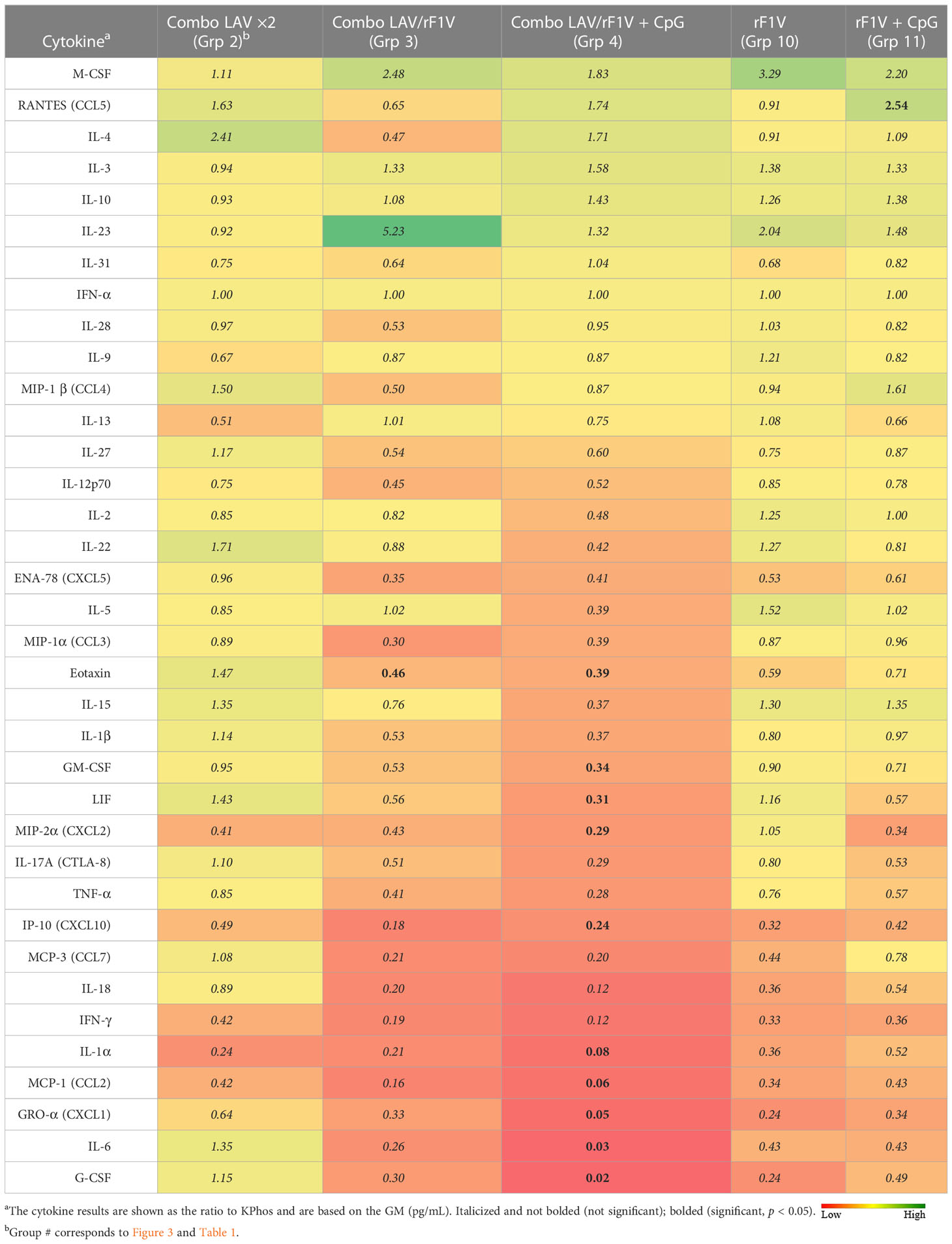
Table 5 Fold change in cytokine levels of spleen homogenates from vaccinated groups relative to the KPhos group.
In an effort to further characterize heterologous plague vaccination approaches, we examined the impact of extended times between the prime and boost vaccine injections along with varying durations between the prime vaccine and subsequent booster dose(s) in the absence of CpG. We also chose to re-evaluate the ability of the CO92 pgm− pPst− LAV to protect mice from the C12 challenge because of its significantly different protein expression profile and the fact that it is an excluded non-select agent strain. In this experiment, the vaccines were administered once only or with one or two additional booster dose(s) given at different times after the first dose, as detailed in Figure 4 (The vaccine regimens provided to the groups are described in Table 6). Specifically, one group received a single dose only (group 2). Single boosters were administered 21 days after the prime dose (group 3), 46 days after the prime (groups 4 and 9), or 90 days after the prime (groups 5, 7, 8, 10, 11). Double boosters were injected on days 46 and 90 after the prime (groups 6 and 12). Figure 4 illustrates the survival rates after the aerosol challenge with a dose of 13 LD50s of C12. Two groups that had been primed with a LAV (CO92 pgm− pPst− or CO92 ΔyscN, groups 7 and 11, respectively) and given one booster of rF1V (without CpG) 90 days after the prime dose had the highest survival rates (37.5%), but survival did not reach significance relative to the KPhos group. A small number of survivors (12.5%) were observed in a heterologous group of mice initially given the CO92 pgm− pPst− LAV and boosted twice, first with CO92 pgm− pPst− at 46 days after the prime and second with rF1V at 90 days after the initial vaccination (group 6). The mice primed with CO92 ΔyscN and boosted on day 46 with the same LAV and on day 90 with rF1V (group 12) had no survivors but had a significantly longer mean TTM than the KPhos controls, with geometric means of 5.0 and 3.8 days, respectively (p = 0.0285). The remaining groups of mice had no survivors and succumbed at the same time post-challenge as the KPhos controls (day 4). Albeit complete protection was not achieved in this study, the results again suggested that heterologous vaccination strategies are superior and that administering one booster injection of rF1V vaccine 3 months after priming with a LAV could be associated with better protection than schedules where the prime and booster doses were spaced more closely in time. Of interest, priming and boosting 90 days later with homologous LAVs (groups 5 and 10) were not protective. Thus, a heterologous LAV/rF1V vaccination scheme appears to be optimal, but further evaluation is needed.
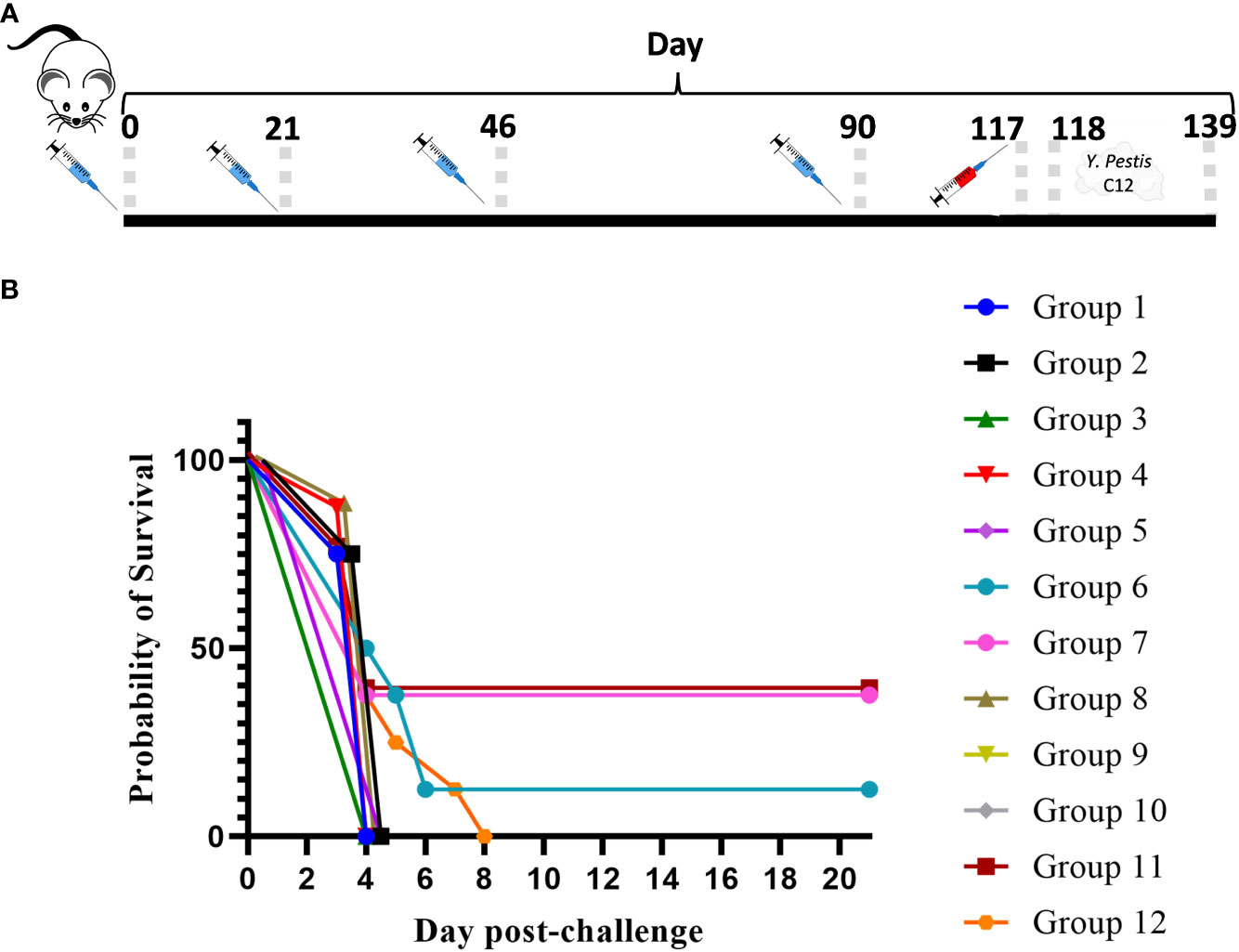
Figure 4 The influence on survival after Yersinia pestis C12 challenge of extending the time between prime and boost doses of the vaccines. The study design, vaccines, and their schedule of administration are detailed in (A) and Table 6. A cohort of mice was euthanized on day 119 in order to collect blood and organ homogenates for immunological assays, and the remainder of the mice were exposed to aerosolized Y. pestis C12 on day 120. The LAV doses contained approximately 1 × 107 CFU (0.86–1.34 × 107 CFU), and 2 µg/dose of the subunit vaccines were delivered. The CO92 pgm− pPst− live vaccine is deleted of the 102-kb pgm locus and cured of the pPst plasmid. Each vaccine group had eight mice. The mice were challenged 28 days after the final vaccination with 1.0 × 106 CFU (13 LD50s) of aerosolized Y. pestis C12 and checked daily for 21 days after challenge (B). NA, not applicable. The vaccine regimens provided to the groups are described in Table 6 embedded into manuscript section 3.3.
Table 6 Vaccination regimens used in the experiment detailed in Figure 4.
Antibody titers to the four antigens were assessed by ELISA on sera collected from a cohort of mice 1 day before the aerosol challenge. The antigens included rF1V, rV, Y. pestis TS CO92, and Y. pestis TS C12. In agreement with the protection data, the two groups (7 and 11) with the greatest survival rates had the highest titers to rF1V and CO92 whole cells but not to C12 whole cells (Table 7), similar to the responses to C12 described in Table 3, and the differences were confirmed statistically with the analyses detailed in Supplementary Table 4. Importantly, groups 7 and 11 did have some of the highest anti-rV titers in this study. These two groups had been primed with either the encapsulated CO92 pgm− pPst− or CO92 ΔyscN LAV and boosted once with rF1V 90 days later. Similarly, mice in groups 6 and 12, which had received prime and boost doses at day 46 of a CO92 LAV (pgm− pPst− or ΔyscN) and a second boost at day 90 with rF1V, and which were partially protected or had an extended TTM, exhibited the second highest antibody responses to rF1V. However, this partial protection was not clearly associated with humoral responses to either the F1+ or F1− whole-cell antigens (Table 7, Supplementary Table 4). The most elevated anti-TS C12 levels were observed in mice given two homologous doses of a LAV or heterologous prime and boost doses of the LAVs 90 days apart, i.e., groups 5, 8, and 10 in Table 7, although the GM anti-TS C12 titers of all 11 vaccine groups were significantly elevated compared with the KPhos group (p = 0.008 to 0.029). The GM Y. pestis TS C12 antibody titer of group 8 (320,000) was significantly greater than that of vaccine groups 2, 3, 6, 7, 9, 11, and 12 (p = 0.008 to 0.04). Apart from group 2 (which received one prime dose only), these groups had received a final dose on day 90 of rF1V. The presence of rF1V in the vaccines appears to mostly blunt the humoral response to Y. pestis TS C12. Generally, the humoral responses to Y. pestis TS C12 were not associated with protection.
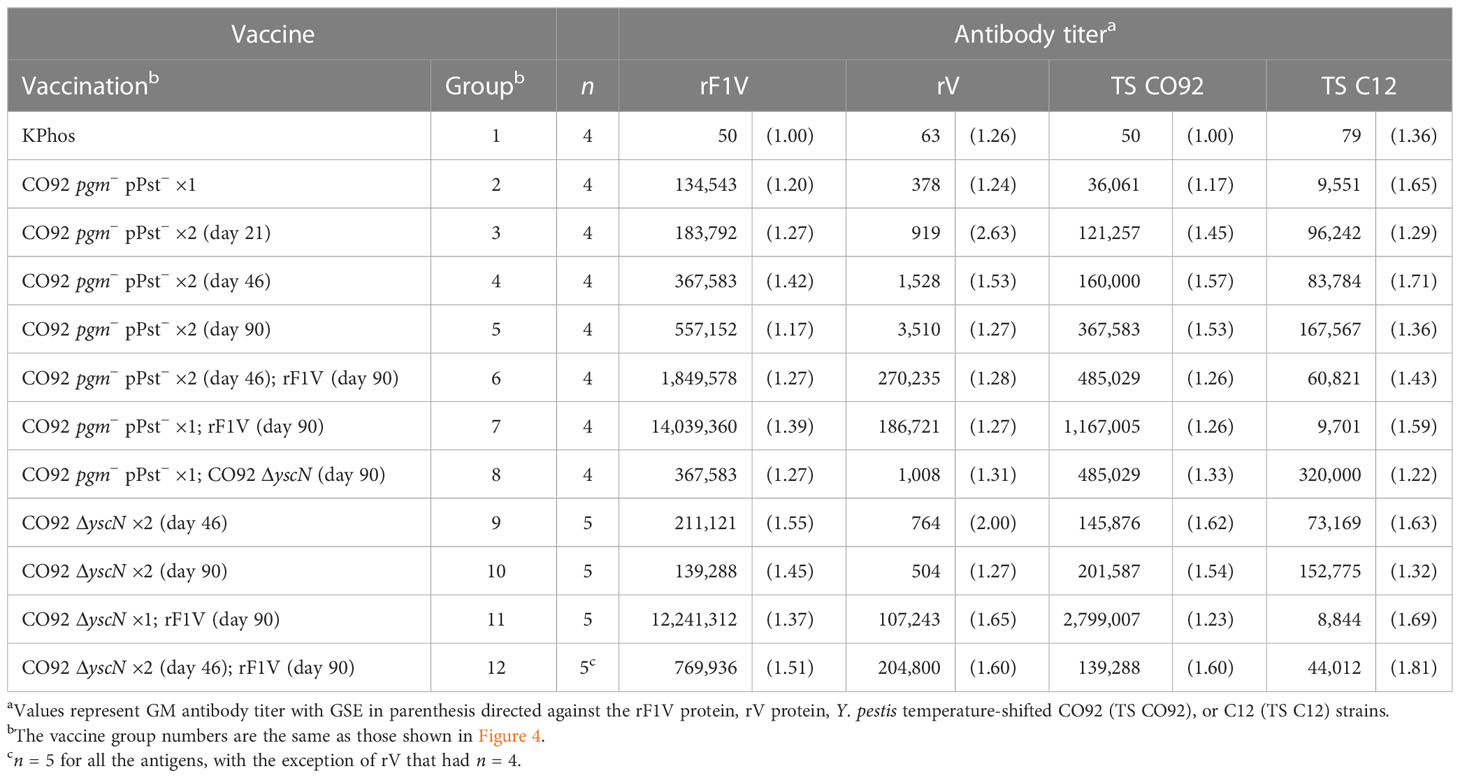
Table 7 Total IgG antibody response against rF1V, rV, Yersinia pestis CO92, or Y. pestis C12.
To assess cellular immune responses, spleens were collected from animals 1 day before the aerosol challenge for ELISpot and Luminex restimulation assays. ELISpot assays for IFN-γ were performed using unstimulated splenocytes or splenocytes stimulated with rF1V or Y. pestis TS C12. The unstimulated cells from the vaccine groups produced ≤15 IFN-γ-producing cells (SFC) per 106 splenocytes (Supplementary Figure 1). In contrast, splenocytes from all the vaccinated mice stimulated with rF1V secreted much higher levels than did the KPhos controls (Figure 5A), p = 0.0034 by pairwise comparison analyses. The greatest levels of IFN-γ were produced by rF1V-stimulated cells from mouse groups 6, 7, 11, and 12 (p < 0.0001). As illustrated for the humoral immune responses, these results again paralleled the differential protective efficacies afforded by the vaccines. Interestingly, as depicted in Figure 5B, Y. pestis TS C12 whole cells elicited IFN-γ maximally from groups 9 to 12 (p < 0.0001 compared with KPhos controls), all of which had been primed with the LAV CO92 ΔyscN and not with the CO92 pgm− pPst− vaccine. We hypothesize that the difference in IFN-γ production may be associated with the expression of V by the CO92 pgm− pPst LAV, as V is linked to immune suppression. Additionally, the altered secretion of the other Yops may also have contributed to the different immune responses observed. The numbers of SFCs from splenocytes of these four groups (Figure 5B) were greater than those produced after stimulation with rF1V (Figure 5A). IFN-γ expression by Y. pestis TS C12-stimulated cells was maximal for groups 11 and 12 (but not statistically different from groups 9 and 10; p = 0.17); both groups 11 and 12 had been primed with CO92 ΔyscN and boosted on day 90 with rF1V. These results contrast with the humoral responses to Y. pestis TS C12, where the inclusion of rF1V reduced antibody titers. In addition, this may reflect a greater role for cell-mediated immunity in protection associated with the ΔyscN LAVs relative to the CO92 pgm− pPst− vaccine.
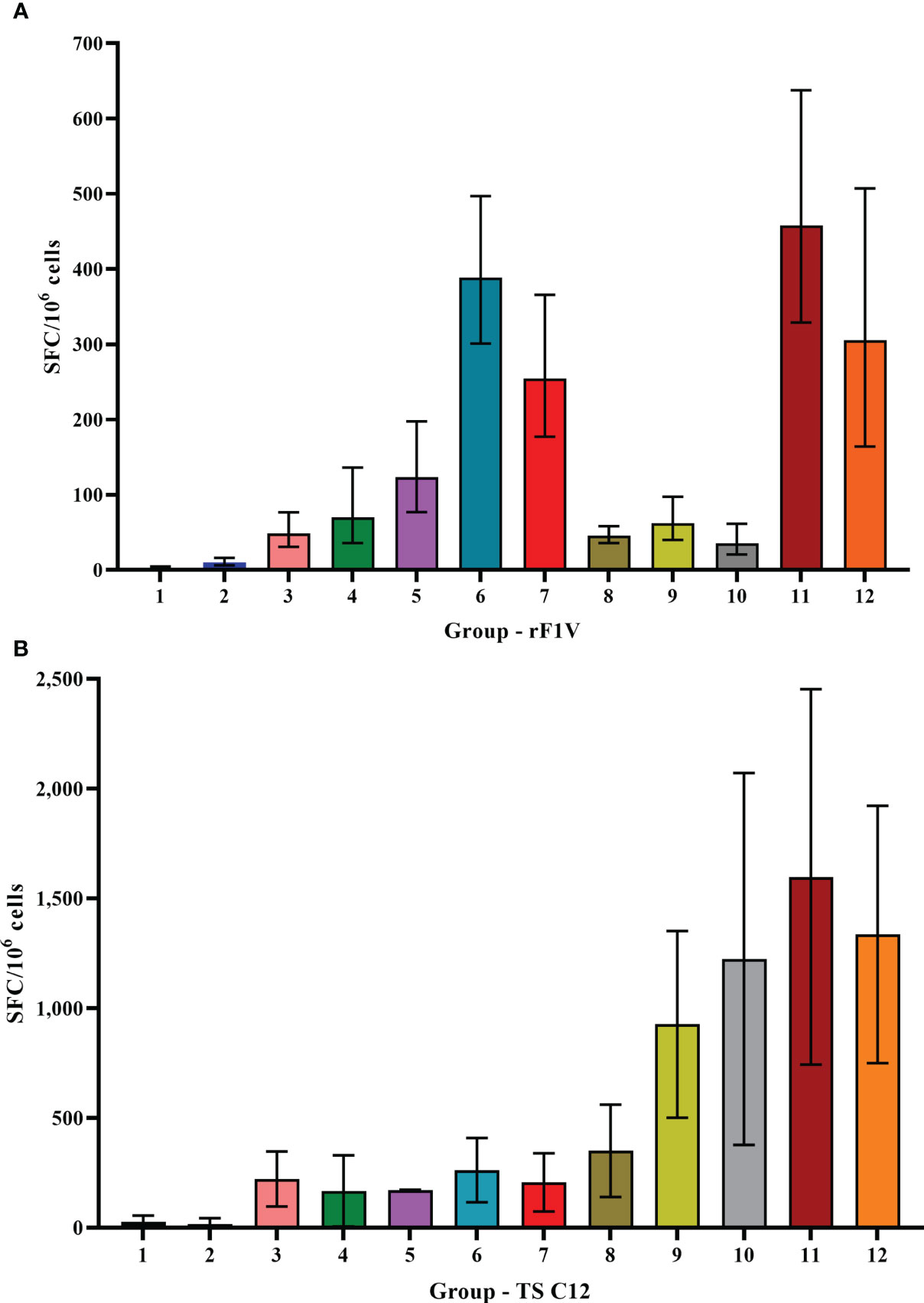
Figure 5 ELISpot assays for IFN-γ-secreting splenocytes collected post-vaccination and detected after stimulation with rF1V (A) or temperature-shifted, killed whole-cell TS C12 (B). The vaccine regimens provided to the groups are described in Table 6 embedded into manuscript section 3.3. SFC, spot-forming unit. The samples were obtained from four or five mice/group and the data were expressed as GM and GSE. Note the difference in the Y-axes ranges.
Cytokine production by stimulated splenocytes from vaccinated groups was analyzed with Luminex bead-based immunoassay. The levels of cytokines produced by splenocytes stimulated with rF1V of the vaccinated groups relative to the KPhos group are illustrated in Table 8 and Supplementary Table 5. Relative to KPhos, all CO92 ΔyscN-vaccinated groups promoted a strong cytokine response, with the most notable upregulation of MIP-1α, IL-2, MCP-3, MIP-1β, IL-3, IL-17A, MIP-2α, and RANTES. The addition of rF1V to CO92 ΔyscN-vaccinated mice drastically enhanced overall cytokine response, while the addition of rF1V to CO92 pgm− pPst−-vaccinated groups only moderately and selectively enhanced some cytokines, notably, MIP-1α, IL-2, MIP-1β, IL-4, and IL-6.
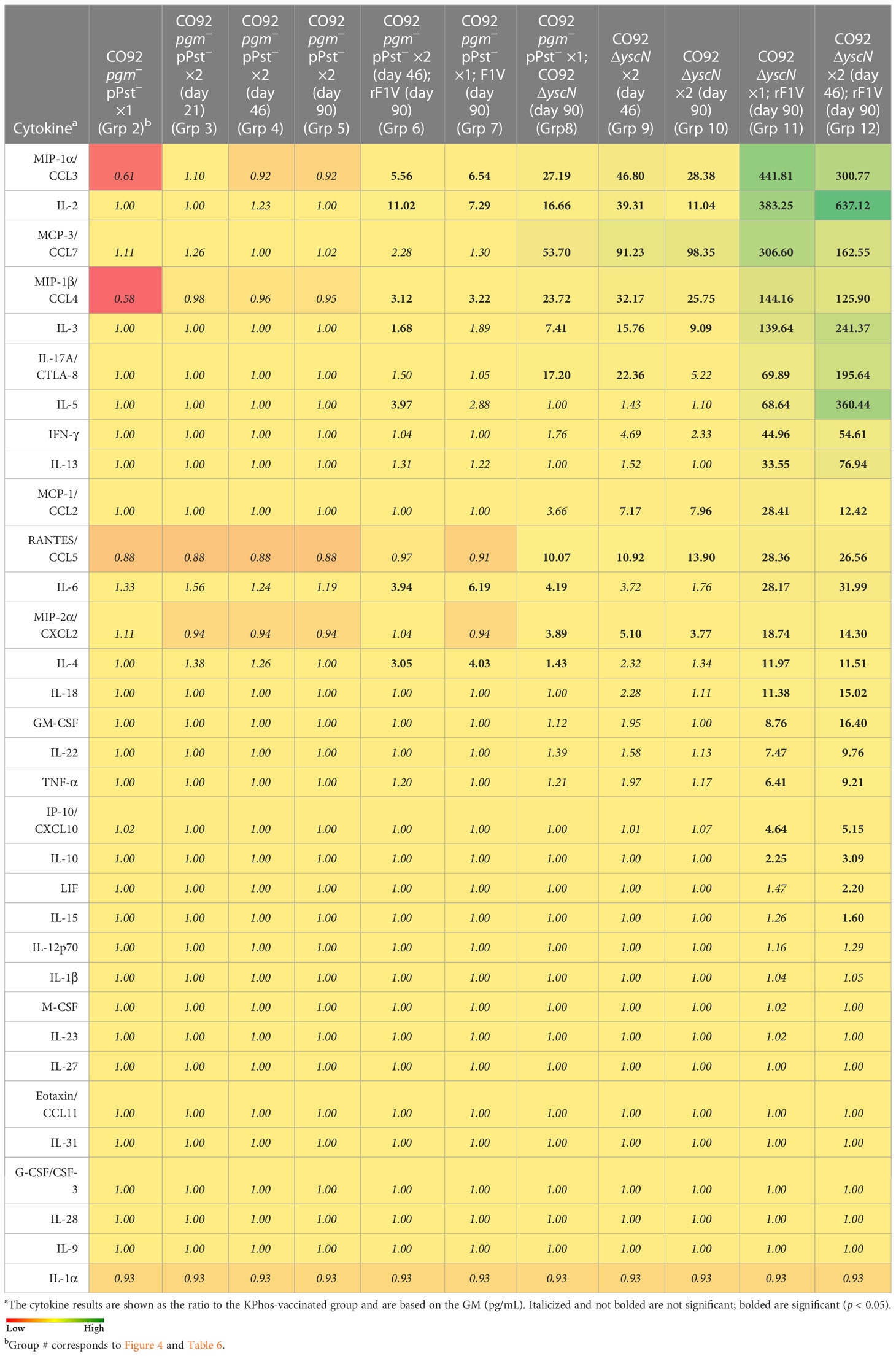
Table 8 Fold change relative to the KPhos-vaccinated group in splenocytes stimulated with rF1V.
Relative cytokine production by splenocytes stimulated with Y. pestis TS C12 showed an overall increased cytokine response for six groups (groups 2–7), all of which had received the CO92 pgm− pPst− LAV with or without rF1V. This increased response was most pronounced for mice vaccinated with a double dose of CO92 pgm− pPst− 46 days apart with or without rF1V (groups 4 and 6) (Table 9, Supplementary Table 6). Substantially greater responses were observed for five groups (groups 8–12), all of which were CO92 ΔyscN vaccinees (Table 9). These results agreed in part with the humoral antibody and ELISpot data and suggest that the elevated antibody responses to rF1V and Y. pestis TS CO92, and the heightened Y. pestis TS C12-stimulated cellular immune responses following CO92 ΔyscN vaccination, may be generally associated with protection. However, the associations between the cell-mediated immune responses and survival rates are not readily apparent, likely due to the low vaccine-mediated protection against the high challenge dose of Y. pestis C12 delivered in this study.
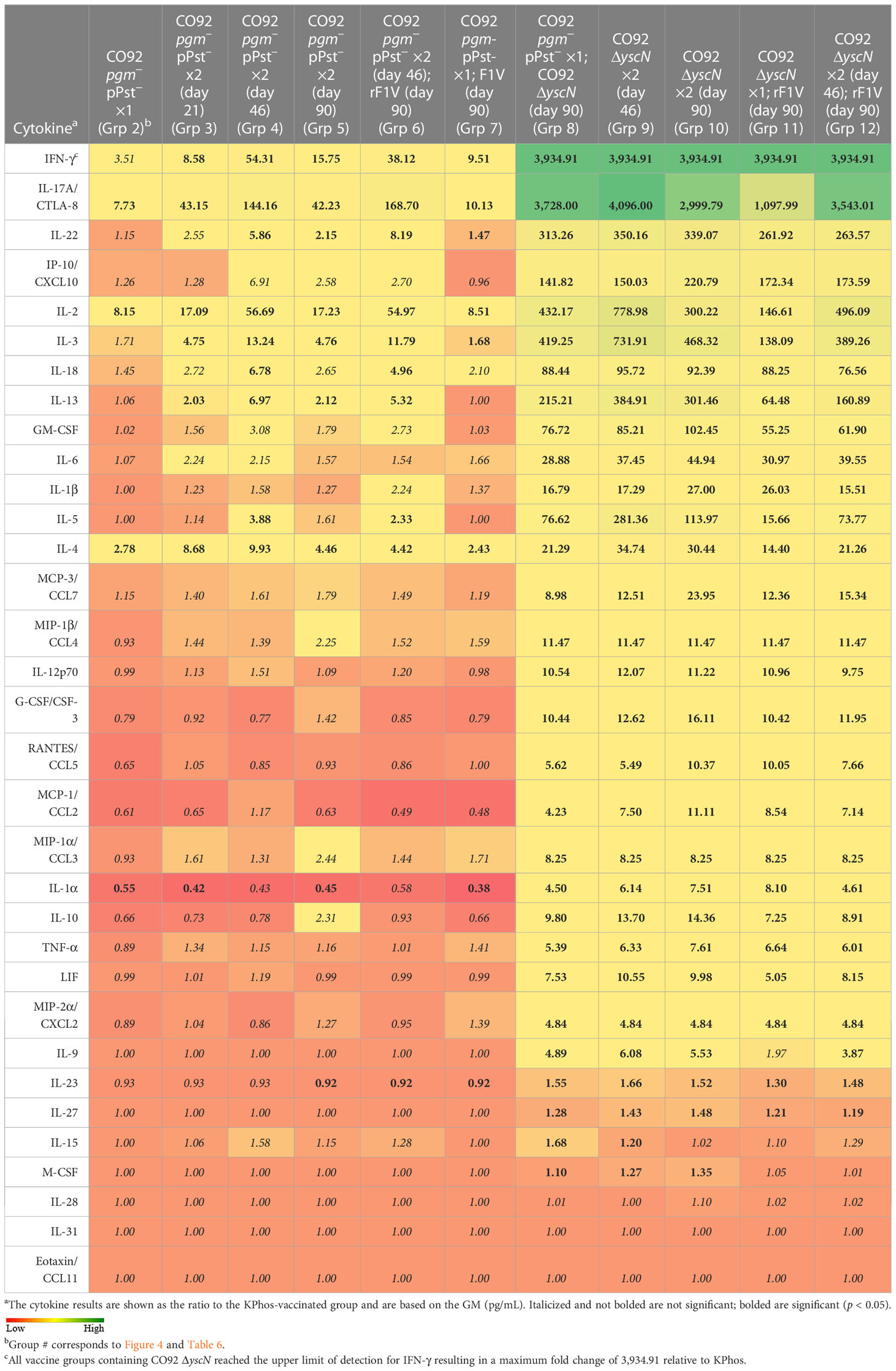
Table 9 Fold change relative to the KPhos-vaccinated group in splenocytes stimulated with TS C12.
Following possible exposure to Y. pestis, the time between infection and manifestation of lethal disease is brief. During that time, an individual needs to be properly diagnosed in order to receive the appropriate therapy; hence, any effort to prolong the window of opportunity for therapeutic intervention can dramatically improve disease outcome. The protection afforded to BALB/c mice against aerosol challenge with Y. pestis C12 by a suboptimal vaccination and delayed post-challenge antibiotic treatment strategy was examined. The mice were either vaccinated twice with mutant CO92 ΔyscN (21 days apart) or administered buffer alone and then challenged 4 weeks after the second vaccine dose with a lethal aerosol dose of Y. pestis C12. They were subsequently treated with 20 mg of streptomycin/kg (or buffer alone) beginning 48 or 60 h after challenge, and the effects on survival were determined, as shown in Figures 6A, B. The vaccination had no effect on the survival of mice after challenge with Y. pestis C12, while the administration of the antibiotic 48 h following challenge was associated with 90% and 100% of the treated (only) and vaccinated mice treated with antibiotics, respectively (Figure 6A). However, increasing the time between challenge and antibiotic treatment to 60 h revealed the enhanced effect on survival of the layered approach. Whereas all the mice treated with 20 mg/kg of streptomycin alone or vaccinated alone succumbed, 80% of the vaccinated and antibiotic-treated mice survived (p = 0.0007 vs. treated and p = 0.0011 vs. vaccinated alone); the median TTM of the treated group was extended compared with the unvaccinated and vaccinated control groups (Figure 6B), i.e., 7.0 days vs. 5.0 days (p = 0.0044) or vs. 5.5 days (p = 0.044), respectively. The benefits accrued from this vaccination–treatment scheme are further illustrated in Figure 6C. To improve the survival of unvaccinated mice treated with antibiotic at a delayed time (60 h vs. 48 h) after challenge, animals were given a higher dose of streptomycin. A total of 20% of mice receiving 40 mg/kg of streptomycin 60 h after challenge survived. In contrast, 70% of the CO92 ΔyscN-vaccinated and antibiotic-treated mice survived, an increase that approached significance on days 7 and 21 (p = 0.070). All of the mice in the two control groups succumbed by day 6, p = 0.0044 vs. CO92 ΔyscN-vaccinated and KPhos control groups compared with vaccinated and antibiotic-treated mice (Figure 6C). The C12 ΔyscN vaccine was evaluated similarly in the context of antibiotic treatment (Figure 7). Vaccination and treatment with 20 mg/kg of streptomycin 60 h post-challenge with Y. pestis C12 elicited significantly better protection against aerosol infection. In addition, the Y. pestis C12 challenge dose was higher in this experiment (1.67 × 106 CFU, 22 LD50s) than that used in the study in Figure 6 (GM 3.62 × 105 CFU, 5 LD50s). Half of the vaccinated and antibiotic-treated mice survived, and none of the vaccinated only or antibiotic-treated only controls survived (p = 0.0325 for survival on days 7 and 21 after challenge). This layered therapy revealed that the combinations of vaccination and post-exposure antibiotic treatment were synergistic in the challenge experiments shown in Figure 6B (synergy score 4.84, p = 0.0013), Figure 6C (synergy score 2.66, p = 0.040), and Figure 7 (synergy score 3.16, p = 0.009). These synergistic effects allowed the treatment to be delayed after exposure to aerosolized Y. pestis and may facilitate a sparing effect on the amount of antibiotic required.
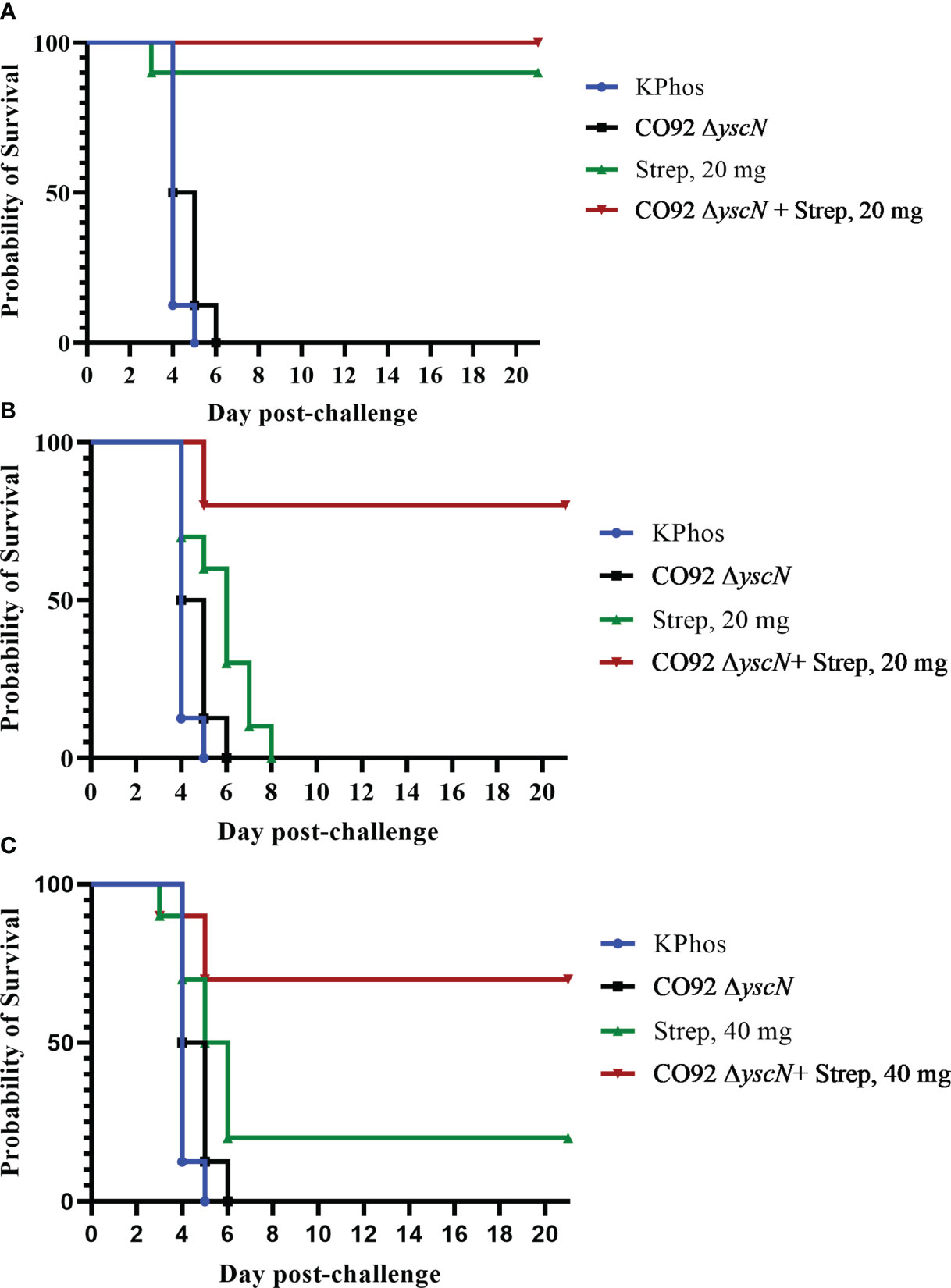
Figure 6 The protection of BALB/c mice against lethal aerosol challenge with Yersinia pestis strain C12 by a vaccination and post-challenge antibiotic treatment strategy: the impact on survival of antibiotic dose and time of treatment. In each experiment, two groups of mice were vaccinated twice (20 days apart) with mutant CO92 ΔyscN (0.92 × 107 CFU and 0.48 × 107 CFU/mouse, respectively), and two groups were unvaccinated. All mice were challenged with aerosolized Y. pestis C12 28 days after the boost dose. The challenge doses were administered in two iterations, with doses of 5.6 × 105 CFU/mouse (6 LD50s) for the vaccinated groups and 3.11 × 105 CFU (4 LD50s) for unvaccinated mice. The mice were treated with either 20 mg/kg of streptomycin starting at 48 h (A) or 60 h (B) after challenge or 40 mg/kg starting at 60 h after challenge (C). n = 8 mice/group (KPhos alone and vaccine alone) or 10 mice/group (streptomycin alone and vaccine + streptomycin).
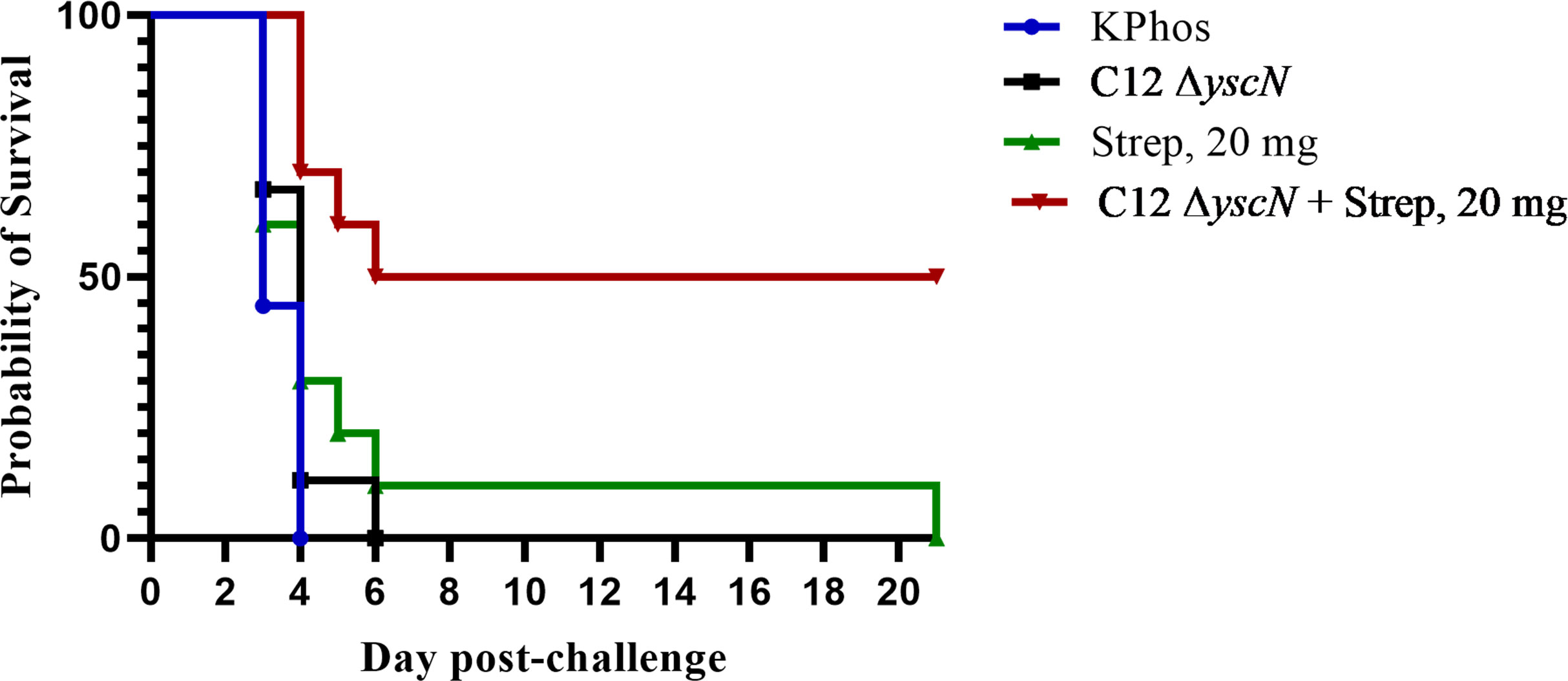
Figure 7 The protection of BALB/c mice against lethal aerosol challenge with Yersinia pestis strain C12 by a vaccination and post-challenge antibiotic treatment strategy. The mice were vaccinated twice (23 days apart) with Y. pestis C12 ΔyscN (0.48 × 107 and 0.70 × 107 CFU for doses 1 and 2, respectively); controls received KPhos alone. The mice were challenged 28 days later with 1.67 × 106 CFU (22 LD50s) of aerosolized Y. pestis C12. They were either untreated or treated with 20 mg/kg of streptomycin 60 h after challenge with Y. pestis strain C12 4 weeks after the boost dose. n = 9 mice/group (KPhos alone and vaccine alone) or 10 mice/group (streptomycin alone and vaccine + streptomycin).
Most wild-type strains of Y. pestis produce an outer capsule composed of a polymer of fraction 1 protein (F1) encoded by the caf1 operon of the pMT1 plasmid. F1 is a highly immunogenic and protective virulence factor that appears to have a role in preventing phagocytosis. However, the primary Y. pestis virulence factors that block uptake by phagocytosis are those encoded by the type 3 secretion system (T3SS) (Du et al., 2002). In addition, strains with mutations preventing F1 synthesis frequently retain virulence in various mammalian models of plague, especially in mouse and guinea pig pneumonic plague models (Drozdov et al., 1995; Friedlander et al., 1995; Welkos et al., 1995; Worsham et al., 1995; Davis et al., 1996; Du and Wang, 2016; Biryukov et al., 2021). These strains have not only been produced experimentally but have also been recovered in natural settings of plague infection (Winter et al., 1960; Anderson et al., 1997). nonencapsulated strains have been isolated in Indonesia and Kazakhstan (Meka-Mechenko, 2003; Eppinger et al., 2012), and while the impact on human populations is not well documented, a nonencapsulated or weakly encapsulated strain may have been associated with a fatal case of plague in the USA in the 1950s (Winter et al., 1960). Thus, the virulence of F1-negative strains has been widely documented, and the findings support the conclusion that new-generation vaccines must protect against infection by nonencapsulated strains (Winter et al., 1960; Drozdov et al., 1995; Friedlander et al., 1995; Welkos et al., 1995; Worsham et al., 1995; Davis et al., 1996; Samoilova et al., 1996; Heath et al., 1998; Powell et al., 2005; Quenee et al., 2008; Sebbane et al., 2009; Sha et al., 2011; Du and Wang, 2016).
Nevertheless, F1-deficient strains are not as virulent as F1+ in certain situations, and their virulence may vary depending on the Y. pestis strain, exposure dose and route, and animal host, i.e., strain, sex, or source (Donavan et al., 1961; Williams and Cavanaugh, 1984; Friedlander et al., 1995; Welkos et al., 2004; Sebbane et al., 2009; Weening et al., 2011). The F1 antigen appears to contribute to Y. pestis virulence in some mammalian models of bubonic and sometimes pneumonic plague (Donavan et al., 1961; Williams and Cavanaugh, 1984; Samoilova et al., 1996; Du et al., 2002; Sha et al., 2011; Weening et al., 2011; Du and Wang, 2016). For example, in BALB/c mice, the LD50 values of the F1+ CO92 and F1− C12 strains are approximately the same by the aerosol route (6.8–7.7 × 104 CFU) (Heine et al., 2007; Biryukov et al., 2021), but CO92 has an approximately five-fold lower LD50 than C12 by the SC route (bubonic model), ≤1.9 CFU vs. 9.1 CFU, respectively (Worsham et al., 1995; Biryukov et al., 2021; Cote et al., 2021). Sha et al. reported similar results with BALB/c mice from a different vendor, and Weening and coworkers observed F1-associated virulence differences in both bubonic and pneumonic models in C57BL/J mice (Sha et al., 2011; Weening et al., 2011). The absence of F1 impacted virulence to a greater extent in C57BL/6 mice than in BALB/c mice, showing the effect of animal background on virulence (Weening et al., 2011). In the outbred CD-1 mouse strain, the SC LD50s of C12 and CO92 differed by 18-fold, i.e., 942 CFU and 52 CFU, respectively; similar findings were observed using outbred Swiss–Webster mice (Sebbane et al., 2009; Biryukov et al., 2021; Cote et al., 2021).
In previous studies, we characterized the protective efficacy of several live attenuated mutant strains derived from Y. pestis strains CO92 or KIM6+ against bubonic and aerosol challenges of BALB/c mice with the wild-type encapsulated CO92 strain. Three of the LAV strains were confirmed to be safe in high doses (Cote et al., 2021), and one dose of either F1+ CO92 mutant (the ΔyscN or pgm− pPst− strain) protected BALB/c mice against infection by subcutaneous or inhalational routes to Y. pestis CO92. Full protection against CO92 in this model required the induction of an immune response to F1 (Cote et al., 2021). Two vaccines (CO92 ΔyscN, alone or combined with C12 ΔyscN) were down-selected for further analyses of protection against other virulent strains and especially those deficient in capsule synthesis. The ΔyscN strains do not secrete the V antigen due to the disrupted T3SS but do retain some V protein associated with the bacterial cells (Bozue et al., 2012), and in the case of the C12-derived vaccine strain, no F1 was produced. Thus, they allowed us to investigate the protection afforded by potentially novel antigens, such as non-capsule surface antigens displayed by the C12 ΔyscN, e.g., Pla, Ail, and LPS (Wang et al., 2020).
The current work extended these studies by characterizing protection afforded by these LAVs, with or without the defined rF1V subunit vaccine, against the challenge with the nonencapsulated and vaccine-resistant C12 strain of Y. pestis. We found that the most protective vaccination scheme, as tested under various conditions and in both bubonic and pneumonic plague models, was heterologous vaccination with a live vaccine (Combo LAV composed of the C12 and CO92 ΔyscN mutants, or the CO92 pgm− pPst− strain) and a separate dose of a protein subunit vaccine. Although one or two identical doses of either ΔyscN mutant alone or combined provided significant protection against bubonic plague, they were poorly protective against pneumonic challenge (Figures 1, 2). With the inclusion of a subunit rF1V vaccine, the heterologous vaccination approach was optimally protective against aerosol challenge regardless of changes in vaccination schedule and challenge conditions. The latter included the spacing between prime vaccine doses and subsequent booster dose(s) which was predicted to optimize antibody response (Castiglione et al., 2012). For instance, heterologous vaccination increased the survival rate or extended the survival time to a greater extent than did two doses of Combo LAV when mice were exposed to a significantly elevated C12 challenge. Additionally, in a heterologous scenario (Figures 3, 4), a different LAV, i.e., the CO92 pgm− pPst− strain, was as efficacious as the Combo LAV, when paired with rF1V booster. This situation requires further examination, but it potentially affords flexibility in the selection of vaccine components.
In efforts to improve the rF1V vaccine, Amemiya et al. and Biryukov et al. showed that the addition of TLR agonist CpG2006 to rF1V augmented vaccine efficacy and immunogenicity (Amemiya et al., 2009; Biryukov et al., 2021). A similarly protective role of CpG in mice vaccinated with LAVs or protein subunit combination vaccines was examined (Figure 3). Our results suggested that CpG enhanced protection afforded by the heterologous Combo LAV and rF1V vaccine against the Y. pestis C12 aerosol challenge (Figures 3B, C). Although CpG might not be required for maximal protection against the bubonic plague model described here, there may be scenarios where the addition of CpG could improve disease outcome (Figure 3A).
The impact of vaccination on accelerating bacterial clearance or inhibition of dissemination in vivo was also determined by quantitatively assessing bacterial loads in tissues 3 days after the aerosol challenge. The organ and blood levels of Y. pestis C12 in mice vaccinated with the heterologous vaccine (Combo LAV prime and rF1V + CpG boost) were the lowest compared with those in the other control and vaccinated groups. There appeared to be a relationship between CFU and protection, i.e., lower CFU load in the lungs at day 3 was associated with vaccines’ protective efficacy, albeit a statistical correlation was precluded due to low numbers of samples. Furthermore, the survival rates of the two groups vaccinated with Combo LAV plus rF1V (+/− CpG) were not significantly different (90% and 50%, respectively), yet the mean TTM post-challenge of the group receiving the CpG immunostimulant was increased significantly compared with that of most other groups.
We also evaluated the associations between the extent of protection and the immune responses elicited by the vaccine. The heterologous vaccination groups with the highest survival rates had the greatest serum titers to rF1V and rV and the CO92 whole-cell antigens, and their splenocytes secreted higher levels of IFN-γ after rF1V stimulation in ELISpot assays. These findings confirm the strong immunogenicity of rF1V in BALB/c mice as demonstrated in our initial vaccine studies using the live attenuated plague strains (Cote et al., 2021); full protection against F1+ Y. pestis CO92 required induction of a strong humoral anti-F1 response. The consistently greater protection against aerosol exposure to Y. pestis C12 of the heterologous, rF1V-containing vaccines compared with homologous single or Combo LAVs (Figures 3B, C, 4) supports a role for rF1V-associated immune responses in vaccine efficacy against C12. Importantly, although the ΔyscN-based vaccines (and in general most live vaccines designed to ameliorate plague) do not generate a robust antibody response against secreted V antigen, there appears to be enough V protein associated with the cell that can act as a prime vaccination against this antigen. Once boosted with rF1V (+/− CpG), there is an appreciable increase in anti-rV titers. These titers are greater than those observed in mice receiving a single dose of rF1V (+/− CpG), but this increase is not statistically significant. This lack of statistical significance in anti-rV titers among groups 3, 4, 10, and 11 (Table 3, Figure 3B) is an important point of discussion as it implies that there are other immunogens that were provided by the LAV prime vaccination that are ultimately increasing vaccine efficacy in the mice (survival of groups 3 and 4 compared with groups 10 and 11 in Figure 3B), and protection could not be correlated with anti-rV titers alone. Accordingly, we continue to explore and characterize the immune response generated by our LAVs in the mouse model.
In a previous study with our LAVs (alone), cell-mediated immunity involving elevated Th17- and Th2-associated cytokines was associated with protection against the encapsulated CO92 strain (Cote et al., 2021). The findings were obtained using splenocytes collected before the aerosol challenge. Although an association with protection against the C12 strain was not clearly observed with stimulated splenocytes collected pre-exposure from the extended boost time study (Figure 4), none of the vaccines included CpG and none were significantly protective in this high-dose challenge study. In contrast, with some exceptions, protection was associated with overall lower expression levels of cytokines in tissue extracts obtained after challenge with C12 in heterologously vaccinated mice relative to KPhos-vaccinated mice. This controlled cytokine response potentially mitigates excessive Th17 cell and neutrophil migration (CXCL1, CXCL2) as well as activation (G-CSF, IL-17A, IL-1β, IL-6, IL-22) which may contribute to reduced inflammation-related destruction of lung tissue (Table 4). In addition, the inability to control the infection may predispose mice to excessive alveolar macrophage and eosinophil activation and recruitment via colony-stimulating factors (G-CSF, M-CSF, GM-CSF). Excessive GM-CSF production by lung epithelium may also be a direct correlate of lung damage and act as a marker of futile efforts to repair and restore epithelial barrier function (Trapnell and Whitsett, 2002; Standiford et al., 2012; Chen et al., 2016; Rosler and Herold, 2016).
Thus, we intend to investigate the protective role for vaccine-associated dampening of the cytokine storm induced by exposure to virulent F1+ and F1− Y. pestis strains. We predict that a fine-tuned modulation between a protective and damaging extent of cytokine expression is required for optimal vaccine efficacy. Cytokine expression which was either excessively high or low was associated with poor protection, as suggested for example by the cytokine profiles of restimulated splenocytes described in Table 9. Of the two most protective vaccine groups (CO92 pgm− pPst− + rF1V and CO92 ΔyscN + rF1V) that afforded the same level of protection, there appeared to be a distinct immune response that induced a relatively low anti-TS C12 antibody response in both groups. Controlled upregulation of IFN-γ, IL-17A, IL-2, and IL-4 in the CO92 pgm− pPst− + rF1V-vaccinated mice appeared to correlate with protection, while controlled upregulation of Th2- (IL-4, IL-5, IL-13) and Th17 (IL-17A, IL-22) along with the pleiotropic IL-2 and IL-3 cytokines was associated with CO92 ΔyscN + rF1V-vaccinated mice. It is important to note that our cytokine data are collected from a single time point and these expression levels and their correlation with protection may differ throughout the course of infection. However, the anti-rF1V and anti-CO92 antibody responses were the highest in the two most protective vaccines and may be used as correlates of protection. The cytokine response against TS C12 was much more pronounced with vaccines that included CO92 ΔyscN relative to CO92 pgm− pPst−, and yet the level of protection was the same. Following rF1V stimulation, the number of IFN-γ-secreting splenocytes after 24 h was relatively high in all rF1V vaccine groups, but after 48 h, the levels of IFN-γ were substantially higher in all CO92 ΔyscN vaccine groups, especially in groups that were also administered rF1V. The short half-life of IFN-γ may be an indicator of this large discrepancy between the two time points for the different vaccine formulations, such that the CO92 pgm− pPst− + rF1V vaccine group produces a strong yet transient response that recedes by 48 h relative to the CO92 ΔyscN + rF1V that appears to be longer in duration and more intense (Lortat-Jacob et al., 1996). Furthermore, TS C12 antigen stimulated a greater IFN-γ response in splenocytes from mouse groups vaccinated with CO92 ΔyscN mutant (+/− rF1V: groups 9–12) and not CO92 pgm− pPst− (+/− rF1V: groups 2–7).
In addition to the enhanced efficacy and potential modularity of our combination vaccine approach against both F1+ and F1− Y. pestis strains, an advantage inherent with the ΔyscN mutant LAVs is their inclusion of antigens some of which are not expressed in other live vaccines, i.e., potential antigens encoded by the chromosomal 102-kb pgm locus, by other Y. pestis chromosomal genes, and/or by plasmids pMT1 (pFra) and pPcP1 (pPst). However, the CO92 pgm− pPst− strain was also highly protective, possibly due in part to an immune response to the secreted T3SS Yops.
The V antigen is an essential component of the Y. pestis T3SS and an indispensable virulence factor of Y. pestis; thus, it is an obvious target for countermeasure development. However, although V-specific immunity has been correlated with protection, the adaptive immune responses to V are complicated and may not be essential to an effective vaccine. In our previous work (Cote et al., 2021), the ΔyscN vaccines did not induce a significant anti-V antibody response, as expected due to the requirement of the YscN ATPase for a functional T3SS (Bozue et al., 2012). In this current work, we noted a significant increase in anti-rV titers when the F1-V subunit vaccine was used as a booster. However, the lack of a robust anti-V response in animals or humans vaccinated with live attenuated F1+ and F1− Y. pestis strains has been documented repeatedly (Williamson et al., 1995; Quenee et al., 2008; Brasiale et al., 2009; Qiu et al., 2010; Bozue et al., 2012; Sun et al., 2014; Demeure et al., 2019; Feodorova et al., 2020; Cote et al., 2021). The basis of this observation has not been fully discerned but may be related to the V-producing strain of live vaccine and possibly the animal host. The potential role of the immune-modulatory activity of V in the suppression of the host immune responses in the context of LAVs remains to be clarified (Leary et al., 1995; Brubaker, 2003; Depaolo et al., 2008; Quenee et al., 2011; Feodorova and Motin, 2012; Daniel et al., 2016; Feodorova et al., 2020). However, our data suggest that a LAV followed by an rF1V booster can overcome the low V titers typically observed with LAVs and this heterologous strategy may represent a superior vaccination strategy.
To maximize protection against infection, we also compared the efficacies of pre-exposure vaccination and post-exposure antibiotic treatment, alone and combined, against inhalational challenge with Y. pestis C12 strains. A layered defense strategy employing vaccination with the ΔyscN mutant of the wild-type or F1-deficient strain, separately or combined, and treatment with streptomycin provided optimal protection and was clearly synergistic (Figures 6, 7). These promising results predict that the “pre–post” strategy will extend the time post-exposure when antibiotic administration must be initiated to be successful. Our ongoing studies focus on assessing the limits of this strategy as well as its impact on vaccination with the heterologous LAV/subunit constructs.
The layered countermeasure strategy offers several attractive advantages to single medical countermeasure strategies. As observed in animal studies, the efficacy of antibiotic treatment of Warfighters exposed to a bolus of aerosolized Y. pestis is predicted to be enhanced in prophylactically vaccinated compared with unvaccinated individuals. Vaccination is also expected to extend the window of time post-exposure during which antibiotics are effective, i.e., before reaching the “point of no return.” Concomitantly, for vaccinees with waning immunity, post-exposure antibiotic treatment could potentially enhance the protective effects of the vaccine. This might be especially important in a biothreat scenario where the limited time before potential exposure allowed only a single administration of a plague vaccine. Finally, strategies employing layered or combination countermeasures afford vaccine antigen/antibiotic dose-sparing and potentially allow the use of second-line antibiotics as needed, e.g., in situations involving naturally or engineered antibiotic-resistant challenge strains (Zauberman et al., 2019; Klimko et al., 2022b).
Additional studies which evaluated combinations of vaccines and therapeutics against critical pathogens have been reported. In a mouse intranasal challenge model of pneumonic plague, Zauberman and coworkers showed that post-exposure vaccination with the live pgm− Y. pestis strain EV76 enhanced the efficacy of antibiotic treatment against the virulent Kim53 strain of Y. pestis. Optimal protection was achieved when the vaccine was administered subcutaneously at the same time as an intranasal virulent Y. pestis challenge, and antibiotic treatment was begun 48 h after challenge (Zauberman et al., 2019). Also, such combination countermeasure strategies are not restricted to one pathogen or model. The potential broad applicability of a similar protection strategy was exemplified in our recent studies with Burkholderia pseudomallei, the etiologic agent of melioidosis (Klimko et al., 2022a). Burkholderia pseudomallei is intrinsically resistant to many commonly used antibiotics, and infections with it are notoriously difficult to eradicate. Effective antibiotic treatment regimens are complex and of long duration. Klimko et al. demonstrated that when used in a layered approach, established vaccine strategies employing recombinant protein subunit conjugates or a LAV strain, and the antibiotic co-trimoxazole, produced high levels of protection in mice.
Further efforts are being pursued to optimize the vaccination and integrated prophylactic/post-exposure countermeasure strategies. These include a determination of the latest time post-exposure to aerosolized bacteria when antibiotic treatment can rescue the mice and the lowest effective dose of antibiotic; the optimal heterologous vaccine composition, order of delivery, and schedule; the most synergistic antibiotic(s) for various vaccine strategies; and, in the context of single vaccination, the longest interval between vaccination and challenge after which the antibiotic treatment remains more effective than in unvaccinated treated individuals. BALB/c mice are an appropriate early model for both pathogenesis research and medical countermeasure development; however, due to differences in LD50 estimations and other inherent differences, their recapitulation of human disease remains equivocal, and thus, follow-on work will also be required in a nonhuman primate model of pneumonic plague. Additionally, plague vaccine candidates have generally been evaluated using only one virulent strain. However, many distinct strains of virulent Y. pestis have been isolated from multiple geographical sources (Vogler et al., 2016), and analyses of vaccines for their performance against such strains are needed.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal studies were approved by the United States Army Medical Research Institute of Infectious Diseases (USAMRIID) IACUC. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was not obtained from the owners for the participation of their animals in this study because the mice were purchased for research.
SB and CC designed and supervised the project. SB, CK, JD, RT, JS, MH, NR, YT, MD, JB, and CC prepared the reagents, treatments, and vaccines and performed the experiments. SB, CK, MD, JQ, DF, SW, and CC performed the data analyses. SB, SW and CC wrote the first draft. SB, JB, SW, and CC revised and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the US Defense Threat Reduction Agency (DTRA) under projects CB10392 and CB10794.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors CC and JB declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army or the Department of Defense Health Agency.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbrio.2023.1240698/full#supplementary-material
Adamovicz J. J., Worsham P. L. (2012). “Laboratory Animal Models of Plague,” in Biodefense: Research Methodology and Animal Models (Boca Raton, FL: CRC Press), 113–145.
Aftalion M., Tidhar A., Vagima Y., Gur D., Zauberman A., Holtzman T., et al. (2023). Rapid Induction of Protective Immunity against Pneumonic Plague by Yersinia pestis Polymeric F1 and LcrV Antigens. Vaccines (Basel) 2, 11. doi: 10.3390/vaccines11030581
Alibek K. (1999). Biohazard: The Chilling True Story of the Largest Covert Biological Weapons Program in the World - Told from the Inside by the Man Who Ran It New York (NY: Random House, Inc).
Amemiya K., Meyers J. L., Rogers T. E., Fast R. L., Bassett A. D., Worsham P. L., et al. (2009). CpG oligodeoxynucleotides augment the murine immune response to the Yersinia pestis F1-V vaccine in bubonic and pneumonic models of plague. Vaccine 27, 2220–2229. doi: 10.1016/j.vaccine.2009.02.016
Amemiya K., Meyers J. L., Trevino S. R., Chanh T. C., Norris S. L., Waag D. M. (2006). Interleukin-12 induces a Th1-like response to Burkholderia mallei and limited protection in BALB/c mice. Vaccine 24, 1413–1420. doi: 10.1016/j.vaccine.2005.09.021
Anderson G. W. Jr., Worsham P. L., Bolt C. R., Andrews G. P., Welkos S. L., Friedlander A. M., et al. (1997). Protection of mice from fatal bubonic and pneumonic plague by passive immunization with monoclonal antibodies against the F1 protein of Yersinia pestis. Am. J. Trop. Med. Hyg. 56, 471–473. doi: 10.4269/ajtmh.1997.56.471
Andrews G. P., Strachan S. T., Benner G. E., Sample A. K., Anderson G. W. Jr., Adamovicz J. J., et al. (1999). Protective efficacy of recombinant Yersinia outer proteins against bubonic plague caused by encapsulated and nonencapsulated Yersinia pestis. Infect. Immun. 67, 1533–1537. doi: 10.1128/IAI.67.3.1533-1537.1999
Anisimov A. P., Dentovskaya S. V., Panfertsev E. A., Svetoch T. E., Kopylov P., Segelke B. W., et al. (2010). Amino acid and structural variability of Yersinia pestis LcrV protein. Infect. Genet. Evol. 10, 137–145. doi: 10.1016/j.meegid.2009.10.003
Baril L., Valles X., Stenseth N. C., Rajerison M., Ratsitorahina M., Pizarro-Cerda J., et al. (2019). Can we make human plague history? A call to action. BMJ Glob Health 4, e001984. doi: 10.1136/bmjgh-2019-001984
Bevins S. N., Chandler J. C., Barrett N., Schmit B. S., Wiscomb G. W., Shriner S. A. (2021). Plague exposure in mamMalian wildlife across the western United States. Vector Borne Zoonotic Dis. 21, 667–674. doi: 10.1089/vbz.2020.2765
Biryukov S., Dankmeyer J. L., Shamsuddin Z., Velez I., Rill N. O., Rosario-Acevedo R., et al. (2021). Impact of Toll-Like Receptor-Specific Agonists on the Host Immune Response to the Yersinia pestis Plague rF1V Vaccine. Front. Immunol. 12, 726416. doi: 10.3389/fimmu.2021.726416
Bozue J., Cote C. K., Chance T., Kugelman J., Kern S. J., Kijek T. K., et al. (2014). A Yersinia pestis tat mutant is attenuated in bubonic and small-aerosol pneumonic challenge models of infection but not as attenuated by intranasal challenge. PloS One 9, e104524. doi: 10.1371/journal.pone.0104524
Bozue J., Cote C. K., Webster W., Bassett A., Tobery S., Little S., et al. (2012). A Yersinia pestis YscN ATPase mutant functions as a live attenuated vaccine against bubonic plague in mice. FEMS Microbiol. Lett. 332, 113–121. doi: 10.1111/j.1574-6968.2012.02583.x
Brasiale V., Nash M., Sinha N., Zudina I., Motin V. (2009). “Correlates of Immunity Elicited by Live Yersinia pestis Vaccine,” in NIH volume 1, Frontiers in Research Totawa (NJ: Humana Press), 473–480.
Brubaker R. R. (2003). Interleukin-10 and inhibition of innate immunity to Yersiniae: roles of Yops and LcrV (V antigen). Infect. Immun. 71, 3673–3681. doi: 10.1128/IAI.71.7.3673-3681.2003
Cabanel N., Bouchier C., Rajerison M., Carniel E. (2018). Plasmid-mediated doxycycline resistance in a Yersinia pestis strain isolated from a rat. Int. J. antimicrobial. agents. 51, 249–254. doi: 10.1016/j.ijantimicag.2017.09.015
Castiglione F., Mantile F., De Berardinis P., Prisco A. (2012). How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math Methods Med. 2012, 842329. doi: 10.1155/2012/842329
Chen G. H., Teitz-Tennenbaum S., Neal L. M., Murdock B. J., Malachowski A. N., Dils A. J., et al. (2016). Local GM-CSF-dependent differentiation and activation of pulmonary dendritic cells and macrophages protect against progressive cryptococcal lung infection in mice. J. Immunol. 196, 1810–1821. doi: 10.4049/jimmunol.1501512
Cote C. K., Biryukov S. S., Klimko C. P., Shoe J. L., Hunter M., Rosario-Acevedo R., et al. (2021). Protection Elicited by Attenuated Live Yersinia pestis Vaccine Strains against Lethal Infection with Virulent Y. pestis. Vaccines (Basel). 16, 9. doi: 10.3390/vaccines9020161
Cowdrey A. E. (1984). "Germ warfare" and public health in the Korean conflict. J. Hist Med. Allied Sci. 39, 153–172. doi: 10.1093/jhmas/39.2.153
Daniel C., Dewitte A., Poiret S., Marceau M., Simonet M., Marceau L., et al. (2019). Polymorphism in the Yersinia LcrV antigen enables immune escape from the protection conferred by an LcrV-secreting Lactococcus lactis in a pseudotuberculosis mouse model. Front. Immunol. 10, 1830. doi: 10.3389/fimmu.2019.01830
Daniel C., Titecat M., Poiret S., Cayet D., Boutillier D., Simonet M., et al. (2016). Characterization of the protective immune response to Yersinia pseudotuberculosis infection in mice vaccinated with an LcrV-secreting strain of Lactococcus lactis. Vaccine 34, 5762–5767. doi: 10.1016/j.vaccine.2016.09.060
Davis K. J., Fritz D. L., Pitt M. L., Welkos S. L., Worsham P. L., Friedlander A. M. (1996). Pathology of experimental pneumonic plague produced by fraction 1-positive and fraction 1-negative Yersinia pestis in African green monkeys (Cercopithecus aethiops). Arch. Pathol. Lab. Med. 120 (2), 156–163.
Demeure C. E., Derbise A., Guillas C., Gerke C., Cauchemez S., Carniel E., et al. (2019). Humoral and cellular immune correlates of protection against bubonic plague by a live Yersinia pseudotuberculosis vaccine. Vaccine 37, 123–129. doi: 10.1016/j.vaccine.2018.11.022
Demidenko E., Miller T. W. (2019). Statistical determination of synergy based on Bliss definition of drugs independence. PloS One 14, e0224137. doi: 10.1371/journal.pone.0224137
Depaolo R. W., Tang F., Kim I., Han M., Levin N., Ciletti N., et al. (2008). Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe 4, 350–361. doi: 10.1016/j.chom.2008.09.004
Donavan J. E., Ham D., Fukui G. M., Surgalla M. J. (1961). Role of the capsule of Pasteurella pestis in bubonic plague in the Guinea pig. J. Infect. Dis. 109, 154–157. doi: 10.1093/infdis/109.2.154
Drozdov I. G., Anisimov A. P., Samoilova S. V., Yezhov I. N., Yeremin S. A., Karlyshev A. V., et al. (1995). Virulent non-capsulate Yersinia pestis variants constructed by insertion mutagenesis. J. Med. Microbiol. 42, 264–268. doi: 10.1099/00222615-42-4-264
Du Y., Rosqvist R., Forsberg A. (2002). Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect. Immun. 70, 1453–1460. doi: 10.1128/IAI.70.3.1453-1460.2002
Du Z., Wang X. (2016). Pathology and pathogenesis of Yersinia pestis. Adv. Exp. Med. Biol. 918, 193–222. doi: 10.1007/978-94-024-0890-4_7
Elvin S. J., Williamson E. D. (2004). Stat 4 but not Stat 6 mediated immune mechanisms are essential in protection against plague. Microb. Pathog. 37, 177–184. doi: 10.1016/j.micpath.2004.06.009
Eppinger M., Radnedge L., Andersen G., Vietri N., Severson G., Mou S., et al. (2012). Novel plasmids and resistance phenotypes in Yersinia pestis: unique plasmid inventory of strain Java 9 mediates high levels of arsenic resistance. PLoS One 7 (3), e32911. doi: 10.1371/journal.pone.0032911
Feodorova V. A., Lyapina A. M., Khizhnyakova M. A., Zaitsev S. S., Saltykov Y. V., Motin V. L. (2020). Yersinia pestis Antigen F1 but Not LcrV Induced Humoral and Cellular Immune Responses in Humans Immunized with Live Plague Vaccine-Comparison of Immunoinformatic and Immunological Approaches. Vaccines (Basel). 19, 8. doi: 10.3390/vaccines8040698
Feodorova V. A., Motin V. L. (2012). Plague vaccines: current developments and future perspectives. Emerg. Microbes Infect. 1 (11), e36. doi: 10.1038/emi.2012.34
Ferber D. M., Brubaker R. R. (1979). Mode of action of pesticin: N-acetylglucosaminidase activity. J. Bacteriol. 139, 495–501. doi: 10.1128/jb.139.2.495-501.1979
Fetherston J. D., Schuetze P., Perry R. D. (1992). Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 kb of chromosomal DNA which is flanked by a repetitive element. Mol. Microbiol 6 (18), 2693–704. doi: 10.1111/j.1365-2958.1992.tb01446.x
Friedlander A. M., Welkos S. L., Worsham P. L., Andrews G. P., Heath D. G., Anderson G. W. Jr., et al. (1995). Relationship between virulence and immunity as revealed in recent studies of the F1 capsule of Yersinia pestis. Clin. Infect. Dis. Suppl 2, S178–S181. doi: 10.1093/clinids/21.supplement_2.s178
Guiyoule A., Gerbaud G., Buchrieser C., Galimand M., Rahalison L., Chanteau S., et al. (2001). Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerging Infect. Dis. 7, 43. doi: 10.3201/eid0701.010106
Heath D. G., Anderson G. W. Jr., Mauro J. M., Welkos S. L., Andrews G. P., Adamovicz J., et al. (1998). Protection against experimental bubonic and pneumonic plague by a recombinant capsular F1-V antigen fusion protein vaccine. Vaccine 16, 1131–1137. doi: 10.1016/S0264-410X(98)80110-2
Heine H. S., Louie A., Sorgel F., Bassett J., Miller L., Sullivan L. J., et al. (2007). Comparison of 2 antibiotics that inhibit protein synthesis for the treatment of infection with Yersinia pestis delivered by aerosol in a mouse model of pneumonic plague. J. Infect. Dis. 196 (5), 782–7. doi: 10.1086/520547
Hinnebusch B. J., Rosso M. L., Schwan T. G., Carniel E. (2002). High-frequency conjugative transfer of antibiotic resistance genes to Yersinia pestis in the flea midgut. Mol. Microbiol. 46, 349–354. doi: 10.1046/j.1365-2958.2002.03159.x
Klimko C. P., Shoe J. L., Rill N. O., Hunter M., Dankmeyer J. L., Talyansky Y., et al. (2022a). Layered and integrated medical countermeasures against Burkholderia pseudomallei infections in C57BL/6 mice. Front. Microbiol. 13, 965572. doi: 10.3389/fmicb.2022.965572
Klimko C. P., Welkos S. L., Shoe J. L., Mou S., Hunter M., Rill N. O., et al. (2022b). Efficacy of Treatment with the Antibiotic Novobiocin against Infection with Bacillus anthracis or Burkholderia pseudomallei. Antibio. (Basel). 23, 11. doi: 10.3390/antibiotics11121685
Kon E., Levy Y., Elia U., Cohen H., Hazan-Halevy I., Aftalion M., et al. (2023). A single-dose F1-based mRNA-LNP vaccine provides protection against the lethal plague bacterium. Sci. Adv. 9, eadg1036. doi: 10.1126/sciadv.adg1036
Leary S. E., Williamson E. D., Griffin K. F., Russell P., Eley S. M., Titball R. W. (1995). Active immunization with recombinant V antigen from Yersinia pestis protects mice against plague. Infect. Immun. 63, 2854–2858. doi: 10.1128/iai.63.8.2854-2858.1995
Lortat-Jacob H., Baltzer F., Grimaud J. A. (1996). Heparin decreases the blood clearance of interferon-gamma and increases its activity by limiting the processing of its carboxyl-terminal sequence. J. Biol. Chem 271 (27), 16139–161343. doi: 10.1074/jbc.271.27.16139
McDonough K. A., Falkow S. (1989). A Yersinia pestis-specific DNA fragment encodes temperature-dependent coagulase and fibrinolysin-associated phenotypes. Mol. Microbiol. 3, 767–775. doi: 10.1111/j.1365-2958.1989.tb00225.x
Meka-Mechenko T. V. (2003). F1-negative natural Y. pestis strains. Adv. Exp. Med. Biol. 529, 379–81. doi: 10.1007/0-306-48416-1_76
Meyer K. F. (1970). Effectiveness of live or killed plague vaccines in man. Bull. World Health Organ 42 (5), 653–666.
Miller N. C., Quenee L. E., Elli D., Ciletti N. A., Schneewind O. (2012). Polymorphisms in the lcrV gene of Yersinia enterocolitica and their effect on plague protective immunity. Infect. Immun. 80, 1572–1582. doi: 10.1128/IAI.05637-11
Mizel S. B., Graff A. H., SrIranganathan N., Ervin S., Lees C. J., Lively M. O., et al. (2009). Flagellin-F1-V fusion protein is an effective plague vaccine in mice and two species of nonhuman primates. Clin. Vaccine Immunol. 16, 21–28. doi: 10.1128/CVI.00333-08
Mueller C. A., Broz P., Muller S. A., Ringler P., Erne-Brand F., Sorg I., et al. (2005). The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 310, 674–676. doi: 10.1126/science.1118476
National Research Council (2011). Guide laboratory animals for the care and use of eighth edition committee for the update of the guide for the care and use of laboratory animals institute for laboratory animal research division on earth and life studies. Washington, DC: The National Academies Press.
Nelson C. A., Meaney-Delman D., Fleck-Derderian S., Cooley K. M., Yu P. A., Mead P. S., et al. (2021). Antimicrobial treatment and prophylaxis of plague: recommendations for naturally acquired infections and bioterrorism response. MMWR Recomm Rep. 70, 1–27. doi: 10.15585/mmwr.rr7003a1
Pendrak M. L., Perry R. D. (1991). Characterization of a hemin-storage locus of Yersinia pestis. Biol. Met. 4, 41–47. doi: 10.1007/BF01135556
Perry R. D., Pendrak M. L., Schuetze P. (1990). Identification and cloning of a hemin storage locus involved in the pigmentation phenotype of Yersinia pestis. J. Bacteriol. 172, 5929–5937. doi: 10.1128/jb.172.10.5929-5937.1990
Pettersson J., Holmstrom A., Hill J., Leary S., Frithz-Lindsten E., von Euler-Matell A., et al. (1999). The V-antigen of Yersinia is surface exposed before target cell contact and involved in virulence protein translocation. Mol. Microbiol. 32, 961–976. doi: 10.1046/j.1365-2958.1999.01408.x
Pitt L. M. (2004). “Non-human primates as a model for pneumonic plague; animal models and correlates of protection for plague,” in Proceedings of the Plague Vaccines Workshop (Gaithersburg, MD, USA). Available at: https://www.fda.gov/cber/minutes/ plague101304t.pdf.
Powell B. S., Andrews G. P., Enama J. T., Jendrek S., Bolt C., Worsham P., et al. (2005). Design and testing for a nontagged F1-V fusion protein as vaccine antigen against bubonic and pneumonic plague. Biotechnol. Prog. 21, 1490–1510. doi: 10.1021/bp050098r
Qiu Y., Liu Y., Qi Z., Wang W., Kou Z., Zhang Q., et al. (2010). Comparison of immunological responses of plague vaccines F1+rV270 and EV76 in Chinese-origin rhesus macaque, Macaca mulatta. Scand. J. Immunol. 72, 425–433. doi: 10.1111/j.1365-3083.2010.02456.x
Quenee L. E., Ciletti N. A., Elli D., Hermanas T. M., Schneewind O. (2011). Prevention of pneumonic plague in mice, rats, Guinea pigs and non-human primates with clinical grade rV10, rV10-2 or F1-V vaccines. Vaccine 29, 6572–6583. doi: 10.1016/j.vaccine.2011.06.119
Quenee L. E., Cornelius C. A., Ciletti N. A., Elli D., Schneewind O. (2008). Yersinia pestis caf1 variants and the limits of plague vaccine protection. Infect. Immun. 76, 2025–2036. doi: 10.1128/IAI.00105-08
Randremanana R., Andrianaivoarimanana V., Nikolay B., Ramasindrazana B., Paireau J., Ten Bosch Q. A., et al. (2019). Epidemiological characteristics of an urban plague epidemic in Madagascar, August-November 2017: an outbreak report. Lancet Infect. Dis. 19, 537–545. doi: 10.1016/S1473-3099(18)30730-8
Roggenkamp A., Geiger A. M., Leitritz L., Kessler A., Heesemann J. (1997). Passive immunity to infection with Yersinia spp. mediated by anti-recombinant V antigen is dependent on polymorphism of V antigen. Infect. Immun. 65, 446–451. doi: 10.1128/iai.65.2.446-451.1997
Rosario-Acevedo R., Biryukov S. S., Bozue J. A., Cote C. K. (2021). Plague prevention and therapy: perspectives on current and future strategies. Biomed. Oct 9 (10), 1421. doi: 10.3390/biomedicines9101421
Rosenzweig J. A., Hendrix E. K., Chopra A. K. (2021). Plague vaccines: new developments in an ongoing search. Appl. Microbiol. Biotechnol. 105, 4931–4941. doi: 10.1007/s00253-021-11389-6
Rosler B., Herold S. (2016). Lung epithelial GM-CSF improves host defense function and epithelial repair in influenza virus pneumonia-a new therapeutic strategy? Mol. Cell Pediatr. 3, 29. doi: 10.1186/s40348-016-0055-5
Samoilova S. V., Samoilova L. V., Yezhov I. N., Drozdov I. G., Anisimov A. P. (1996). Virulence of pPst+ and pPst- strains of Yersinia pestis for Guinea-pigs. J. Med. Microbiol. 45, 440–444. doi: 10.1099/00222615-45-6-440
Sebbane F., Jarrett C., Gardner D., Long D., Hinnebusch B. J. (2009). The Yersinia pestis caf1M1A1 fimbrial capsule operon promotes transmission by flea bite in a mouse model of bubonic plague. Infect. Immun. 77, 1222–1229. doi: 10.1128/IAI.00950-08
Sha J., Endsley J. J., Kirtley M. L., Foltz S. M., Huante M. B., Erova T. E., et al. (2011). Characterization of an F1 deletion mutant of Yersinia pestis CO92, pathogenic role of F1 antigen in bubonic and pneumonic plague, and evaluation of sensitivity and specificity of F1 antigen capture-based dipsticks. J. Clin. Microbiol. 49, 1708–1715. doi: 10.1128/JCM.00064-11
Smiley S. T. (2008). Current challenges in the development of vaccines for pneumonic plague. Expert Rev. Vaccines 7, 209–221. doi: 10.1586/14760584.7.2.209
Sodeinde O. A., Goguen J. D. (1988). Genetic analysis of the 9.5-kilobase virulence plasmid of Yersinia pestis. Infect. Immun. 56, 2743–2748. doi: 10.1128/iai.56.10.2743-2748.1988
Standiford L. R., Standiford T. J., Newstead M. J., Zeng X., Ballinger M. N., Kovach M. A., et al. (2012). TLR4-dependent GM-CSF protects against lung injury in Gram-negative bacterial pneumonia. Am. J. Physiol. Lung Cell Mol. Physiol. 302, L447–L454. doi: 10.1152/ajplung.00415.2010
Stepanov A. V., Marinin L. I., Vorob'ev A. A. (1999). Aerosol vaccination against dangerous infectious diseases. Vestn Ross Akad Med. Nauk. (8), 47–54.
Sun W. (2016). Plague vaccines: status and future. Adv. Exp. Med. Biol. 918, 313–360. doi: 10.1007/978-94-024-0890-4_12
Sun W., Sanapala S., Henderson J. C., Sam S., Olinzock J., Trent M. S., et al. (2014). LcrV delivered via type III secretion system of live attenuated Yersinia pseudotuberculosis enhances immunogenicity against pneumonic plague. Infect. Immun. 82, 4390–4404. doi: 10.1128/IAI.02173-14
Swietnicki W., Carmany D., Retford M., Guelta M., Dorsey R., Bozue J., et al. (2011). Identification of small-molecule inhibitors of Yersinia pestis Type III secretion system YscN ATPase. PloS One 6, e19716. doi: 10.1371/journal.pone.0019716
Titball R. W., Williamson E. D. (2004). Yersinia pestis (plague) vaccines. Expert Opin. Biol. Ther. 4, 965–973. doi: 10.1517/14712598.4.6.965
Trapnell B. C., Whitsett J. A. (2002). Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol. 64, 775–802. doi: 10.1146/annurev.physiol.64.090601.113847
Trevino S. R., Klimko C. P., Reed M. C., Aponte-Cuadrado M. J., Hunter M., Shoe J. L., et al. (2018). Disease progression in mice exposed to low-doses of aerosolized clinical isolates of Burkholderia pseudomallei. PloS One 13, e0208277. doi: 10.1371/journal.pone.0208277
Verma S. K., Tuteja U. (2016). Plague vaccine development: current research and future trends. Front. Immunol. 7, 602. doi: 10.3389/fimmu.2016.00602
Vogler A. J., Keim P., Wagner D. M. (2016). A review of methods for subtyping Yersinia pestis: From phenotypes to whole genome sequencing. Infect. Genet. Evol. 37, 21–36. doi: 10.1016/j.meegid.2015.10.024
Wang X., Singh A., Sun W. (2020). Protection and Safety Evaluation of Live Constructions Derived from the Pgm– and pPCP1– Yersinia pestis Strain. Vaccines 8, 1–14. doi: 10.3390/vaccines8010095
Wang X., Zhang X., Zhou D., Yang R. (2013). Live-attenuated Yersinia pestis vaccines. Expert Rev. Vaccines 12, 677–686. doi: 10.1586/erv.13.42
Weening E. H., Cathelyn J. S., Kaufman G., Lawrenz M. B., Price P., Goldman W. E., et al. (2011). The dependence of the Yersinia pestis capsule on pathogenesis is influenced by the mouse background. Infect. Immun. 79, 644–652. doi: 10.1128/IAI.00981-10
Welkos S. L., Andrews G. P., Lindler L. E., Snellings N. J., Strachan S. D. (2004). Mu dI1(Ap lac) mutagenesis of Yersinia pestis plasmid pFra and identification of temperature-regulated loci associated with virulence. Plasmid 51, 1–11. doi: 10.1016/j.plasmid.2003.09.003
Welkos S. L., Davis K. M., Pitt L. M., Worsham P. L., Freidlander A. M. (1995). Studies on the contribution of the F1 capsule-associated plasmid pFra to the virulence of Yersinia pestis. Contrib Microbiol. Immunol. 13, 299–305.
Welkos S. L., Friedlander A. M., Davis K. J. (1997). Studies on the role of plasminogen activator in systemic infection by virulent Yersinia pestis strain C092. Microb. Pathog. 23, 211–223. doi: 10.1006/mpat.1997.0154
Welkos S., Pitt M. L., Martinez M., Friedlander A., Vogel P., Tammariello R. (2002). Determination of the virulence of the pigmentation-deficient and pigmentation-/plasminogen activator-deficient strains of Yersinia pestis in non-human primate and mouse models of pneumonic plague. Vaccine 20, 2206–2214. doi: 10.1016/S0264-410X(02)00119-6
Williams J. E., Cavanaugh D. C. (1984). Potential for rat plague from nonencapsulated variants of the plague bacillus (Yersinia pestis). Experientia 40, 739–740. doi: 10.1007/BF01949752
Williams P., Wallace D. (1989). Unit 731: Japan's Secret Biological Warfare in World War II (New York, NY: The Free Press).
Williamson E. D., Eley S. M., Griffin K. F., Green M., Russell P., Leary S. E., et al. (1995). A new improved sub-unit vaccine for plague: the basis of protection. FEMS Immunol. Med. Microbiol. 12, 223–230. doi: 10.1111/j.1574-695X.1995.tb00196.x
Williamson E. D., Oyston P. C. (2013). Protecting against plague: towards a next-generation vaccine. Clin. Exp. Immunol. 172, 1–8. doi: 10.1111/cei.12044
Winter C. C., Cherry W. B., Moody M. D. (1960). An unusual strain of Pasteurella pestis isolated from a fatal human case of plague. Bull. World Health Organ. 23 (2), 408–409.
Worsham P. L., McGovern T. W., Vietri N. J., Bozue J. (2018). “Plague,” in Medical Aspects of Biological Warfare (Ft. Sam Houston, TX: The Borden Institute), 247–284.
Worsham P. L., Stein M. P., Welkos S. L. (1995). Construction of defined F1 negative mutants of virulent Yersinia pestis. Contrib Microbiol. Immunol. 13, 325–328.
Keywords: plague, Yersinia pestis, vaccine, nonencapsulated, mice, bubonic, pneumonic
Citation: Biryukov SS, Klimko CP, Dankmeyer JL, Toothman RG, Shoe JL, Hunter M, Rill NO, Talyansky Y, Davies ML, Qiu J, Fetterer DP, Bozue JA, Welkos SL and Cote CK (2023) Live attenuated vaccines and layered defense strategies to combat infections caused by nonencapsulated Yersinia pestis. Front. Bacteriol. 2:1240698. doi: 10.3389/fbrio.2023.1240698
Received: 15 June 2023; Accepted: 31 July 2023;
Published: 22 September 2023.
Edited by:
Myron Christodoulides, University of Southampton, United KingdomReviewed by:
Emanuelle Mamroud, Israel Institute for Biological Research (IIBR), IsraelCopyright © 2023 Biryukov, Klimko, Dankmeyer, Toothman, Shoe, Hunter, Rill, Talyansky, Davies, Qiu, Fetterer, Bozue, Welkos and Cote. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher K. Cote, Y2hyaXN0b3BoZXIuay5jb3RlLmNpdkBoZWFsdGgubWls
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.