 Luke P. Malone
Luke P. Malone Serena M. Best
Serena M. Best Ruth E. Cameron
Ruth E. Cameron- Department of Materials Science and Metallurgy, Cambridge Centre for Medical Materials, University of Cambridge, Cambridge, United Kingdom
Additive manufacturing and electrospinning are widely used to create degradable biomedical components. This work presents important new data showing that the temperature used in accelerated tests has a significant impact on the degradation process in amorphous 3D printed poly-l-lactic acid (PLLA) fibres. Samples (c. 100
1 Introduction
Additive manufacturing has the potential to revolutionise medical device fabrication. The range of potential applications is considerable, encompassing everything from drug delivery devices (Bácskay et al., 2022), to stents (Guerra et al., 2018), screws (MacLeod et al., 2020), meshes (Ren et al., 2022) and 3D printed organs (George et al., 2023). Much research focuses on the concept of personalised device fabrication, producing devices that perfectly match the individual patient requirements (Nagarajan et al., 2018). This in turn could pave the way to a reduction in recovery time, material wastage or device cost (Goyanes et al., 2015; Qamar et al., 2019). Similarly, electrospinning has been used to fabricate scaffolds for a variety of biomedical applications including cartilage repair (Castilho et al., 2019), bone tissue regeneration (Lee et al., 2013) and tendons or ligaments (Hochleitner et al., 2018). The deposited scaffold architectures are highly controllable, forming structures that can mimic the native tissue environment (Kai et al., 2014).
Poly-L-lactic acid (PLLA) is an example of a widely used man-made poly (
One of the major advantages of PLLA as a biomedical material is its ability to degrade in-vivo in a controlled manner over a long time frame. The degradation produces harmless by-products that can be processed naturally via the Cori cycle (da Silva et al., 2018). However, this specific advantage (its long degradation time), is also a major drawback for studying the material within a research setting. PLLA requires several years to completely degrade within the body or similar in vitro conditions. As a result, many researchers use accelerated degradation testing to speed up the process. Accelerated degradation testing of poly (
The degradation pathway of PLLA is commonly characterised as a bulk degradation process, with the diffusion of fluid into the structure faster than the degradation rate of the polymer backbone (Pierre and Chiellini, 1987; Deb and Di Silvio, 2009; von Burkersroda et al., 2002). Initially, water or fluid is absorbed into the material. Several studies have suggested that within a semi-crystalline microstructure, the amorphous regions are selectively degraded first (Fischer et al., 1973; Li et al., 1990; Pistner et al., 1993; Tsuji and Ikada, 1997; Tsuji and Ikada, 1998; Tsuji et al., 2000; Chye et al., 2005), with the eventual degradation of crystalline domains shown to proceed from the edges inwards (Fischer et al., 1973).
The impact of manufacturing technique on the degradation rate needs to be carefully considered (Vaid et al., 2021). Chausse et al. previously investigated the degradation of 3D printed PLLA stents (starting strut thickness 120.9
Following the initial degradation of the polymer chain backbone, the diffusion of degradation products out of the structure is slow. Only when the molecular weight of the formed fragments and the surrounding material are sufficiently small, can they diffuse out of the structure, marking the onset of mass loss and pH change (Göpferich, 1997). In the time between the initial polymer chain degradation, but prior to the diffusion of the molecular weight fragments out of the structure, the degradation products can catalyse the reaction via autocatalysis (leading to heterogeneous bulk degradation) (Vert et al., 1991). However, autocatalysis has not been reported in all degradation studies and is strongly linked to the material starting thickness and the product’s diffusion path to the sample exterior. Multiple studies have found that with an increase in sample thickness, the degradation rate increases (Grizzi et al., 1995; Lu et al., 1999; Witt and Kissel, 2001). With an increase in sample thickness, the diffusion path for the release of degradation products is greater, and their build-up can then autocatalyse the reaction (Fu et al., 2000). However, the critical thickness at which autocatalysis becomes significant is highly dependent upon the starting polymer and manufacturing technique. Vaid et al. reported no evidence of autocatalysis in PLA fibres (starting material contained less than 2% D-isomer) 10–15
This study aims to investigate how 100
2 Materials and methods
2.1 Materials
PLLA (2500HP) NatureWorks pellets were extruded using a Noztek extruder at
2.2 Hydration study
To investigate the effect of fluid immersion on the glass transition, a short hydration study involving the as printed PLLA was conducted. Samples were immersed for 3 days in fluid at either
2.3 Degradation testing
After printing, the PVA was peeled off. Samples were then weighed and split into groups based on their degradation time and temperature. There were three samples for each degradation time and temperature, with the total starting group mass approximately the same. Samples were placed into 7 mL individual bijou tubes with phosphate buffer saline (PBS) pipetted on top in a sample mass to fluid ratio of 1 mg:125 mL. The bijou tubes were placed into one of three temperature-controlled environments (
After the allocated degradation time, samples were filtered, washed twice in deionised water and vacuum dried for 3 days.
2.4 Sample characterisation
2.4.1 pH change
Following degradation and removal from the temperature controlled environment, the fluid pH was measured three times. The error in the pH measurement was determined by the resolution of the probe.
2.4.2 Mass loss
After 3 days of vacuum drying, samples were weighed three times to find their final dry mass. The percentage mass loss during degradation was calculated using Eq. (1).
where
The percentage error for each time and temperature was calculated by taking the standard deviation in mass loss for each sample and then using standard error propagation rules to combine all samples of the same degradation time and temperature.
2.4.3 Molecular weight
Gel-permeation chromatography (GPC) was conducted on a 1,260 Infinity II Multi-Detector GPC/SEC System at the University of Manchester, supported by the Henry Royce Institute for Advanced Materials. 10–15 mg of a dried sample was dissolved in chloroform (3 mg/mL). The dissolved samples were filtered through a 0.2
2.4.4 X-ray diffraction
Wide-angle X-ray scattering (WAXS) scans on dried samples were undertaken using a Bruker D8 Advance diffractometer with a Cu-K
To validate the computational model, PLLA was mixed with the diffraction standard corundum in three different ratios. The corundum was supplied by MetPrep (Alpha 1.0 micron category number 159980). Across all three ratios, the model was able to correctly predict the final corundum to polymer ratio within
2.4.5 Thermal properties
Thermal properties were measured using a TA Instruments differential scanning calorimetry (DSC) 2,500. The DSC was run in batches, with all degraded material from a single fluid temperature run at the same time. Before each batch was run, a sample of as printed material was run. The initial material characterisation values and associated errors were calculated based upon the average and standard deviation of this data. For every sample, 2–5 mg of dried material was sealed in an aluminium pan, and DSC was conducted using a
2.4.6 Mechanical properties
Tensile mechanical testing was undertaken using a Tinius Olsen 1ST universal tensile testing machine. A 25 N load cell with self-tightening grips was used. Sample gauge length was measured and approximately 20 mm. The strain rate was 0.001
Sample cross section was recorded using optical microscopy in reflection mode. Samples were sectioned into three pieces. A ×10 objective lens was used to find sample width and thickness for individual lines prior to mechanical testing. The cross sectional area was approximately rectangular in shape, and the formula for area of a rectangle was used to calculate the cross sectional area (Roper et al., 2021).
3 Results
3.1 Initial material characterisation
Prior to degradation testing, the initial molecular weight, X-ray diffraction scattering, thermal properties and mechanical properties of the as printed PLLA fibres were characterised (Figure 1).
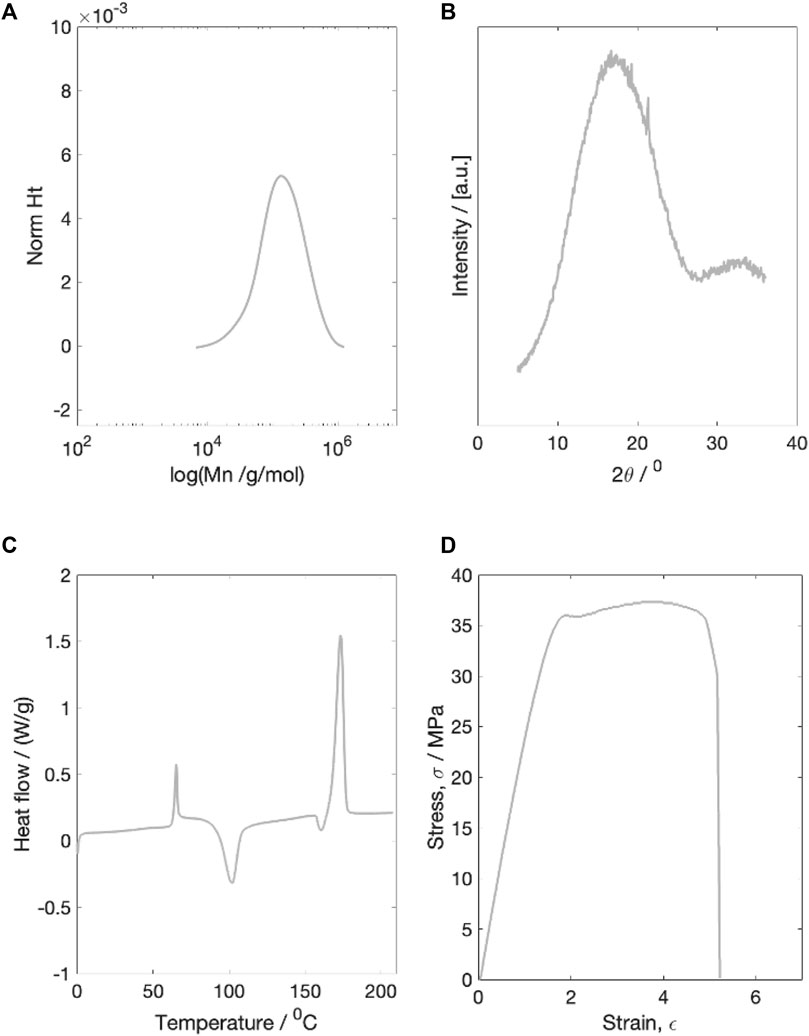
Figure 1. Example characterisation plots of the dried initial material. (A) molecular weight characterisation (B) X-ray diffraction characterisation (C) Thermal properties characterisation (D) Example stress-strain plot for a single dried starting sample. All four tests were run on dried material.
3.1.1 Molecular weight
The dry number average starting molecular weight (Mn) was measured to be 100,598 g/mol. The starting PDI was 1.79
3.1.2 X-ray diffraction
The initial dry material was completely amorphous, with a single broad WAXS peak and a calculated crystallinity of 4.5
3.1.3 Thermal properties
Thermal characterisation of the initial dry material revealed an average Tg of 61.5
3.1.4 Mechanical properties
The average dry starting Young’s modulus was measured to be 2.00
3.2 Hydration study
The DSC curves for samples hydrated in PBS for 3 days at
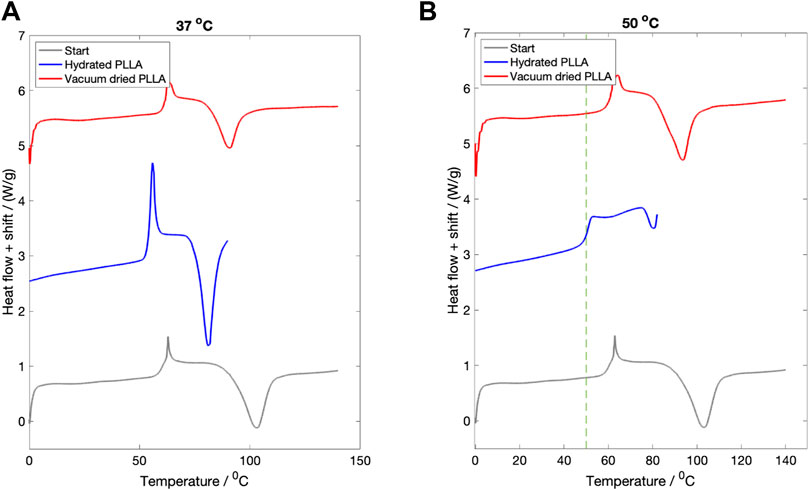
Figure 2. The effect of fluid immersion for 3 days on the recorded thermal properties. (A) Hydration at
The same three DSC curves for PLLA hydrated at
The study was not undertaken at
3.3 Degradation study
3.3.1 Degraded sample appearance
The changes to sample appearance after degradation and sample drying, are shown in Figure 3. At
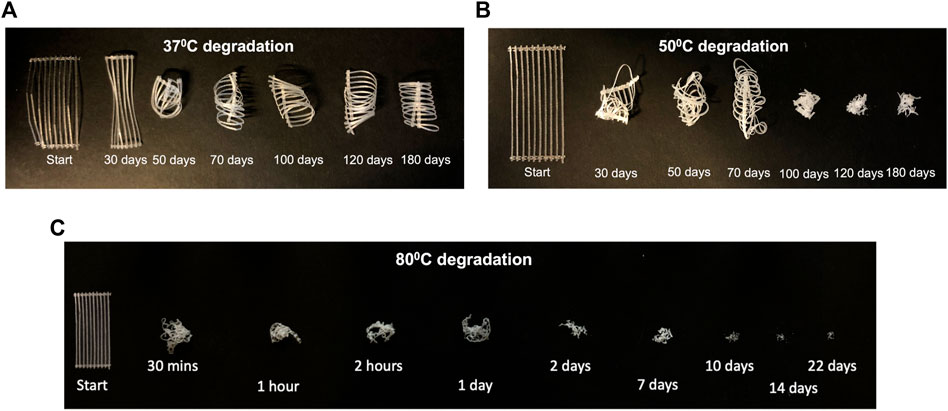
Figure 3. Change in sample appearance after drying for samples degraded at (A)
3.3.2 Mass loss and pH change
The percentage mass loss and pH change are shown in Figure 4. At
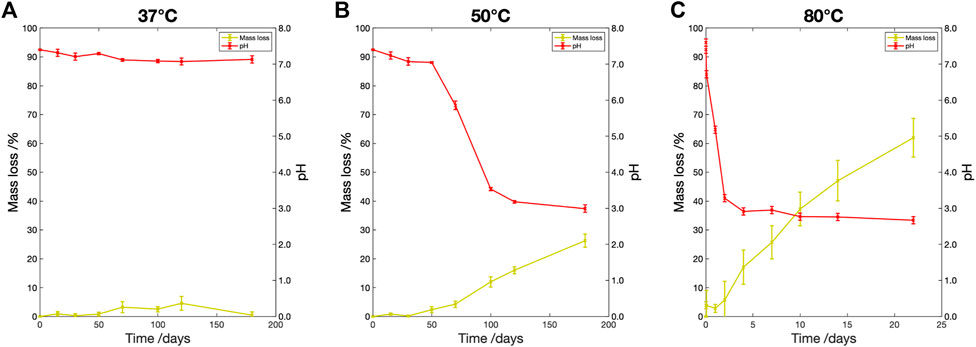
Figure 4. Percentage mass loss (yellow) and pH change (red) vs. degradation time for samples degraded in PBS at (A)
At
At
3.3.3 Molecular weight
Across all temperatures, there was a reduction in number average molecular weight (Mn) with time (Figure 5). Samples degraded at
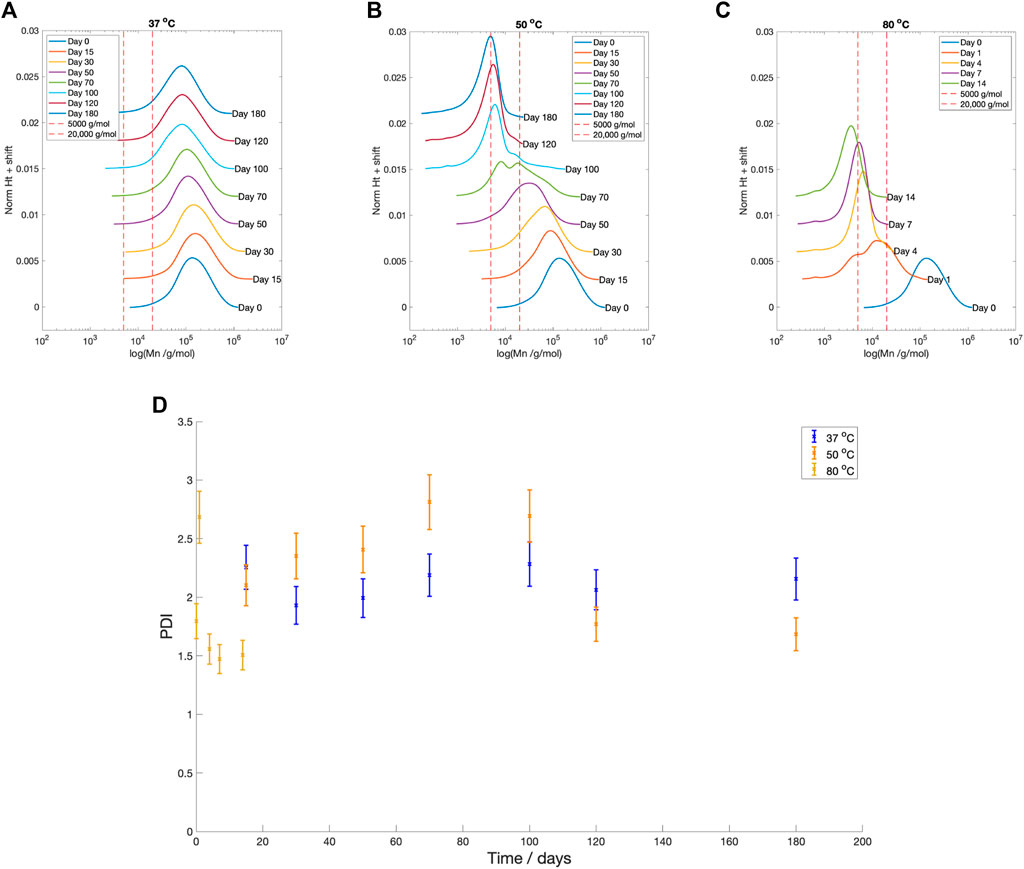
Figure 5. GPC profiles at degradation time for PLLA degraded in PBS at (A)
3.3.4 X-ray diffraction
X-ray diffraction profile of samples degraded at each fluid temperature are plotted in Figure 6. After 6 months degradation at
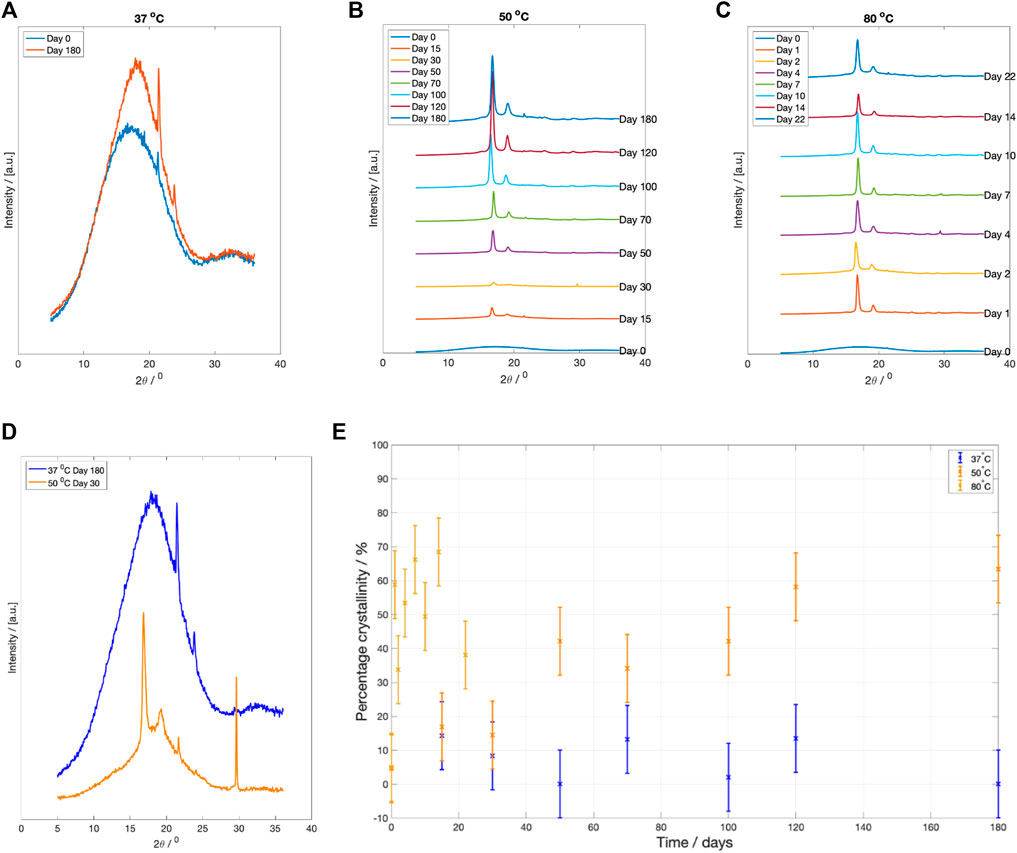
Figure 6. XRD scattering results at specific degradation time points after drying. (A) WAXS pattern for PLLA degraded at
3.3.5 Crystallinity
The change in crystallinity, estimated using Rietveld refinement, is plotted in Figure 6E. There was little recorded change in sample crystallinity for PLLA degraded at
3.3.6 Thermal properties
The change in thermal properties of the dried degraded samples was investigated using DSC. For degradation at
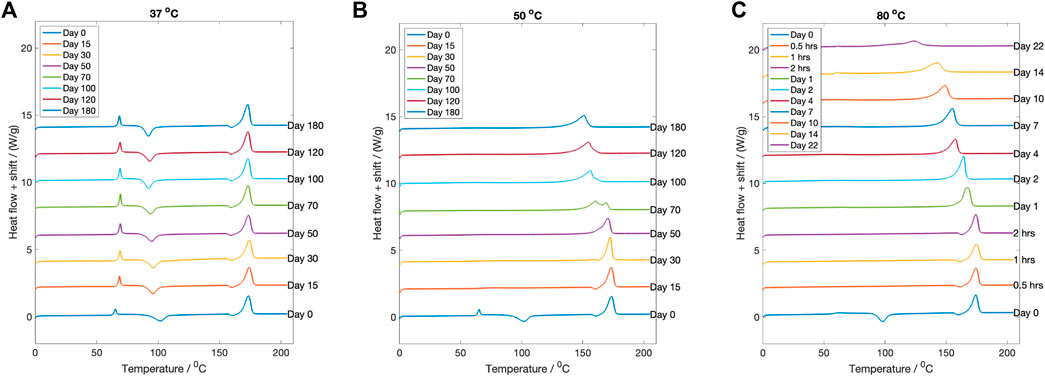
Figure 7. The DSC curves for each fluid temperature at every degradation time point. (A)
Conversely, for samples degraded at
Samples degraded at
3.3.7 Mechanical properties
The change in Young’s modulus, yield stress and ultimate tensile strength (UTS) for dried samples degraded at
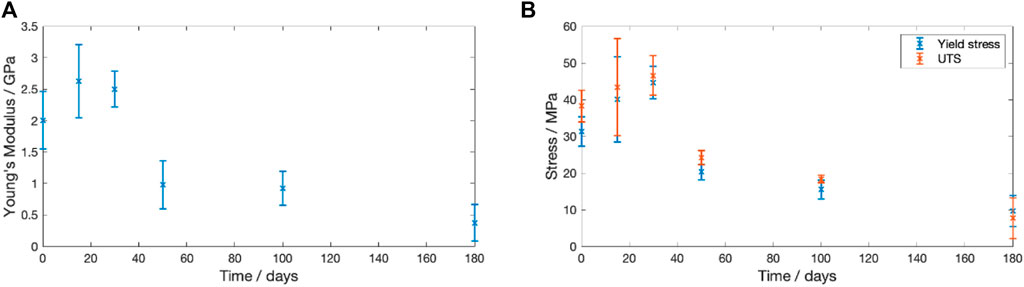
Figure 8. Change in the recorded mechanical properties for PLLA degraded in PBS at
4 Discussion
4.1 Three-stage degradation model
When the data from the pH and mass loss experiments were combined with the molecular weight data, across all fluid temperatures, a three-stage degradation process could be identified (Figure 9). Previous studies have subdivided the degradation of poly(
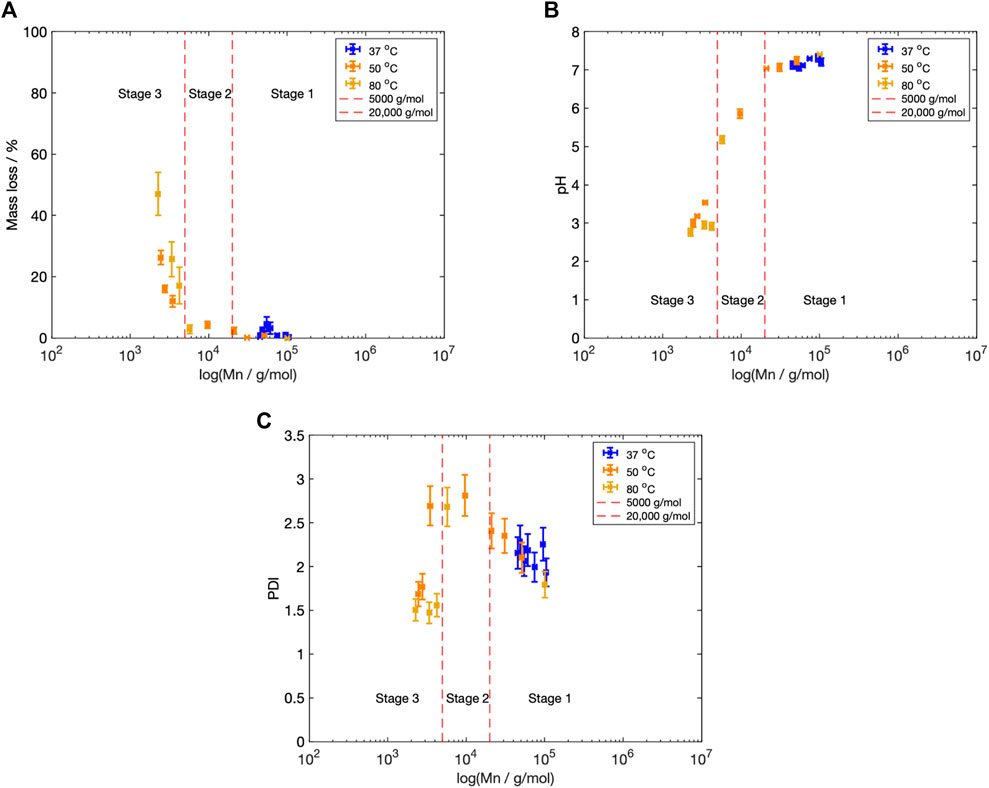
Figure 9. (A) Mn vs. mass loss across all three fluid temperatures,
The Mn boundaries between each stage are approximate; however, the overall categorisation was based on trends across the entire data set and previous literature. The boundary between Stage 1 and 2 was determined by the increase in the PDI from 2.40 to greater than 2.60 (Figure 5D), as well as the reduction in fluid pH (Figure 4), once the Mn fell below 20,000 g/mol (Figure 5). Weir et al. also reported the onset of mass loss for 0.8 mm annealed PLLA once the sample molecular weight fell below 20,000 (Weir et al., 2004a). The boundary between Stage 2 and 3 was based upon the drop in the PDI from 2.60 to below 2.00 (Figure 5D), alongside the increase in mass loss (Figure 4) that occurred when Mn shifted to less than 5,000 g/mol (Figure 5). Grizzi et al. similarly reported the onset of significant mass loss once the molecular weight was less than 7,000 g/mol, for 0.3 mm thick compression moulded amorphous poly(D-L lactic acid) (Grizzi et al., 1995; Sevim and Pan, 2018).
Stage 1: Mn
Stage 2: 5,000
Stage 3: Mn
4.2 Molecular weight changes in Stage 1 and the effect on mechanical properties
4.2.1 Equation to represent molecular weight evolution with time in Stage 1
Two models have been proposed to represent the reduction in Mn, prior to the onset of mass loss (Stage 1). Anderson et al. suggested a statistical approach with the molecular weight following an inverse relationship with degradation time, t (Eq. (2)):
where
The model assumes that no autocatalysis has occurred. Pitt and Gu developed an alternative model to account for the autocatalysis of the carboxylic acid end groups generated during the degradation (Pitt et al., 1981). Their model predicted an exponential relationship with degradation time, t (Eq. (3)):
Neither model can be applied to samples degraded at
Table 1 shows that at
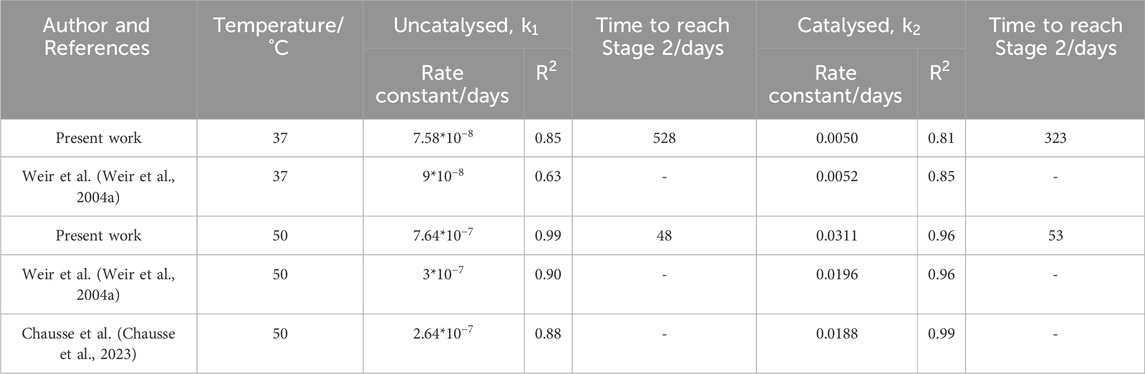
Table 1. Rate constants and accompanying
Using the degradation models developed by Anderson et al. and Pitt and Gu respectively, the time to reach a molecular weight of 20,000 g/mol was determined (Table 1). This marks the boundary between Stage 1 and 2 and the approximate onset of mass loss and pH changes (Figure 9).
4.2.2 The dependency of mechanical properties on molecular weight in Stage 1
The reduction in Mn during Stage 1, was accompanied by a change in mechanical properties at
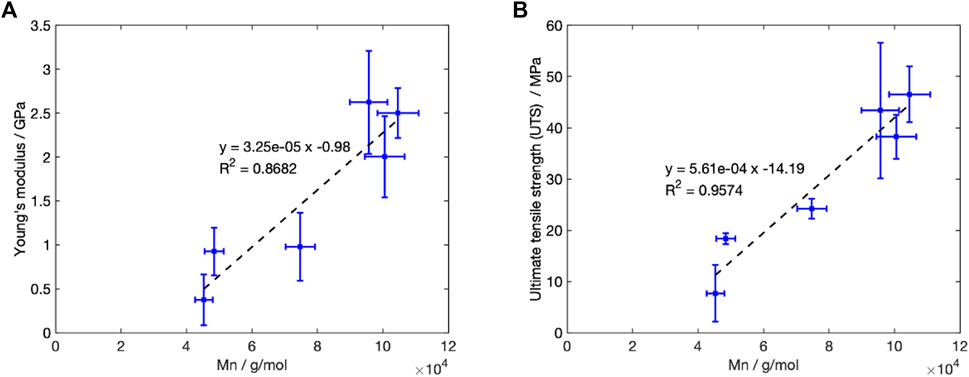
Figure 10. Change in the recorded mechanical properties with Mn for degradation at
4.3 Mass and molecular weight changes in Stage 3
4.3.1 Equation to represent mass change with time in Stage 3
In Stage 3, oligomer and short-chain monomers were predicted to move via diffusion from the sample into the surrounding fluid. This diffusion was assumed to obey Fick’s diffusion laws. As such, Stefan’s approximation predicting the change in mass over time was fitted (Eq. (4)) (Moliner et al., 2020).
Where
A constant saturation mass, diffusion length and coefficient are assumed in Eq. (4).
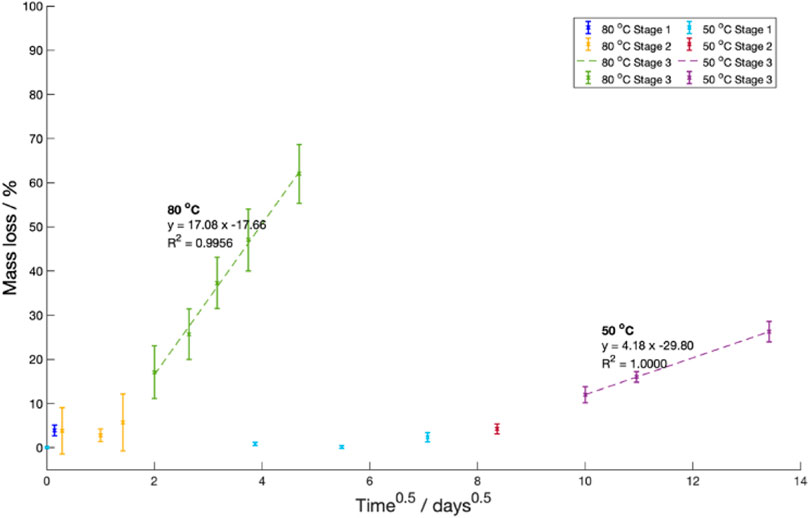
Figure 11. Mass loss against the square route of the degradation time at
4.3.2 Evolution of molecular weight with time in Stage 3
Tsuji et al. previously found that the molecular weight of crystalline residues decreased with a linear time dependency (Tsuji and Ikarashi, 2004a; Tsuji and Ikarashi, 2004b). They manufactured their starting crystalline residues by degrading as-cast PLLA 100
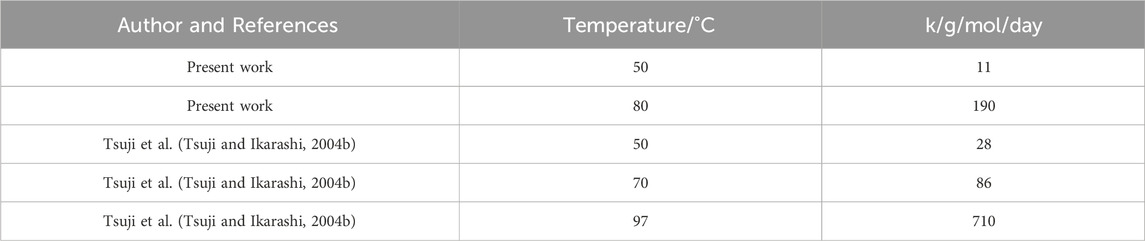
Table 2. Linear rate constant during Stage 3 degradation. For the present work data, k is calculated from Day 100 onwards at
There was good matching between the calculated rate constant at
4.4 Overall degradation pathway as a function of temperature
In Figures 5B,C, the development of multi-modal peaks during Stage 2 at
In Figure 9, the overall degradation was divided into three-stages. Samples degraded at
4.4.1 Degradation at
Above Tg, changes to polymer orientation or crystallinnity can occur. For samples degraded at
4.4.2 Degradation at
Conversely, for samples degraded at
During the 180 days degradation there were significant changes to the mechanical properties. Figure 8 shows that during the first 30 days of degradation there was an increase in the Young’s Modulus and UTS. However, the PLLA transitioned from elastic-plastic behaviour to brittle behaviour. The recorded behaviour matches that reported by Pan et al. who found that physical ageing of 380–400
4.4.3 Degradation at
For degradation occurring at
This reduction in Tg when immersed in fluid, meant the samples followed the behaviour recorded at
It is important to consider whether the Tg of samples degraded at
4.4.4 Extrapolation from
This study has shown a clear difference between the degradation behaviour at different fluid temperatures. When degraded samples with a similar Mn at different temperatures are compared, significant differences between the material microstructures were found. After 30 days degradation at
To determine whether extrapolation from
where k is the rate constant, A is a constant,
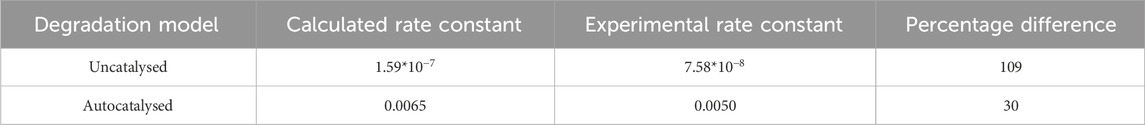
Table 3. Extrapolation using the Arrhenius equation and the experimentally measured
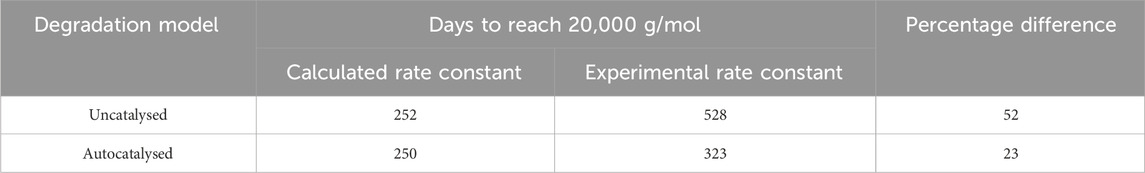
Table 4. The time to reach Stage 2, at
The values in Tables 3 and 4 show that at best, there is a 30% difference between the calculated rate constant and experimental rate constant, which leads to a 23% difference in time to reach the onset of mass loss. Based on this data, it appears that Arrhenius temperature extrapolation is not a suitable method for predicting the lifespan of amorphous PLLA at
The results gathered from this study suggest that 3D printed PLLA degrading at
5 Conclusion
This work investigated the degradation pathway of 100
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
LM: Investigation, Methodology, Writing–original draft, Writing–review and editing. SB: Supervision, Writing–original draft, Writing–review and editing. RC: Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Engineering and Physical Sciences Research Council (EPSRC) grants EP/R513180/1, EP/T517847/1. This work was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1 and EP/P025498/1.
Acknowledgments
The authors thank Giulio Lampronti for his assistance in collecting and analysing the X-ray diffraction data. We thank the Henry Royce Institute for Advance Materials at the University of Manchester for data collection using the 1260 Infinity II Multi-Detector GPC/SEC System.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2024.1419654/full#supplementary-material
References
Agrawal, C. M., Huang, D., Schmitz, J. P., and Athanasiou, K. A. (1997). Elevated temperature degradation of a 50:50 copolymer of PLA-PGA. Tissue Eng. 3, 345–352. doi:10.1089/ten.1997.3.345
Alexy, R. D., and Levi, D. S. (2013). Materials and manufacturing technologies available for production of a pediatric bioabsorbable stent. BioMed Res. Int. 2013, 1–11. doi:10.1155/2013/137985
Ali, Z. A., Gao, R., Kimura, T., Onuma, Y., Kereiakes, D. J., Ellis, S. G., et al. (2018). Three-year outcomes with the absorb bioresorbable scaffold: individual-patient-data meta-analysis from the ABSORB randomized trials. Circulation 137, 464–479. doi:10.1161/CIRCULATIONAHA.117.031843
Anderson, J. M. (1995). “Perspectives on the in vivo responses of biodegradable polymers,” in Biomedical applications of synthetic biodegradable polymers. Editor J. O. Hollinger (Boca Raton, FL, USA: CRC Press), 223–233.
Auras, R. A., Lim, L. T., Selke, S. E. M., Tsuji, H., Lim, L. T., and Tsuji, H. (2010). Poly(Lactic acid): synthesis, structures, properties, processing, and applications. Incorporated: John Wiley and Sons.
Bácskay, I., Ujhelyi, Z., Fehér, P., and Arany, P. (2022). The evolution of the 3D-printed drug delivery systems: a review. Pharmaceutics 14, 1312. doi:10.3390/pharmaceutics14071312
Bergsma, J. E., Rozema, F. R., Bos, R. R. M., Boering, G., Joziasse, C. A. P., and Pennings, A. J. (1995). In vitro predegradation at elevated temperatures of poly(lactide). J. Mater. Sci. Mater. Med. 6, 642–646. doi:10.1007/BF00123445
Castilho, M., Mouser, V., Chen, M., Malda, J., and Ito, K. (2019). Bi-layered micro-fibre reinforced hydrogels for articular cartilage regeneration. Acta Biomater. 95, 297–306. doi:10.1016/j.actbio.2019.06.030
Chausse, V., Iglesias, C., Bou-Petit, E., Ginebra, M. P., and Pegueroles, M. (2023). Chemical vs thermal accelerated hydrolytic degradation of 3D-printed PLLA/PLCL bioresorbable stents: characterization and influence of sterilization. Polym. Test. 117, 107817. doi:10.1016/j.polymertesting.2022.107817
Chye, J. L. S., Ooi, C. P., Hong, E. W. S., and Chiang, F. B. Y. (2005). Effect of isothermal annealing on the hydrolytic degradation rate of poly(lactide-co-glycolide) (PLGA). Biomaterials 26, 2827–2833. doi:10.1016/j.biomaterials.2004.08.031
Cui, L., Imre, B., Tátraaljai, D., and Pukánszky, B. (2020). Physical ageing of Poly(Lactic acid): factors and consequences for practice. Polymer 186, 122014. doi:10.1016/j.polymer.2019.122014
da Silva, D., Kaduri, M., Poley, M., Adir, O., Krinsky, N., Shainsky-Roitman, J., et al. (2018). Biocompatibility, biodegradation and excretion of polylactic acid (PLA) in medical implants and theranostic systems. Chem. Eng. J. 340, 9–14. doi:10.1016/j.cej.2018.01.010
Deb, S. (2009). “2 - degradable polymers and polymer composites for tissue engineering,” in Cellular Response to biomaterials (woodhead publishing). Editor L. Di Silvio (Cambridge, United Kingdom: Woodhead Publishing Series in Biomaterials), 28–60. doi:10.1533/9781845695477.1.28
de Tayrac, R., Chentouf, S., Garreau, H., Braud, C., Guiraud, I., Boudeville, P., et al. (2008). In vitro degradation and in vivo biocompatibility of poly(lactic acid) mesh for soft tissue reinforcement in vaginal surgery. J. Biomed. Mater. Res. Part B Appl. Biomaterials 85B, 529–536. doi:10.1002/jbm.b.30976
Ding, L., Davidchack, R. L., and Pan, J. (2012). A molecular dynamics study of Young’s modulus change of semi-crystalline polymers during degradation by chain scissions. J. Mech. Behav. Biomed. Mater. 5, 224–230. doi:10.1016/j.jmbbm.2011.09.002
Duek, E. A. R., Zavaglia, C. A. C., and Belangero, W. D. (1999). In vitro study of poly(lactic acid) pin degradation. Polymer 40, 6465–6473. doi:10.1016/S0032-3861(98)00846-5
Eyal, A. M., and Canari, R. (1995). pH dependence of carboxylic and mineral acid extraction by amine-based extractants: effects of pKa, amine basicity, and diluent properties. Industrial Eng. Chem. Res. 34, 1789–1798. doi:10.1021/ie00044a030
Fischer, E. W., Sterzel, H. J., and Wegner, G. (1973). Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reactions. Kolloid-Z, U. Z. Polym., 11. doi:10.1007/BF01498927
Fu, K., Pack, D. W., Klibanov, A. M., and Langer, R. (2000). Visual evidence of acidic environment within degrading poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm. Res. 17, 100–106. doi:10.1023/A:1007582911958
George, A. S. H., George, D. A. S., Baskar, D. T., and Shahul, A. (2023). 3D printed organs: a new frontier in medical technology. Partners Univers. Int. Innovation J. 1, 187–208. doi:10.5281/zenodo.8076965
Goyanes, A., Buanz, A. B. M., Hatton, G. B., Gaisford, S., and Basit, A. W. (2015). 3D printing of modified-release aminosalicylate (4-ASA and 5-ASA) tablets. Eur. J. Pharm. Biopharm. 89, 157–162. doi:10.1016/j.ejpb.2014.12.003
Grizzi, I., Garreau, P., Li, S., and Vert, M. (1995). Hydrolytic degradation of devices based on poly(dl-lactic acid) size-dependence. Biomaterials 16, 305–311. doi:10.1016/0142-9612(95)93258-F
Guerra, A. J., Cano, P., Rabionet, M., Puig, T., and Ciurana, J. (2018). 3D-Printed PCL/PLA composite stents: towards a new solution to cardiovascular problems. Materials 11, 1679. doi:10.3390/ma11091679
Hochleitner, G., Chen, F., Blum, C., Dalton, P. D., Amsden, B., and Groll, J. (2018). Melt electrowriting below the critical translation speed to fabricate crimped elastomer scaffolds with non-linear extension behaviour mimicking that of ligaments and tendons. Acta Biomater. 72, 110–120. doi:10.1016/j.actbio.2018.03.023
Hu, Q., Zhang, R., Zhang, H., Yang, D., Liu, S., Song, Z., et al. (2021). Topological structure design and fabrication of biocompatible PLA/TPU/ADM mesh with appropriate elasticity for hernia repair. Macromol. Biosci. 21, 2000423. doi:10.1002/mabi.202000423
Jumat, M. A., Chevallier, P., Mantovani, D., Copes, F., Razak, S. I. A., and Saidin, S. (2021). Three-dimensional printed biodegradable poly(l-lactic acid)/poly(d-lactic acid) scaffold as an intervention of biomedical substitute. Polymer-Plastics Technol. Mater. 60, 1005–1015. doi:10.1080/25740881.2021.1876879
Kai, D., Liow, S. S., and Loh, X. J. (2014). Biodegradable polymers for electrospinning: towards biomedical applications. Mater. Sci. Eng. C 45, 659–670. doi:10.1016/j.msec.2014.04.051
Lee, H., Ahn, S., Choi, H., Cho, D., and Kim, G. (2013). Fabrication, characterization, and in vitro biological activities of melt-electrospun PLA micro/nanofibers for bone tissue regeneration. J. Mater. Chem. B 1, 3670–3677. doi:10.1039/C3TB20424C
Li, S. (1999). Hydrolytic degradation characteristics of aliphatic polyesters derived from lactic and glycolic acids. J. Biomed. Mater. Res. 48, 342–353. doi:10.1002/(SICI)1097-4636(1999)48:3⟨342::AID-JBM20⟩3.0.CO;2-7
Li, S., Garreau, H., and Vert, M. (1990). Structure-property relationships in the case of the degradation of massive poly(alpha-hydroxy acids) in aqueous media. J. Mater. Sci. Mater. Med. 1, 198–206. doi:10.1007/BF00701077
Limsukon, W., Rubino, M., Rabnawaz, M., Lim, L. T., and Auras, R. (2023). Hydrolytic degradation of poly(lactic acid): unraveling correlations between temperature and the three phase structures. Polym. Degrad. Stab. 217, 110537. doi:10.1016/j.polymdegradstab.2023.110537
Lotz, B., Li, G., Chen, X., and Puiggali, J. (2017). Crystal polymorphism of polylactides and poly(Pro-alt-CO): the metastable beta and gamma phases. Formation of homochiral PLLA phases in the PLLA/PDLA blends. Polymer 115, 204–210. doi:10.1016/j.polymer.2017.03.018
Lu, L., Garcia, C. A., and Mikos, A. G. (1999). In vitro degradation of thin poly(DL-lactic-co-glycolic acid) films. J. Biomed. Mater. Res. 46, 236–244. doi:10.1002/(SICI)1097-4636(199908)46:2⟨236::AID-JBM13⟩3.0.CO;2-F
MacLeod, A., Patterson, M., MacTear, K., and Gill, H. S. (2020). 3D printed locking osteosynthesis screw threads have comparable strength to machined or hand-tapped screw threads. J. Orthop. Res. 38, 1559–1565. doi:10.1002/jor.24712
McCormick, C. (2018). “1 - overview of cardiovascular stent designs,” in Functionalised cardiovascular stents. Editors J. G. Wall, H. Podbielska, and M. Wawrzyńska (Duxford, United Kingdom: Woodhead Publishing), 3–26. doi:10.1016/B978-0-08-100496-8.00001-9
Middleton, J. C., and Tipton, A. J. (2000). Synthetic biodegradable polymers as orthopedic devices. Biomaterials 21, 2335–2346. doi:10.1016/S0142-9612(00)00101-0
Moliner, C., Finocchio, E., Arato, E., Ramis, G., and Lagazzo, A. (2020). Influence of the degradation medium on water uptake, morphology, and chemical structure of poly(lactic acid)-sisal bio-composites. Materials 13, 3974. doi:10.3390/ma13183974
Nagarajan, N., Dupret-Bories, A., Karabulut, E., Zorlutuna, P., and Vrana, N. E. (2018). Enabling personalized implant and controllable biosystem development through 3D printing. Biotechnol. Adv. 36, 521–533. doi:10.1016/j.biotechadv.2018.02.004
Pan, P., Liang, Z., Zhu, B., Dong, T., and Inoue, Y. (2008). Roles of physical aging on crystallization kinetics and induction period of poly(l-lactide). Macromolecules 41, 8011–8019. doi:10.1021/ma801436f
Pan, P., Zhu, B., and Inoue, Y. (2007). Enthalpy relaxation and embrittlement of poly(l-lactide) during physical aging. Macromolecules 40, 9664–9671. doi:10.1021/ma071737c
Pantani, R., and Sorrentino, A. (2013). Influence of crystallinity on the biodegradation rate of injection-moulded poly(lactic acid) samples in controlled composting conditions. Polym. Degrad. Stab. 98, 1089–1096. doi:10.1016/j.polymdegradstab.2013.01.005
Passerini, N., and Craig, D. Q. M. (2001). An investigation into the effects of residual water on the glass transition temperature of polylactide microspheres using modulated temperature DSC. J. Control. Release 73, 111–115. doi:10.1016/S0168-3659(01)00245-0
Pierre, T. S., and Chiellini, E. (1987). Review: biodegradability of synthetic polymers for medical and pharmaceutical applications: Part 2—backbone hydrolysis. J. Bioact. Compatible Polym. 2, 4–30. doi:10.1177/088391158700200101
Pistner, H., Bendi, D. R., Mühling, J., and Reuther, J. F. (1993). Poly (l-lactide): a long-term degradation study in vivo: Part III. Analytical characterization. Biomaterials 14, 291–298. doi:10.1016/0142-9612(93)90121-H
Pitt, G. G., Gratzl, M. M., Kimmel, G. L., Surles, J., and Sohindler, A. (1981). Aliphatic polyesters II. The degradation of poly (DL-lactide), poly (ε-caprolactone), and their copolymers in vivo. Biomaterials 2, 215–220. doi:10.1016/0142-9612(81)90060-0
Qamar, N., Abbas, N., Irfan, M., Hussain, A., Arshad, M. S., Latif, S., et al. (2019). Personalized 3D printed ciprofloxacin impregnated meshes for the management of hernia. J. Drug Deliv. Sci. Technol. 53, 101164. doi:10.1016/j.jddst.2019.101164
Ren, J., Murray, R., Wong, C. S., Qin, J., Chen, M., Totsika, M., et al. (2022). Development of 3D printed biodegradable mesh with antimicrobial properties for pelvic organ prolapse. Polymers 14, 763. doi:10.3390/polym14040763
Roper, D. M., Kwon, K. A., Best, S. M., and Cameron, R. E. (2021). The 3D printing of freestanding PLLA thin layers and improving first layer consistency through the introduction of sacrificial PVA. Appl. Sci. 11, 6320. doi:10.3390/app11146320
Roper, D. M., Kwon, K. A., Malone, L. P., Best, S. M., and Cameron, R. E. (2022). Induced birefringence in 3D printing: concealing information optically within printed objects. Adv. Mater. Technol. 7, 2200139. doi:10.1002/admt.202200139
Ru, J. F., Yang, S. G., Zhou, D., Yin, H. M., Lei, J., and Li, Z. M. (2016). Dominant β-form of poly(l-lactic acid) obtained directly from melt under shear and pressure fields. Macromolecules 49, 3826–3837. doi:10.1021/acs.macromol.6b00595
Sakamoto, A., Jinnouchi, H., Torii, S., Virmani, R., and Finn, A. V. (2018). Understanding the impact of stent and scaffold material and strut design on coronary artery thrombosis from the basic and clinical points of view. Bioengineering 5, 71. doi:10.3390/bioengineering5030071
Sevim, K., and Pan, J. (2018). A model for hydrolytic degradation and erosion of biodegradable polymers. Acta Biomater. 66, 192–199. doi:10.1016/j.actbio.2017.11.023
Siemann, U. (1985). The influence of water on the glass transition of poly(dl-lactic acid). Thermochim. Acta 85, 513–516. doi:10.1016/0040-6031(85)85633-1
Singh, D., Babbar, A., Jain, V., Gupta, D., Saxena, S., and Dwibedi, V. (2019). Synthesis, characterization, and bioactivity investigation of biomimetic biodegradable PLA scaffold fabricated by fused filament fabrication process. J. Braz. Soc. Mech. Sci. Eng. 41, 121. doi:10.1007/s40430-019-1625-y
Song, Z., Ma, Y., Xia, G., Wang, Y., Kapadia, W., Sun, Z., et al. (2017). In vitro and in vivo combined antibacterial effect of levofloxacin/silver co-loaded electrospun fibrous membranes. J. Mater. Chem. B 5, 7632–7643. doi:10.1039/C7TB01243H
Tamai, H., Igaki, K., Kyo, E., Kosuga, K., Kawashima, A., Matsui, S., et al. (2000). Initial and 6-month results of biodegradable poly-l-lactic acid coronary stents in humans. Circulation 102, 399–404. doi:10.1161/01.cir.102.4.399
Tsuji, H. (2002). Autocatalytic hydrolysis of amorphous-made polylactides: effects of l-lactide content, tacticity, and enantiomeric polymer blending. Polym. (Guilford) 43, 1789–1796. doi:10.1016/S0032-3861(01)00752-2
Tsuji, H., and Ikada, Y. (1997). Blends of crystalline and amorphous poly(lactide). III. Hydrolysis of solution-cast blend films. J. Appl. Polym. Sci. 63, 855–863. doi:10.1002/(sici)1097-4628(19970214)63:7<855::aid-app5>3.3.co;2-4
Tsuji, H., and Ikada, Y. (1998). Properties and morphology of poly(L-lactide). II. Hydrolysis in alkaline solution. J. Polym. Sci. Part A Polym. Chem. 36, 59–66. doi:10.1002/(SICI)1099-0518(19980115)36:1⟨59::AID-POLA9⟩3.0.CO;2-X
Tsuji, H., and Ikarashi, K. (2004a). In vitro hydrolysis of poly(l-lactide) crystalline residues as extended-chain crystallites. Part I: long-term hydrolysis in phosphate-buffered solution at 37°C. Biomaterials 25, 5449–5455. doi:10.1016/j.biomaterials.2003.12.053
Tsuji, H., and Ikarashi, K. (2004b). In vitro hydrolysis of poly(l-lactide) crystalline residues as extended-chain crystallites: II. Effects of hydrolysis temperature. Biomacromolecules 5, 1021–1028. doi:10.1021/bm034523l
Tsuji, H., Ikarashi, K., and Fukuda, N. (2004). Poly(l-lactide): XII. Formation, growth, and morphology of crystalline residues as extended-chain crystallites through hydrolysis of poly(l-lactide) films in phosphate-buffered solution. Polym. Degrad. Stab. 84, 515–523. doi:10.1016/j.polymdegradstab.2004.01.010
Tsuji, H., Mizuno, A., and Ikada, Y. (2000). Properties and morphology of poly(L-lactide). III. Effects of initial crystallinity on long-term in vitro hydrolysis of high molecular weight poly(L-lactide) film in phosphate-buffered solution. J. Appl. Polym. Sci. 77, 1452–1464. doi:10.1002/1097-4628(20000815)77:7⟨1452::AID-APP7⟩3.0.CO;2-S
Tsuji, H., and Suzuyoshi, K. (2002). Environmental degradation of biodegradable polyesters 1. Poly(ε-caprolactone), poly[(R)-3-hydroxybutyrate], and poly(L-lactide) films in controlled static seawater. Polym. Degrad. Stab. 75, 347–355. doi:10.1016/S0141-3910(01)00240-3
Vaid, R., Yildirim, E., Pasquinelli, M. A., and King, M. W. (2021). Hydrolytic degradation of polylactic acid fibers as a function of pH and exposure time. Molecules 26, 7554. doi:10.3390/molecules26247554
Vasanthan, N., and Ly, O. (2009). Effect of microstructure on hydrolytic degradation studies of poly (l-lactic acid) by FTIR spectroscopy and differential scanning calorimetry. Polym. Degrad. Stab. 94, 1364–1372. doi:10.1016/j.polymdegradstab.2009.05.015
Vert, M., Li, S., and Garreau, H. (1991). More about the degradation of LA/GA-derived matrices in aqueous media. J. Control. release 16, 15–26. doi:10.1016/0168-3659(91)90027-B
von Burkersroda, F., Schedl, L., and Göpferich, A. (2002). Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials 23, 4221–4231. doi:10.1016/S0142-9612(02)00170-9
von Recum, H. A., Cleek, R. L., Eskin, S. G., and Mikos, A. G. (1995). Degradation of polydispersed poly(l-lactic acid) to modulate lactic acid release. Biomaterials 16, 441–447. doi:10.1016/0142-9612(95)98816-W
Vyavahare, O., Ng, D., and Hsu, S. L. (2014). Analysis of structural rearrangements of poly(lactic acid) in the presence of water. J. Phys. Chem. B 118, 4185–4193. doi:10.1021/jp500219j
Wang, X., Li, G., Liu, Y., Yu, W., and Sun, Q. (2017). Biocompatibility of biological material polylactic acid with stem cells from human exfoliated deciduous teeth. Biomed. Rep. 6, 519–524. doi:10.3892/br.2017.881
Wasanasuk, K., Tashiro, K., Hanesaka, M., Ohhara, T., Kurihara, K., Kuroki, R., et al. (2011). Crystal structure analysis of poly(l-lactic acid) α form on the basis of the 2-dimensional wide-angle synchrotron X-ray and neutron diffraction measurements. Macromolecules 44, 6441–6452. doi:10.1021/ma2006624
Weir, N. A., Buchanan, F. J., and Dickson, G. R. (2004b). Degradation of poly-L-lactide. Part 1: in vitro and in vivo physiological temperature degradation. Proc. Institution Mech. Eng. Part H, J. Eng. Med. 218, 307–319. doi:10.1243/0954411041932782
Weir, N. A., Buchanan, F. J., Orr, J. F., Farrar, D. F., and Dickson, G. R. (2004a). Degradation of poly-L-lactide. Part 2: increased temperature accelerated degradation. Proc. Institution Mech. Eng. Part H J. Eng. Med. 218, 321–330. doi:10.1243/0954411041932809
Witt, C., and Kissel, T. (2001). Morphological characterization of microspheres, films and implants prepared from poly(lactide-co-glycolide) and ABA triblock copolymers: is the erosion controlled by degradation, swelling or diffusion? Eur. J. Pharm. Biopharm. 51, 171–181. doi:10.1016/S0939-6411(01)00130-8
Yoon, S. D., Kwon, Y. S., and Lee, K. S. (2017). Biodegradation and biocompatibility of poly L-lactic acid implantable mesh. Int. Neurourol. J. 21, S48–S54. doi:10.5213/inj.1734882.441
Keywords: poly-l-lactic acid, degradation, biodegradable polymers, crystallinity, molecular weight, 3D printing, electrospinning
Citation: Malone LP, Best SM and Cameron RE (2024) Accelerated degradation testing impacts the degradation processes in 3D printed amorphous PLLA. Front. Bioeng. Biotechnol. 12:1419654. doi: 10.3389/fbioe.2024.1419654
Received: 18 April 2024; Accepted: 18 June 2024;
Published: 05 July 2024.
Edited by:
Eliza L. S. Fong, National University of Singapore, SingaporeReviewed by:
Ajay Devidas Padsalgikar, Biolinq Inc, United StatesLuis A. Diaz-Gomez, Universidade de Santiago de Compostela, Spain
Copyright © 2024 Malone, Best and Cameron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luke P. Malone, bHBtMzlAY2FtLmFjLnVr