 Ran-Ran Su
Ran-Ran Su Bi-Qiong Pan
Bi-Qiong Pan Xia-Lin Zheng
Xia-Lin Zheng Xiao-Yun Wang
Xiao-Yun Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 22 February 2024
Sec. Industrial Biotechnology
Volume 12 - 2024 | https://doi.org/10.3389/fbioe.2024.1340168
This article is part of the Research Topic Bioconversion of Insect Resources for Sustainability View all 6 articles
The intestinal bacteria of longhorn beetles would be ideal targets for pest control and lignocellulosic resources by destroying or exploiting their cellulose-degrading function. This article aims to investigate the diversity and community structure of intestinal bacteria the oligophagous longhorn beetle Glenea cantor. Additionally, it seeks to identify the presence of lignocellulose-degrading bacteria in the gut, and explore their role in consuming host kapok trees Bombax malabaricum. In this study, the bacterial community from G. cantor was examined by Illumina sequencing of 16S ribosomal RNA (rRNA) targeting the V3 and V4 regions. A total of 563,201 valid sequences and 814 OTUs were obtained. The dominant phyla were Proteobacteria, and the dominant genera were Acinetobacter and Lactococcus. The analysis of microbial diversity revealed a high bacterial diversity in the samples, with the gut bacteria playing a crucial role in the physiological activities of the host, particularly, 9 genera of intestinal bacteria with cellulose degradation function were found, highlighting their vital role in cellulose degradation. Five strains of cellulose-degrading bacteria, belonging to the genus Pseudomonas, were obtained from the intestinal tract of G. cantor larvae using traditional isolation and culture techniques as well as 16S rDNA sequencing. Among these strains, A4 exhibited a cellulase activity of 94.42 ± 0.42 U/mL, while A5 displayed the highest filter paper enzyme activity of 127.46 ± 3.54 U/mL. These results offered valuable insights into potential targets for pest control through internal attack digestion and cellulose-degrading bacteria in longhorn beetles.
Glenea cantor Fabricius (Coleoptera: Cerambycidae: Lamiinae) is widely distributed in Vietnam and southern China (Lu et al., 2007). Under suitable conditions, it can reproduce round without dormancy and diapause all year (Lu et al., 2011). Glenea cantor is oligophagous and can cause serious damage to Bombax malabaricum, an important landscaping tree species (Wu et al., 2022). The females laid eggs in slots on the branches with weak growth vigor (Lai et al., 2008). The methods for controlling these beetles mainly include manual trapping and killing of adults, timely removal of dead branches and debris to reduce the insect population and use chemical control (Zhong et al., 2011). Due to its extensive egg production, high hatching rate and strong reproductive ability (Lai et al., 2008; Lu et al., 2013; Dong et al., 2017), it is an ideal model for the study of management and efficient cellulose degradation in longhorn beetles. As an oligophagous pest, the integrated measurement of G. cantor could be developed by breaching its internal digestion system to achieve (Tokuda, 2019). As a member of Cerambycidae, the wood degradation mechanism of G. cantor would also contribute to research on other devastating longhorn beetles, such as Monochamus alternatus and M. galloprovincialis, the vectors of Bursaphelenchus xylophilus, a pine wood nematode that causes pine wilt disease (Alves et al., 2016; Guo et al., 2020).
The cellulose degradation of longhorn beetles is related to their structure of host plants and intestinal cellulase and hemicellulase activities (Luo et al., 2019). The structure of longhorn beetles’ host plant wood is composed of varying amounts of lignin, cellulose and hemicellulose arranged in a certain pattern (Pauchet et al., 2014). Longhorn beetles inhabit wood and can digest cellulose and hemicellulose, which are the primary constituents of wood (Li et al., 2020a). The wood consumption habits and characteristics of the longhorn beetle derive from digestive enzymes, which may be from endogenous enzymes or/and intestinal flora (Willis et al., 2010; Guan et al., 2011; Li et al., 2020c). Firstly, several endogenous cellulase genes had been cloned from different beetles, including the yellow-spotted longhorn beetle Psacothea hilaris (Sugimura et al., 2003), the mulberry longhorn beetle Apriona germari, Bateocera horsfieldi (Wei et al., 2006a; Wei et al., 2006b; Xia et al., 2013), M. alternatus (Li et al., 2020b) and Mesosa myops (Liu et al., 2015). Secondly, the symbiotic microorganisms of insects, such as bacteria and fungi, are traditionally believed to produce cellulase and other digestive enzymes for nutrients (Shelomi and Chen, 2020; Li et al., 2021). Insect growth and development were affected by microorganisms that directly or indirectly participate in their metabolism (Ayayee et al., 2016). To some extent, the intestinal microbial function of insects were determined by the structure of their intestinal microbial community (Hooper and Gordon, 2001). As a result, the study of gut microbiota has emerged as a novel field concerning the ecological and functional dynamics of the intestinal microbial habitats (Jang and Kikuchi, 2020).
The methods of gut microbiota research include the culture-based method, culture-independent method, and Amplicon-based taxonomic identification, and so on (Romero et al., 2019). A range of bacteria from the insect gut have been identified using traditional isolation and culture techniques (O'Sullivan, 2000). However, culture-dependent bacterial isolation methods may introduce bias in the characterization of microbial communities, as not all bacteria can be cultured in the laboratory (Brock, 1987). 16S rRNA technology has become a widely recognized method which can reflect the dynamic changes of intestinal microbial ecological community structure, and it provides a more accurate means of detecting and identifying both known and unknown bacteria in intestinal flora (Munoz-Benavent et al., 2021). This technology overcomes the complex problems of traditional methods such as morphological examination, isolation and culture, and has sufficient variation to differentiate between most bacterial species (Nguyen et al., 2016). By constructing a library based on 16S rRNA sequencing to analyze the structure and function of intestinal bacteria, we learned that bacteria occupy a dominant position in the intestinal microbiota of beetles, and the dominant flora is relatively stable. For instance, the dominant phyla of Stromatium barbatum were Actinomycetes, Proteobacteria, and Firmicutes (Yadav et al., 2022). Similarly, Cacosceles newmannii exhibited dominance of Proteobacteria (Javal et al., 2023), while Monochamus saltuarius mainly harbored Proteobacteria and Firmicutes (Ge et al., 2021). Additionally, the main intestinal microorganisms of Anoplophora glabripennis consisted of the phyla Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes (Schloss et al., 2006). Thus, the dominant bacterial phyla in the intestinal bacteria of longhorn beetles consist primarily of Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. Although 16S rRNA gene analysis is a valuable approach to study difficult-to-cultivate microorganisms, it overcomes the limitations of traditional isolation methods. It can effectively detect and identify both known and unknown bacteria in the intestinal flora (Munoz-Benavent et al., 2021). However, it is not without its challenges. The relatively accuracy is low in identifying bacterial species. Additionally, the dominant populations may mask the presence of rare microbial communities. To obtain a more comprehensive understanding of insect gut microbiota, long-term studies have demonstrated that a combination of culture-dependent and high-throughput sequencing technologies is necessary (Wang et al., 2020).
The larvae of longhorn beetles exhibit a remarkable capacity for cellulose and lignin degradation, with their intestinal microorganisms playing a vital role in facilitating this process. Therefore, extensive research had been conducted on the screening and functional analysis of cellulose-degrading bacteria in longhorn beetles. The types of intestinal cellulose-degrading bacteria varied depending on the species of longhorn beetles, their hosts and habitats. For instance, the intestinal tract of Batocera lineaolata larvae contained strains belonging to the genus Ochrobactrum and Raoultella, which exhibited high cellulase production (Yang et al., 2021). Similarly, Bacillus subtilis, derived from the gut of Aromia bungii larvae, with high cellulase activity, had the ability to enhance the protein content of broiler chickens and improve their intestinal microflora (Su et al., 2015). The cellulose-degrading bacteria derived from the pre-midgut of Apriona germari larvae belong to the genus Cellulamonas and Bacillus subtilis (He et al., 2001; Xie et al., 2020). Longhorn beetles are a diverse group of insects, with approximately 35,000 known species worldwide. In China, more than 2,000 species have been reported. These beetles mainly feed on woody plants (Jin et al., 2019). They develop intestinal microorganisms that acquired the ability to break down cellulose and lignin, giving them a competitive advantage with long-term domestication of host plants (Dar et al., 2022; Yadav et al., 2022; Xie et al., 2023). Despite the abundance of longhorn beetles, there is insufficient research on their cellulose degradation mechanisms.
The ability of longhorn beetles to degrade cellulose primarily depends on cellulase. Cellulase production was carried out by symbiotic bacteria in the gut, such as Batocera lineaolata (Yang et al., 2021), Aromia bungii (Su et al., 2015), and Apriona germari (Xie et al., 2020). Additionally, longhorn beetles themselves produce proteins encoded by their own cellulase genes, for example, Batocera horsfieldi (Mei et al., 2016), Monochamus alternatus (Li et al., 2020c), and Anoplophora malasiaca (Chang et al., 2012). In some cases, they may produce cellulase using both mechanisms, for instance, Apriona germari (Wei et al., 2006a; Wei et al., 2006b; Xie et al., 2020). Cellulase with endoglucanase activity, cellobiase activity, and filter paper enzyme activity mainly existed in midgut of G. cantor (Yang et al., 2011). However, it was unknown that the source of its enzyme production originated from intestinal microorganisms, the beetles themselves, or both. From this perspective, the comparative 16S rRNA sequencing method was used to obtain gut bacterial diversity analysis of the larval gut and frass directly. Cellulose-degrading bacteria were screened and identified by traditional isolation culture and 16S rDNA bacterial identification technology, which derived from the gut of G. cantor. These results would contribute to the bacterial community structure and diversity, the function of the intestinal bacteria, the digestive mechanism and the possible gut bacteria targets for developing novel control method in G. cantor.
The complexity of cellulose structure (Tsegaye et al., 2019) and the low activity of cellulose-degrading enzymes in nature pose challenges in efficiently degrading and utilizing cellulose resources in practical applications (Zhu and Pan, 2022). Consequently, the high-value utilization of cellulose as a resource is limited. This study aims to analyze the structure and function of the intestinal microbial community and screen target strains in G. cantor larvae. The findings provide a foundation for understanding the mechanism of insect cellulose degradation and offer valuable insights for utilizing insect intestinal microorganisms in cellulose degradation, based on the cellulose-degrading properties of insect-derived cellulases.
Glenea cantor was collected from kapok trees in Qingxiu Mountain (22°12′∼23°32′N, 107°45′∼108°51′E), Nanning, China. When an adult came out of the wood, the male and female adults were placed in a glass bottle for mating, and a small section of coarse kapok, about 4 cm in diameter and 6 cm in length, was provided as food. The cuticle with the eggs were cut out with a knife and cultured in a petri dish containing moist cotton. The eggs were incubated until the larvae reached the fourth instar. G. cantor were reared under controlled conditions with an indoor temperature of 25°C ± 1°C, a relative humidity of 70% ± 5% and a photoperiod of 14L: 10D.
The fourth instar larvae of G. cantor with similar body weight were randomly divided into three groups. Fresh frass samples from larvae were collected daily at 7 P.M. for a duration of 6 days. These frass samples were then stored at −20°C before DNA extraction. The larval frass samples were labeled as GcLF. The detailed information of insects and frass samples was listed in Supplementary Table S1 for reference. Five larvae were grouped together as a repetition and transferred to a moist, sterile culture dish. They were starved for 48 h to empty the intestinal food residue. The larvae were washed with sterile water and subsequently treated with 75% alcohol for surface sterilization, then rinsed with sterile water, repeated the process three times. Under the sterile conditions, the larvae of G. cantor were dissected by removing the head and cutting along the back line of the cuticle in PBS solution. The intestines were then extracted from the body using tweezers. The surface of the intestines was carefully stripped of any irrelevant material. The extracted intestines were placed in a 1.5 mL centrifuge tube containing PBS buffer and frozen in liquid nitrogen. Finally, the samples were stored at −80°C. The larval gut of G. cantor was labeled as GcLG.
DNA samples were extracted using the HiPure Tissue DNA Kits and HiPure Stool DNA Kits (Magen, Guangzhou, China) following the manufacturer’s protocols. The concentration of DNA was assessed by Nanodrop 2000C (Thermo Scientific, Waltham, MA, USA), and purity was monitored by 1% agarose gel electrophoresis. DNA was diluted to a concentration of 100 ng/μL with sterile water. For PCR amplification, the V3-V4 region of the bacterial 16S rRNA gene was performed using the primers (341F: CCTACGGGNGGCWGCAG; 806R: GGACTACHVGGGTATC TAAT) (Guo et al., 2017). PCR reactions were performed in triplicate using a 50 μL mixture containing 5 μL of 10 × KOD Buffer, 5 μL of 2 mM dNTPs, 3 μL of 25 mM MgSO4, 1.5 μL of each primer (10 μM), 1 μL of KOD Polymerase, and 100 ng of template DNA.
Amplicons were extracted from 2% agarose gels, purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, U.S.) and quantified by ABI StepOnePlus Real-Time PCR System (Life Technologies, Foster City, USA). The purified amplicons were pooled in equimolar and paired-end sequencing (PE250) on an Illumina platform according to the standard protocols. FASTP was used to excluded low-quality adapters and reads which contained more than 10% of unknown nucleotides and less than 50% of bases with quality (Q-value) > 20 (Chen et al., 2018). Then the achieved paired end clean reads were merged as raw tags using FLSAH (version 1.2.11) with a minimum overlap of 10 bp and a mismatch error rate of 2% (Magoč and Salzberg, 2011). Raw tags were filtered by QIIME (version 1.9.1) pipeline to obtain the high-quality clean tags (Caporaso et al., 2010). Finally, the effective tags were obtained by removing all chimeric tags which were found by reference-based chimera checking via UCHIME algorithm by searching against the reference database (version r20110519, http://drive5.com/uchime/uchime_download.html, accessed in July 2020) (Edgar et al., 2011).
The effective tags were clustered into operational taxonomic units (OTUs) of ≥ 97% similarity using UPARSE (version 9.2.64) (Edgar, 2013). Between groups Venn analysis of achieved OTUs was performed in the R project Venn Diagram package (version 1.6.16) (Chen and Boutros, 2011). The tag sequence with highest abundance was selected as representative sequence within each cluster and were classified into organisms by a naive Bayesian model with the RDP classifier (version 2.2) based on Greengene database (version gg_13_5) (DeSantis et al., 2006; Wang et al., 2007). The stacked bar plot of the community composition was visualized in R project ggplot2 package (version 2.2.1) (Wickham and Chang, 2008). Chao1, Simpson and all other alpha diversity index were also calculated in QIIME (Caporaso et al., 2010). The comparison of alpha index between groups was calculated by Welch’s t-test and Wilcoxon rank test in the R project Vegan package (version 2.5.3) (Dixon, 2003). The KEGG pathway analysis of the OTUs was inferred using Tax4Fun (version 1.0) and PICRUSt2 (version 2.1.4) (Langille et al., 2013; Aßhauer et al., 2015).
The intestinal tract of the fourth-instar larvae of G. cantor were close to neutral, and pH 7 should be selected when screening intestinal bacteria based on our pH measurement of different intestinal sections (Figure 1; Supplementary Table S2). Ten fourth-instar larvae with a similar size were selected and subjected to a 48-hour starvation period to remove any remaining food residues from their intestines. The dissection process was carried out under the super clean workbench after 30 min of UV treatment. Initially, the larvae were cleansed by immersing them in sterile water for 30 s, followed by a 1-minute soak in 75% alcohol to sterilize their body surfaces. This was followed by another 10-second rinse in sterile water. The process was repeated three times. During the dissection, the epidermis of the larvae was carefully cut along the top line by the sterilized scissors. The intestine was then gently extracted using tweezers, while ensuring the removal of any fat or other substances attached to its surface. The obtained intestinal tract was placed in a 2.0 mL centrifuge tube containing 500 μL of sterile water. It was thoroughly ground using a grinding pestle, and sterile water was added to achieve a final volume of 1 mL. Subsequently, 50% glycerol was added, and the resulting solution was mixed with the intestinal bacteria solution in a 1:1 ratio. This mixture was then stored in a −80°C refrigerator as the stock solution of longhorn beetles’ larval intestines for future use.
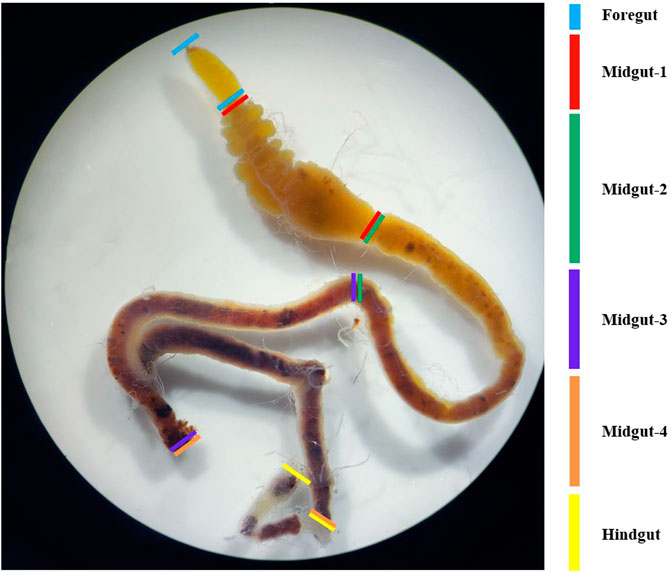
FIGURE 1. Sectional morphology of the intestinal tract of the fourth instar of G. cantor. Note: To determine the pH of each tissue in the intestinal tract of G. cantor, fourth-instar larvae were collected and dissected to obtain the complete intestinal tract on a dissection plate. The intestinal tract contents were squeezed out on a wide range of pH test paper, and the preliminary pH were estimated by comparing the color of the test paper with the color chart. For accurate measurement, precision pH test paper was used based on the preliminary results (Supplementary Table S2).
For enrichment purposes, 1 mL of each gut homogenate was separately inoculated into conical flasks containing 100 mL of the enrichment medium (All components of the culture medium were listed in Supplementary Table S3). The inoculated culture media were incubated at 37°C and 200 r/min for 48 h. The cultured bacterial solution was serially diluted in sterilized water from 10−3 to 10−8 dilution. Then, 100 μL of each dilution was spread in three duplicates on the Congo red cellulose Sodium medium plates. The sterile water control group 1 and empty medium control group 2 were added. The plates were incubated under aerobic conditions at 37°C for 48 h in case of cellulose-degrading bacteria. The cellulose degradation circles were observed in the plates. Next, the bacterial colonies, which produced the cellulose degradation circles, were picked and transferred to LB solid medium plates for purification. The isolated bacteria were purified through the repeated streaking methods, codified and preserved for further examination. The purified single colonies were incubated overnight at 37°C and 200 r/min. Finally, they were mixed with 50% glycerol in a 1:1 ratio and stored it at −80 °C for future use.
The preserved strains were diluted to a concentration of 10−5 and spread on a medium containing Congo red cellulose sodium. Four sets of biological replicates were prepared for each strain and cultured upside down at 37°C for 48 h. Subsequently, colonies from each group were selected from the solid medium of sodium carboxymethyl cellulose. From each plate, 3 colonies were picked and a total of 5 plates were used in each group. The selected strains were then cultured at 37°C for 48 h. To evaluate the hydrolysis zone, the strains on the petri dish were first stained with a 1% Congo red staining solution for 30 min, followed by decolorization with a 1 mol/L NaCl solution for another 30 min. After decolorization, the colony diameter of the hydrolysis zone was measured and recorded using an electronic vernier caliper. The ability of cellulose-degrading bacteria to degrade cellulose can be compared by the ratio of the diameter of the transparent circle (D) to the diameter of the colony (d). The larger the ratio of D/d, the stronger the ability of the corresponding strain to degrade cellulose, and vice versa.
The strains that produced the cellulose degradation circles should be inoculated into the enzyme-producing medium with a 1% inoculum. The cultivation should be carried out for 48 h at a temperature of 37°C and a speed of 180 r/min. After cultivation, the mixture should be centrifuged at 5000 r/min at 4°C for 15 min. The resulting supernatant is the crude enzyme solution, which should be stored at 4°C for future use.
The Filter Paper Activity (FPA) was measured using the DNSA method (Miller, 1959). The strips of qualitative filter paper measuring 1 cm × 6 cm were cut and placed at the bottom of a test tube. Next, 1.5 mL of Hac-NaAc buffer solution (pH = 4.8) and 1 mL of crude enzyme solution were added. The mixture was then incubated in a constant temperature water bath at 50°C for 30 min. After that, 2.5 mL of DNS reagent was added and the solution was boiled for 5 min. Subsequently, the mixture was diluted to 10 mL using distilled water and the OD value was measured at a wavelength of 540 nm. The enzyme activity unit (IU) was defined as follows: 1 mL of crude enzyme solution produced 1 μg of reducing sugar within 1 min.
Cellulase activity was measured using the DNSA method (Miller, 1959). To perform the measurement, 1 mL of prepared crude enzyme solution was added to a test tube. Then, 1.5 mL of Hac-NaAc buffer solution with 1% CMC-Na was added to the same test tube. The mixture was thoroughly mixed and kept at 50°C for 30 min. After that, DNS solution (2.5 mL) was added to each test tube and shaken well. The test tubes were then placed in a boiling water bath and boiled to stop the enzymatic reaction for 5 min. The absorbance value was measured at a wavelength of 540 nm after quickly cooling down the mixture and adjusting the volume to 10 mL. The obtained value was substituted into the glucose calibration curve to calculate the corresponding value A.
The enzyme activity of FPA/CMCA was calculated using the following formula:
Note: The value A is obtained by substituting the measured absorbance value into the standard curve equation (Supplementary Figure S1, see the attachment for standard curve preparation and Supplementary Table S4). Here, n represents the dilution ratio of the solution, 1000 is the conversion factor from mg to μg, t represents the color development time, and v represents the volume of the crude enzyme solution.
The morphological identification and physiological characteristics of cellulose-degrading bacteria can be determined by observing and recording the color, shape, and texture of a single colony under an optical microscope after plate culture. The identification of bacterial cell morphology follows the methods outlined in the “Bergey’s Bacteria Identification Manual,” which includes identifying bacterial colony color, bacterium shape, size, and distinguishing between Gram-negative and Gram-positive bacteria. Molecular identification of cellulose-degrading bacteria involves extracting the total DNA of each bacteria using a bacterial DNA extraction kit. Universal bacterial primers 27F (AGTTTGATCMTGGCTCAG) and 1492R (GGTTACCTTGTTACGACTT) are then used to amplify bacterial 16S rDNA. Subsequently, 16S rDNA sequencing is performed to construct a phylogenetic tree and perform cluster analysis to identify the species of cellulose-degrading bacteria. The 16S rDNA gene sequence of the cellulose-degrading bacteria strains found in the larval gut of G. cantor was compared with known 16S rDNA sequences in the NCBI database (https://www.ncbi.nlm.nih.gov/) using BLAST. The highly similar and reliable sequences were downloaded for clustering analysis. The obtained sequences and target gene sequences were analyzed by MEGA 11.0 to construct a Neighbor-joining phylogenetic tree. The bootstrap analysis was set to 1000 repetitions to calculate the support rate of each branch.
Through Illumina HiSeq of 16S rRNA gene amplicon sequencing, 643, 626 raw pair end reads were obtained from 6 samples (Supplementary Data Sheet S1). After quality filtering and chimera removal, 563,201 (87.5%) effective tags remained for analysis. These tags were clustered into 814 OTUs at 97% sequence identity. The average value of common and unique OTUs between the two groups of GcLG and GcLF were analyzed and viewed by a Venn diagram (Figure 2). There were 248 OTUs shared between each component in the group, which accounted for 47.88% and 45.59% of the total number of OTUs in each sample, respectively. Additionally, there were 270 and 296 unique OTUs within each group, accounting for 52.12% and 54.41% of the total number of OTUs in the sample.
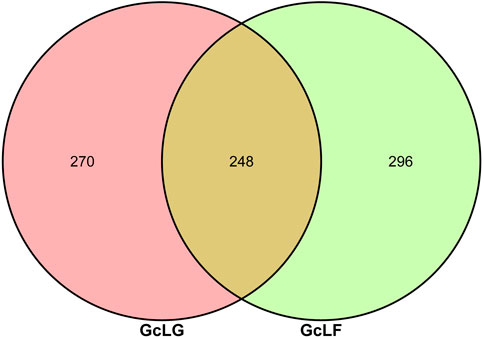
FIGURE 2. Veen diagram of OTUs of bacteria in intestinal and frass samples from the larva of G. cantor. Note: GcLG: intestinal sample; GcLF: frass sample.
Bacterial alpha diversity indexes, including ace, chao, Simpson, Shannon and sobs were compared between larval gut and frass samples (GcLG and GcLF) in G. cantor (Table 1). The species abundance and diversity of GcLF were higher than that of GcLG, but the difference between GcLG and GcLF were not significant, which indicated that the dominant bacteria of larval gut may be transferred to the feces through the digestive tract. Therefore, the analysis of fresh fecal bacteria may reflect larval intestinal status.
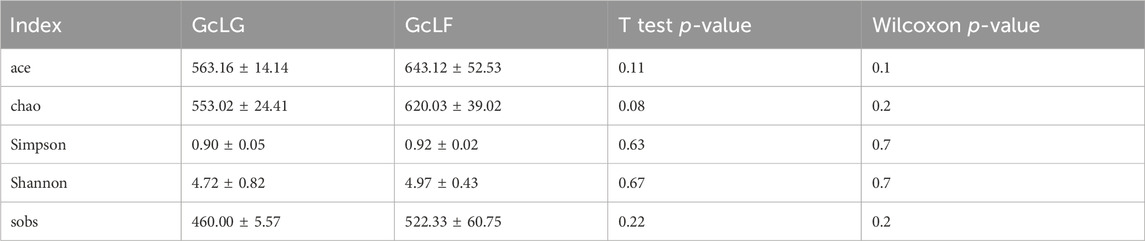
TABLE 1. Bacterial alpha diversity in larval gut and frass samples (GcLG and GcLF) in G. cantor.
Bacterial community composition were analyzed at phylum, class and genus level which only the relative abundance of top 10 species was listed in larval gut and frass samples (GcLG and GcLF) of G. cantor (Figure 3). The six most abundant phyla were Proteobacteria, Firmicutes, Cyanobacteria, Bacteroidetes, Actinobacteria and Patescibacteria in gut samples. The first four phyla accounted for 96% of all the reads. Similarly, the four most abundant phyla of frass samples were Proteobacteria, Bacteroidetes, Patescibacteria, and Actinobacteria, which account for 99% of all the reads. Overall, Proteobacteria was the most common phylum, accounting for an average of 71% of all the reads (Figure 3A). At class level, Gammaproteobacteria was the most abundant with 50% and 71% of reads in larval gut and frass samples (Figure 3B). At genus level, Lactococcus and Enterococcus were the two most abundant of larval gut samples, accounting for 17% and 3% of all reads. In contrast, Acinetobacter and Ochrobactrum were the two most abundant of larval frass samples, making up 42% and 5% of all reads (Figure 3C).
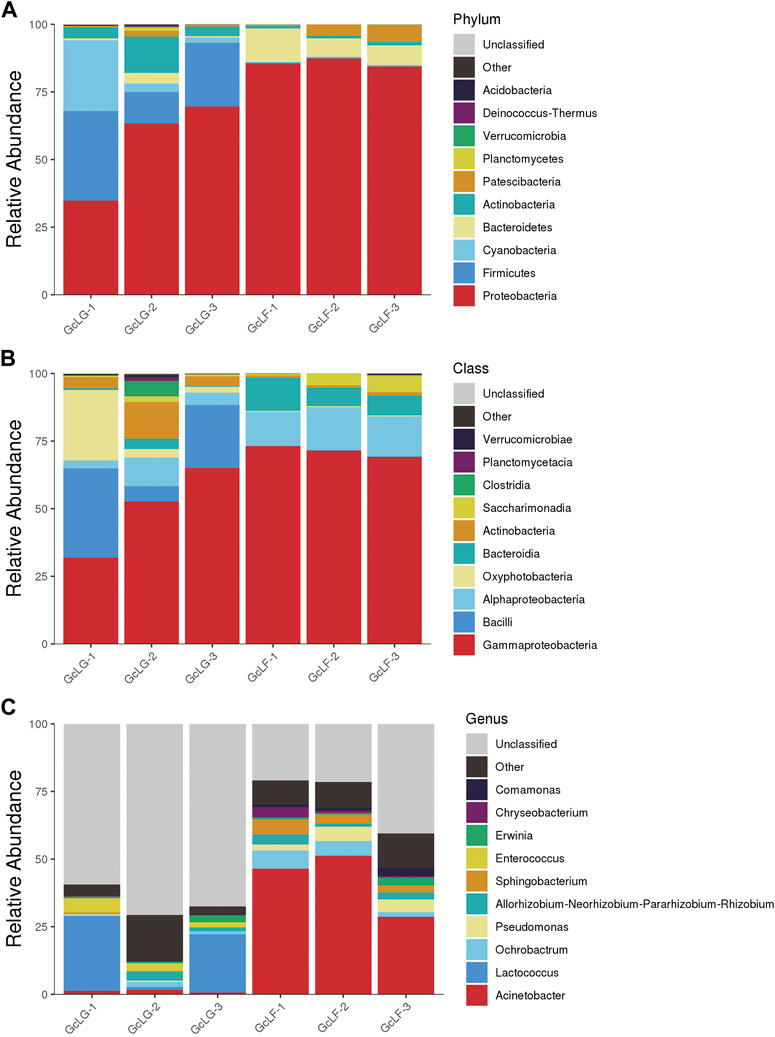
FIGURE 3. Bacterial community composition in larval gut and frass samples in G. cantor. Note: (A) Bacterial community composition at phylum level; (B) Bacterial community composition at class level; (C) Bacterial community composition at genus level. Only the relative abundance of top 10 species were listed at relative levels. “Other” indicates the relative abundance of species that not included but could be identified at relative level; thus “unclassified” indicates species that could not be identified.
The distribution of functional genes in the metabolic pathways of the gut and larval fecal microbial communities of G. cantor were shown in Figure 4. The bacterial flora of the gut and feces primarily contributed to metabolic activities, genetic information processing, and environmental information processing (Figure 4A). Upon averaging, the functions can be ranked from high to low according to the relative abundance ratio (greater than 1%): Carbohydrate Metabolism (14.02%), Membrane Transport (12.96%), Amino Acid Metabolism (10.94%), Signal Transduction (7.54%), Metabolism of Cofactors and Vitamins (6.72%), Energy Metabolism (6.41%), Nucleotide Metabolism (5.41%), Translation (4.60%), Replication and Repair (4.34%), Lipid Metabolism (3.61%), Xenobiotics Biodegradation and Metabolism (3.36%), Infectious Diseases (3.29%), Glycan Biosynthesis and Metabolism (2.83%), Enzyme Families (2.76%), Metabolism of Other Amino Acids (2.55%), Folding, Sorting and Degradation (2.32%), Cell Motility (2.25%), Cell Growth and Death (1.37%) (Figure 4B). The results indicated that the intestinal bacterial flora of G. cantor was mainly involved in carbohydrate metabolism, membrane transport, and amino acid metabolism, as well as partial lipid, nucleotide metabolism and biodegradation of the host. In carbohydrate metabolism, the bacterial flora primarily participated in starch and sucrose metabolism, amino sugar and nucleotide sugar metabolism, and pyruvate metabolism. Additionally, they played a role in arginine and proline metabolism, glycine, serine and threonine metabolism, and phenylalanine, tyrosine and tryptophan biosynthesis in Amino Acid Metabolism. Furthermore, the bacterial flora was involved in the membrane transport, particularly ABC transporters and bacterial secretion system (Figure 4C). There was no significant difference in the abundance of these metabolic functions between the larval intestine and larval frass. The biodegradation function indicated that intestinal bacterial flora participated in the degradation of certain substances (such as starch and sucrose, amino sugar, nucleotide sugar and amino acid) of the host in these metabolic pathways.
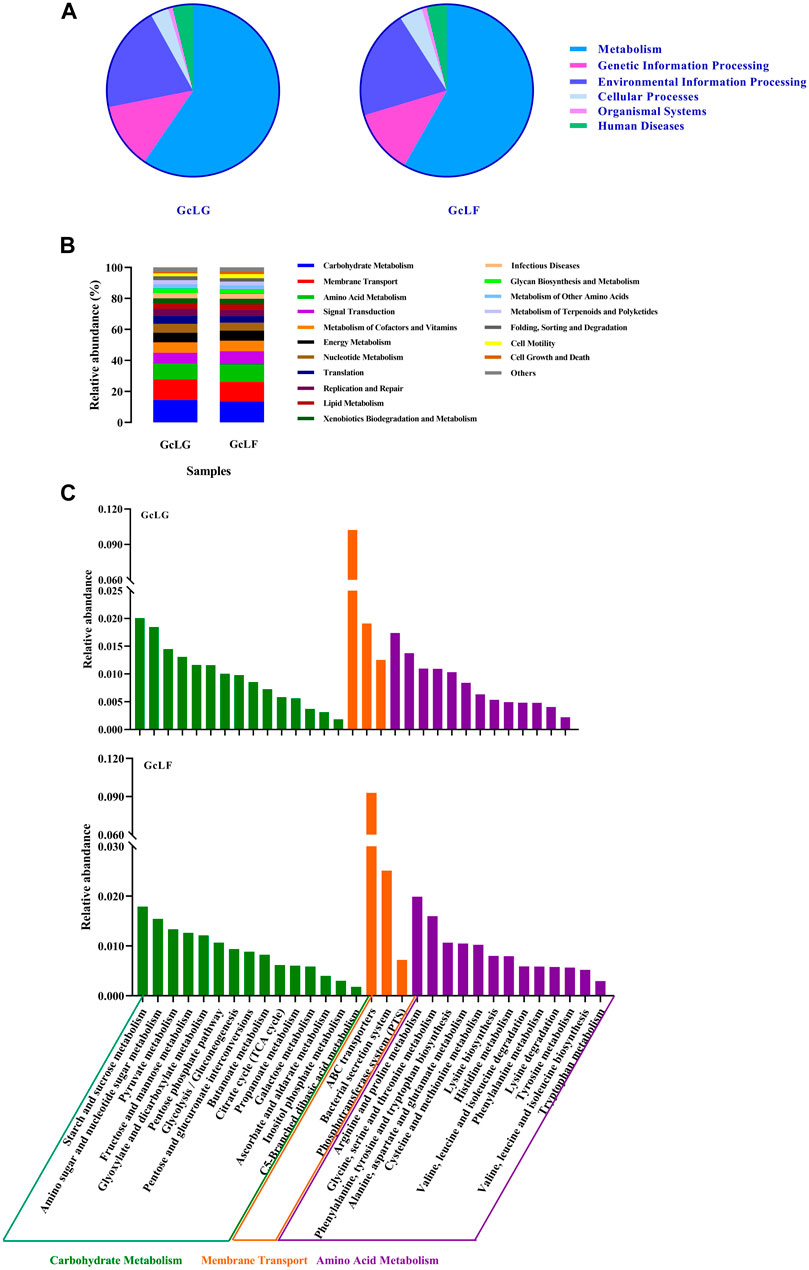
FIGURE 4. The KEGG function analysis of intestinal and fecal in G. cantor larvae. Note: GcLG: intestinal sample; GcLF: frass sample. (A) The pie chart of KEGG function analysis based on Tax4Fun algorithms. (B) The stacked diagram of KEGG functional analysis based on PICRUST2 algorithms. The diagram is only for metabolic pathways whose relative abundance was greater than 1%. (C) The results of KEGG functional analysis in Carbohydrate Metabolism, Membrane Transport and Amino Acid Metabolism based on Tax4Fun algorithms.
Based on the results of OTUs’ annotation of species, the lignocellulose-degrading bacteria were found at the genus level, as shown in Table 2. Cellulose-degrading bacteria were found at the genus level belonging to 12 genera. Among them, the abundance information ratio of cellulose-degrading bacteria belonging to 4 genera, namely Microbacterium, Streptomyces, Lachnospiraceae_NK4A136_group and Gordonia, were higher in the gut compared to fecal samples. On the other hand, the abundance information of 8 genera of cellulose-degrading bacteria, including Chryseobacterium, Stenotrophomonas, Flavobacterium, Cellvibrio, Acinetobacter, Paenibacillus, Pseudomonas and Sphingobacterium, were lower than that in fecal samples. Notably, the abundance of Chryseobacterium, Acinetobacter, and Stenotrophomonas in fecal samples was much greater than that in intestinal samples, and their GcLF/GcLG values were 41.69, 34.72, and 22.51, respectively.
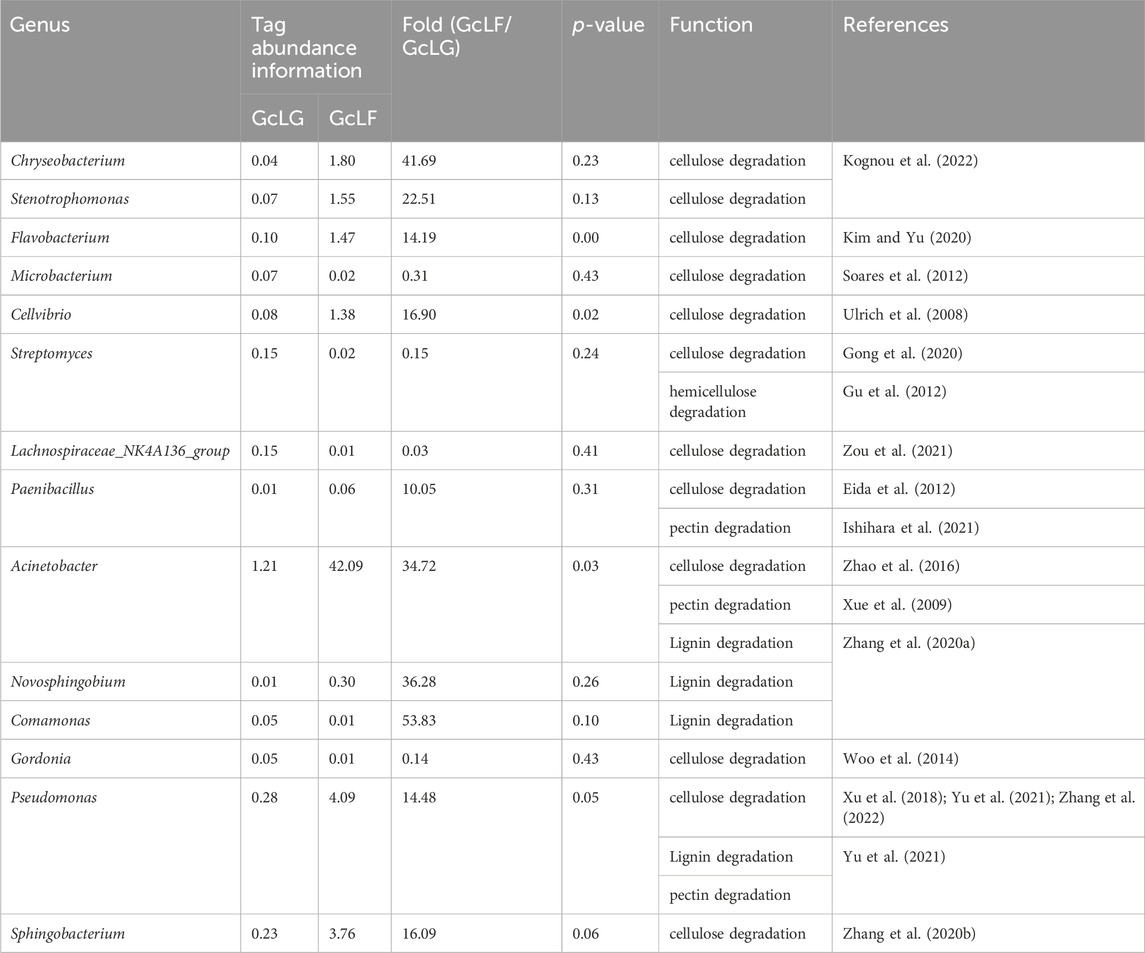
TABLE 2. At the genus level, bacterial with lignocellulose decomposition ability obtained in larval gut and frass samples (GcLG and GcLF) in G. cantor.
The intestinal microorganisms of the G. cantor fourth-instar larvae were screened and cultivated for cellulose, and 5 strains of cellulose-degrading bacteria were isolated, named A1, A2, A3, A4 and A5 respectively (Figure 5). Each colony degraded cellulose and produced a transparent circle, colony diameter and transparent circle diameter were shown in the Table 3. A3 has the strongest ability to degrade cellulose, significantly higher than A1, A4 and A5, but not significantly different from A2, and A5 has the weakest ability to degrade cellulose.
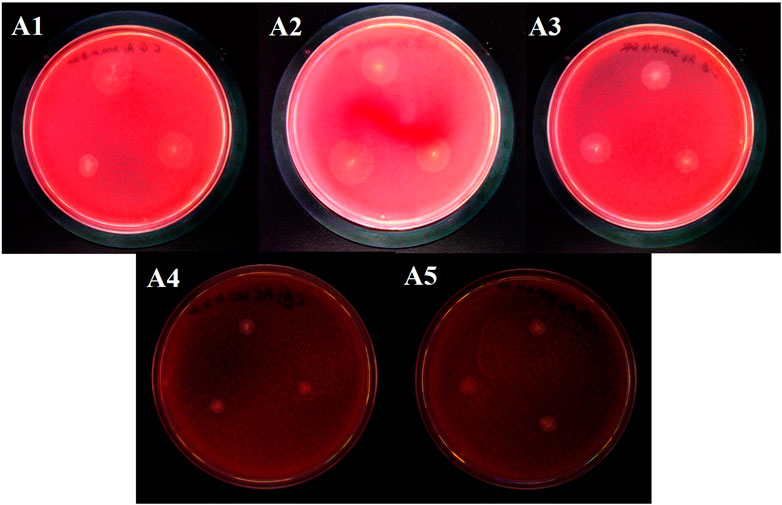
FIGURE 5. Cellulose-degrading bacteria derived from the intestinal tract of G. cantor.

TABLE 3. Colony diameters and cellulose degradation circles diameters of cellulose degrading bacteria derived from intestinal tract of G. cantor.
The cellulase activity (CMCA) and filter paper enzyme activity (FPA) were measured using the DNSA method (Table 4). Among the 5 cellulose degrading bacteria strains, A3 and A4 had the highest cellulase activity measuring 94.09 ± 1.10 U/mL and 94.42 ± 0.42 U/mL, respectively. And the statistical analysis showed no significant difference compared to A1 and A2, while A5 had the lowest cellulase activity. The filter paper enzyme activity of A5 was the highest (127.46 ± 3.54 U/mL), but there was no significant difference compared to A1, A2 and A3. The filter paper enzyme activity of A4 was the lowest, and there was no difference between the A1, A2 and A3.
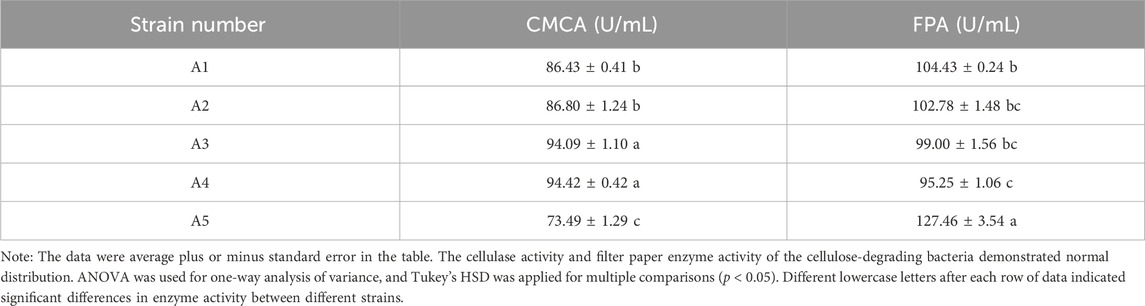
TABLE 4. Cellulase activity and filter paper activity of cellulose-degrading bacteria strains of G. cantor.
All 5 cellulose degrading bacterial colonies were circular, white, opaque with protrusions, moist surfaces, and intact edges. No differences were observed in terms of colony morphology. Gram staining was performed on them separately, and the results were all pink (Supplementary Figure S2), indicating that they were Gram negative bacteria. Under the microscope, the strains appeared rod-shaped. DNA of five cellulose-degrading bacteria were extracted and used as a template for PCR amplification with 27F and 1492R as primers. The agarose gel electrophoresis results were shown in Supplementary Figure S3. The PCR product bands were about 1500 bp (Supplementary Data Sheet S2). By comparing the 16S rDNA sequence of five strains with that of NCBI strain, 1000 bootstraps were applied to the phylogenetic tree, it was found that five cellulose-degrading bacteria belonging to Pseudomonas (Figure 6). Based on the morphology and Gram staining results, 5 strains of cellulose-degrading bacteria were identified as Pseudomonas aeruginosa.
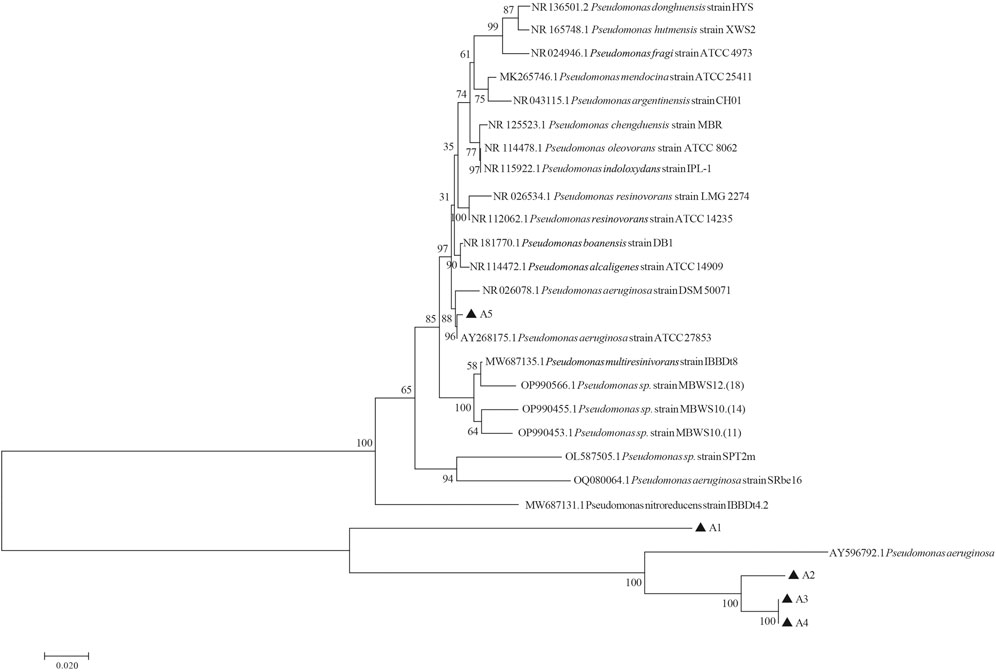
FIGURE 6. Phylogenetic tree of cellulose-degrading bacteria derived from the intestinal tract of G. cantor larvae constructed based on 16S rDNA.
The gut of G. cantor larvae exhibited a diverse and complex bacterial community and Proteobacteria was the most dominant phylum (Figure 3A). This phylum is widely recognized as the most common in insect gut bacteria, and similar findings have been reported in other studies. The dominant intestinal flora of M. alternatus and A. chinensis larvae also consisted of Proteobacteria (Rizzi et al., 2013; Hu et al., 2017). Similarly, the dominant bacterial phylum in A. glabripennis larvae was Proteobacteria, and it remained unaffected by the host plant (Scully et al., 2014). This suggests that different beetles may share common dominant bacteria. We hypothesize that certain genera, such as cellulose-degrading bacteria, may be associated with cellulose-degrading similarities observed in wood-eating insects like Proteobacteria. For instance, the cellulose-degrading bacteria Ochrobactrum, found in the gut of B. horsfieldi larvae and Spodoptera frugiperda larvae, both belong to the Proteobacteria phylum (Li et al., 2020b; Yang et al., 2021). The dominant bacteria genera were different between the larval gut and larval frass (Figure 3C), indicating that bacterial structure of G. cantor changed after food digestion in gut. Additionally, Lactococcus and Enterococcus were the two most abundant of larval gut samples. Some cellulolytic Enterococcus strains were found in the eri silkworm larvae, Samia ricini (MsangoSoko et al., 2021; Unban et al., 2022), since Enterobacteriales have been observed to assist in the breakdown of plant cell wall compounds, including pectin (Shil et al., 2014; Blankenchip et al., 2018). Therefore, Enterococcus may have more important role in cellulose degradation in G. cantor (Robert and Bernalier-Donadille, 2003).
The function of bacterial flora was mainly involved in carbohydrate metabolism and amino acid metabolism, partial lipid, nucleotide metabolism and biodegradation in the host (Figure 4B). Hence, it was speculated that apart from endogenous cellulase genes (Su et al., 2021), intestinal bacteria also play a significant role in the kapok-degraded cellulose feeding of beetles. In carbohydrate metabolism, the bacterial flora primarily participated in starch and sucrose metabolism, amino sugar and nucleotide sugar metabolism, and pyruvate metabolism (Figure 4C). The results indicated that the intestinal flora of beetle larvae plays a significant role in carbohydrate metabolism, so it was speculated that beetle gut system may modulate the composition and function of the flora to serve nutrient metabolic needs and aid in food digestion. Furthermore, insects can be capable of regulating their own intestinal microorganisms and acquiring beneficial bacteria to facilitate food digestion (Mira and Moran, 2002).
The functional prediction results revealed bacteria with cellulose-degrading abilities, which serve as a basis for subsequent screening of cellulose-degrading bacteria. 12 genera of bacteria reported cellulose degrading bacteria were found in G. cantor (Table 2), which is significant to their intestinal digestion mechanism. Based on the functional prediction results of isolation and identification experiments, five strains of cellulose-degrading bacteria were obtained (Figure 5). In particular, the abundance information ratio of cellulose-degrading bacteria, belonging to 4 genera of Microbacterium, Streptomyces, Lachnospiraceae_NK4A136_group, and Gordonia, were higher in the intestinal tract compared to fecal samples (Table 2). Microbacterium had been found in the intestines of other insects, such as Stromatium barbatum (Fabr.) and Zootermopsis angusticollis (Wenzel et al., 2002; Yadav et al., 2022). However, the successful isolation and culture of Microbacterium, Streptomyces, Lachnospiraceae_NK4A136_group, and Gordonia were derived from the external environment, such as soil (Soares et al., 2012; Woo et al., 2014; Gong et al., 2020; Zou et al., 2021), which were relatively rare from the gut. The results indicated that the cellulose-degrading bacteria in G. cantor which belonging to Microbacterium, Streptomyces, Lachnospiraceae_NK4A136_group, and Gordonia of the gut may not be easily cultured in the environment. Five strains of culturable cellulose-degrading bacteria were obtained from the intestinal tract of longhorn beetles by screening cellulose-derived bacteria (Figure 5). Combining their morphological and physiological characteristics, the strains were identified as P. aeruginosa (Figure 6), which belongs to the genus Pseudomonas. This finding was consistent with the predicted function of cellulose-degrading bacteria in the results of intestinal bacterial diversity analysis. Other common cellulose-degrading bacteria include Bacillus (Li et al., 2020b; Kumawat et al., 2021; Zhang et al., 2021), Klebsiella (Dar et al., 2021), Enterococcus (MsangoSoko et al., 2021), and Pseudomonas sp. (Zhang et al., 2022).
The laccase, produced by Pseudomonas, had the capability to disintegrate the crystal structure of cellulose, unblock lignin on cellulose and hemicellulose, and enhance the ability to degrade cellulose (Zhang et al., 2007). P. aeruginosa was capable of producing alkaline cellulase. Previous studies had revealed that the main function of this enzyme was to break down the crystalline structure of cellulose, facilitate the release of amorphous cellulose and enhance the saccharification of fiber in an alkaline environment. Additionally, a portion of the cellulose was converted into simple sugars (Lu et al., 2017). Pseudomonas sp. may play a role in the degradation of secondary metabolites in host plant, which were associated with defense substances specific to the host plant. For instance, P. aeruginosa had demonstrated the capability to degrade linalool, which was isolated from Pagiophloeus tsushimanus (Qiao et al., 2023). Additionally, Pseudomonas sp. was obtained from the gut of mountain pine beetle Dendroctonus ponderosae and had shown the ability to degrade terpenes (Adams et al., 2013). Pseudomonas sp. had exhibited the capacity to degrade α-pinene and displayed resistance to high levels of α-pinene which isolated from the gut of the red turpentine beetle D. valens LeConte (Scolytinae) (Xu et al., 2016). 3-hexanone, decanal, nonanal and para-xylene (p-xylene) had an attractive effect on both male and female adults of G. cantor, which were compounds from the kapok (Dong, 2021). Additionally, Pseudomonas could somehow support the monophagous feeding habit of Brassicogethes matronalis (Teoh et al., 2021). It is hypothesized that this kapok consuming activity may be also attributed to intestinal bacteria in G. cantor. Further research is needed to investigate whether the P. aeruginosa from G. cantor plays a role in the degradation of these secondary metabolites.
The screening of cellulose-degrading bacteria yielded only 5 target strains in the intestinal tract of G. cantor. This number was significantly lower compared to the 12 genera of cellulose-degrading bacteria identified in the OTUs annotation results of 16S rDNA sequencing. These findings suggest that obtaining the desired target strain of cellulose-degrading bacteria from the intestinal source may be challenging due to various factors such as temperature, aerobic/anaerobic conditions, pH value, special nutrients, and microbial interactions in the environment. However, it is important to acknowledge that the study has certain limitations.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving animals were reviewed and approved by Ethics Review Committee on animal Experiments of Guangxi University.
R-RS: Writing–original draft, Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization. B-QP: Methodology, Validation, Writing–review and editing. Y-XL: Validation, Writing–review and editing. X-LZ: Conceptualization, Supervision, Writing–review and editing. WL: Conceptualization, Project administration, Resources, Supervision, Writing–review and editing. X-YW: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Software, Supervision, Validation, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by High-level Talents Introduction Project of Guangxi University (China), grant number A3310051008; Scientific Research and Technology Development Program Project of Guangxi Forestry Administration (China), 2023GXLK39.
We thank Zhongyan Huang and Guanxin Wu for the help during the experiments and thank Huili Ouyang for her help in revising the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2024.1340168/full#supplementary-material
Adams, A. S., Aylward, F. O., Adams, S. M., Erbilgin, N., Aukema, B. H., Currie, C. R., et al. (2013). Mountain pine beetles colonizing historical and naive host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl. Environ. Microbiol. 79 (11), 3468–3475. doi:10.1128/AEM.00068-13
Alves, M., Pereira, A., Matos, P., Henriques, J., Vicente, C., Aikawa, T., et al. (2016). Bacterial community associated to the pine wilt disease insect vectors Monochamus galloprovincialis and Monochamus alternatus. Sci. Rep. 6 (1), 23908. doi:10.1038/srep23908
Ayayee, P. A., Larsen, T., Rosa, C., Felton, G. W., Ferry, J. G., and Hoover, K. (2016). Essential amino acid supplementation by gut microbes of a wood-feeding cerambycid. Environ. Entomol. 45 (1), 66–73. doi:10.1093/ee/nvv153
Aßhauer, K. P., Wemheuer, B., Daniel, R., and Meinicke, P. (2015). Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31 (17), 2882–2884. doi:10.1093/bioinformatics/btv287
Blankenchip, C. L., Michels, D. E., Braker, H. E., and Goffredi, S. K. (2018). Diet breadth and exploitation of exotic plants shift the core microbiome of Cephaloleia, a group of tropical herbivorous beetles. PeerJ 6, e4793. doi:10.7717/peerj.4793
Brock, T. D. (1987). The study of microorganisms in situ: progress and problems. Symposium Soc. General Microbiol. 41, 1–17. doi:10.12691/jaem-2-4-11
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7 (5), 335–336. doi:10.1038/nmeth.f.303
Chang, C. J., Wu, C. P., Lu, S. C., Chao, A. L., Ho, T. H. D., Yu, S. M., et al. (2012). A novel exo-cellulase from white spotted longhorn beetle (Anoplophora malasiaca). Insect Biochem. Mol. Biol. 42 (9), 629–636. doi:10.1016/j.ibmb.2012.05.002
Chen, H., and Boutros, P. C. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinforma. 12 (1), 35–37. doi:10.1186/1471-2105-12-35
Chen, S. F., Zhou, Y. Q., Chen, Y. R., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), 884–890. doi:10.1093/bioinformatics/bty560
Dar, M. A., Shaikh, A. F., Pawar, K. D., Xie, R., Sun, J., Kandasamy, S., et al. (2021). Evaluation of cellulose degrading bacteria isolated from the gut-system of cotton bollworm, Helicoverpa armigera and their potential values in biomass conversion. PeerJ 9, e11254. doi:10.7717/peerj.11254
Dar, M. A., Xie, R., Pandit, R. S., Danso, B., Dong, C., and Sun, J. (2022). Exploring the region-wise diversity and functions of symbiotic bacteria in the gut system of wood-feeding termite, Coptotermes formosanus, toward the degradation of cellulose, hemicellulose, and organic dyes. Insect Sci. 29 (5), 1414–1432. doi:10.1111/1744-7917.13012
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72 (7), 5069–5072. doi:10.1128/AEM.03006-05
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14 (6), 927–930. doi:10.1111/j.1654-1103.2003.tb02228.x
Dong, Z. S. (2021). Behavior and mechanism on oviposition selection of Glenea cantor Fabricius. PhD dissertation: Guangxi University. (In Chinese). doi:10.27034/d.cnki.ggxiu.2021.000044
Dong, Z. S., Zhang, Y. J., Lai, K. P., Zheng, X. L., Wang, Q., and Lu, W. (2017). Biological characteristics of Glenea cantor Fabricius (Coleoptera: Cerambycidae). J. Environ. Entomology 39, 1313–1318. (In Chinese). doi:10.3969/j.issn.1674-0858.2017.06.17
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10 (10), 996–998. doi:10.1038/NMETH.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27 (16), 2194–2200. doi:10.1093/bioinformatics/btr381
Eida, M. F., Nagaoka, T., Wasaki, J., and Kouno, K. (2012). Isolation and characterization of cellulose-decomposing bacteria inhabiting sawdust and coffee residue composts. Microbes Environ. 27 (3), 226–233. doi:10.1264/jsme2.ME11299
Ge, S. X., Shi, F. M., Pei, J. H., Hou, Z. H., Zong, S. X., and Ren, L. L. (2021). Gut bacteria associated with Monochamus saltuarius (Coleoptera: Cerambycidae) and their possible roles in host plant adaptations. Front. Microbiol. 12, 687211. doi:10.3389/fmicb.2021.687211
Gong, X. J., Zou, H. T., Qian, C. R., Yu, Y., Hao, Y. B., Li, L., et al. (2020). Construction of in situ degradation bacteria of corn straw and analysis of its degradation efficiency. Ann. Microbiol. 70 (1), 62–15. doi:10.1186/s13213-020-01601-9
Gu, W. J., Xu, Y. Q., Xu, P. Z., Xie, K. Z., Lu, Y. S., Tang, S. H., et al. (2012). Screening and identification of hemicellulose degrading microorganisms in acid soil. Acta Microbiol. Sin. 52 (10), 1251–1259. (In Chinese). doi:10.13343/j.cnki.wsxb.2012.10.004
Guan, N., Mi, H. Z., Du, Q. S., Zhang, S. H., and Huang, R. B. (2011). Research on Glenea cantor cellulase characteristics. Guangxi Sci. 18 (03), 261–263+268. doi:10.13656/j.cnki.gxkx.2011.03.032
Guo, M., Wu, F., Hao, G., Qi, Q., Li, R., Li, N., et al. (2017). Bacillus subtilis improves immunity and disease resistance in rabbits. Front. Immunol. 8, 354. doi:10.3389/fimmu.2017.00354
Guo, Y. J., Lin, Q. N., Chen, L. Y., Carballar-Lejarazú, R., Zhang, A. S., Shao, E. S., et al. (2020). Characterization of bacterial communities associated with the pinewood nematode insect vector Monochamus alternatus Hope and the host tree Pinus massoniana. BMC genomics 21 (1), 337–413. doi:10.1186/s12864-020-6718-6
He, Z. B., Yin, Y. P., Cao, Y. Q., Dong, Y. M., and Zhang, W. (2001). “Study on the Apriona germari (Hope) larvae’s intestinal bacterial flora, Acta Microbiol. Sin. 06, 741–744. Wei sheng wu xue bao = Acta microbiologica Sinica, (In Chinese). doi:10.13343/j.cnki.wsxb.2001.06.015
Hooper, L. V., and Gordon, J. I. (2001). Commensal host-bacterial relationships in the gut. Science 292 (5519), 1115–1118. doi:10.1126/science.1058709
Hu, X., Li, M., Raffa, K. F., Luo, Q. Y., Fu, H. J., Wu, S. Q., et al. (2017). Bacterial communities associated with the pine wilt disease vector Monochamus alternatus (Coleoptera: Cerambycidae) during different larval instars. J. Insect Sci. 17 (6), 115. doi:10.1093/jisesa/iex089
Ishihara, M., Kikkawa, M., Shimizu, K., Suzuki, H., and Honda, H. (2021). Simple stain-free screening method for pectinolytic microorganisms under alkalophilic conditions. Biotechnol. Lett. 43 (9), 1905–1911. doi:10.1007/s10529-021-03162-6
Jang, S., and Kikuchi, Y. (2020). Impact of the insect gut microbiota on ecology, evolution, and industry. Curr. Opin. Insect Sci. 41, 33–39. doi:10.1016/j.cois.2020.06.004
Javal, M., Terblanche, J. S., Benoit, L., Conlong, D. E., Lloyd, J. R., Smit, C., et al. (2023). Does host plant drive variation in microbial gut communities in a recently shifted pest? Microb. Ecol. 86 (1), 636–646. doi:10.1007/s00248-022-02100-x
Jin, M. X., Liu, X. H., Yan, Y. Y., Xie, G. A., Yu, A. L., and Tu, Y. G. (2019). Advances in research on gut content of longhorned beetles. China Plant Prot. 39 (12), 23–27+36. In Chinese.
Kim, H., and Yu, S. M. (2020). Flavobacterium nackdongense sp. nov., a cellulose-degrading bacterium isolated from sediment. Archives Microbiol. 202 (3), 591–595. doi:10.1007/s00203-019-01770-5
Kognou, A. L. M., Chio, C., Khatiwada, J. R., Shrestha, S., Chen, X. T., Han, S. H., et al. (2022). Characterization of cellulose-degrading bacteria isolated from soil and the optimization of their culture conditions for cellulase production. Appl. Biochem. Biotechnol., 1–23. doi:10.1007/s12010-022-04002-7
Kumawat, P. K., Sahoo, A., Sarkar, S., and Kumar, S. (2021). Isolation and characterization of anaerobic bacteria with fiber degradation potential from faeces of Boselaphus tragocamelus grazing on semi arid Indian conditions. Archives Microbiol. 203, 5105–5116. doi:10.1007/s00203-021-02477-2
Lai, K. P., Lu, W., Liu, D. X., Luo, Z. R., and Gao, P. Y. (2008). The larval instars and stadia of the longhorn beetle Glenea cantor. Chin. J. Appl. Entomology 45, 138–140. (In Chinese).
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31 (9), 814–821. doi:10.1038/nbt.2676
Li, H., Young, S. E., Poulsen, M., and Currie, C. R. (2021). Symbiont-Mediated Digestion of plant biomass in fungus-farming insects. Annu. Rev. Entomology 66, 297–316. doi:10.1146/annurev-ento-040920-061140
Li, H. J., Young, S. E., Poulsen, M., and Currie, C. R. (2020a). Symbiont-mediated digestion of plant biomass in fungus-farming insects. Annu. Rev. Entomology 66 (1), 297–316. doi:10.1146/annurev-ento-040920-061140
Li, H. W., Yang, X. J., Xiang, Y. Z., Lin, L. B., and Zhang, Q. L. (2020b). Isolation and identification of the intestinal bacteria, and screening of the cellulolytic bacteria, of Spodoptera frugiperda (Lepidoptera: nocturid) larvae. Chin. J. Appl. Entomology 57 (03), 608–616. (In Chinese). doi:10.7679/j.issn.2095-1353.2020.061
Li, Y., Chen, H., Chu, X., Ma, Q., Liang, G., Wu, S., et al. (2020c). Molecular cloning and expression analysis of the endogenous cellulase gene MaCel1 in Monochamus alternatus. Forests 11 (12), 1372. doi:10.3390/f11121372
Liu, J., Song, K. Q., Teng, H. J., Zhang, B., Li, W. Z., Xue, H. J., et al. (2015). Endogenous cellulolytic enzyme systems in the longhorn beetle Mesosa myops (Insecta: Coleoptera) studied by transcriptomic analysis. Acta Biochimica Biophysica Sinica 47 (9), 741–748. doi:10.1093/abbs/gmv070
Lu, W., Wang, Q., Tian, M. Y., He, X. Z., Zeng, X. L., and Zhong, Y. X. (2007). Mate location and recognition in Glenea cantor (Fabr.) (Coleoptera: Cerambycidae: Lamiinae): roles of host plant health, female sex pheromone, and vision. Environ. Entomol. 36 (4), 864–870. doi:10.1603/0046-225X(2007)36[864:MLARIG]2.0.CO;2
Lu, W., Wang, Q., Tian, M. Y., Xu, J., Lv, J., Wei, S. G., et al. (2013). Reproductive traits of Glenea cantor (Coleoptera: Cerambycidae: Lamiinae). J. Econ. Entomology 106 (1), 215–220. doi:10.1603/EC12251
Lu, W., Wang, Q., Tian, M. Y., Xu, J., and Qin, A. Z. (2011). Phenology and laboratory rearing procedures of an Asian longicorn beetle Glenea cantor (Coleoptera: Cerambycidae: Lamiinae). J. Econ. Entomology 104, 509–516. doi:10.1603/EC10345
Lu, Z. L., Lu, Q., Chen, Y., Wu, R. Z., Huang, J., Chen, X. L., et al. (2017). Study on the degradation of bagasse cellulose by Pseudomonas aeruginosa alkaline cellulase. China Meeting, 160–162. (In Chinese) Available at: https://kns.cnki.net/kcms2/article/abstract?v=2F6201taHdcJ_YimeZKpqnPJMkyabyuUkrIskD4Q3dfPgxlDae2VJ8lMF4sxdJB6p8jfdUOnv9JhLXMhksyXgoXq_NlDj7HwyQxsJKvtbThb03vCwoWdzQc6jS4268hUTihKZWuAiuSpQShmA6ubyQ==&uniplatform=NZKPT&language=CHS.
Luo, C. B., Li, Y. Q., Chen, Y., Fu, C., Nong, X., and Yang, Y. J. (2019). Degradation of bamboo lignocellulose by bamboo snout beetle Cyrtotrachelus buqueti in vivo and vitro: efficiency and mechanism. Biotechnol. Biofuels 12 (1), 75–14. doi:10.1186/s13068-019-1406-y
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27 (21), 2957–2963. doi:10.1093/bioinformatics/btr507
Mei, H. Z., Xia, D. G., Zhao, Q. L., Zhang, G. Z., Qiu, Z. Y., Qian, P., et al. (2016). Molecular cloning, expression, purification and characterization of a novel cellulase gene (Bh-EGaseI) in the beetle Batocera horsfieldi. Gene 576 (1), 45–51. doi:10.1016/j.gene.2015.09.057
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428. doi:10.1021/ac60147a030
Mira, A., and Moran, N. A. (2002). Estimating population size and transmission bottlenecks in maternally transmitted endosymbiotic bacteria. Microb. Ecol. 44, 137–143. doi:10.1007/s00248-002-0012-9
MsangoSoko, K., Bhattacharya, R., Ramakrishnan, B., Sharma, K., and Subramanian, S. (2021). Cellulolytic activity of gut bacteria isolated from the eri silkworm larvae, Samia ricini (Lepidoptera: saturniidae). Int. J. Trop. Insect Sci. 41, 2785–2794. doi:10.1007/s42690-021-00459-x
Munoz-Benavent, M., Perez-Cobas, A. E., Garcia-Ferris, C., Moya, A., and Latorre, A. (2021). Insects’ potential: understanding the functional role of their gut microbiome. J. Pharm. Biomed. Analysis 194, 113787. doi:10.1016/j.jpba.2020.113787
Nguyen, N. P., Warnow, T., Pop, M., and White, B. (2016). A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2, 16004. doi:10.1038/npjbiofilms.2016.4
O'Sullivan, D. J. (2000). Methods for analysis of the intestinal microflora. Curr. Issues Intestinal Microbiol. 1 (2), 39–50. Available at: https://www.caister.com/backlist/ciim/v/v1/04.pdf.
Pauchet, Y., Kirsch, R., Giraud, S., Vogel, H., and Heckel, D. G. (2014). Identification and characterization of plant cell wall degrading enzymes from three glycoside hydrolase families in the cerambycid beetle Apriona japonica. Insect Biochem. Mol. Biol. 49, 1–13. doi:10.1016/j.ibmb.2014.03.004
Qiao, H., Zhu, H., Li, H., Chen, H. J., Li, S. Y., Chen, C., et al. (2023). Isolation and characterization of gut bacteria associated with the degradation of host-specific terpenoids in Pagiophloeus tsushimanus (Coleoptera: Curculionidae) larvae. J. Insect Sci. 23 (2), 14–19. doi:10.1093/jisesa/iead019
Rizzi, A., Crotti, E., Borruso, L., Jucker, C., Lupi, D., Colombo, M., et al. (2013). Characterization of the bacterial community associated with larvae and adults of Anoplophora chinensis collected in Italy by culture and culture-independent methods. BioMed Res. Int. 2013 (1), 1–12. doi:10.1155/2013/420287
Robert, C., and Bernalier-Donadille, A. (2003). The cellulolytic microflora of the human colon: evidence of microcrystalline cellulose-degrading bacteria in methane-excreting subjects. FEMS Microbiol. Ecol. 46 (1), 81–89. doi:10.1016/S0168-6496(03)00207-1
Romero, S., Nastasa, A., Chapman, A., Kwong, W. K., and Foster, L. J. (2019). The honey bee gut microbiota: strategies for study and characterization. Insect Mol. Biol. 28 (4), 455–472. doi:10.1111/imb.12567
Schloss, P. D., Delalibera, I., Handelsman, J. O., and Raffa, K. F. (2006). Bacteria associated with the guts of two wood-boring beetles: Anoplophora glabripennis and Saperda vestita (Cerambycidae). Environ. Entomol. 35 (3), 625–629. doi:10.1603/0046-225X-35.3.625
Scully, E. D., Geib, S. M., Carlson, J. E., Tien, M., McKenna, D., and Hoover, K. (2014). Functional genomics and microbiome profiling of the Asian longhorned beetle (Anoplophora glabripennis) reveal insights into the digestive physiology and nutritional ecology of wood feeding beetles. BMC genomics 15 (1), 1096–1121. doi:10.1186/1471-2164-15-1096
Shelomi, M., and Chen, M. J. (2020). Culturing-enriched metabarcoding analysis of the Oryctes rhinoceros gut microbiome. Insects 11 (11), 782. doi:10.3390/insects11110782
Shil, R. K., Mojumder, S., Sadida, F. F., Uddin, M., and Sikdar, D. (2014). Isolation and identification of cellulolytic bacteria from the gut of three phytophagus insect species. Braz. Archives Biol. Technol. 57657, 927–932. doi:10.1590/S1516-8913201402620
Soares, F. L., Melo, I. S., Dias, A. C. F., and Andreote, F. D. (2012). Cellulolytic bacteria from soils in harsh environments. World J. Microbiol. Biotechnol. 28 (5), 2195–2203. doi:10.1007/s11274-012-1025-2
Su, L. J., Gao, X. H., Wang, S. L., Song, A. D., and Wang, W. B. (2015). Screening of cellulose degradative bacteria in the gut of Aromia bungii Faldermann larvae and its impact on broilers' production performance. Feed Ind. 36 (07), 38–43. (In Chinese). doi:10.13302/j.cnki.fi.2015.07.009
Su, R. R., Huang, Z. Y., Qin, C. W., Zheng, X. L., Lu, W., and Wang, X. Y. (2021). Evaluation of reference genes in Glenea cantor (Fabricius) by using qRT-PCR. Genes 12 (12), 1984. doi:10.3390/genes12121984
Sugimura, M., Watanabe, H., Lo, N., and Saito, H. (2003). Purification, characterization, cDNA cloning and nucleotide sequencing of a cellulase from the yellow-spotted longicorn beetle, Psacothea hilaris. Eur. J. Biochem. 270 (16), 3455–3460. doi:10.1046/j.1432-1033.2003.03735.x
Teoh, M. C., Furusawa, G., and Veera, S. G. (2021). Multifaceted interactions between the pseudomonads and insects: mechanisms and prospects. Archives Microbiol. 203, 1891–1915. doi:10.1007/s00203-021-02230-9
Tokuda, G. (2019). Plant cell wall degradation in insects: recent progress on endogenous enzymes revealed by multi-omics technologies. Adv. Insect Physiology 57, 97–136. doi:10.1016/bs.aiip.2019.08.001
Tsegaye, B., Balomajumder, C., and Roy, P. (2019). Isolation and characterization of novel lignolytic, cellulolytic, and hemicellulolytic bacteria from wood-feeding termite Cryptotermes brevis. Int. Microbiol. 22, 29–39. doi:10.1007/s10123-018-0024-z
Ulrich, A., Klimke, G., and Wirth, S. (2008). Diversity and activity of cellulose-decomposing bacteria, isolated from a sandy and a loamy soil after long-term manure application. Microb. Ecol. 55 (3), 512–522. doi:10.1007/s00248-007-9296-0
Unban, K., Klongklaew, A., Kodchasee, P., Pamueangmun, P., Shetty, K., and Khanongnuch, C. (2022). Enterococci as dominant xylose utilizing lactic acid bacteria in Eri silkworm midgut and the potential use of Enterococcus hirae as probiotic for eri culture. Insects 13 (2), 136. doi:10.3390/insects13020136
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73 (16), 5261–5267. doi:10.1128/AEM.00062-07
Wang, S., Wang, L., Fan, X., Yu, C., Feng, L., and Yi, L. (2020). An insight into diversity and functionalities of gut microbiota in insects. Curr. Microbiol. 77, 1976–1986. doi:10.1007/s00284-020-02084-2
Wei, Y. D., Lee, K. S., Gui, Z. Z., Yoon, H. J., Kim, I., Je, Y. H., et al. (2006a). N-linked glycosylation of a beetle (Apriona germari) cellulase Ag-EGase II is necessary for enzymatic activity. Insect Biochem. Mol. Biol. 36 (6), 435–441. doi:10.1016/j.ibmb.2006.03.007
Wei, Y. D., Lee, K. S., Gui, Z. Z., Yoon, H. J., Kim, I., Zhang, G. Z., et al. (2006b). Molecular cloning, expression, and enzymatic activity of a novel endogenous cellulase from the mulberry longicorn beetle, Apriona germari. Comp. Biochem. Physiology Part B Biochem. Mol. Biol. 145 (2), 220–229. doi:10.1016/j.cbpb.2006.07.007
Wenzel, M., Schönig, I., Berchtold, M., Kämpfer, P., and König, H. (2002). Aerobic and facultatively anaerobic cellulolytic bacteria from the gut of the termite Zootermopsis angusticollis. J. Appl. Microbiol. 92 (1), 32–40. doi:10.1046/j.1365-2672.2002.01502.x
Wickham, H., and Chang, W. (2008). ggplot2: an implementation of the Grammar of Graphics. R package version 0.7, Available at: http://CRAN.R-project.org/package=ggplot2.
Willis, J. D., Oppert, C., and Jurat-Fuentes, J. L. (2010). Methods for discovery and characterization of cellulolytic enzymes from insects. Insect Sci. 17 (3), 184–198. doi:10.1111/j.1744-7917.2010.01322.x
Woo, H. L., Hazen, T. C., Simmons, B. A., and DeAngelis, K. M. (2014). Enzyme activities of aerobic lignocellulolytic bacteria isolated from wet tropical forest soils. Syst. Appl. Microbiol. 37 (1), 60–67. doi:10.1016/j.syapm.2013.10.001
Wu, G. X., Su, R. R., Ouyang, H. L., Zheng, X. L., Lu, W., and Wang, X. Y. (2022). Antennal transcriptome analysis and identification of olfactory genes in Glenea cantor Fabricius (Cerambycidae: Lamiinae). Insects 13 (6), 553. doi:10.3390/insects13060553
Xia, D. G., Wei, Y. D., Zhang, G. Z., Zhao, Q. L., Zhang, Y. S., Xiang, Z. H., et al. (2013). cDNA cloning, expression, and enzymatic activity of a novel endogenous cellulase from the beetle Batocera horsfieldi. Gene 514 (1), 62–68. doi:10.1016/j.gene.2012.08.044
Xie, A. L. C., Yang, Y. X., Wang, X. L., and Chen, Y. L. (2020). Cloning endo-glucanase gene of cellulolytic microorganisms from Apriona germari and its expression in Lactobacillus. Chin. J. Animal Nutr. 32 (03), 1344–1352. doi:10.3969/j.issn.1006-267x.2020.03042
Xie, R., Dong, C., Wang, S., Danso, B., Dar, M. A., Pandit, R. S., et al. (2023). Host-specific diversity of culturable bacteria in the gut systems of fungus-growing termites and their potential functions towards lignocellulose bioconversion. Insects 14 (4), 403. doi:10.3390/insects14040403
Xu, L. P., Jiang, Z., Ge, Y. L., and Sun, H. W. (2018). Bacterial community on the corn straw in suihua area of heilongjiang province by Illumina miseq sequencing. Sci. Technol. Food Industry (23), 105–110. (In Chinese). doi:10.13386/j.issn1002-0306.2018.23.019
Xu, L. T., Lu, M., and Sun, J. H. (2016). Invasive bark beetle-associated microbes degrade a host defensive monoterpene. Insect Sci. 23 (2), 183–190. doi:10.1111/1744-7917.12255
Xue, W. W., Xue, Y. C., Zhai, Q. M., and Zheng, L. J. (2009). Isolation, screening and representation of bacterium strains for degrading Apocynum vertetum L. pectin. Ind. Microbiol. 39 (2), 55–58. (In Chinese) https://kns.cnki.net/kcms/detail/detail.aspx?FileName=GYWS200902018&DbName=CJFQ2009.
Yadav, D. S., Ranade, Y., Sawant, I., Ghule, S., and Mhaske, S. (2022). Isolation, identification and functional characterisation of bacteria associated with gut of wood feeding Stromatium barbatum (Fabr.) (Coleoptera: Cerambycidae) larvae. Int. J. Trop. Insect Sci. 42 (3), 2603–2616. doi:10.1007/s42690-022-00789-4
Yang, D. F., Guan, N., Mi, H. Z., Du, Q. S., Zhang, S. S., and Huang, R. B. (2011). Research on Glenea cantor cellulase characteristics. Guangxi Sci. 18, 4. (In Chinese). doi:10.13656/j.cnki.gxkx.2011.03.032
Yang, Z. D., Fang, X. Y., Duan, H. J., Zhu, L. H., and Feng, S. Y. (2021). Isolation and identification of cellulolytic bacteria and optimization of enzymatic production conditionsin the intestinal tract of the larva of Batocera lineolate (Chaevroat). Biot. Resour. 43 (02), 153–159. (In Chinese). doi:10.14188/j.ajsh.2021.02.007
Yu, S. T., Mao, S. R., Hu, A. T., Lu, Z. X., Zhu, H. Y., Kong, L. Y., et al. (2021). Study on screening of microorganisms for improving tobacco leaf quality and their effects. J. Nanjing Agric. Univ. 44 (4), 766–777. doi:10.7685/jnau.202011008
Zhang, K., Su, Z. P., Xu, Y., Li, Q. L., Wang, Y., Liu, L., et al. (2020a). Isolation and identification of cellulose-degrading bacteria in the posterior intestine. Biot. Resour. 42 (02), 228–233. doi:10.14188/j.ajsh.2020.02.010
Zhang, X., Xu, C., and Wang, H. (2007). Pretreatment of bamboo residues with Coriolus versicolor for enzymatic hydrolysis. J. Biosci. Bioeng. 104 (2), 149–151. doi:10.1263/jbb.104.149
Zhang, X. P., Wang, X., Sun, J., and Li, S. L. (2022). Isolation and identification of s Pseudomonas strain and its application potential in rape straw composting in Qinghai, China. Acta Agric. Zhejiangensis 34 (02), 343–351. doi:10.3969/j.issn.1004-1524.2022.02.16
Zhang, X. Y., Yang, T., Wu, H., Li, M. Y., Huang, X., Li, J., et al. (2020b). Isolation, identification, and functional characteristics of cultural bacteria in waterlogged bamboo slips unearthed from Tuzishan site in Yiyang, Hunan Province. Chin. J. Appl. Environ. Biol. (06), 1418–1425. (In Chinese). doi:10.19675/j.cnki.1006-687x.2019.11052
Zhang, Y. L., Lu, B. Q., Yang, F., Tu, Y., Jiang, F. Y. D., Qi, K. X., et al. (2021). Isolation, identification and functional analysis of intestinal microorganisms of Brontispa longissimi Gestro. Chin. J. Trop. Crops 42 (04), 1066–1070. (In Chinese) Available at: https://kns.cnki.net/kcms/detail/46.1019.S.20200727.1649.004.html.
Zhao, X. H., Li, H. B., Ding, A. H., Zhou, G. Z., Sun, Y. J., and Zhang, D. Y. (2016). Preparing and characterizing Fe3O4@ cellulose nanocomposites for effective isolation of cellulose-decomposing microorganisms. Mater. Lett. 163, 154–157. doi:10.1016/j.matlet.2015.10.061
Zhong, P. S., Tang, Y. J., and Zhang, S. S. (2011). Investigations on the diseases and insect pests of the ancient and famous trees of west lake scenic area in Huizhou and suggestions for their protection. J. South. Agric. 42 (4), 412–414. (In Chinese).
Zhu, J. Y., and Pan, X. (2022). Efficient sugar production from plant biomass: current status, challenges, and future directions. Renew. Sustain. Energy Rev. 164, 112583. doi:10.1016/j.rser.2022.112583
Keywords: gut bacterial community, Glenea cantor, illumina MiSeq 16S rRNA sequencing, lignocellulose degradation, cellulose degrading bacteria
Citation: Su R-R, Pan B-Q, Luo Y-X, Zheng X-L, Lu W and Wang X-Y (2024) Characterization of bacterial diversity and screening of cellulose-degrading bacteria in the gut system of Glenea cantor (Fabricius) larvae. Front. Bioeng. Biotechnol. 12:1340168. doi: 10.3389/fbioe.2024.1340168
Received: 17 November 2023; Accepted: 01 February 2024;
Published: 22 February 2024.
Edited by:
Jun Wang, Jiangsu University of Science and Technology, ChinaCopyright © 2024 Su, Pan, Luo, Zheng, Lu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Yun Wang, d3h5ODc3MUAxNjMuY29t, eGlhb3l1bndhbmdAZ3h1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.