 Min-Chao Jiang
Min-Chao Jiang Wei-Li Hsu
Wei-Li Hsu Ching-Yu Tseng2
Ching-Yu Tseng2 Na-Sheng Lin
Na-Sheng Lin Yau-Heiu Hsu
Yau-Heiu Hsu Chung-Chi Hu
Chung-Chi Hu- 1PhD Program in Microbial Genomics, National Chung Hsing University and Academia Sinica, Taichung, Taiwan
- 2Graduate Institute of Microbiology and Public Health, College of Veterinary Medicine, National Chung Hsing University, Taichung, Taiwan
- 3Institute of Plant and Microbial Biology, Academia Sinica, Taipei, Taiwan
- 4Graduate Institute of Biotechnology, National Chung Hsing University, Taichung, Taiwan
- 5Advanced Plant Biotechnology Center, National Chung Hsing University, Taichung, Taiwan
Plants offer a promising platform for cost-effective production of biologically active therapeutic glycoproteins. In previous studies, we have developed a plant expression system based on Bamboo mosaic virus (BaMV) by incorporating secretory signals and an affinity tag, which resulted in notably enhanced yields of soluble and secreted fusion glycoproteins (FGs) in Nicotiana benthamiana. However, the presence of fusion tags on recombinant glycoproteins is undesirable for biomedical applications. This study aimed to develop a refined expression system that can efficiently produce tag-free glycoproteins in plants, with enhanced efficacy of mature interferon gamma (mIFNγ) against viruses. To accommodate the specific requirement of different target proteins, three enzymatically or chemically cleavable linkers were provided in this renovated BaMV-based expression system. We demonstrated that Tobacco etch virus (TEV) protease could process the specific cleavage site (LTEV) of the fusion protein, designated as SSExtHis(SP)10LTEV-mIFNγ, with optimal efficiency under biocompatible conditions to generate tag-free mIFNγ glycoproteins. The TEV protease and secretory-affinity tag could be effectively removed from the target mIFNγ glycoproteins through Ni2+-NTA chromatography. In addition, the result of an antiviral assay showed that the tag-free mIFNγ glycoproteins exhibited enhanced biological properties against Sindbis virus, with comparable antiviral activity of the commercialized HEK293-expressed hIFNγ. Thus, the improved BaMV-based expression system developed in this study may provide an alternative strategy for producing tag-free therapeutic glycoproteins intended for biomedical applications.
1 Introduction
Harnessing recombinant proteins for biomedical applications is among the central goals of molecular bioengineering. To achieve this, scientists have developed diverse expression systems, using organisms like animals, yeasts, and bacteria, to efficiently produce a range of therapeutic proteins (Berlec and Strukelj, 2013; Wells and Robinson, 2017). Plants may serve as alternative biofactories for producing therapeutic proteins due to their unique advantages in high safety, rapid scaling-up, and cost-efficient production (Daniell et al., 2001; Schillberg et al., 2019). In addition, plant-made pharmaceuticals (PMPs) allow for eukaryotic post-translational modification (PTM), which is essential for protein stability, maturation, and biological function (Walsh and Jefferis, 2006; Schneider et al., 2014; Liu and Timko, 2022). However, one major drawback associated with PMP production is the lack of efficient downstream purification processes for achieving biomedical acceptance.
To overcome challenges in downstream processing of plant-produced recombinant proteins, the use of various fusion tags has emerged as a valuable strategy. For example, a small ubiquitin-related modifier (bdSUMO) derived from Brachypodium distachyon (Islam et al., 2019; Islam et al., 2020), an elastin-like polypeptide (ELP) (Conley et al., 2009; Kaldis et al., 2013; Marin Viegas et al., 2022), and hydrophobins (HFBI) derived from Trichoderma reesei (Joensuu et al., 2010; Reuter et al., 2014) have been used to significantly improve productivity in plant-based systems. The combination of secretory signal (SS) and a plant-specific glycomodule tag, hydroxyproline (Hyp)-O-glycosylated peptide (HypGP) consisting of a repeated “Ser-Pro” motif, has resulted in a 16-fold increase in the overall yield of the tagged protein when transiently expressed in inoculated N. benthamiana leaves (Dolan et al., 2014). Such tags have been used for boosting secretion, which could dramatically promote the production of secreted recombinant proteins in various plant cell culture systems, including tobacco BY2 suspension cells (Xu et al., 2007; Xu et al., 2010; Zhang et al., 2016), green microalga Chlamydomonas reinhardtii (Ramos-Martinez et al., 2017), and tobacco hairy root cultures (Zhang et al., 2019). In particular, the use of secretory plant cell culture systems is applicable in large-scale and continuous production of therapeutic glycoproteins in compliance with the current good manufacturing practice (CGMP) regulations.
In our previous study, by using an overexpression system based on Bamboo mosaic virus (BaMV), we incorporated a novel secretory signal, SSExt, derived from N. benthamiana extensin protein (Jiang et al., 2020) to transport the mature interferon gamma (mIFNγ) protein via the secretory pathway for maintaining intact post-translational modification. Additional fusion of the HypGP tag, (SP)10, which contains 10 repeats of the “Ser-Pro” motif of hydroxyproline-O-glycosylated peptides, to the C-terminus of mIFNγ dramatically enhanced the solubility and secretion of the fusion glycoprotein (FG), designated as SSExtmIFNγ(SP)10. This strategy has simplified the downstream purification process for scaled-up production of FG in N. benthamiana plants (Jiang et al., 2020). It was noted that the fusion protein SSExtmIFNγ(SP)10 was partially O- or N-glycosylated and post-translationally modified to various forms such as monomeric mIFNγ (Mγ, 16 kDa), monomeric monoglycosylated mIFNγ (1N-MG, 18 kDa), and monomeric diglycosylated mIFNγ (2N-MG, 20 kDa), which then underwent self-assembly into the dimeric glycosylated mIFNγ (DG, 32–40 kDa). However, the fusion tags on SSExtmIFNγ(SP)10 proteins were still not completely processed through the secretory pathway, as confirmed by immunoblotting with specific antibodies. These FG forms would possibly affect their biological activity due to their undesirable protein folding conformations.
One solution to increase the efficiency in the processing of fusion proteins (FPs) is through the insertion of cleavable peptides in between fusion tags and recombinant proteins, such as the motifs recognized by Staphylococcus aureus sortase A (SrtA) (Ton-That et al., 1999; Tsukiji and Nagamune, 2009), Tobacco etch virus (TEV) protease, enterokinase, or factor Xa (Arnau et al., 2006; Waugh, 2011). TEV protease was widely applied in the removal of fusion tags from FPs since it exhibits high enzymatic activity even at low temperature (4°C) (Arnau et al., 2006). Due to steric occlusion between the fusion partner and target protein, some FPs, particularly membranous ones, could not be cleaved by proteases even in the presence of the specific cleavage site (Waugh, 2011). For such membranous FPs, a new sequence-specific nickel-assisted cleavage (SNAC) tag has been developed as a chemically cleavable linker in FPs, which could efficiently remove the fusion partner by the Ni2+ ion under biocompatible conditions (Dang et al., 2019). Apparently, different target proteins may pose different challenges for processing, and there is still need for development of alternative strategies to process the fusion tags to accommodate the different requirements of diverse recombinant proteins.
In this work, we intended to develop a BaMV-based expression system with different cleavable fusion tags to suit the specific needs of target proteins and tested the applicability of the system for obtaining tag-free mIFNγ glycoproteins suitable for biomedical usages. We introduced three different enzymatically or chemically cleavable linkers mentioned above, designated as LSrtA, LTEV, and LSNAC, in between the combinational secretory-affinity tag (SSExtHis(SP)10) and mIFNγ in the BaMV-based overexpression system. The results revealed that the fusion glycoprotein, designated as SSExtHis(SP)10LTEV-mIFNγ, expressed by the BaMV-based vector in N. benthamiana, was successfully produced, and the tags were proteolytically cleaved by TEV protease with optimal efficiency. A series of purification procedures were established for the efficient production of tag-free mIFNγ glycoproteins, which were further verified to exhibit improved biological activity against Sindbis virus.
2 Materials and methods
2.1 Construction of BaMV-based expression cassettes for FG processing
The improved BaMV-based expression vector developed in this study was modified from a previously constructed chimeric BaMV vector, pKB19 (Muthamilselvan et al., 2016; Jiang et al., 2019), which harbors BaMV RNA replicase, the silencing suppressor P19 of Tomato bushy stunt virus (TBSV) (519 bp, GenBank Accession No. AJ288926) under the control of a dual constitutive 35S promoter of Cauliflower mosaic virus (CaMV), and an Agrobacterium nopaline synthase (nos) terminator (Figure 1A). In addition, the SSExt derived from the N. benthamiana extensin protein with the fusion of the secretory booster, (SP)10, was incorporated into the expression vector to generate the secretory expression vector, pKB19SSExtHis(SP)10, which was further used to construct the plasmid pKB19SSExtHis(SP)10-mIFNγ for the production of human mIFNγ glycoproteins (amino acid positions 21–166, GenBank accession number AY121833.1) with enhanced solubility and secretion (Jiang et al., 2020). However, such fusion tags may affect the conformation and biological activity of the recombinant mIFNγ glycoproteins by hampering the proper folding. Thus, in this study, we have improved the design of the BaMV-based protein expression system by the insertion of cleavable linkers, including LSrtA, LTEV, and LSNAC, positioned in between the combinational secretory-affinity tag SSExt His(SP)10 [containing a 6X His-tag between SSExt and (SP)10] and mIFNγ to generate plasmids designated as pKB19SSExtHis(SP)10LSrtA-mIFNγ, pKB19SSExtHis(SP)10LTEV-mIFNγ, and pKB19SSExtHis(SP)10LSNAC-mIFNγ, respectively (Figure 1A). The construction process of the above plasmids is described in brief as follows. The fragment of 6X His-tag and (SP)10LSrtA-mIFNγ were synthesized by overlapping PCR with specific primers, including F-XbaI-LPETG-mIFNγ, F-(SP)10-XbaI-LPETG, F-MluI-(SP)10, and F-MluI-6XHis-GG-(SP)10, individually plus R-SpeI-TGA-mIFNγ (Supplementary Table S1). The amplified PCR fragment of His(SP)10LSrtA-mIFNγ, containing 6X His-tag at the N-terminus, was digested with MluI and SpeI and ligated into the respective sites in the pKB19 expression vector to generate the chimeric plasmid pKB19His(SP)10LSrtA-mIFNγ. The SSExt signal was constructed by primer extension of two mutually complementary primers, F-MluI-SSExt and R-MluI-SSExt, followed by digestion with MluI. The digested product of the SSExt fragment was cloned into pKB19His(SP)10LSrtA-mIFNγ to generate the final chimeric plasmid pKB19SSExtHis(SP)10LSrtA-mIFNγ. The fragments of LTEV-mIFNγ and LSNAC-mIFNγ were amplified with the mIFNγ gene as a template using gene-specific forward primers, namely, F-XbaI-ENLYFQG-mIFNγ and F-XbaI-GSHHW-mIFNγ, respectively, plus the reverse primer R-SpeI-TGA-mIFNγ (Supplementary Table S1). The amplified PCR product fragments of LTEV-mIFNγ and LSNAC-mIFNγ were digested with XbaI and SpeI and ligated into the pKB19SSExtHis(SP)10 secretory expression vector cut with cognate enzymes to generate the chimeric plasmids pKB19SSExtHis(SP)10LTEV-mIFNγ and pKB19SSExt His(SP)10LSNAC-mIFNγ, respectively.
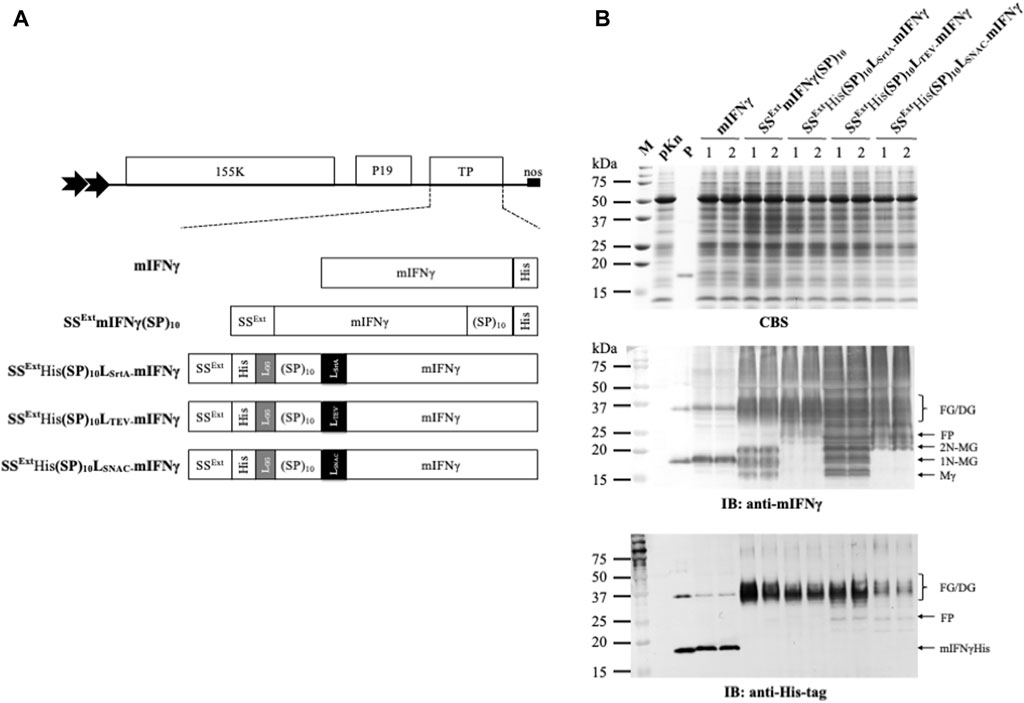
FIGURE 1. Effect of different cleavable linkers on the transient expression of various fusion Glyco-mIFNγ (FGs) in N. benthamiana. (A) Schematic representation of the renovated BaMV-based secretory expression cassettes, with the cleavable linker (LSrtA, LTEV, or LSNAC) inserted between the combinational secretory-affinity tag (SSExtHis(SP)10) and mIFNγ. A short peptide, LGG (denoted by the gray boxes) (Chen et al., 2013), was used as a linker between SSExt-His and (SP)10. The transcription of the BaMV-based vector supplemented with the RNA silencing suppressor, P19, was under the control of a dual 35S promoter from the Cauliflower mosaic virus (denoted by the double arrowheads) and a nopaline synthase (nos) terminator. (B) Analysis of FG expression in infiltrated leaves at 5 days post-infiltration (5 dpi). Total proteins were extracted from infiltrated N. benthamiana leaves, separated by electrophoresis through a 12% polyacrylamide gel containing 1% SDS (SDS-PAGE), followed by visualization with Coomassie blue staining (CBS) and immunoblotting (IB) analysis with alkaline phosphatase (AP)-conjugated antibodies specific to mIFNγ or His-tag. M, marker; pKn, vector only; P, purified mIFNγ protein derived from E. coli as a positive control; FG, SSExtHis(SP)10L-mIFNγ glycoprotein; FP, SSExtHis(SP)10L-mIFNγ protein; Mγ, monomeric mIFNγ; 1N-MG, M monoglycosylated mIFNγ; 2N-MG, M diglycosylated mIFNγ; DG, dimeric mIFNγ glycoprotein.
2.2 Transient expression of FGs in N. benthamiana plants
The transient expression of recombinant proteins in N. benthamiana was achieved using Agrobacterium-mediated infiltration. A. tumefaciens (PGV3850 strain) harboring the expression constructs of different FGs were cultured, harvested by centrifugation at 12,000 rpm for 1 min, and recovered in agro-infiltration buffer (10 mM MES and 10 mM MgCl2, pH 5.5) to reach appropriate OD600 for each construct. The culture solutions were then infiltrated into 6-week-old N. benthamiana plants by a syringe- or vacuum-infiltration process (Jiang et al., 2020). The infiltrated plants were individually maintained in the culture room at 28°C for 16-h light/8-h dark intervals.
2.3 Immunoblotting analysis
The infiltrated leaves were harvested at 5 days post-infiltration (dpi), and the total proteins extracted with 1:2.5 (w/v) protein extraction buffer (50 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 10 mM KCl, 1 mM EDTA, 20% glycerol, 10% β-mercaptoethanol, and 2% SDS). The extracted total proteins were analyzed by electrophoresis through a 12% polyacrylamide gel containing 1% sodium dodecyl sulfate (SDS-PAGE), followed by visualization using Coomassie blue staining (CBS) and analysis by immunoblotting (IB). The rabbit primary antibodies against His-tag antibodies (1:5000 dilution) or mIFNγ (1:5000 dilution) were used in IB for the determination of FGs or mIFNγ glycoprotein accumulations, as described previously (Jiang et al., 2019).
2.4 Separation of soluble FGs from N. benthamiana leaf homogenates
For isolation of the soluble FGs (SSExtHis(SP)10LSrtA-mIFNγ or SSExtHis(SP)10LTEV-mIFNγ), the infiltrated leaves were harvested at 5 dpi. Leaf homogenates were prepared with the use of 1:2 (w/v) extraction buffer A [50 mM Tris–HCl, pH 7.6, 120 mM KCl, 15 mM MgCl2, 0.1 mM PMSF, 20% glycerol, 0.1% β-mercaptoethanol, and one tablet of cOmplete™ EDTA-free proteinase inhibitor cocktail (Roche Life Science, Penzberg, Germany)] (Osman and Buck, 1996). The homogenized extracts were filtered through a layer of Miracloth and centrifuged at 30,000 × g for 30 min to separate the pellet (designated P30) and supernatant (designated S30) fractions. To remove the abundant RuBisCO protein contaminations, the S30 fraction was treated with ultrapure acetic acids to achieve a pH value of 5.1 and then subjected to centrifugation at 30,000 × g for 30 min to obtain the P30-treated (P30t) and S30-treated (S30t) fractions as described previously (Park et al., 2015; Jiang et al., 2020). After the removal of most RuBisCO protein contaminations, the pH of soluble FGs in the S30t fraction was adjusted to 7.0 by adding 1 M NaOH to avoid protein degradation.
2.5 Preliminary purification of soluble FGs in a Ni2+-NTA column
To purify the FGs (SSExtHis(SP)10LSrtA-mIFNγ or SSExtHis(SP)10LTEV-mIFNγ glycoproteins) from the S30t fraction, the soluble FGs were vacuum-filtered through a 0.22-μm Millipore membrane and applied to a Ni2+-NTA column according to the manufacturer’s instructions. All fractions were collected and brought to equal volume with protein sample buffer (with 2% SDS) and assayed by SDS-PAGE, followed by visualization with CBS and IB with specific antiserum. The fractions containing the target FGs were pooled and subjected to further processing.
2.6 Proteolytic cleavage of SSExtHis(SP)10LSrtA-mIFNγ using sortase A
For the removal of the LSrtA-linked secretory-affinity tag (SSExtHis(SP)10) from the target protein, 1 μg of FG [SSExtHis(SP)10LSrtA-mIFNγ] was treated with SrtA, produced in the laboratory from E. coli harboring the SrtA overexpressing plasmid purchased from Addgene (Watertown, MA, United States). The multifunctional SrtA exhibits cysteine protease, protein ligase, and transpeptidase activities, specifically recognizing the LPET/G motif (the “/” sign denotes the cleavage site) in proteins (Ton-That et al., 1999; Tsukiji and Nagamune, 2009). To determine the optimal reaction condition, two-fold diluted SrtA (from 4 to 0.25 μg/μL) was incubated with 1 μg of the purified SSExtHis(SP)10LSrtA-mIFNγ in reaction buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM CaCl2, and 4 mM β-mercaptoethanol) at 28°C overnight. The processed proteins were subjected to further purification as described below.
2.7 Proteolytic cleavage of SSExt (SP)10LTEV-mIFNγ using TEV protease
To remove the LTEV-linked tag, 10 mg of SSExtHis(SP)10LTEV-mIFNγ was treated with 0.2 mg of TEV protease as described previously (Kapust and Waugh, 1999). The TEV protease exhibits high cysteine protease activity under a wide range of reaction conditions (pH, salt, and temperature), which recognizes the specific ENLUFQG/S motif. The TEV protease concentrations were diluted either two-fold from 4 to 0.25 μg/μL or five-fold from 1 to 0.008 μg/μL to determine the optimal reaction condition. Each diluted TEV protease was added to 1 μg of the purified SSExtHis(SP)10LTEV-mIFNγ in reaction buffer (50 mM Tris-HCl, pH 8.0, 5 mM DTT, and 0.5 mM EDTA) at 4°C overnight. The processed proteins were subjected to further purification to extract the target protein.
2.8 Further purification of target FGs by chromatography through a second Ni2+-NTA and Superdex™ 200 pg (S200) column
The above protease-treated FGs in the reaction mixture were passed through a second Ni2+-NTA agarose column for three times, and the target mIFNγ glycoproteins were collected in the unbound fractions (flow-through). The Ni2+-NTA agarose was washed once with 10 mL washing buffer (20 mM 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 20 mM imidazole), and the 6x His-tagged proteases (sortase A or TEV protease) bound to the Ni2+-NTA agarose were subsequently eluted for recycling with 10 mL of elution buffer A (20 mM 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 100 mM imidazole) and 10 mL of elution buffer B (20 mM 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 300 mM imidazole). The flow-through fractions containing tag-free mIFNγ glycoproteins were collected and then loaded on a HiLoad™ 16/60 Superdex™ 200 pg (S200) using an FPLC system (AKTA Purifier, GE Healthcare, IL, United States) in S200 buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 5 mM β-mercaptoethanol) at a flow rate of 0.5–1 mL/min. The FG-containing fractions were pooled and further concentrated using a 10-kDa NMWL Centricon filter (GE Healthcare, IL, US). The concentrated mIFNγ glycoproteins were dialyzed with PBS buffer for further biological activity assay.
2.9 Protein quantification
The quantification of all purified proteins was confirmed by using different methods, as followed by a previous study (Jiang et al., 2019; Jiang et al., 2020), including Bradford colorimetric assay, ELISA, and Coomassie blue staining (CBS) following SDS-PAGE.
2.10 Infection of Sindbis virus in cells treated with interferon–gamma (IFN-γ) variants
To investigate the biological activity of recombinant IFN-γ variants, an anti-SINV assay was performed, as described previously (Jiang et al., 2020), using the reporter Sindbis virus carrying an enhanced green fluorescent protein expression cassette (designated as SINV-eGFP) kindly provided by Professor Lih-Hwa Hwang (Graduate Institute of Microbiology and Immunology, National Yang-Ming University, Taipei, Taiwan). In brief, HEK293 cells (2.5 × 105) were seeded in a 24-well plate 8 h prior to treatment. The cells were washed twice with PBS and treated with DMEM (Mock), the extract from healthy leaves of N. benthamiana as the negative control (NC), or five-fold serial dilutions (from 50 ng/mL to 0.08 ng/mL) of commercial mIFN-γ (Accession No. CAA31639) produced from E. coli as a positive control (PC-IFN-γ), another PC-IFN-γ (Accession No. NP_000610.2) produced from HEK293 (R&D Systems, Inc., MN, United States), or three IFN-γ variants (SSmIFNγ(SP)10, SSHis(SP)10LTEV-mIFNγ, and Tag-free mIFNγ) produced from infiltrated N. benthamiana for 12 h at 37°C with 5% CO2 (Busnadiego et al., 2020). Subsequently, the cell monolayer was infected with SINV-eGFP at a multiplicity of infection (MOI) of 1 (Tseng et al., 2015; Jiang et al., 2020). At 24 h post-infection (hpi), the levels of eGFP signals and viral proteins were detected by fluorescence microscopy and IB analysis with specific rabbit primary antibodies. The inhibition ratio of SINV was further determined by ELISA with eGFP-specific antibodies.
2.11 Statistical analysis
Statistical analysis was performed using the standard Student’s t-test (SPSS version 20, IBM Corp, Armonk, NY, United States). Mean expression ratio (%) and standard deviation (SD) from three independent experiments with technical triplicates are presented. p values < 0.001 were considered significant.
3 Results
3.1 Design of a cleavable-tag protein expression system based on the BaMV vector
Previous studies have developed several effective fusion tags to increase the productivity and ease of downstream processing (Xu et al., 2007; Xu et al., 2010; Zhang et al., 2016; Ramos-Martinez et al., 2017; Zhang et al., 2019). However, the presence of fusion tags may negatively influence the biological functions of the target proteins, and an effective approach for the removal of such tags is highly desirable. In this study, we designed a series of BaMV-based viral vectors in which cleavable linkers, including LSrtA, LTEV, and LSNAC, were inserted between the N-terminal SSExtHis(SP)10 tag and mIFNγ, as shown in Figure 1A. By using such a BaMV-based secretory expression system, we pursue the possibility of efficient production of near-native recombinant glycoproteins without any foreign fusion tags. To compare the processing efficiency of various cleavable linkers in our BaMV-based vectors, these constructions were infiltrated into N. benthamiana leaves through Agrobacterium-mediated inoculation, and the expression profiles were further examined. The previously reported constructs of pKB19mIFNγ (Jiang et al., 2019) and pKB19SSExtmIFNγ(SP)10 (Jiang et al., 2020) were also included in the experiment, serving as the bases for comparison.
To investigate whether inserted cleavable linkers affect the expression of mIFNγ fusion glycoproteins in the BaMV-based expression system, total proteins extracted from the infiltrated N. benthamiana leaf samples were examined at 5 dpi by IB analysis with specific antibodies against mIFNγ or His-tag (Figure 1B). The result of IB analysis using anti-His-tag antibodies revealed the presence of unprocessed SSExtHis(SP)10L-mIFNγ protein (FP, Figure 1B) and SSExtHis(SP)10L-mIFNγ glycoprotein (FG, Figure 1B) with relative molecular masses (Mr) of 25 kDa and 32–40 kDa, respectively, which are similar to those observed in the control sample SSExtmIFNγ(SP)10. The product of FGs (32–40 kDa) was possibly produced by the partial Hyp-O-glycosylation of FPs (25 kDa) via the N. benthamiana secretory pathway, as observed previously (Dolan et al., 2014; Jiang et al., 2020). By IB analysis with mIFNγ-specific antibodies, three extra protein bands were detected in leaf extracts of SSExtHis(SP)10LTEV-mIFNγ, with apparent Mr of 16 kDa (Mγ), 18 kDa (1N-MG), and 20 kDa (2N-MG) (indicated at the right of the panel, Figure 1B), as compared to those expressing SSExtHis(SP)10LSrtA-mIFNγ or SSExtHis(SP)10LSNAC-mIFNγ. These extra protein banding patterns were also observed in the control sample SSExtmIFNγ(SP)10 (Jiang et al., 2020), which also exhibit the same molecular weight, indicating SSExtHis(SP)10LTEV-mIFNγ glycoproteins might be post-translationally modified by the proteolytic cleavage of the combinational secretory-affinity tag, SSExtHis(SP)10, with varying degrees of N-glycosylation (including 0N, 1N, and 2N glycosylation, corresponding to the molecular weights of 16 kDa, 18 kDa, and 20 kDa, respectively). It also implied that plant endogenous protease activity could partially digest the FGs to form the various cleaved mIFNγ glycoproteins during the secretory process.
The overall accumulation of various mIFNγ forms was further quantified by ELISA. The result revealed the average levels of TPs produced in leaves infiltrated with A. tumefaciens harboring constructs for the expression of SSExtHis(SP)10LsrtA-mIFNγ and SSExtHis(SP)10LTEV-mIFNγ accumulated up to 595 ± 60.1 and 688 ± 5.9 μg/g fresh weight, accounting for 9.8% and 10.8% of the TSP, respectively (Table 1), which are significantly higher than those observed in the control groups. The result indicated that the fusion of combinational secretory-affinity tag (SSExtHis(SP)10LsrtA or SSExtHis(SP)10LTEV) could further improve the yield of TPs. However, the expression of SSExtHis(SP)10LSNAC-mIFNγ glycoprotein in infiltrated N. benthamiana leaves was lower than that of other constructs (Figure 1B; Table 1), and thus it was excluded in the following experiments.

TABLE 1. Expression levels of TPs as quantified by ELISA.
3.2 Proteolytic cleavage of SSExtHis(SP)10LSrtA-mIFNγ using sortase A
To determine the feasibility of SrtA in removing the combinational secretory-affinity tag from the SSExtHis(SP)10LsrtA-mIFNγ fusion glycoproteins (FGs), the following experiment was performed. Ni2+-NTA column-purified FGs (32–40 kDa) and SrtA (17.4 kDa) were collected, examined by SDS-PAGE (Supplementary Figure S1), and quantified for their total soluble protein (TSP) levels by using the Bradford colorimetric assay (Sigma-Aldrich, St. Louis, MO, United States). To determine the optimal protein-to-enzyme ratio, the purified FGs (1 μg, SSExtHis(SP)10LSrtA-mIFNγ) were incubated with various concentrations of SrtA (two-fold serial dilutions from 4 to 0.25 μg/μL). Subsequently, each reaction was analyzed by SDS-PAGE, and the FGs or various cleaved mIFNγ forms were visualized by CBS and IB analysis with specific antibodies against mIFNγ or His-tag. As shown in the CBS-stained gel, the cleaved mIFNγ forms with Mγ (16 kDa), 1N-MG (18 kDa), and 2N-MG (20 kDa) were partially released from FGs (SSExtHis(SP)10LSrtA-mIFNγ) following SrtA digestion (Figure 2A). Additionally, the dimeric and heterogeneous mIFNγ glycoproteins (32–40 kDa, DG) were also observed, which might be generated from the subsequent dimerization of two cleaved mIFNγ monomers with different degrees of N-glycosylation (Sareneva et al., 1994; Sareneva et al., 1995). The result of IB analysis with mIFNγ-specific antibodies showed that the maximal efficiency for the removal of combinational secretory-affinity tag was approximately 50%, with high doses (2 and 4 μg/μL) of SrtA digestion. Analysis with His-tag-specific antibodies further verified that the cleavage of FGs was not complete (Figure 2). Based on the above observations, a 1:2 ratio of FG to enzyme was chosen for SrtA-mediated cleavage. The cleaved combinational secretory-affinity tag was subsequently separated from the target proteins by passing through an Ni2+-NTA column. However, analysis of the eluted fraction from 300 mM imidazole treatment revealed that the majority of the processed mIFNγ glycoproteins (32–40 kDa), including Mγ (16 kDa), 1N-MG (18 kDa), and 2N-MG (20 kDa), were still accompanied with SrtA (Figures 2B, IB panel, lane 4). This observation indicated that SrtA-mediated digestion could not efficiently process the FGs (SSExtHis(SP)10LSrtA-mIFNγ) and that the contamination of the combinational secretory-affinity tag and SrtA may pose a threat to the downstream process of target proteins.
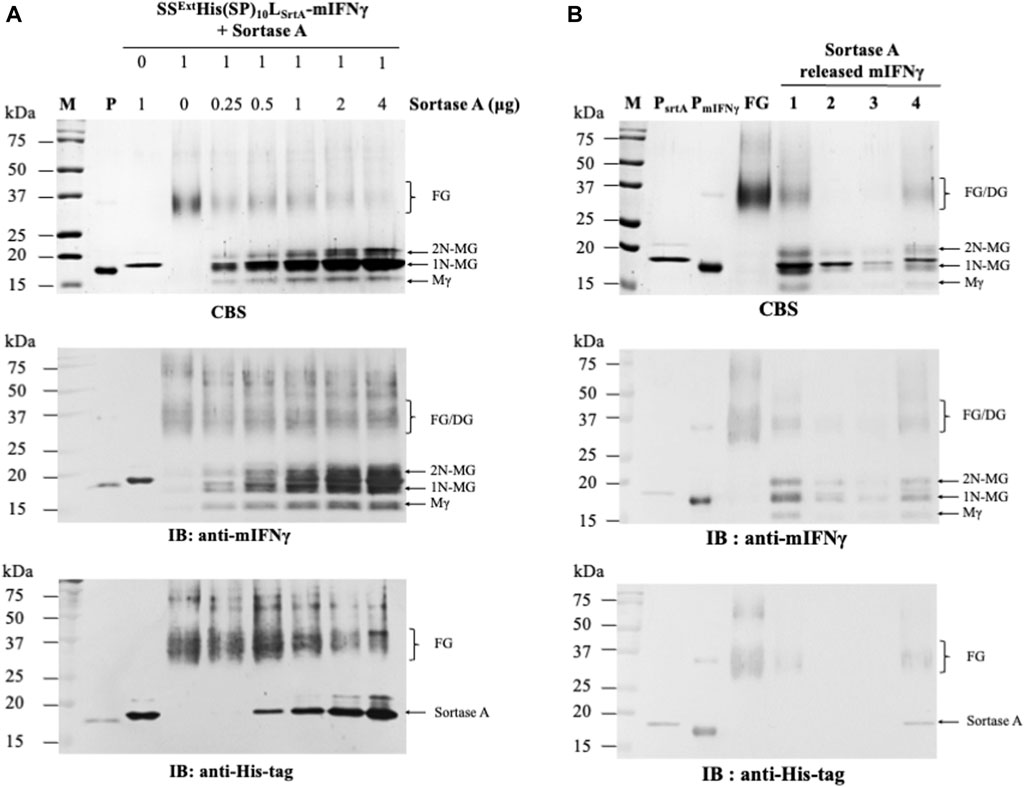
FIGURE 2. Processing of SSExtHis(SP)10LSrtA-mIFNγ glycoproteins by SrtA protease. (A) Optimization of the reaction condition. To determine the optimal protein-to-enzyme ratio, the purified FGs (1 μg, SSExtHis(SP)10LSrtA-mIFNγ) were incubated with different concentrations of SrtA, as indicated in the panel, at 28°C overnight. Subsequently, each reaction was analyzed by SDS-PAGE and visualized with CBS and IB analysis with antibodies specific to mIFNγ or His-tag. (B) Purification of SrtA-released mIFNγ from SSExtHis(SP)10LSrtA-mIFNγ glycoproteins by the Ni2+-NTA column. Each fraction was collected and assayed by SDS-PAGE, followed by CBS and IB analysis with specific antibodies, as indicated at the bottom of the panel. M, marker; P, positive control, purified mIFNγ protein derived from E. coli; FG, SSExtHis(SP)10LSrtAmIFNγ glycoprotein; lane 1, total reaction product containing released mIFNγ glycoproteins from FGs (1 μg, SSExtHis(SP)10LSrtA-mIFNγ); lane 2, flow-through fraction; lane 3, eluted fraction using 25 mM imidazole; lane 4, eluted fraction using 300 mM imidazole. The positions of different forms of mIFNγ or sortase A are indicated on the right of each panel. Mγ, M mIFNγ; 1N-MG, M monoglycosylated mIFNγ; 2N-MG, M diglycosylated mIFNγ; DG, dimeric mIFNγ glycoprotein.
3.3 Proteolytic cleavage of SSExtHis(SP)10LTEV-mIFNγ using TEV protease
To test the applicability of LTEV linker and TEV protease in the BaMV-based system, experiments similar to those described above were performed. Both SSExtHis(SP)10LTEV-mIFNγ and TEV proteins were also initially purified by the Ni2+-NTA column. Analysis of the eluates revealed that the FGs (32–40 kDa) could be efficiently obtained, accompanied with a few processed mIFNγ forms, including Mγ (16 kDa), 1N-MG (18 kDa), and 2N-MG (20 kDa) (Supplementary Figure S2).
To determine the optimal reaction condition, the purified SSExtHis(SP)10LTEV-mIFNγ glycoproteins (1 μg) were treated with different concentrations of TEV protease (two-fold serial dilutions ranging from 4 to 0.25 μg/μL) at 4°C overnight. Subsequently, each reaction was analyzed by SDS-PAGE, and the FGs or the cleaved mIFNγ forms were visualized by CBS and IB. The result of IB analysis with His-tag-specific antibodies (Figure 3A, the bottom panel) showed that the SSExtHis(SP)10 tag was completely removed by TEV protease from FGs (SSExtHis(SP)10LTEV-mIFNγ). To further fine-tune the reaction condition for the removal of the SSExtHis(SP)10 tag from FGs, the purified SSExtHis(SP)10LTEV-mIFNγ glycoproteins (1 μg) were further incubated with five-fold serial dilutions (ranging from 1 μg/μL to 0.008 μg/μL) of TEV protease. Analysis of the digestion result by IB with His-tag-specific antibodies revealed that FGs could be efficiently digested by using 0.2 μg/μL of TEV protease (Figure 3B, the bottom panel). Consequently, a 1:0.2 ratio of FG to enzyme was chosen for TEV protease-mediated cleavage. As expected, following the processing of the fusion tag, the monomeric polypeptide subunit of mIFNγ glycoproteins would assemble into dimeric and heterogeneous mIFNγ glycoproteins (32–40 kDa, DG), as shown by IB analysis with anti-mIFNγ-specific antibodies (Figures 3A, B, the middle panel). These findings indicated that TEV protease may serve as an efficient process in the production of target proteins containing LTEV-linked fusion tags in the BaMV-based expression vector.
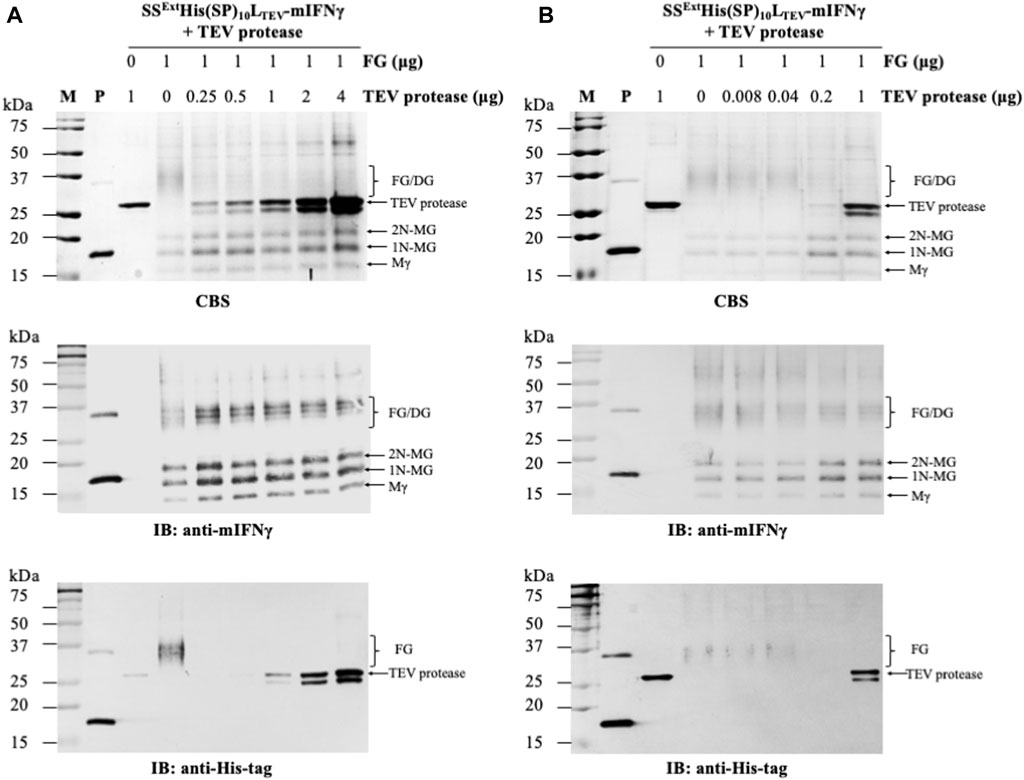
FIGURE 3. Processing of SSExtHis(SP)10LTEV-mIFNγ glycoproteins by TEV protease. To determine the optimal protein-to-enzyme ratio, the purified FGs (1 μg, SSExtHis(SP)10LSrtA-mIFNγ) were initially incubated with 2-fold serial dilutions of TEV proteinase (starting from 4 μg/μL) (A) and further fine-tuned with 5-fold serial dilutions of TEV proteinase (starting from 1 μg/μL) (B), as indicated in the panel, at 4°C overnight. Subsequently, each reaction was assayed by SDS-PAGE, followed by CBS and IB analysis, as described above. M, marker; P, positive control, purified mIFNγ protein derived from E. coli; FG, SSExtHis(SP)10LTEV-mIFNγ glycoprotein. The positions of different forms of mIFNγ are indicated on the right of each panel. Mγ, M mIFNγ; 1N-MG, M monoglycosylated mIFNγ; 2N-MG, M diglycosylated mIFNγ; DG, dimeric mIFNγ glycoprotein.
Subsequently, we developed a protocol for obtaining tag-free recombinant glycoproteins from plant-made SSExtHis(SP)10LTEV-mIFNγ, as illustrated in Figure 4A. To examine whether this protocol could be efficiently scaled up for processing the FGs into tag-free mIFNγ, 10 mg of SSExtHis(SP)10LTEV-mIFNγ was incubated with TEV protease (2 mg/mL) in a dialysis tube (MWCO 12–14 kDa) overnight at 4°C. After incubation, the released mIFNγ glycoproteins and SSExtHis(SP)10 tag were separated by passing through a second Ni2+-NTA column. Analysis of each fraction by SDS-PAGE and IB with mIFNγ-specific antibodies revealed that the released mIFNγ glycoproteins, including Mγ (16 kDa), 1N-MG (18 kDa), 2N-MG (20 kDa), and DG (32–40 kDa), were efficiently recovered by a low concentration of imidazole (25 mM) (Figure 4B, the middle panel, lane 3). The result of IB analysis with His-tag-specific antibodies confirmed that the combinational secretory-affinity tag was efficiently removed from FGs (Figure 4B, the bottom panel, lane 3). Additionally, TEV protease (Mr of 27 kDa) was found in the fractions eluted with 300 mM imidazole (Figure 4B, lane 4), indicating that TEV protease could be separated from the products and recovered for recycling through the Ni2+-NTA column. These results showed that the use of TEV protease-mediated processing could be scaled up to efficiently remove the SSExtHis(SP)10 tag from SSExtHis(SP)10LTEV-mIFNγ. The observation also suggested that this system could be used as a means to mass-produce near-native mIFNγ glycoproteins and other recombinant therapeutic proteins. To further improve the purity of tag-free mIFNγ, the collected fractions were pooled and applied to gel filtration chromatography (Superdex™ 200 pg, S200). Each step-fraction was collected, and the protein contents were confirmed again by SDS-PAGE stained with CBS and IB with mIFNγ-specific antibodies (Figure 4C). The results indicated that tag-free mIFNγ could be efficiently obtained. The final yield was estimated to be approximately 102 ± 3 mg/kg fresh tissue weight (FW) with a purity higher than 98%, which is comparatively higher than that for SSExtmIFNγ(SP)10 (94 ± 7 mg/kg FW), as reported previously (Jiang et al., 2020).
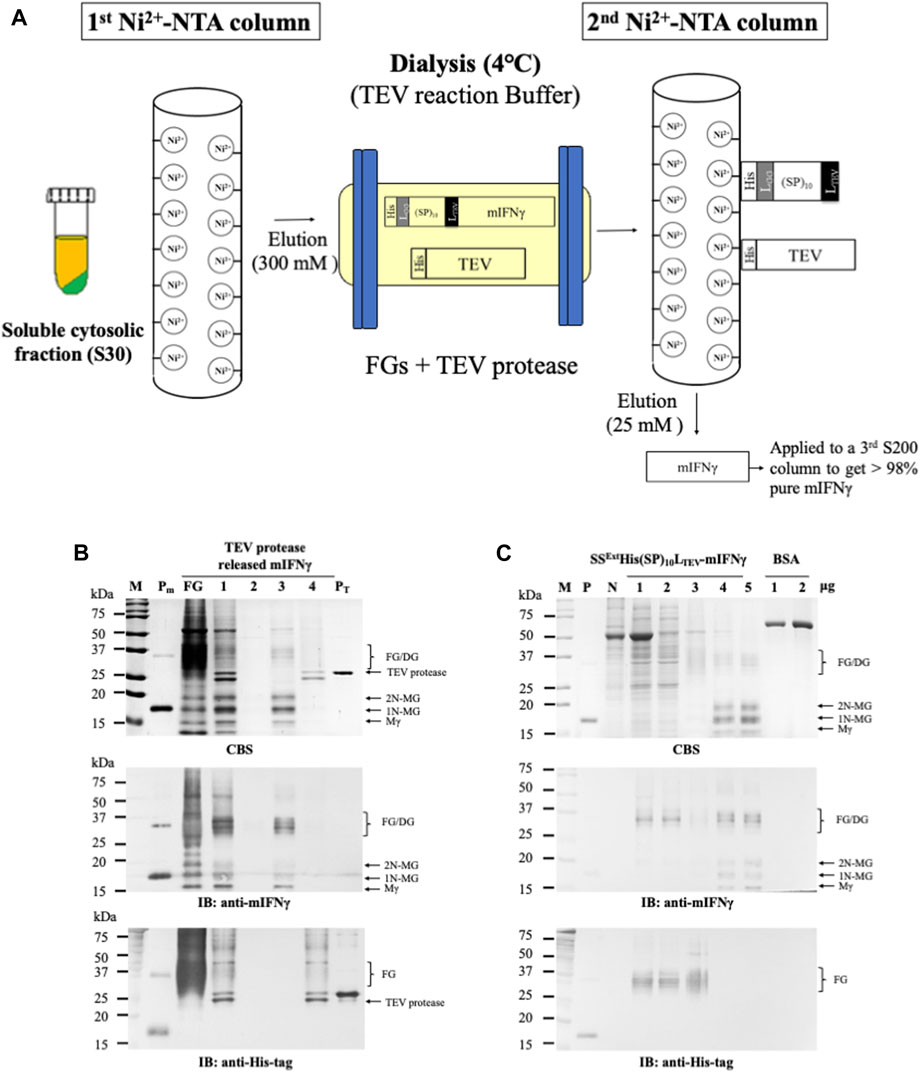
FIGURE 4. Purification of TEV protease-released mIFNγ from SSExtHis(SP)10LTEV-mIFNγ glycoproteins by Ni2+-NTA and gel filtration chromatography. (A) Schematic representation of the process for purification of tag-free mIFNγ glycoproteins. Briefly, the target fusion proteins containing His-tags were initially purified through an immobilized metal affinity chromatography (IMAC) column. Following incubation with TEV protease in a dialysis tubing overnight, the released tag-free mIFNγ glycoproteins were further purified through a second IMAC column. (B) Analysis of the released mIFNγ glycoproteins. The FGs (10 mg, SSExtHis(SP)10LTEV-mIFNγ) were processed using TEV protease (2 mg)-mediated cleavage, followed by purification through a Ni2+-NTA column. Each fraction was collected and assayed by SDS-PAGE, followed by CBS and IB analysis, as described above. Lane 1, total reaction content containing FGs; lane 2, flow-through; lane 3, eluted fraction of tag-free mIFNγ glycoproteins using 25 mM imidazole; lane 4, eluted fraction of TEV protease using 300 mM imidazole (lane 4). (C) SDS-PAGE analysis of each fraction from the three-step chromatography purification process. Each fraction was collected and assayed by SDS-PAGE, followed by CBS and IB analysis, as described above. Lane 1, soluble fraction following centrifugation (S30); lane 2, S30 treated with acetic acid (S30t); lane 3, eluted fraction from first Ni2+-NTA purification; lane 4, eluted fraction from the second Ni2+-NTA purification; lane 5, products of the third gel filtration purification. M, marker; P, positive control, purified mIFNγ protein derived from E. coli; FG, SSExtHis(SP)10LTEV-mIFNγ glycoproteins; Mγ, M mIFNγ; 1N-MG, M monoglycosylated mIFNγ; 2N-MG, M diglycosylated mIFNγ; DG, dimeric mIFNγ glycoprotein; BSA, bovine serum albumin as a protein loading standard.
3.4 Efficacy of IFN-γ variants against chimeric Sindbis virus
The inhibitory effect of recombinant IFN-γ has been demonstrated against several viruses (Parvez et al., 2006; Wang et al., 2014; Busnadiego et al., 2020) and cancer cells (Razaghi et al., 2017; Shen et al., 2018). Moreover, as presented in our previous study, plant-made SSExtmIFN(SP)10 was shown to induce antiviral activity in a reporter Sindbis virus expressing eGFP (SINV-eGFP) and reduce influenza virus (H1N1 strain) replication to a similar extent, as achieved by the commercial mIFNγ produced in E. coli (Jiang et al., 2020). To evaluate the efficacy of the IFNγ variants, including SSExtmIFNγ(SP)10, SSExtHis(SP)10LTEV-mIFNγ, and tag-free mIFNγ purified from N. benthamiana, the recombinant proteins were prepared in PBS buffer and the concentrations of each sample were further confirmed by SDS-PAGE stained with CBS and IB with mIFNγ-specific or His-tag-specific antibodies (Figure 5). The commercial mIFNγ produced in E. coli or mammalian cells, designated PC-mIFNγ (E. coli) and PC-IFNγ (HEK-293), respectively, were used as the positive controls. The antiviral effect was then evaluated using the SINV-eGFP reporter virus as a target pathogen. HEK293 cells were cultured in DMEM with IFN-γ variants at various concentrations, which were 5-fold serially diluted from 50 to 0.08 ng/mL for 12 h, followed by challenging with the SINV-eGFP. The cells cultured in DMEM treated with PBS buffer were used as the mock treatment group (Mock), whereas those treated with the total protein extract of healthy Nicotiana benthamiana served as the negative control (NC). At 24 h post-infection (hpi), the eGFP fluorescence, representing the overall infection status, was examined and recorded by fluorescent microscopy. As shown in Figure 6A, upon treatment with 2–50 ng IFNγ variants, the eGFP fluorescence signals were obviously decreased and mostly confined to a single-cell level, as compared to those in the Mock and NC (50 ng) groups. It is worth noting that cells treated with the tag-free mIFNγ variant at low doses (0.08 ng or 0.4 ng) had lower eGFP signals, demonstrating the antiviral efficacy of tag-free mIFNγ. The results of eGFP fluorescence intensity measurements and IB analysis showed that treatment with the tag-free mIFNγ variant at the lowest dose (0.08 ng) significantly decreased the level of eGFP (p-values <0.001), which is comparable to the effect of commercial PC-IFNγ (HEK293). Treatments with three IFNγ variants at medium (2 ng) or high (50 ng) doses reduced the eGFP levels in a dose-dependent manner (Figure 6B; Supplementary Figure S3). Among them, the tag-free mIFNγ was observed to have a higher efficacy in decreasing SINV-eGFP infection as compared to those of variants with fusion tags, including SSExtmIFNγ(SP)10 and SSExtHis(SP)10LTEV-mIFNγ (a significant difference with p-values <0.001). To corroborate this result, the inhibition ratio of SINV-eGFP infection was determined by ELISA for quantification of eGFP levels, and the half-maximal inhibitory concentration (IC50) of each recombinant protein was estimated. As shown in Figure 7; Table 2, tag-free mIFNγ exhibited a higher inhibitory effect on SINV-eGFP infection, which was similar to that of commercial PC-IFNγ (HEK293), with IC50 values of 2.5–2.9 ng/mL. In contrast, the IC50 values of SSExtmIFNγ(SP)10 and SSExtHis(SP)10LTEV-mIFNγ were 7.9 and 37.2 ng/mL, respectively, which were notably higher than that of the tag-free protein. The lower IC50 value and similar efficacy comparable with commercial IFNγ (HEK-293) protein indicated that plant-made tag-free mIFNγ exhibits satisfactory antiviral activity, which also suggested that these IFNγ glycoproteins may maintain the native folding suitable for inhibiting viruses.
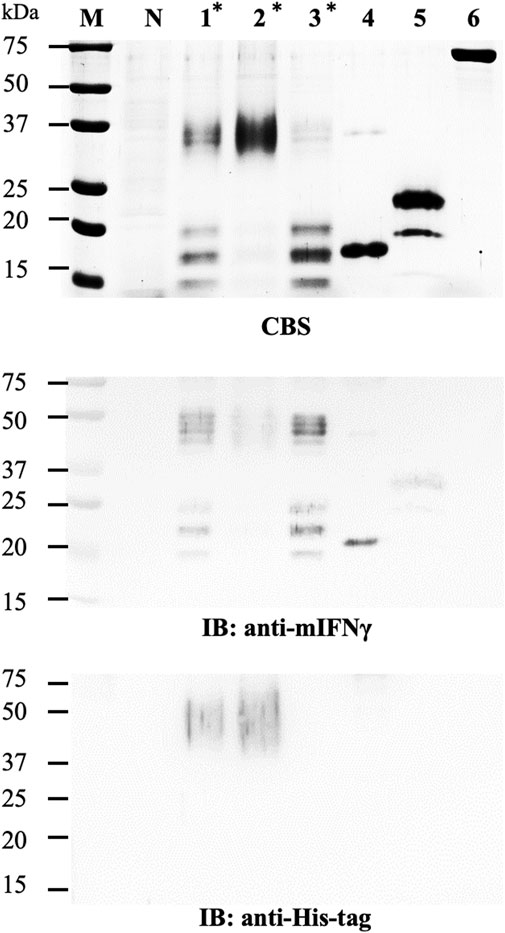
FIGURE 5. Comparison of various hIFNγ variants. Each IFNγ variant (1 μg) was analyzed by SDS-PAGE, followed by CBS and IB analysis, as described above. Lane 1, fusion protein SSExtmIFNγ(SP)10 from N. benthamiana leaf homogenates; lane 2, fusion SSExtHis(SP)10LTEV-mIFNγ glycoproteins; lane 3, tag-free mIFNγ glycoprotein; lane 4, commercial E. coli expressed IFNγ; lane 5, HEK293-expressed IFNγ; lane 6, BSA as the protein loading standard. M, marker; *, mIFNγ variants collected following the three-step purification protocol described above.
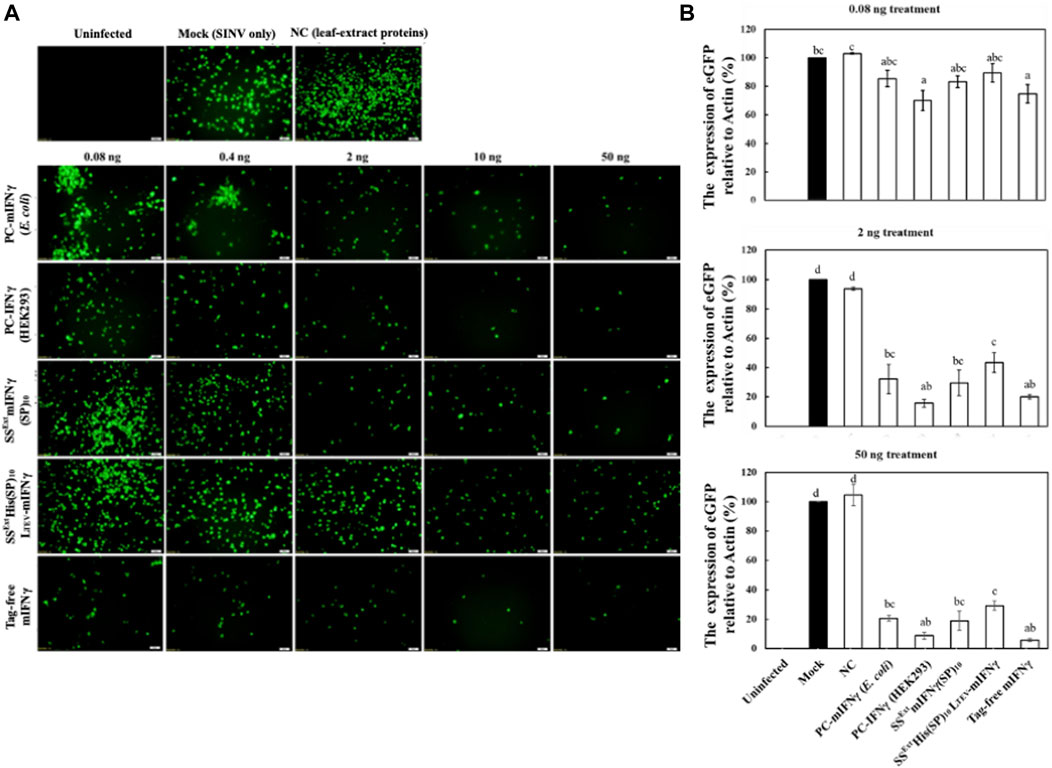
FIGURE 6. Effect of fusion tags on biological activity of mIFNγ against SINV. HEK293-T cells were left uninfected or pre-treated with DMEM (Mock), 50 ng of leaf-extract proteins as the negative control (NC), different concentrations of commercially available IFNγ proteins as positive controls (PCs), designated as PC-mIFNγ (produced from E. coli), PC-IFNγ (produced from HEK293), or various recombinant proteins produced from plants, including SSExtmIFNγ(SP)10, SSExtHis(SP)10LTEV-mIFNγ, or tag-free mIFNγ for 12 h. Subsequently, cells were infected with the reporter virus, SINV-eGFP, at an MOI of 1 for 24 h. The accumulated eGFP signals were directly observed by fluorescence microscopy (A) (scale bar = 50 μm) and quantified by densitometry (B). The eGFP level in DMEM (Mock)-treated cells was arbitrarily set as 100%, and relative protein accumulation levels in other samples were estimated. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc multiple comparison analysis. The p-value of <0.05 was considered significantly different, as denoted by different letters.
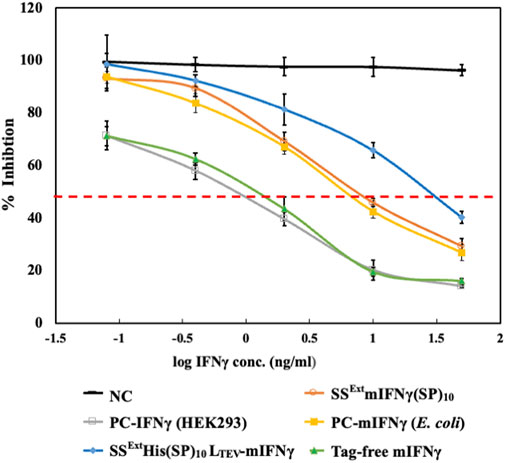
FIGURE 7. Relative anti-SINV activity of various interferon gamma variants. The relative inhibition ratio (%) of SINV in cells treated with different samples was estimated using the quantification of the eGFP level by ELISA. The dashed line in red indicates 50% of the relative inhibition. The half-maximal inhibitory concentration (IC50) of each sample is represented in Table 2.
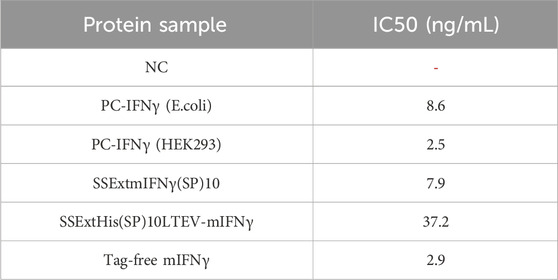
TABLE 2. Half-maximal inhibitory concentration (IC50) of each sample.
4 Discussion
The safety and productivity of therapeutic proteins are among the major considerations for the biomedical application of plant-made pharmaceuticals. In this study, we have developed an improved BaMV-based expression system using the combinational secretory-affinity tag attached to the mIFNγ protein through various cleavable linkers (LSrtA, LTEV, and LSNAC) and tested the efficiency for the removal of fusion tags from FGs. We demonstrated that the TEV protease effectively processed LTEV-linked FGs to release tag-free mIFNγ glycoproteins, containing Mγ (16 kDa), 1N-MG (18 kDa), and 2N-MG (20 kDa), which would undergo self-assembly to form the biologically active DG form (32–40 kDa). Both the cleaved combinational secretory-affinity tag and TEV protease were efficiently removed through a second immobilized metal affinity and gel filtration chromatography (IMAC) for obtaining the tag-free forms of mIFNγ glycoproteins. In addition, the antiviral activity of mIFNγ glycoproteins was verified and shown to exhibit similar or higher biological activities against the Sindbis virus as compared to those of the commercial HEK293-expressed hIFNγ or tagged versions of mIFNγ. These observations highlight the potential of this refined BaMV-based expression vector as an efficient system for the production of plant-made biopharmaceutical candidates with antiviral therapeutic efficacy.
Recently, several groups have successfully established plant-based protein secretion systems based on HypGP tag techniques (Xu et al., 2007; Xu et al., 2010; Zhang et al., 2016; Ramos-Martinez et al., 2017; Zhang et al., 2019). The expression of secreted fusion proteins either in inoculated plant leaves or suspension cell culture by fusion of the combinational secretory peptides, including plant-derived SS and “Ser-Pro” motif repeats to target proteins, provides feasible production for large-scale downstream processing. Additional fusion of affinity His-tag to the target protein is a useful technique for the efficient purification of target proteins (Wilken and Nikolov, 2012). However, there has been no attempt in these previous studies to remove the combinational secretory-affinity tag from FGs through enzymatic or chemical cleavage.
Our previous study showed that the proteolytic cleavage of N- and C-terminal fusions of SSExtmIFNγ(SP)10 through plant secretory processes may convert the FGs into the maturation forms of mIFNγ glycoproteins, including Mγ (16 kDa), 1N-MG (18 kDa), 2N-MG (20 kDa), and DG (32–40 kDa) (Jiang et al., 2020). However, the native processing activity of the plant secretory system was not sufficient to produce tag-free mIFNγ efficiently. In this study, it was noted that the SSExtmIFNγ(SP)10 containing 6X His-tag at the C-terminus was still detected by His-tag-specific antibodies (Figure 1B, the bottom panel), indicating that FGs were only partially processed. Therefore, various cleavable linkers, including LSrtA, LTEV, and LSNAC, were taken into consideration for the efficient production of tag-free mIFNγ in N. benthamiana through the BaMV-based vector in this study. We showed that all three cleavable linkers allowed the successful expression of FGs by the renovated BaMV-based vector, with partial O-glycosylated proteins of estimated molecular masses ranging from 32 to 40 kDa (Figure 1B, the bottom panel). The yield is similar to those observed in N. benthamiana expressing SStab (SP)32-EGFP (Dolan et al., 2014) and in tobacco hairy root expressing SStab (SP)32-EGFP (Zhang et al., 2019). Among these constructs with different inserted linkers, the expression level of SSExtHis(SP)10LSNAC-mIFNγ was lower than those of SSExtHis(SP)10LSrtA-mIFNγ and SSExtHis(SP)10LTEV-mIFNγ (Figure 1B, the bottom panel), suggesting that different cleavable linkers may affect the expression, conformational flexibility, and/or stability of FGs. It is worth noting that SSExtHis(SP)10LTEV-mIFNγ glycoproteins were partially cleaved to Mγ (16 kDa), 1N-MG (18 kDa), and 2N-MG (20 kDa) by endogenous proteases in plants, possibly through the secretory pathway, which was in agreement with our previous result for SSExtmIFNγ(SP)10 expressed from the secretory BaMV-based vector (Jiang et al., 2020). Similar observations of this proteolytic process have also been reported (Zhang et al., 2011; Puchol Tarazona et al., 2021). It has been shown that plant endogenous Kex2p-like serine protease or additional unknown protease may cleave the GMCSF-L-GFP protein (FP) in vivo to release the individual protein through the plant secretory pathway (Zhang et al., 2011). Additionally, two apoplastic-abundant subtilisin-like serine proteases, NbSBT1 and NbSBT2, were recently identified to be responsible for the major proteolytic processes on recombinant IgG1 glycoproteins during protein secretion in N. benthamiana (Puchol Tarazona et al., 2021). Although these reports demonstrated that the fusion tags on target proteins may be automatically removed by endogenous proteolytic processing, especially through the secretory pathways, the efficiencies seem to require further improvement for industrial-scale production. Furthermore, the yield of the target proteins may be affected by the addition of different fusion tags. To ensure the efficient production of tag-free target proteins, various chemically or enzymatically cleavable linkers are used in the expression constructs. The above observations indicated that it is necessary to test the effects on yield and cleavage efficiency of such linkers for optimization of protein expression systems. For this purpose, the BaMV-based tag-free expression vector system developed in this study provides three cleavable linkers in the construct that may exert different effects on different target proteins and thus could be conveniently tested and optimized for different needs.
Fusion tags are often used to increase the efficiency of downstream production since they can be helpful for enhancing the stability and solubility of target proteins as well as their purification processes (Arnau et al., 2006; Yadav et al., 2016; Liu and Timko, 2022). The use of a combinational secretory-affinity tag in this study was not only for the isolation of soluble FGs in the S30t fraction but also the successful recovery of pure SSExtHis(SP)10LSrtA-mIFNγ and SSExtHis(SP)10LTEV-mIFNγ glycoproteins after the primary Ni2+-NTA purification (Supplementary Figure S1, 2). Although the fusion technology is a powerful tool for the efficient production of many recombinant proteins, previous studies have shown that fusion tags would affect the biological function and immunogenicity of recombinant proteins through interference with the formation of native structures, especially the dimer/oligomer conformation (Gaberc-Porekar et al., 1999; Wu and Filutowicz, 1999; Yadav et al., 2016). The removal of fusion tags from FGs using enzymatic or chemical cleavage is thus a critical procedure for producing therapeutic proteins for biomedical use (Yadav et al., 2016; Dang et al., 2019; Islam et al., 2019). The enzyme-mediated proteolytic cleavage assays in this study showed that the TEV protease could near-completely remove the combinational affinity tag from FGs (Figure 3) as compared to those of SrtA-mediated cleavage (Figure 2). Additionally, under biocompatible conditions at 4°C overnight, TEV protease was active to cleave the FGs to release C-terminal mIFNγ glycoproteins, including Mγ (16 kDa), 1N-MG (18 kDa) and 2N-MG (20 kDa), resulting in a biologically active and heterogeneous DG form (32–40 kDa) with multiple bands by self-assembly. Subsequently, the combinational secretory-affinity tag and TEV protease could be efficiently removed through a second Ni2+-NTA column to obtain tag-free mIFNγ glycoproteins (Figure 4B, lane 3). After further gel filtration purification, tag-free mIFNγ glycoproteins could be obtained successfully, with a higher yield as compared to that of SSExtmIFNγ(SP)10 glycoproteins reported previously (Jiang et al., 2020). The result demonstrated that the combinational fusion tags, either SSExtHis(SP)10LSrtA or SSExtHis(SP)10LTEV, N-terminally fused to mIFNγ was advantageous to the increase of productivity for downstream purification processes.
The current procedure for the production of tag-free mIFNγ might be too tedious for further industrial applications. A streamlined process might be developed as a one-column-for-all format as shown in Supplementary Figure S4: the plant-made fusion proteins are initially bound on the Ni2+-NTA column and separated from the non-target proteins. Following in-column cleavage of the fusion protein with TEV protease, which also contains the 6X His-tag, the tag-free mIFNγ target protein is then eluted and collected in pure form. In the final elution step, the TEV containing 6X-His-tag may be collected and reused in the subsequent processes. In this streamlined procedure, only one Ni2+-NTA column is used, and the TEV cleavage step is performed in the column, which may significantly reduce the required resources and processes as compared to the procedure shown in Figure 4A.
Glycosylation is one of the major post-translational modifications of proteins that occurs during ER and Golgi processing within eukaryotic cells, resulting in the addition of different glycans on transported proteins (Faye et al., 2005; Gomord et al., 2010; Liu and Timko, 2022). The native mIFNγ has two glycosylation sites which are known to be required for resistance to granulocyte proteases, cathepsin G, and elastase and play an important role in increasing hIFNγ half-life in human blood (Abiri et al., 2016; Cao et al., 2022). In our previous study, glycosylation of target protein in plant cells has been achieved by fusing plant-specific SS peptide to a recombinant hIFNγ, leading to successful production of glycoprotein with diverse complex-type N-glycans (Jiang et al., 2020). It has been reported that the degrees of similarity in glycosylation patterns between native hIFNγ and those produced in different expression systems are in the order of native hIFNγ (HEK293 cell) > mammalian cells (CHO) > plant cells (N. benthamiana) > insect cells (Sf9) > yeast (Pichia pastoris) > prokaryotic cells (E. coli) (Abiri et al., 2016). In this study, plant-made mIFNγ glycoproteins without undesired tags were shown to induce a higher antivirus activity against SINV-eGFP, as compared to those of the other fusion variants, including SSExtmIFNγ(SP)10, SSExtHis(SP)10LTEV-mIFNγ, and PC-mIFNγ (E. coli) (Figure 6; Figure 7; Table 2). This observation indicated that glycosylated, tag-free mIFNγ could enhance therapeutic efficacy similar to those induced by HEK293- and CHO-expressed hIFNγ with a complex-type glycan that exhibits higher levels of therapeutic efficacy against ovarian cancer cells as compared to its non-glycosylated form (Razaghi et al., 2017).
5 Conclusion
In this study, we have renovated the BaMV-based vector system for successful removal of the combinational secretory-affinity tag from FGs for the production of tag-free glycoproteins in N. benthamiana. We demonstrated that, through fusing the combinational secretory-affinity (SSExtHis(SP)10) tag and TEV-cleavable peptide (LTEV) to the N-terminal of the mIFNγ protein, rapid accumulation of soluble SSExtHis(SP)10LTEV-mIFNγ glycoproteins in N. benthamiana could be achieved. Our study also revealed that TEV protease may efficiently remove the fusion tag from SSExtHis(SP)10LTEV-mIFNγ glycoproteins under biocompatible conditions, releasing C-terminus mIFNγ that underwent self-assembly to biologically active forms with enhanced antivirus activity. Through providing three different cleavable fusion linkers to suit the requirement of different target proteins, the renovated BaMV-based expression system developed in this study offers an alternative strategy for obtaining tag-free therapeutic proteins in compliance with biomedical applications.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
M-CJ: methodology, conceptualization, data curation, formal analysis, investigation, and writing–original draft. W-LH: conceptualization, methodology, writing–review and editing, and investigation. C-YT: data curation, methodology, and writing–review and editing. N-SL: conceptualization, methodology, writing–review and editing, and funding acquisition. Y-HH: methodology, writing–review and editing, funding acquisition, and project administration. C-CH: conceptualization, methodology, project administration, and writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article. This work was financially supported in part by the Advanced Plant and Food Crop Biotechnology Center from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan and by the Ministry of Science and Technology, Taiwan (MOST 111-2313-B-005-051 and MOST 111-2313-B-005-004).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2023.1341340/full#supplementary-material
References
Abiri, R., Valdiani, A., Maziah, M., Shaharuddin, N. A., Sahebi, M., Yusof, Z. N., et al. (2016). A critical review of the concept of transgenic plants: insights into pharmaceutical biotechnology and molecular farming. Curr. Issues Mol. Biol. 18, 21–42. doi:10.21775/cimb.018.021
Arnau, J., Lauritzen, C., Petersen, G. E., and Pedersen, J. (2006). Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr. Purif. 48, 1–13. doi:10.1016/j.pep.2005.12.002
Berlec, A., and Strukelj, B. (2013). Current state and recent advances in biopharmaceutical production in Escherichia coli, yeasts and mammalian cells. J. Ind. Microbiol. Biotechnol. 40, 257–274. doi:10.1007/s10295-013-1235-0
Busnadiego, I., Fernbach, S., Pohl, M. O., Karakus, U., Huber, M., Trkola, A., et al. (2020). Antiviral activity of type I, II, and III interferons counterbalances ACE2 inducibility and restricts SARS-CoV-2. MBio 11, e01928–e01920. doi:10.1128/mBio.01928-20
Cao, L., Zhang, L., Zhang, X., Liu, J., Jia, M. A., Zhang, J., et al. (2022). Types of interferons and their expression in plant systems. J. Interferon Cytokine Res. 42, 62–71. doi:10.1089/jir.2021.0148
Chen, X., Zaro, J. L., and Shen, W. C. (2013). Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 65, 1357–1369. doi:10.1016/j.addr.2012.09.039
Conley, A. J., Joensuu, J. J., Jevnikar, A. M., Menassa, R., and Brandle, J. E. (2009). Optimization of elastin-like polypeptide fusions for expression and purification of recombinant proteins in plants. Biotechnol. Bioeng. 103, 562–573. doi:10.1002/bit.22278
Dang, B., Mravic, M., Hu, H., Schmidt, N., Mensa, B., and DeGrado, W. F. (2019). SNAC-tag for sequence-specific chemical protein cleavage. Nat. Methods 16, 319–322. doi:10.1038/s41592-019-0357-3
Daniell, H., Streatfield, S. J., and Wycoff, K. (2001). Medical molecular farming: production of antibodies, biopharmaceuticals and edible vaccines in plants. Trends Plant Sci. 6, 219–226. doi:10.1016/s1360-1385(01)01922-7
Dolan, M. C., Wu, D., Cramer, C. L., and Xu, J. F. (2014). Hydroxyproline-O-glycosylated peptide tags enhance recombinant protein yields in tobacco transient expression. Process Biochem. 49, 490–495. doi:10.1016/j.procbio.2013.12.010
Faye, L., Boulaflous, A., Benchabane, M., Gomord, V., and Michaud, D. (2005). Protein modifications in the plant secretory pathway: current status and practical implications in molecular pharming. Vaccine 23, 1770–1778. doi:10.1016/j.vaccine.2004.11.003
Gaberc-Porekar, V., Menart, V., Jevsevar, S., Vidensek, A., and Stalc, A. (1999). Histidines in affinity tags and surface clusters for immobilized metal-ion affinity chromatography of trimeric tumor necrosis factor α. J. Chromatogr. A 852, 117–128. doi:10.1016/s0021-9673(99)00374-x
Gomord, V., Fitchette, A. C., Menu-Bouaouiche, L., Saint-Jore-Dupas, C., Plasson, C., Michaud, D., et al. (2010). Plant-specific glycosylation patterns in the context of therapeutic protein production. Plant Biotechnol. J. 8, 564–587. doi:10.1111/j.1467-7652.2009.00497.x
Islam, M. R., Choi, S., Muthamilselvan, T., Shin, K., and Hwang, I. (2020). In vivo removal of N-terminal fusion domains from recombinant target proteins produced in nicotiana benthamiana. Front. Plant Sci. 11, 440. doi:10.3389/fpls.2020.00440
Islam, M. R., Kwak, J. W., Lee, J. S., Hong, S. W., Khan, M. R. I., Lee, Y., et al. (2019). Cost-effective production of tag-less recombinant protein in Nicotiana benthamiana. Plant Biotechnol. J. 17, 1094–1105. doi:10.1111/pbi.13040
Jiang, M. C., Hu, C. C., Hsu, W. L., Hsu, T. L., Lin, N. S., and Hsu, Y. H. (2020). Fusion of a novel native signal peptide enhanced the secretion and solubility of bioactive human interferon gamma glycoproteins in nicotiana benthamiana using the Bamboo mosaic virus-based expression system. Front. plant Sci. 11, 594758. doi:10.3389/fpls.2020.594758
Jiang, M. C., Hu, C. C., Lin, N. S., and Hsu, Y. H. (2019). Production of human IFNγ protein in nicotiana benthamiana plant through an enhanced expression system based on Bamboo mosaic virus. Viruses 11, 509. doi:10.3390/v11060509
Joensuu, J. J., Conley, A. J., Lienemann, M., Brandle, J. E., Linder, M. B., and Menassa, R. (2010). Hydrophobin fusions for high-level transient protein expression and purification in Nicotiana benthamiana. Plant Physiol. 152, 622–633. doi:10.1104/pp.109.149021
Kaldis, A., Ahmad, A., Reid, A., McGarvey, B., Brandle, J., Ma, S., et al. (2013). High-level production of human interleukin-10 fusions in tobacco cell suspension cultures. Plant Biotechnol. J. 11, 535–545. doi:10.1111/pbi.12041
Kapust, R. B., and Waugh, D. S. (1999). Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8, 1668–1674. doi:10.1110/ps.8.8.1668
Liu, H., and Timko, M. P. (2022). Improving protein quantity and quality-the next level of plant molecular farming. Int. J. Mol. Sci. 23, 1326. doi:10.3390/ijms23031326
Marin Viegas, V. S., Ocampo, C. G., Restucci, F. E., Vignolles, F., Mazzini, F. N., Candreva, A. M., et al. (2022). Synthesis of single-chain antibody fragment fused to the elastin-like polypeptide in Nicotiana benthamiana and its application in affinity precipitation of difficult to produce proteins. Biotechnol. Bioeng. 119, 2505–2517. doi:10.1002/bit.28158
Muthamilselvan, T., Lee, C. W., Cho, Y. H., Wu, F. C., Hu, C. C., Liang, Y. C., et al. (2016). A transgenic plant cell-suspension system for expression of epitopes on chimeric Bamboo mosaic virus particles. Plant Biotechnol. J. 14, 231–239. doi:10.1111/pbi.12377
Osman, T. A., and Buck, K. W. (1996). Complete replication in vitro of tobacco mosaic virus RNA by a template-dependent, membrane-bound RNA polymerase. J. Virol. 70, 6227–6234. doi:10.1128/JVI.70.9.6227-6234.1996
Park, S. R., Lim, C. Y., Kim, D. S., and Ko, K. (2015). Optimization of ammonium sulfate concentration for purification of colorectal cancer vaccine candidate recombinant protein ga733-FcK isolated from plants. Front. Plant Sci. 6, 1040. doi:10.3389/fpls.2015.01040
Parvez, M. K., Sehgal, D., Sarin, S. K., Basir, S. F., and Jameel, S. (2006). Inhibition of hepatitis B virus DNA replicative intermediate forms by recombinant interferon-γ. World J. Gastroenterol. 12, 3006–3014. doi:10.3748/wjg.v12.i19.3006
Puchol Tarazona, A. A., Maresch, D., Grill, A., Bakalarz, J., Torres Acosta, J. A., Castilho, A., et al. (2021). Identification of two subtilisin-like serine proteases engaged in the degradation of recombinant proteins in Nicotiana benthamiana. FEBS Lett. 595, 379–388. doi:10.1002/1873-3468.14014
Ramos-Martinez, E. M., Fimognari, L., and Sakuragi, Y. (2017). High-yield secretion of recombinant proteins from the microalga Chlamydomonas reinhardtii. Plant Biotechnol. J. 15, 1214–1224. doi:10.1111/pbi.12710
Razaghi, A., Villacrés, C., Jung, V., Mashkour, N., Butler, M., Owens, L., et al. (2017). Improved therapeutic efficacy of mammalian expressed-recombinant interferon gamma against ovarian cancer cells. Exp. Cell Res. 359, 20–29. doi:10.1016/j.yexcr.2017.08.014
Reuter, L. J., Bailey, M. J., Joensuu, J. J., and Ritala, A. (2014). Scale-up of hydrophobin-assisted recombinant protein production in tobacco BY-2 suspension cells. Plant Biotechnol. J. 12, 402–410. doi:10.1111/pbi.12147
Sareneva, T., Pirhonen, J., Cantell, K., and Julkunen, I. (1995). N-glycosylation of human interferon-γ: glycans at Asn-25 are critical for protease resistance. Biochem. J. 308 (1), 9–14. doi:10.1042/bj3080009
Sareneva, T., Pirhonen, J., Cantell, K., Kalkkinen, N., and Julkunen, I. (1994). Role of N-glycosylation in the synthesis, dimerization and secretion of human interferon-γ. Biochem. J. 303 (3), 831–840. doi:10.1042/bj3030831
Schillberg, S., Raven, N., Spiegel, H., Rasche, S., and Buntru, M. (2019). Critical analysis of the commercial potential of plants for the production of recombinant proteins. Front. Plant Sci. 10, 720. doi:10.3389/fpls.2019.00720
Schneider, J. D., Marillonnet, S., Castilho, A., Gruber, C., Werner, S., Mach, L., et al. (2014). Oligomerization status influences subcellular deposition and glycosylation of recombinant butyrylcholinesterase in <scp>N</scp>icotiana benthamiana. Plant Biotechnol. J. 12, 832–839. doi:10.1111/pbi.12184
Shen, J., Xiao, Z., Zhao, Q., Li, M., Wu, X., Zhang, L., et al. (2018). Anti-cancer therapy with TNFα and IFNγ: a comprehensive review. Cell Prolif. 51, e12441. doi:10.1111/cpr.12441
Ton-That, H., Liu, G., Mazmanian, S. K., Faull, K. F., and Schneewind, O. (1999). Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. U. S. A. 96, 12424–12429. doi:10.1073/pnas.96.22.12424
Tseng, Y. Y., Lin, F. Y., Cheng, S. F., Tscharke, D., Chulakasian, S., Chou, C. C., et al. (2015). Functional analysis of the short isoform of orf virus protein OV20.0. J. Virol. 89, 4966–4979. doi:10.1128/JVI.03714-14
Tsukiji, S., and Nagamune, T. (2009). Sortase-mediated ligation: a gift from Gram-positive bacteria to protein engineering. Chembiochem 10, 787–798. doi:10.1002/cbic.200800724
Walsh, G., and Jefferis, R. (2006). Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 24, 1241–1252. doi:10.1038/nbt1252
Wang, D., Ren, H., Xu, J. W., Sun, P. D., and Fang, X. D. (2014). Expression, purification and characterization of human interferon-gamma in Pichia pastoris. Mol. Med. Rep. 9, 715–719. doi:10.3892/mmr.2013.1812
Waugh, D. S. (2011). An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 80, 283–293. doi:10.1016/j.pep.2011.08.005
Wells, E., and Robinson, A. S. (2017). Cellular engineering for therapeutic protein production: product quality, host modification, and process improvement. Biotechnol. J. 12. doi:10.1002/biot.201600105
Wilken, L. R., and Nikolov, Z. L. (2012). Recovery and purification of plant-made recombinant proteins. Biotechnol. Adv. 30, 419–433. doi:10.1016/j.biotechadv.2011.07.020
Wu, J., and Filutowicz, M. (1999). Hexahistidine (His6)-tag dependent protein dimerization: a cautionary tale. Acta. Biochim. Pol. 46, 591–599. doi:10.18388/abp.1999_4131
Xu, J., Okada, S., Tan, L., Goodrum, K. J., Kopchick, J. J., and Kieliszewski, M. J. (2010). Human growth hormone expressed in tobacco cells as an arabinogalactan-protein fusion glycoprotein has a prolonged serum life. Transgenic Res. 19, 849–867. doi:10.1007/s11248-010-9367-8
Xu, J., Tan, L., Goodrum, K. J., and Kieliszewski, M. J. (2007). High-yields and extended serum half-life of human interferon α2b expressed in tobacco cells as arabinogalactan-protein fusions. Biotechnol. Bioeng. 97, 997–1008. doi:10.1002/bit.21407
Yadav, D. K., Yadav, N., Yadav, S., Haque, S., and Tuteja, N. (2016). An insight into fusion technology aiding efficient recombinant protein production for functional proteomics. Arch. Biochem. Biophys. 612, 57–77. doi:10.1016/j.abb.2016.10.012
Zhang, B., Rapolu, M., Huang, L., and Su, W. W. (2011). Coordinate expression of multiple proteins in plant cells by exploiting endogenous kex2p-like protease activity. Plant Biotechnol. J. 9, 970–981. doi:10.1111/j.1467-7652.2011.00607.x
Zhang, N., Dolan, M., Wu, D., Phillips, G. C., and Xu, J. (2016). Dramatic secretion of recombinant protein expressed in tobacco cells with a designer glycopeptide tag is highly impacted by medium composition. Plant Cell Rep. 35, 2513–2522. doi:10.1007/s00299-016-2051-6
Zhang, N., Wright, T., Wang, X., Karki, U., Savary, B. J., and Xu, J. (2019). Engineering 'designer' glycomodules for boosting recombinant protein secretion in tobacco hairy root culture and studying hydroxyproline-O-glycosylation process in plants. Plant Biotechnol. J. 17, 1130–1141. doi:10.1111/pbi.13043
Keywords: viral vector, therapeutic protein, Bamboo mosaic virus (BaMV), cleavable peptide, tag-free IFNγ, antivirus activity
Citation: Jiang M-C, Hsu W-L, Tseng C-Y, Lin N-S, Hsu Y-H and Hu C-C (2024) Development of a tag-free plant-made interferon gamma production system with improved therapeutic efficacy against viruses. Front. Bioeng. Biotechnol. 11:1341340. doi: 10.3389/fbioe.2023.1341340
Received: 20 November 2023; Accepted: 21 December 2023;
Published: 11 January 2024.
Edited by:
Zhen Fang, Jiangsu University, ChinaCopyright © 2024 Jiang, Hsu, Tseng, Lin, Hsu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yau-Heiu Hsu, eWhoc3VAbmNodS5lZHUudHc=; Chung-Chi Hu, Y2NodUBkcmFnb24ubmNodS5lZHUudHc=