
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 14 December 2023
Sec. Industrial Biotechnology
Volume 11 - 2023 | https://doi.org/10.3389/fbioe.2023.1291245
 Suzanne F. Dagher1Asmita Vaishnav2
Suzanne F. Dagher1Asmita Vaishnav2 Christopher B. Stanley3
Christopher B. Stanley3 Flora Meilleur4,5Brian F. P. Edwards2
Flora Meilleur4,5Brian F. P. Edwards2 José M. Bruno-Bárcena1*
José M. Bruno-Bárcena1*Hamamotoa (Sporobolomyces) singularis codes for an industrially important membrane bound ß-hexosyltransferase (BHT), (BglA, UniprotKB: Q564N5) that has applications in the production of natural fibers such as galacto-oligosaccharides (GOS) and natural sugars found in human milk. When heterologously expressed by Komagataella phaffii GS115, BHT is found both membrane bound and soluble secreted into the culture medium. In silico structural predictions and crystal structures support a glycosylated homodimeric enzyme and the presence of an intrinsically disordered region (IDR) with membrane binding potential within its novel N-terminal region (1–110 amino acids). Additional in silico analysis showed that the IDR may not be essential for stable homodimerization. Thus, we performed progressive deletion analyses targeting segments within the suspected disordered region, to determine the N-terminal disorder region’s impact on the ratio of membrane-bound to secreted soluble enzyme and its contribution to enzyme activity. The ratio of the soluble secreted to membrane-bound enzyme shifted from 40% to 53% after the disordered N-terminal region was completely removed, while the specific activity was unaffected. Furthermore, functional analysis of each glycosylation site found within the C-terminal domain revealed reduced total secreted protein activity by 58%–97% in both the presence and absence of the IDR, indicating that glycosylation at all four locations is required by the host for the secretion of active enzyme and independent of the removed disordered N-terminal region. Overall, the data provides evidence that the disordered region only partially influences the secretion and membrane localization of BHT.
Hamamotoa (Sporobolomyces) singularis (H. singularis) expresses, under inducible conditions, a unique extracellular membrane-bound glycosylated ß-hexosyltransferase (BHT) (Gorin et al., 1964a; Gorin et al., 1964b). This distinctive membrane-bound enzyme has maintained biological interest for the last 60 years and is known by many names (BglA, UniprotKB: Q564N5) (Phaff and Carmo-Sousa, 1962; Gorin et al., 1964a; Gorin et al., 1964b; Gorin et al., 1964b; Blakely and MacKenzie, 1969; Ishikawa et al., 2005). In nature, H. singularis BHT functions as a glucosyl hydrolase that catalyzes the hydrolysis of cellobiose ß-(1–4) glycosidic linkages. Interestingly, BHT also has ß-galactosidase activity, demonstrated by its ability to cleave lactose (β-D-galactopyranosyl-(1→4)-α-D-glucopyranose). However, unlike ß-galactosidases, H. singularis BHT simultaneously carries out hydrolase and galactosyl transferase activities, converting lactose (independent of initial lactose concentration) to galacto-oligosaccharides (GOS) without extracellular accumulation of galactose (Gorin et al., 1964a; Gorin et al., 1964b; Blakely and MacKenzie, 1969) and to natural sugars found in human milk (Arnold et al., 2021b). These fibers are regarded as prebiotic components that have physiological effects on the make-up and functioning of the gut microbiota, thereby benefiting the health of the host (Torres et al., 2010; Azcárate-Peril et al., 2011; Bruno-Barcena and Azcarate-Peril, 2015; Monteagudo-Mera et al., 2016; Arnold et al., 2018; Panesar et al., 2018; Watson et al., 2019; Whittington et al., 2019; Arnold et al., 2021a; Arnold et al., 2021b). Due to its numerous health benefits, GOS are widely utilized as functional food additives on a global scale (Spherix Consulting Inc, 2010).
The 594-residue polypeptide that makes up the enzyme (BHT) has two distinct regions: the C-terminal domain, which is homologous to other glycosyl hydrolase family 1 (GH1) members, and the N-terminal section (residues 1–110), which is unique to BHT and has no homology to any known glycosyl transferases or β-glucosidases (Dagher et al., 2013; Dagher and Bruno-Bárcena, 2016). We previously showed the presence of an active 1–22 signal sequence with a membrane anchor signature inside the 110 N-terminal region using in silico analysis and subsequent functional studies using K. phaffii GS115 for secretion (Dagher and Bruno-Bárcena, 2016). In that study, the 1–22 signal sequence was replaced with MFα, which resulted in a 10-fold increase in the amount of secreted catalytically active rBHT in the culture broth compared to expression of full-length rBHT which remained membrane-bound. Surprisingly, the bulk of rBHT remained affixed to the K. phaffii GS115 membrane rather than being fully transferred to the medium (Dagher et al., 2013; Dagher and Bruno-Bárcena, 2016).
In silico analysis also revealed that the N-terminal region comprises regions of low complexity that have yet to be defined and characterized (Dagher et al., 2013; Dagher and Bruno-Bárcena, 2016). Furthermore, the crystal structures solved by Uehara et al., 2020 (HsBglA, PDB: 6M4E) (Uehara et al., 2020) and in this study (BHT, PDB: 7L74) showed a potential for an intrinsically disordered region (IDR) within the N-terminus. IDRs are flexible and extended protein segments known to lack organized secondary structure under physiological conditions. However, their biological function depends on this unstructured state (Uversky, 2019). Intrinsically disordered proteins (IDPs) exist in interchanging conformations rather than adapting well-defined structures as previously reviewed (Uversky, 2019). This is consistent with IDRs’ functional advantages and ability to fold in response to partner contact or in a template-dependent manner (Darling and Uversky, 2018).
It has been demonstrated that proteins with large stretches of IDRs are essential elements for membrane interactions because these flexible areas allow for protein-protein or protein-lipid interactions, great selectivity and low affinities for key components of signal transduction cascades (Cornish et al., 2020). Additionally, membrane attachment constricts the protein’s search space, consequently membrane localization can increase the effective concentration while simultaneously acting as a steric barrier to prevent interactions from occurring in solution (Cornish et al., 2020).
Proteome-wide investigations have shown connections between IDRs and several post-translational modifications (PTMs), including acetylation, methylation, and glycosylation (Gao and Xu, 2012). Previous studies by us and others in which E. coli was unable to express active rBHT suggested the critical importance of PTMs for appropriate folding and/or enzymatic activity (Ishikawa et al., 2005; Dagher and Bruno-Bárcena, 2016). One of the most important post-translational modifications of proteins is glycosylation, which primarily involves the attachment of glycans to the nitrogen atom of asparagine residues (N-linked) or to the hydroxyl oxygen of serine, threonine, or tyrosine residues (O-linked). Other important post-translational modifications of proteins include C-mannosylation, phospho-serine glycosylation, and glypiation (formation of GPI anchors) (Prabakaran et al., 2012; Darling and Uversky, 2018). In K. phaffii GS115, N-glycans form high-mannose-type heterogeneous oligosaccharides beginning with the addition of the core unit Glc3Man9GlcNAc2 (Glc = glucose; GlcNAc = N-acetylglucosamine; Man = mannose) at asparagine in the recognition sequence Asn-X-Ser/Thr X≠P (Bretthauer and Castellino, 1999). N-glycosylation has been shown to influence enzymatic activity, stability, and cell surface expression as previously reviewed (Ge et al., 2018).
Further investigations into the intricacies of the structure of this enzyme are therefore needed to provide suggestions on how to enhance soluble secretion of rBHT. In this study we conducted a detailed kinetic analysis of rBHT variants lacking progressive portions of the IDR, in comparison to the full-length enzyme. To evaluate the impacts on protein secretion and enzyme activity, this study looked at modifications in the IDR length, N-glycosylation sites, and dimer stability. The results provide insight into the dynamics of the IDR related to enzyme secretion and localization of active rBHT generated by K. phaffii GS115.
The bacterial and K. phaffii GS115 strains used in this study are shown in Table 1. Bacteria were grown at 37°C in Luria-Bertani (LB) medium with antibiotic ampicillin (100 μg/mL) (Thermo Fisher Scientific). Growth and maintenance of GS115 (Invitrogen Life Technologies, Thermo Fisher Scientific) was described previously (Dagher and Bruno-Bárcena, 2016). E. coli XL1-Blue was used as the cloning host (Agilent Technologies, Thermo Fisher Scientific). The plasmid pPIC9 (Invitrogen Life Technologies, Thermo Fisher Scientific) was used as cloning vector containing codon optimized Bht (rBht sequences) (GenBank accession number JF29828).
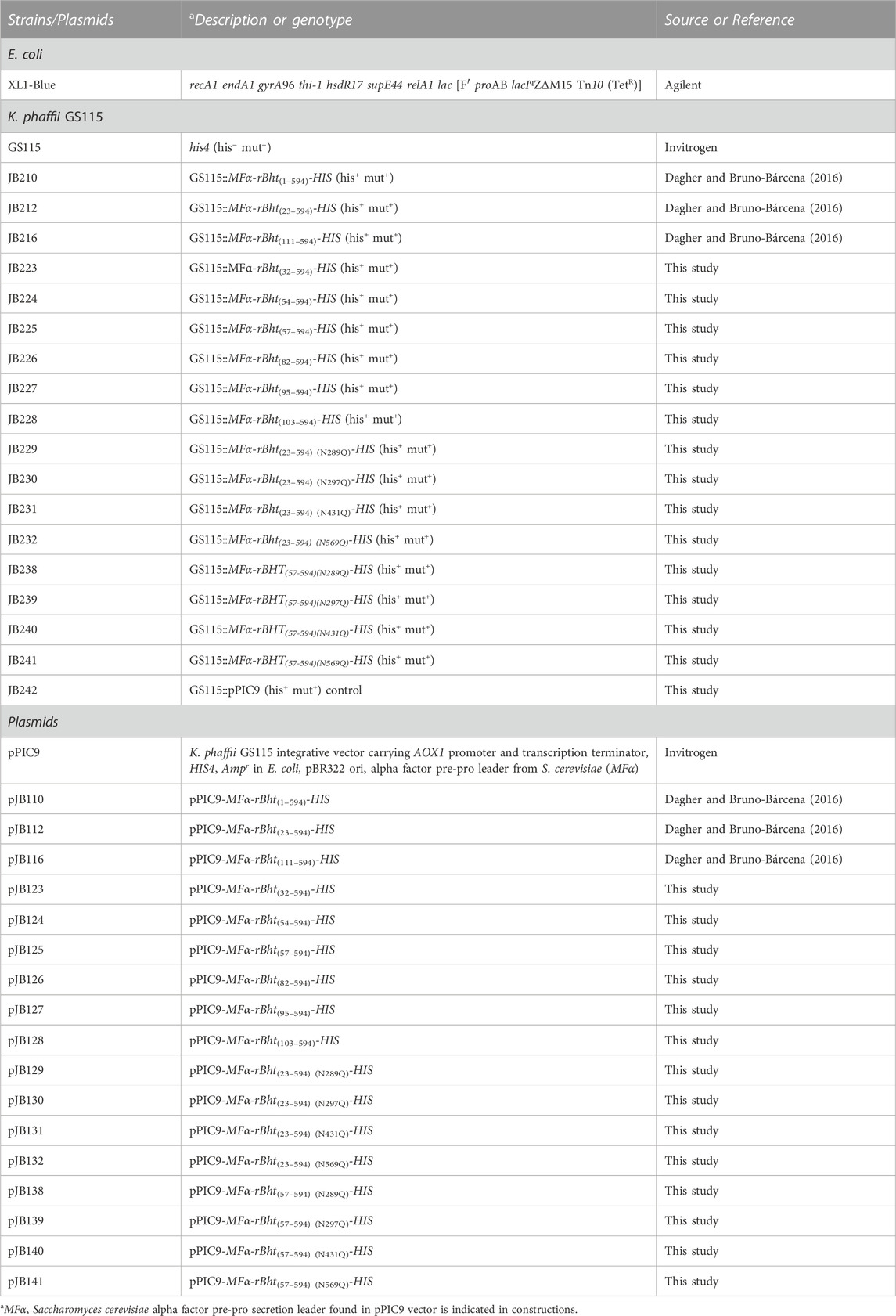
TABLE 1. Strains and plasmids used in this study.
All molecular biology protocols were carried out as previously described (Dagher and Bruno-Bárcena, 2016). Briefly, expression by K. phaffii GS115 was achieved by homologous integration of DNA fragments bearing rBht sequences, for example, coding for mutations and truncations.
The truncated rBht sequences were generated by PCR amplification using pJB110 (pPIC9-MFα-rBht(1–594)-HIS) as template. Primers were purchased from Integrated DNA Technologies (IDT Coralville, IA, USA) (listed in Table 2). When appropriate, the primers included restriction sites to facilitate cloning (Table 2). Briefly, primer pairs for sequences coding for the N-terminal truncated rBht sequences encoding protein sizes included the following: 23–594 (primers: JBB7/JBB5), 32–594 (primers: JBB21/JBB5), 54–594 (primers: JBB22/JBB5), 57–594 (primers: JBB23/JBB5), 82–594 (primers: JBB24/JBB5), 95–594 (primers: JBB25/JBB5) and 103–594 (primers: JBB26/JBB5). PCR amplicons were digested with XhoI-NotI and cloned into pPIC9 (Invitrogen Life Technologies, Thermo Fisher Scientific) generating pJB112 (pPIC9-MFα-rBht(23–594)-HIS), pJB123 (pPIC9-MFα-rBht(32–594)-HIS), pJB124 (pPIC9-MFα-rBht(54–594)-HIS), pJB125 (pPIC9-MFα-rBht(57–594)-HIS), pJB126 (pPIC9-MFα-rBht(82–594)-HIS), pJB127 (pPIC9-MFα-rBht(95–594)-HIS) and pJB128 (pPIC9-MFα-rBht(103–594)-HIS) respectively.
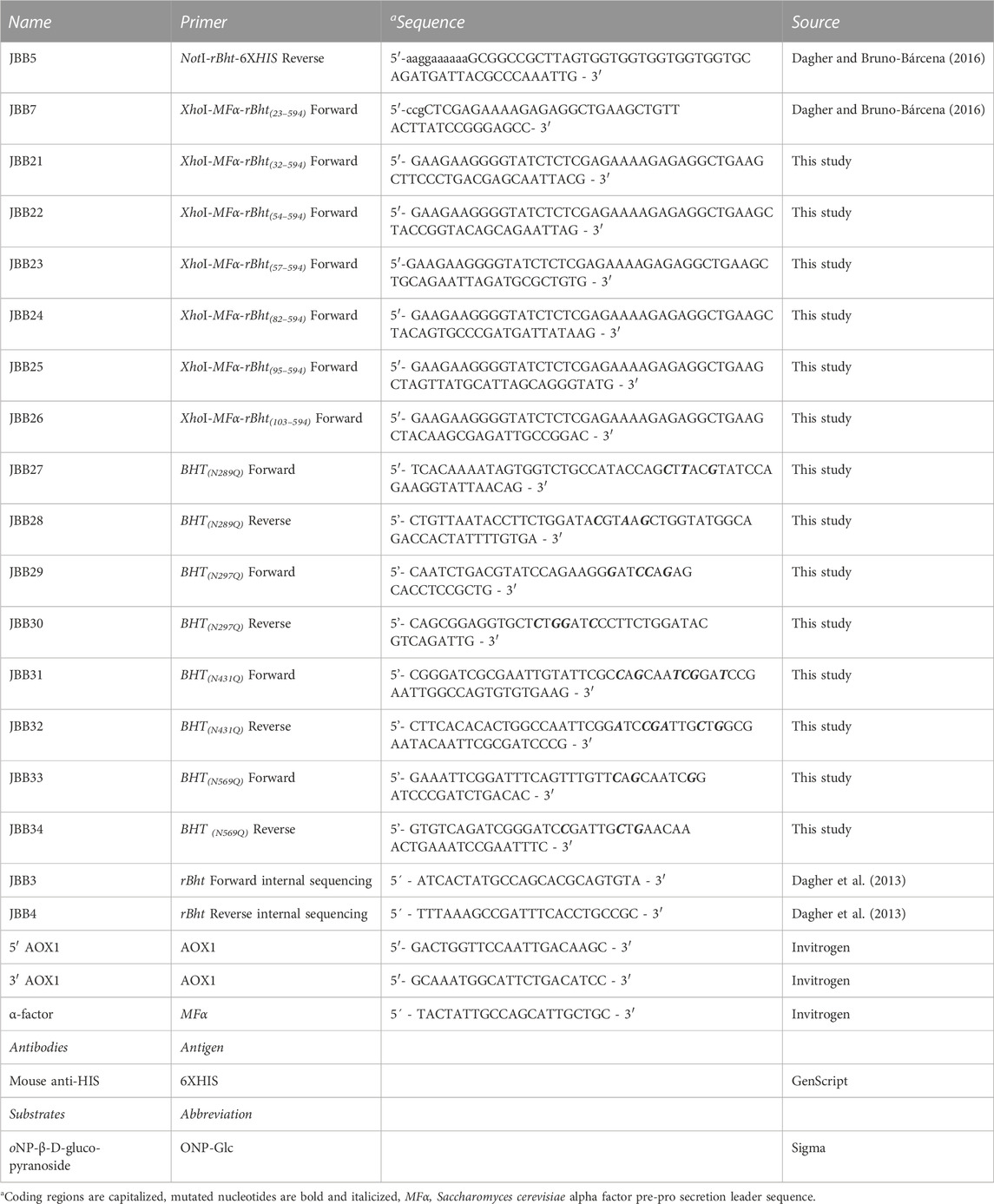
TABLE 2. Primers, antibodies, and substrates used in this study.
Site directed mutagenesis was performed using complementary oligonucleotides designed to incorporate the desired base changes using QuickChange site directed mutagenesis kit (Agilent Technologies Santa Clara, CA, USA) according to manufacturer’s instructions. The generated variants include single amino acid exchanges replacing asparagine for glutamine in putative N-glycosylation sites using as template pJB112 (pPIC9-MFα-rBht(23–594)-HIS). The residues modified include positions; N289Q (primers: JBB27/JBB28) to generate pJB129 (pPIC9-MFα-rBht(23–594) (N289Q)-HIS), N297Q (primers: JBB29/JBB30) to generate pJB130 (pPIC9-MFα-rBht(23–594) (N297Q)-HIS), N431Q (primers: JBB31/JBB32) to generate pJB131 (pPIC9-MFα-rBht(23–594) (N431Q)-HIS), and N569Q (primers: JBB33/JBB34)) to generate pJB132 (pPIC9-MFα-rBht(23–594) (N569Q)-HIS) (Dagher and Bruno-Bárcena, 2016) (Table 1). Next, the set of primers JBB23/JBB5 were used on each sequence to obtain plasmids pJB138 (pPIC9-MFα-rBht(57–594) (N289Q)-HIS), pJB139 (pPIC9-MFα-rBht(57–594) (N297Q)-HIS), pJB140 (pPIC9-MFα-rBht(57–594) (N431Q)-HIS) and pJB141 (pPIC9-MFα-rBht(57–594) (N569Q)-HIS).
As described above PCR amplicons were digested with XhoI-NotI and cloned into pPIC9 (Invitrogen Life Technologies, Thermo Fisher Scientific). DNA fragments from restriction enzyme digests were purified from agarose gels using QIAquick gel extraction kit (Qiagen, Hilden, Germany). All mutations were confirmed with restriction digests for detecting restriction sites in primers and by Sanger sequencing performed by the Azenta Life Sciences (USA) using primers JBB3, JBB4, 5′ AOX1, 3’ AOX1 and α-factor (Table 2).
K. phaffii GS115 was transformed with linearized plasmids as per the Invitrogen Pichia Expression Kit manual (Invitrogen, USA). Plasmid integration and Mut+ phenotype in histidine positive colonies was confirmed by sequencing PCR products generated by primers 5′ AOX1 and 3’ AOX1(Invitrogen Pichia expression kit). Single copy integration was confirmed as previously described (Dagher and Bruno-Bárcena, 2016).
Expression and purification have been described previously (Dagher and Bruno-Bárcena, 2016). Briefly, filtered culture media was purified using the ÄKTApurifier and HISTrap™ HP Nickel column (GE Healthcare, Life sciences). The purified proteins were quantified by Bradford protein assay (Thermo Fisher Scientific) (Bradford, 1976).
Proteins were analyzed by SDS-PAGE using 10% resolving gels and visualized using Coomassie and silver stains (Bio-Rad, Hercules, CA). Immunoblots were probed with 1:10,000 dilution of anti-HIS antibody (GenScript, Piscataway, NJ) followed by 1:10,000 dilution of alkaline phosphatase conjugated goat anti-mouse antibody (GenScript, Piscataway, NJ). Detection was carried out with 1-Step™ NBT/BCIP Substrate Solution according to manufacturer’s instructions (Thermo Fisher Scientific).
Hydrolysis of o-nitrophenyl-β-D-glucopyranoside (ONP-Glc) was followed by measurement of absorbance at 405 nm for determination of β-glucosidase activity using the methods described previously (Dagher and Bruno-Bárcena, 2016). Briefly, cells were harvested by centrifugation (5,000 g at 4°C), to separate soluble rBHT from membrane bound rBHT. The cells were then washed two times with 50 mM phosphate-citrate buffer (pH 5). Assays on soluble secreted and membrane bound rBHT were performed in a 50 mM phosphate-citrate buffer under optimal temperature of 42°C and optimal pH 5 for 10 min. Reactions were stopped by the addition of an equal volume of 0.25 M sodium carbonate and the absorbance was measured at 405 nm.
The Michaelis-Menten constants (Km and Vmax) of 0.3 µg rBHT (at 42°C) were determined by varying ONP-Glc from 0 to 10.4 mM in 50 mM phosphate-citrate buffer (pH 5) and measuring the initial reaction rate at 20°C, 30°C, 42°C, and 55°C. The kinetic constants at each temperature were determined with OriginPro 7.5 using nonlinear regression of the Hill equation with a Hill coefficient of 1.
Secondary structure consensus prediction of BHT was performed at the PSIPRED server (protein structure prediction) (Jones, 1999; Cozzetto et al., 2016; Buchan and Jones, 2019) and at the NPS@server (network protein sequence analysis) (Combet et al., 2000). The signal sequence was predicted using the SignalP 5.0 algorithm (Almagro Armenteros et al., 2019). Protein disorder was predicted using the consensus of six methods, Dispred3 (Jones and Cozzetto, 2015), Phyre2 (Kelley et al., 2015), IUPred2A (Meszaros et al., 2018), PONDR-VSL2 (Peng et al., 2005) and GlobPlot (prediction of protein disorder and globularity) (Jones, 1999; Linding et al., 2003), PHYRE2 (Kelley et al., 2015). Domain boundaries were predicted using the DomPred server (Bryson et al., 2007) and Pfam version 32.0 (El-Gebali et al., 2018).
BHT N- and O-glycosylation site prediction was performed at the GlycoEP server (Chauhan et al., 2013).
BHT phosphorylation site prediction was performed using DEPP (Disorder enhanced phosphorylation predictor), also known as DisPhos1.3 (http://www.dabi.temple.edu/disphos/) (Iakoucheva et al., 2004) and NetPhosYeast1.0 (http://www.cbs.dtu.dk/services/NetPhosYeast/) (Ingrell et al., 2007).
Structural figures and structural superimpositions were generated in PyMOL (http://www.schrodinger.com/pymol/) (Schrodinger, 2022). A homodimer is present in the crystal asymmetric unit; however, the monomer was considered for structural analysis.
rBHT(23–594)-HIS was further purified by gel filtration chromatography on a Sephacryl S-300 (GE Healthcare, Life Sciences) column equilibrated with 100 mM Tris pH 7.5, 200 mM sodium chloride, 1 mM dithiothreitol to reduce aggregates and concentrated to 6 mg/mL using Amicon® Ultra 15, molecular weight cut-off 10,000 (Millipore Sigma) in 10 mM HEPES pH 7.5. Protein concentrations were determined by Lowry method (Lowry et al., 1951) using bovine serum albumin as a standard. The crystals were grown by vapor diffusion using the sitting drop method. The crystals were grown using a crystallization solution made by mixing 1 µL (10 μg/μL) purified protein with 1 µL of precipitant solution (35% polyethylene glycol 4k, 0.1 M HEPES pH 7.5, 0.2 M calcium chloride) and equilibrating the drop against 0.5 mL of the precipitant at 22°C–23°C. Crystals usually appeared in less than a week. Prior to data collection, the crystals were soaked for 10 min in a cryoprotectant solution (35% polyethylene glycol 4k, 0.1 M HEPES pH 7.5, 0.2 M calcium chloride, 20% ethylene glycol) and then immediately flash-vitrified in liquid nitrogen.
Single crystal diffraction data were collected at the Life Sciences Collaborative Access Team facility (Advanced Photon Source sector 21, Argonne National Laboratory (Lemont, IL, USA) on beamline 21G (Table 3). The data covered 360° in 0.5-degree increments. The frames were integrated with XDS (Kabsch, 2010) and scaled with Aimless (Evans and Murshudov, 2013) in AutoProc (Vonrhein et al., 2011). The structure was solved by molecular replacement in PHENIX (Adams et al., 2010) using a homology model generated by RaptorX (Källberg et al., 2012). The structure was rebuilt and refined with PHENIX and then optimized with PDB-REDO (Joosten et al., 2014). Coot (Emsley et al., 2010) was used to add and to optimize individual residues, posttranslational modifications and ligands.
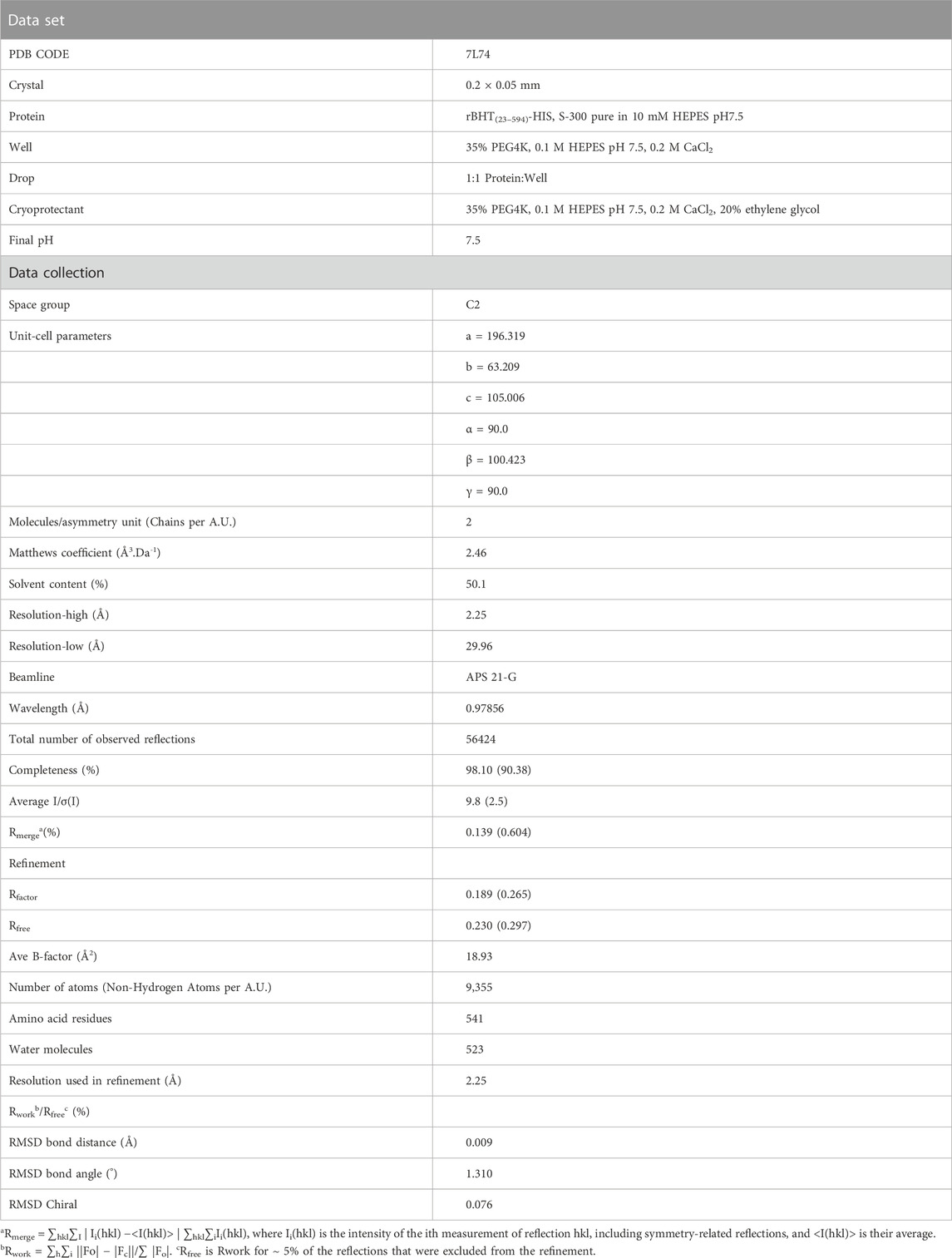
TABLE 3. Data collection and refinement statistics of the structure of rBHT(23–594)-HIS at pH 7.5.
When expressed by K. phaffii GS115, the rBHT variant (rBHT(23–594)-6XHIS) is functionally independent of its location either associated with the membrane or soluble (Dagher et al., 2013; Dagher and Bruno-Bárcena, 2016). To gain insights into the structure-function characteristics, we used soluble rBHT(23–594)-6XHIS to solve the crystal structure. Data processing and refinement statistics are presented in Table 3 and the final model was deposited in the Protein Data Bank (PDB: 7L74). The structure was solved by molecular replacement at a resolution of 2.25 Å and the asymmetric unit contains two molecules of rBHT(23–594)-6XHIS and the value Vm was estimated to be 2.46 Å3.Da-1.
rBHT folds into two domains, the N-domain, and the C-domain. The structure for enzymatic activity in the C-terminal region is composed of a sugar-binding catalytic domain organized in a (α/β)8 TIM barrel that stretches from residue 116 to residue 547 (BHT, PDB: 7L74). Eight parallel β-strands comprise the core BHT (α/β)8 barrel, which is coupled to eight external α-helices that is common to Glycoside Hydrolase Family I (GH1) members (Henrissat et al., 1995; Glycoside Hydrolase Family, 2012).
The initial portion of the N-domain (residues 23–53) upstream of the carboxy GH1 domain, lacked electron density and could not be modelled indicating the presence of a potential N-terminal intrinsically disordered region (IDR). Additional support for the IDR within the N-terminal domain comes from a previously solved crystal structure (HsBglA, PDB: 6M4E) (Uehara et al., 2020). HsBglA and rBHT crystal structures show minor structural differences with an RMSD across all Cα pairs of 0.22 Å.
The relevance of the disordered regions in membrane lipid association and interactions of membrane associated proteins can only be understood by examining the properties of the interacting environment (Mohammad et al., 2019; Cornish et al., 2020; Csizmadia et al., 2021). This can be complicated by the multiple interactions and functions exhibited by disordered regions (Theillet et al., 2013; Uversky, 2019) and their ability to fold upon contact in a template-dependent manner or with specific ligand partners (Bürgi et al., 2016).
To make the IDR structure accessible for systematic analysis, IDR boundaries and PTM predictions needed to be made to reveal incomplete regions, which is particularly important for IDR analysis as described and shown below (Figure 1). This compelled us to perform a series of in silico structural predictions by using five available prediction tools over the full length of BHT (Figure 1). By combining different disorder predictors we expect to reinforce the reliability of the predicted regions since they use different definitions of disorder (Lieutaud et al., 2016). For example, PSIPRED and Globplot methods were employed to strengthen the lack of secondary structure and globular domains in the IDR region. Upon comparison, the disorder datasets derived from Phyre2, IUPred2A, DISOPRED3, Globplot Disorder and PONDR indicate probable disorder boundaries throughout the unique 1–110 N-terminal region and a common overlapping boundary at residues 52–53 (Figure 1), in agreement with PDB: 7L74 N-terminal boundary lacking electron density.
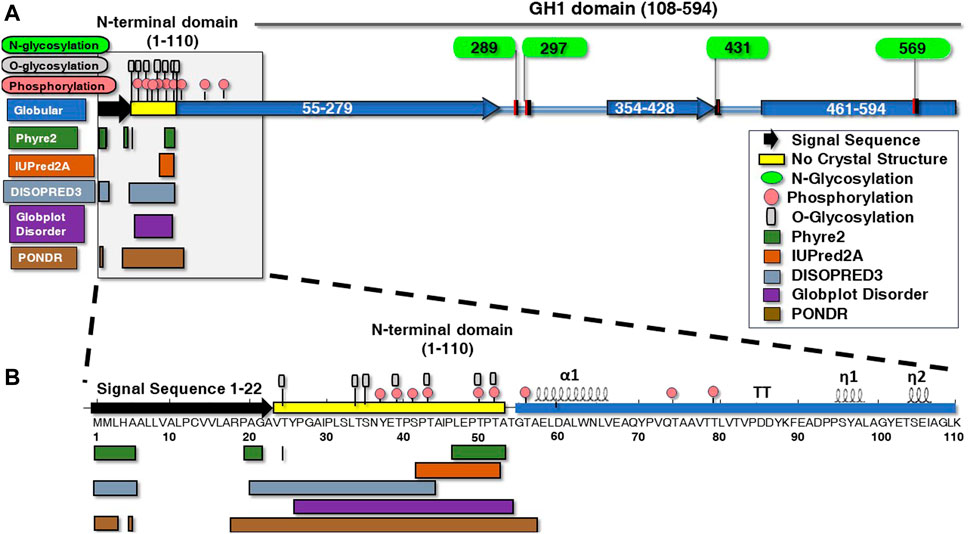
FIGURE 1. Structural posttranslational modifications and disordered versus ordered secondary motifs of ß-hexosyltransferase from H. singularis. (A) BHT protein glycosylation, phosphorylation and secondary structures were predicted in the amino terminus using various algorithms. Depicted are the structural elements, conserved regions, and functional domains of BHT using PSIPRED and Globplot Globular prediction tools (blue rectangles). GlycoEP displays N-glycosylation (green ovals) (N289, N297, N431, N569). (B) The N-terminal domain (1–110) has been expanded along with predicted glycosylation, phosphorylation, and secondary structures. Numbers indicate BHT amino acid residue number. The secondary structure elements of BHT shown above the amino acid residues was generated with ENDscript (Robert and Gouet, 2014) (https://endscript.ibcp.fr). Disordered regions within the N-terminal domain were predicted using algorithms Phyre2 (1–5, 20–21, 24, 47–53), IUPRED2A (42–52), DISOPRED3 (1–5, 21–43) Globplot Disorder (26–54), and PONDR (1–3, 5, 18–57). Phosphorylation servers DisPhos1.3 and NetPhosYeast1.0 predicted phosphorylation sites within the disordered region (pink circles) (37, 39, 41, 43, 50, 52). GlycoEP displays O-glycosylation (gray rectangles) (24, 34, 35, 39, 43, 50, 52), while no N-glycosylation or C-mannosylation sites were predicted.
IDRs often contain a substantial degree of post-translational modifications (PTMs) such as phosphorylation, glycosylation, ubiquitination, acylation, and others that mediate potential interactions with high specificity (van der Lee et al., 2014; Cornish et al., 2020). For instance, phosphorylation can stabilize the tertiary structural organization of the IDR while enhancing and stabilizing its binding to the protein’s ligand (Gsponer et al., 2008; Nishi et al., 2011). Bioinformatic analysis has suggested that this function is tunable by PTMs and correlated with a high content of serine, threonine, glutamine and asparagine (Chuang et al., 2020).
The BHT IDR (resides 23–53) is composed of 29.1% serine and threonine residues that may act as potential phosphorylation/O-glycosylation sites. This finding is significant since phosphorylation is thought to function as an electrostatic switch by reducing the net charge thereby reducing membrane interactions (Aivazian and Stern, 2000; Cornish et al., 2020). O-linked glycosylation is also predicted to populate the BHT IDR (T24, T34, S35, T39, T43, T50, T52) (Figure 1B) where it likely functions along with phosphorylation (Y37, T39, S41, T43, T50, T52) to protect the region from proteolysis (Nishikawa et al., 2010; Prates et al., 2018).
It is known that disordered proteins often display a compositional bias toward polar residues and depleted of hydrophobic amino acids (Uversky, 2019). The disorder promoting residues are known to include aspartic acid, methionine, lysine, arginine, serine, glutamine, glycine, alanine, proline, and glutamic acid and commonly found on the surface of proteins (Theillet et al., 2013; Uversky, 2019). The BHT disordered region (residues 23–53) is made up of 3.2% valine, 9.7% alanine, 6.5% isoleucine, 9.7% leucine, 6.5% tyrosine, 3.2% glycine, 22.6% proline, 3.2% asparagine, 6.5% glutamic acid, 9.7% serine, and 19.4% threonine (Figure 1B). Surprisingly, hydrophobic amino acids make up 35.6% of the BHT IDR (Figure 1B).
Biologically important disordered regions have also been known as N-terminal fusion carriers to promote protein folding, act in folding quality control and thus enhance protein solubility. Our approach was to utilize the in silico analysis (Figure 1B) to perform progressive and selective deletions of the predicted IDR and to determine their impact on soluble secretion of catalytically active rBHT. Predictions were also made by the algorithm DisPhos1.3 (DEPP) that uses disorder information to help improve and discriminate between phosphorylation and non-phosphorylation sites (Materials and Methods). Furthermore, the accuracy of DEPP reaches 76.0±0.3%, 81.3±0.3% and 83.3±0.3% for serine, threonine, and tyrosine respectively (Iakoucheva et al., 2004; Ingrell et al., 2007).
The high percentage of serine and threonine (29.1%) residues in the IDR (Figure 1B) provided the opportunity for phosphorylation as well as o-glycosylation and formed the basis for our deletion analysis strategy within the IDR. Most putative phosphorylation and o-glycosylation sites are “nested” between residues 32–54. Therefore, the first truncation involved the removal of amino acids 1–31 (rBHT(32–594)-HIS), prior to the nest of putative PTM sites. While removal of amino acids 1–53 (rBHT(54–594)-HIS) completely removed the putative PTM nest. The 1st phosphorylation site found in the crystal structure (T56) was also removed rBHT(57–594)-HIS. The final 2 phosphorylation sites (T74 and T79) and α1 alpha helix were eliminated by removal of residues 1–81 (rBHT(82–594)-HIS). Finally, removal of residues 1 to 94 (rBHT(95–594)-HIS), and 1 to 102 (rBHT(103–594)-HIS) eliminated the β-turn (TT) and 310 helix random coil (η1), respectively. Removal of the entire unique N-terminal residues 1–110 (rBHT(111–594)-HIS) was previously described (Dagher and Bruno-Bárcena, 2016). A schematic representation of the complete rBHT and rBHT-truncated variants is illustrated in Figure 2A.
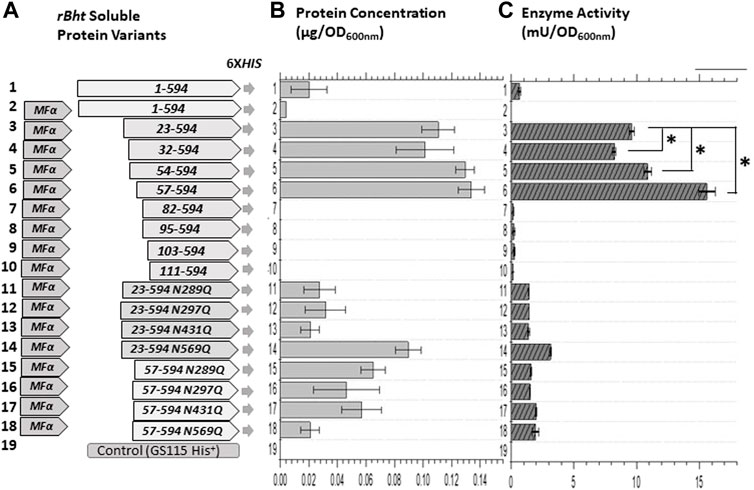
FIGURE 2. Enzyme activity comparisons of K. phaffii GS115 strains carrying sequences of rBht-HIS under the AOX1 promoter. (A) Graphic representations of chimeric genes generated containing combinations of leader domains and ORFs of rBht-HIS sequences. Specific tags, mutations and deletions are indicated. (B) Protein concentration of soluble secreted protein normalized for the final culture (OD600nm). (C) Enzymatic activity of the secreted protein normalized for the final culture (OD600nm). The following recombinant strains were compared: row 1, GS115::rBht(1–594)-HIS; row 2, GS115::MFα-rBht(1–594)-HIS; row 3, GS115::MFα-rBht(23–594)-HIS; row 4, GS115::MFα-rBht(32–594)-HIS; row 5, GS115::MFα-rBht(54–594)-HIS; row 6,GS115::MFα-rBht(57–594)-HIS; row 7, GS115::MFα-rBht(82–594)-HIS; row 8, GS115::MFα-rBht(95–594)-HIS; row 9, GS115::MFα-rBht(103–594)-HIS; row 10, GS115::MFα-rBht(111–594)-HIS; row 11, GS115::MFα-rBht(23–594) (N289Q)-HIS; row 12, GS115::MFα-rBht(23–594) (N297Q)-HIS; row 13, GS115::MFα-rBht(23–594) (N431Q)-HIS; row 14, GS115::MFα-rBht(23–594) (N569Q)-HIS; row 15, GS115::MFα-rBht(57–594) (N289Q)-HIS; row 16, GS115::MFα-rBht(57–594) (N297Q)-HIS; row 17, GS115::MFα-rBht(57–594) (N431Q)-HIS; row 18, GS115::MFα-rBht(57–594) (N569Q)-HIS; row 20, GS115 (His+) control. Error bars represent standard deviations from the means of three replicates. A single asterisk indicates a statistically significant increase was observed between two activity values (p < 0.05).
Methanol induced protein expression of each variant by K. phaffii GS115, for both membrane associated and soluble enzymes were evaluated as previously described (Dagher and Bruno-Bárcena, 2016). The presence of rBHT truncated protein variants in the broth (Figure 2B) was initially inspected by Coomassie stained SDS-PAGE (Figure 3A) followed by Western blot analysis (Figure 3B). Variants rBHT(23–594)-HIS, rBHT(32–594)-HIS, rBHT(54–594)-HIS and rBHT(57–594)-HIS were clearly detectable by Coomassie stain (Figure 3A) and Western blot (Figure 3B). While removal of additional fragments within the unique 110 region (rBHT(82–594)-HIS, rBHT(95–594)-HIS, rBHT(103–594)-HIS, and rBHT(111–594)-HIS) did not render detectable activity or protein using Coomassie stain or Western blot, indicating residues downstream of 57 (non-IDR regions) were important for processing secreted protein. In agreement with previous results, full length rBHT(1–594)-HIS variant was barely visible by Western blot (Dagher and Bruno-Bárcena, 2016). K. phaffii GS115 transformed with an empty pPIC9 vector was utilized as the negative control.
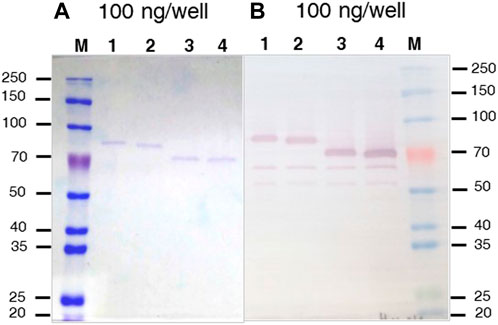
FIGURE 3. Coomassie stained SDS-PAGE and Western blot of rBHT-HIS deletion variants (A) SDS-PAGE (10%) stained with Coomassie blue. (B) Western blot exposed to anti-HIS antiserum of separated proteins. The Figures show protein cell free extracts (soluble secreted proteins) of K. phaffii GS115 expressing different recombinant BHT constructs generated by; lane 1, GS115::MFα-rBht(23–594)-HIS; lane 2, GS115::MFα-rBht(32–594)-HIS; lane 3, GS115::αMF-rBht(54–594)-HIS, lane 4, GS115::αMF-rBht(57–594)-HIS. Equal amounts (100 ng) were loaded in each lane to aid in the comparison. M indicates lane containing the molecular weight protein markers (Thomas Scientific, Swedesboro, NJ) and (kDa) shown to the left and right of the panels.
Most notable result was an approximately 30 kDa mobility shift on SDS-PAGE between rBHT(32–594)-HIS and rBHT(54–594)-HIS (Figure 3), possibly due to the deletion of the PTM nest containing predicted O-glycosylation (Figure 1B, GlycoEP) at positions (T34, S35, T39, T43, T50, T52) or phosphorylation (Figure 1B, DisPhos1.3) sites and surrounding acidic residues (Y37 (LTSNYETPS), T39 (SNYETPSPT), S41 (YETPSPTAI), T43 (TPSPTAIPL), T50 (PLEPTPTAT), T52 (EPTPTATGT)), known to retard proteins on SDS-PAGE (Lee et al., 2019).
An additional feature tested was the ability to drive secretion from predominantly membrane associated to soluble form. The secreted enzymatic activity associated with the membrane remained constant for rBHT(23–594)-HIS, rBHT(32–594)-HIS and rBHT(54–594)-HIS and rBHT(57–594)-HIS and no significant differences in ratio of soluble secreted versus membrane associated enzyme activity were observed for variants rBHT(23–594)-HIS, rBHT(32–594)-HIS and rBHT(54–594)-HIS. However, while rBHT(57–594)-HIS variant’s activity found associated with the membrane remained relatively constant, the ratio of secreted versus membrane associated enzyme activity increased between 25% and 40% (Table 4) when compared to variants rBHT(23–594)-HIS, rBHT(32–594)-HIS and rBHT(54–594)-HIS.
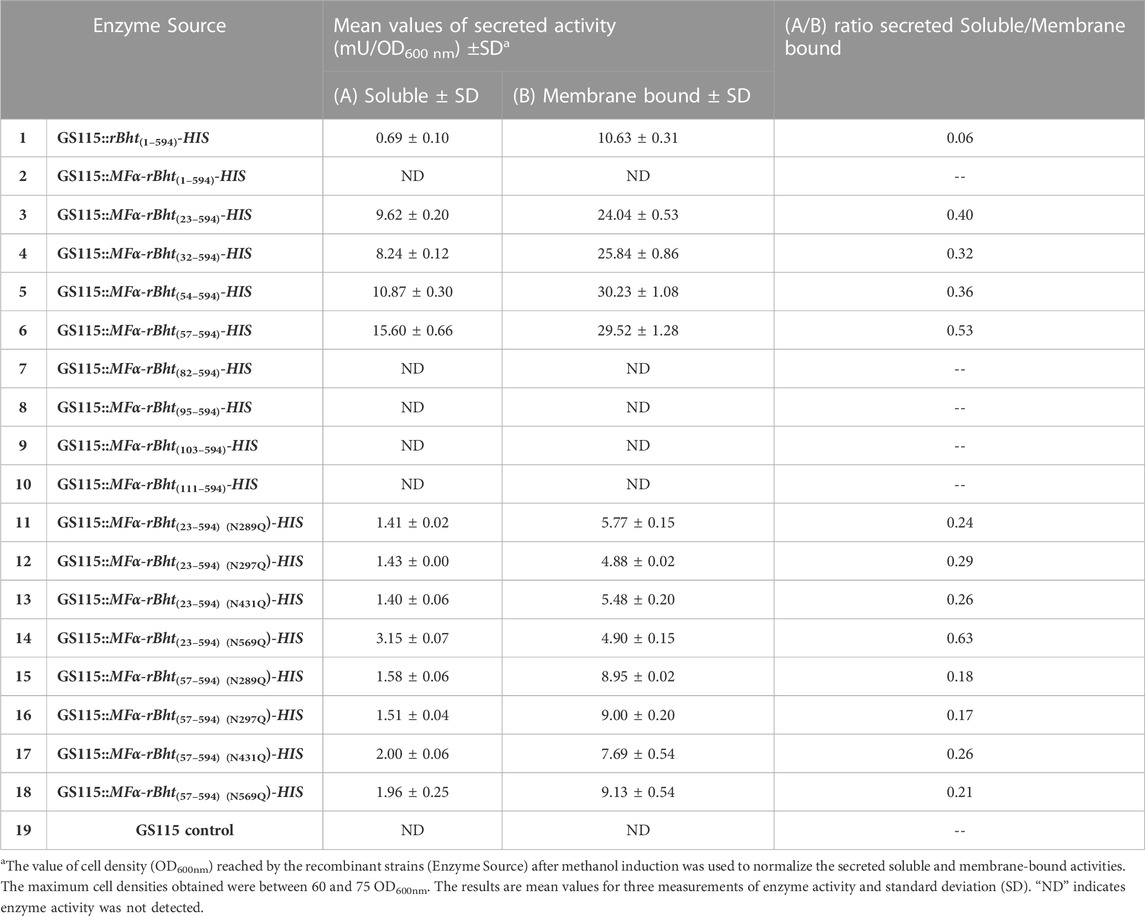
TABLE 4. Normalized enzyme activity comparison of (A) soluble versus (B) membrane bound secreted protein variants.
Under our experimental conditions, the results showed undetectable amounts of soluble, or membrane associated active protein when residues downstream of the IDR were removed (Table 4). To further evaluate whether bioactive rBHT(82–594)-HIS, rBHT(95–594)-HIS and rBHT(103–594)-HIS variants, were produced and secreted in low amounts, inductions of the corresponding cell lines were performed, and culture broth was concentrated 100-fold. However, neither soluble nor cell-associated rBHT from those variants had any enzymatic activity. This may be caused by ineffective secretion or destruction of the protein molecules that were not secreted.
The combined results strengthen a new finding that the BHT IDR by itself is not directly responsible for enzymatic activity or membrane interactions (Table 4).
Since the initial analysis of truncated variants revealed greater rBHT(57–594)-HIS titers (Table 4), we investigated if the structural organization exhibits kinetic biases or, more specifically, whether the IDR more broadly affects protein kinetics.
Active soluble secreted rBHT variants were functionally assessed by conventional kinetic measurements after being purified to homogeneity utilizing a C-terminal 6xHistidine epitope and Nickel affinity chromatography. Examination of the isolated rBHT variants by SDS-PAGE separation under reducing conditions showed the proteins were essentially homogenous (data not shown).
The kinetic parameters of each active secreted soluble variant, rBHT(23–594)-HIS, rBHT(32–594)-HIS, rBHT(54–594)-HIS, and rBHT(57–594)-HIS were investigated. To obtain a full kinetic picture, an important parameter evaluated was the impact of temperature on enzymatic activity. Therefore, assays were performed at the optimum temperature for rBHT(23–594)-HIS of 42°C (Dagher and Bruno-Bárcena, 2016), below 42°C (20°C and 30°C) and above 42°C (55°C) (Figure 4). The results of the respective kcat/km for all four truncated enzyme variants indicate a temperature optimum of 42°C (Figure 4). At each temperature tested all enzyme-truncated variants maintain a similar affinity for the substrate ONP-Glc (Km) and turnover activity (kcat) therefore confirming that truncations within the IDR do not play a role in the catalytic integrity of rBHT.
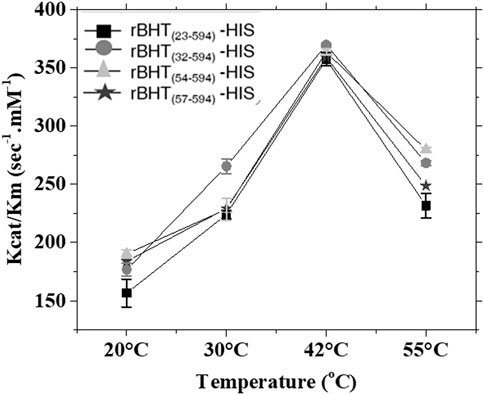
FIGURE 4. Enzyme kinetic parameters for rBHT variants tested at 20°C, 30°C, 42°C and 55°C. Enzyme assays depicted as kcat/km versus temperature were carried out in the presence of 0.3 µg rBHT(23–594)-HIS, rBHT(32–594)-HIS, rBHT(54–594)-HIS and rBHT(57–594)-HIS over a range of ONP-Glc substrate concentrations (0.08–10.4 mM). Assays were described under “Methods.” Km and kcat were calculated from initial velocities of ONP-Glc cleavage using the Hill equation with a Hill coefficient of 1. The values are the average of three independent measurements ± Standard Deviation (SD).
Proteome-wide investigations have revealed relationships between IDRs and several PTMs, including acetylation, methylation, and glycosylation (Dunker et al., 2013). Extensive in silico analysis revealed there are only 4 predicted N-linked glycosylation sites out of a possible 19, and none were discovered in the 110-residue N-terminus that contains the IDR (Figure 1). Investigations have indicated that glycosites are found mostly in structured sections, some distance from the disordered stretches, which is consistent with our findings (Singh et al., 2018; Goutham et al., 2020). All four N-glycosylation sites are in highly conserved glycosylation consensus sites (Asn-X-Ser/Thr X≠Pro) and nearby residues in the crystal structures of rBHT(23–594)-HIS (BHT, PDB: 7L74 and HsBglA, PMB: 6M4E), indicating a high likelihood of functionally relevant glycosylation at those positions N289LTY, N297STS, N431QSD, and N569QSD.
We examined each of the four N-glycosylation sites of BHT to determine if they had any functional importance. By employing rBht(23–594)-HIS as a template for site-directed mutagenesis, the asparagine residues (N289, N297, N431 and N569) were separately changed to glutamine residues to abrogate glycosylation, as explained in Materials and Methods. The results showed significant reductions of secreted soluble enzyme activities up to (90%, 95% and 97%) from three variants GS115::MFα-rBht(23–594) (N431Q)-HIS, GS115::MFα-rBht(23–594) (N289Q)-HIS and GS115::MFα-rBht(23–594) (N297Q)-HIS when compared to non-mutated variant GS115::MFα-rBht(23–594)-HIS activity, respectively. When compared to the GS115::MFα-rBht(23–594)-HIS version, the GS115::MFα-rBht(23–594) (N569Q)-HIS variant demonstrated a less pronounced activity loss of 67% and an enhanced ratio of secreted to cell membrane associated activity from 0.40 to 0.63 (Table 4). When compared to the parent strain GS115::MFα-rBht(23–594)-HIS, cell membrane associated activity for the strains GS115::MFα-rBht(23–594) (N289Q)-HIS, GS115::MFα-rBht(23–594) (N297Q)-HIS, GS115::MFα-rBht(23–594) (N431Q)-HIS, and GS115::MFα-rBht(23–594) (N569Q)-HIS, decreased by up to 81%, 95%, 84%, and 75%, respectively (Table 4). When the complete IDR is removed (GS115::MFα-rBht(57–594) (N569Q)-HIS), similar outcomes are shown. Notably, membrane-bound related activity was markedly reduced but not eliminated, indicating that glycosylation affects cell membrane localization and secretion.
We purified each N-glycosylation mutant by Ni-chromatography and confirmed that the variants had similar specific activities as the non-mutated forms (Table 4). Therefore, altering the N-glycosylation site interfered with secretion but did not alter the activity of the variants.
rBHT was crystallized in the C2 space group with two molecules per asymmetric unit, suggesting a possible dimer (Table 3). The same dimer is found in 6M4E, though in 6M4E a crystallographic axis of 2-fold symmetry runs through the dimer leading to only one molecule per asymmetric unit (Uehara et al., 2020). To distinguish between significant crystal interface interactions and artifacts of crystal packing (Krissinel, 2010) we used the program PISA (Protein Interfaces, Surfaces, and Assemblies) which calculates interface stability and entropy of dissociation to identify stable chemical contacts (Krissinel and Henrick, 2007). Analysis of PDB: 7L74 revealed a buried surface area of 7,803.5Å2 between molecules A and B. The interface between molecules A and B has the largest negative ΔiG (−21.3 kcal/mol) so it is predicted to be the strongest and with dissociation energy ΔGdiss (23.8 kcal/mol) and corresponded with the experimental results shown below. Residues in Loops C (yellow) and D (orange) contribute to most of the protein-protein interaction formed between the monomers (Figure 5). The residues involved in this interaction include hydrogen bond formation and salt bridges as shown in Figure 5B and depicted as yellow, orange, and gray sticks (Figure 5A).

FIGURE 5. Structural organization of rBht(23–594)-HIS dimer interface. (A) A ribbon representation of rBht(23–594)-HIS dimer highlighting loops (A; blue, B; green, C; yellow, D; orange) surrounding the active site is shown on the left. The inset on the right shows the dimer interface in greater detail with predicted salt bridges and H-bonds as black dashed lines. (B) Types of interface bonds and distances. The TRIS molecules occupying the −1 subsite is represented as a magenta stick. NAG are shown as green sticks. Ca2+ ion is represented as a black sphere. The figure was produced using PyMOL (https://pymol.org/2/) (Schrodinger, 2022). PDBePISA (https://www.ebi.ac.uk/pdbe/pisa/picite.html) was used to predict interface bonds and distances (Krissinel and Henrick, 2007).
To gain further insights into the oligomerization properties of BHT, we performed small X-ray scattering (SAXS) analysis (Figure 6A). Guinier and P(r) analysis was performed using PRIMUS and GNOM, respectively (Svergun, 1992; Konarev et al., 2003). Dmax values were manually chosen in GNOM to optimize the P(r) calculation (Figure 6B). These Dmax values are approximate to ∼±2–3 Å. Molecular mass were calculated using the method described by Rambo and Tainer (Rambo and Tainer, 2013). The data are presented in Figure 6C. The molecular mass determined from SAXS (∼169 KDa) confirmed that rBHT forms a dimer in solution (Figure 6C). The Rg and Dmax of the dimer in solution are 39 Å and 124 Å, respectively. As stated above, the X-ray crystallographic structures (PDB: 7L74, 6M4E, 6M4F and 6M55) also suggest that rBHT forms a dimer. The Rg and Dmax of the PDB: 7L74 crystallographic dimer (molecule A and molecule B) calculated using Crysol are 34 Å and 110 Å, respectively (Svergun et al., 1995). These values are in agreement with the experimental SAXS data. The disordered N-terminus led to a more expanded dimer in solution, and we conclude that rBHT(23–594)-HIS likely functions as a dimer. The SAXS experimental data have been deposited in the Small-Angle Scattering Biological Data Bank (SASBDB) (https://www.sasbdb.org/) under accession codes SASDN57 for rBHT(23–594)-HIS, 1 mg/mL and SASDN67 for rBHT(23–594)-HIS, 4 mg/mL.
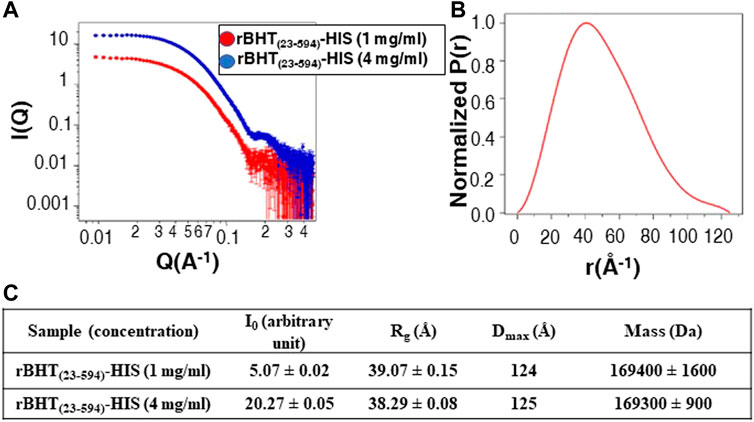
FIGURE 6. SAXS data for rBHT(23–594)-HIS at 1 mg/mL and 4 mg/mL. (A) SAXS data are shown on a log–log plot (left). I(Q) is in arbitrary units. (B) P(r) curve calculated from the SAXS data are normalized to a maximum height of 1.0. (C) Solution scattering parameters zero-angle intensity I0, radius of gyration Rg, and maximum dimension Dmax and SAXS-calculated molecular weight for rBHT(23–594)-HIS at 1 mg/mL and 4 mg/mL.
The dimer conformation was further validated in solution by size exclusion chromatography (SEC). The variants rBHT(23–594)-HIS, rBHT(32–594)-HIS, rBHT(54–594)-HIS, and rBHT(57–594)-HIS subjected to SEC showed the rBHT variants eluted as a single peak having a retention time of 12.5 min with calculated Mw 150 kDa demonstrating rBHT(23–594)-HIS, rBHT(32–594)-HIS, rBHT(54–594)-HIS, and rBHT(57–594)-HIS all exist as dimers in solution (Data not shown). The confirmation of dimer formation of rBHT(23–594)-HIS, rBHT(32–594)-HIS, rBHT(54–594)-HIS, and rBHT(57–594)-HIS, suggested that the unstructured regions spanning residues 23–56 are not involved in dimerization. Based on these analysis the IDR of rBHT does not appear to be essential for dimer formation or secretion of active enzyme.
Here, we investigated whether the presence of the distinctive N-terminal intrinsically disordered region (IDR) and/or putative posttranslational modifications in the GH1 C-terminal domain affect the amount of secreted active BHT. These results confirm that the rBHT IDR is not essential for activity or drive protein-membrane interactions.
Because the crystal structure did not provide any observable electron density at the N-terminal residues 23 to 53, IUPRED2A was used to predict the C-terminal border of the disordered region at amino acid 56. Deletion variants were generated based on the expected disordered portions until all 56 N-terminal residues were removed. Although native BHT is a membrane-associated protein, all rBHT variations partitioned between soluble secreted and cell membrane associated forms. Furthermore, soluble secreted enzyme variants that were shorted up to residue 56 displayed comparable catalytic properties. Although additional N-terminal deletions variants were not detected, it is possible that their removal affected rBHT’s stability or secretory pathway. Disordered regions can be discriminated from ordered ones based on the amino acid sequence (Garner et al., 1998; Darling and Uversky, 2018) and in most cases, disordered proteins are less evolutionarily conserved but rather their disordered structure has been maintained (Brown et al., 2011). Previously reviewed data indicated that low sequence complexity, high net charge, and low concentration of hydrophobic residues are a hallmark of disordered protein regions employed for interactions with lipid bilayers (Theillet et al., 2013; Cornish et al., 2020). However, a significant elemental preference for disorder-promoting residues reported in classical IDRs is called into doubt by the high proportion of hydrophobic residues in the BHT IDR (35.6%), placing it within the category of molecular recognition features (MoRFs) (Theillet et al., 2013; Yan et al., 2015).
Beyond the secretion signal sequences chosen, several other factors also govern protein secretion. For instance, the release of heterologous proteins depends on N-glycosylation, a post-translational modification involved in protein folding in the ER (Skropeta, 2009). It was therefore crucial to conduct additional research on the relationship between rBHT N-glycosylation and enzymatic properties to assess the stability, activity, and even secretion of the enzyme. However, not all polypeptides with predicted N-glycosylated sequons are glycosylated in vivo. Finding the locations of the N-linked glycosylation sites in the C-terminal GH1 domain was made easier by solving the crystal structure of rBHT(23–594)-HIS. No N-glycosylation sites were predicted in the IDR region, even though algorithms were useful at predicting O-glycosylation sites within the IDR (Figure 1). In this study, in vivo analyses were primarily used to evaluate the impact of eliminating a putative glycosylation site on expression, secretion, and activity. Although it appears that glycosylation is not necessary for enzymatic activity, the significant decrease in overall protein secretion observed for each of the four variants suggested that glycosylation may provide protection by increasing protein stability, shielding exposed hydrophobic surfaces, reducing proteolysis, and even increasing solubility.
When associated to the membrane, BHT must be conformationally flexible, whereas when unconnected to the membrane, it must be stable. Given that rBHT homodimer activity and stability when expressed by K. phaffii GS115 are independent of the N-terminal 56 amino acids, it is possible that elements (PTMs) in addition to unique amino acids within the catalytic domain may serve as a handle for specific catalytic advantages in preserving the active enzyme.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.wwpdb.org/, 7L74 https://www.sasbdb.org/, Small-Angle Scattering Biological Data Bank (SASBDB) under accession codes SASDN57 and SASDN67 (https://www.sasbdb.org/).
SD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. AV: Data curation, Investigation, Writing–review and editing. CS: Formal Analysis, Investigation, Writing–review and editing. FM: Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing–review and editing. BE: Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing–review and editing. JB-B: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Department of Plant and Microbial Biology, the Office of Research Commercialization, and the Chancellor’s Innovation Fund (1,108) (2018-2092 to JB-B) at North Carolina State University; Protein crystallization and data generation used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357; Use of the Life Sciences Collaborative Access Team (LS-CAT) Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (085P1000817 to BE).
The authors gratefully acknowledge M. Andrea Azcarate-Peril for reviewing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
BHT, native ß–hexosyltransferase; rBht, optimized coding sequence; rBHT, expressed recombinantly; Glc, glucose; GlcNAc, N-acetylglucosamine; Man, mannose; Asn, asparagine; Ser, serine; Thr, threonine; MFα, mating factor α; GH1, glycosyl hydrolase 1; PTM, post-translation modifications; SD, Standard Deviation; ONP-Glc, oNP-β-D-glucopyranoside.
Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66 (Pt 2), 213–221. doi:10.1107/S0907444909052925
Aivazian, D., and Stern, L. J. (2000). Phosphorylation of T cell receptor ζ is regulated by a lipid dependent folding transition. Nat. Struct. Biol. 7 (11), 1023–1026. doi:10.1038/80930
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37 (4), 420–423. doi:10.1038/s41587-019-0036-z
Arnold, J. W., Roach, J., Fabela, S., Moorfield, E., Ding, S., Blue, E., et al. (2021a). The pleiotropic effects of prebiotic galacto-oligosaccharides on the aging gut. Microbiome 9 (1), 31. doi:10.1186/s40168-020-00980-0
Arnold, J. W., Simpson, J. B., Roach, J., Bruno-Barcena, J. M., and Azcarate-Peril, M. A. (2018). Prebiotics for lactose intolerance: variability in galacto-oligosaccharide utilization by intestinal lactobacillus rhamnosus. Nutrients 10 (10), 1517. doi:10.3390/nu10101517
Arnold, J. W., Whittington, H. D., Dagher, S. F., Roach, J., Azcarate-Peril, M. A., and Bruno-Barcena, J. M. (2021b). Safety and modulatory effects of humanized galacto-oligosaccharides on the gut microbiome. Front. Nutr. 8, 640100. doi:10.3389/fnut.2021.640100
Azcárate-Peril, M. A., Sikes, M., and Bruno-Bárcena, J. M. (2011). The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am. J. Physiology-Gastrointestinal Liver Physiology 301 (3), G401–G424. doi:10.1152/ajpgi.00110.2011
Blakely, J. A., and MacKenzie, S. L. (1969). Purification and properties of a ß-hexosidase from Sporobolomyces singularis. Can. J. Biochem. 47 (11), 1021–1025. doi:10.1139/o69-164
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 (1-2), 248–254. doi:10.1016/0003-2697(76)90527-3
Bretthauer, R. K., and Castellino, F. J. (1999). Glycosylation of Pichia pastoris derived proteins. Biotechnol. Appl. Biochem. 30 (3), 193–200. doi:10.1111/j.1470-8744.1999.tb00770.x
Brown, C. J., Johnson, A. K., Dunker, A. K., and Daughdrill, G. W. (2011). Evolution and disorder. Curr. Opin. Struct. Biol. 21 (3), 441–446. doi:10.1016/j.sbi.2011.02.005
Bruno-Barcena, J. M., and Azcarate-Peril, M. A. (2015). Galacto-oligosaccharides and colorectal cancer: feeding our intestinal probiome. J. Funct. Foods 12, 92–108. doi:10.1016/j.jff.2014.10.029
Bryson, K., Cozzetto, D., and Jones, D. T. (2007). Computer-assisted protein domain boundary prediction using the DomPred server. Curr. Protein Pept. Sci. 8 (2), 181–188. doi:10.2174/138920307780363415
Buchan, D. W. A., and Jones, D. T. (2019). The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 47 (W1), W402–W407. doi:10.1093/nar/gkz297
Bürgi, J., Xue, B., Uversky, V. N., and van der Goot, F. G. (2016). Intrinsic disorder in transmembrane proteins: roles in signaling and topology prediction. PLOS ONE 11 (7), e0158594. doi:10.1371/journal.pone.0158594
Chauhan, J. S., Rao, A., and Raghava, G. P. S. (2013). In silico platform for prediction of N-O- and C-glycosites in eukaryotic protein sequences. PLOS ONE 8 (6), e67008. doi:10.1371/journal.pone.0067008
Chuang, C.-N., Woo, T.-T., Tsai, S.-Y., Li, W.-C., Chen, C.-L., Liu, H.-C., et al. (2020). Intrinsic disorder codes for leaps of protein expression.
Combet, C., Blanchet, C., Geourjon, C., and Deleage, G. (2000). NPS@: network protein sequence analysis. Trends Biochem. Sci. 25 (3), 147–150. doi:10.1016/s0968-0004(99)01540-6
Cornish, J., Chamberlain, S. G., Owen, D., and Mott, H. R. (2020). Intrinsically disordered proteins and membranes: a marriage of convenience for cell signalling? Biochem. Soc. Trans. 48 (6), 2669–2689. doi:10.1042/BST20200467
Cozzetto, D., Minneci, F., Currant, H., and Jones, D. T. (2016). FFPred 3: feature-based function prediction for all Gene Ontology domains. Sci. Rep. 6, 31865. doi:10.1038/srep31865
Csizmadia, G., Erdős, G., Tordai, H., Padányi, R., Tosatto, S., Dosztányi, Z., et al. (2021). The MemMoRF database for recognizing disordered protein regions interacting with cellular membranes. Nucleic Acids Res. 49 (D1), D355–D360. doi:10.1093/nar/gkaa954
Dagher, S. F., Azcarate-Peril, M. A., and Bruno-Bárcena, J. M. (2013). Heterologous expression of a bioactive ß-hexosyltransferase, an enzyme producer of prebiotics, from Sporobolomyces singularis. Appl. Environ. Microbiol. 79 (4), 1241–1249. doi:10.1128/aem.03491-12
Dagher, S. F., and Bruno-Bárcena, J. M. (2016). A novel N-terminal region of the membrane β-hexosyltransferase: its role in secretion of soluble protein by Pichia pastoris. Microbiology 162 (1), 23–34. doi:10.1099/mic.0.000211
Darling, A. L., and Uversky, V. N. (2018). Intrinsic disorder and posttranslational modifications: the darker side of the biological dark matter. Front. Genet. 9 (158), 158. doi:10.3389/fgene.2018.00158
Dunker, A. K., Babu, M. M., Barbar, E., Blackledge, M., Bondos, S. E., Dosztányi, Z., et al. (2013). What’s in a name? Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 1 (1), e24157. doi:10.4161/idp.24157
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2018). The Pfam protein families database in 2019. Nucleic Acids Res. 47 (D1), D427–D432. doi:10.1093/nar/gky995
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of coot. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66 (Pt 4), 486–501. doi:10.1107/S0907444910007493
Evans, P. R., and Murshudov, G. N. (2013). How good are my data and what is the resolution? Acta Crystallogr. Sect. D. 69 (7), 1204–1214. doi:10.1107/S0907444913000061
Gao, J., and Xu, D. (2012). Correlation between posttranslational modification and intrinsic disorder in protein. Pac Symp. Biocomput, 94–103. doi:10.1142/9789814366496_0010
Garner, E., Cannon, P., Romero, P., Obradovic, Z., and Dunker, A. K. (1998). Predicting disordered regions from amino acid sequence: common themes despite differing structural characterization. Genome Inf. Ser. Workshop Genome Inf. 9, 201–213. doi:10.11234/gi1990.9.201
Ge, F., Zhu, L., Aang, A., Song, P., Li, W., Tao, Y., et al. (2018). Recent advances in enhanced enzyme activity, thermostability and secretion by N-glycosylation regulation in yeast. Biotechnol. Lett. 40 (5), 847–854. doi:10.1007/s10529-018-2526-3
Glycoside Hydrolase Family (2012). Available at: http://www.cazypedia.org/index.php?title=Glycoside_Hydrolase_Family_1&oldid=7911.
Gorin, P. A. J., Phaff, H. J., and Spencer, J. F. T. (1964a). Structures of galactosyl-lactose and galactobiosyl-lactose produced from lactose by Sporobolomyces singularis. Can. J. Chem. 42 (6), 1341–1344. doi:10.1139/v64-206
Gorin, P. A. J., Spencer, J. F. T., and Phaff, H. J. (1964b). Synthesis of β-galacto-β-gluco-pyranosyl disaccharides by Sporobolomyces singularis. Can. J. Chem. 42 (10), 2307–2317. doi:10.1139/v64-338
Goutham, S., Kumari, I., Pally, D., Singh, A., Ghosh, S., Akhter, Y., et al. (2020). Mutually exclusive locales for N-linked glycans and disorder in human glycoproteins. Sci. Rep. 10 (1), 6040. doi:10.1038/s41598-020-61427-y
Gsponer, J., Futschik, M. E., Teichmann, S. A., and Babu, M. M. (2008). Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science 322 (5906), 1365–1368. doi:10.1126/science.1163581
Henrissat, B., Callebaut, I., Fabrega, S., Lehn, P., Mornon, J. P., and Davies, G. (1995). Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc. Natl. Acad. Sci. U. S. A. 92 (15), 7090–7094. doi:10.1073/pnas.92.15.7090
Iakoucheva, L. M., Radivojac, P., Brown, C. J., O'Connor, T. R., Sikes, J. G., Obradovic, Z., et al. (2004). The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32 (3), 1037–1049. doi:10.1093/nar/gkh253
Ingrell, C. R., Miller, M. L., Jensen, O. N., and Blom, N. (2007). NetPhosYeast: prediction of protein phosphorylation sites in yeast. Bioinformatics 23 (7), 895–897. doi:10.1093/bioinformatics/btm020
Ishikawa, E., Sakai, T., Ikemura, H., Matsumoto, K., and Abe, H. (2005). Identification, cloning, and characterization of a Sporobolomyces singularis β-galactosidase-like enzyme involved in galacto-oligosaccharide production. J. Biosci. Bioeng. 99 (4), 331–339. doi:10.1263/jbb.99.331
Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrices 1 1Edited by G. Von Heijne. J. Mol. Biol. 292 (2), 195–202. doi:10.1006/jmbi.1999.3091
Jones, D. T., and Cozzetto, D. (2015). DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31 (6), 857–863. doi:10.1093/bioinformatics/btu744
Joosten, R. P., Long, F., Murshudov, G. N., and Perrakis, A. (2014). The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1 (Pt 4), 213–220. doi:10.1107/S2052252514009324
Källberg, M., Wang, H., Wang, S., Peng, J., Wang, Z., Lu, H., et al. (2012). Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7 (8), 1511–1522. doi:10.1038/nprot.2012.085
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi:10.1038/nprot.2015.053
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003). PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282. doi:10.1107/s0021889803012779
Krissinel, E. (2010). Crystal contacts as nature's docking solutions. J. Comput. Chem. 31 (1), 133–143. doi:10.1002/jcc.21303
Krissinel, E., and Henrick, K. (2007). Inference of macromolecular Assemblies from crystalline state. J. Mol. Biol. 372 (3), 774–797. doi:10.1016/j.jmb.2007.05.022
Lee, C.-R., Park, Y.-H., Min, H., Kim, Y.-R., and Seok, Y.-J. (2019). Determination of protein phosphorylation by polyacrylamide gel electrophoresis. J. Microbiol. 57 (2), 93–100. doi:10.1007/s12275-019-9021-y
Lieutaud, P., Ferron, F., Uversky, A. V., Kurgan, L., Uversky, V. N., and Longhi, S. (2016). How disordered is my protein and what is its disorder for? A guide through the "dark side" of the protein universe. Intrinsically Disord. proteins 4 (1), e1259708. doi:10.1080/21690707.2016.1259708
Linding, R., Russell, R. B., Neduva, V., and Gibson, T. J. (2003). GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 31 (13), 3701–3708. doi:10.1093/nar/gkg519
Lowry, O. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J. (1951). Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193 (1), 265–275. doi:10.1016/s0021-9258(19)52451-6
Meszaros, B., Erdos, G., and Dosztanyi, Z. (2018). IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46 (W1), W329–w337. doi:10.1093/nar/gky384
Mohammad, I.-L., Mateos, B., and Pons, M. (2019). The disordered boundary of the cell: emerging properties of membrane-bound intrinsically disordered proteins. Biomol. Concepts 10 (1), 25–36. doi:10.1515/bmc-2019-0003
Monteagudo-Mera, A., Arthur, J. C., Jobin, C., Keku, T., Bruno-Barcena, J. M., and Azcarate-Peril, M. A. (2016). High purity galacto-oligosaccharides enhance specific Bifidobacterium species and their metabolic activity in the mouse gut microbiome. Benef. Microbes 7 (2), 247–264. doi:10.3920/BM2015.0114
Nishi, H., Hashimoto, K., and Panchenko, A. R. (2011). Phosphorylation in protein-protein binding: effect on stability and function. Structure 19 (12), 1807–1815. doi:10.1016/j.str.2011.09.021
Nishikawa, I., Nakajima, Y., Ito, M., Fukuchi, S., Homma, K., and Nishikawa, K. (2010). Computational prediction of O-linked glycosylation sites that preferentially map on intrinsically disordered regions of extracellular proteins. Int. J. Mol. Sci. 11 (12), 4991–5008. doi:10.3390/ijms11124991
Panesar, P. S., Kaur, R., Singh, R. S., and Kennedy, J. F. (2018). Biocatalytic strategies in the production of galacto-oligosaccharides and its global status. Int. J. Biol. Macromol. 111, 667–679. doi:10.1016/j.ijbiomac.2018.01.062
Peng, K., Vucetic, S., Radivojac, P., Brown, C. J., Dunker, A. K., and Obradovic, Z. (2005). Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform Comput. Biol. 3 (1), 35–60. doi:10.1142/s0219720005000886
Phaff, H. J., and Carmo-Sousa, L. D. (1962). Four new species of yeast isolated from insect frass in bark of Tsuga heterophylla (Raf.) Sargent. Ant. Van Leeuwenhoek 28, 193–207. doi:10.1007/bf02538734
Prabakaran, S., Lippens, G., Steen, H., and Gunawardena, J. (2012). Post-translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 4 (6), 565–583. doi:10.1002/wsbm.1185
Prates, E. T., Guan, X., Li, Y., Wang, X., Chaffey, P. K., Skaf, M. S., et al. (2018). The impact of O-glycan chemistry on the stability of intrinsically disordered proteins. Chem. Sci. 9 (15), 3710–3715. doi:10.1039/C7SC05016J
Rambo, R. P., and Tainer, J. A. (2013). Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496 (7446), 477–481. doi:10.1038/nature12070
Robert, X., and Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42 (W1), W320–W324. doi:10.1093/nar/gku316
Singh, A., Kumari, I., Pally, D., Goutham, S., Ghosh, S., Akhter, Y., et al. (2018). 'Mutually exclusive locales for N-linked glycans and disorder in glycoproteins. bioRxiv, 443143. doi:10.1101/443143
Skropeta, D. (2009). The effect of individual N-glycans on enzyme activity. Bioorg Med. Chem. 17 (7), 2645–2653. doi:10.1016/j.bmc.2009.02.037
Spencer, J. F. T., de Spencer, A. L. R., and Laluce, C. (2002). Non-conventional yeasts. Appl. Microbiol. Biotechnol. 58 (2), 147–156. doi:10.1007/s00253-001-0834-2
Spherix Consulting Inc (2010). Generally Recognized as Safe (GRAS) determination for the use of galacto-oligosaccharides in foods and infants’ formulas.
Svergun, D., Barberato, C., and Koch, M. H. J. (1995). CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28 (6), 768–773. doi:10.1107/S0021889895007047
Svergun, D. I. (1992). Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25 (4), 495–503. doi:10.1107/S0021889892001663
Theillet, F.-X., Kalmar, L., Tompa, P., Han, K.-H., Selenko, P., Dunker, A. K., et al. (2013). The alphabet of intrinsic disorder: I. Act like a Pro: on the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord. proteins 1 (1), e24360. doi:10.4161/idp.24360
Torres, D. P. M., Gonçalves, M. D. F., Teixeira, J. A., and Rodrigues, L. R. (2010). Galacto-oligosaccharides: production, properties, applications, and significance as prebiotics. Compr. Rev. Food Sci. Food Saf. 9 (5), 438–454. doi:10.1111/j.1541-4337.2010.00119.x
Uehara, R., Iwamoto, R., Aoki, S., Yoshizawa, T., Takano, K., Matsumura, H., et al. (2020). Crystal structure of a GH1 β-glucosidase from Hamamotoa singularis. Protein Sci. 29 (9), 2000–2008. doi:10.1002/pro.3916
Uversky, V. N. (2019). Intrinsically disordered proteins and their “mysterious” (Meta)Physics. Front. Phys. 7 (10). doi:10.3389/fphy.2019.00010
van der Lee, R., Buljan, M., Lang, B., Weatheritt, R. J., Daughdrill, G. W., Dunker, A. K., et al. (2014). Classification of intrinsically disordered regions and proteins. Chem. Rev. 114 (13), 6589–6631. doi:10.1021/cr400525m
Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., et al. (2011). Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D. Biol. Crystallogr. 67 (Pt 4), 293–302. doi:10.1107/S0907444911007773
Watson, V. E., Jacob, M. E., Bruno-Bárcena, J. M., Amirsultan, S., Stauffer, S. H., Píqueras, V. O., et al. (2019). Influence of the intestinal microbiota on disease susceptibility in kittens with experimentally-induced carriage of atypical enteropathogenic Escherichia coli. Veterinary Microbiol. 231, 197–206. doi:10.1016/j.vetmic.2019.03.020
Whittington, H. D., Dagher, S. F., and Bruno-Bárcena, J. M. (2019). “Production and conservation of starter cultures: from “backslopping” to controlled fermentations,” in How fermented foods feed a healthy gut microbiota: a nutrition continuum. Editors M. A. Azcarate-Peril, R. R. Arnold, and J. M. Bruno-Bárcena (Cham: Springer International Publishing), 125–138.
Keywords: disorder, expression, kinetics, mutagenesis, transglycosylation, Hamamotoa singularis
Citation: Dagher SF, Vaishnav A, Stanley CB, Meilleur F, Edwards BFP and Bruno-Bárcena JM (2023) Structural analysis and functional evaluation of the disordered ß–hexosyltransferase region from Hamamotoa (Sporobolomyces) singularis. Front. Bioeng. Biotechnol. 11:1291245. doi: 10.3389/fbioe.2023.1291245
Received: 08 September 2023; Accepted: 16 November 2023;
Published: 14 December 2023.
Edited by:
Hyun-Dong Shin, Bereum Co., Ltd., Republic of KoreaReviewed by:
Long Liu, Jiangnan University, ChinaCopyright © 2023 Dagher, Vaishnav, Stanley, Meilleur, Edwards and Bruno-Bárcena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José M. Bruno-Bárcena, amJiYXJjZW5AbmNzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.