 Christopher J. Bridgeman
Christopher J. Bridgeman Shrey A. Shah
Shrey A. Shah Robert S. Oakes
Robert S. Oakes Christopher M. Jewell
Christopher M. Jewell
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 27 June 2023
Sec. Tissue Engineering and Regenerative Medicine
Volume 11 - 2023 | https://doi.org/10.3389/fbioe.2023.1184938
This article is part of the Research Topic Advanced Materials and Models in the Establishment and Repair of Pathological Microenvironment View all 11 articles
Biomaterials allow for the precision control over the combination and release of cargo needed to engineer cell outcomes. These capabilities are particularly attractive as new candidate therapies to treat autoimmune diseases, conditions where dysfunctional immune cells create pathogenic tissue environments during attack of self-molecules termed self-antigens. Here we extend past studies showing combinations of a small molecule immunomodulator co-delivered with self-antigen induces antigen-specific regulatory T cells. In particular, we sought to elucidate how different ratios of these components loaded in degradable polymer particles shape the antigen presenting cell (APC) -T cell interactions that drive differentiation of T cells toward either inflammatory or regulatory phenotypes. Using rapamycin (rapa) as a modulatory cue and myelin self-peptide (myelin oligodendrocyte glycoprotein- MOG) – self-antigen attacked during multiple sclerosis (MS), we integrate these components into polymer particles over a range of ratios and concentrations without altering the physicochemical properties of the particles. Using primary cell co-cultures, we show that while all ratios of rapa:MOG significantly decreased expression of co-stimulation molecules on dendritic cells (DCs), these levels were insensitive to the specific ratio. During co-culture with primary T cell receptor transgenic T cells, we demonstrate that the ratio of rapa:MOG controls the expansion and differentiation of these cells. In particular, at shorter time points, higher ratios induce regulatory T cells most efficiently, while at longer time points the processes are not sensitive to the specific ratio. We also found corresponding changes in gene expression and inflammatory cytokine secretion during these times. The in vitro results in this study contribute to in vitro regulatory T cell expansion techniques, as well as provide insight into future studies to explore other modulatory effects of rapa such as induction of maintenance or survival cues.
Multiple sclerosis (MS) is an autoimmune disease whereby dysfunctional immune cells attack myelin that insulates neurons in the central nervous system leading to loss of motor control. In MS, innate and adaptive immune cells drive a proinflammatory state that leads to degradation of the myelin protein matrix (MPM) (Dendrou, Fugger and Friese, 2015). Although the pathogenesis of MS is incompletely understood, both the innate and the adaptive immune cell compartments contribute to disease progression. For example, antigen-specific B cells contribute to disease through the generation of myelin recognizing auto-antibodies (Arneth, 2019). Antigen presenting cells (APCs) such as dendritic cells (DCs) also initiate auto-activation of self-reactive T cells through the presentation of MPM-derived peptide antigen to myelin-specific CD4+ and CD8+ T cells (Gandhi, Laroni and Weiner, 2010; Weissert, 2013; Saxena et al., 2019). Once activated, these T cells drive disease through inflammatory cytokine production and targeted killing of host tissue expressing myelin antigens. Although therapies exist for MS, these require lifelong treatment and none are curative (Huang, Chen and Zhang, 2017). Further, even the most advanced therapies – such as monoclonal antibodies – do not distinguish between healthy and dysfunctional immune cells (Torkildsen, Myhr and Bø, 2016; Hauser and Cree, 2020). Thus, there is a need for new treatments that selectively control autoreactive immune cells while leaving the remainder of the immune system intact. Regulatory CD4+ T cells (TREG) are one target of particular interest that attenuate unwanted inflammation through the release of anti-inflammatory cytokines in response to self-antigens (Danikowski, Jayaraman and Prabhakar, 2017; Kimura, 2020). Indeed, ex vivo expanded TREG have shown promise in clinical trials for treatment of autoimmune diseases such as type 1 diabetes and Crohn’s disease (Mosanya and Isaacs, 2019; Romano et al., 2019). However, these TREG are polyclonal, and not necessarily specific to the disease-causing self-antigen. Alternatively, the expansion of disease specific TREG may improve the potency and efficacy of these engineered cells. One emerging strategy to treat MS relies on the use of biomaterials to control the delivery of the TREG promoting small molecule drug rapamycin (rapa) with MPM peptides to drive the expansion of antigen-specific TREG that would protect against myelin degradation. This strategy fundamentally relies on the use of polymer microparticles (MPs) to control the release of these cues for myelin specific TREG expansion in vivo (Tostanoski et al., 2016).
Polymer particle delivery strategies are an exciting area of research due to their ability to control drug delivery parameters, such as controlled release, drug co-localization, and tissue targeting (Andorko, Hess and Jewell, 2015; Tostanoski, 2016; Bookstaver et al., 2018). Poly(lactic-co-glycolic acid) (PLGA), for example, is a biodegradable polymer of particular interest due to its biocompatibility and degradation into non-toxic byproducts (Blasi, 2019; Su et al., 2021). Additionally, these particles are versatile in their ability to encapsulate a variety of immune cargos including proteins, peptides, and nucleic acids (Danhier et al., 2012). We and others have used poly(lactic-co-glycolide) (PLGA) microparticles loaded with myelin self-peptides, including myelin oligodendrocyte glycoprotein (MOG) and the mechanistic target of rapamycin (mTOR) inhibitor rapa to re-condition the microenvironment of lymph nodes (LNs) to promote antigen-specific TREG (Tostanoski et al., 2016; Gammon et al., 2023). However, it is unclear how specific combinations and concentrations of these signals could further shape the response of APCs and T cells.
The mTOR metabolic pathway has been shown to critically regulate cell functions such as proliferation, and metabolism (März et al., 2013; Mukhopadhyay et al., 2016; Yoo et al., 2017). While rapa has historically served a role of an important immunosuppressant (Saunders, Metcalfe and Nicholson, 2001; Yellen et al., 2011; Hester et al., 2012), over the last decade its ability to facilitate TREG expansion and survival has been extensively explored (Marín Morales et al., 2019; Sato et al., 2021; Vakrakou et al., 2022). Additionally, rapa encapsulation in microparticle and nanoparticle systems for antigen-specific TREG induction has been studied in multiple pre-clinical models of autoimmune diseases (Maldonado et al., 2015; Tostanoski, 2016; Gammon et al., 2023). However, more mechanistic studies are needed to deconvolute how specific ratios of encapsulated rapa:MOG fundamentally shape the APC-T cell signaling axis to promote antigen-specific TREG. This paper seeks to bridge this knowledge gap by using validated in vitro assays to test how specific ratios of rapa and MOG regulate APC activation, and subsequent antigen-specific TREG polarization (Figure 1A). In particular, MPs were synthesized with specific ratios of rapa and MOG (Figure 1B) to test their effect on primary APCs and subsequent polarization of antigen-specific T cells to TREG. Using this library, we show all particles containing rapa reduced the activation of stimulated DCs, but these effects were insensitive to the ratio of rapa:MOG. Interestingly, however, the expansion of MOG-specific T cells could be maximized by tuning the rapa:MOG ratio. These results provide additional insight into design parameters for polarizing T cells toward TREG for antigen-specific autoimmune therapies.
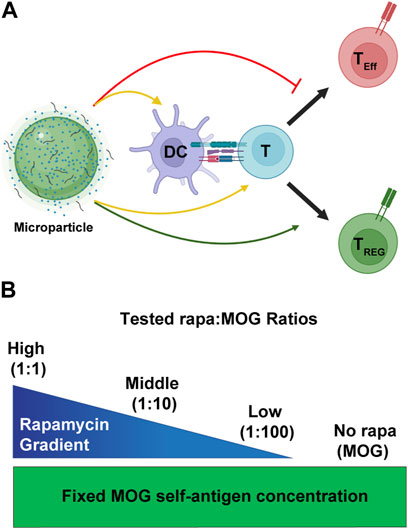
FIGURE 1. Overview schematic of MP treatment strategy. (A) MPs release immune cues that act on both DCs and T cells (yellow arrows). These cues modulate the interaction of DCs and T cells to promote generation of TREG (green arrow) and not inflammatory effector T cells (TEff). (B) To test the effect of rapa:MOG ratio, quantity of MOG peptide encapsulated in MPs was kept constant while rapa quantity was titrated. Created with BioRender.
MOG peptide (MOG35-55, MEVGWYRSPFSRVVHLYRNGK) was synthesized by Genscript (Piscataway, NJ, United States) with >98% purity. Rapamycin was purchased from LC Laboratories (Woburn, MA, United States). 50:50 poly(lactic-co-glycolide) (PLGA) was purchased from LACTEL Absorbable Polymers. High molecular weight poly(vinyl alcohol) (PVA) was purchased from Alpha Aesar (Tewksbury, MA, United States). Dichloromethane (DCM) was purchased from Sigma Aldrich (St. Louis, MO, United States). Dimethyl sulfoxide (DMSO), micro-bicinchoninic acid (mBCA) assay, and eBioscience FoxP3 Fixation/Permeation kit were purchased from Thermo Fisher Scientific (Waltham, MA, United States). Antibodies for flow cytometry, including BV605-CD11c, v450-CD25, PE-T-bet, PE-CD40, PE-Cy-7-CD4, PE-Cy-7-CD86, APC-FoxP3, APC-CD80 were purchased from BD Biosciences. Zombie NIR Fixable Viability Kit was purchased from BioLegend.
MPs were synthesized as previously described using a water-in-oil-in-water double emulsion technique (Tostanoski et al., 2016). Briefly, 500 µL of aqueous phase was prepared by dissolving 2.2 mg of MOG into water. An organic phase was prepared by first dissolving PLGA at 16 mg/mL in DCM for 1 h (PLGA-DCM). For rapa-loaded MPs, lyophilized rapa was dissolved in a PLGA-DCM aliquot to 0.22 mg/mL, and then titrated 1:10 in PLGA-DCM. Inner emulsion was formed by sonicating the aqueous phase with organic phase at 12 W for 30 s. This emulsion was then homogenized with 40 mL of 2% w/v PVA solution for 3 min at 16,000 RPM to form final emulsions. MP solutions were then stirred overnight to evaporate DCM. MPs were then poured through a 40 μm cell strainer, and centrifuged for 5 min at 5000g. After aspiration of supernatant, MPs were washed 3x with 1 mL of water (resuspend, spin 5 min 5000g). MPs were resuspended in a final volume of 1 mL of water.
MP size and distribution were calculated using dynamic laser scattering (DLS) (Horiba LA-950). Rapa and MOG loading were quantified by dissolving a known mass of dried MPs in DMSO. MOG concentration was determined using a mBCA assay according to the manufacturer’s instructions. Rapa concentration was determined by measuring ultraviolet/visible (UV/Vis) spectroscopy values and comparing to a standard curve of known concentrations. Loading efficiency was calculated by comparing measured quantities to initial loading quantities.
All studies involving animals were carried out under the supervision of the University of Maryland Institutional Animal Care and Use Committee (IACUC) in compliance with local, state, and federal guidelines. To test the effect of rapa:MOG in MPs on DC activation, CD11c+ DCs were harvested from the spleens of female C57Bl/6J mice (Jackson Laboratories, United States) using a bead based CD11c positive selection kit according to manufacturer’s instructions (Miltenyi, United States). DCs were plated at 100,000 cells per well in a 96 well flat bottom plate in 100 µL of RPMI + L-Glutamine media supplemented with 10% fetal bovine serum, 55 µM β-mercaptoethanol, 1x non-essential amino acid, 10 mM HEPES, and 2 x Pen/Strep. Cells were then immediately treated with MP formulations matched to treat each well with 15 µg of MPs. MOG (no rapa) MPs, and empty MPs were used to control for the effect of MOG and MP effect respectively. To assess the effect of rapa on DC activation, cells were stimulated with 0.25 μg/mL lipopolysaccharide (LPS). DCs were cultured at 37°C for 24 h at 5% CO2. DCs were then sequentially Fc blocked for 20 min, washed, stained in NIR viability dye, and stained with anti-CD80, anti-CD86, and anti-CD40 antibodies for flow cytometry on a Beckman Coulter CytoFLEX. Median fluorescence intensity of markers was used to determine activation.
To test the effect of rapa:MOG on antigen-specific T cell proliferation, transgenic 2D2 mice that express a MOG specific T cell receptor (TCR) were used (Jackson Laboratories). DCs were first harvested as described, and treated with MP formulations. After 24 h, CD4+ T cells were isolated from 2D2 mouse spleens using a bead based negative selection kit (Stem Cell, United States). T cells were added in a 3:1 ratio to wells containing DCs (300,000 T cells per well). At 48 h post T cell addition, T cells were sequentially Fc blocked for 20 min, washed, and then stained in NIR viability dye for 20 min. Cells were then washed and stained with antibodies for anti-CD4 and anti-CD25. After 1 h, cells were washed and placed in fixation buffer. Cells were then intracellularly stained with antibodies for the expression of forkhead box P3 (FoxP3) and T-box 21 (T-bet) transcription factors. Cells were analyzed for the expression of these markers using a Beckman Coulter CytoFLEX.
To test the effect of rapa:MOG MPs on CD4+ T cell gene expression, real time quantitative polymerase chain reaction (RT-qPCR) was used to look at the expression of pro-inflammatory and regulatory genes. DCs and CD4+ T cells were co-cultured as described. Twenty-four hours following T cell addition to DCs, the non-adherent T cells were removed, pelleted, and lysed using RNA lysis buffer (Zymo Research). After lysis, RNA isolation was performed followed by RNA quantification using a NanoDrop 2000c (Fisher Scientific). Complementary DNA (cDNA) amplification was then performed on 200 ng of RNA from each sample using the SuperScript IV VILO Master Mix kit and following the manufacturer’s instructions. cDNA samples were then stored at −80°C until RT-qPCR analysis. For RT-qPCR analysis, 1 µL of probe, 10 µL of Master Mix, 4 µL of cDNA, and 5 µL of molecular grade water were combined in a 96 well RT-qPCR reaction plate. T cell expression of interferon gamma (IFN-ɣ) was tested against control probes for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and beta-actin (ACTB). RT-qPCR and determination of cycle threshold (Ct) values were performed by a QuantStudio 7 Flex Real Time PCR System (Thermo Fisher Scientific). Delta Ct (ΔCt) was determined as previously described (Schmittgen and Livak, 2008). ΔCt values for cells treated with rapa:MOG were divided by the MOG (no rapa) ΔCt value to determine fold difference. Values are plotted as -log2(ΔCt).
Flow cytometry gating and analysis was performed in FlowJo (v 10.8.1). All statistical analyses were performed using GraphPad Prism (v 9.4.1). To test significant difference between two groups, an unpaired t-test was used. To test significance between multiple groups, an analysis of variance (ANOVA) was used followed by a Tukey post-hoc test for multiple comparisons. Significance was defined if p values were less than 0.05. Graphs were made using GraphPad Prism software.
An established double emulsion synthesis procedure was used to prepare particles with defined ratios of rapa:MOG (Figure 2A; Supplementary Figure S1) (Gammon et al., 2017; Gosselin, Tostanoski and Jewell, 2017). This process resulted in particles with direct control over the quantity of loaded rapa (Figure 2A) and peptide (Figure 2B). We observed a slightly larger standard deviation in the MOG loading into MPs co-loaded with high and middle quantities of rapa which could be attributed to charge interactions between hydrophobic rapa and PLGA, and hydrophilic MOG. This hypothesis is supported by our data showing that rapa had a significantly higher loading efficiency (87%) compared to MOG (74%) (Figure 2C). MP size was determined using dynamic laser scattering. These measurements revealed that MP diameter was not impacted by the specific rapa:MOG composition (Figure 2D). However, MPs compositions containing cargo were modestly larger than empty MPs. There was no significant difference between MPs loaded with rapa:MOG relative to MOG (no rapa) MPs; MP size was consistent with values previously reported from our lab (Tostanoski et al., 2016; Gosselin et al., 2021).
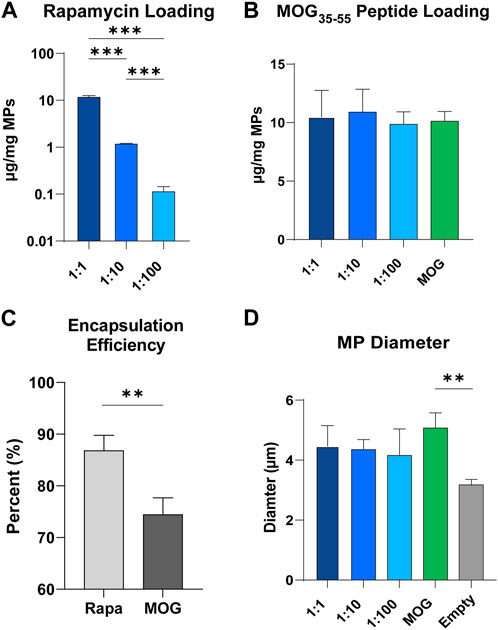
FIGURE 2. Characterization of MPs. (A) Quantity of rapa-loaded into MPs as measured through UV/Vis spectroscopy. N = 3 for each group. An ANOVA with a Tukey multiple comparisons post-hoc test was used to compare each group. Statistical significance is indicated by a (*). ***p < 0.001. (B) Quantity of MOG peptide loaded into MPs as measured through mBCA. (C) Loading efficiency of rapa and MOG into PLGA microparticles. An unpaired t-test was used to compare groups. Statistical significance is indicated by a (*). **p < 0.01. (D) MP size quantified through DLS. An ANOVA with a Tukey multiple comparisons post-hoc test was used to compare each group. Statistical significance is indicated by a (*). **p < 0.01.
To study the modulatory effect of rapa:MOG ratio, primary derived wildtype splenic DCs were isolated, activated using LPS, then and treated with MPs (Figure 3A). After 24 h, DC expression of activation markers was characterized using flow cytometry. Live cell population was determined using a fixable viability stain, followed by positive selection of DCs based on their expression of CD11c (Figure 3B). Quantification of live cells showed that rapa:MOG MP treatment did not significantly impact cell viability compared to controls (Figure 3C). CD11c+ DCs were then analyzed for their expression of co-stimulation molecules CD86 (Figure 3D) and CD80 (Figure 3E) which play an important role in DC-T cell engagement. All groups treated with MPs containing both rapa and MOG exhibited reduced expression of CD86 and CD80 relative to control samples pulsed with LPS, including empty MPs. However, these reductions were equivalent irrespective to the specific rapa:MOG ratio in the MPs. This indicates rapa:MOG MP treatment modulates DC CD86 and CD80 signaling under highly inflammatory conditions, as opposed to complete attenuation of cell activity, and that this modulation is relative insensitive to ratio. As this assay was designed to test the effect of rapa:MOG ratio directly on DCs, we next wanted to test the effect of rapa:MOG treatment in the context of both DCs and T cells.
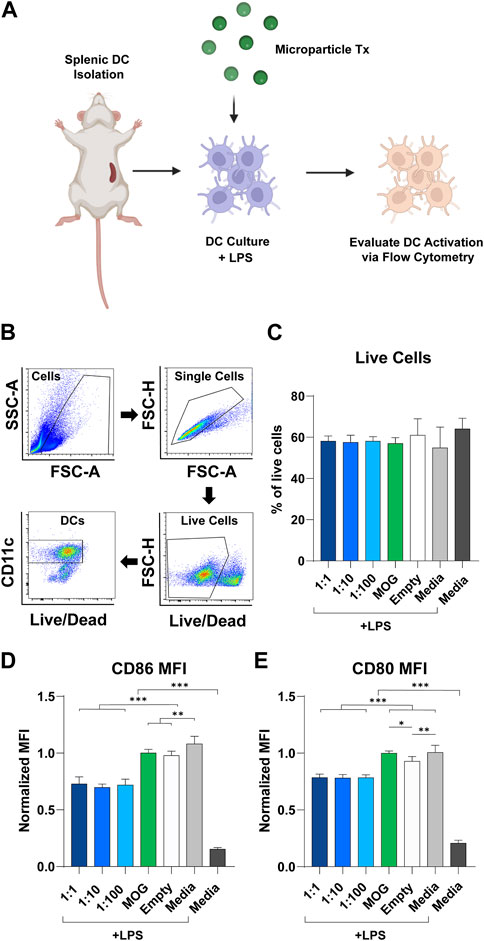
FIGURE 3. Effect of rapa:MOG MP treatment (Tx) on DC expression of activation receptors. (A) Overview schematic of experiment design. Primary derived DCs were harvested from mice and cultured with MP treatment for 24 h. DCs were then evaluated via flow cytometry for their expression of surface receptors. (B) Representative flow cytometry gating strategy. (C) Rapa:MOG MP treatment did not significantly reduce DC viability. (D) CD86 and (E) CD80 co-stimulation molecule expression on DCs was significantly lower compared to controls, but not within different rapa:MOG treatments. This study was completed in triplicate. For each study, N = 3. An ANOVA with a Tukey multiple comparisons post-hoc test was used to compare each group. Statistical significance is indicated by a (*). *p < 0.05, **p < 0.01, ***p < 0.001. Created with BioRender.
Next, we tested if the ratio of rapa:MOG in MPs differentially expands antigen-specific TREG in vitro. DCs were cultured with each MP formulation for 24 h. Transgenic CD4+ T cells expressing a MOG specific TCR were then isolated from 2D2 mice and co-cultured with the DCs (Figure 4A). After 96 h, cells were stained for viability and T cell phenotype markers to characterize their polarization as a function of ratio (Figure 4B). Increasing the ratio of rapa:MOG loaded into MPs did not decrease CD4+ T cell viability (Figure 4C). MOG (no rapa) MP treated cells on average had a higher trend in CD4+ T cell count compared to rapa:MOG treatments, but this was not significant (Figure 4D). This is likely due to the absence of the restraining effect of rapa in the control group, leading to more rapid T cell expansion. However, at this 96-hour expansion timepoint, polarization of TREG (CD4+/CD25+/FoxP3+) percentage was not dependent on the specific ratio of rapa:MOG (Figure 4E). This result was also observed in our analysis of TREG count confirming that rapa:MOG treatment increased polarization of TREG independent of ratio (Figure 4F).
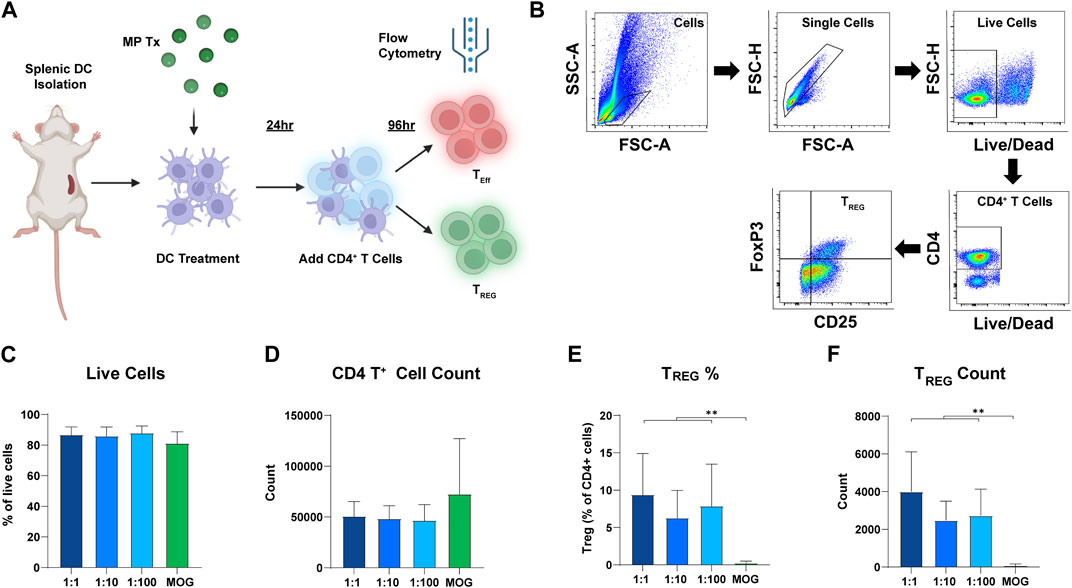
FIGURE 4. Results of DC-CD4 T cell co-culture experiment after 72 h. (A) Overview schematic of co-culture assay. (B) Representative gating strategy. (C) Viability was not significantly different between groups. (D) CD4+ T cell count was not significantly different between groups. (E) TREG percentage of CD4+ T cells was significantly increased compared to MOG (no rapa) control. (F) Similarly, TREG count confirmed that the number of TREG was significantly higher compared to the MOG (no rapa) control. This study was completed in triplicate. For each study, N = 3. An ANOVA with a Tukey multiple comparisons post-hoc test was used to compare each group. Statistical significance is indicated by a (*). **p < 0.01. Created with BioRender.
Since T cell polarization and expansion is a dynamic process, we hypothesized that differences in rapa:MOG polarization may be distinguishable at earlier timepoints of T cell-APC engagement. To test this hypothesis, we analyzed T cells after 48 h of co-culture instead of 96 h (Figure 5A). Interestingly, we observed a significant increase in T cell counts of low rapa:MOG treated cells compared to MOG (no rapa) treated cells (Figure 5B). Similar to the 96-h timepoint, rapa:MOG MP treatment significantly expanded TREG across all tested ratios. Excitingly, however, there was a significant dependence on the ratio of rapa:MOG for the polarization of TREG percentage (Figure 5C). Since we observed this ratio dependent trend in TREG, we hypothesized that there would also be a decrease in functional inflammatory profile of T cells. First, we used intra-cellular antibody staining to assess the transcription T-bet which is associated with inflammatory T cell subtypes, and found that rapa:MOG treatment significantly reduced T-bet expression (Figure 5D). We also used RT-qPCR to assess T cell gene expression for a key functional cytokine produced during inflammation, IFN-ɣ, and found that the highest rapa:MOG treatment led to the lowest expression of the IFN-ɣ gene (Figure 5E). Taken together, these results indicate MPs loaded with a high ratio of rapa:MOG MP play an important role in polarizing antigen-specific T cell differentiation to TREG and correspondingly decreasing expression of inflammatory markers.
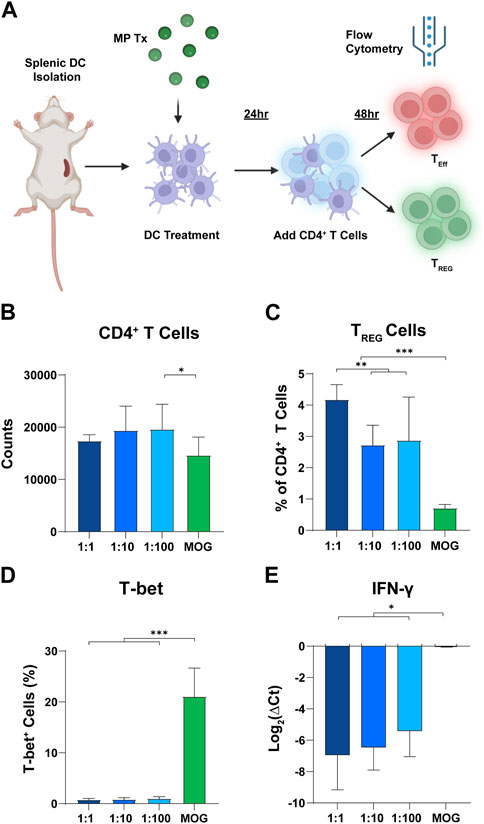
FIGURE 5. High rapa:MOG promotes greater expansion of TREG after 48 h. (A) Overview schematic of co-culture assay. (B) There was no significant difference in T cell count between rapa:MOG treated groups. (C) High ratio of rapa:MOG induced the highest percentage of TREG. All groups induced significant increase in TREG percentage over MOG (no rapa) control. (D) Rapa:MOG polarized cells away from expression of the inflammatory transcription factor T-bet. (E) RT-qPCR analysis showed rapa:MOG treated cells had significantly lower expression of the gene for the inflammatory cytokine IFN-ɣ. This study was completed in triplicate. An ANOVA with a Tukey multiple comparisons post-hoc test was used to compare each group. Statistical significance is indicated by a (*). *p < 0.05, **p < 0.01, ***p < 0.001. Created with BioRender.
Biomaterial enabled strategies for drug delivery are gaining increasing interest due to their ability to precisely control release parameters such as time, location, and concentration. Particularly, rapa delivery through both microparticle and nanoparticle formulations in vivo has shown significant immune engineering promise across multiple species models in the study of chronic inflammation, transplant rejection, and auto-immune disorders (Fan et al., 2019; Fisher et al., 2019; Nguyen et al., 2022). Although many biomaterial vehicles may be used to facilitate rapa delivery, much work continues to rely on the use of PLGA due to its versatility and extensive use in U.S. Food and Drug Administration (FDA) approved products (E. Emerson et al., 2020). More recently, the addition of specific self-antigens to rapa delivery strategies has shown promise in directing the formation of antigen-specific TREG. The work in this paper builds off of previous studies that have shown that co-encapsulation of self-antigen (MOG) and a modulatory cue (rapa) can promote the polarization of antigen-specific regulatory T cells for treatment of pre-clinical model of MS (experimental autoimmune encephalomyelitis) (Tostanoski et al., 2016). Specifically, this paper studies how the ratio of rapa and MOG co-localized in MPs controls the activation of APCs and the formation of TREG in vitro.
Diffusion limited MPs rely on their size for retention within the LN in order to facilitate the direct release of cues to the tissue (Jewell, Bustamante López and Irvine, 2011). Although the quantity of rapa was decreased by over 100-fold, particles retained previously reported engineered diameter that would allow for future in vivo testing (Jewell, Bustamante López and Irvine, 2011). Additionally, particle size plays an important role in phagocytic uptake, which could have implications for in vitro TREG expansion therapies. Interestingly, both the hydrophilic MOG cargo, and hydrophobic rapa cargo had an influence on MP size. However, our results showed that particles maintained a diameter that would permit phagocytosis.
APC co-stimulation is an important requirement for T cell activation and polarization into either an inflammatory or anti-inflammatory phenotype. However, rapa is a potent mTOR inhibitor that has been previously linked to reduced expression of co-stimulation receptors on APCs (Carey, Gammon and Jewell, 2021; Gosselin et al., 2021). Thus, it is important to understand if high ratios of rapa:MOG have a deleterious effect on DC viability and co-stimulation. Our study focused on the expression of the co-stimulatory molecules CD86 and CD80 which are the main ligands for the T cell activation receptor CD28 which is needed to promote T cell survival. The results of our experiments showed that DC expression of co-stimulation receptors was insensitive to rapa:MOG treatment. This suggests that both the low and high ratio of rapa:MOG exerted similar modulatory effects on DCs after 24 h.
To understand the effect of rapa:MOG ratio on the interaction of DCs and antigen-specific T cells, we co-cultured and treated primary derived DCs and primary derived transgenic MOG recognizing T cells. As the transgenic T cells only express one receptor, groups without MOG antigen had low viability and were excluded from analysis. Rapa is a well-known immunomodulator that drives proliferation of TREG, and our experiment showed that groups treated with MPs containing rapa indeed adopted a CD25+ FoxP3+ regulatory phenotype. Although the ratio of rapa:MOG did not significantly change the percentage of polarized TREG after 96 h, we did observe significant ratio dependent changes after 48 h. The lack of significant difference between rapa:MOG treatments at later timepoints could potentially be attributed to the aggressive expansion kinetics of transgenic T cells engineered to recognize specific self-antigens. Alternatively, the half-life of rapa is approximately 60 h and may elicit its strongest effect at earlier timepoints. Additionally, T cells would not have undergone as many divisions at earlier timepoints, and thus the concentration of rapa per cell could have a more evident effect-potentially as a function of cell metabolism (Zhang et al., 2019). However, these ratio dependent differences were only observed for the percentage of TREG, and not for T-bet transcription factor expression or IFN-ɣ gene expression. Despite the highly constrained nature of this study, these results lay important groundwork for interrogating the role of rapa and self-antigen in directing anti-inflammatory responses at initial stages of treatment.
Although this study focused on testing how rapa:MOG ratios shape TREG induction, it has been shown that rapa can have modulatory effects beyond TREG induction such as in the generation of “memory” like phenotypes. Future work should seek to understand how the ratio of rapa and self-antigen modulate the adoption of memory cells which could be useful in engineering long lasting tolerizing T cell responses for treatment of auto-immune diseases. Although the concept of regulatory memory is continually evolving, metabolic reprogramming of T cells is known to play a critical role in cell fate. Interestingly, our results showed rapa:MOG led to a trend in lower T cell counts at 96 h, but actually resulted in a significant increase in the low ratio group at 48 h. This suggests that the MOG and rapa work synergistically to modulate cell functions such as metabolism and proliferation. Thus, future studies should attempt to mechanistically differentiate antigen-specific TREG responses from non-antigen-specific responses which potentially carries important implications for in vitro TREG expansion applications. Finally, the in vitro culture conditions assayed here do not fully recapitulate the complexity of the immune microenvironment. Thus, future studies should test how rapa:MOG ratios can shape efficacy or immune memory under in vivo conditions.
Our work here showed that the ratio of rapa:MOG encapsulated in polymer MPs does not differentially alter the engineered properties of MPs for future in vivo intra-LN translation. Rapa:MOG ratio did not modulate APC expression of costimulatory receptors involved in T cell activation highlighting DC tolerance to rapa. Interestingly, the ratio of rapa:MOG differentially drives the expansion of TREG, and anti-inflammatory T cell profile of CD4+ T cells.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by University of Maryland Institutional Animal Care and Use Committee (IACUC).
CB, SS, RO, and CJ designed the experiments. CB and CJ wrote the manuscript. CB and SS performed all experiments. CB characterized all MP formulations and performed all in vitro cell studies. CB and SS performed RT-qPCR experiments. RO and SS contributed to RT-qPCR experiment design, and data analysis. All authors contributed to the article and approved the submitted version.
This data was supported in part by NIH R01 AI169686.
The authors acknowledge the University of Maryland Department of Laboratory Animal Resources staff for their support with animal husbandry. The authors acknowledge support from University of Maryland Bioworkshop core facility for use of its QuantStudio 7 Flex Real-Time PCR System, and NanoDrop 2000c Spectrophotometer.
CJ and RO are employees of the VA Maryland Health Care System. The reviews reported here do not reflect the views of the VA or the United States Government. CJ has an equity position with Cartesian Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The view reported here do not reflect those of the United States Government or Department of Veteran Affairs.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2023.1184938/full#supplementary-material
Andorko, J. I., Hess, K. L., and Jewell, C. M. (2015). Harnessing biomaterials to engineer the lymph node microenvironment for immunity or tolerance. AAPS J. 17 (2), 323–338. doi:10.1208/s12248-014-9708-2
Arneth, B. M. (2019). Impact of B cells to the pathophysiology of multiple sclerosis. J. Neuroinflammation 16 (1), 128. doi:10.1186/s12974-019-1517-1
Blasi, P. (2019). Poly(lactic acid)/poly(lactic-co-glycolic acid)-based microparticles: An overview. J. Pharm. Investigation 49 (4), 337–346. doi:10.1007/s40005-019-00453-z
Bookstaver, M. L., Tsai, S. J., Bromberg, J. S., and Jewell, C. M. (2018). Improving vaccine and immunotherapy design using biomaterials. Trends Immunol. 39 (2), 135–150. doi:10.1016/j.it.2017.10.002
Carey, S. T., Gammon, J. M., and Jewell, C. M. (2021). Biomaterial-enabled induction of pancreatic-specific regulatory T cells through distinct signal transduction pathways. Drug Deliv. Transl. Res. 11 (6), 2468–2481. doi:10.1007/s13346-021-01075-5
Danhier, F., Ansorena, E., Silva, J. M., Coco, R., Le Breton, A., and Préat, V. (2012). PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 161 (2), 505–522. doi:10.1016/j.jconrel.2012.01.043
Danikowski, K. M., Jayaraman, S., and Prabhakar, B. S. (2017). Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflammation 14 (1), 117. doi:10.1186/s12974-017-0892-8
Dendrou, C. A., Fugger, L., and Friese, M. A. (2015). Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15 (9), 545–558. doi:10.1038/nri3871
Emerson, E., Hiremath, S. C., and Weaver, J. D. (2020). Biomaterial-based approaches to engineering immune tolerance. Biomaterials Sci. 8 (24), 7014–7032. doi:10.1039/D0BM01171A
Fan, Y., Zheng, X., Ali, Y., Berggren, P. O., and Loo, S. C. J. (2019). Local release of rapamycin by microparticles delays islet rejection within the anterior chamber of the eye. Sci. Rep. 9 (1), 3918. doi:10.1038/s41598-019-40404-0
Fisher, J. D., Balmert, S. C., Zhang, W., Schweizer, R., Schnider, J. T., Komatsu, C., et al. (2019). Treg-inducing microparticles promote donor-specific tolerance in experimental vascularized composite allotransplantation. Proc. Natl. Acad. Sci. 116 (51), 25784–25789. doi:10.1073/pnas.1910701116
Gammon, J. M., Carey, S. T., Saxena, V., Eppler, H. B., Tsai, S. J., Paluskievicz, C., et al. (2023). Engineering the lymph node environment promotes antigen-specific efficacy in type 1 diabetes and islet transplantation. Nat. Commun. 14 (1), 681. doi:10.1038/s41467-023-36225-5
Gammon, J. M., Gosselin, E. A., Tostanoski, L. H., Chiu, Y. C., Zeng, X., Zeng, Q., et al. (2017). Low-dose controlled release of mTOR inhibitors maintains T cell plasticity and promotes central memory T cells. J. Control. Release 263, 151–161. doi:10.1016/j.jconrel.2017.02.034
Gandhi, R., Laroni, A., and Weiner, H. L. (2010). Role of the innate immune system in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 221 (1), 7–14. doi:10.1016/j.jneuroim.2009.10.015
Gosselin, E. A., Noshin, M., Black, S. K., and Jewell, C. M. (2021). Impact of excipients on stability of polymer microparticles for autoimmune therapy. Front. Bioeng. Biotechnol. 8, 609577. doi:10.3389/fbioe.2020.609577
Gosselin, E. A., Tostanoski, L. H., and Jewell, C. M. (2017). Controlled release of second generation mTOR inhibitors to restrain inflammation in primary immune cells. AAPS J. 19 (4), 1175–1185. doi:10.1208/s12248-017-0089-1
Hauser, S. L., and Cree, B. A. C. (2020) ‘Treatment of multiple sclerosis: A review’, Am. J. Med., 133(12), pp. 1380–1390. doi:10.1016/j.amjmed.2020.05.049
Hester, J., Schiopu, A., Nadig, S., and Wood, K. (2012). Low-Dose rapamycin treatment increases the ability of human regulatory T cells to inhibit transplant arteriosclerosis in vivo. Am. J. Transplant. 12 (8), 2008–2016. doi:10.1111/j.1600-6143.2012.04065.x
Huang, W.-J., Chen, W.-W., and Zhang, X. (2017). Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 13 (6), 3163–3166. doi:10.3892/etm.2017.4410
Jewell, C. M., Bustamante López, S. C., and Irvine, D. J. (2011). In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc. Natl. Acad. Sci. 108 (38), 15745–15750. doi:10.1073/pnas.1105200108
Kimura, K. (2020). Regulatory T cells in multiple sclerosis. Clin. Exp. Neuroimmunol. 11 (3), 148–155. doi:10.1111/cen3.12591
Maldonado, R. A., LaMothe, R. A., Ferrari, J. D., Zhang, A. H., Rossi, R. J., Kolte, P. N., et al. (2015). Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc. Natl. Acad. Sci. 112 (2), E156–E165. doi:10.1073/pnas.1408686111
Marín Morales, J. M., Münch, N., Peter, K., Freund, D., Oelschlägel, U., Hölig, K., et al. (2019). Automated clinical grade expansion of regulatory T cells in a fully closed system. Front. Immunol. 10, 38. doi:10.3389/fimmu.2019.00038
März, A. M., Fabian, A. K., Kozany, C., Bracher, A., and Hausch, F. (2013). Large FK506-binding proteins shape the pharmacology of rapamycin. Mol. Cell. Biol. 33 (7), 1357–1367. doi:10.1128/MCB.00678-12
Mosanya, C. H., and Isaacs, J. D. (2019). Tolerising cellular therapies: What is their promise for autoimmune disease? Ann. Rheumatic Dis. 78 (3), 297–310. doi:10.1136/annrheumdis-2018-214024
Mukhopadhyay, S., Frias, M. A., Chatterjee, A., Yellen, P., and Foster, D. A. (2016). The enigma of rapamycin dosage. Mol. Cancer Ther. 15 (3), 347–353. doi:10.1158/1535-7163.MCT-15-0720
Nguyen, T. L., Choi, Y., Im, J., Shin, H., Phan, N. M., Kim, M. K., et al. (2022). Immunosuppressive biomaterial-based therapeutic vaccine to treat multiple sclerosis via re-establishing immune tolerance. Nat. Commun. 13 (1), 7449. doi:10.1038/s41467-022-35263-9
Romano, M., Fanelli, G., Albany, C. J., Giganti, G., and Lombardi, G. (2019). Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front. Immunol. 10. doi:10.3389/fimmu.2019.00043
Sato, Y., Keino, H., Nakayama, M., Kano, M., and Okada, A. A. (2021). Effect of in vivo expansion of regulatory T cells with IL-2/anti-IL-2 antibody complex plus rapamycin on experimental autoimmune uveoretinitis. Ocular Immunol. Inflamm. 29 (7–8), 1520–1529. doi:10.1080/09273948.2020.1757119
Saunders, R. N., Metcalfe, M. S., and Nicholson, M. L. (2001). Rapamycin in transplantation: A review of the evidence. Kidney Int. 59 (1), 3–16. doi:10.1046/j.1523-1755.2001.00460.x
Saxena, V., Li, L., Paluskievicz, C., Kasinath, V., Bean, A., Abdi, R., et al. (2019). Role of lymph node stroma and microenvironment in T cell tolerance. Immunol. Rev. 292 (1), 9–23. doi:10.1111/imr.12799
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3 (6), 1101–1108. doi:10.1038/nprot.2008.73
Su, Y., Zhang, B., Sun, R., Liu, W., Zhu, Q., Zhang, X., et al. (2021). PLGA-Based biodegradable microspheres in drug delivery: Recent advances in research and application. Drug Deliv. 28 (1), 1397–1418. doi:10.1080/10717544.2021.1938756
Torkildsen, Ø., Myhr, K.-M., and Bø, L. (2016). Disease-modifying treatments for multiple sclerosis – A review of approved medications. Eur. J. Neurology 23 (S1), 18–27. doi:10.1111/ene.12883
Tostanoski, L. H., Chiu, Y. C., Gammon, J., Simon, T., Andorko, J., Bromberg, J., et al. (2016). Reprogramming the local lymph node microenvironment promotes tolerance that is systemic and antigen specific. Cell. Rep. 16 (11), 2940–2952. doi:10.1016/j.celrep.2016.08.033
Tostanoski, L. H., Gosselin, E. A., and Jewell, C. M. (2016). Engineering tolerance using biomaterials to target and control antigen presenting cells. Discov. Med. 21 (117), 403–410.
Vakrakou, A. G., Alexaki, A., Brinia, M. E., Anagnostouli, M., Stefanis, L., and Stathopoulos, P. (2022). The mTOR signaling pathway in multiple sclerosis; from animal models to human data. Int. J. Mol. Sci. 23 (15), 8077. doi:10.3390/ijms23158077
Weissert, R. (2013). The immune pathogenesis of multiple sclerosis. J. Neuroimmune Pharmacol. 8 (4), 857–866. doi:10.1007/s11481-013-9467-3
Yellen, P., Saqcena, M., Salloum, D., Feng, J., Preda, A., Xu, L., et al. (2011). High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell. Cycle 10 (22), 3948–3956. doi:10.4161/cc.10.22.18124
Yoo, Y. J., Kim, H., Park, S. R., and Yoon, Y. J. (2017). An overview of rapamycin: From discovery to future perspectives. J. Industrial Microbiol. Biotechnol. 44 (4–5), 537–553. doi:10.1007/s10295-016-1834-7
Keywords: microparticle and nanoparticle, autoimmunity, mTOR, antigen-specificity, tolerance, biomaterial, vaccine, immunotherapy
Citation: Bridgeman CJ, Shah SA, Oakes RS and Jewell CM (2023) Dissecting regulatory T cell expansion using polymer microparticles presenting defined ratios of self-antigen and regulatory cues. Front. Bioeng. Biotechnol. 11:1184938. doi: 10.3389/fbioe.2023.1184938
Received: 12 March 2023; Accepted: 12 June 2023;
Published: 27 June 2023.
Edited by:
Zuhao Li, Jilin University, ChinaReviewed by:
Matjaž Jeras, University of Ljubljana, SloveniaCopyright © 2023 Bridgeman, Shah, Oakes and Jewell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher M. Jewell, Y21qZXdlbGxAdW1kLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.