
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol., 20 June 2022
Sec. Synthetic Biology
Volume 10 - 2022 | https://doi.org/10.3389/fbioe.2022.936914
This article is part of the Research TopicExploration of Highly Active Enzymes, Performance Enhancement and Enzymatic Processing TechniquesView all 7 articles
 Yang Shen
1,2
†
Yang Shen
1,2
†
 Yulu Wang
2
†
Yulu Wang
2
† Xue Wei2Boting Wen2
Xue Wei2Boting Wen2
 Shujun Liu
2Huishuang Tan3Jingjian Zhang4
Shujun Liu
2Huishuang Tan3Jingjian Zhang4
 Shuli Shao
1*
Shuli Shao
1*
 Fengjiao Xin
2*
Fengjiao Xin
2*The human gut microbiota play essential roles in metabolism and human health, especially by enzymatically utilizing dietary fiber that the host cannot directly digest and releasing functional components including short-chain fatty acids (SCFAs) and hydroxycinnamic acids (e.g., ferulic acid). In our previous study, seven potential feruloyl esterase (FAE) genes were identified from the gut microbiota. In the current work, one of the genes encoding a novel FAE (DfFAE) from Dorea formicigenerans of Firmicutes was bacterially expressed, purified and characterized. The 30.5 kDa type-A DfFAE has an optimum pH and temperature of 8.4 and 40 °C, respectively, exhibiting a higher substrate specificity toward short-chain acyl-ester substrate (pNPA). The AlphaFold2 based ab initio structural modeling revealed a five α-helices cap domain that shaped an unusually narrow and deep active site pocket containing a specific substrate access tunnel in DfFAE. Furthermore, rational design strategy was subjected to the active site pocket in an aim of improving its enzymatic activities. The mutants V252A, N156A, W255A, P149A, and P186A showed 1.8 to 5.7-fold increase in catalytic efficiency toward pNPA, while W255A also exhibited altered substrate preference toward long-chain substrate pNPO (45.5-fold). This study highlighted an unusual active site architecture in DfFAE that influenced its substrate selectivity and illustrated the applicability of rational design for enhanced enzymatic properties.
The concept of dietary fiber and prebiotics has long been strongly promoted by the nutritional community. These dietary components resist breakdown in the human small intestine and are utilized by gut microbes in the colon that live in symbiosis with the body (Slavin, 2013). The activities of gut microbiota profoundly influence the metabolism and human health (Eckburg et al., 2005; Sonnenburg et al., 2010). Xylan, the second most abundant saccharides in plant kingdom, is an important constituent of dietary fiber and prebiotic supplements. Structurally, the backbone of xylan commonly consists of β-1,4-linked D-xylopyranose residues that are often substituted with a variety of side chains, including glucuronyl groups, acetyl groups and arabinosyl groups. Ferulic acid or other hydroxycinnamic acids, are additionally estified to the C-5 position of some arabinosyl moieties, which may further crosslink to lignin or neighboring xylan chains by forming diferulate, thus increasing the recalcitrance of plant saccharides to enzymatic hydrolysis in the colon of humans (Scheller and Ulvskov, 2010).
Ferulic acid or feruloyl esterases (FAEs, EC 3.1.1.73) are a subgroup of carboxylic ester hydrolases, which catalyze the cleavage of the ester bond between arabinofuranose and hydroxycinnamic acid. Removing the cross-linked ferulic acid side chain is necessary for the efficient degradation of feruloylated xylan polymers (Williamson et al., 1998; Faulds, 2010; Scheller and Ulvskov, 2010). Hence, FAEs possess wide biotechnological application in the biofuel, food and pharmaceutical industries (Oliveira et al., 2019). FAEs belong to Carbohydrate Esterase Family 1 (CE1) and initially categorized into four subclasses (A, B, C and D) according to their substrate specificity and sequence similarity. In general, FAEs display a common catalytic mechanism involving the Ser-His-Asp catalytic triad, and adopt the structure of canonical α/β hydrolase fold, which are usually composed of the core domain (also known as the catalytic domain) containing the catalytic triad and the cap domain positioning atop the core domain (Oliveira et al., 2019). The cap domain confines the active site cavity and its conformation influences substrate recognition and catalytic properties in different FAEs (Bauer et al., 2020).
FAEs are mostly found in microorganisms, but also in edible mushrooms (Wang et al., 2014) and plants (de O Buanafina et al., 2019). FAEs of fungal origin, such as Aspergillus niger (Hermoso et al., 2004), have been extensively studied. Recently, FAEs of gut symbiotic bacteria have gained increasing attention. An increasing number of FAEs have been identified from Bifidobacteria (Raimondi et al., 2015; Fritsch et al., 2017), Lactobacillus (Lai et al., 2009), or Bacteroides (Wefers et al., 2017), but they display distinct catalytic features with respect to properties such as substrate specificities and optimal reaction conditions. These gut microbial-derived FAEs are the main pathway for the release of hydroxycinnamic acids from dietary fiber, which are released in the form of free acids and absorbed into the circulatory system for action (Andreasen et al., 2001; Faulds, 2010). Hydroxycinnamic acids play a positive role in human health due to their excellent antioxidant, anti-inflammatory, anti-diabetes, anti-cancer and neuroprotective capacities (El-Seedi et al., 2012). Thus, the discovery and characterization of novel gut-derived FAEs is of great importance to explain the intestinal hydrolysis and releasing mechanism of dietary hydroxycinnamic acids on behalf of the human health.
In our previous work, seven potential FAE genes were identified from in vitro fermentation of human fecal slurry using metagenomic sequencing (Chen et al., 2020) in which one FAE from Alistipes shahii (AsFAE) was characterized (Wei et al., 2021). Here, a novel FAE from Dorea formicigenerans (DfFAE) of Firmicutes was bacterially expressed and purified to assess its enzymatical properties. DfFAE belongs to type-A FAE and has a higher preference for hydrolyzing short-chain ester substrate (p-nitrophenyl acetate, pNPA), which shows superior catalytic activity than AsFAE. To understand the structural basis of DfFAE catalytic properties, an ab initio modeling using AlphaFold2 was generated, which highlighted a relatively narrow and deep substrate binding pocket including a specific substrate access tunnel. Then site-specific mutagenesis and kinetic studies were carried out on residues around the active site tunnel, which identified five mutants with improved catalytic efficiency and/or broadened substrate preference. In silico analysis shed light on the possible molecular mechanisms underlying improved enzymatic properties. Collectively, this study presented the biochemical, structural and rational design studies of a novel FAE derived from Dorea formicigenerans, sharpening our mechanistic understanding of diverse mode of action in various FAEs.
The vector pET-28a (+) for plasmid construction was obtained from Novagen (Madison, WI, United States). The Escherichia coli (E. coli) Trans1 (T1) strain purchased from TransGen Biotech (Beijing, China) was used to recombinant plasmids amplification, and the E. coli T7 Express strain purchased from Biomed (Beijing, China) was used to recombinant proteins expression.
A series of substrates used in this experiment: p-nitrophenyl acetate (pNPA), p-nitrophenyl butyrate (pNPB), and p-nitrophenyl octanoate (pNPO) were purchased from Sigma-Aldrich (St. Louis, MO, United States), and p-nitrophenyl trans-ferulate (pNPF) was purchased from MREDA (Columbia, MD, United States); methyl ferulate (MFA), methyl sinapinate (MSA), methyl caffeate (MCA) and methyl p-coumarate (MpCA) were purchased from TCI (Shanghai, China); the standards of ferulic acid (FA), sinapic acid (SA), caffeic acid (CA) and p-coumaric acid (pCA) were purchased from Sigma-Aldrich (St. Louis, MO, United States).
The sequence similarity searches were carried out using the Blastp search engine from the National Center for Biotechnology Information (NCBI) database in conjunction with the PDB database. The SingnalP 5.0 was applied to predict the sequence of potential signal peptides. ClustalX version 2.0 (Larkin et al., 2007) and ESPript version 3.0 (Robert and Gouet, 2014) were used to align multiple sequences. The phylogenetic tree was built with the maximum likelihood estimation by MEGA version 11.0 (Tamura et al., 2021).
The accession number of Dffae sequence is WP_117657856.1 in the NCBI database. The cDNA sequence after E. coli preference codon optimization was synthesized (BGI-write, Beijing, China), and restriction endonuclease sites, BamH I and Xho I (Thermo Fisher Scientific, Hudson, NH, United States), were added at both ends of the sequence. Then the target fragment was inserted into the restriction enzyme-digested pET-28a (+) plasmid and transmitted into E. coli T7 for expression.
The mutagenesis were carried out standard Quikchange PCR procedure. pET28a-Dffae was used as the template and specific primers were used for site-directed mutagenesis. The original template was digested by Dpn I enzyme (Thermo Fisher Scientific, Hudson, NH, United States) and then transformed into E. coli T1 for amplification. Clones successfully mutated were verified by DNA sequencing.
E. coli T7 cells containing Dffae and its mutants were cultured in Luria-Bertani (LB) medium adding 100 μg ml−1 kanamycin at 37 °C, and shaked at 220 rpm for 3–4 h. When OD600 came to 0.8, expression of protein was induced by 0.2 mM isopropyl thiogalactoside (IPTG) at 16°C for 12 h. The cultures were centrifuged at 5,000 rpm and resuspended in 50 mM Tris-HCl (pH 8.0) buffer supplemented with 300 mM NaCl, 10 mM MgCl2, 1 mM phenylmethanesulfonyl fluoride (PMSF), 5 mg L−1 lysozyme, 0.03 mg L−1 DNase I. High pressure homogenization was used to lyse the cells, and the cell fragment supernatant was loaded on affinity chromatography (Ni2+-NTA, Qiagen, Hilden, Germany) eluted with 300 mM imidazole. The proteins were further purified using anion exchange column chromatography (Source 15Q, GE Healthcare Life Sciences, Issaquah, WA, United States) and gel filtration chromatography (Superdex™ 200 Increase 10/300 GL column, GE Healthcare Life Sciences, Issaquah, WA, United States). Ultimately, the purified DfFAE was analyzed by 12% SDS-PAGE and quantified by Bradford protein quantification kit (Solarbio, Beijing, China), and then stored at -80 °C for later use.
The basic biochemical properties of DfFAE were measured using a spectrophotometer (SpectraMax 190, Molecular Devices) with pNPA (dissolved in DMSO) as the substrate. The reactant mixture (total volume was 0.3 ml) included 0.3 mM pNPA and 0.3 μM DfFAE, and 2.5% Ttrion X-100 was added to all buffer solutions to stabilize the substrate in these experiments. The pH optima was determined at 40°C by reacting in 10 mM different buffers, including phosphate-citrate buffer (pH 4.0 and 5.0), K2HPO4-KH2PO4 buffer (pH 6.0, 6.6 and 7.0) and Tris-HCl buffer (pH 7.5, 8.0, 8.4 and 8.9). For pH stability measurements, the residual activity was detected at 40°C and pH 8.4, after incubating DfFAE in different pH buffers (pH 4.0–8.9) at 4°C for 4 h. The temperature optima was determined at different temperatures of 4 °C and 20–60°C (10°C intervals) in 10 mM Tris-HCl buffer (pH 8.4). The temperature stability was assayed by incubating DfFAE in the optimum pH buffer at different temperatures of 4°C and 20–60°C (10°C intervals) for 0.25, 0.75, 1, 2 and 4 h, and the residual activity was detected under the optimum pH and temperature conditions. To investigate the effects of metal ions and chemicals, DfFAE was incubated in 10 mM Tris-HCl buffer (pH 8.4) containing different metal ions and detergents at a final concentration of 1 mM (w/v) and 1% (v/v), respectively, for 15 min at room temperature. Then the residual activity was measured under the optimum pH and temperature conditions. The enzymatic activity without additives was defined as 100%. Three replicate experiments were performed for each reaction, and all data were shown as averages with the standard deviation.
The kinetic properties of DfFAE and its mutants were assayed in 10 mM Tris-HCl buffer (pH 8.4) containing 2.5% Triton X-100 at 40°C, using different concentrations (0.25–2 mM) of pNPA, pNPB and pNPO as substrates. The amount of produced pNP was detected at 405 nm and calculated based on the pNP standard curve. The initial rate of pNP release was plotted against substrate concentration, and the Michaelis-Menten constant (K m) and maximum reaction velocity (V max) were calculated by nonlinear regression analysis using Graphpad Prism eight software. The turnover number (k cat) was calculated as V max divided by the corresponding enzymatic concentration.
MFA, MSA, MCA and MpCA were used as substrates to verify the feruloyl esterase activity of DfFAE. The reaction mixture contained 1.25 mM substrate, 10 mM Tris-HCl (pH 8.4) and 2.5% Triton X-100. The reaction was initiated by adding 8 μM DfFAE at 40°C for 1 h, followed by the addition of equal volume of methanol to terminate the reaction. After being filtered through an organic membranefilter with a pore size of 0.22 μm, the samples were loaded onto the column and HPLC analysis was performed. The Agilent ZORBAX SB-C18 (4.6 × 250 mm, 5 μm) column was used with mobile phase A (water-formic acid, 99:1, v/v) and mobile phase B (methanol-formic acid, 99:1, v/v). The elution procedure was as follows: the total time was set to 25 min, at a flow rate of 0.5 ml/min, 0–9.5 min, 40% mobile phase B; 9.5–19 min, 85% mobile phase B; and 19–25 min returned to 40% mobile phase B. The column was finally equilibrated by the first-step conditions of the elution procedure. The product profiles were monitored at 323 nm and identified by comparing their retention periods to the standards’ retention times.
The structural model of DfFAE was constructed using the open-source program AlphaFold2 (https://github.com/deepmind/alphafold). The program returned a total of five results, which were ranked as rank0-4 in descending order of accuracy, and the best prediction rank0 was selected for the following analysis. 98% of the residues under this model had pLDDT (predicted Local Distance Difference Test) scores greater than 80, hence results were generally reliable. Molecular dockings were carried out using the open-source program AutoDock_Vina (https://vina.scripps.edu/). The ligands (pNPA, pNPB and pNPO) structures were downloaded from NCBI PubChem Database (https://pubchem.ncbi.nlm.nih.gov/). The ligand-free pdb files of DfFAE and ligands were converted into pdbqt files using MGLTools (http://mgltools.scripps.edu/), a grid box of 50 × 50 × 50 Å3 was generated to cover the active site. The dock program with default parameters generated the top 20 best affinity results for each ligand, and the reasonable conformation with the lowest binding energy was selected for subsequent analysis. The structural comparison, visualization and homology modeling of W255A mutant were performed with PyMOL 2.3 software.
The full-length nucleotide sequence of Dffae is 825 bp and composed of 274 amino acids, which was defined as the α/β hydrolase in the NCBI protein database (accession number: WP_117657856.1). The theoretical molecular weight and isoelectric point were estimated as 30.5 kDa and 5.82, respectively, according to Expasy website (https://web.expasy.org/protparam/). The SignalP (Almagro Armenteros et al., 2019) analysis indicated that DfFAE lacks the possible signal peptide and thus might be a cytoplasmic protein. A BlastP search against the Protein Data Bank (PDB) database showed that DfFAE had the highest sequence homology of 29.66% with a thermophilic esterase TtEst from Thermogutta terrifontis (PDB code: 4UHD) (Figure 1A). 16 characterized FAE sequences that were classified as A, B, C and D subtypes were used for phylogenetic tree analysis with DfFAE, containing both eukaryotic and prokaryotic origins. The results showed that these sequences were not clustered strictly in accordance with the FAE classification, and DfFAE is adjacent to type A FAEs of prokaryotic origin (Figure 1B). Sequence alignment of DfFAE with TtEst and AsFAE (sequence identity of 19.5%) identified three amino acids (Ser100, Asp223, and His251) as the putative catalytic triad of DfFAE. Ser100 is situated in the pentapeptide GXSXG consensus motif, which is a typical feature of esterases (Figure 1A).
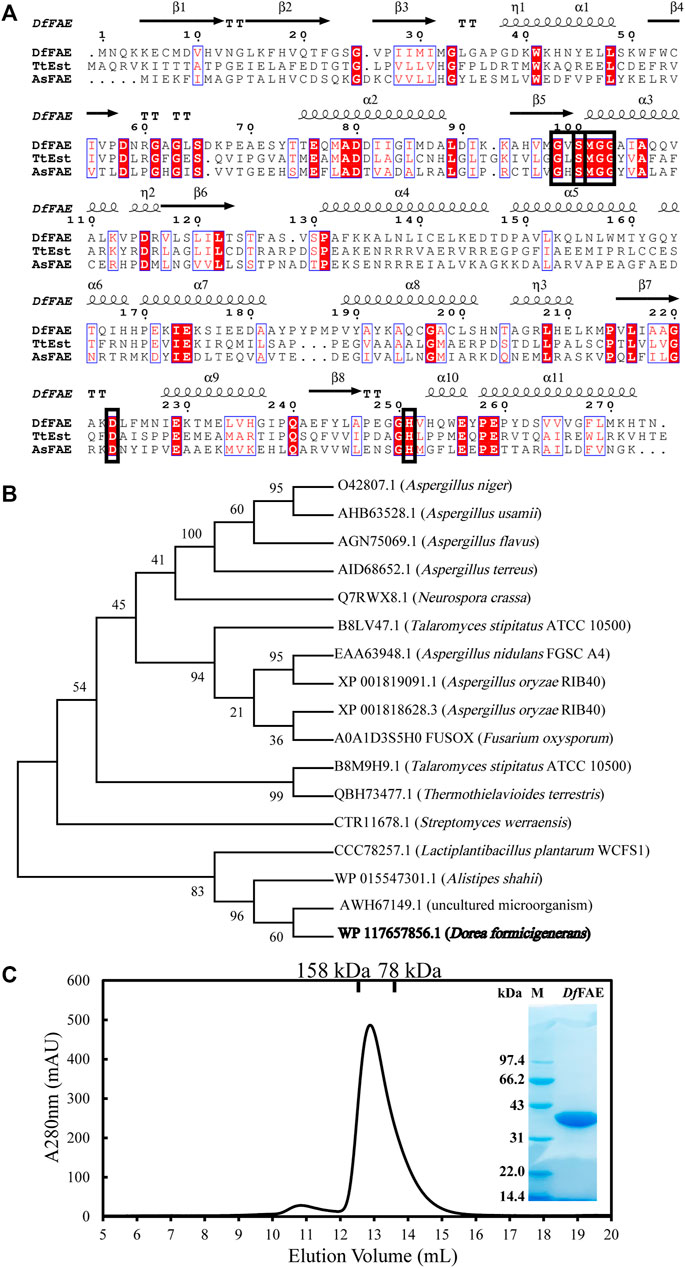
FIGURE 1. Sequence analysis and protein expression and purification of DfFAE. (A) Amino acid sequence alignment of DfFAE, TsEst and AsFAE. The ClustalX 2.0 tool was used to align the sequences, and the ESpript 3.0 program was used to display them. The alpha helices and beta strands are displayed above the sequences denoted as α and β, respectively. The red background denotes highly conserved residues. The black box denotes the highly conserved catalytic triad and pentapeptide GXSXG motif (B) Phylogenetic tree analysis of DfFAE and other classified FAEs. The sequences in the phylogenetic tree are, from top to bottom, AnFAEA (O42807.1) of Aspergillus niger; AuFaeA (AHB63528.1) of Aspergillus usamii; AfFaeA (AGN75069.1) of Aspergillus flavus; AtFaeA (AID68652.1) of Aspergillus terreus; NcFaeD-3.544 (Q7RWX8.1) of Neurospora crassa; TsFaeC (B8LV47.1) of Talaromyces stipitatus; AN1772.2 (EAA63948.1) of Aspergillus nidulans; AoFaeB (XP_001818628.3) and AoFaeC (XP_001819091.1) of Aspergillus oryzae; FoFaeC (A0A1D3S5H0_FUSOX) of Fusarium oxysporum; SwFAED (CTR11678.1) of Streptomyces werraensis; LP_0796 (CCC78257.1) of Lactobacillus plantarum; AsFAE (WP_015547301.1) of Alistipes Shahii; FAE-Xuan (AWH67149.1) of Soil Metagenomic Library; DfFAE is shown in bold (C) SEC and SDS-PAGE analysis of the purified DfFAE. The elution volume is 12.9 ml, and the molecular mass markers are indicated above: 75 kDa standard protein corresponds to conalbumin, and 158 kDa standard protein corresponds to aldolase. The SDS-PAGE gel was stained with Coomassie blue. M represents the molecular weight marker of protein standards.
To confirm the putative function, the recombinant DfFAE was overexpressed in E. coli T7 host cells and purified using Ni2+-NTA affinity column, followed by Source 15Q ion exchange and Superdex 200 gel filtration chromatographies. An obvious band at 34 kDa can be seen with 12% SDS-PAGE analysis, consistent with the theoretical molecular weight of DfFAE plus the N-terminal His6-tag, thrombin cutting site and T7-tag sequences from pET-28a (+) vector (34.1 kDa). The size exclusion chromatography (SEC) analysis showed that the apparent MW of the protein in solution was about 117 kDa, suggesting that the recombinant DfFAE is primarily trimeric and purified to homogeneity (Figure 1C).
To verify its feruloyl esterase activity, DfFAE was assayed using four model hydroxycinnamoyl ester substrates, MFA, MSA, MCA and MpCA. As shown in Figure 2, DfFAE can partially hydrolyze MFA, MSA and MpCA. However no detectable activity toward MCA was observed using the same enzyme load (a final concentration of 8 μM), indicating DfFAE can be classified as a type-A FAE.
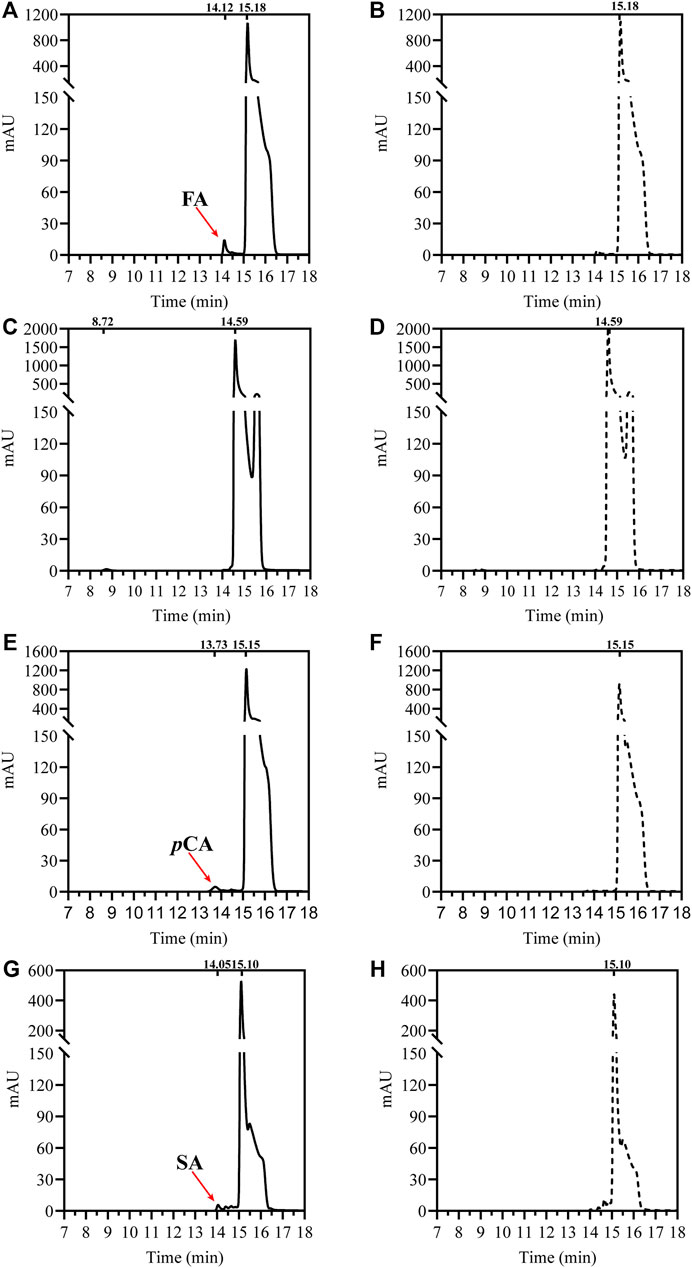
FIGURE 2. HPLC analysis of DfFAE-catalyzed hydrolysis of four model substrates of FAEs. (A) The product profile of DfFAE-catalyzed hydrolysis of MFA; (B) the blank control group of MFA without adding DfFAE; (C) The product profile of DfFAE-catalyzed hydrolysis of MCA; (D) the blank control group of MCA without adding DfFAE; (E) The product profile of DfFAE-catalyzed hydrolysis of MpCA; (F) the blank control group of MpCA without adding DfFAE; (G) The product profile of DfFAE-catalyzed hydrolysis of MSA; (H) the blank control group of MSA without adding DfFAE. The purified DfFAE (8 μM) was incubated with 1.25 mM substrates in 10 mM Tris-HCl buffer (pH 8.4) at 40 °C for 1 h. The positions of hydroxycinnamic acids and their esters detected in the assays are also indicated. The chromatograms were recorded at 323 nm.
To further analyze the substrate preference of DfFAE, the enzymatic activity was measured spectrophotometrically by hydrolysis of four p-nitrophenyl esters with different chain lengths, including pNPA (C2), pNPB (C4), pNPO (C8) and pNPF as substrates. The results indicated that DfFAE was most active toward short-chain substrate pNPA, while its hydrolysis activity toward pNPB and pNPO decreased to 81 and 51%, respectively, compared with that of pNPA. Under the same experimental conditions, the hydrolysis of long-chain pNPF by DfFAE could not be detected, suggesting DfFAE had a substrate preference toward short-chain esters (Figure 3A).
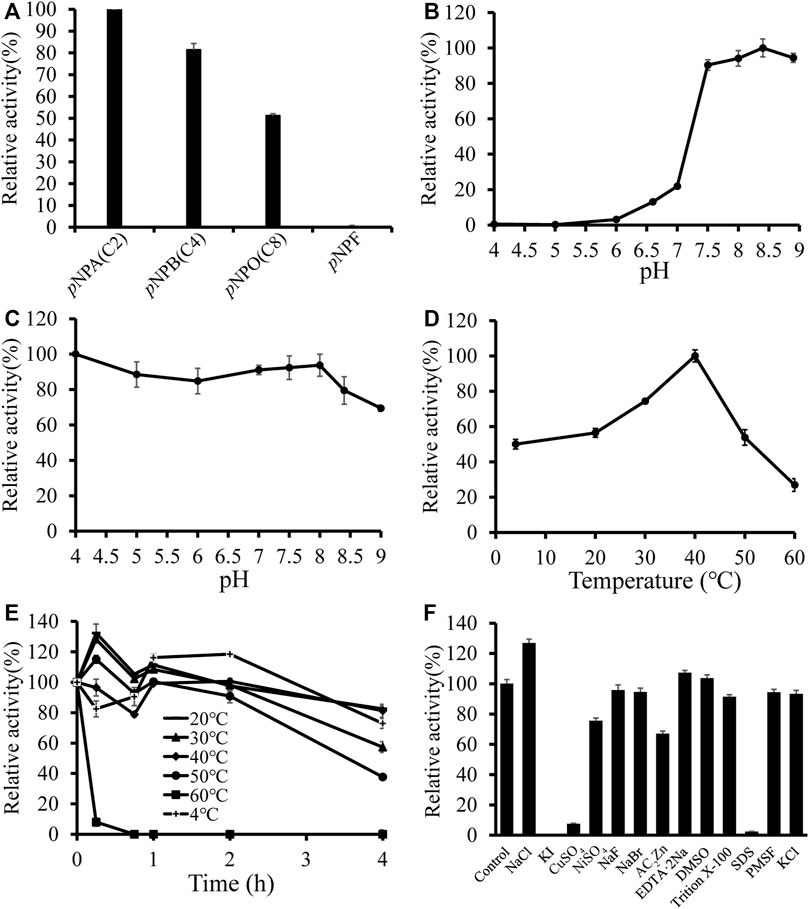
FIGURE 3. Biochemical characterization of DfFAE. (A) Substrate preference of DfFAE was measured with p-nitrophenyl esters. The observed maximum activity was defined as 100% (B–C) Effect of pH on DfFAE activity (B) and stability (C). (D–E) Effect of temperature on DfFAE activity (D) and stability (E). 4 °C, plus sign (+); 20 °C, minus sign (-); 30 °C, solid triangle (▲); 40 °C, solid tilted square (◆); 50 °C, solid circle (●); 60 °C, solid box (■). The activity measured without preincubation is defined as 100% (F) Effect of various metal ions and detergents on DfFAE activity. The activity of the blank control group was taken as 100%. The error bars indicated the standard deviations obtained from three independent experiments.
Then the enzymatic characteristics of DfFAE were assayed using the more preferred substrate pNPA. DfFAE has an optimal pH of 8.4 and maintains higher activity at alkaline conditions, almost 90% of its maximum activity. The enzyme became almost inactive when the pH decreased below 6.0 (Figure 3B). The pH stability was assessed by incubating DfFAE at 4 °C in different pH buffers for 4 h before measured the activity in the optimum conditions. DfFAE was stable over a broad pH range from 4.0 to 8.0 with more than 80% residual activity maintained (Figure 3C). Furthermore, the esterase activity of DfFAE was tested over a temperature range of 4–60°C, showing that DfFAE exhibiting an optimum temperature of 40°C and remaining at 54 and 27% of its maximal activity at 50 and 60°C, respectively (Figure 3D). For thermal stability, DfFAE was stable below 50°C, while its activity drastically decreased after incubation at 60°C for 15 min (Figure 3E). At a final concentration of 1 mM (w/v) or 1% (v/v), the effect of metal ions and different detergents on DfFAE activity was also investigated (Figure 3F). It can be observed that the addition of KI completely inactivated DfFAE, while CuSO4 and SDS significantly reduced the enzymatic activity to 8 and 2%, respectively. NiSO4 (76%) and ZnAC2 (67%) showed a mild inhibitory effect, while activity could be significantly promoted by 1 mM NaCl (127%). The other substances showed no significant effect on DfFAE enzymatic activity.
We further determined the kinetic properties of DfFAE toward pNPA, pNPB and pNPO. The results are summarized in Table 1. DfFAE showed a higher substrate preference for hydrolyzing pNPA on which it exhibited the highest catalytic efficiency with a k cat/K m value of 7.73 s−1mM−1, almost 3.5- and 21.8-fold higher than that of pNPB (2.23 s−1mM−1) and pNPO (0.35 s−1mM−1), respectively. The hydrolytic activity of DfFAE toward pNPA is comparable or much higher than other gut-derived FAEs, including BiFae1A (k cat/K m value of 11.9 s−1mM−1) (Wefers et al., 2017), BoCE1 (k cat/K m value of 0.5 s−1mM−1) (Kmezik et al., 2020) and AsFAE (k cat/K m value of 0.84 s−1mM−1) (Wei et al., 2021). Nonetheless, DfFAE showed a slightly higher affinity toward pNPB (K m value of 0.36 mM) compared with pNPA and pNPO (K m values of 0.57 and 0.53 mM, respectively), indicating the three substrates are easily accessible to the active site region.
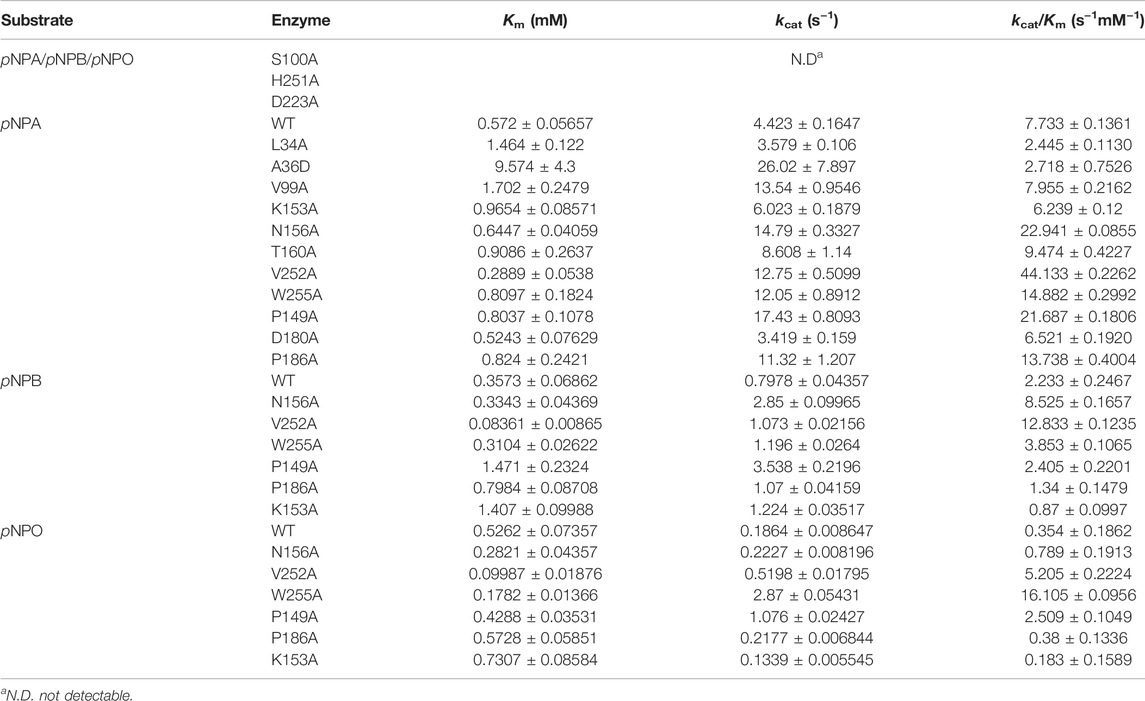
TABLE 1. Kinetic parameters of wild-type DfFAE and its mutants.
To further illustrate the structural basis for the catalytic properties of DfFAE, we attempted to crystallize it but unfortunately failed after multiple rounds of trying. Therefore, we used AlphaFold2, a newly introduced structure prediction program with unprecedented accuracy, to build the 3D-structure of DfFAE based on its amino acid sequence. The pLDDT values under the best model were greater than 80 for 98% of the residues and less than 50 for only the top two residues, indicating that the model was generally plausible. The overall structure of DfFAE contains a typical α/β hydrolase fold core domain composed of a central eight-stranded β-sheet flanked by six helices, and a cap domain formed by α4-α8 helices positioned atop the core domain (Figure 4A). The active site region responsible for substrate binding and catalysis is located between the core and cap domains. The entrance of the active site pocket is formed by two flexible surface loops between α4-α5 (Asp146-Pro149) and α7-α8 (Ala182-Val189) from the cap domain. The catalytic Ser100 is located at the nucleophilic elbow between the β5 sheet and the α3 helix, lying within the hydrogen-bond distance (4.7 Å) with the general base His251 which further interacts with Asp223 to form a charge relay system (Figure 4B). The entire catalytic triad is encapsulated inside the active site pocket and less exposed to solvent. Leu34 and Met101 potentially contribute to the formation of the oxyanion hole according to the sequence alignment (Figure 1A).
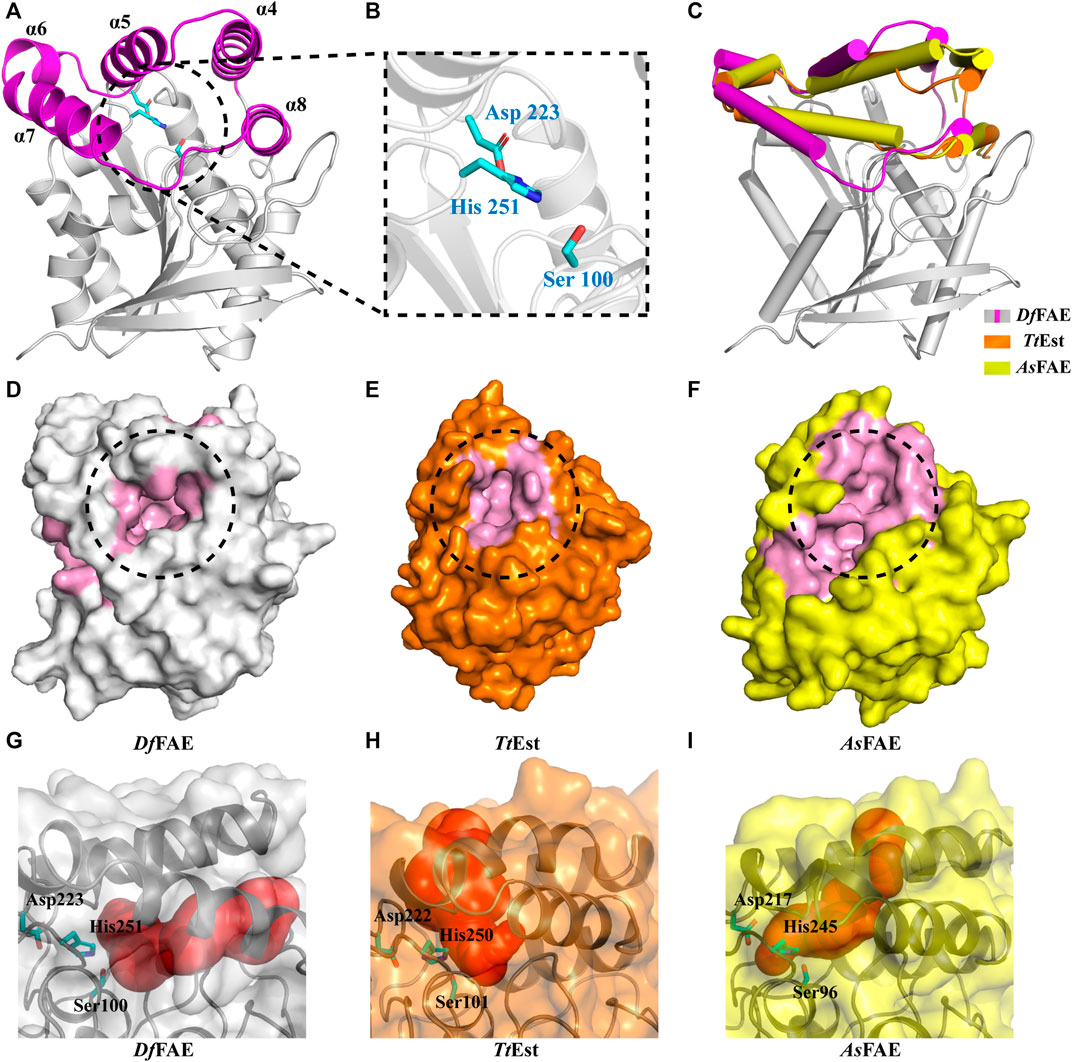
FIGURE 4. (A) Overall structure of DfFAE. The magenta and gray colors represent the core and cap domains, respectively. (B) Close-up views of the catalytic triad. Residues in the catalytic triad are represented as cyan sticks (C) Comparison of the cap domain of DfFAE (magenta and gray), TtEst (orange) and AsFAE (yellow). (D–E) Surface representation of the active site pocket of DfFAE (D, gray), TtEst (E, orange) and AsFAE (F, yellow). The active site pocket region is marked in pink, and the entrance is highlighted by black dotted circle (G–I) DfFAE, TtEst and AsFAE substrate access tunnel conformations calculated by CAEVER 3.0, shown as red spheres of different radius.
The cap domain is highly variable across different esterase structures and affects the conformation of the active site pocket (Goldstone et al., 2010; Bains et al., 2011; Wefers et al., 2017). Structural superimposition of DfFAE with homologous TtEst and AsFAE revealed considerable variation in the orientation of the cap domain, leading to a significant difference in the size of active site pocket and the position of its entrance (Figures 4C–F). Notably, DfFAE features a relatively close and deep pocket in which the entrance is partially covered by the connecting loop between α7 and α8, three residues longer than that in TtEst and AsFAE (Figure 1A; Supplementary Figure S1). The DoGSiteScorer analysis (Volkamer et al., 2010; Volkamer et al., 2012) of the size of active site pocket indicated that DfFAE has the largest pocket with the volume of 761.92 Å3, while the volumes of AsFAE and TtEst active site pocket are calculated as 701.82 Å3 and 514.05 Å3, respectively. These observations suggest DfFAE possess an active site pocket with narrow entrance but large internal cavity to accommodate long-chain substrates, and may explain why DfFAE has the ability to catalyze the hydrolysis of pNPO, while the other two esterases do not (Sayer et al., 2015; Wei et al., 2021).
To more intuitively illustrate the conformational differences of the active site pocket of DfFAE and its homologous proteins, the potential substrate access tunnel which refers to a pathway connecting the protein outer surface toward the buried catalytic center was identified using CAVER 3.0 (Chovancova et al., 2012; Kokkonen et al., 2019). The substrate access tunnel of DfFAE is formed at the interface of a four-helix bundle composed of α4, α5, α7 and α8 helices from the cap domain with a length of 22 Å and an average radius of 2.63 Å (Figures 4G; Supplementary Figures S2A). In contrast, the tunnel entrance of TtEst and AsFAE is positioned in the middle of the top two helices (α4-α5) from the cap domain, and the overall tunnel is nearly vertical to that of DfFAE (Figures 4H, I; Supplementary Figures S2B, C). These results indicated that the three structurally similar homologous esterases have totally different active site topology.
Rational design provides an effective tool to modulate the biocatalytic properties of enzymes. In previous studies, rational design strategies have been successfully applied to several FAEs with improved enzymatic activity or thermostability by modifying the active site (Antonopoulou et al., 2018; Liu et al., 2021; Yang et al., 2022). DfFAE features a narrow active site pocket including a substrate access tunnel deeply into the protein, which may reduce the entry of the large carboxyl portion of the ester substrate to the catalytic residues. Hence this study aims to engineer the active site for enhanced catalytic efficiency of DfFAE. In order to identify the residues that likely contribute to substrate binding, the predicted DfFAE structure was subjected to molecular docking by Autodock_vina tool (Trott and Olson, 2010) with pNPA, pNPB and pNPO. All three pNP-esters showed the similar mode of binding into the active site pocket (Figures 5A–C). The distance between the catalytic Ser100 and the carbonyl carbon atom of each substrate is approximately 3.6 Å. In the docking conformation of pNPA that is most preferred by DfFAE, the nitro group oxygen makes a salt bridge to Lys153 and also a hydrogen bond to Asn156 on helix α5 from cap domain, while the acyl group oxygen is stabilized by hydrogen bonds to Leu34 main chain and Ser100 side chain in line for catalysis. Residues within 5 Å of pNPA (Ala36, Val99, Val252 and Trp255 from the core domain and Thr160 from the cap domain) are expected to form hydrophobic interactions (Figure 5D). All these residues are not conserved according to sequence alignment (Figure 1A) and were selected for mutagenesis. In addition, Pro149, Asp180 and Pro186 from the cap domain occupied the entrance to the substrate access tunnel and were mutated to Ala in an aim to open up the catalytic pocket (Figure 5E).
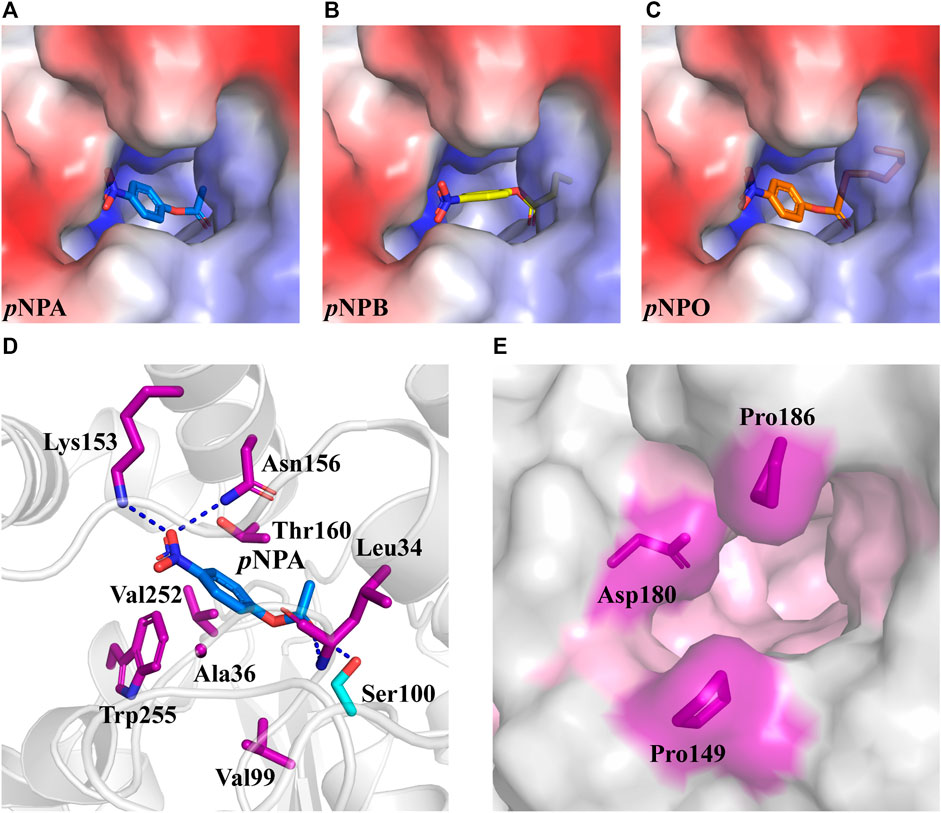
FIGURE 5. The optimal conformation of pNPA, pNPB and pNPO docking in DfFAE. (A–C) DfFAE is shown as surface representation, colored by electrostatic potentials (positive, blue; negative, red). pNPA (A, blue), pNPB (B, yellow) and pNPO (C, orange) are shown as sticks. (D) Cartoon representation of pNPA binding to the active site of DfFAE. The catalytic Ser100 is shown as a cyan stick, and other interacting residues are shown as purple sticks. Hydrogen bonding forces are shown as blue dashed lines (E) Surface representation of mutation sites at the entrance of binding pocket. The binding pocket area colored in pink, mutation site residues are shown as purple sticks.
All mutants were overexpressed in E. coli T7 and purified by Ni2+-NTA and ion exchange chromatographies. Then kinetic experiments were performed to evaluate the role of each residue (Figure 6A; Table 1). Mutation of the catalytic triad Ser100, His251 and Asp223 resulted in the complete elimination of enzymatic activity, confirming their essential roles in acyl ester hydrolysis. In the case of residues involved in substrate recognition, V252A displayed the highest catalytic efficiency toward pNPA with both decreased K m and increased k cat, which was 5.71-fold higher (k cat /K m value of 44.13 s−1mM−1) than the wild type (k cat /K m value of 7.73 s−1mM−1). Similarly, N156A and W255A showed a slightly decreased K m but an elevated k cat values, resulting in enhanced catalytic efficiency compared to the wild type (k cat /K m value of 22.94 s−1mM−1 and 14.88 s−1mM−1, respectively). These results suggested that Ala replacement of residues with bulky side chain possibly leads to relieving the steric hindrance and increasing the internal space of active site pocket which could provide additional degree of freedom for substrate positioning and accelerate substrate turnover (k cat). In contrast, Ala36 mutated to a large Asp showed a K m value increased to 16.74-fold but a k cat /K m value decreased to 0.35-fold. Leu34 is a likely candidate to stabilize the oxyanion hole and its mutation reduced the enzymatic activity remarkably. V99A, T160A and K153A showed little effects on DfFAE catalytic efficiency but an apparent decrease in substrate binding affinity, suggesting the three residues may play a role in the substrate binding and catalysis process. Alternatively, in the case of amino acids involved in substrate entry, the catalytic efficiency of P149A and P186A for pNPA hydrolysis increased by about 3-fold and 2-fold, respectively, while D180A showed a negligible effect on DfFAE activity, indicating that Pro149 and Pro186 play an essential role in gating the substrate access tunnel.
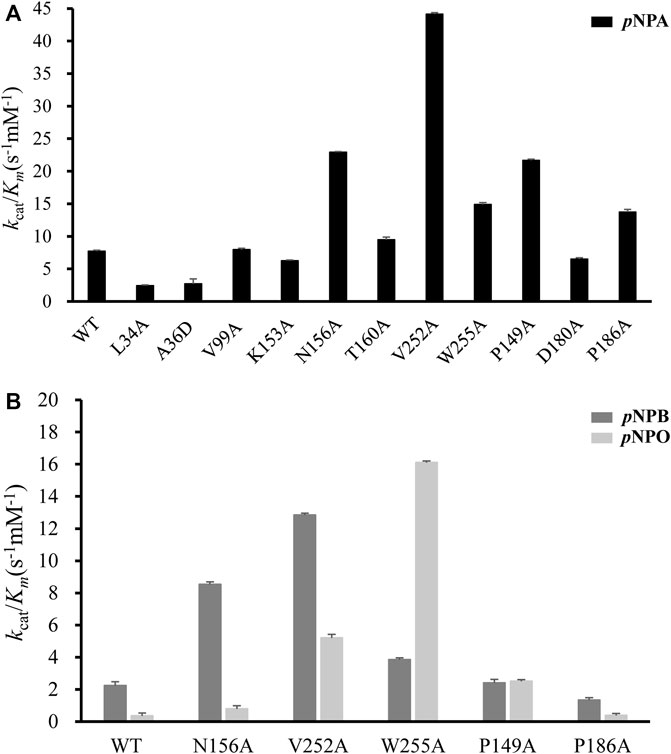
FIGURE 6. Comparison of the catalytic efficiency (represented by k cat/K m) measured for wild-type DfFAE and its mutants. (A) The k cat/K m values on pNPA for WT and all mutants (B) The k cat/K m values of WT and some mutants on pNPB and pNPO. The error bars indicated the standard errors from three independent experiments.
Subsequently, five mutants V252A, N156A, P149A, P186A, and W255A which had higher activities toward pNPA were selected to further determine the kinetic parameters toward pNPB and pNPO substrates. In particular, the k cat /K m values of V252A were respectively 5.75- and 14.7-fold higher toward pNPB and pNPO than that of the wild-type, and the k cat /K m values of N156A toward pNPB and pNPO enhanced 3.69- and 2.23-fold, respectively, compared with the wild-type. On the other hand, mutation of Pro149 resulted in 7.09-fold higher catalytic efficiency toward pNPO but have a minor influence on pNPB, while P186A performed poorly on both substrates. Interestingly, we observed W255A gained a higher preference toward pNPO with 45.5-fold increase in catalytic efficiency (k cat /K m value of 16.11 s−1mM−1) compared with the wild-type, while its activities toward pNPA and pNPB were 1.92- and 1.73-fold, respectively. To determine the mechanisms underlying the influences of W255A on DfFAE activity, we reanalyzed the structure of active site pocket in DfFAE and observed that besides the main substrate access tunnel there was still a side part in the internal space that is partitioned by the bulky side chain of Trp255 (Supplementary Figure S3; Figures 7A, C). Substitution of Trp255 to small Ala broadened internal cavity and possibly provided a new route for long-chain substrate easy access to the catalytic center (Figures 7B, D). The binding energy of pNPO in the new route is -7.0 kcal/mol, the same as the main substrate access tunnel (-7.0 kcal/mol). Therefore, these results indicated that modification of residues inside tunnel (Val252, Asn156 and Trp255) play a more vital role in improving DfFAE activity and broadening substrate profile for ester hydrolysis compared with residues gating the tunnel (Pro149, Asp180 and Pro186).
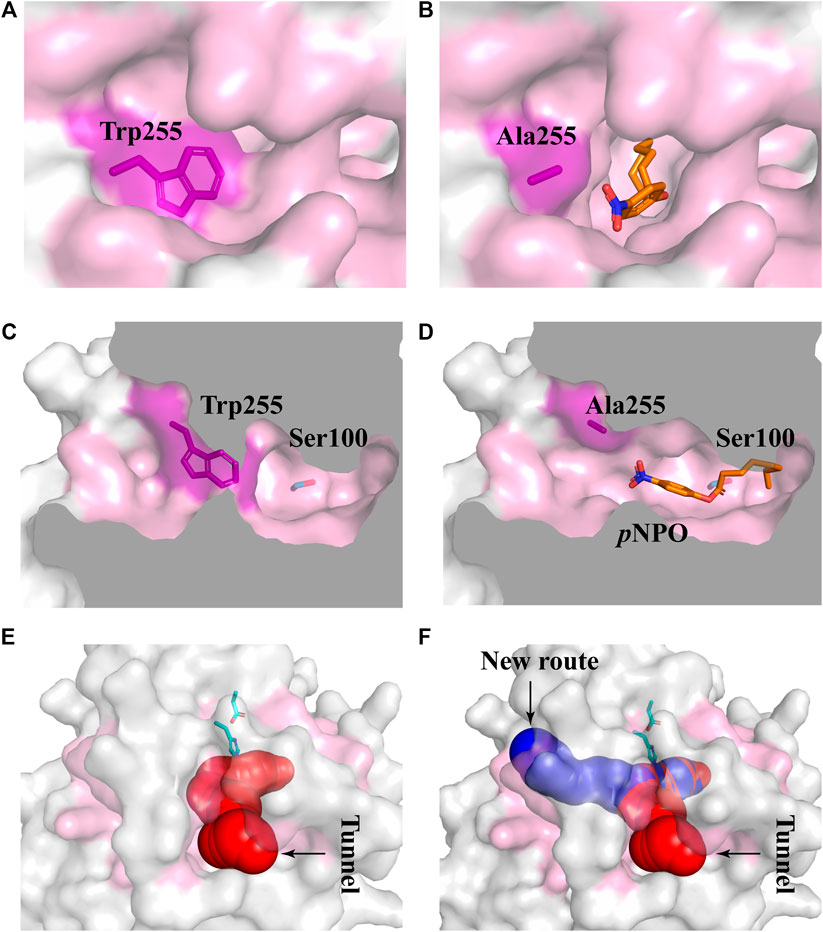
FIGURE 7. Structural differences in the substrate access tunnel of wild-type DfFAE and W255A mutant. (A–B) The entrance of the new route of W255A (B) and the main substrate access tunnel of WT (A). (C–D) Cutaway view of the new route of W255A (D) and the main substrate access tunnel of WT (C). (E–F) Comparison of the substrate access tunnel conformation of W255A (F) calculated by CAVER 3.0 and that of WT (E). The binding pocket is colored by pink. The ligand pNPO is shown as orange stick, the new route and the main substrate access tunnel are shown as blue and red, respectively.
In conclusion, this study characterized a novel type-A FAE from gut-derived bacteria D. formicigenerans (DfFAE). DfFAE showed a higher preference for short-chain esters, possibly due to its narrow and relatively closed active site pocket, including a deep substrate access tunnel. Structure-guided mutagenesis within the active site identified the mutants inside the cavity play a more vital role in enhancing catalytic efficiency toward pNP-esters by expanding the internal space of the tunnel. Moreover, W255A created a possible new route for substrate entry and showed a higher preference toward long-chain pNPO. Collectively, these results revealed the molecular determinant for how the substrate preference of DfFAE is influenced by its active site architecture and the applicability of the rational design strategy for obtaining enhanced biocatalysts, which can be referenced by other FAEs and gut-derived enzymes.
The original contributions presented in the study are included in the article/Supplementary Material further inquiries can be directed to the corresponding authors.
FX, YW, and XW designed the experiment. YS performed most of the experiments. YS and YW analyzed the data and wrote the manuscript. BW provided DfFAE sequence. XW, BW, SL, and JZ assisted in experiments and data analysis. HT contributed to DfFAE structural modeling by AlphaFold2. FX and SS supervised the whole project.
This study was supported by the Financial Fund of Institute of Food Science, Technology, Nutrition and Health (Cangzhou), CAAS (CAAS-IFSTNH-CZ-2022-06), the Central Public-interest Scientific Institution Basal Research Fund (S2020JBKY-13), and the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2021-IFST-SN202107).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank School of Life Sciences of Tsinghua University for providing technical support for structural prediction computations.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2022.936914/full#supplementary-material
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol. 37, 420–423. doi:10.1038/s41587-019-0036-z
Andreasen, M. F., Kroon, P. A., Williamson, G., and Garcia-Conesa, M.-T. (2001). Esterase Activity Able to Hydrolyze Dietary Antioxidant Hydroxycinnamates Is Distributed along the Intestine of Mammals. J. Agric. Food Chem. 49, 5679–5684. doi:10.1021/jf010668c
Antonopoulou, I., Hunt, C., Cerullo, G., Varriale, S., Gerogianni, A., Faraco, V., et al. (2018). Tailoring the Specificity of the Type C Feruloyl Esterase FoFaeC from Fusarium Oxysporum towards Methyl Sinapate by Rational Redesign Based on Small Molecule Docking Simulations. PLoS One 13, e0198127. doi:10.1371/journal.pone.0198127
Bains, J., Kaufman, L., Farnell, B., and Boulanger, M. J. (2011). A Product Analog Bound Form of 3-Oxoadipate-Enol-Lactamase (Pad) Reveals a Multifunctional Role for the Divergent Cap Domain. J. Mol. Biol. 406, 649–658. doi:10.1016/j.jmb.2011.01.007
Bauer, T. L., Buchholz, P. C. F., and Pleiss, J. (2020). The Modular Structure of α/β‐hydrolases. Febs J. 287, 1035–1053. doi:10.1111/febs.15071
Chen, M., Liu, S., Imam, K. M. S. U., Sun, L., Wang, Y., Gu, T., et al. (2020). The Effect of Xylooligosaccharide, Xylan, and Whole Wheat Bran on the Human Gut Bacteria. Front. Microbiol. 11, 568457. doi:10.3389/fmicb.2020.568457
Chovancova, E., Pavelka, A., Benes, P., Strnad, O., Brezovsky, J., Kozlikova, B., et al. (2012). CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 8, e1002708. doi:10.1371/journal.pcbi.1002708
de O Buanafina, M. M., Fernanda Buanafina, M., Laremore, T., Shearer, E. A., and Fescemyer, H. W. (2019). Characterization of Feruloyl Esterases in Maize Pollen. Planta 250, 2063–2082. doi:10.1007/s00425-019-03288-y
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., et al. (2005). Diversity of the Human Intestinal Microbial Flora. Science 308, 1635. doi:10.1126/science.1110591
El-Seedi, H. R., El-Said, A. M. A., Khalifa, S. A. M., Göransson, U., Bohlin, L., Borg-Karlson, A.-K., et al. (2012). Biosynthesis, Natural Sources, Dietary Intake, Pharmacokinetic Properties, and Biological Activities of Hydroxycinnamic Acids. J. Agric. Food Chem. 60, 10877–10895. doi:10.1021/jf301807g
Faulds, C. B. (2010). What Can Feruloyl Esterases Do for Us? Phytochem. Rev. 9, 121–132. doi:10.1007/s11101-009-9156-2
Fritsch, C., Jänsch, A., Ehrmann, M. A., Toelstede, S., and Vogel, R. F. (2017). Characterization of Cinnamoyl Esterases from Different Lactobacilli and Bifidobacteria. Curr. Microbiol. 74, 247–256. doi:10.1007/s00284-016-1182-x
Goldstone, D. C., Villas-Bôas, S. G., Till, M., Kelly, W. J., Attwood, G. T., and Arcus, V. L. (2010). Structural and Functional Characterization of a Promiscuous Feruloyl Esterase (Est1E) from the Rumen Bacterium Butyrivibrio Proteoclasticus. Proteins 78, 1457–1469. doi:10.1002/prot.22662
Hermoso, J. A., Sanz-Aparicio, J., Molina, R., Juge, N., González, R., and Faulds, C. B. (2004). The Crystal Structure of Feruloyl Esterase A from Aspergillus niger Suggests Evolutive Functional Convergence in Feruloyl Esterase Family. J. Mol. Biol. 338, 495–506. doi:10.1016/j.jmb.2004.03.003
Kmezik, C., Bonzom, C., Olsson, L., Mazurkewich, S., and Larsbrink, J. (2020). Multimodular Fused Acetyl-Feruloyl Esterases from Soil and Gut Bacteroidetes Improve Xylanase Depolymerization of Recalcitrant Biomass. Biotechnol. Biofuels 13, 60. doi:10.1186/s13068-020-01698-9
Kokkonen, P., Bednar, D., Pinto, G., Prokop, Z., and Damborsky, J. (2019). Engineering Enzyme Access Tunnels. Biotechnol. Adv. 37, 107386. doi:10.1016/j.biotechadv.2019.04.008
Lai, K. K., Lorca, G. L., and Gonzalez, C. F. (2009). Biochemical Properties of Two Cinnamoyl Esterases Purified from a Lactobacillus Johnsonii Strain Isolated from Stool Samples of Diabetes-Resistant Rats. Appl. Environ. Microbiol. 75, 5018–5024. doi:10.1128/AEM.02837-08
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X Version 2.0. Bioinformatics 23, 2947–2948. doi:10.1093/bioinformatics/btm404
Liu, S., Soomro, L., Wei, X., Yuan, X., Gu, T., Li, Z., et al. (2021). Directed Evolution of Feruloyl Esterase from Lactobacillus Acidophilus and its Application for Ferulic Acid Production. Bioresour. Technol. 332, 124967. doi:10.1016/j.biortech.2021.124967
Oliveira, D. M., Mota, T. R., Oliva, B., Segato, F., Marchiosi, R., Ferrarese-Filho, O., et al. (2019). Feruloyl Esterases: Biocatalysts to Overcome Biomass Recalcitrance and for the Production of Bioactive Compounds. Bioresour. Technol. 278, 408–423. doi:10.1016/j.biortech.2019.01.064
Raimondi, S., Anighoro, A., Quartieri, A., Amaretti, A., Tomás-Barberán, F. A., Rastelli, G., et al. (2015). Role of Bifidobacteria in the Hydrolysis of Chlorogenic Acid. Microbiologyopen 4, 41–52. doi:10.1002/mbo3.219
Robert, X., and Gouet, P. (2014). Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 42, W320–W324. doi:10.1093/nar/gku316
Sayer, C., Isupov, M. N., Bonch-Osmolovskaya, E., and Littlechild, J. A. (2015). Structural Studies of a Thermophilic Esterase from a New Planctomycetes species,Thermogutta Terrifontis. FEBS J. 282, 2846–2857. doi:10.1111/febs.13326
Scheller, H. V., and Ulvskov, P. (2010). Hemicelluloses. Annu. Rev. Plant Biol. 61, 263–289. doi:10.1146/annurev-arplant-042809-112315
Slavin, J. (2013). Fiber and Prebiotics: Mechanisms and Health Benefits. Nutrients 5, 1417–1435. doi:10.3390/nu5041417
Sonnenburg, E. D., Zheng, H., Joglekar, P., Higginbottom, S. K., Firbank, S. J., Bolam, D. N., et al. (2010). Specificity of Polysaccharide Use in Intestinal bacteroides Species Determines Diet-Induced Microbiota Alterations. Cell 141, 1241–1252. doi:10.1016/j.cell.2010.05.005
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 38, 3022–3027. doi:10.1093/molbev/msab120
Trott, O., and Olson, A. J. (2009). AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31, 455. doi:10.1002/jcc.21334
Volkamer, A., Griewel, A., Grombacher, T., and Rarey, M. (2010). Analyzing the Topology of Active Sites: On the Prediction of Pockets and Subpockets. J. Chem. Inf. Model. 50, 2041–2052. doi:10.1021/ci100241y
Volkamer, A., Kuhn, D., Grombacher, T., Rippmann, F., and Rarey, M. (2012). Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 52, 360–372. doi:10.1021/ci200454v
Wang, L., Ma, Z., Du, F., Wang, H., and Ng, T. B. (2014). Feruloyl Esterase from the Edible Mushroom Panus Giganteus: A Potential Dietary Supplement. J. Agric. Food Chem. 62, 7822–7827. doi:10.1021/jf405654u
Wefers, D., Cavalcante, J. J. V., Schendel, R. R., Deveryshetty, J., Wang, K., Wawrzak, Z., et al. (2017). Biochemical and Structural Analyses of Two Cryptic Esterases in bacteroides Intestinalis and Their Synergistic Activities with Cognate Xylanases. J. Mol. Biol. 429, 2509–2527. doi:10.1016/j.jmb.2017.06.017
Wei, X., Wang, Y.-l., Wen, B.-t., Liu, S.-j., Wang, L., Sun, L., et al. (2021). The α-Helical Cap Domain of a Novel Esterase from Gut Alistipes Shahii Shaping the Substrate-Binding Pocket. J. Agric. Food Chem. 69, 6064–6072. doi:10.1021/acs.jafc.1c00940
Williamson, G., Kroon, P. A., and Faulds, C. B. (1998). Hairy Plant Polysaccharides: a Close Shave with Microbial Esterases. Microbiology 144, 2011–2023. doi:10.1099/00221287-144-8-2011
Keywords: feruloyl esterase, gut microbiota, Dorea formicigenerans, AlphaFold2, rational design, substrate access tunnel
Citation: Shen Y, Wang Y, Wei X, Wen B, Liu S, Tan H, Zhang J, Shao S and Xin F (2022) Engineering the Active Site Pocket to Enhance the Catalytic Efficiency of a Novel Feruloyl Esterase Derived From Human Intestinal Bacteria Dorea formicigenerans. Front. Bioeng. Biotechnol. 10:936914. doi: 10.3389/fbioe.2022.936914
Received: 05 May 2022; Accepted: 01 June 2022;
Published: 20 June 2022.
Edited by:
Zheng Guo, Aarhus University, DenmarkCopyright © 2022 Shen, Wang, Wei, Wen, Liu, Tan, Zhang, Shao and Xin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengjiao Xin, MjAwMmhvbmd6aGkzMEAxNjMuY29t; Shuli Shao, c2hzaGwzMkAxNjMuY29t
† These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.