 Wenfang Wang1
Wenfang Wang1 Yinhua Lu
Yinhua Lu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Bioeng. Biotechnol. , 13 July 2021
Sec. Synthetic Biology
Volume 9 - 2021 | https://doi.org/10.3389/fbioe.2021.692797
This article is part of the Research Topic Mining, Deciphering and Engineering of Bacterial Natural Products View all 6 articles
Microbial natural products (NPs) are a major source of pharmacological agents. Most NPs are synthesized from specific biosynthetic gene clusters (BGCs). With the rapid increase of sequenced microbial genomes, large numbers of NP BGCs have been discovered, regarded as a treasure trove of novel bioactive compounds. However, many NP BGCs are silent in native hosts under laboratory conditions. In order to explore their therapeutic potential, a main route is to activate these silent NP BGCs in heterologous hosts. To this end, the first step is to accurately and efficiently capture these BGCs. In the past decades, a large number of effective technologies for cloning NP BGCs have been established, which has greatly promoted drug discovery research. Herein, we describe recent advances in strategies for BGC cloning, with a focus on the preparation of high-molecular-weight DNA fragment, selection and optimization of vectors used for carrying large-size DNA, and methods for assembling targeted DNA fragment and appropriate vector. The future direction into novel, universal, and high-efficiency methods for cloning NP BGCs is also prospected.
Natural products (NPs) produced by microbes are a major source of pharmacological agents and industrially useful compounds. With the spread of drug-resistant pathogens rendering widely used antibiotics ineffective, the discovery of new NPs has become an urgent necessity (Atanasov et al., 2021). The development of next-generation sequencing technology has led to the genomes of a vast array of culturable microorganisms being sequenced in recent years. Through analysis of sequenced microbial genomes, a remarkably large number of orphan biosynthetic gene clusters (BGCs) have been discovered, which represent a treasure trove of novel bioactive compounds with potential pharmacological relevance (Kang and Kim, 2021). However, translating these putative BGCs into specialized compounds is a challenge, as the majority of NP BGCs are either poorly or not at all expressed in native hosts under defined conditions (Li et al., 2021). Further, it has been estimated that over 99% of environmental microbes are recalcitrant to culture under laboratory conditions (Daniel, 2005). Now, metagenomics has emerged as a strategic approach to explore uncultivated microbes from environment (Daniel, 2005), which also revealed the presence of a vast array of NP BGCs. In addition, to facilitate the exploration of NP sources from uncultured microbes, many innovative techniques for targeted or high-throughput cultivation of novel microorganisms are emerging rapidly. Nevertheless, further development of cultivation technologies is still required (Lewis et al., 2021).
In the past decades, efforts have been committed to explore this treasure trove and a number of efficient strategies for activating silent gene clusters have been developed, among which the heterologous expression of NP BGCs has been most widely used (Kang and Kim, 2021). An advantage of this strategy is that once a novel metabolite appears in the surrogate host cell wherein the BGC has been introduced, it can be ascribed to the gene cluster with a high degree of confidence (Hu et al., 2016). A prerequisite for heterologous expression is to clone the target BGC into a suitable vector. Traditional library construction method is sequence-independent and has been proven to be efficient for cloning NP BGCs. Recently, it has been successfully employed for cloning NP BGCs larger than 150 kb using the bacterial artificial chromosome (BAC) (Zhang L. et al., 2017; Hashimoto et al., 2018; Sun et al., 2018). However, it requires considerable screening, which is time-consuming and laborious. In order to directly clone the target BGCs, researchers have developed a variety of DNA cloning or assembly methods, including in vitro DNA assembly (restriction enzyme-mediated assembly, recombination-based assembly, enzyme-independent DNA assembly), as well as in vivo direct cloning methods, such as Red/ET-mediated recombination in Escherichia coli and transformation-associated recombination (TAR) cloning in Saccharomyces cerevisiae (Abbasi et al., 2020; Lin et al., 2020).
The previously reported in vitro, in vivo, or even vitro/vivo hybrid technologies for cloning large DNA fragments have distinct mechanisms, advantages, as well as drawbacks. However, regardless of the methods employed, it is necessary to prepare high-quality and high-molecular-weight DNA as well as to select suitable vectors. Further, efficient strategies for assembling large DNA fragments and vectors are required (Figure 1). The cloned BGCs usually have to be refactored in order to become more compatible with the heterologous host. In this review, we will focus on recent developments of the process for high-molecular-weight DNA fragment preparation, vectors used for carrying large-size DNA and methods for assembling target BGCs and vectors, and have a prospect on novel, universal and high-efficiency cloning methods for large-size DNA.
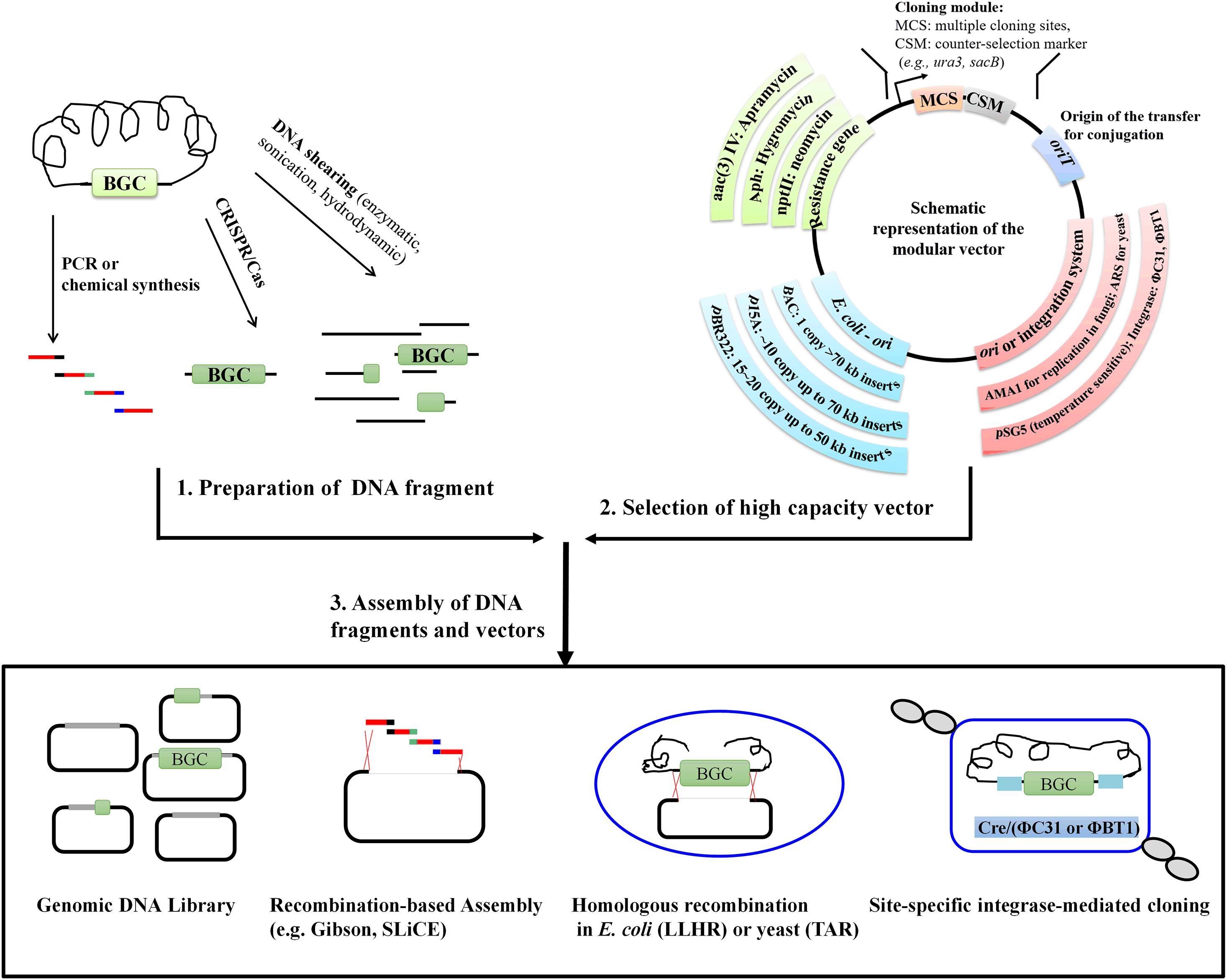
Figure 1. The general workflow for the cloning of natural product BGCs.
Microbial NP BGCs normally range from 10 to 150 kb in length. Thus, methods for preparing high-quality and high-molecular-weight DNA are critical for successful cloning of intact BGCs. Fundamentally, genomic DNA extraction from microorganisms mainly involves three steps: (1) lysis of the cell wall or membrane using chemical disruption [e.g., SDS (sodium dodecyl sulfate)], enzymatic lysis (e.g., lysozyme and proteinase K), or physical disruption (e.g., manually grinding, sonication); (2) removal of all other unwanted cell components including cell wall debris, proteins, polysaccharides, and other metabolic substances by CTAB (cetyltrimethylammonium bromide) and/or phenol-chloroform; (3) recovery of the pure genomic DNA by ethanol precipitation, spin column-based technique, or magnetic bead-based strategy (Park, 2007; Varma et al., 2007). Integrity of genomic DNA may be mainly affected by the mechanical shearing and endogenous DNases. In these methods, proteinase K, phenol, and EDTA could suppress DNase activity, to a certain extent, enhancing the integrity of genomic DNA (Varma et al., 2007). To prevent mechanical shearing of DNA, microbial cells (e.g., protoplasts) can be embedded in low-melting-point agarose gels in the form of plugs, resulting in the preparation of megabase-sized DNA (Zhang M. et al., 2012). However, this process takes 3 days, and the operation is complex. A novel method for genomic DNA extraction, which involves cell grinding in liquid nitrogen, lysis with SDS-base buffer, and purification using carboxylated magnetic beads, was recently developed. Using this method, up to 80 kb DNA fragments could be prepared rapidly (∼1 h) and efficiently (Mayjonade et al., 2016). By fine-tuning three critical parameters, including the grinding duration and vibrational frequency, as well as lysis temperature and duration, the sizes of genomic DNA fragments ranging from 79 to 145 kb can be obtained (Penouilh-Suzette et al., 2020). More detailed information of the methods for genome DNA extraction can refer to a recent review (Gomez-Acata et al., 2019). However, no method can be universally applicable to all microorganisms. In general, researchers need to modify or blend different methods to obtain DNA of desired quality (Varma et al., 2007).
In the case where the genome sequence information of native hosts is under-characterized (e.g., environmental DNA), target BGCs can only be obtained through the construction of a genomic DNA library and subsequent screening via PCR or the identification of corresponding products via heterologous expression. Currently, three methods are available for DNA fragmentation for the construction of large-sized fragment libraries, including enzymatic digestion, sonication, and hydrodynamic shearing (Ignatov et al., 2019). Among them, sonication and hydrodynamic methods randomly disrupt the genome, which may cause the shearing of intact BGCs into different segments (Ignatov et al., 2019). So, enzymatic digestion is the most widely used for DNA fragmentation of library construction.
Currently, more and more microbial genome sequences are being published. For cloning small- to mid-sized BGCs, long-amplicon PCR could be used to amplify BGCs fragments, and then entire BGCs were obtained by DNA assembly (Greunke et al., 2018). However, as the length of NP BGCs increases, the probability of mutations introduced by PCR also increases. Fortunately, the development of genome editing tool CRISPR-Cas (clustered regularly interspaced short palindromic repeat-CRISPR-associated protein) system has made it possible to isolate the exact sequence of target BGCs (Lee et al., 2015; Jiang and Zhu, 2016). With the aid of Cas9 endonuclease, DNA segment of desired sizes can be obtained through generating the double strand breaks (DSBs) at specific sites within the genome guided by sgRNA. For example, bacterial cultures (e.g., Escherichia coli) was embedded in a low-melting-point agarose gel plug, treated with lysozyme and proteinase K, and subsequently washed to remove cellular components, leaving behind genomic DNA. Finally, the plug was transferred into cleavage buffer containing Cas9 and corresponding sgRNA pairs, which were designed to target genome segments of different lengths (50, 75, 100, 150, 200 kb). Clear DNA bands at the expected lengths were observed using pulsed-field gel electrophoresis (PFGE) assessment (Jiang et al., 2015). Recently, CISMR (CRISPR-mediated isolation of specific megabase-sized regions of the genome), which enables the targeted isolation of contiguous megabase-sized segments of the mouse genome, has also been developed by improving in vitro CRISPR specificity with the aid of both Target Finder and ZiFIT Targeter software to design 17 base sgRNA other than traditional 20 base target sequences (Bennett-Baker and Mueller, 2017). Further, a highly sensitive novel method for the simultaneous separation and concentration of high-molecular-weight DNA fragments was established by optimizing the formulation of viscoelastic liquids and engineering a capillary system. It was successfully used to isolate a 31.5 kb DNA fragment from the complicated 450 Mb Medicago truncatula genome with the aid of Cas9 cleavage (Milon et al., 2019).
The quality of DNA fragments can be analyzed via fragment analyzer or horizontal agarose gel electrophoresis. DNA fragments of desired sizes can be separated and extracted through multiple rounds of PFGE with different ramped pulse times (Clos and Zander-Dinse, 2019). Regardless of the cloning method used, sufficient amounts of DNA fragments are indispensable. Therefore, the preparation of high-quality and high-molecular-weight DNA fragments is recognized as a critical step in gene cluster cloning (Sapojnikova et al., 2017).
Given that most NP BGCs are of relatively large in length, appropriate vector systems capable of carrying the entire gene cluster as well as shuffling these genetic segments between different hosts are necessary. Since the first generation of general cloning vectors was introduced in 1973, a variety of high-capacity vectors have been developed so far (Bajpai, 2014). Despite the dazzling choice of commercial and other available vectors, cloning vector selection can be determined by several key criteria, such as the BGC size and GC content, vector copy number, host compatibility of different vectors, selection markers, and multiple cloning sites. Several types of high-capacity vectors are available for cloning large DNA fragments, including cosmid and artificial chromosomes, such as the fungal artificial chromosome (FAC), yeast artificial chromosome (YAC), bacterial artificial chromosome (BAC), and P1 phage artificial chromosome (PAC) (Monaco and Larin, 1994; Bajpai, 2014; Bok et al., 2015; Clevenger et al., 2017).
Cosmid vectors, the first generation of high-capacity vectors used in genome research, are hybrids of plasmid and phage λ vector. As such, the cosmid vector encodes cos sequences required for packaging large fragments into the λ capsid and propagates their DNA as a virus or plasmid in the host cell. Since the possibility for cloning large fragments in cosmid vectors was first confirmed in 1979, they have been widely used for the construction of genomic libraries of various biological species, including Drosophila, mice, and humans. The construction of cosmid library is relatively simple and has been widely applied for cloning various NP BGCs. However, as the cosmid library requires tedious screening, it is necessary to combine high-throughput screening and sequencing methods. For example, the anisomycin BGC from Streptomyces hygrospinosus was identified using a bioactivity-guided high-throughput method for cosmid library screening (Zheng et al., 2017). Recently, CONKAT-seq (co-occurrence network analysis of targeted sequences) was used to uncover the potential of the rare BGCs from millions of cosmid clones harboring metagenomic DNA inserts (Libis et al., 2019).
Cosmid vectors can accommodate up to approximately 40 kb of DNA. They are multicopy plasmids in E. coli that facilitate DNA isolation and in vitro manipulation. However, loss of inserted DNA sometimes occurs within cosmid clones, which may be indicative of sequences that are instability in E. coli or of the transcription/translation products of the sequences are toxic to E. coli, particularly at a high copy number.
An F factor cosmid (fosmid), which contains a replicon derived from F factor and exists at a low/single copy number in E. coli, is more stable than its conventional cosmid counterpart (Kim et al., 1992). In addition, fosmid has an inducible oriV replication start point for high copy propagation, if necessary. Recently, a fosmid library containing 10,656 clones of metagenomic DNA isolated from the ATII (the Red Sea brine pool, Atlantis II Deep) lower convective layer (LCL) was functionally screened, and the products of two putative NP BGCs were detected to exhibit antibacterial and anticancer effects (Ziko et al., 2019). Typically, a cosmid or fosmid vector can only accept relatively small BGCs (up to 45 kb), which greatly hampers their application in cloning large NP BGCs.
To address the limitation of cosmids, artificial chromosomal vectors, including YAC, PAC, BAC, and FAC, which harbor the carrying capacity of 100∼350 kb, have been used for cloning NP BGCs.
YAC vectors contain two copies of yeast telomeres for chromosomal stability, a yeast centromere for segregation, a yeast ARS (autonomously replicating sequence) for replication, and appropriate markers for the selection of recombinant molecules (Burke et al., 1987). YAC provides the largest DNA insert capacity among all cloning vector types. Exogenous DNA fragments with sizes up to several hundred kilobase pairs or even as much as 2 Mb can be cloned into YAC vectors (Monaco and Larin, 1994). A cornerstone of the Human Genome Project (HGP) is the cloning of large chromosomal fragments using YAC vectors. However, problems are frequently observed during the use of YAC clones, including chimerics, deletions, and rearrangements. Furthermore, the isolation of YAC clones is challenging because of large sizes (Ramsay, 1994). As a result, each YAC clone must be carefully analyzed to ensure that no DNA rearrangements occur. In addition, the YAC system is established from eukaryotes and mainly used to study eukaryotic genomes, in which randomly distributed ARS sequences of 20∼30 kb, while being rarely in prokaryotic genomes (Stinchcomb et al., 1980).
The first PAC vector pCYPAC1 combining the characteristics of P1-phage and F factor was developed in 1994 (Loannou et al., 1994). It can be efficiently transformed into E. coli via electrotransformation. Foreign DNA inserted in the PAC exhibits almost no chimerism or rearrangement. The PAC vector can carry DNA fragments of up to approximately 300 kb. The recombinant PAC can stably exist as a single copy and propagate efficiently. To facilitate the use of PAC vectors in Streptomyces strains, the ΦC31 attP-int elements required for chromosomal integration in Streptomcyes was incorporated into a pCYPAC1-derivative vector (Ioannou et al., 1996), generating so-called ESAC (E. coli–Streptomyces artificial chromosome, pESAC) vectors. Using these pESAC vectors, up to 140 kb segments of Actinomyces DNA can be cloned and introduced into genetically accessible Streptomyces lividans via protoplast transformation, stably maintaining the vector in an integrative form (Sosio et al., 2000). Using PAC vector pESAC13 (a derivative of pESAC) harboring an oriT site, which allows for conjugal transfer instead of time-consuming protoplast transformation, a genomic library of Streptomyces tsukubaensis was generated, and the entire 83.5 kb FK506 (tacrolimus) gene cluster was then identified (Jones et al., 2013). The PAC library of Stretomyces sp. PCS3-D2 was also constructed and analyzed in silico. Two clones containing 130 and 140 kb DNA inserts were identified to harbor Type I and Type III PKS (polyketide synthase) gene clusters, respectively (Bayot Custodio and Alcantara, 2019). The positive rates of recombinant clones containing DNA inserts can be greatly improved by introducing the sacB or URA3 gene into PAC vectors as counter-screening markers, which can catalyze the production of toxicants in the presence of sucrose or 5-fluoroorotic acid (5-FOA), respectively (Noskov et al., 2003; Tang X. et al., 2015).
In 1992, the first BAC vector pBAC108L was constructed based on the well-studied E. coli F factor. This BAC vector retained the oriS, repE, parA, and parB of the F factor for replication and copy number control, while also harboring a chloramphenicol resistance marker as well as the bacteriophage λ cosN and Pl loxP sites for specific cleavage by terminase and Cre enzymes, respectively. This BAC vector has been reported to carry human genomic DNA fragments approaching 300 kb (Shizuya et al., 1992). Further, it enables the cloning of large-sized DNA fragments from complex genomic sources into bacteria, where they remain stable and are easily manipulated. However, normally, only 10–50% of the clones carry DNA inserts, depending upon the batch of the vector and insert DNA used (Shizuya et al., 1992). To facilitate the screening of positive clones, the pBeloBAC11 BAC vector contains an additional component, β-galactosidase (encoded by lacZ), which allows clones with DNA inserts to be readily identified based on an X-gal color change. Additionally, the plndigoBAC vector displays a much faster and deeper X-gal color change as a result of a point mutation in the 3’ end of lacZ (Shizuya and Kouros-Mehr, 2001). Various BAC vectors, such as pStreptoBAC and pSBAC, have been extensively used for library construction with the purpose of cloning target large-sized NP BGCs (Sosio et al., 2000; Martinez et al., 2004; Miao et al., 2005; Liu et al., 2009). These BAC vectors harbor two replication origins. One is ori that is essential for the initiation of single-copy replication in E. coli, which is crucial for stability when large DNA fragments were inserted. The other is oriV, which can be induced to increase DNA yield.
Thus far, when compared to YAC and PAC, the BAC vectors are more commonly employed for NP BGC cloning. When the genomic sequence information of BGCs is unknown (e.g., metagenome), BAC-based library construction strategies for NP discovery are always employed. Recently, using this strategy, several large NP BGCs, such as an aminopolyol polyketide BGC over 150 kb, and a quinolidomicin BGC over 200 kb, have been successfully cloned and heterologously expressed (Zhang L. et al., 2017; Hashimoto et al., 2018; Sun et al., 2018). However, due to the low positive rates, laborious screening is necessary (Lin et al., 2020). Therefore, high-throughput screening methods have received considerable attention. Recently, the MAPLE (Microfluidic automated plasmid library enrichment) method, which combines BAC libraries with single-cell droplet microfluidic techniques for discovering functional biosynthetic pathways, was developed. Using MAPLE, a type I PKS gene cluster from an Antarctic soil metagenome was isolated and sequenced (Xu et al., 2020). In addition, when the genome sequence is available, the pSBAC vector can be inserted into the flanking regions of target BGCs within the chromosome in advance and the entire target BGCs can then be captured into pSBAC through specific restriction enzyme digestion and self-ligation. Using this method, the meridamycin (MER, ∼95 kb), tautomycetin (TMC, ∼80 kb), pikromycin (PIK, ∼60 kb), and daptomycin (DPT, ∼65 kb) BGCs have been successfully cloned (Liu et al., 2009; Nah et al., 2015; Pyeon et al., 2017; Choi et al., 2019). However, a major drawback is the problematic identification of naturally existing unique restriction enzyme recognition sites on both sides of the target BGCs. Therefore, artificial insertion of a specific DNA sequence into the genome via homologous recombination (HR) is a prerequisite, limiting the application of this method in intractable strains.
Besides bacterial strains (especially actinomycetes), fungi are also prolific producers of NPs. However, despite the abundance of available fungal genome data that encode a large number of NP BGCs, the majority of them are silent in laboratory growth conditions and most fungi are not genetically amendable. To efficiently discover fungal NPs, Bok and colleagues created a novel Aspergillus/E. coli shuttle FAC expression vector, which is modified from the BAC vector via inserting the fungal autonomously replicating element AMA1 (Bok et al., 2015). Using FAC and metabolomic scoring (MS), 56 recombinant FACs containing uncharacterized BGCs from diverse fungal species were constructed and expressed in Aspergillus nidulans. Finally, 15 new metabolites were discovered and assigned with confidence to their BGCs (Clevenger et al., 2017). It could be anticipated that the development of FAC will facilitate NP research of fungi in the future.
With the rapid development of synthetic biology, standardized and orthogonal vectors, which follow uniform and modular standards, have been developed. They enable the rapid and easy exchange of modules and boost the interoperability of genetic devices among different users (Martinez-Garcia et al., 2020). However, within the field of specialized NP synthetic biology, even though there are multifarious vectors for large DNA fragment cloning, few such standard vectors have been constructed. It is well known that the size (from a few kb to more than 100 kb) of NP BGCs, the genomic GC content, and the repeat or similar sequence in the PKS or NRPS (non-ribosomal peptide synthase) genes can affect the choice of vectors for BGC cloning (Aubry et al., 2019). Thus, vectors that are flexible and adapted to various assembly methods are preferred. Recently, a suite of standardized, orthogonal integration vectors based on three site-specific integration systems (ΦBT1, ΦC31, and VWB), four antibiotic resistance genes (conferring resistance against apramycin, spectinomycin, thiostrepton, and ampicillin, respectively), and 14 promoters were constructed in order to characterize heterologous genes in Streptomyces species. However, these vectors were mainly used for monocistronic gene expression (Phelan et al., 2017). A set of 12 standardized and modular (three different resistance markers and four orthogonal integration systems) vectors based on model SEVA plasmids were designed to allow for the assembly of NP BGCs through various cloning methods in Streptomyces species (Aubry et al., 2019). In these vectors, the FLP (flippase) recombination system was also incorporated for the recycling of antibiotic markers and for reducing unwanted homologous recombination when several vectors are used simultaneously (Aubry et al., 2019). It can be expected that through the modularization and orthogonalization of key vector elements, including orthogonal integration systems, origins of replication, antibiotic selection markers, and a variety of cargoes with specific applications, a suitable vector can be quickly designed to efficiently assemble or clone large DNA fragments. It is worth to note that so far, many laboratories have designed and constructed a large number of multifarious vectors according to their own needs. To further promote NP research, laboratories should make their vectors freely available to other research groups.
High fidelity, effective and seamless assembly of large DNA fragments and appropriate vectors is the pivotal step for obtaining entire NP BGCs for heterologous expression. With the rapid development of synthetic biology, various DNA cloning and assembly methods have been established and successfully utilized for cloning NP BGCs. Depending on the experimental setting, assembly methods can be divided into two categories: in vitro and in vivo DNA assembly (Juhas and Ajioka, 2017; Li et al., 2017; Aubry et al., 2019; Kang and Kim, 2021).
In vitro cloning and assembly approaches include three main types: (1) restriction enzyme-mediated methods, e.g., BioBrick, Golden Gate, and MASTER ligation (Engler et al., 2009; Chen et al., 2013); (2) recombination-based assembly methods, such as Gibson assembly (Gibson et al., 2009), ligase cycling reaction (LCR) (Schlichting et al., 2019), direct pathway cloning (DiPaC) (Greunke et al., 2018), and DNA assembly methods based on the use of site-specific integrases (e.g., ΦC31, ΦBT1) (Li et al., 2017); (3) enzyme-independent DNA assembly, including enzyme-free cloning (EFC) and twin primer non-enzymatic DNA assembly (TPA) (Liang et al., 2017). NP BGCs can also be directly captured using several in vivo methods, including the use of Red/ET system-mediated cloning tools, such as linear-linear homologous recombination (LLHR) or linear-circular-homologous recombination (LCHR) (Fu et al., 2012), exonuclease combined with Red/ET (ExoCET) (Wang et al., 2018), transformation-associated recombination (TAR) cloning method based on the natural recombination capability of S. cerevisiae (Noskov et al., 2011; Kouprina et al., 2020), and site-specific recombination (SSR) system-based tools (Du et al., 2015; Gao et al., 2020).
The classic method for DNA assembly is via the use of enzymes for the cutting and ligation of DNA fragments and vectors. However, these will leave scars at the restriction site. To address this problem, type IIs restriction enzymes (e.g., BbsI, BsaI, and BpiI), which cut outside of the recognition sites and generate single-stranded DNA overhangs, are employed. The DNA overhangs can be appropriately designed to guide the corresponding DNA fragments for ligation in a designated order. This method was named Golden Gate (Figure 2A), which reflects the concept of modular assembly (Mitchell et al., 2015). Recently, it was employed for refactoring carotenoid biosynthetic pathways. In particular, each biosynthetic gene equipped with different promoters and terminators was assembled, resulting in various expression cassettes. A library containing 96 combinatorial refactored carotenoid pathways was then successfully generated by assembling these cassettes (Ren et al., 2017). Based on type IIs restriction enzymes, a Golden Gate shuffling method was developed, which can achieve the assembly of at least nine DNA fragments in a single step with high efficiency (90%) (Engler et al., 2009). A similar method named MASTER (methylation-assisted tailorable ends rational) ligation based on MspJI, a specific type IIs endonuclease, was also developed for sequence-independent hierarchical DNA assembly. Using the MspJI-mediated method, the blue-colored antibiotic actinorhodin (ACT) BGC (29 kb) from Streptomyces coelicolor was successfully assembled and expressed in a fast-growing Streptomyces sp. (Chen et al., 2013). To be appropriate for Golden Gate cloning, special care should be taken to ensure that the type IIs restriction site is present in opposite orientation at the ends of the vector and DNA fragments but absent in internal sequences (Marillonnet and Grutzner, 2020). However, type IIs enzymes are relatively rare, and thus few options are available. Usually, internal type IIs restriction sites should be removed by silent mutations. In addition, the number of DNA fragments that can be simultaneously assembled is still limited (Schmid-Burgk et al., 2013; Marillonnet and Grutzner, 2020).
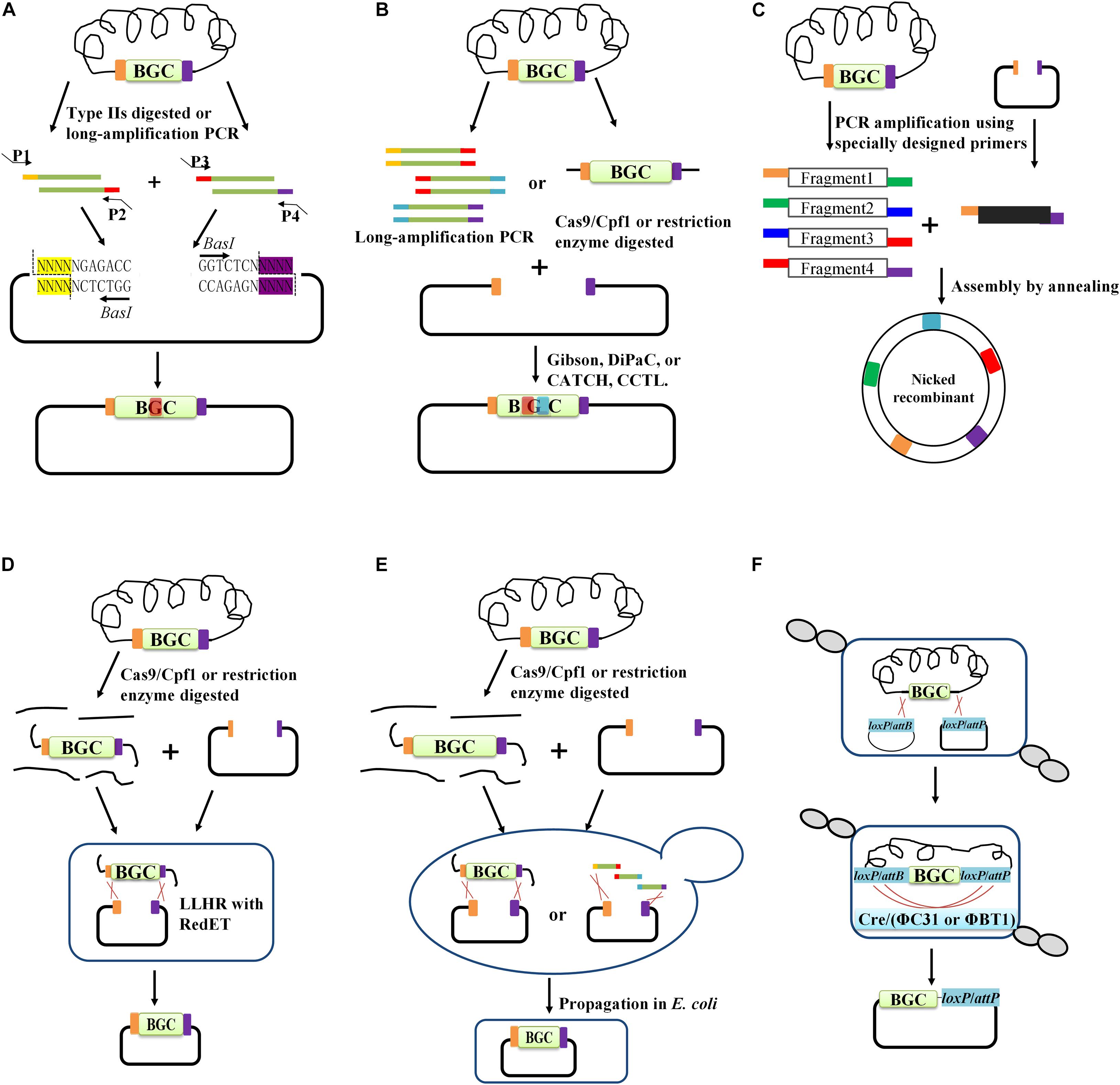
Figure 2. The main cloning methods for BGC capturing. In vitro DNA assembly method including: (A) restriction enzyme-mediated digestion and ligation (e.g., Golden gate), (B) recombination-based assembly methods (e.g., Gibson assembly, DiPaC, CATCH, and CCTL), (C) enzyme-independent DNA assemble method. In vivo assembly approaches including: (D) phage-recombinase-mediated HR in E. coli, (E) TAR cloning in yeast. (F) Site-specific integrase (e.g., Cre/loxP, ΦC31, or ΦBT1) mediated cloning.
Although traditional restriction cutting and ligation methods are still widely used, their low efficiency and enzyme site-dependence do not meet the increasing demand for assembling large DNA fragments. Thus, recombination-based assembly methods (Figure 2B) based on the existence of short homologous regions at the extremities of DNA fragments and vectors are attracting more attention. These methods include ligation-independent cloning (LIC) (Aslanidis and de Jong, 1990), sequence and ligation-independent cloning (SLiC) (Li and Elledge, 2012), seamless ligation cloning extract (SliCE) (Zhang Y. et al., 2012), circular polymerase extension cloning (CPEC) (Quan and Tian, 2014), Gibson assembly, and Cas9-associated targeting of chromosome segments (CATCH) (Jiang et al., 2015) and so on. Recombination-based DNA assembly usually employs one to three enzymes in the in vitro reaction, wherein DNA polymerase, exonuclease, and ligase are the most commonly used. Hereafter, we provide a brief introduction to the above-mentioned assembly methods.
Ligation-independent cloning mediates the assembly between a DNA fragment and a PCR-amplified vector with a 12-nt tail complementary to the DNA fragment’s end. It does not require the use of restriction enzymes and T4 DNA ligase. The 3′-terminal sequence can be removed via the (3′→5′) exonuclease activity of T4 DNA polymerase, leading to DNA fragments with a 5′-overhang (10–12 nt in length), which results in annealing and circularization between vector molecules and DNA fragments mediated by the 10–12-nt cohesive ends (Aslanidis and de Jong, 1990). Based on this method, SLIC was developed, which can achieve the assembly of multiple DNA fragments in a single reaction by combining in vitro HR and single-strand annealing. SLIC is more efficient at very low DNA concentrations, especially in the presence of the HR protein RecA (Li and Elledge, 2007). SLiCE is a highly cost-effective method, which utilizes cell extracts from E. coli with overexpression of the λ phage Red recombination system for DNA assembly in vitro. This method provides an effective strategy for directional seamless DNA cloning from BAC or complex genomes (Zhang Y. et al., 2012).
The Gibson assembly method uses three commercial enzymes (T5 exonuclease, Phusion DNA polymerase, and Taq DNA ligase) for the assembly of DNA fragments with short homologous ends in vitro. Unlike the T4 DNA polymerase in LIC, which produces a 5’-overhang, T5 exonuclease chews back the homologous ends to generate 3’-overhangs, which anneal to each other, followed by Phusion DNA polymerase and Taq DNA ligase, which fill the gap and covalently link the fragments, respectively. However, Gibson assembly cannot be efficiently employed for the assembly of DNA fragments with high GC content due to high vector self-ligation background. Recently, a modified Gibson assembly method was developed by adding a pair of universal terminal overhangs with high AT content (21 bp) to the ends of the BAC vector, greatly reducing vector self-ligation (Li L. et al., 2015). Using this method, a 67 kb pristinamycin II (PII) BGC from Streptomyces pristinaespiralis was hierarchically assembled from 15 PCR-amplified fragments (Li L. et al., 2015). In addition, a T5 exonuclease-mediated DNA assembly (TEDA) method was established, in which homologous ends were treated with T5 exonuclease alone. After annealing, the reaction sample was transformed into E. coli to repair the gap and form a phosphodiester bond to link the fragment and vector with the endogenous DNA repair enzymes. The results indicated that the cloning efficiency of TEDA was higher than that of the traditional Gibson assembly tool (Xia et al., 2019). The development of CRISPR technology has greatly promoted recombination-mediated DNA assembly. A novel DNA assembly method named CATCH was developed by combining the in vitro CRISPR/Cas9 endonuclease-mediated genome treatment and Gibson assembly, which could achieve the direct cloning of large bacterial genomic segments (up to 100 kb) (Jiang et al., 2015). Using this tool, the 78-kb bacillaene BGC from Bacillus subtilis was cloned into a BAC vector at a ∼12% positive rate. In addition, the 36 kb jadomycin BGC from Streptomyces venezuelae and the 32 kb chlortetracycline BGC from Streptomyces aureofaciens, were also successfully captured with a ∼90% positive rate, highlighting the versatility of CATCH for cloning large BGCs (Jiang et al., 2015). It should be noted that these recombination-based methods (e.g., Gibson assembly, CATCH) might be inefficient when homologous regions of the fragment extremities have complicated DNA sequences, such as secondary hairpin structure formation or high GC content (Casini et al., 2014; Li L. et al., 2015).
Unlike Cas9 which introduces double-strand breaks (DSBs) and produces blunt ends, Cpf1 (Cas12a) cleaves target DNA and produces sticky overhangs, which makes Cpf1 an alternative tool for DNA assembly in vitro (Lei et al., 2017). Recently, a method named CCTL (Cpf1-assisted Cutting and Taq DNA ligase mediated Ligation) was developed, with the 8-nt sticky end produced by Cpf1 cleavage for ligation instead of homologous sequences in recombination-based methods, which therefore make the CCTL is suitable for cloning complicated DNA sequences (Lei et al., 2017). However, the requirement of specific PAM limits its application. To address this limitation, the PAM specificity of Cas12a was expanded via specific structure-guided mutagenesis and two engineered Cas12a variant EP15 and EP16 were obtained, which increased the targeting range by fourfold. Based on this modified Cas12a, the iCOPE (improved Cas12a-assisted one-pot DNA Editing) method was developed, which can avoid many of the DNA sequence constraints (Wang et al., 2019).
In addition to the methods described above, various in vitro novel DNA assembly methods for BGCs cloning have been designed and established. These include the assembly of fragment ends after PCR (AFEAP cloning) (Zeng et al., 2017), versatile genetic assembly system (VEGAS) (Mitchell et al., 2015), uracil-specific excision reagent cloning (USER) (Salomonsen et al., 2014), LCR (de Kok et al., 2014), DNA assembly with thermostable exonuclease and ligase (DATEL) (Jin et al., 2016), overlap extension PCR and recombination (OEPR Cloning) (Liu C. J. et al., 2017), direct pathway cloning (DiPaC) (Greunke et al., 2018), as well as DiPaC combination of SLIC cloning (D’Agostino and Gulder, 2018), which provide alternatives for large fragment cloning under different experimental conditions.
Enzyme-mediated DNA assembly methods are efficient and straightforward. As mentioned above, Golden Gate assembly is robust and suitable for assembling over 15 DNA fragments with high efficiency and fidelity. However, due to limited commercially available Type IIs endonucleases, it is not always possible to find suitable restriction enzymes that avoid the naturally occurring Type IIs sites within BGCs (Liang et al., 2017). Therefore, additional efforts are needed to modify the sequences of BGCs in order to eliminate the undesired cut sites. Gibson assembly is versatile, but its efficiency and fidelity drop sharply when the number of fragments is more than four. Furthermore, essential components such as promoters, ribosomal binding sites, and terminators are notoriously difficult for Gibson assembly because of their secondary structures (Liang et al., 2017). Enzyme-independent DNA assembly methods can realize DNA assembly without enzymes, which saves costs and is especially suitable for high-throughput settings. These approaches mainly include enzyme-free cloning (EFC), polymerase incomplete primer extension (PIPE), and Twin primer non-enzymatic DNA assembly (TPA).
A highly efficient EFC procedure for DNA assembly was previously established, utilizing tailed PCR primer sets to generate complementary staggered overhangs on both fragments and vectors via a denaturation-hybridization reaction (Tillett and Neilan, 1999). This approach enables directional cloning in a ligase-free manner. Therefore, it is not constrained by the requirement for appropriate enzyme sites. However, this method is mainly used for the assembly of two DNA fragments, and its efficiency is low. TPA for the efficient assembly of multiple PCR fragments was recently developed (Figure 2C), allowing for the successful construction of a 31 kb plasmid harboring an n-butanol production pathway (∼26 kb) from five fragments with ∼50% fidelity (Liang et al., 2017). TPA cloning is also seamless and sequence-independent, and its performance rivals even the best in vitro assembly methods. Although these enzyme-free cloning tools provide a number of advantages over other cloning strategies, they still have limitations. For example, these methods usually require a number of specially designed primers, and the assembly capability as well as fidelity drop sharply with increasing fragment size (Tillett and Neilan, 1999; Yuan et al., 2016; Liang et al., 2017; Richter et al., 2019).
In vitro assembly methods provide flexible cloning of DNA fragments, wherein the DNA fragments can be produced through multiple rounds of PCR or direct chemical synthesis. However, random mutations cannot be entirely ruled out. In addition, incorrect pairing of DNA fragments during assembly may also cause unanticipated mutations, especially in the PKS or NRPS genes, which contain numerous repeat sequences. In vivo DNA cloning methods for direct capture of the target DNA fragment, which are based on the strong homologous recombination ability of E. coli expressing the Red/ET system or that of yeast, have previously been developed. They represent an alternative strategy for BGC cloning. These methods mainly include phage recombinase-mediated homologous recombination cloning in E. coli such as LLHR (Figure 2D), transformation-associated recombination-mediated cloning (TAR) in yeast (Figure 2E) and site-specific recombination (SSR)-mediated cloning in Streptomyces (Figure 2F; Li et al., 2017; Abbasi et al., 2020; Kang and Kim, 2021).
The endogenous HR system in E. coli is mainly mediated by the chromosome-encoded recombinases RecA/RecBCD (Abbasi et al., 2020). Many cloning strategies based on the endogenous HR have been created, such as in vivo cloning (IVC), in which PCR products containing terminal sequences identical to the two terminals of the linearized vector were co-transfected into E. coli to incorporate PCR fragments into the vector via the high HR ability of E. coli (Oliner et al., 1993). However, due to its strong exonuclease activity, the RecBCD complex can rapidly degrade exogenous linear DNA molecules. The PCR products and linear vector can be introduced and stably maintained only in RecBCD-deficient E. coli strains (Abbasi et al., 2020). In addition, RecA-dependent recombination requires a much longer homologous region (approximately 500 bp). To develop a more efficient and reliable HR system in E. coli, Red/ET recombineering was developed, which depends on phage-recombinases, either RecE/RecT from the Rac prophage or Redα/Redβ from the λ phage (Zhang et al., 1998). RecE and Redα are 5′→3′ ATP-independent exonucleases, while RecT and Redβ are DNA annealing proteins. Another protein, Redγ, identified only in the λ phage, was found to significantly promote the recombination efficiency of Redα/Redβ. It was later identified as an inhibitor of the RecB subunit of RecBCD. This protein protects linear DNA from degradation by endogenous nucleases (Abbasi et al., 2020).
Red/ET recombineering has been established as an efficient in vivo homologous recombination strategy for E. coli (Wang et al., 2016). This technology was first used to reconstitute an entire 43 kb myxochromide BGC from two overlapping cosmids (Wenzel et al., 2005). Subsequently, it has been widely applied for the cloning of a variety of NP BGCs ranging from 11 to 106 kb from different microbes, including Streptomyces, Sorangium, and Cystobacter (Binz et al., 2008; Lesic and Rahme, 2008; Wang et al., 2018). In these cases, the reconstitution process was mediated by very short homologous regions (usually 40–50 bp) between a replicative circular vector and a linear DNA molecule, and was therefore termed “linear-circular homologous recombination (LCHR).” However, the approach utilizing Redαβ or the truncated version of RecET is inefficient at mediating homologous recombination between two linear DNA molecules, which hampers its use for direct cloning of target BGCs (Fu et al., 2012).
Fu et al. (2012) discovered that full-length RecE along with RecT considerably increased the efficiency of recombination between two linear DNA molecules (a linearized target DNA fragment and a PCR-amplified linear vector backbone flanked with homology arms to the target DNA). Using this LLHR (linear–linear homologous recombination) approach, ten large NRPS and PKS BGCs (with sizes from 10 to 37 kb) from the genomic DNA of Photorhabdus luminescens were directly cloned into linear expression vectors in a one-step recombination event. However, they failed to direct clone the intact 106 kb salinomycin gene cluster from the genome of Streptomyces albus using LLHR. Finally, the group successfully cloned three fragments of salinomycin BGC using LLHR separately and assembled them into a complete one (Yin et al., 2015).
To improve the performance for direct cloning of large-sized (>50 kb) DNA segments from complex genomes such as mammalian genomes, which are three orders of magnitude larger than bacterial genomes, exonuclease (in vitro) combined with RecET recombination (in vivo) (ExoCET) was developed (Wang et al., 2018). For the in vitro assembly, several exonucleases including T4 polymerase (T4 pol), T5 exonuclease, T7 exonuclease, DNA polymerase I Klenow fragment, T7 DNA polymerase, λ exonuclease, Exonuclease III, and Phusion DNA polymerase, were tested. The 3’ exonuclease activity of T4 polymerase was selected due to it having the highest efficiency and fidelity (Wang et al., 2018). After exonuclease chew-back, the target DNA fragment and the vector were annealed together via the homology arm (about 80 bp) and were then transformed into E. coli for in vivo HR via Red/ET. This concerted action of T4 pol and Red/ET is believed to be more proficient for the direct cloning of long DNA regions than either T4 pol or Red/ET alone (Wang et al., 2018). ExoCET is generally applicable to a broader range of direct cloning with respect to size (up to 106 kb) and genome complexity (Wang et al., 2018). It should be noted that, in order to ensure a high efficiency for the LLHR-mediated cloning method, genomic DNA must be cleaved by unique restriction enzymes near the 5’ and 3’ ends of target BGCs. However, it is not always easy to find appropriate restriction enzyme cutting sites. With the advent of the programmable CRISPR/Cas9 system, which is able to recognize and cut DNA sequences near target BGCs to easily release linear DNA fragments, this limitation could be overcome (Lee et al., 2015; Wang et al., 2018). With improved Red/ET technology and rapidly growing microbial genome sequence data in public databases, a variety of complete NP BGCs have been cloned directly from microbial genomic DNA via LLHR (Table 1).
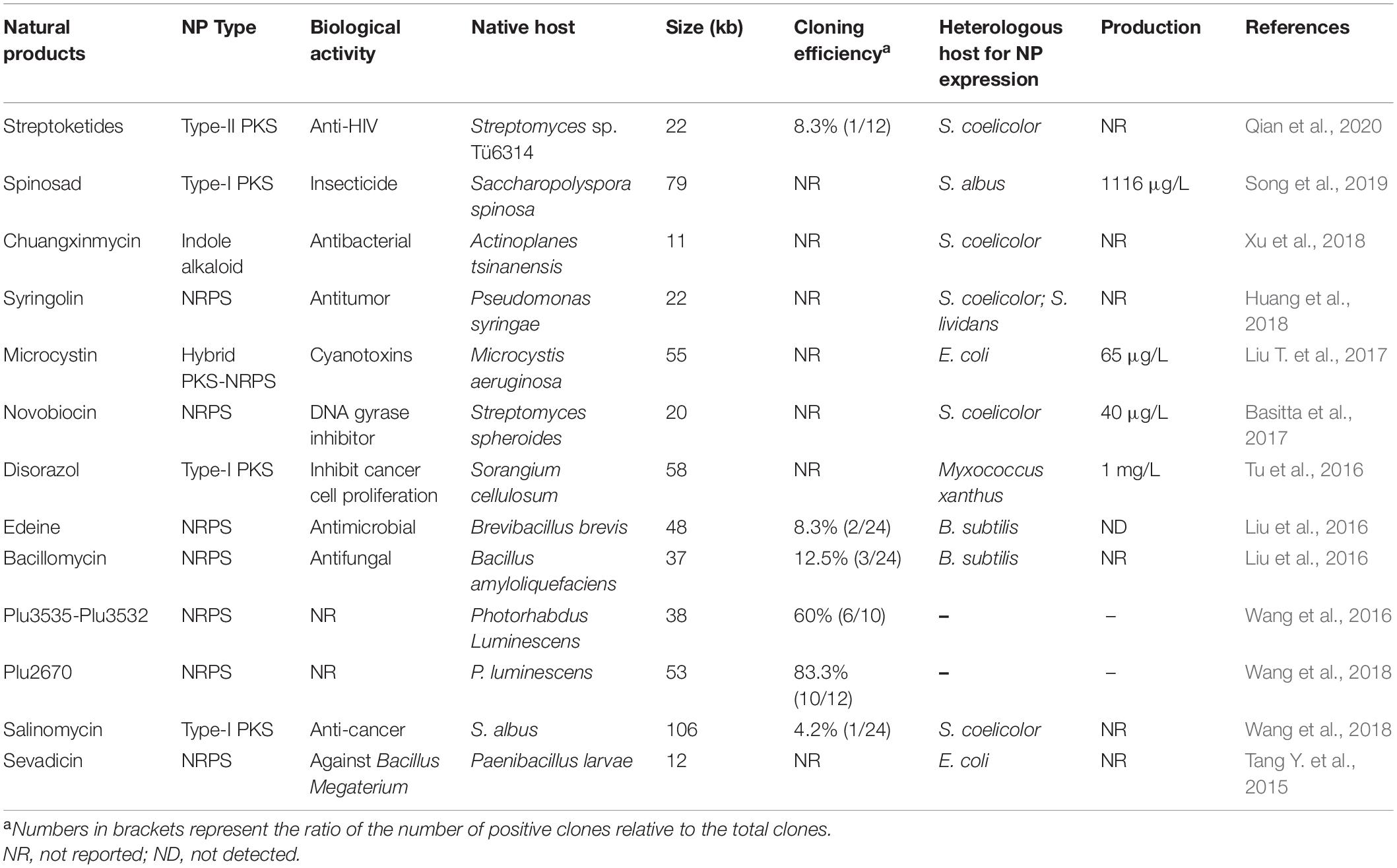
Table 1. Examples of BGCs cloned by Red/ET recombination (from 2015 to present).
The assembly of two DNA molecules containing homologous sequences via recombination in yeast was first demonstrated by Kunes et al. (1985). A couple of years later, a convenient method for plasmid construction using this in vivo bimolecular recombination reaction was developed (Ma et al., 1987). Motivated by this method, a transformation-associated recombination (TAR) strategy in yeast based on this approach was later introduced, allowing for the selective isolation of large genomic regions from complex genomic DNA (Larionov et al., 1997; Noskov et al., 2002).
Transformation-associated recombination was initially been used to isolate large regions of mammalian genomic DNA in the 1990s (Larionov et al., 1997). The propagation of TAR-generated DNA constructs depends on ARS-like sequences, which can function as an origin of replication in yeast. The ARS sequences are frequently and randomly distributed throughout all eukaryotic genomes per 20–30 kb on average (Stinchcomb et al., 1980). Chromosomal regions with high G + C content are poor in ARS-like sequences, and ARS frequency might be reduced in prokaryotic genomes, which precludes their isolation via the standard TAR method. Noskov et al. (2003) inserted ARS into the TAR vector, using HIS3 as a positive selection marker and URA3 as a negative marker. The modified TAR cloning system enables the isolation of genomic regions lacking yeast ARS-like sequences (e.g., bacterial genome DNA) and eliminates the high vector recircularization background caused by end-joining during yeast transformation (Noskov et al., 2003). This modified TAR cloning method was further extended to capture microbial NP BGCs by constructing the yeast-E. coli–Streptomyces tri-shuttle vector pTARa. Using pTARa, multiple BGCs were directly cloned or reassembled from environmental DNA (eDNA) libraries (Kim et al., 2010). In contrast to pTARa that harbors oriV, pCAP01, a novel capture vector equipped with a pUC ori, can maintain multiple copies without induction and remained stable even when carrying > 50 kb inserts (Yamanaka et al., 2012, 2014). Using the pCAP101 vector, a 67 kb silent NRPS BGC responsible for the biosynthesis of taromycin from the marine actinomycete Saccharomonospora sp. CNQ-490 was successfully captured and activated in S. coelicolor M1146 (Yamanaka et al., 2014). However, the construction process of pCAP01-based capture plasmids is tedious and time-consuming. It involves the assembly of a pair of 1-kb capture arms into pCAP01, overlapping with the flanking regions of target BGCs. Larson et al. (2017) streamlined this procedure by employing a fully synthetic 360 bp capture arm, which reduced the duration of the cloning process and opened the door for high-throughput applications. Using this modified TAR method, a 54 kb cosmomycin BGC from Streptomyces sp. CNT-302 was successfully captured (Larson et al., 2017). The range of heterologous hosts compatible with the TAR platform was expanded to the Gram-positive Bacillus subtilis with low G + C content by replacing the high G + C content Streptomyces element in pCAP01 with the Bacillus element (Bourgouin et al., 1990) to yield the yeast-E. coli-B. subtilis tri-shuttle vector pCAPB1. Using pCAPB1, the surfactin BGC was successfully cloned from B. subtilis 1779 (Li Y. et al., 2015). Later, a TAR vector pCAP05 was constructed by introducing an RK2 replicon. It replicates at a low copy number in a wide range of Gram-negative bacteria via the oriV and trfA gene, which determine host range and copy number (Scott et al., 2003; Zhang J. J. et al., 2017). Using pCAP05, the violacein BGC (∼8 kb) from marine bacterium Pseudoalteromonas luteoviolacea was cloned and expressed in Pseudomonas putida and Agrobacterium tumefaciens (Zhang J. J. et al., 2017). Overall, TAR has been widely employed for BGC cloning, leading to the identification of many novel NPs (Alberti et al., 2019; Kouprina and Larionov, 2019; Table 2).
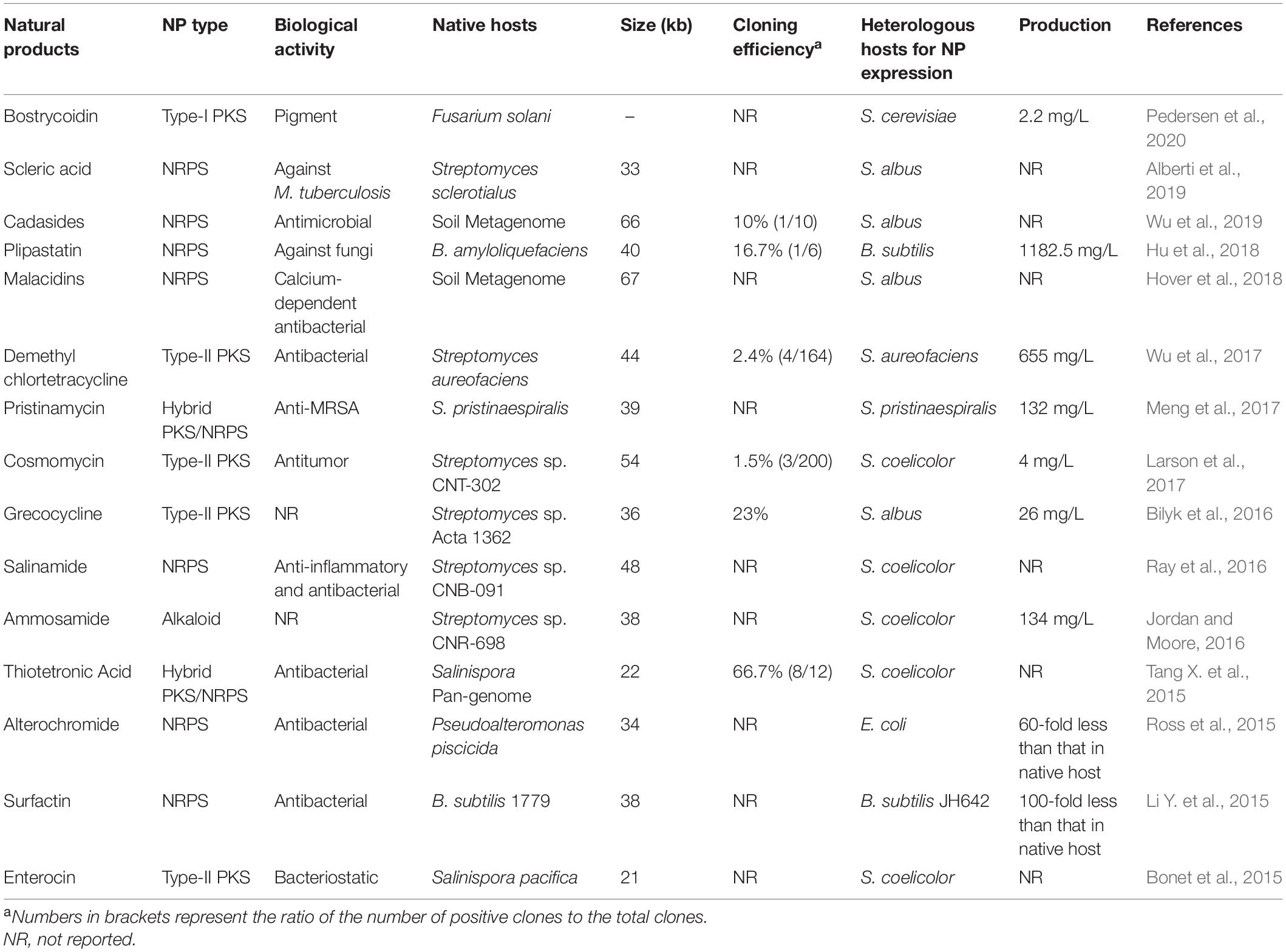
Table 2. Examples of BGCs cloned by TAR cloning (from 2015 to present).
Although TAR cloning can be used to directly clone NP BGCs of interest, the method exhibits very low cloning efficiency (0.5–2%) due to vector recircularization via end joining in yeast, which leads to time-consuming screening of hundreds of clones. Thus, two different strategies have been introduced to increase the positive rates (Lee et al., 2015; Tang X. et al., 2015). The first one is to use a counter-selection marker for colony selection. Tang X. et al. (2015) introduced the URA3 gene under a strong pADH1promoter into pCAP01 in order to generate pCAP03, which allows for convenient screening against recircularization in the presence of 5-FOA. Using pCAP03, a 26 kb thiolactomycin BGC from Salinispora pacifica was captured at a positive rate of 75%, and a 33 kb genome locus containing the thiotetroamide BGC (∼29 kb) was cloned at a positive rate of 20% (Tang X. et al., 2015). The second strategy is to use the RNA-guided Cas9 endonuclease to cleave chromosomal DNA (Lee et al., 2015). Homologous recombination has been reported as more efficient when the linearized capturing vector hooks (homology arms) are located closer to the ends of the target DNA sequences (Kouprina et al., 2006). Although unique restriction enzymes can be theoretically obtained to cleave near the 5’ and 3’ ends of target DNA, it is always challenging to find suitable cutting sites. The programmable CRISPR/Cas9 system was used to precisely cleave both sides of the target DNA, significantly improving TAR cloning efficiency by up to 32% (Lee et al., 2015). Currently, capturing target chromosomal regions requires the screening of less than a dozen transformants. It is conceivable that TAR cloning, combined with a counter-selection marker and the CRISPR/Cas9 system, will further accelerate the direct cloning of microbial NP BGCs. So far, TAR cloning is the only available method for selectively capture chromosomal segments up to 300 kb from complex genomes (Kouprina and Larionov, 2016). Collective examples for the direct cloning of NP BGCs by TAR are summarized in Table 2.
In addition to DNA cloning and assembly methods based on homologous recombination in E. coli or yeast, there are other in vivo cloning systems based on site-specific recombination (SSR), which consist of a specialized recombinase and its target sites. There are two evolutionarily distinct site-specific recombinases with different recombination mechanisms, including tyrosine recombinases (e.g., Cre recombinase) and serine integrases (e.g., ΦC31 and ΦBT1 integrase) (Fogg et al., 2014).
Generally, bacteriophage-derived serine integrases bind to specific 40–60 bp DNA sites (so-called attachment sites derived from the phage attP and cognate bacterial chromosome attB) and bring these sites together, cut and then rejoin the sites to yield the recombinant product (Grindley et al., 2006). Site-specific serine integration systems have been mainly used to integrate foreign DNA constructs into the attB site of prokaryotes, eukaryotes, or archaea chromosomes for the production of stable engineered strains. Integrases are capable of promoting efficient genomic integration of large NP BGCs (>100 kb) via attP × attB unidirectional recombination (Myronovskyi and Luzhetskyy, 2013). Based on this SSR system, a novel strategy for cloning large BGCs was devised in Streptomyces based on the ΦBT1 integrase (Du et al., 2015). First, the paired ΦBT1 integration sites attB/attP and the replicative plasmid pKC1139 are individually introduced on either side of the target BGC via two single crossover recombination events. Thereafter, the ΦBT1 recombinase is expressed, which mediates the cleavage of the two paired integration sites, resulting in circularization of the target BGC in pKC1139. Recombinant clones containing the target BGC are then extracted and transferred into E. coli for recovery. Using this strategy, the actinorhodin BGC (25 kb) from S. coelicolor, the napsamycin BGC (45 kb), and the daptomycin BGC (157 kb) from Streptomyces roseosporus were successfully isolated with high efficiency greater than 80% (Du et al., 2015). The entire 34 kb neomycin BGC from Streptomyces fradiae CGMCC 4.576 was similarly cloned using the ΦBT1 integration system (Zheng et al., 2019).
The Cre enzyme, as well as Flp and Dre recombinases, belongs to the tyrosine recombinase family. Cre recombinase can specifically and efficiently catalyze recombination between two specific 34-bp sites called loxP. The Cre/loxP system is effective in both bacterial and eukaryotic cells. Cre-mediated recombination results in the excision of the intervening DNA segment and produces a circular DNA molecule if two loxP sites in the DNA strand are in the same orientation. Therefore, when a cloning vector backbone is included in the intervening DNA, the circularized DNA molecule can replicate as a plasmid. Using this “Cre/loxP plus BAC” strategy, the 32 kb T3SS (type 3 secretion system) gene cluster from Photorhabdus luminescens and the 78 kb siderophore BGC from A. tumefaciens were successfully cloned (Hu et al., 2016).
However, as described above, SSR-mediated cloning methods require the initial integration of specific sites into the chromosome in advance. Therefore, they cannot be employed in difficult-to-manipulate organisms. Recently, a robust BGC cloning method named CAPTURE (Cas12a-assisted precise targeted cloning using in vivo Cre-loxP recombination) was developed by combining in vitro Cas12a-based treatment of genome and in vivo Cre-loxp recombination. This method could achieve direct NP BGC cloning with high efficiency (Enghiad et al., 2021). The microbial genome was purified and digested by the Cas12 protein to release the target BGC and then mixed with two PCR-amplified vector elements in a T4 DNA polymerase exo + fill-in DNA assembly reaction to join the three fragments into a linear DNA product. Finally, the linear DNA assembly products were transformed into E. coli expressing Cre recombinase for in vivo Cre-loxp circularization. This method avoids pre-insertion sites at both ends of the BGCs in the genome that are difficult to manipulate genetically. In addition, each PCR amplified vector element only contains one loxP site and does not carry the selection marker and the origin of replication, which could eliminate vector recircularization. Using CAPTURE, 47 NP BGCs ranging from 10 to 113 kb from both Actinomyces and Bacilli were directly cloned with up to 100% efficiency. Heterologous expression of the cloned BGCs led to the discovery of 15 previously uncharacterized NPs (Enghiad et al., 2021).
Exploring new antibiotics to combat against emerging drug resistance as well as the identification of new lead drugs for the treatment of various diseases are of utmost necessity. Thus, mining of NPs will continue to play an indispensable role in the drug discovery field. Traditionally, NP BGCs of interest are often cloned by construction of genomic DNA libraries using cosmids, fosmids, or artificial chromosomes. These methods are sequence-independent and have been proven to be efficient for cloning NP BGCs. However, these conventional methods are not suitable for the large-scale and high-throughput discovery of novel natural agents due to the requirement of extensive screening. With the availability of an increasing number of bacterial genome sequences and progress in genetic manipulation techniques, a variety of approaches for the direct cloning of large-sized BGCs from chromosomes have been developed. After carefully preparing high-quality large DNA fragments harboring putative BGCs and selecting appropriate vectors, these BGCs can be assembled or directly cloned with high efficiency in vitro or in vivo (Tables 1–3). Upon cloning, BGCs can be introduced into suitable microbial hosts for heterologous expression and subsequent identification of the corresponding products.
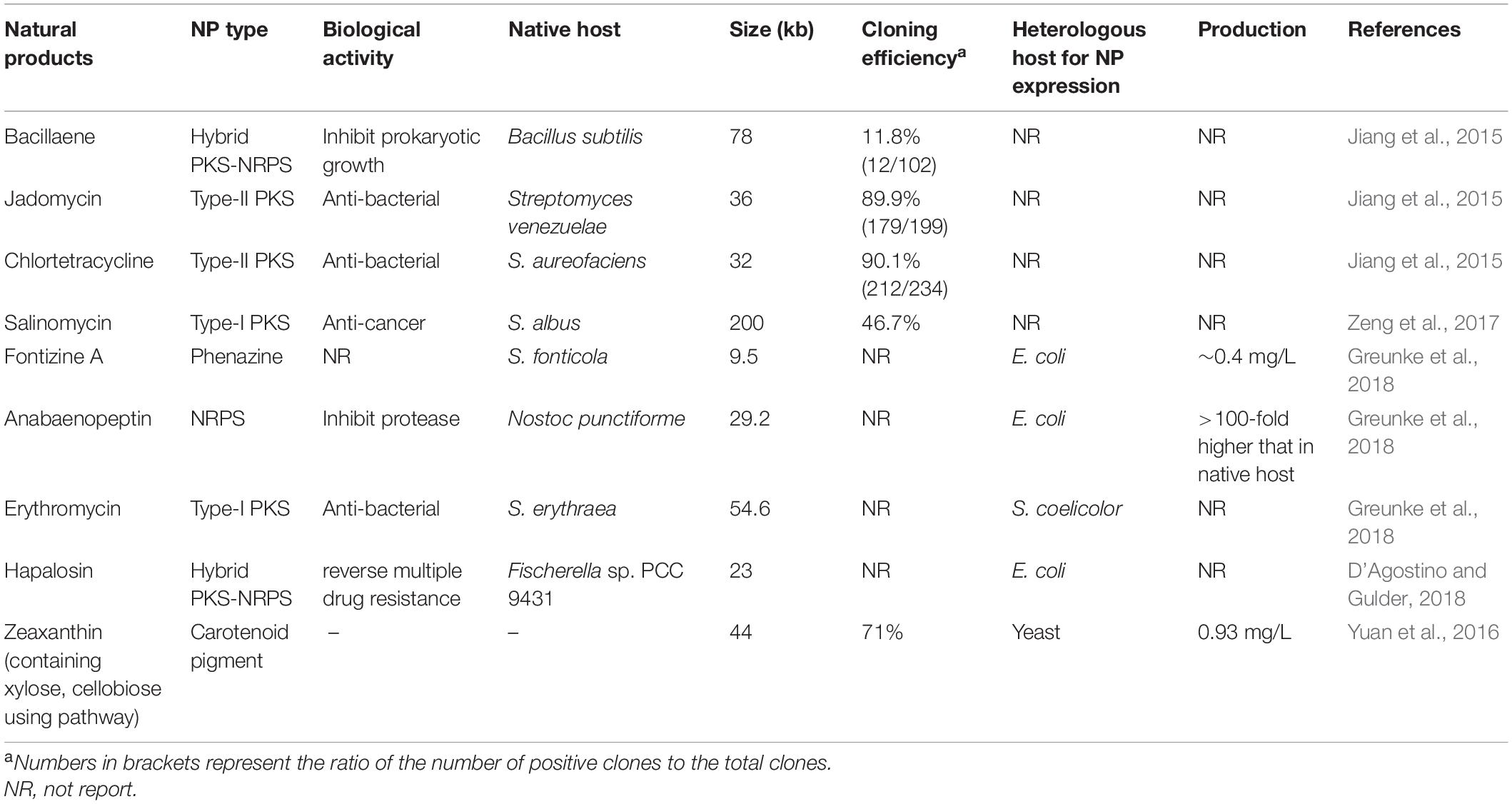
Table 3. Examples of BGCs cloned via in vitro assembly (from 2015 to present).
The aforementioned methods differ in both mechanism and cloning scale, providing effective means to meet different needs. The development of in vivo, in vitro, or even in vivo/in vitro hybrid strategies, especially those employing Cas9 or Cas12a cleavage, has greatly facilitated the cloning or assembly of microbial NP BGCs. It is therefore expected that these methodologies will greatly improve genome mining efforts that precede the discovery of novel compounds. However, to our knowledge, a universal approach suitable for all experimental situations is still lacking. Therefore, the combination of different cloning approaches, and the establishment of novel, easy-to-use, highly efficient, and accurate cloning methods remain a necessity.
WW and GZ wrote the draft. YL edited the manuscript. All the authors contributed to the article and approved the submitted version.
This work was supported by the National Key Research and Development Program (2019YFA0905400 and 2018YFA0903700) and the National Natural Science Foundation of China (31770088 and 31970083).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbasi, M. N., Fu, J., Bian, X., Wang, H., Zhang, Y., and Li, A. (2020). Recombineering for genetic engineering of natural product biosynthetic pathways. Trends Biotechnol. 38, 715–728. doi: 10.1016/j.tibtech.2019.12.018
Alberti, F., Leng, D. J., Wilkening, I., Song, L., Tosin, M., and Corre, C. (2019). Triggering the expression of a silent gene cluster from genetically intractable bacteria results in scleric acid discovery. Chem. Sci. 10, 453–463. doi: 10.1039/c8sc03814g
Aslanidis, C., and de Jong, P. J. (1990). Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074. doi: 10.1093/nar/18.20.6069
Atanasov, A. G., Zotchev, S. B., Dirsch, V. M., and Supuran, T. C. (2021). Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug. Discov. 20, 200–216. doi: 10.1038/s41573-020-00114-z
Aubry, C., Pernodet, J. L., and Lautru, S. (2019). Modular and integrative vectors for synthetic biology applications in Streptomyces spp. Appl. Environ. Microbiol. 85:e00485-19. doi: 10.1128/AEM.00485-19
Basitta, P., Westrich, L., Rösch, M., Kulik, A., Gust, B., and Apel, A. K. (2017). AGOS: a plug-and-play method for the assembly of artificial gene operons into functional biosynthetic gene clusters. ACS Chem. Biol. 6, 817–825. doi: 10.1021/acssynbio.6b00319
Bayot Custodio, A., and Alcantara, E. P. (2019). Identification of polyketide synthase gene clusters in a phage P1-derived artificial chromosome library of a Philippine strain of Streptomyces sp. PCS3-D2. Asia Pac. J. Mol. Biol. Biotechnol. 27, 56–63. doi: 10.35118/apjmbb.2019.027.2.08
Bennett-Baker, P. E., and Mueller, J. L. (2017). CRISPR-mediated isolation of specific megabase segments of genomic DNA. Nucleic Acids Res. 45:e165. doi: 10.1093/nar/gkx749
Bilyk, O., Sekurova, O. N., Zotchev, S. B., and Luzhetskyy, A. (2016). Cloning and heterologous expression of the grecocycline biosynthetic gene cluster. PLoS One 11:e0158682. doi: 10.1371/journal.pone.0158682
Binz, T. M., Wenzel, S. C., Schnell, H. J., Bechthold, A., and Muller, R. (2008). Heterologous expression and genetic engineering of the phenalinolactone biosynthetic gene cluster by using red/ET recombineering. Chembiochem 9, 447–454. doi: 10.1002/cbic.200700549
Bok, J. W., Ye, R., Clevenger, K. D., Mead, D., Wagner, M., Krerowicz, A., et al. (2015). Fungal artificial chromosomes for mining of the fungal secondary metabolome. BMC Genom. 16:343. doi: 10.1186/s12864-015-1561-x
Bonet, B., Teufel, R., Crusemann, M., Ziemert, N., and Moore, B. S. (2015). Direct capture and heterologous expression of Salinispora natural product genes for the biosynthesis of enterocin. J. Nat. Prod. 78, 539–542. doi: 10.1021/np500664q
Bourgouin, C., Delecluse, A., de la Torre, F., and Szulmajster, J. (1990). Transfer of the toxin protein genes of Bacillus sphaericus into Bacillus thuringiensis subsp. israelensis and their expression. Appl. Environ. Microbiol. 56, 340–344. doi: 10.1128/AEM.56.2.340-344.1990
Burke, D. T., Carle, G. F., and Olson, M. V. (1987). Cloning of large segments of exogenous DNA into yeast by means of artificial chromosome vectors. Science 236, 806–812. doi: 10.1126/science.3033825
Casini, A., MacDonald, J. T., De Jonghe, J., Christodoulou, G., Freemont, P. S., Baldwin, G. S., et al. (2014). One-pot DNA construction for synthetic biology: the modular Overlap-Directed Assembly with Linkers (MODAL) strategy. Nucleic Acids Res. 42:e7. doi: 10.1093/nar/gkt915
Chen, W. H., Qin, Z. J., Wang, J., and Zhao, G. P. (2013). The MASTER (methylation-assisted tailorable ends rational) ligation method for seamless DNA assembly. Nucleic Acids Res. 41:e93. doi: 10.1093/nar/gkt122
Choi, S., Nah, H. J., Choi, S., and Kim, E. S. (2019). Heterologous expression of daptomycin biosynthetic gene cluster via Streptomyces artificial chromosome vector system. J. Microbiol. Biotechnol. 29, 1931–1937. doi: 10.4014/jmb.1909.09022
Clevenger, K. D., Bok, J. W., Ye, R., Miley, G. P., Verdan, M. H., Velk, T., et al. (2017). A scalable platform to identify fungal secondary metabolites and their gene clusters. Nat. Chem. Biol. 13, 895–901. doi: 10.1038/nchembio.2408
Clos, J., and Zander-Dinse, D. (2019). Cosmid library construction and functional cloning. Methods Mol. Biol. 1971, 123–140. doi: 10.1007/978-1-4939-9210-2_6
D’Agostino, P. M., and Gulder, T. A. M. (2018). Direct pathway cloning combined with sequence- and ligation-independent cloning for fast biosynthetic gene cluster refactoring and heterologous expression. ACS Synth. Biol. 7, 1702–1708. doi: 10.1021/acssynbio.8b00151
Daniel, R. (2005). The metagenomics of soil. Nat. Rev. Microbiol. 3, 470–478. doi: 10.1038/nrmicro1160
de Kok, S., Stanton, L. H., Slaby, T., Durot, M., Holmes, V. F., Patel, K. G., et al. (2014). Rapid and reliable DNA assembly via ligase cycling reaction. ACS Chem. Biol. 3, 97–106. doi: 10.1021/sb4001992
Du, D., Wang, L., Tian, Y., Liu, H., Tan, H., and Niu, G. (2015). Genome engineering and direct cloning of antibiotic gene clusters via phage φbT1 integrase-mediated site-specific recombination in Streptomyces. Sci. Rep. 5:8740. doi: 10.1038/srep08740
Enghiad, B., Huang, C., Guo, F., Jiang, G., Wang, B., Tabatabaei, S. K., et al. (2021). Cas12a-assisted precise targeted cloning using in vivo Cre-lox recombination. Nat. Commun. 12:1171. doi: 10.1038/s41467-021-21275-4
Engler, C., Gruetzner, R., Kandzia, R., and Marillonnet, S. (2009). Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One 4:e5553. doi: 10.1371/journal.pone.0005553
Fogg, P. C., Colloms, S., Rosser, S., Stark, M., and Smith, M. C. (2014). New applications for phage integrases. J. Mol. Biol. 426, 2703–2716. doi: 10.1016/j.jmb.2014.05.014
Fu, J., Bian, X., Hu, S., Wang, H., Huang, F., Seibert, P. M., et al. (2012). Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 30, 440–448. doi: 10.1038/nbt.2183
Gao, H., Taylor, G., Evans, S. K., Fogg, P. C. M., and Smith, M. C. M. (2020). Application of serine integrases for secondary metabolite pathway assembly in Streptomyces. Synth. Syst. Biotechnol. 5, 111–119. doi: 10.1016/j.synbio.2020.05.006
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. doi: 10.1038/nmeth.1318
Gomez-Acata, E. S., Centeno, C. M., and Falcon, L. I. (2019). Methods for extracting ‘omes from microbialites. J. Microbiol. Methods 160, 1–10. doi: 10.1016/j.mimet.2019.02.014
Greunke, C., Duell, E. R., D’Agostino, P. M., Glöckle, A., Lamm, K., and Gulder, T. A. M. (2018). Direct Pathway Cloning (DiPaC) to unlock natural product biosynthetic potential. Metab. Eng. 47, 334–345. doi: 10.1016/j.ymben.2018.03.010
Grindley, N. D., Whiteson, K. L., and Rice, P. A. (2006). Mechanisms of site-specific recombination. Annu. Rev. Biochem. 75, 567–605. doi: 10.1146/annurev.biochem.73.011303.073908
Hashimoto, T., Hashimoto, J., Kozone, I., Amagai, K., Kawahara, T., Takahashi, S., et al. (2018). Biosynthesis of quinolidomicin, the largest known macrolide of terrestrial origin: identification and heterologous expression of a biosynthetic gene cluster over 200 kb. Org. Lett. 20, 7996–7999. doi: 10.1021/acs.orglett.8b03570
Hover, B. M., Kim, S. H., Katz, M., Charlop-Powers, Z., Owen, J. G., Ternei, M. A., et al. (2018). Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 3, 415–422. doi: 10.1038/s41564-018-0110-1
Hu, S., Liu, Z., Zhang, X., Zhang, G., Xie, Y., Ding, X., et al. (2016). “Cre/loxP plus BAC”: a strategy for direct cloning of large DNA fragment and its applications in Photorhabdus luminescens and Agrobacterium tumefaciens. Sci. Rep. 6:29087. doi: 10.1038/srep29087
Hu, Y., Nan, F., Maina, S. W., Guo, J., Wu, S., and Xin, Z. (2018). Clone of plipastatin biosynthetic gene cluster by transformation-associated recombination technique and high efficient expression in model organism Bacillus subtilis. J. Biotechnol. 288, 1–8. doi: 10.1016/j.jbiotec.2018.10.006
Huang, F., Tang, J., He, L., Ding, X., Huang, S., Zhang, Y., et al. (2018). Heterologous expression and antitumor activity analysis of syringolin from Pseudomonas syringae pv. syringae B728a. Microb. Cell Fact. 17:31. doi: 10.1186/s12934-018-0859-1
Ignatov, K. B., Blagodatskikh, K. A., Shcherbo, D. S., Kramarova, T. V., Monakhova, Y. A., and Kramarov, V. M. (2019). Fragmentation through polymerization (FTP): a new method to fragment DNA for next-generation sequencing. PLoS One 14:e0210374. doi: 10.1371/journal.pone.0210374
Ioannou, P. A., de, P. J., and Jong (1996). Current Protocols in Human Genetics. Hoboken, NJ: Wiley.
Jiang, W., Zhao, X., Gabrieli, T., Lou, C., Ebenstein, Y., and Zhu, T. F. (2015). Cas9-Assisted targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat. Commun. 6:8101. doi: 10.1038/ncomms9101
Jiang, W., and Zhu, T. F. (2016). Targeted isolation and cloning of 100-kb microbial genomic sequences by Cas9-assisted targeting of chromosome segments. Nat. Protoc. 11, 960–975. doi: 10.1038/nprot.2016.055
Jin, P., Ding, W., Du, G., Chen, J., and Kang, Z. (2016). DATEL: a scarless and sequence-independent DNA assembly method using thermostable exonucleases and ligase. ACS Chem. Biol. 5, 1028–1032. doi: 10.1021/acssynbio.6b00078
Jones, A. C., Gust, B., Kulik, A., Heide, L., Buttner, M. J., and Bibb, M. J. (2013). Phage P1-derived artificial chromosomes facilitate heterologous expression of the FK506 gene cluster. PLoS One 8:e69319. doi: 10.1371/journal.pone.0069319
Jordan, P. A., and Moore, B. S. (2016). Biosynthetic pathway connects cryptic ribosomally synthesized posttranslationally modified peptide genes with pyrroloquinoline alkaloids. Cell Chem. Biol. 23, 1504–1514. doi: 10.1016/j.chembiol.2016.10.009
Juhas, M., and Ajioka, J. W. (2017). High molecular weight DNA assembly in vivo for synthetic biology applications. Crit. Rev. Biotechnol. 37, 277–286. doi: 10.3109/07388551.2016.1141394
Kang, H. S., and Kim, E. S. (2021). Recent advances in heterologous expression of natural product biosynthetic gene clusters in Streptomyces hosts. Curr. Opin. Biotechnol. 69, 118–127. doi: 10.1016/j.copbio.2020.12.016
Kim, J. H., Feng, Z., Bauer, J. D., Kallifidas, D., Calle, P. Y., and Brady, S. F. (2010). Cloning large natural product gene clusters from the environment: piecing environmental DNA gene clusters back together with TAR. Biopolymers 93, 833–844. doi: 10.1002/bip.21450
Kim, U. J., Shizuya, H., de Jong, P. J., Birren, B., and Simon, M. I. (1992). Stable propagation of cosmid sized human DNA inserts in an F factor based vector. Nucleic Acids Res. 20, 1083–1085. doi: 10.1093/nar/20.5.1083
Kouprina, N., and Larionov, V. (2016). Transformation-associated recombination (TAR) cloning for genomics studies and synthetic biology. Chromosoma 125, 621–632. doi: 10.1007/s00412-016-0588-3
Kouprina, N., and Larionov, V. (2019). TAR cloning: perspectives for functional genomics, biomedicine, and biotechnology. Mol. Ther. Methods Clin. Dev. 14, 16–26. doi: 10.1016/j.omtm.2019.05.006
Kouprina, N., Noskov, V. N., and Larionov, V. (2006). Selective isolation of large chromosomal regions by transformation-associated recombination cloning for structural and functional analysis of mammalian genomes. Methods Mol. Biol. 349, 85–101. doi: 10.1385/1-59745-158-4
Kouprina, N., Noskov, V. N., and Larionov, V. (2020). Selective isolation of large segments from individual microbial genomes and environmental DNA samples using transformation-associated recombination cloning in yeast. Nat. Protocols 15, 734–749. doi: 10.1038/s41596-019-0280-1
Kunes, S., Botstein, D., and Fox, M. S. (1985). Transformation of yeast with linearized plasmid DNA: formation of inverted dimers and recombinant plasmid products. J. Mol. Biol. 184, 375–387. doi: 10.1016/0022-2836(85)90288-8
Larionov, V., Kouprina, N., Solomon, G., Barrett, J. C., and Resnick, M. A. (1997). Direct isolation of human brca2 gene by transformation-associated recombination in yeast. Proc. Natl. Acad. Sci. U S A. 94, 7384–7387. doi: 10.1073/pnas.94.14.7384
Larson, C. B., Crusemann, M., and Moore, B. S. (2017). PCR-independent method of transformation-associated recombination reveals the cosmomycin biosynthetic gene cluster in an ocean Streptomycete. J. Nat. Prod. 80, 1200–1204. doi: 10.1021/acs.jnatprod.6b01121
Lee, N. C. O., Larionov, V., and Kouprina, N. (2015). Highly efficient CRISPR/Cas9-mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Res. 43:e55. doi: 10.1093/nar/gkv112
Lei, C., Li, S. Y., Liu, J. K., Zheng, X., Zhao, G. P., and Wang, J. (2017). The CCTL (Cpf1-assisted cutting and Taq DNA ligase-assisted Ligation) method for efficient editing of large DNA constructs in vitro. Nucleic Acids Res. 45:e74. doi: 10.1093/nar/gkx018
Lesic, B., and Rahme, L. G. (2008). Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol. Biol. 9:20. doi: 10.1186/1471-2199-9-20
Lewis, W. H., Tahon, G., Geesink, P., Sousa, D. Z., and Ettema, T. J. G. (2021). Innovations to culturing the uncultured microbial majority. Nat. Rev. Microbiol. 19, 225–240. doi: 10.1038/s41579-020-00458-8
Li, L., Jiang, W. H., and Lu, Y. H. (2017). New strategies and approaches for engineering biosynthetic gene clusters of microbial natural products. Biotechnol. Adv. 35, 936–949. doi: 10.1016/j.biotechadv.2017.03.007
Li, L., Maclntyre, L. W., and Brady, S. F. (2021). Refactoring biosynthetic gene clusters for heterologous production of microbial natural products. Curr. Opin. Biotechnol. 69, 145–152. doi: 10.1016/j.copbio.2020.12.011
Li, L., Zhao, Y., Ruan, L., Yang, S., Ge, M., Jiang, W., et al. (2015). A stepwise increase in pristinamycin II biosynthesis by Streptomyces pristinaespiralis through combinatorial metabolic engineering. Metab. Eng. 29, 12–25. doi: 10.1016/j.ymben.2015.02.001
Li, M. Z., and Elledge, S. J. (2007). Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256. doi: 10.1038/nmeth1010
Li, M. Z., and Elledge, S. J. (2012). SLIC: a method for sequence- and ligation-independent cloning. Methods Mol. Biol. 852, 51–59. doi: 10.1007/978-1-61779-564-0_5
Li, Y., Li, Z., Yamanaka, K., Xu, Y., Zhang, W., Vlamakis, H., et al. (2015). Directed natural product biosynthesis gene cluster capture and expression in the model bacterium Bacillus subtilis. Sci. Rep. 5:9383. doi: 10.1038/srep09383
Liang, J., Liu, Z., Low, X. Z., Ang, E. L., and Zhao, H. (2017). Twin-primer non-enzymatic DNA assembly: an efficient and accurate multi-part DNA assembly method. Nucleic Acids Res. 45:e94. doi: 10.1093/nar/gkx132
Libis, V., Antonovsky, N., Zhang, M., Shang, Z., Montiel, D., Maniko, J., et al. (2019). Uncovering the biosynthetic potential of rare metagenomic DNA using co-occurrence network analysis of targeted sequences. Nat. Commun. 10:3848. doi: 10.1038/s41467-019-11658-z
Lin, Z., Nielsen, J., and Liu, Z. (2020). Bioprospecting through cloning of whole natural product biosynthetic gene clusters. Front. Bioeng. Biotechnol. 8:526. doi: 10.3389/fbioe.2020.00526
Liu, C. J., Jiang, H., Wu, L., Zhu, L. Y., Meng, E., and Zhang, D. Y. (2017). OEPR cloning: an efficient and seamless cloning strategy for large- and multi-fragments. Sci. Rep. 7:44648. doi: 10.1038/srep44648
Liu, H., Jiang, H., Haltli, B., Kulowski, K., Muszynska, E., Feng, X., et al. (2009). Rapid cloning and heterologous expression of the meridamycin biosynthetic gene cluster using a versatile Escherichia coli-Streptomyces artificial chromosome vector, pSBAC. J. Nat. Prod. 72, 389–395. doi: 10.1021/np8006149
Liu, Q., Shen, Q., Bian, X., Chen, H., Fu, J., Wang, H., et al. (2016). Simple and rapid direct cloning and heterologous expression of natural product biosynthetic gene cluster in Bacillus subtilis via Red/ET recombineering. Sci. Rep. 6:34623. doi: 10.1038/srep34623
Liu, T., Mazmouz, R., Ongley, S. E., Chau, R., Pickford, R., Woodhouse, J. N., et al. (2017). Directing the heterologous production of specific cyanobacterial toxin variants. ACS Chem. Biol. 12, 2021–2029. doi: 10.1021/acschembio.7b00181
Loannou, P. A., Amemiya, C. T., Garnes, J., Kroisel, P. M., Shizuya, H., Chen, C., et al. (1994). A new bacteriophage P1–derived vector for the propagation of large human DNA fragments. Nat. Genet. 6, 84–89. doi: 10.1038/ng0194-84
Ma, H., Kunes, S., Schatz, P. J., and Botstein, D. (1987). Plasmid construction by homologous recombination in yeast. Gene 58, 201–216. doi: 10.1016/0378-1119(87)90376-3
Marillonnet, S., and Grutzner, R. (2020). Synthetic DNA assembly using golden gate cloning and the hierarchical modular cloning pipeline. Curr. Protoc. Mol. Biol. 130:e115. doi: 10.1002/cpmb.115
Martinez, A., Kolvek, S. J., Yip, C. L. T., Hopke, J., Brown, K. A., et al. (2004). Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl. Environ. Microbiol. 70, 2452–2463. doi: 10.1128/AEM.70.4.2452-2463.2004
Martinez-Garcia, E., Goni-Moreno, A., Bartley, B., McLaughlin, J., Sanchez-Sampedro, L., Pascual Del, H., et al. (2020). SEVA 3.0: an update of the standard european vector architecture for enabling portability of genetic constructs among diverse bacterial hosts. Nucleic Acids Res. 48, D1164–D1170. doi: 10.1093/nar/gkz1024
Mayjonade, B., Gouzy, J., Donnadieu, C., Pouilly, N., Marande, W., Callot, C., et al. (2016). Extraction of high-molecular-weight genomic DNA for long-read sequencing of single molecules. Biotechniques 61, 203–205. doi: 10.2144/000114460
Meng, J., Feng, R., Zheng, G., Ge, M., Mast, Y., Wohlleben, W., et al. (2017). Improvement of pristinamycin I (PI) production in Streptomyces pristinaespiralis by metabolic engineering approaches. Synth. Syst. Biotechnol. 2, 130–136. doi: 10.1016/j.synbio.2017.06.001
Miao, V., Coeffet-LeGal, M. F., Brian, P., Brost, R., Penn, J., Whiting, A., et al. (2005). Daptomycin biosynthesis in Streptomyces roseosporus: cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology 151, 1507–1523. doi: 10.1099/mic.0.27757-0
Milon, N., Chantry-Darmon, C., Satge, C., Fustier, M. A., Cauet, S., Moreau, S., et al. (2019). muLAS technology for DNA isolation coupled to Cas9-assisted targeting for sequencing and assembly of a 30 kb region in plant genome. Nucleic Acids Res. 47, 8050–8060. doi: 10.1093/nar/gkz632
Mitchell, L. A., Chuang, J., Agmon, N., Khunsriraksakul, C., Phillips, N. A., Cai, Y., et al. (2015). Versatile genetic assembly system (VEGAS) to assemble pathways for expression in S. cerevisiae. Nucleic Acids Res. 43, 6620–6630. doi: 10.1093/nar/gkv466
Monaco, A. P., and Larin, Z. (1994). YACs, BACs, PACs and MACs: artificial chromosomes as research tools. Trends Biotechnol. 12, 280–286. doi: 10.1016/0167-7799(94)90140-6
Myronovskyi, M., and Luzhetskyy, A. (2013). Genome engineering in actinomycetes using site-specific recombinases. Appl. Microbiol. Biotechnol. 97, 4701–4712. doi: 10.1007/s00253-013-4866-1
Nah, H. J., Woo, M. W., Choi, S. S., and Kim, E. S. (2015). Precise cloning and tandem integration of large polyketide biosynthetic gene cluster using Streptomyces artificial chromosome system. Microb. Cell Fact. 14:140. doi: 10.1186/s12934-015-0325-2
Noskov, V., Kouprina, N., Leem, S. H., Koriabine, M., Barrett, J. C., and Larionov, V. (2002). A genetic system for direct selection of gene-positive clones during recombinational cloning in yeast. Nucleic Acids Res. 30:E8. doi: 10.1093/nar/30.2.e8
Noskov, V. N., Chuang, R. Y., Gibson, D. G., Leem, S. H., Larionov, V., and Kouprina, N. (2011). Isolation of circular yeast artificial chromosomes for synthetic biology and functional genomics studies. Nat. Protoc. 6, 89–96. doi: 10.1038/nprot.2010.174
Noskov, V. N., Kouprina, N., Leem, S. H., Ouspenski, I., Barrett, C., and Larionov, V. (2003). A general cloning system to selectively isolate any eukaryotic or prokaryotic genomic region in yeast. BMC Genomics 4:16. doi: 10.1186/1471-2164-4-16
Oliner, J. D., Kinzler, K. W., and Vogelstein, B. (1993). In vivo cloning of PCR products in E. coli. Nucleic Acids Res. 21, 5192–5197. doi: 10.1093/nar/21.22.5192
Park, D. (2007). Genomic DNA isolation from different biological materials. Methods Mol. Biol. 353, 3–13. doi: 10.1385/1-59745-229-7:3
Pedersen, T. B., Nielsen, M. R., Kristensen, S. B., Spedtsberg, E. M. L., Yasmine, W., Matthiesen, R., et al. (2020). Heterologous expression of the core genes in the complex fusarubin gene cluster of Fusarium Solani. Int. J. Mol. Sci. 21:7601. doi: 10.3390/ijms21207601
Penouilh-Suzette, C., Fourre, S., Besnard, G., Godiard, L., and Pecrix, Y. (2020). A simple method for high molecular-weight genomic DNA extraction suitable for long-read sequencing from spores of an obligate biotroph oomycete. J. Microbiol. Methods 178:106054. doi: 10.1016/j.mimet.2020.106054
Phelan, R. M., Sachs, D., Petkiewicz, S. J., Barajas, J. F., Blake-Hedges, J. M., Thompson, M. G., et al. (2017). Development of next generation synthetic biology tools for use in Streptomyces venezuelae. ACS Chem. Biol. 6, 159–166. doi: 10.1021/acssynbio.6b00202
Pyeon, H. R., Nah, H. J., Kang, S. H., Choi, S. S., and Kim, E. S. (2017). Heterologous expression of pikromycin biosynthetic gene cluster using Streptomyces artificial chromosome system. Microb. Cell Fact. 16:96. doi: 10.1186/s12934-017-0708-7
Qian, Z., Bruhn, T., D’Agostino, P. M., Herrmann, A., Haslbeck, M., Antal, N., et al. (2020). Discovery of the Streptoketides by direct cloning and rapid heterologous expression of a cryptic PKS II gene cluster from Streptomyces sp. Tü 6314. J. Org. Chem. 85, 664–673. doi: 10.1021/acs.joc.9b02741
Quan, J., and Tian, J. (2014). Circular p polymerase extension cloning. Methods Mol. Biol. 1116, 103–117. doi: 10.1007/978-1-62703-764-8_8
Ramsay, M. (1994). Yeast artificial chromosome cloning. Mol. Biotechnol. 1, 181–201. doi: 10.1007/BF02921558
Ray, L., Yamanaka, K., and Moore, B. S. (2016). A peptidyl-transesterifying type I thioesterase in salinamide biosynthesis. Angew. Chem. Int. Ed. Engl. 55, 364–367. doi: 10.1002/anie.201508576
Ren, H., Hu, P., and Zhao, H. (2017). A plug-and-play pathway refactoring workflow for natural product research in Escherichia coli and Saccharomyces cerevisiae. Biotechnol. Bioeng. 114, 1847–1854. doi: 10.1002/bit.26309
Richter, D., Bayer, K., Toesko, T., and Schuster, S. (2019). ZeBRalpha a universal, multi-fragment DNA-assembly-system with minimal hands-on time requirement. Sci. Rep. 9:2980. doi: 10.1038/s41598-019-39768-0
Ross, A. C., Gulland, L. E. S., Dorrestein, P. C., and Moore, B. S. (2015). Targeted capture and heterologous expression of the pseudoalteromonas alterochromide gene cluster in Escherichia coil represents a promising natural product exploratory platform. ACS Chem. Biol. 4, 414–420. doi: 10.1021/sb500280q
Salomonsen, B., Mortensen, U. H., and Halkier, B. A. (2014). USER-derived cloning methods and their primer design. Methods Mol. Biol. 1116, 59–72. doi: 10.1007/978-1-62703-764-8_5
Sapojnikova, N., Asatiani, N., Kartvelishvili, T., Asanishvili, L., Zinkevich, V., Bogdarina, I., et al. (2017). A comparison of DNA fragmentation methods - applications for the biochip technology. J. Biotechnol. 256, 1–5. doi: 10.1016/j.jbiotec.2017.06.1202
Schlichting, N., Reinhardt, F., Jager, S., Schmidt, M., and Kabisch, J. (2019). Optimization of the experimental parameters of the ligase cycling reaction. Synth. Biol. 4:ysz020. doi: 10.1093/synbio/ysz020
Schmid-Burgk, J. L., Schmidt, T., Kaiser, V., Honing, K., and Hornung, V. (2013). A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat. Biotechnol. 31, 76–81. doi: 10.1038/nbt.2460
Scott, H. N., Laible, P. D., and Hanson, D. K. (2003). Sequences of versatile broad-host-range vectors of the RK2 family. Plasmid 50, 74–79. doi: 10.1016/s0147-619x(03)00030-1
Shizuya, H., Birren, B., Kim, U. J., Mancino, V., Slepak, T., Tachiiri, Y., et al. (1992). Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. U S A. 89, 8794–8797. doi: 10.13/pnas.89.18.8794
Shizuya, H., and Kouros-Mehr, H. (2001). The development and applications of the bacterial artificial chromosome cloning system. Keio J. Med. 50, 26–30. doi: 10.2302/kjm.50.26
Song, C., Luan, J., Cui, Q., Duan, Q., Li, Z., Gao, Y., et al. (2019). Enhanced heterologous spinosad production from a 79-kb synthetic multioperon assembly. ACS Chem. Biol. 8, 137–147. doi: 10.1021/acssynbio.8b00402
Sosio, M., Giusino, F., Cappellano, C., Bossi, E., Puglia, A. M., and Donadio, S. (2000). Artificial chromosomes for antibiotic-producing actinomycetes. Nat. Biotechnol. 18, 343–345. doi: 10.1038/73810
Stinchcomb, D. T., Thomas, M., Kelly, J., Selker, E., and Davis, R. W. (1980). Eukaryotic DNA segments capable of autonomous replication in yeast. Proc. Natl. Acad. Sci. U S A. 77, 4559–4563. doi: 10.1073/pnas.77.8.4559
Sun, F., Xu, S., Jiang, F., and Liu, W. (2018). Genomic-driven discovery of an amidinohydrolase involved in the biosynthesis of mediomycin A. Appl. Microbiol. Biotechnol. 102, 2225–2234. doi: 10.1007/s00253-017-8729-z
Tang, X., Li, J., Millán-Aguiñaga, N., Zhang, J. J., O’Neill, E. C., Ugalde, J. A., et al. (2015). Identification of thiotetronic acid antibiotic biosynthetic pathways by target-directed genome mining. ACS Chem. Biol. 10, 2841–2849. doi: 10.1021/acschembio.5b00658
Tang, Y., Frewert, S., Harmrolfs, K., Herrmann, J., Karmann, L., Kazmaier, U., et al. (2015). Heterologous expression of an orphan NRPS gene cluster from Paenibacillus larvae in Escherichia coli revealed production of sevadicin. J. Biotechnol. 194, 112–114. doi: 10.1016/j.jbiotec.2014.12.008
Tillett, D., and Neilan, B. A. (1999). Enzyme-free cloning: a rapid method to clone PCR products independent of vector restriction enzyme sites. Nucleic Acids Res. 27:e26. doi: 10.1093/nar/27.19.e26
Tu, Q., Herrmann, J., Hu, S., Raju, R., Bian, X., Zhang, Y., et al. (2016). Genetic engineering and heterologous expression of the disorazol biosynthetic gene cluster via Red/ET recombineering. Sci. Rep. 6:21066. doi: 10.1038/srep21066
Varma, A., Padh, H., and Shrivastava, N. (2007). Plant genomic DNA isolation: an art or a science. Biotechnol. J. 2, 386–392. doi: 10.1002/biot.200600195
Wang, H., Li, Z., Jia, R., Hou, Y., Yin, J., Bian, X., et al. (2016). RecET direct cloning and red alpha beta recombineering of biosynthetic gene clusters, large operons or single genes for heterologous expression. Nat. Protoc. 11, 1175–1190. doi: 10.1038/nprot.2016.054
Wang, H., Li, Z., Jia, R., Yin, J., Li, A., Xia, L., et al. (2018). ExoCET: exonuclease in vitro assembly combined with RecET recombination for highly efficient direct DNA cloning from complex genomes. Nucleic Acids Res. 46:e28. doi: 10.1093/nar/gkx1249
Wang, L., Wang, H., Liu, H., Zhao, Q., Liu, B., Wang, L., et al. (2019). Improved CRISPR-Cas12a-assisted one-pot DNA editing method enables seamless DNA editing. Biotechnol. Bioeng. 116, 1463–1474. doi: 10.1002/bit.26938
Wenzel, S. C., Gross, F., Zhang, Y., Fu, J., Stewart, A. F., and Muller, R. (2005). Heterologous expression of a myxobacterial natural products assembly line in pseudomonads via red/ET recombineering. Chem. Biol. 12, 349–356. doi: 10.1016/j.chembiol.2004.12.012
Wu, C., Shang, Z., Lemetre, C., Ternei, M. A., and Brady, S. F. (2019). Cadasides, calcium-dependent acidic lipopeptides from the soil metagenome that are active against multidrug-resistant bacteria. J. Am. Chem. Soc. 141, 3910–3919. doi: 10.1021/jacs.8b12087
Wu, N., Huang, H., Min, T., and Hu, H. (2017). TAR cloning and integrated overexpression of 6-demethylchlortetracycline biosynthetic gene cluster in Streptomyces aureofaciens. Acta Biochim. Biophys. Sin. 49, 1129–1134. doi: 10.1093/abbs/gmx110
Xia, Y., Li, K., Li, J., Wang, T., Gu, L., and Xun, L. (2019). T5 exonuclease-dependent assembly offers a low-cost method for efficient cloning and site-directed mutagenesis. Nucleic Acids Res. 47:e15. doi: 10.1093/nar/gky1169
Xu, P., Modavi, C., Demaree, B., Twigg, F., Liang, B., Sun, C., et al. (2020). Microfluidic automated plasmid library enrichment for biosynthetic gene cluster discovery. Nucleic Acids Res. 48:e48. doi: 10.1093/nar/gkaa131
Xu, X., Zhou, H., Liu, Y., Liu, X., Fu, J., Li, A., et al. (2018). Heterologous expression guides identification of the biosynthetic gene cluster of chuangxinmycin, an indole alkaloid antibiotic. J. Nat. Prod. 81, 1060–1064. doi: 10.1021/acs.jnatprod.7b00835
Yamanaka, K., Reynolds, K. A., Kersten, R. D., Ryan, K. S., Gonzalez, D. J., Nizet, V., et al. (2014). Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. U S A. 111, 1957–1962. doi: 10.1073/pnas.1319584111
Yamanaka, K., Ryan, K. S., Gulder, T. A., Hughes, C. C., and Moore, B. S. (2012). Flavoenzyme-catalyzed atropo-selective N,C-bipyrrole homocoupling in marinopyrrole biosynthesis. J. Am. Chem. Soc. 134, 12434–12437. doi: 10.1021/ja305670f
Yin, J., Hoffmann, M., Bian, X., Tu, Q., Yan, F., Xia, L., et al. (2015). Direct cloning and heterologous expression of the salinomycin biosynthetic gene cluster from Streptomyces albus DSM41398 in Streptomyces coelicolor A3(2). Sci. Rep. 5:15081. doi: 10.1038/srep15081
Yuan, Y., Andersen, E., and Zhao, H. (2016). Flexible and versatile strategy for the construction of large biochemical pathways. ACS Chem. Biol. 5, 46–52. doi: 10.1021/acssynbio.5b00117
Zeng, F., Zang, J., Zhang, S., Hao, Z., Dong, J., and Lin, Y. (2017). AFEAP cloning: a precise and efficient method for large DNA sequence assembly. BMC Biotechnol. 17:81. doi: 10.1186/s12896-017-0394-x
Zhang, J. J., Tang, X., Zhang, M., Nguyen, D., and Moore, B. S. (2017). Broad-host-range expression reveals native and host regulatory elements that influence heterologous antibiotic production in Gram-negative bacteria. mBio 8:e01291-17. doi: 10.1128/mBio.01291-17
Zhang, L., Hashimoto, T., Qin, B., Hashimoto, J., Kozone, I., Kawahara, T., et al. (2017). Characterization of giant modular PKSs provides insight into genetic mechanism for structural diversification of aminopolyol polyketides. Angew Chem. Int. Ed. Engl. 56, 1740–1745. doi: 10.1002/anie.201611371
Zhang, M., Zhang, Y., Scheuring, C. F., Wu, C. C., Dong, J. J., and Zhang, H. B. (2012). Preparation of megabase-sized DNA from a variety of organisms using the nuclei method for advanced genomics research. Nat. Protoc. 7, 467–478. doi: 10.1038/nprot.2011.455
Zhang, Y., Buchholz, F., Muyrers, J. P., and Stewart, A. F. (1998). A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 20, 123–128. doi: 10.1038/2417
Zhang, Y., Werling, U., and Edelmann, W. (2012). SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 40:e55. doi: 10.1093/nar/gkr1288
Zheng, J., Li, Y., Guan, H., Zhang, J., and Tan, H. (2019). Enhancement of neomycin production by engineering the entire biosynthetic gene cluster and feeding key precursors in Streptomyces fradiae CGMCC 4.576. Appl. Microbiol. Biotechnol. 103, 2263–2275. doi: 10.1007/s00253-018-09597-8
Zheng, X., Cheng, Q., Yao, F., Wang, X., Kong, L., Cao, B., et al. (2017). Biosynthesis of the pyrrolidine protein synthesis inhibitor anisomycin involves novel gene ensemble and cryptic biosynthetic steps. Proc. Natl. Acad. Sci. U S A. 114, 4135–4140. doi: 10.1073/pnas.1701361114
Keywords: natural product, biosynthetic gene cluster (BGC), high capacity vector, DNA assembly, direct cloning
Citation: Wang W, Zheng G and Lu Y (2021) Recent Advances in Strategies for the Cloning of Natural Product Biosynthetic Gene Clusters. Front. Bioeng. Biotechnol. 9:692797. doi: 10.3389/fbioe.2021.692797
Received: 09 April 2021; Accepted: 18 June 2021;
Published: 13 July 2021.
Edited by:
Xu-Ming Mao, Zhejiang University, ChinaReviewed by:
Eung-Soo Kim, Inha University, South KoreaCopyright © 2021 Wang, Zheng and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yinhua Lu, eWhsdUBzaG51LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.