 Sandhya Mishra
Sandhya Mishra Ziqiu Lin1,2
Ziqiu Lin1,2 Pankaj Bhatt
Pankaj Bhatt Shaohua Chen
Shaohua Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Bioeng. Biotechnol. , 10 February 2021
Sec. Bioprocess Engineering
Volume 9 - 2021 | https://doi.org/10.3389/fbioe.2021.632059
This article is part of the Research Topic Advanced Bioremediation Technologies and Processes for the Treatment of Synthetic Organic Compounds (SOCs) View all 14 articles
Global environmental contamination with a complex mixture of xenobiotics has become a major environmental issue worldwide. Many xenobiotic compounds severely impact the environment due to their high toxicity, prolonged persistence, and limited biodegradability. Microbial-assisted degradation of xenobiotic compounds is considered to be the most effective and beneficial approach. Microorganisms have remarkable catabolic potential, with genes, enzymes, and degradation pathways implicated in the process of biodegradation. A number of microbes, including Alcaligenes, Cellulosimicrobium, Microbacterium, Micrococcus, Methanospirillum, Aeromonas, Sphingobium, Flavobacterium, Rhodococcus, Aspergillus, Penecillium, Trichoderma, Streptomyces, Rhodotorula, Candida, and Aureobasidium, have been isolated and characterized, and have shown exceptional biodegradation potential for a variety of xenobiotic contaminants from soil/water environments. Microorganisms potentially utilize xenobiotic contaminants as carbon or nitrogen sources to sustain their growth and metabolic activities. Diverse microbial populations survive in harsh contaminated environments, exhibiting a significant biodegradation potential to degrade and transform pollutants. However, the study of such microbial populations requires a more advanced and multifaceted approach. Currently, multiple advanced approaches, including metagenomics, proteomics, transcriptomics, and metabolomics, are successfully employed for the characterization of pollutant-degrading microorganisms, their metabolic machinery, novel proteins, and catabolic genes involved in the degradation process. These technologies are highly sophisticated, and efficient for obtaining information about the genetic diversity and community structures of microorganisms. Advanced molecular technologies used for the characterization of complex microbial communities give an in-depth understanding of their structural and functional aspects, and help to resolve issues related to the biodegradation potential of microorganisms. This review article discusses the biodegradation potential of microorganisms and provides insights into recent advances and omics approaches employed for the specific characterization of xenobiotic-degrading microorganisms from contaminated environments.
The environment is everything that naturally surrounds us and affects our daily lives on Earth. A safe and healthy environment is essential for the existence of life on this planet. However, in the era of advanced industrialization and urbanization, various anthropogenic activities are largely responsible for the introduction of toxic and hazardous pollutants such as environmental xenobiotics (Embrandiri et al., 2016; Malla et al., 2018; Bhatt et al., 2020a; Rodriguez et al., 2020). Xenobiotics are chemical substances not naturally produced or expected to be present within organisms. The term “xenobiotic” is usually used in the context of environmental pollutants to refer to synthetic compounds produced in large volumes for industrial, agricultural, and domestic use (Embrandiri et al., 2016; Atashgahi et al., 2018; Dinka, 2018). There is growing public concern over the wide range of xenobiotic compounds being introduced, deliberately or accidentally, into the environment, which involves high potential risk to humans and animals (Jacob and Cherian, 2013; Hashmi et al., 2017; Zhu et al., 2017; Dinka, 2018). Environmental xenobiotics include pesticides, polycyclic aromatic hydrocarbons (PAHs), pharmaceutical active compounds (PhACs), personal-care products (PCPs), phenolics, chlorinated compounds, and other industrial chemicals. Their increasing frequency in different environmental compartments has raised concerns about their potential adverse effects (Crinnion, 2010; Kim et al., 2013; Embrandiri et al., 2016; Tsaboula et al., 2016; Dhakal et al., 2017). Their toxicity results in unprecedented health hazards and risks to environmental safety and security (Godheja et al., 2016; Dovrak et al., 2017; Burgos-Aceves et al., 2018; Ravindra and Haq, 2019). Once xenobiotics are released into the environment, they can bioaccumulate within the food chain due to their high affinity toward organic substances, and produce toxic adverse effects toward natural ecosystems, humans, and animals (Pedersen et al., 2003; Iovdijova and Bencko, 2010; Maurya, 2016). Consequently, they can cause severe chronic effects such as respiratory tract infections, damage to the immune system, pulmonary bronchitis, dysfunction of the nervous system, disruption of the endocrine system, behavioral and developmental disorders, and carcinogenic and mutagenic effects (Sajid et al., 2015; Zhu et al., 2017; Dinka, 2018; Catron et al., 2019; Mishra et al., 2019; Bertotto et al., 2020). Thus, xenobiotic contamination represents a persistent anthropogenic threat and raises serious environmental concerns. Various physical and chemical treatment methods such as coagulation, filtration, adsorption, chemical precipitation, electrolysis ozonation, etc. have been used for the degradation and detoxification of such xenobiotic compounds, but not all these methods are very useful due to their high cost, waste-disposal problem and generation of toxic by-products that are sometimes more hazardous than the parent compound. In contrast, the biological remediation method, “bioremediation,” is a widely accepted clean-up strategy for the degradation of xenobiotics from contaminated environments without producing harmful products (Paul et al., 2005; Perelo, 2010). Bioremediation involves the metabolic capabilities of microorganisms in the removal of pollutants and thus, is the most suitable and promising technology these days (Gilliespie and Philp, 2013; Azubuike et al., 2016).
Microbial remediation of xenobiotic compounds is regarded as a superficial, proficient, economically feasible approach that uses a wide range of microorganisms to consume organic pollutants as carbon or nitrogen supplements to sustain their developmental activities (Chen et al., 2013; Mahmoud, 2016; Arora et al., 2018; Ortiz-Hernandez et al., 2018; Siles and Margesin, 2018; Zhan et al., 2018; Bhatt et al., 2020b). Microorganisms are ubiquitous in nature, and diverse microbial communities thrive in natural and extreme stress environments, including soil, water, the human gut, hydrothermal vents, acid mine runoff, and oil reservoirs (Cycoń and Piotrowska-Seget, 2016; Jalowiecki et al., 2016; Ding et al., 2017; Aguinga et al., 2018; Wang Y. F. et al., 2018; Zierer et al., 2018; Delegan et al., 2019; Arora, 2020; Shekhar et al., 2020). Microbial populations exhibit potential for the remediation of any contaminated environment because of their genetic diversity and functionality (Chen et al., 2015; Bastida et al., 2016; Bhatt and Barh, 2018; Dangi et al., 2018). Therefore, the study of microbial population existing in contaminated environments provides a significant knowledge of specific microbial characteristics that improve degradation rates. However, effective implementation of microbial remediation strategies needs advanced technical approaches, which provide an in-depth understanding about the dynamics aspects of microbial activity and survival under stressed environment (Rastogi and Sani, 2011; Lima-Morales et al., 2016; Mao et al., 2019). The development in molecular, biotechnological, bioinformatics and system biology tools pertaining to bio-remedial problems have provided gene level mechanisms of bioremediation (Ahmad and Ahmad, 2014; Aora and Bar, 2014; Singh D. P. et al., 2018; Jaiswal and Shukla, 2020; Nkongolo and Kotha, 2020; Wolf et al., 2020). Moreover, the direct study of microorganisms in a contaminated environment including the whole microbial population granted a new frontier of the scientific community to share the knowledge of the uncultured microbial world (Zepeda et al., 2015; Zhao Q. et al., 2017; Panigrahi et al., 2019; Yan et al., 2020). The development of advance molecular tools and a better understanding of microbial metabolic and genetic structures and functions have accelerated encroachment in recombinant engineering techniques to enhanced bioremediation for removal of environmental pollutants (Ram et al., 2005; Temperton and Giovannoni, 2012; Singh V. et al., 2018; Stein et al., 2018; Delegan et al., 2019; Marco and Abram, 2019; Puckett et al., 2020). Soil is the most dynamic environment for the enormous microbial population of immense diversity. It has been estimated that one gram of soil approximately contains 109 bacterial cells, but only <1% of these may be culturable in the laboratory (Rossello-Mora and Amann, 2001). Culture based identification of diverse microbial population in a contaminated environment, is a challenging task, which is limited to fast-growing microbial diversity (Gillbride et al., 2006). Thus, modern culture-independent molecular techniques represent a feasible approach to unrevealing the diversity and functional dynamics of microbial population in contaminated environments. Moreover, the advanced innovation in molecular tools and techniques provides new insights and changes the traditional research trend in the field of bioremediation (Malik et al., 2008; Shah et al., 2011; Devarapalli and Kumavnath, 2015; Mahmoud, 2016; Biswas and Sarkar, 2018; Shakya et al., 2019). Omics technologies are the result of advanced molecular techniques, which involved direct characterization of genome structure of microorganisms, devoid of their culture sample (Segata et al., 2013; Biswas and Sarkar, 2018; Jaiswal et al., 2019; Yu K. et al., 2019). Therefore, the applications of modern molecular techniques like metagenomic, transcriptomic, proteomic generates relevant information on genes and proteins expression levels in whole microbial communities under contaminated environments attempted to unravel the mechanism of microbial degradation and successful execution of bioremediation (Keller and Hettich, 2009; Yang, 2013; Malla et al., 2018; Bharagava et al., 2019; Marco and Abram, 2019; Rodriguez et al., 2020). These methods are comparatively efficient, quicker, and accurate, which overcome the limitations of conventional molecular techniques. It explores the advanced microbial degradation mechanism of xenobiotics, their metabolic activities, genetic regulation and molecular-biology aspects (Cycoń et al., 2017; Gutierrez et al., 2018; Gutleben et al., 2018; Mishra et al., 2020). Hence, this review article highlights the biodegradation potential of microorganisms and provides insights into recent advance methods of “omics” technologies employed in microbial degradation and remediation purpose of xenobiotics and their perspectives in modern biological research.
The application of microorganisms in removing xenobiotics from soil, water or sediments through complete transformation or mineralization into harmless end products like CO2 and H2O is a basic concept of bioremediation strategy (Ortiz et al., 2013; Singh et al., 2016). Different microorganisms including bacteria (Pseudomonas, Alcaligenes, Cellulosimicrobium, Microbacterium, Micrococcus, Methanospirillum, Aeromonas, Bacillus, Sphingobium, Flavobacterium, and Rhodococcus), fungi (Aspergillus, Penecillium, Trichoderma, and Fusarium), and yeasts (Pichia, Rhodotorula, Candida, Aureobasidium, and Exophiala) have been reported to be involved in the efficient biodegradation of xenobiotic compounds from contaminated soil/water environments, due to their exceptional bioremediation potential (Sathishkumar et al., 2008; Nzila, 2013; Sunita et al., 2013; Zhao Q. et al., 2017; Bharadwaj, 2018; Yang J. et al., 2018; Yang T. et al., 2018; Yu Y. et al., 2019; Bhatt et al., 2020c). The biodegradation ability of microbes is greatly influenced by interactive ecological factors including soil, salinity, temperature, carbon source, moisture content, pH, nitrogen sources, inoculums concentration, etc. (Megharaj and Naidu, 2010; Wu et al., 2014; Bhatt et al., 2019). Microorganisms harbor remarkable catabolic potential, genes, enzymes, and degradation pathways implicated in the process of bioremediation, which might be responsible in the evolution of novel traits and characters (Widada et al., 2002; Scholer et al., 2017; Yan et al., 2018; Zhu et al., 2020). Moreover, microbial plasmids believed to be responsible for the continuous progression, evolution, and distribution of novel biodegradable genes/enzymes (Zhang et al., 2016; Jeffries et al., 2019). These novel genes/enzymes have endowed microorganism's biodegradation capability to remove or detoxify a wide variety of environmental pollutants due to their inheritance horizontal gene transfer property (Singh V. et al., 2018; Jaiswal et al., 2019; Li et al., 2019; Phale et al., 2019; French et al., 2020). The microbial remediation process can be further improved via successful application of genome editing and biochemical techniques that modify existing strains and result in the development of a genetically modified organism capable of simultaneously degrading several xenobiotics (Shanker et al., 2011; Zhang et al., 2016; Hussain et al., 2018; Janssen and Stucki, 2020). The advancement of genetic manipulation technology gives more clear information and explores future prospects of bioremediation of xenobiotics through highly proficient microorganisms (Sayler and Ripp, 2000; Shapiro et al., 2018; Wong, 2018; Liu et al., 2019). The chemical structures of several xenobiotic compounds are presented in Figure 1.
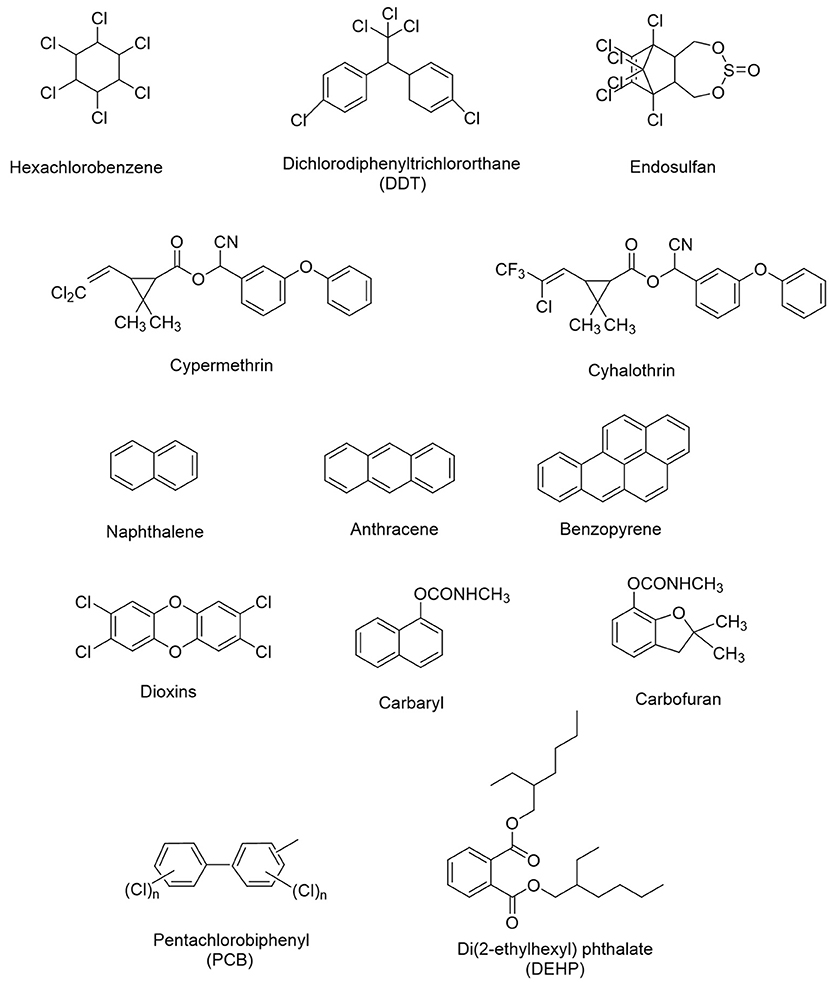
Figure 1. Chemical structures of several xenobiotic compounds.
Synthetic pesticides are the major example of xenobiotics especially the organochlorine pesticides (OCPs) that used extensively worldwide for a long period of time in agriculture as well as in insect control program. Several OCPs such as aldrin, dieldrin, dichloro diphenyl trichloro ethane (DDT), benzene hexachloride (BHC), and hexachlorocyclohexane are highly toxic in nature due to their stability and bioaccumulative property (Aktar et al., 2009; Jayaraj et al., 2016; Awasthi and Awasthi, 2019; Pang et al., 2020). Lindane (γ-hexachlorocyclohexane) is one of the highly toxic organochlorine xenobiotic compound, which is well-studied for its microbial biodegradation through physical (soil structure, carbon and oxygen gradients, pH, and temperature) and chemical (dechlorination, dehydroxylation, and dehydrogenation) interactions (Kaur and Kaur, 2016; Bashir et al., 2018; Zhang et al., 2021). The increasing concentration of lindane residues into the environment imposes severe health hazards such as carcinogenicity, mutagenicity, endocrine disruption, and immune-suppression diseases into the humans and other organisms (Cuozzo et al., 2017; Zhang et al., 2020). Bacterial strains genera such as Bacillus, Burkholderia, Pseudomonas, Kocuria, Archromobacter, Sphingomonas, Chromohalobacter, demonstrated lindane biodegradation under axenic as well as anoxic conditions via dehydrogenation, dehydrochlorination, and hydroxylation, results in complete degradation or mineralization (Giri et al., 2014; Cuozzo et al., 2017; Wang W. et al., 2018; Nagata et al., 2019; Zhang et al., 2020). A simplified catabolic pathway of lindane is presented in Figure 2.
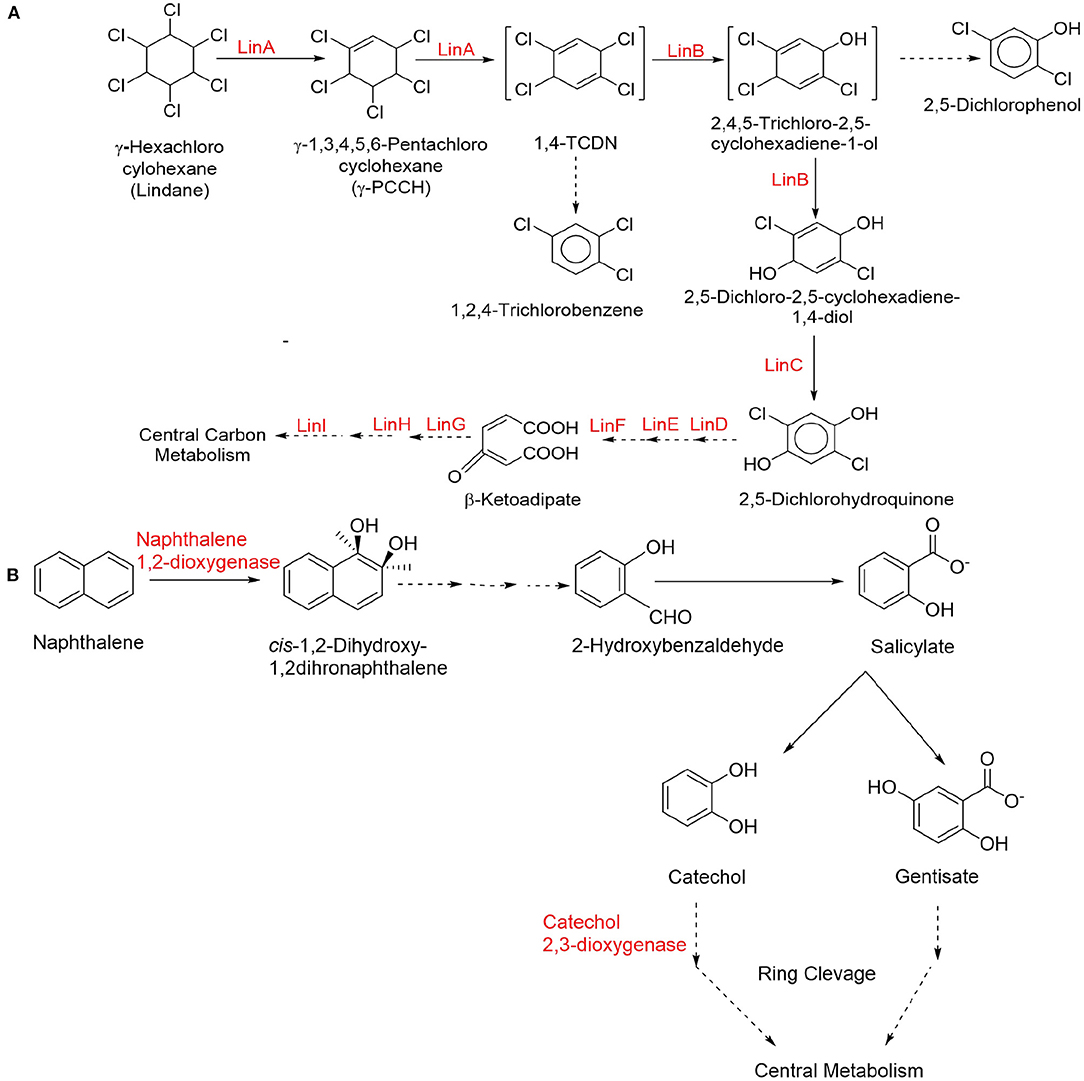
Figure 2. Simplified catabolic degradation pathways of organochlorine and polycyclic aromatic hydrocarbon (PAH). Compounds (A) lindane (organochlorine) (Endo et al., 2005; Geueke et al., 2013) and (B) naphthalene (PAH) (Kiyohara et al., 1994; Dutta et al., 1998; Bamforth and Singleton, 2005; Baboshin et al., 2008).
Pyrethroids are broad spectrum pesticide mainly used against agricultural and household pests. Cypermethrin, cyhalothrin, deltamethrin, cyfluthrin, bifenthrin are the common example of synthetic pyrethroids (Chen et al., 2012, 2014; Bhatt et al., 2019; Zhan et al., 2020). These pesticides are highly toxic and persistent and can cause molecular toxicity, neurotoxicity, and reproductive toxicity (Sharma et al., 2018; Bhatt et al., 2019; Gammon et al., 2019). One of the pyrethroid i.e., cypermethrin can cross the blood-brain barrier and induces neurotoxicity and motor deficits (Singh et al., 2012). The persistence of these pesticides in the environment poses a severe threat to humans and other non-target terrestrial and aquatic organisms (Burns and Pastoor, 2018; Ullah et al., 2018; Lu et al., 2019). Microbial strains such as Acinetobacter, Trichoderma, Roultella, Pseudomonas, Cunninghamella, and Bacillus have been reported for their efficient degradation of broad spectrum pesticides like cypermethrin and other pyrethroid pesticides through pyrethroid hydrolases (Cycoń and Piotrowska-Seget, 2016; Zhan et al., 2018; Bhatt et al., 2019; Chen and Zhan, 2019). Co-metabolism exhibiting strains include Flavobacterium, Sphingomonas, Arthrobacter, Azotobacter, Achromobacter, Microbacterium, Brevibacterium, Rhodococcus, Trichoderma, and Aspergillus demonstrated their pollutant degradation capability, exclusive of pollutant consumption as an energy resource (Nzila, 2013).
Polychlorinated bis-phenyles (PCBs) are classified as persistent organic pollutants with high toxicity (Lallas, 2001; Pathiraja et al., 2019). They have been linked to chronic effects in humans including immune system damage, decreased pulmonary function, bronchitis, and interference with hormones leading to cancer (Schecter et al., 2006). Microbial degradation of polychlorinated bis-phenyles (PCBs) is a promising remediation technology involving two major pathways: aerobic degradation and anaerobic dehalogination (Abraham et al., 2002; Pathiraja et al., 2019). Bacterial strains Pseudomonas, Rhosocossus, Comamonas, Burkholderia, and Bacillus have been characterized for the oxidative degradation of PCBs through ring cleavage resulting into a common by product of chlorbenzoic acid (Anyasi and Atagana, 2011; Jing et al., 2018).
Polycyclic aromatic hydrocarbons (PAHs) are potent environmental contaminants and xenobiotics that are widely distributed in the environment due to the incomplete combustion of organic matter. PAHs have moderate to high acute toxicity to aquatic life and birds (Abdel-Shafy and Mansour, 2016; Pandey et al., 2017). Some PAHs such as Anthracene, benzo(a)pyrene, phenanthrene, and naphthalene are well-known to produce harmful biological effects such as genotoxicity, mutagenicity, and carcinogenicity and therefore pose a serious threat to the human health (Kim et al., 2013; Lin et al., 2020). Microorganisms belonging to Sphingomonas, Sphingobium, and Novosphingobium have been found as efficient degrader of PAHs (Lee et al., 2016b; Fida et al., 2017; Auti et al., 2019). Rhodococcus, Cunninghamella, Pleurotus ostreatus, Oscillatoria, Agmenellum quadriplicatum, Brevibacterium, and Nocardiodes have been widely shown to metabolize particularly naphthalene and phenanthrene (Ghosal et al., 2016; Siles and Margesin, 2018). Ortega-Gonzalez et al. (2015) demonstrated that Amycolaptosis sp. Poz14 degraded 100% of naphthalene and 37.87% of anthracene within 45 days. A PAH-degrading marine bacterium Cycloclasticus sp. has been isolated from sea sediments capable to breakdown xenobiotic naphthalene, pyrene, phenanthrene, and other aromatic hydrocarbons into their supplementary products through enzymatic pathways (Wang W. et al., 2018). A simplified catabolic pathway of naphthalene is presented in Figure 2.
Phthalates or esters of phthalic acids are synthetic xenobiotic chemicals, which are extensively used as plasticizers added to polyvinyl chloride to improve its flexibility and hardness (Crinnion, 2010; Singh and Li, 2011; Przybylinska and Wyszkowski, 2016). Phthalates are readily released into the environment and create a risk of exposure for humans and other living organisms due their endocrine disrupting behavior (Przybylinska and Wyszkowski, 2016). They cause infertility, reproductive and developmental toxicity in humans and animals (Singh and Li, 2011; Przybylinska and Wyszkowski, 2016). Aerobic and anaerobic microbial degradation of xenobiotic phthalic acid, and isophthalaic acid considered as the most effective means of their removal from the environment (Junghare et al., 2019; Boll et al., 2020). Di-2(ethyehxyl) phthalate (DEHP) is the most common member of phthalates, which are extensively used as plasticizer in plastics and disposable medical materials (Singh and Li, 2011). DEHP is best known as an endocrine disrupter and can produce neural, heptotoxic, cardiotoxic, and carcinogenic effects on humans and animals (Rowdhwal and Chen, 2018). Arthrobacter, Pseudomonas, Gordonia, Providencia, Acinetobacter, Microbacterium, and Rhodococuus have been identified as efficient DEHP degrading bacteria (Nahurira et al., 2017; Yang T. et al., 2018).
In addition, the microbial consortium gained greater attention in bioremediation rather than pure microbial monocultures. The consortia cultures are better equipped in terms of metabolic and pollutant removal capability owing to their constant revelation of contaminant and promising mutual relationship with other available strains (Patowary et al., 2016). Interestingly, microbial consortia can alleviate the metabolic limitations of single microbial culture and enhance biodegradation process by their miscellaneous assemblage of bacterial populations employed with extensive degradation potential (Zafra et al., 2016; Li et al., 2020).
There are several revolutionary advanced molecular practices, including genomics, metagenomics, proteomics, transcriptomics, and metabolomics, which deliver deeper insights into microbial activities with respect to their genes, proteins, mRNA expression levels, enzymes and metabolic pathways with changing environments (Figure 1). The integrated approach of these multiple technologies in the field of bioremediation is termed as the “omics approach,” used for the undeviating characterization of biological macromolecules, and their specific genetic and molecular structures and function mechanisms in a set of microorganisms/microbial communities (Desai et al., 2009; Yang, 2013; Godheja et al., 2014; Franzosa et al., 2015; Chandran et al., 2020). The application of omics technologies provides comprehensive insights into microbial populations, their mechanisms of interaction with pollutants, metabolic activities, and genetic-regulation and molecular-biology aspects (Akinsanya et al., 2015; Kaul et al., 2016; Misra et al., 2018; Marco and Abram, 2019; Huang et al., 2021). Moreover, these approaches can broaden our knowledge of the so-called “viable but non-culturable (VBNC)” bacteria and their potentially novel pathways for degrading environmental pollutants (Oliver, 2010; Bodor et al., 2020). It is believed that these uncultured bacteria may play an important role in the biodegradation of environmental pollutant. However, little is known about the VBNC bacteria as these bacteria cannot be cultivated on conventional media and are very different from cells (Su et al., 2013). VBNC cells exhibit metabolic and respiratory activities and may perform transcription and gene expression, which allows them to recover culturability (Oliver et al., 2005; Zhao X. et al., 2017). The VBNC bacteria could be resuscitated in favorable conditions by an autoinducer (AI-2) (Bari et al., 2013) or resuscitated promoting factor (Rpf) (Li et al., 2015). The VBNC bacterial cells are detected on the basis of their viability. Several culture-independent molecular-based methods such as denaturing and temperature gradient gel electrophoresis (DGGE/TGGE), fluorescent in situ hybridization (FISH), terminal restriction fragment length polymorphism (T-RFLP), fatty acid methyl ester (FAME), and next-generation sequencing (NGS) technology are used for obtaining important information about the structural composition and genetic diversity of unculturable microorganisms (Zhao X. et al., 2017; Bodor et al., 2020). Selective gene amplification is an emerging approach to detect viable cells. Zhong et al. (2016) developed a real-time fluorescence LAMP technique combined with PMA (propidium monoazide), a high-affinity photolysis DNA nucleic acid dye applied for the detection of VBNC V. parahaemolyticus. Thus, the combined use of these advanced molecular technologies with bioinformatic approaches increases understanding and brings in a new era of unrevealed soil microbial communities, as well as their associated mechanisms of biodegradation for their future applications in bioremediation (Figure 3; Mocalli and Benedetti, 2010; Kumar et al., 2016; Dangi et al., 2018; Pandey et al., 2019; Pinu et al., 2019).
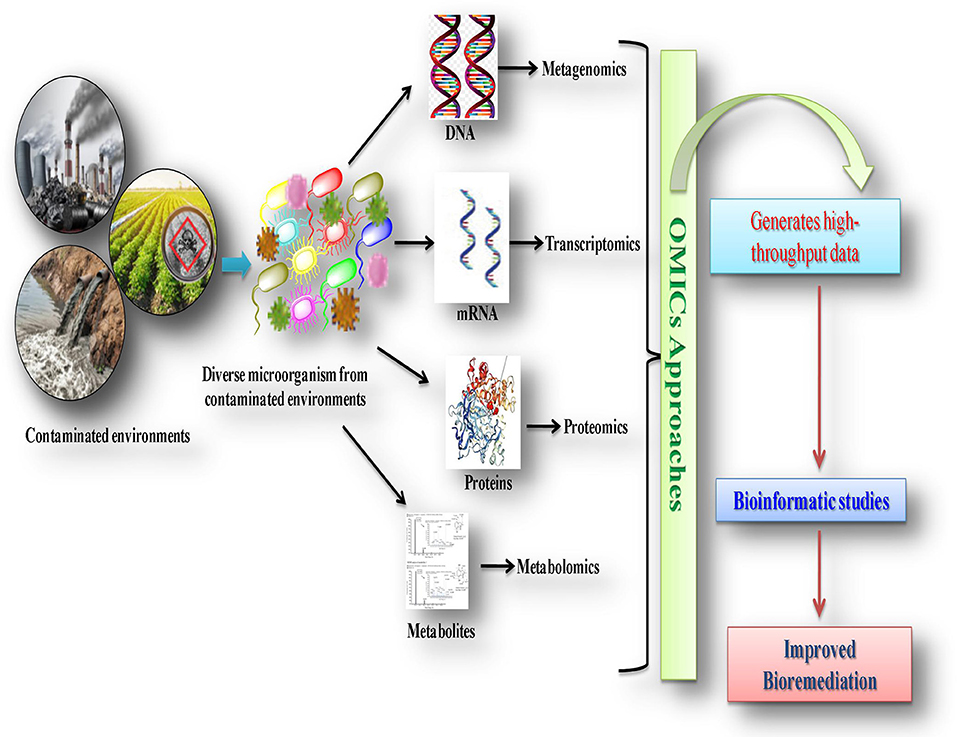
Figure 3. Graphical presentation of integrated approach of advanced technologies in biodegradation of xenobiotic compounds.
Genomics and metagenomics are powerful tools for analyzing microbial communities at the genomic level from various contaminated environments. This technology gives a new array to environmental microbiologists for understanding unculturable microbiota with a genetic variability of microbial communities (Devarapalli and Kumavnath, 2015; Zhu et al., 2018; Awasthi et al., 2020). It gives more details about the particular degradation potential of microbial communities, as it directly entails the whole-genome sequence from environmental samples (Table 1). Metagenomic studies unblock traditional ways of uncultured microorganisms and explore their genetic advantage in the process of bioremediation (Rahimi et al., 2018; Nascimento et al., 2020). Complete genome-sequence data of some important microbial strains, including Pseudomonas aeruginosa KT2440, Shewanella oneidensis MR-1, Deinococcus indicus R1, and Dehalococcoides mccartyi WBC-2 have already been given, which is pertinent to successful bioremediation (http://www.tigr.org). The new genes can tell too much about the degradation capability and substrate specificity.
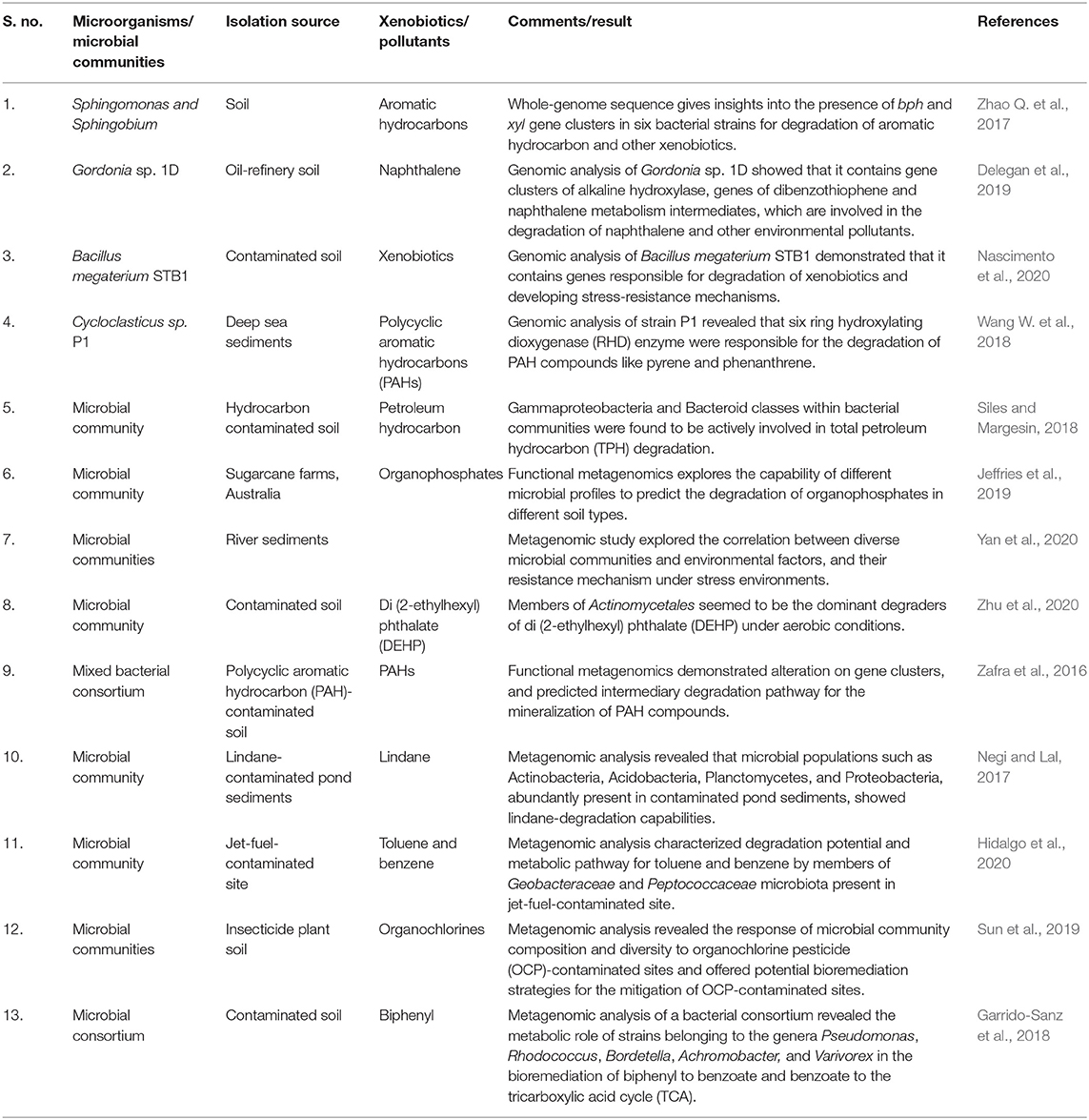
Table 1. Microorganisms and microbial communities using genomic and metagenomic approaches in biodegradation.
Current metagenomic practices allowed for identifying the whole-genome structure of microorganisms and specifying particular genes that are attributed to encode degradative enzymes for the mineralization of xenobiotics (Zafra et al., 2016; Zhu et al., 2020). Thus, metagenomics clearly highlights the crucial role of novel genes in connecting the entire microbial population with functional diversity and structural identity. Metagenomics involves the manufacturing of metagenomic libraries that include (I) production of the proper size of DNA fragments, and ligation of these fragments into a suitable cloning vector; (II) further recombinant vectors introduced into an appropriate bacterium cloning host; (III) clones that harbor specific characters, functions, or sequences were screened for libraries (Figure 3). Moreover, the screening of metagenomic libraries can be performed by two processes, i.e., sequence-driven analysis using high-throughput sequencing, and functional analysis using phenotypic expressions (Handelsman, 2004). However, recent sequence-based metagenome analyses (such as SOLiD system of Applied Biosystems, Roche 454 sequencing) are performed without the construction of cloned libraries (Kumar et al., 2020).
Function-driven metagenomics is a potent method for studying the functional aspect of genes. It is widely used for discovering novel genes with desired functions or exploring the sequence diversity of protein families (Taupp et al., 2011; Lam et al., 2015). A function-driven analysis involves the construction and screening of metagenomic libraries to identify novel enzymes (Chakraborthy and Das, 2016; Kumar et al., 2020). Using functional metagenomics, many novel antibiotic-resistant genes were identified from environmental sources (Ngara and Zhang, 2018). The majority of metagenome-derived hydrolytic enzymes, mainly esterases and glycoside hydrolases, have been characterized biochemically and mainly originated from functional metagenomics (Steele et al., 2009; Taupp et al., 2011). A novel functional screening method, a metagenome extract thin layer chromatography (META) system, was developed by Rabausch et al. (2013) for the rapid detection of glycosyltransferase (GT) and other flavonoid-modifying enzymes from metagenomic clone libraries. It involves the screening of 38,000 clones from two different metagenomic libraries and allowed for the identification of two novel UDP glycosyltransferase (UGT) genes. Bouhajja et al. (2017) utilized function-based screening of metagenomic libraries to explore the diversity of genes and microorganisms involved in the monooxygenase-mediated toluene degradation in a hydrocarbon-polluted sediment sample.
Metagenomic approaches offer broad and reliable microbial identification on the species and strain levels, but they are much costlier methods, and the most challenging part is data investigation, which requires all short DNA sequences to pair together to assemble the final genome structure (Bragg and Tyson, 2014). The involvement of indigenous soil microorganisms in the degradation of PAHs was undertaken by Zafra et al. (2016) through a metagenomic approach. This study demonstrated the biodegradation efficiency of microbial consortia with their degradative enzymes and metabolites generated during the remediation process. The microbial-community dynamics of refined- and crude-petroleum-contaminated soil by next-generation sequencing (NGS), and their capability to degrade hydrocarbons and plant-growth-promotion potential through in-silico analysis was investigated by Auti et al. (2019). In this study, 16S rRNA amplicon sequencing on the Illumina MiSeq platform and PICRUSt revealed that both types of soil contained microbial communities with excellent metabolic potential for petroleum hydrocarbon (PHC) degradation. Using KEGG orthology, the abundance of functional genes involved in hydrocarbon degradation showed the presence of 61 enzyme-encoding genes, such as alkane monooxygenase, alcohol dehydrogenase, and aldehyde dehydrogenase (Auti et al., 2019). 16S rDNA or 16S rRNA gene sequencing has led to evolutionary insights into the phylogenetic and taxonomic identification of microorganisms. The 16S rRNA gene consists of several highly conserved regions interleaved with variable regions in all microorganisms and thus is highly suited as a target gene for sequencing DNA (Fuks et al., 2018; Gursoy and Can, 2019). The bacterial 16S rRNA gene generally contains nine “hypervaiable regions” that demonstrate the considerable sequence diversity of bacterial species and can be used for species identification. The 16 S rRNA gene sequence similarity between two strains provides a simple yet robust criterion for the identification of newly isolated strains, whereas phylogenetic analyses can be used to elucidate the overall evolutionary relationship between related taxa (Johnson et al., 2019). Thus, 16S rRNA gene sequencing analysis is a highly recommended cost-effective technique for the phylogenetic resolution and taxonomic profiling of microbial communities (Auti et al., 2019).
The NGS approach has completely changed microbial-community analysis, as it provides comparative details in terms of temporal and spatial data (Hidalgo et al., 2020). There are several NGS technologies, including Illumina, Ion Torrent, SOLiD, and 454 (Caporaso et al., 2012; Liu et al., 2012; Knief, 2014; Salipante et al., 2014; Machado et al., 2019). These are high-throughput sequencing techniques of ribosomal genes that quantify community structures and functions at a higher resolution, e.g., 16S rRNA in prokaryotes, and 5S or 18S rRNA genes, or the internal-transcribe-spacer (ITS) region in eukaryotes (Luo et al., 2012). The effectiveness of such NGS technologies in analyzing microbial communities from diverse environments was elucidated, validated, and documented in many studies (Brown et al., 2013; Shokralla et al., 2014; Zhou et al., 2015; Niu et al., 2016; Scholer et al., 2017). In addition, PacBio (Pacific Biosciences) and Oxford Nanopore are highly advanced, reliable, and accurate third-generation sequencing platforms applied to microbial community analysis (Lu et al., 2016; Chandran et al., 2020). Oxford Nanopore Technologies has launched a portable MinION USB nanopore that does not rely on DNA replication and has the advantage of reading full-length molecules in real time. PacBio RS II, the first commercialized genomic sequencer, developed by Pacific Biosciences, uses single-molecule, real-time (SMRT) technology and is able to sequence single DNA molecules in real time without PCR amplification (Wagner et al., 2016; Nakano et al., 2017). The complete genome sequence of atrazine-degrading Arthrobacter sp. ZXY-2 (Zhao X. et al., 2017) and organophosphate-degrading Sphingobium fuliginis ATCC 27552 (Azam et al., 2019) was analyzed using the PacBio RSII sequencing platform to gain more insight into the genetic basis and unravel its degradation potential.
Jeffries et al. (2019) performed functional metagenomic studies in pesticide-contaminated soil to explore the degradation rates of organophosphorus xenobiotic compounds. Their study demonstrated that two distinct soil groups had different functional and taxonomic profiles, and predicted biodegradation potential in rapidly and slowly degrading soil clusters. Burkholderia, Acidomicrobium, Koribacter, and Bradyrhizobium were most abundantly present in rapidly degrading clusters, whereas Singulisphaera, Solibacter, and Desulfomonile were in slowly degrading clusters. The degradation assays of organophosphorus also suggested that slow-degradation clusters had significantly higher abundances of virulence genes and metabolic pathways for fatty acids and carbohydrates. In contrast, rapid-degradation clusters contain more abundant genes related to the transposable elements, membrane transport, and nutrient cycling of nitrogen and phosphorus enzymes potentially involved in phosphorus metabolism. Moreover, rapid-degradation soils also showed a higher abundance of genes encoding phosphodiesterase enzymes, which cleave phosphodiester bonds present in organophosphorus and play a major role in pesticide degradation. Overall, this study gives an overall framework of metagenomic approaches to predict the microbial degradation of xenobiotic organophosphorus compounds.
Metagenomic analysis of a complex community of lindane-contaminated pond sediment was conducted by Negi and Lal (2017) through comparative genomics. The results of this study revealed genomic variation present in pond sediment with degradative enzymes (hydrolases, isomerases, lyases, and oxidoreductases) involved in the biodegradation of hexachlorocyclohexane and chlorobenzene (ko00361), and other xenobiotic compounds. Cellulomonas, Micrococcus, Nocardioides, Kribbella, Isoptericola, Clavibacter, Gutenbergia, Streptomyces, Sanguibacter, and Kineococcus were found to be the most dominating genera present in the aromatic-hydrocarbon-contaminated pond sediment. Genes involved in lindane metabolization, enriched with sequences for linA and linB, were also found in the pond-sediment metagenome.
Whole-metagenome sequencing of e-waste-contaminated microbial populations was conducted by Salam and Verma (2019), who demonstrated that the functional diversity and structural composition of microorganisms significantly changes due to the detrimental impact of e-waste. Denaturing gel gradient electrophoresis (DGGE) community-profiling results revealed that bacterial groups such as Proteobacteria, Firmicutes, Bacteroidetes, and Chloroflexi were decreased. Zhu et al. (2020) explored microbial assemblage and functional genes potentially involved in upstream and downstream phthalate degradation in soil via a metagenomic approach. Results indicated that bacterial taxon Actinobacteria (Pimelobacter, Nocardioides, Gordonia, Nocardia, Rhodococcus, and Mycobacterium) was a major degrader under aerobic conditions, and bacterial taxa Proteobacteria (Ramlibacter and Burkholderia), Acidobacteria, and Bacteroidetes were involved under anaerobic conditions. The members of Geobacteraceae and Peptococcaceae microbiota present in the jet-fuel-contaminated site could be exploited for their remarkable metabolic potential for the mitigation of toluene and benzene, as exposed by metagenomic analysis (Hidalgo et al., 2020).
Transcriptomics is a remarkable tool that addresses the division of genes transcribed in any organism known as the transcriptome. It provides functional insight links involving the genome, proteome, and cellular phenotype by studying their mRNA transcriptional profiles, directly extracted from individual microbes or microbial communities (Singh and Nagaraj, 2006; McGrath et al., 2008; Bashiardes et al., 2016). Significant changes were seen in gene-expression level and their regulation in microbial communities under stressful environments for their survival. Thus, transcriptomics and metatranscriptomics provide deep analysis of a genome wide range of differently expressed genes, either of the individual cell or the entire microbial community at a specific time (Table 2; Li et al., 2014; He et al., 2015; Shakya et al., 2019). RNA seq and DNA microarrays are significantly powerful technologies to determine the mRNA expression level of every gene (Diaz, 2004). GeoChip uses key enzymes or genes to spot various microbe-mediated mechanisms for biogeochemical cycles, resistance mechanism for heavy metals, and degradation pathways of xenobiotics (He et al., 2010; Xiong et al., 2010; Xie et al., 2011). Similarly, DNA- and RNA-SIP (Stable Isotope Probing) technologies are also valuable to uncover the microbial taxa and catabolic genes that are important for the bioremediation of polluted environments (Lueders, 2015).
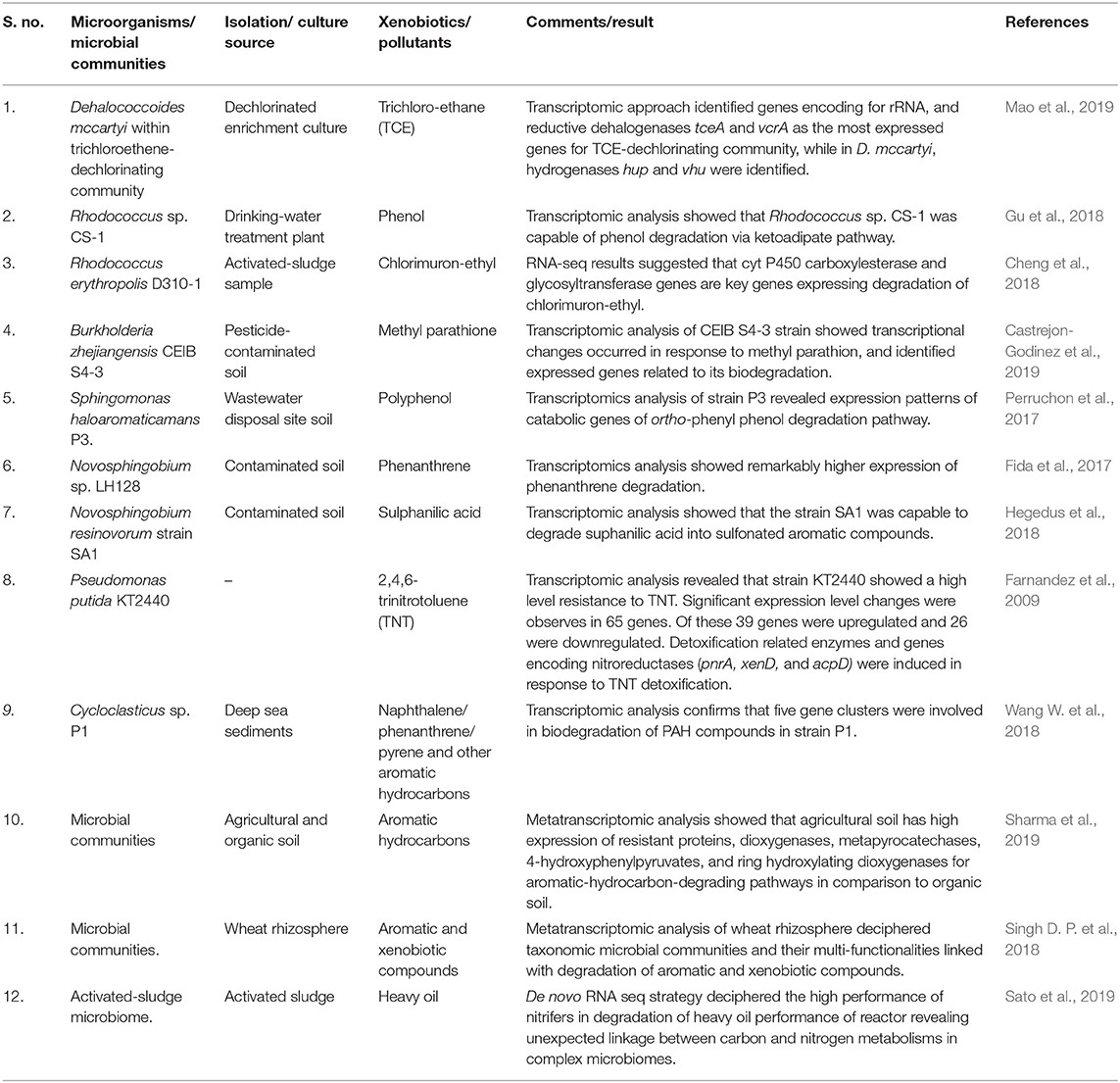
Table 2. Microorganisms and microbial communities using transcriptomic and metatranscriptomic approaches in biodegradation.
Dual RNA-seq transcriptional profile is a better approach to understand the basic nature and mechanism of differently expressed genes in the host and symbiotic microbes at a time (Kaul et al., 2016). RNA seq allows for the detection of more differently expressed genes than a microarray alone does. Thus, recent advancements and developments in microarrays, RNA seq technology, transcriptomics, and metatranscriptomics revealed unexpected microbial diversity in aquatic and terrestrial environments with their synergistic relationships with humans, animals, plants, and other microorganisms (Perez-Losada et al., 2015; White et al., 2016; Moniruzzaman et al., 2017; Berg et al., 2018; Crump et al., 2018).
RNA seq technology is considered more efficient than traditional microarray platforms in gene expression profiling as it provides a wider quantitative range of expression level changes compared to microarrays (Roh et al., 2010; Shakya et al., 2019). The microarray technique requires a lot of effort and money to prepare custom-made microarrays. Furthermore, the target genes to be analyzed are limited in number and cannot cover the whole set of genes in the community. In contrast, a number of kits are now commercially available to carry out RNA-seq analysis, whereby a whole set of genes in the community can be quantitatively analyzed. Therefore, many studies are now performed by RNA-seq technology.
Lima-Morales et al. (2016) investigated the microbial organization and catabolic gene diversity of three types of contaminated soil under continuous long-term pollutant stress with benzene and benzene/toluene/ethylene/xylene (BTEX) to identify shifts in community structure and the prevalence of key genes for catabolic pathways. Moreover, de novo transcriptome synthesis gives new insights into and reveals basic information about non-model species without a genome reference. Hydrocarbon-degrading bacterium Achromobacter sp. was isolated from seawater, and indicated that the upregulation of enzymes such as dehydrogenases and monooxygenases, and novel genes associated with fatty acid metabolism is responsible for its enormous capability for hydrocarbon degradation and survival (Hong et al., 2016).
Metatranscriptomic analysis of the wheat rhizosphere identified dominant bacterial communities of diverse taxonomic phyla, including Acidobacteria, Cyanobacteria, Bacteroidetes, Steptophyta, Ascomycota, and Firmicutes, having functional roles in the degradation of various xenobiotic pollutants (Singh D. P. et al., 2018). Multiple enzymes such as isomerases, oxygenases, decarboxylases, reductases, kinases, and inner membrane translocators were identified that were associated with 21 different pathways for benzoates, aromatic amines, phenols, bisphenols, and other xenobiotics (Singh et al., 2016). An et al. (2020) elucidated the study of the transcriptome for the characterization of hexaconazole degrading strain Sphingobacterium multivorum, obtained from activated sludge. This strain was capable of degrading 85.6% hexaconazole in just 6 days and of generating three different metabolites, M1, M2, and M3, recognized as (2-(2,4-dichlorophenyl)-1-(1H-1,2,4-triazol-1-yl)hexane-2,5diol), (2-(2,4-dichlorophenyl) hexane-1,2-diol), and (1H-1,2,4-triazol), respectively. The results of transcriptome sequencing revealed the presence of 864 differential genes in which dehydrogenases, aldehydes, monooxygenases, and RND and AC transporters were upregulated. The M1 metabolite was perhaps generated due to the participation of monooxygenases.
Genomic and transcriptomic approaches were used by Sengupta et al. (2019) for gaining mechanistic insight into 4-nitrophenol (4-NP) degrading bacterium Rhodococcus sp. strain BUPNP1. This study identified a catabolic 43 gene cluster named nph that harbors not only mandatory genes for the breakdown of 4-NP into acetyl co-A and succinate by nitrocatechol, but also for other diverse aromatic compounds. An integrated approach of metagenomics and metatranscriptomics revealed the metabolic capabilities and synergistic relationship between Sphingomonas spp., Pusillimonas sp., and Pseudomonas sp. in the degradation of bisphenol A (BPA) (Yu K. et al., 2019).
Metatranscriptomic analysis of this interaction model demonstrated genes encoding the transcription of 1,2-bis(4-hydroxyphenyl)-2-propanol (1-BP) into 4-hydroxybenzaldehyde (4-HBD) and 4-hydroxy-acetophenone (4-HAP) via 3,4-dihydroxybenzoate (3,4-DHB) and 3-oxoadipate (3-ODP), respectively, to the tricarboxylic acid cycle (TCA)cycle. Marcelino et al. (2019) identified fungal species and subspecies in a mixed community by using metatranscriptomics. This study suggested a strain-level discrepancy between the Cryptococcus fungal species and their in-situ mock communities. Thus, transcriptomic analysis provides a large amount of gene information about the potential function of microbial communities in adaptation and survival in extreme environments (Singh D. P. et al., 2018; Mao et al., 2019; Marcelino et al., 2019).
Proteins are crucial effectors of biological responses, stabler than RNAs in living organisms, and possibly confer an integral view of biological function expressed in situ; the term proteomics is put forward to study the entire set of proteins expressed in an organism (Ram et al., 2005; Singh, 2006; Hettich et al., 2013). Thus, proteomics has emerged as an interesting and fruitful technology to study protein expression (post-translational modifications, protein turnover, proteolysis, and changes in the corresponding gene expression) of the microbial world (Keller and Hettich, 2009; Aslam et al., 2017). Proteomics is a promising aspect of omic technologies in the field of microbiology, allowing for investigating the complete protein profile obtained in a straight line from a composite microbial population in a contaminated environment (Williams et al., 2013; Arsene-Ploetze et al., 2015; Wang et al., 2016). However, metaproteomics is used to decipher crucial information regarding the protein profiling of two diverse ecological units (Arsene-Ploetze et al., 2015). Proteomics has been used to identify microbial communities/microorganisms in various ecosystems including soil and sediment, activated sludge, marine and groundwater sediment, acid mine biofilms, and wastewater plants, as illustrated in Table 3 (Williams et al., 2013; Colatriano et al., 2015; Grob et al., 2015; Bastida et al., 2016; Jagadeesh et al., 2017). These studies revealed secret information related to protein synthesis, gene-expression stability, mRNA turnover, and protein–protein interaction networks in microbial communities in stress environments (Aslam et al., 2017).
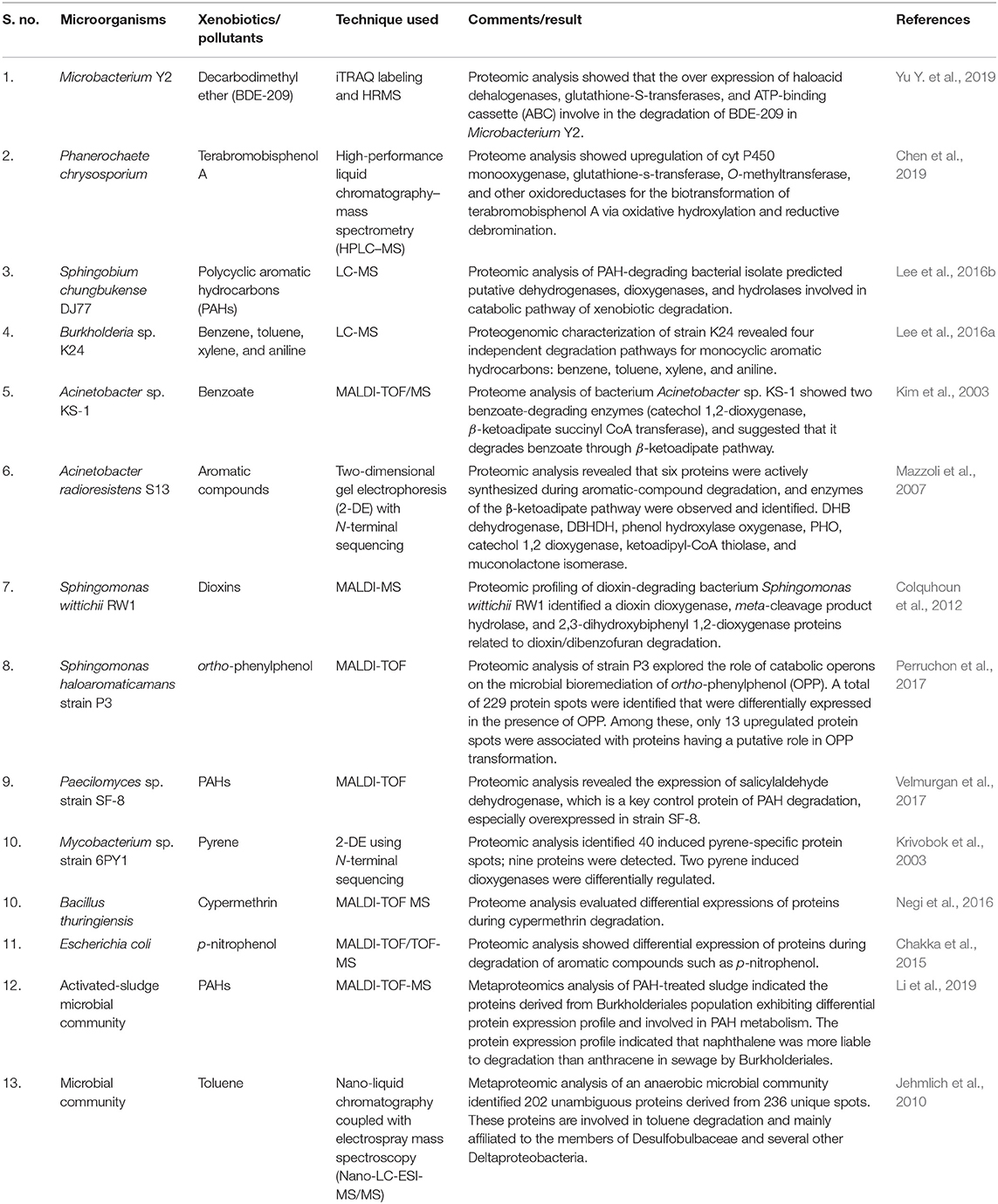
Table 3. Microorganisms or microbial communities using proteomic and metaproteomic approaches in biodegradation.
The inclusion of a proteomic approach helps to identify related enzymes and their metabolic pathways in the bioremediation of xenobiotics from various contaminated sites (Kim et al., 2004; Liu et al., 2017; Wei et al., 2017). Basically, there are four primary steps that involve proteomic analysis: (1) preparation of a biological sample; (2) extraction and separation of proteins by using two-dimensional gel electrophoresis (2D-GE); (3) protein gel images are examined by means of image-analysis software such as ImageMaster 2D or PDQuest; and (4) proteins are identified by using mass spectroscopy (MS)/MALDI-TOF/MS or LC-MS (Yates et al., 2009; Chakka et al., 2015; Velmurgan et al., 2017).
A combined protein profile of 20 PAH-induced proteins was studied by proteomics in Mycobacterium vanbaalenii PYR-1 grown in a PAH-supplemented culture medium (Kim et al., 2004). PAH exposure of five different compounds, i.e., pyrene, pyerene-4,5-quinone, phenanthrene, anthracene, and fluoranthere causes variation in protein composition, showing upregulation of multiple proteins for PAH treatment compared to an uninduced control sample. Several PAH-induced proteins were identified by LC-MS, including a catalase-peroxidase, a putative monooxygenase, a dioxygenase, a small subunit of naphthalene-inducible dioxygenase, and aldehyde dehydrogenase. The metaproteomic approach was employed by Bastida et al. (2016) to illustrate changes in metabolic activities during compost-treated bioremediation with the help of differential protein expressions in hydrocarbon-polluted soil. Metaproteomic analysis indicated that Sphingomonadales and uncultured bacteria are responsible for the degradation of hydrocarbons in compost-treated soil due to the higher expression of catabolic enzymes such as 2-hydroxymuconic semialdehyde, cis-dihydrodiol dehydrogenase, and catechol 2,3-dioxygenase, dioxygenases involved in the first oxygenation step of aromatic rings. Moreover, biphenyl-2,3-diol 1,2-dioxygenase, estradiol dioxygenase, and naphthalene 1,2-dioxygenase were identified in compost-treated samples. By using metaproteomics, this study explored the functional and phylogenetic relationship of contaminated soil, and the microbial key players involved in compost-assisted bioremediation.
Another study, undertaken by Vandera et al. (2015) demonstrated the comparative proteomic analysis of Arthrobacter phenanivorans Sphe3 on aromatic compounds phenanthrene and phthalates. The proteomic approach confirmed the involvement of several proteins in aromatic-substrate degradation by identifying those mediating the initial ring hydroxylation and ring cleavage of phenanthrene to phthalate. This study also revealed the presence of both the ortho- and the meta- cleavage pathway for the degradation of these aromatic compounds, and it also identified all proteins that take part in these pathways and are highly upregulated upon phthalate growth in comparison to phenanthrene growth.
Proteomic analysis of pyrene-degrading bacterium Achromobacter xylosoxidans PY4 was performed by Nzila et al. (2018), who identified a total of 1,094 proteins. Among these, 95 proteins were detected in glucose supplementation, and 612 proteins were detected in the presence of pyrene. Furthermore, 25 upregulated proteins were found to be involved in stress response and the progression of genetic information. Two upregulated proteins, 4-hydroxyphenylpyruvate dioxygenase and homogentisate 1,2-dioxygenase, are implicated in the lower degradation pathway of pyrene. Enzyme 4-hydroxyphenylpyruvate dioxygenase may catalyze the conversion of 2-hydroxybenzalpyruvic acid (metabolite of pyrene) to homogentisate. Homogentisate 1,2-dioxygenase is involved in the incorporation of 2 oxygen atoms to produce 4-maleyacetoacetate, which is an intermediate in several metabolic pathways.
Lee et al. (2016b)performed proteomic analysis of PAH-degrading bacterial isolate Sphingobium chungbukense DJ77. This strain exhibited outstanding degradation capability for various aromatic compounds. This study demonstrated the degradation of three xenobiotics compounds, i.e., phenanthrene, naphthalene, and biphenyls (PNB), and their associated proteins was analyzed by 2-DE and MALDI-TOF/MS analysis. During PNB biodegradation by bacterial cells, an alteration was observed in protein expression to cope with the stress condition. Comparative analysis of 2-DE results revealed that the intensity of 10 protein spots changes identically upon exposure to these xenobiotics in strain DJ77 (Lee et al., 2016b). Among these ten, five upregulated proteins with multiple functionalities were identified as putative dihydrodiol dehydrogenase (BphB), which catalyzes the NAD+-dependent oxidation of trans-dihydrodiols; 2,3-dihydrobisphenyl 1,2-dioxygenase (PhnQ), which cleaves the aromatic ring; and 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate hydrolase (BphD), which degrades biphenyls and polychlorinated biphenyls. A part of the initial diverse catabolism of PNB by BphB, PhnQ, and BphD converged into the same catechol degradation branch. Now, catechol is first transformed into a ring-cleaved product, i.e., 2-hydroxymuconic semialdehyde by catechol 2,3-dioxygenase (PhnE). Moreover, it is assumed that this ring-cleaved product (2-hydroxymuconic semialdehyde) would be further degraded by 2-hydroxymuconic semialdehyde hydrolase (PhnD), and acetaldehyde dehydrogenase (PhnI) into a compound that can enter into the TCA cycle. Hence, these upregulated proteins, dehydrogenase, dioxygenase, and hydrolase, are involved in the catabolic degradation pathway of xenobiotics. The detection of intermediates from 2,3-dihydroxy-biphenyl degradation to pyruvate and acetyl-CoA by LC/MS analysis showed that ring-cleavage products entered the TCA cycle and were mineralized in strain DJ77. It was also suggested that strain DJ77 could completely degrade a wide range of PAHs via multiple catabolic pathways (Lee et al., 2016b).
The biodegradation mechanism of tetrabromobis-phenol A (TBBPA) was investigated in Phanerochaete chrysosporium by using a proteomic approach. iTRAQ quantitative analysis identified a total of 2,724 proteins in three biological samples. Compared to control TBBPA, stress caused 148 differentially expressed proteins in P. chrysosporium, among which 90 proteins were upregulated and 58 proteins were downregulated. The upregulation of cytochrome p450 monooxygenase, glutathione-S-transferase, O-methyltransferase, and other oxidoreductases is responsible for the biotransformation of TBBPA via oxidative hydroxylation and reductive debromination (Chen et al., 2019).
A biodegradation study of endocrine-disrupting compound 4-n-nonylphenol (4-n-NP) by filamentous fungus Metarhizium robertsii was investigated by Szewczyk et al. (2014) with proteomic analysis. This suggested that the main biodegradation mechanism involves the consecutive oxidation of the alkyl chain and benzene, which consequently results in the complete decomposition of the 4-n-NP compound. Proteomic profiling explored the involvement of nitro-reductase-like proteins related to the oxidation–reduction and ROS defense systems, and mainly engaged group of proteins in the removal of 4-n-NP. Proteomic data obtained in this study could not clearly explain the mechanism of 4-n-NP biodegradation in the tested fungal strain, but allowed for the formulation of hypotheses that the over-expressed enzymes in the cultures with 4-n-NP could play a role in xenobiotic removal and the biodegradation process.
Bioremediation of decabromodiphenyl ether (BDE-209) was explored in Microbacterium Y2 in a polluted water-sediment system through proteomics (Yu Y. et al., 2019). Proteomic analysis showed that the overexpression of haloacid dehalogenases, glutathione s-transferase, and ATP-binding cassette (ABC) transporter might occupy important roles in BDE-209 biotransformation. Moreover, heat-shock proteins (HSPs), ribonuclease E, oligoribonuclease (Orn), and ribosomal proteins were activated to counter the BDE-209 toxicity. Thus, it is suggested that these proteins are implicated in microbial degradation, antioxidative stress, and glycolysis (Yu Y. et al., 2019).
Metabolomics is a well-established recent scientific technology, attributed toward the study of naturally occurring low-molecular weight (<1,000 Da) organic metabolites (organic acids; pyruvate, lactate, malate, formate, fatty acid-like acetate, etc.) inside a tissue, cell, or biofluid (Johnson et al., 2011; Malla et al., 2018; Withers et al., 2020). Metabolomics explores the relationships between organisms and the environment, such as organismal responses to abiotic stressors, including both natural factors such as temperature, and anthropogenic factors such as pollution, to investigate biotic–biotic interactions such as infections, and metabolic responses (Lindon et al., 2006; Griffiths, 2007; Mallick et al., 2019).
The combined use of metabolomics with these applications details the authentic collection of chemical outputs and inputs, which arbitrate the exchange of resources between the community and its host (Table 4; Theriot et al., 2014; Wang Y. F. et al., 2018). Metabolomic studies in environmental sciences have been directed toward understanding changes in the concentration of metabolites associated with exposing model organisms to toxic compounds, such as xenobiotics (Parisi et al., 2009; Seo et al., 2009).
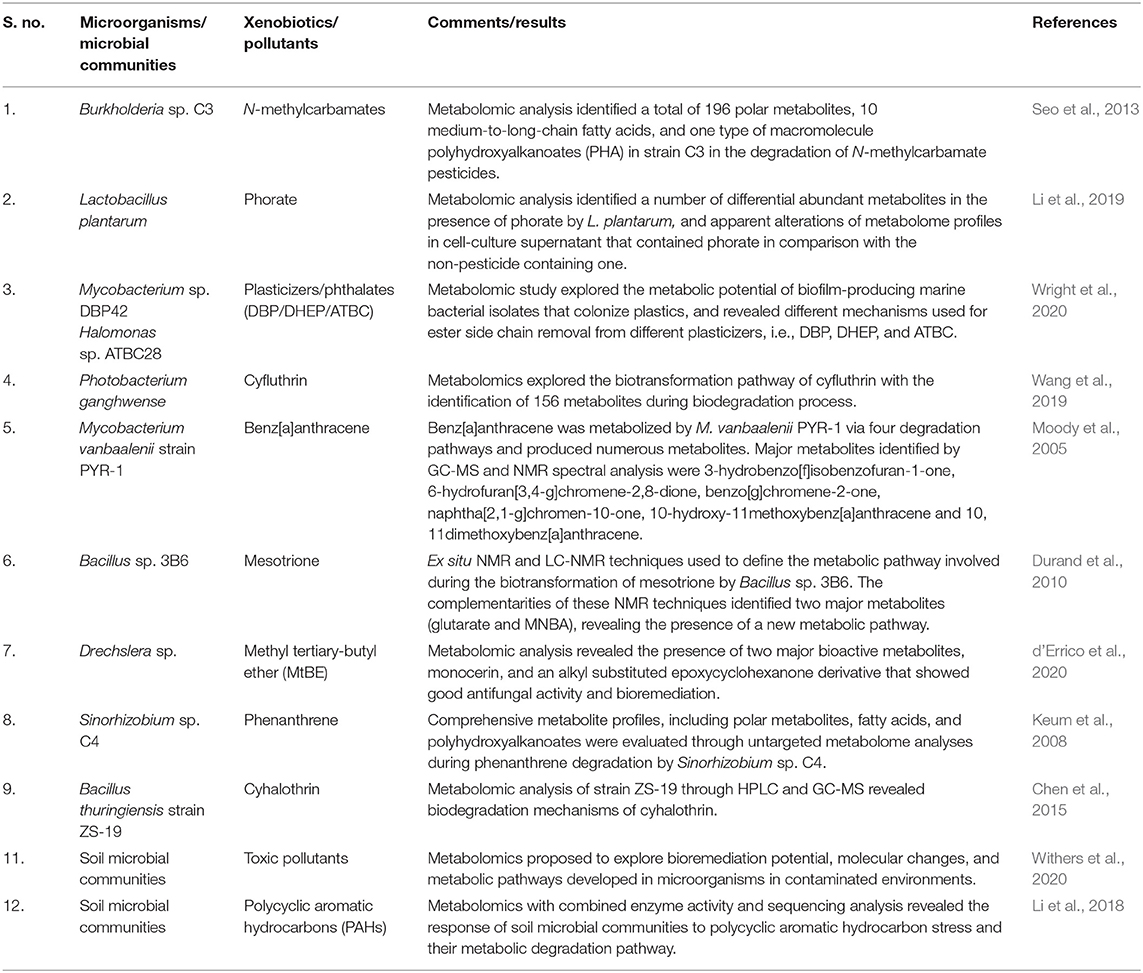
Table 4. Microorganisms or microbial communities using metabolomic approaches in biodegradation.
Metabolomics approach was utilized to investigate the degradation mechanism of carbaryl and other N-methyl carbamates pesticides in Burkholderia sp. strain C3 (Seo et al., 2013). Metabolomes are dynamic and responsive to nutrient and environmental changes. The results of this study showed the metabolic adaptation of Burkholderia sp. C3 to carbaryl in comparison with glucose and nutrient broth. The metabolic changes were notably associated with the biosynthesis and metabolism of amino acids, sugars, PAH lipids, and cofactors. Differential metabolome analysis in response to different substrates identified 196 polar metabolites, 10 fatty acids, and 1 macromolecule (PHA) in this strain, and confirmed up-production metabolites in the pentose phosphate pathway, cysteine metabolism, amino acids, and disaccharides (Seo et al., 2013). Thus, this metabolomic study could provide detailed insights into bacterial adaptation to different metabolic networks, and the metabolism of toxic pesticides and chemicals.
Environmental pollutants cause alterations in microbial communities, which consequently changes biochemical and metabolic functions in soil microorganisms. Microbial degradation of cyfluthrin by Photobacterium ganghwense was investigated via a comparative metabolic approach (Wang et al., 2019). Metabolomics explored the biotransformation pathway of cyfluthrin with the identification of 156 metabolites during the biodegradation process. Recently, on the basis of interactions of indigenous soil microorganisms to PAH-contaminated soil, Li et al. (2018) elucidated that the majority of microbial metabolic functions were adversely affected to cope with PAH pollution. This study includes the combined study of enzyme activity and sequencing analysis with metabolomics, which further exposed the specific inhibition of soil metabolic pathways associated with carbohydrates, amino acids, and fatty acids due to microbial-community shifting under PAH stress.
Soil metabolomics is an effective approach to reveal the complex molecular networks and metabolic pathways operating in the soil microbial community. This approach can also be used to find biomarkers of soil contamination (Jones et al., 2014). High-throughput sequencing and soil metabolomics investigated the differential structures and functions of soil bacterial communities in the pepper rhizosphere and bulk soil under plastic greenhouse vegetable cultivation (PGVC) (Song et al., 2020). A total of 245 metabolites were identified, among which 11 differential metabolites were detected between rhizosphere and bulk soil, including organic acids and sugars that were positively and negatively correlated with the relative abundances of the differential bacteria. A starch and sucrose metabolic pathway was the most differentially expressed pathway in rhizospheric soil. The main functional genes participating in this pathway were predicted to be down regulated in rhizosphere soil. Sugar and organic acids as the main plant-root exudates in the rhizosphere, and they are also the main drivers of the shift in soil microbial community in the rhizosphere. These plant-root exudates act as an energy source to soil microbes, thus benefiting their growth (Kuzyakov and Blagodatskaya, 2015; Shi et al., 2015). Linear discriminant analysis (LDA) effect size (LEfSe) analysis showed that bacterial phyla of Proteobacteria and Bacteroidetes were significantly higher in rhizosphere soil, benefitting plant growth. Thus, the relationship between soil metabolites and microbial communities guides the regulation of plant rhizoprocesses through soil amendments to increase plant growth.
Durand et al. (2010) conducted metabolic analysis of Bacillus sp. to characterize the metabolic pathway for the biodegradation of mesotrione, a herbicide. Analysis was carried out by using LC-NMR and LC-MS, and the result of these instrumental analyses was the identification of six metabolites, of which the structures of four metabolites were suggested. Szewczyk et al. (2017) performed metabolic analysis of fungal strain Metarhizium brunneum ARSEF 2017 to predict a biodegradation-pathway metabolic background for the removal of ametryn, an s-triazene herbicide. Qualitative LC-MS/MS metabolomic analysis of ametryn biodegradation resulted in the generation of four metabolites, i.e., 2-hydroxy atrazine, ethyl hydroxylated ametryn, S-demethylated ametryn, and diethyl ametryn.
Wright et al. (2020) evaluated the metabolomic characterization of two potent marine bacterial isolates, Mycobacterium sp. DBP42 and Halomonas sp. ATBC28, capable of the degradation of phthalate and plasticizers such as ATBC, DBP, and DEHP. That study presented the molecular analysis of metabolites generated during biodegradation. A metabolomic study confirmed that DBP and ATBC were degraded through the sequential removal of ester side chains, and generated monobutyl phthalate and phthalate in the case of DBP degradation, and citrate in the case of ATBC degradation in Mycobacterium sp. However, DEHP degradation did not follow the same pathway as that observed for DBP and ATBC. It was suggested that DEHP degradation is initiated through hydroxylation of the ester side chain by monooxygenase, and may occur via the β-oxidation of fatty acid side chains directly on the DEHP molecule. Moreover, in comparison with Mycobacterium sp., Halomonas sp. did not confirm any detectable degradation intermediates for the degradation of plasticizers and phthalate, but it harbored an array of enzymes suggested to be responsible for the degradation of other aromatic compounds. Therefore, metabolomic analyses demonstrated changes that occur in the composition of metabolites, aiding to fully understand the shifts mechanisms of metabolites during the microbial degradation or mineralization of environmental pollutants (Lindon et al., 2006; Keum et al., 2008; d'Errico et al., 2020).
Bioinformatic technology developed a new array of computational technologies that uses both information technology and biological sciences. This modern technology seeks information from multiple high-throughput biological techniques, and keeps all biological data, helping to investigate and decide the relationship among organic molecules, including macromolecular sequences, biochemical and metabolic pathways, protein expressions, metabolites, and structures (Fulekar and Geetha, 2008; Cooper et al., 2018; Dangi et al., 2018; Greene, 2018; Shekhar et al., 2020). Enormous amounts of data are generated from DNA, RNA, and protein sequences that need to be accurately executed; thus, bioinformatics has led to finding the best possible way to analyze such huge amounts of biological data via specific computational tools (Aora and Bar, 2014; Bhatt et al., 2021). Therefore, bioinformatic-associated tools are very important to understand the bioremediation of toxic pollutants. Bioinformatics provides superior information regarding the cellular, molecular, and genetical bases of xenobiotic degradation and detoxification (Kumar et al., 2016; Huang et al., 2020). There are a number of bioinformatic tools and applications that are available to use for biodegradation studies, as listed in Table 5.
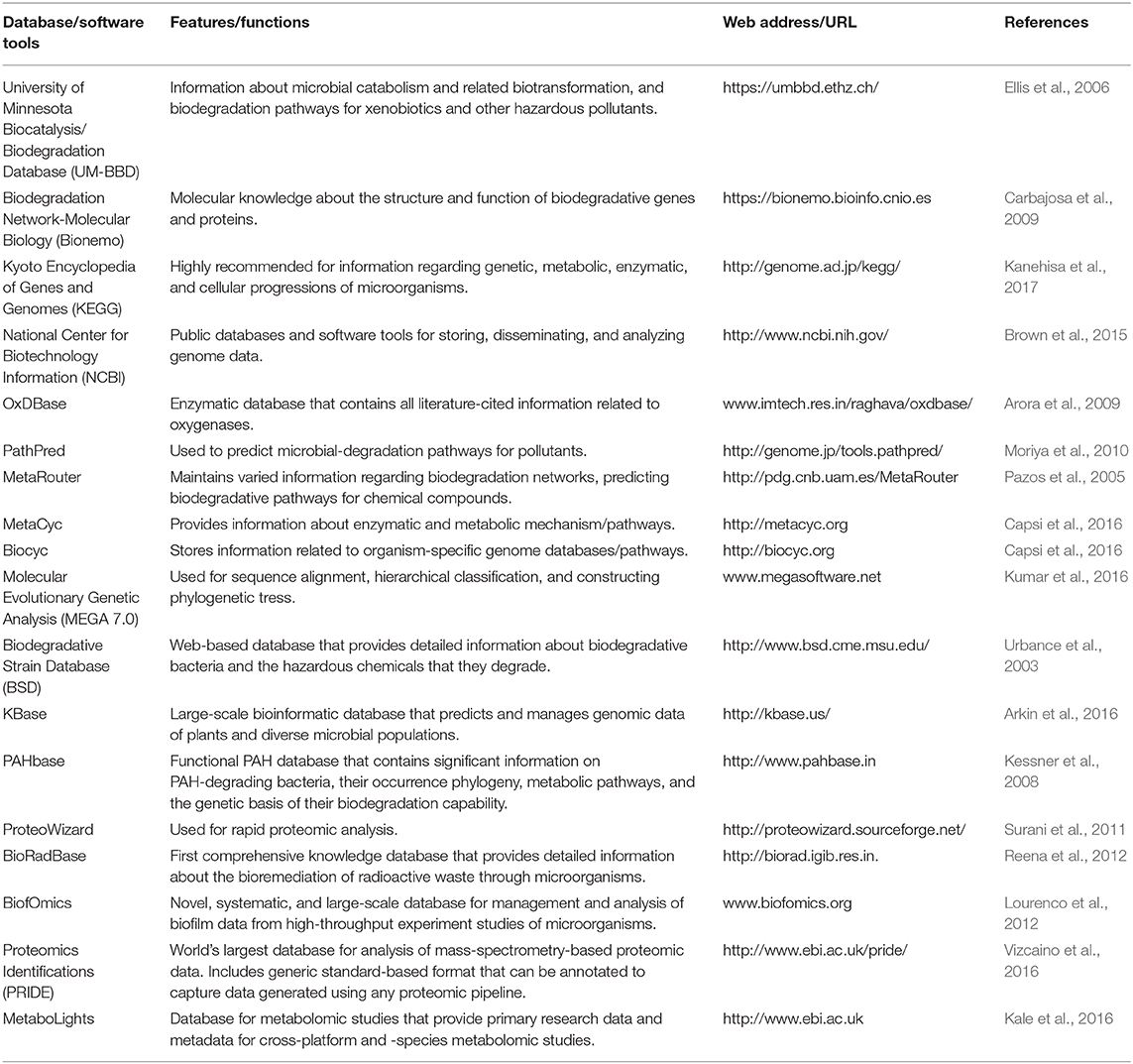
Table 5. Bioinformatic databases and software tools used in biodegradation studies.
MetaRouter is one of such application that is freely open and a modular architecture to a variety of consumers from any place in a safe and secure manner, just by connecting to an Internet server (Pazos et al., 2005). For the analysis of biodegradation studies, many bioinformatic resources are exclusively available. The University of Minnesota Biocatalysts/Biodegradation Database (UM-BBD) was introduced in 1995 and contains information regarding microbial catabolism and related biotransformation, biodegradation reactions, catabolic enzymes, and pathways for xenobiotics and other hazardous pollutants of various microorganisms. This database is connected to several other databases, such as BRENDA, ENZYME, ExPASy, and NCBI, to collect and store information related to gene structure and enzymes that take part in the biodegradation of environmental contaminants (Ellis et al., 2006). Genomic sequences of microorganisms with competent and efficient degradation abilities could be easily investigated via another widely used database, the National Center for Biotechnology Information (NCBI). It gives a detailed and complete pipeline for annotations, and comprehensive analysis of more than 6,000 microbial genomes (Brown et al., 2015). PRIDE (Vizcaino et al., 2016) is the world's largest data repository of mass-spectroscopy-based proteomic data, and MetaboLights (Kale et al., 2016) is a database for metabolomic experiments and derived information. The GenBank database is freely available in NCBI, and it provides most up-to-date and comprehensive DNA sequence information (Benson et al., 2013). Recently, there have been a number of databases such as CAMERA, MG-RAST, and IMG/M that were developed and employed for the analysis and in-depth understanding of diverse microbial populations, metabolic reconstruction, taxonomic affiliations, and their inter- and intra-relationship networks. Another database, Bionemo, developed by the structural computational-biology group at the Spanish National Cancer Research Center, gives information related to specific genes and proteins that take part in biodegradation reactions and metabolic pathways (Carbajosa et al., 2009). It provides insights into sequences, domains, protein structures, and regulatory elements, and transcription factors for their respective genes. Integrated bioinformatic approaches are employed for the metagenomic characterization of the soil microbial communities of different soil sites by using MetaPhlAn, KEGG, XLSX, and LEfSe bioinformatic databases to reveal the ancestral and functional characterization of diverse soil microbial populations (Arora et al., 2009; Xu et al., 2014; Kumar et al., 2016). The degradation or detoxification of xenobiotic pollutants through microbial communities is a highly considered and proficient remediation technology, and there is no single resource accessible that provides all the information with reference to environmental contaminants, microorganisms, and their bioremediation potentialities. Thus, these databases coalescing the detailed information about the nature of pollutants, their metabolic pathways, bioremediation microorganisms, catabolic genes, enzymes, and protein-expression profiles would be a significant tool to open up a new vista and enlighten future research science in the field of bioremediation.
Microbial communities have great potential to mediate the successful biodegradation process of xenobiotic-contaminated soil/water environments. However, the greater part of mainstream microorganisms involved in bioremediation are still undefined because not all organisms in nature could be cultured under in vitro environments, but reside in viable-but-non-culturable (VBNC) environments. Thus, to explore the hidden knowledge of these VBNC organisms, recent advanced practices and sophisticated up-to-date technologies are highly desirable to understand the genetic and molecular biology of microorganisms. Newly developed molecular techniques offer promising approaches to address the in-depth characterization of microbial communities from molecule to gene. Recent omics technologies such as metagenomics, transcriptomics, and proteomics are helpful in obtaining information about nucleic acids, enzymes, catabolic genes, plasmids, and metabolic machineries and metabolites generated during the biodegradation process. However, the solitary employment of any individual omics technology is not sufficient to explore or illustrate secret information regarding microbial-remediation practices. Therefore, an interdisciplinary application of multiple omics studies highlights the perspectives of system biology for providing an integrative understanding between genes, proteins, and environmental factors responsible for the whole microbial-degradation process, and gives a new array of novel technologies, such as genome-editing and next-generation-sequencing tools CRISPR-Cas9, TALEN, and ZFNs, which are potent gene-editing tools that design microbes with specific degradation-function genes and provide unique insights into microbial remediation. Moreover, the successful execution of omics technologies could not be possible without the use of bioinformatic tools. The establishment of informative genomic and proteomic databases has been revolutionized by bioinformatics, which facilitates broad information about cellular- and metabolic-mechanism pathways for environmental pollutants. Hence, the involvement of these advanced technologies in the biological sciences shows the way to next-level research in the bioremediation potential of microorganisms, and exploits their capability to remove xenobiotic contamination.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Conceptualization and writing—original draft preparation: SM. Writing—review and editing: ZL, SP, WZ, PB, and SC. Supervision, funding acquisition, and project administration: SC. All authors contributed to the article and approved the submitted version.
This study was funded by the Key-Area Research and Development Program of Guangdong Province, China (2018B020206001), the National Natural Science Foundation of China (31401763), and the Guangdong Special Branch Plan for Young Talent with Scientific and Technological Innovation, China (2017TQ04N026).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdel-Shafy, H., and Mansour, M. S. M. (2016). A review on polycyclic aromatic hydrocarbons: source, environmental impact, effect on human health and remediation. Egyp. J. Petroleum 25, 107–123. doi: 10.1016/j.ejpe.2015.03.011
Abraham, W. R., Nogales, B., Golyshin, P. N., Piper, D. H., and Timmis, K. N. (2002). Polychlorinated biphenyl-degrading microbial communities in soil and sediments. Curr. Opin. Microbiol. 5, 246–253. doi: 10.1016/S1369-5274(02)00323-5
Aguinga, O. E., McMahon, A., White, K. N., Dean, A. P., and Pittman, J. K. (2018). Microbial community shifts in response to acid mine drainage pollution within a natural wetland ecosystem. Front. Microbiol. 9:1445. doi: 10.3389/fmicb.2018.01445
Ahmad, A., and Ahmad, I. (2014). “Recent advances in the field of bioremediation”, in Biotechnology Vol. 11: Biodegradation and Bioremediation, ed M. Ahmad (India: Stadium Press LLC), 1–42.
Akinsanya, M. A., Goh, J. K., Lim, S. P., and Ting, A. Y. (2015). Metagenomic study of endophytic bacteria in Aloe vera using next-generation technology. Genom. Data 6, 159–163. doi: 10.1016/j.gdata.2015.09.004
Aktar, W., Sengupta, D., and Chowdhary, A. (2009). Impact of pesticides use in agriculture: their benefits and hazards. Interdiscip. Toxicol. 2, 1–12. doi: 10.2478/v10102-009-0001-7
An, X., Tian, C., Xu, J., Dong, F., Liu, X., Wu, X., et al. (2020). Characterization of hexaconazole-degrading strain Sphingobacterium multivorum and analysis of transcriptome for biodegradation mechanism. Sci. Total Environ. 722:37171. doi: 10.1016/j.scitotenv.2020.137171
Anyasi, R., and Atagana, H. (2011). Biological remediation of polychlorinated bisphenyls (PCB) in the environment by microorganisms and plants. Afr. J. Biotechnol. 10, 18916–18938. doi: 10.5897/AJB10.557
Aora, P. K., and Bar, H. (2014). Integration of bioinformatics to biodegradation. Biol. Procedure Online 16:8. doi: 10.1186/1480-9222-16-8
Arkin, A. P., Stevens, R. L., and Cottingham, R. W. (2016). The DOE system biology knowledgebase (KBase). BioRxiv 12, 1–21. doi: 10.1101/096354
Arora, P. K. (2020). Bacilli-mediated degradation of xenobiotic compounds and heavy metals. Front. Bioeng. Biotech. 8:570307. doi: 10.3389/fbioe.2020.570307
Arora, P. K., Kumar, M., Chauhan, A., Raghava, G. P., and Jain, R. K. (2009). OxDBase: a database of oxygenases involved in biodegradation. BMC Res. Notes 2:67. doi: 10.1186/1756-0500-2-67
Arora, P. K., Srivastava, A., Garg, S. K., and Singh, V. P. (2018). Recent advances in degradation of chloronitrophenols. Bioresour. Technol. 250, 902–909. doi: 10.1016/j.biortech.2017.12.007
Arsene-Ploetze, F., Bertin, P. N., and Carapito, C. (2015). Proteomic tools to decipher microbial community structure and functioning. Environ. Sci. Pollut. Res. 22, 13599–13612. doi: 10.1007/s11356-014-3898-0
Aslam, B., Basit, M., Nisar, M. A., Khurshid, M., and Rasool, M. H. (2017). Proteomics: technologies and their applications. J. Chromatogr. Sci. 55, 182–196. doi: 10.1093/chromsci/bmw167
Atashgahi, S., Shetty, S. A., Hauke, S., and de Vos, W. M. (2018). Flux, impact and fate of halogenated xenobiotic compounds in the Gut. Front. Physiol. 9:888. doi: 10.3389/fphys.2018.00888
Auti, A. M., Narwade, N. P., Deshpande, N. M., and Dhotre, D. P. (2019). Microbiome and imputed metagenome study of crude and refined petroleum-oil contaminated soils: potential for hydrocarbon degradation and plant-growth promotion. J. Biosci. 44:114. doi: 10.1007/s12038-019-9936-9
Awasthi, K. R., and Awasthi, M. S. (2019). Health and environmental effects of organochlorine pesticides in developing countries. Open Access J. Environ. Soil Sci. 2, 206–208. doi: 10.32474/OAJESS.2019.02.000135
Awasthi, M. K., Ravindran, B., Sarsaiya, S., Chen, H., Wainaina, S., Singh, E., et al. (2020). Metagenomics for taxonomy profiling: tools and approaches. Bioengineered 11, 356–374. doi: 10.1080/21655979.2020.1736238
Azam, S., Parthasarthy, S., Singh, C., Kumar, S., and Siddavattam, D. (2019). Genome organization and adaptive potential of archetypal organophosphate degrading Sphingobium fuliginis ATCC 27551. Genome Biol. Evol. 11, 2557–2562. doi: 10.1093/gbe/evz189
Azubuike, C. C., Chikere, C. B., and Okpokwasili, G. C. (2016). Bioremediation techniques- classification based on site of application: principle, advantages, limitations, and prospects. World. J. Microbiol. Biotechnol. 32, 180–180. doi: 10.1007/s11274-016-2137-x
Baboshin, M., Akimov, V., Baskunov, B., Born, T. L., Khan, S. U., and Golovleva, L. (2008). Conversion of polycyclic aromatic hydrocarbons by Sphingomonas sp. VKM B-2434. Biodegradation 19, 567–576. doi: 10.1007/s10532-007-9162-2
Bamforth, S. M., and Singleton, I. (2005). Bioremediation of polycyclic aromatic hydrocarbons: current knowledge and future directions. J. Chemical Technol. Biotechnol. 80, 723–736. doi: 10.1002/jctb.1276
Bari, S. M. N., Roky, M. K., Mohiuddin, M., Kamruzzaman, M., Mekalanos, J. J., and Faruque, S. M. (2013). Quorum-sensing autoinducers resuscitate dormant Vibrio cholera in environmental water samples. Proc. Natl. Acad. Sci. U.S.A. 110, 9926–9931. doi: 10.1073/pnas.1307697110
Bashiardes, S., Zilberman-Schapira, G., and Elinav, E. (2016). Use of metatranscriptomics in microbe research. Bioinf. Biol. Insights. 10, 19–25. doi: 10.4137/BBI.S34610
Bashir, S., Kuntze, K., Vogt, C., and Nijenhuis, I. (2018). Anaerobic biotransformation of hexachlorocyclohexane isomers by Dehalococcoides species and an enrichment culture. Biodegradation 29, 409–418. doi: 10.1007/s10532-018-9838-9
Bastida, F., Jehmlich, N., Lima, K., Moris, B. E., Richnow, H. H., Hernandez, T., et al. (2016). The ecological and physiological responses of the microbial community from a semiarid soil to hydrocarbon contamination and its bioremediation using compost amendment. J. Proteomic. 135, 162–169. doi: 10.1016/j.jprot.2015.07.023
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., et al. (2013). GenBank. Nucleic Acids Res. 41, D32–D37. doi: 10.1093/nar/gkt1030
Berg, C., Dupont, C. L., Asplund-Samuelsson, J., Celepli, N. A., Eiler, A., and Allen, A. E. (2018). Dissection of microbial community functions during a cyanobacterial bloom in the Baltic sea via metatranscriptomic. Front. Mar. Sci. 5:55. doi: 10.3389/fmars.2018.00055
Bertotto, L. B., Catron, T. R., and Tal, T. (2020). Exploring interactions between xenobiotics, microbiota and neurotoxicity in zebrafish. Neuro. Toxicol. 76, 235–244. doi: 10.1016/j.neuro.2019.11.008
Bharadwaj, A. (2018). “Bioremediation of xenobiotics: an eco-friendly cleanup approach”, in Green Chemistry in Environmental Sustainability and Chemical Education, eds V. S. Parmar, P. Malhotra, and D. Mathur (Singapore: Springer Nature), 1–13. doi: 10.1007/978-981-10-8390-7_1
Bharagava, R. N., Purchase, D., Saxena, G., and Mulla, S. I. (2019). “Application of metagenomic in microbial bioremediation of pollutants: from genomic to environmental cleanup”, in Microbial Diversity in the Genomic Era, eds S. Das and H. Dask (Elsevier; Academia Press), 459–477. doi: 10.1016/B978-0-12-814849-5.00026-5
Bhatt, P., and Barh, A. (2018). “Bioinformatic tools to study the soil microorganism: an in silico approach for sustainable agriculture”, in In-silico Approach for Sustainable Agriculture, eds D. K. Choudhary, M. Kumar, R. Prasad, and V. Kumar (Singapore: Springer), 169–182. doi: 10.1007/978-981-13-0347-0_10
Bhatt, P., Gangola, S., Bhandari, G., Zhang, W., Maithani, D., Mishra, S., et al. (2020a). New insights into the degradation of synthetic pollutants in contaminated environments. Chemosphere 259:128827. doi: 10.1016/j.chemosphere.2020.128827
Bhatt, P., Huang, Y., Hui, Z., and Chen, S. (2019). Insights into microbial applications for the biodegradation of pyrethroid insecticides. Front. Microbiol. 10:177. doi: 10.3389/fmicb.2019.01778
Bhatt, P., Joshi, T., Bhatt, K., Zhang, W., Huang, Y., and Chen, S. (2020b). Binding interaction of glyphosate with glyphosate oxidoreductase and C–P lyase: molecular docking and molecular dynamics simulation studies. J. Hazard. Mater. doi: 10.1016/j.jhazmat.2020.124927
Bhatt, P., Rene, E. R., Kumar, A. J., Zhang, W., and Chen, S. (2020c). Binding interaction of allethrin with esterase: bioremediation potential and mechanism. Bioresour. Technol. 315:123845. doi: 10.1016/j.biortech.2020.123845
Bhatt, P., Zhao, X., Huang, Y., Zhang, W., and Chen, S. (2021). Characterization of the role of esterases in the biodegradation of organophosphate, carbamate and pyrethroid pesticide. J. Hazard. Mater. 411:125026. doi: 10.1016/j.jhazmat.202.125026
Biswas, R., and Sarkar, A. (2018). “Omics' tools in soil microbiology-the state of art”, in Advances in Soil Microbiology: Recent Trends and Future Prospect, Microorganism for Sustainability, ed T. K. Adhya (Cham: Springer Nature), 35–64. doi: 10.1007/978-981-10-6178-3_3
Bodor, A., Bounedjoum, N., Vincze, E. G., Kis, A. E., Laczi, K., Bende, G., et al. (2020). Challenges of unculturable bacteria: environmental perspectives. Rev. Environ. Sci. Biotechnol. 19, 1–22. doi: 10.1007/s11157-020-09522-4
Boll, M., Geiger, R., Jughare, M., and Schink, B. (2020). Microbial degradation of phthalates: biochemistry and environmental implications. Environ. Microbiol. Rep. 12, 3–15. doi: 10.1111/1758-2229.12787
Bouhajja, E., McGuire, M., Liles, M. R., Bataille, G., Agathos, S. N., and George, I. F. (2017). Identification of novel toluene monooxygenase genes in a hydrocarbon-polluted sediments using sequence- and function-based screening of metagenomic libraries. Appl. Microbiol. Biotechnol. 101, 797–808. doi: 10.1007/s00253-016-7934-5
Bragg, L., and Tyson, G. (2014). “Metagenomics using next-generation sequencing,” in Environmental Microbiology Methods and Protocols, eds I. T. Paulsen and A. J. Holmes (Totowa, NJ: Humana Press), 183–201. doi: 10.1007/978-1-62703-712-9_15
Brown, G. R., Hem, V., and Katz, K. S. (2015). Gene: a gene-centered information resources at NCBI. Nucleic Acids Res. 43, D36–D42. doi: 10.1093/nar/gku1055
Brown, S. P., Callaham, M. A., Oliver, A. K., and Jumpponen, A. (2013). Deep ion torrent sequencing identifies soil fungal community shifts after frequent prescribed fires in a southeastern US forest ecosystem. FEMS Microbiol. Ecol. 86, 557–566. doi: 10.1111/1574-6941.12181
Burgos-Aceves, M. A., Cohen, A., Smith, Y., and Faggio, C. (2018). MicroRNAs and their role on fish oxidative stress during xenobiotic environmental exposure. Ecotoxicol. Environ. Saf. 148, 995–1000. doi: 10.1016/j.ecoenv.2017.12.001
Burns, C. J., and Pastoor, T. P. (2018). Pyrethroid epidemiology: a quality based review. Crit. Rev. Toxicol. 48, 297–311. doi: 10.1080/10408444.2017.1423463
Caporaso, J. G., Lauber, C. L., and Walters, W. A. (2012). Ultra-high-throughtput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Capsi, R., Billington, R., and Ferrer, L. (2016). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathways/genome databases. Nucleic Acids Res. 44, D471–D480. doi: 10.1093/nar/gkv1164
Carbajosa, G., Trigo, A., Valencia, A., and Cases, I. (2009). Bionemo: molecular information on biodegradation on the metabolism. Nucleic Acids Res. 37, D598–D602. doi: 10.1093/nar/gkn864
Castrejon-Godinez, M. L., Ortiz-Hernandez, M. L., Salazar, E., Encaracion, S., Mussali-Galante, P., Tovar-Sanchez, E., et al. (2019). Transcriptional analysis reveals the metabolic state of Burkholderia zhejiangensis CEIB S4-3 during methyl parathion degradation. Peer J. 7:e6822. doi: 10.7717/peerj.6822
Catron, T. R., Gaballah, S., and Tal, T. (2019). Using Zebra fish to investigate interactions between xenobiotics and microbiota. Curr. Pharmacol. Rep. 5, 468–480. doi: 10.1007/s40495-019-00203-7
Chakka, D., Gudla, R., Madikonda, A. K., Pandeeti, E. V. P., Partasarathy, S., Nandavaram, A., et al. (2015). The organophosphate degradation (opd) island-born esterase-induced metabolic diversion in Escherichia coli and its influence on p-nitrophenol degradation. J. Biol. Chem. 290, 29920–29930. doi: 10.1074/jbc.M115.661249
Chakraborthy, J., and Das, S. (2016). Molecular perspectives and recent advances in microbial remediation of persistent organic pollutants. Environ. Sci. Pollut. Res. 23, 16883–16903. doi: 10.1007/s11356-016-6887-7
Chandran, H., Meena, M., and Sharma, K. (2020). Microbial biodiversity and bioremediation assessment through omics approaches. Front. Environ. Chem. 1:570326. doi: 10.3389/fenvc.2020.570326
Chen, S., Chang, C., Deng, Y., An, S., Dong, Y. H., Zhou, J., et al. (2014). Fenpropathrin biodegradation pathway in Bacillus sp. DG-02 and its potential for bioremediation of pyrethroid-contaminated soils. J. Agric. Food Chem. 62, 2147–2157. doi: 10.1021/jf404908j
Chen, S., Deng, Y., Chang, C., Lee, J., Cheng, Y., Cui, Z., et al. (2015). Pathway and kinetics of cyhalothrin biodegradation by Bacillus thuringiensis strain ZS-19. Sci. Rep. 5:8784. doi: 10.1038/srep08784
Chen, S., Dong, Y. H., Chang, C., Deng, Y., Zhang, X. F., Zhong, G., et al. (2013). Characterization of a novel cyfluthrin-degrading bacterial strain Brevibacteriu maureum and its biochemical degradation pathway. Bioresour. Technol. 132, 16–23. doi: 10.1016/j.biortech.2013.01.002
Chen, S., Geng, P., Xiao, Y., and Hu, M. (2012). Bioremediation of β-cypermethrin and 3-phenoxybenzaldehyde contaminated soils using Streptomyces aureus HP-S-01. Appl. Microbiol. Biotechnol. 94, 505–515. doi: 10.1007/s00253-011-3640-5
Chen, S., and Zhan, H. (2019). “Biodegradation of synthetic pyrethroids insecticides,” in Microbial Metabolism of Xenobiotic Compounds, ed P. K. Arora (Springer Nature), 229–244. doi: 10.1007/978-981-13-7462-3_11
Chen, Z., Yin, H., Peng, H., Lu, G., Liu, Z., and Dang, Z. (2019). Identification of novel pathways for biotransformation of tertrabromobisphenol A by Phanerochaete chrysosporium combined with mechanism analysis at proteome level. Sci. Total Environ. 659, 1352–1362. doi: 10.1016/j.scitotenv.2018.12.446
Cheng, Y., Zang, H., Wang, H., Li, D., and Li, C. (2018). Global transcriptomic analysis of Rhocococcus erythopolis D310-1 in responding to chlorimuron-ethyl. Ecotoxicol. Environ. Saf. 157, 111–120. doi: 10.1016/j.ecoenv.2018.03.074
Colatriano, D., Ramachandran, A., Yergeau, E., Maranger, R., Gelinas, Y., and Walsh, D. A. (2015). Metaproteomics of aquatic microbial communities in a deep and stratified estuary. Proteomics 15, 3566–3579. doi: 10.1002/pmic.201500079
Colquhoun, D. R., Hartmann, E. M., and Halden, R. U. (2012). Proteomic profiling of the dioxin degrading bacterium Sphingomonas wittichii RW. J. Biomed. Biotechnol. 9:408690. doi: 10.1155/2012/408690
Cooper, L. A., Stringer, A. M., and Wade, J. T. (2018). Determining the specificity of cascade binding interference, and primed adaptation in-vivo in the Escherichia coli type IE CRISPR-Cas system. mBio 9:e02100-17. doi: 10.1128/mBio.02100-17
Crinnion, W. J. (2010). Toxic effects of the easily avoidable phthalates and parabens. Alternative Med. Rev. 15, 190–196.
Crump, B. C., Wojahn, J. M., Tomas, F., and Mueller, R. S. (2018). Metatranscriptomics and amplicon sequencing reveal mutualism in seagrass microiome. Front. Miocrobiol. 9:388. doi: 10.3389/fmicb.2018.00388
Cuozzo, S. A., Sineli, P. E., Davila Costa, J., and Tortella, G. (2017). Streptomyces sp. is a powerful biotechnological tool for the biodegradation of HCH isomers: biochemical and molecular basis. Crit. Rev. Biotechnol. 38, 719–728. doi: 10.1080/07388551.2017.1398133
Cycoń, M., Mrozik, A., and Piotrowska-Seget, Z. (2017). Bioaugmentation as a strategy for the remediation of pesticide-polluted soil: a review. Chemosphere 171, 52–71. doi: 10.1016/j.chemosphere.2016.12.129
Cycoń, M., and Piotrowska-Seget, Z. (2016). Pyrethroid-degrading microorganisms and their potential for the bioremediation of contaminated soils: a review. Front. Microbiol. 7:1463. doi: 10.3389/fmicb.2016.01463
Dangi, A. K., Sharma, B., Hill, R. T., and Shukla, P. (2018). Bioremediation through microbes: systems biology and metabolic engineering approach. Crit. Rev. Biotechnol. 39, 79–98. doi: 10.1080/07388551.2018.1500997
Delegan, Y. A., Valentovich, L. N., Shafieva, S. M., Ganbarov, K. G., Filonov, A. E., and Vainstein, M. B. (2019). Characterization and genome analysis of highly efficient thermotolerant oil-degrading bacterium Gordonia sp. 1D. Folia Microbiol. 64, 41–48. doi: 10.1007/s12223-018-0623-2
d'Errico, G., Aloj, V., Flematti, G. R., Sivasithamparam, K., and Worth, C. M. (2020). Metabolites of Drechslera sp. endophyte with potential as biocontrol and bioremediation agent. Nat. Prod. Res. 11, 1–9. doi: 10.1080/14786419.2020.1737058
Desai, C., Pathak, H., and Madawar, D. (2009). Advances in molecular and “omics” technologies to gauge microbial communities and bioremediation at xenobiotic/anthropogen contaminated sites. Bioresour. Technol. 101, 1558–1569. doi: 10.1016/j.biortech.2009.10.080
Devarapalli, P., and Kumavnath, R. N. (2015). “Metagenomics-a technological drift in bioremediation,” in Advances in Bioremediation of Wastewater and Polluted Soil, ed N. Shiomi (Rijeka: Intech Open), 73–91. doi: 10.5772/60749
Dhakal, K., Gadupadi, G. S., and Robertson, L. W. (2017). Sources and toxicities of phenolic polychlorinated biphenyls (OH-PCBs). PCBs Risk Eval. Environ. Protect. 25, 16277–16290. doi: 10.1007/s11356-017-9694-x
Diaz, E. (2004). Bacterial degradation of aromatic pollutants: a paradigm of metabolic versatility. Int. Microbiol. 7, 173–180.
Ding, J., Yu, Z., Wang, H., Jian, H., Leng, H., and Xiao, X. (2017). Microbial community structure of deep sea hydrothermal vents on the ultraslow spreading southwest Indian ridge. Front. Microbiol. 8:1012. doi: 10.3389/fmicb.2017.01012
Dinka, D. D. (2018). Environmental xenobiotics and their adverse health impacts-a general review. J. Environ. Poll. Human Health. 6, 77–88. doi: 10.12691/jephh-6-3-1
Dovrak, P., Nikel, P. I., Damborsky, J., and de Lorenzo, V. (2017). Bioremediation 3.0: engineering pollutant-removing bacteria in the times of systemic biology. Biotechnol. Adv. 35, 845–866. doi: 10.1016/j.biotechadv.2017.08.001
Durand, S., Sancelme, M., Besse-Hoggan, P., and Combourieu, B. (2010). Biodegradation pathway of mesotrione: complementaries of NMR, LC-NMR, and LC-MS for qualitative and quantitative metabolomic profiling. Chemosphere 81, 372–380. doi: 10.1016/j.chemosphere.2010.07.017
Dutta, T. K., Selifonov, S. A., and Gunsalus, I. C. (1998). Oxidation of methyl-substituted napthalenes: pathways in a versatile Sphingomonas paucimobilis strain. Appl. Environ. Microbiol. 64, 1884–1889. doi: 10.1128/AEM.64.5.1884-1889.1998
Ellis, L. B. M., Roe, D., and Wackett, L. P. (2006). The University of Minnesota biocatalyst/biodegradation database: the first decade. Nucleic Acid Res. 34, D517–D521. doi: 10.1093/nar/gkj076
Embrandiri, A., Kiyasudeen, S. K., Rupani, P. F., and Ibrahim, M. H. (2016). “Environmental xenobiotics and its effect on natural ecosystem”, in Plant Response to Xenobiotic, eds A. Singh, S. M. Prasad, and R. P. Singh (Singapore: Springer), 1–18. doi: 10.1007/978-981-10-2860-1_1
Endo, R., Kamakura, M., Miyauchi, K., Fukuda, M., Ohtsubo, Y., Tsuda, M., et al. (2005). Identification and characterization of genes involved in the downstream degradation pathway of γ-hexachlorocyclohexane in Sphingomonas paucimobilus UT26. J. Bacteriol. 187, 847–853. doi: 10.1128/JB.187.3.847-853.2005
Farnandez, M., Duque, E., Pizarro-Tobias, P., van Dillewijn, P., Wittich, R. M., and Ramos, J. L. (2009). Microbial responses to xenobiotic compounds. Identification of genes that allow Pseudomonas putida KT2440 to cope with 2,4,6-trinitrotoluene. Microb. Biotechnol. 2, 287–294. doi: 10.1111/j.1751-7915.2009.00085.x
Fida, T. T., Moreno-Forero, S. K., Breugelmans, P., Heipieper, H. J., Roling, W. F. M., and Springael, D. (2017). Physiological and transcriptome response of the polycyclic aromatic hydrocarbon degrading Novosphingobium sp. LH128 after inoculation in soil. Environ. Sci. Technol. 51, 1570–1579. doi: 10.1021/acs.est.6b03822
Franzosa, E. A., Hsu, T., Sirota-Madi, A., Shafquat, A., Abu-Ali, G., Morgan, X. C., et al. (2015). Sequencing and beyond: integrating molecular ‘omics for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372. doi: 10.1038/nrmicro3451
French, K. E., Zhou, Z., and Terry, N. (2020). Horizontal ‘gene drives’ harness indigenous bacteria for bioremediation. Sci. Rep. 10:15091. doi: 10.1038/s41598-020-72138-9
Fuks, G., Elgert, M., Amir, A., Zeisel, A., Turnbaugh, P. J., Soen, Y., et al. (2018). Combining 16S rRNA gene variable regions enables high resolution microbial community profiling. Microbiome 6:17. doi: 10.1186/s40168-017-0396-x
Fulekar, M. H., and Geetha, M. (2008). Bioremediation of chloropyrifos by Pseudomonas aeruginosa using scale up technique. J. Appl. Biosci. 12, 657–660.
Gammon, D. W., Liu, Z., Chandrasekaran, A., El-Naggar, S. F., Kuryshev, Y. A., and Jackson, S. (2019). Pyrethroid neurotoxicity studies with bifenthrin indicate a mixed type I/II mode of action. Pest Manage. Sci. 75, 1190–1197. doi: 10.1002/ps.5300
Garrido-Sanz, D., Manzano, J., Martin, M., Redondo-Nieto, M., and Rivillia, R. (2018). Metagenomic analysis of a biphenyl-degrading soil bacterial consortium reveals the metabolic roles of specific populations. Front. Microbiol. 9:232. doi: 10.3389/fmicb.2018.00232
Geueke, B., Garg, N., Ghosh, S., Fleischmann, T., Holliger, C., Lal, R., et al. (2013). Metabolomics of hexachlorocyclohexane (HCH) transformation: ratio of LinA to LinB determines metabolic fate of HCH isomers. Environ. Microbiol. 15, 1040–1049. doi: 10.1111/1462-2920.12009
Ghosal, D., Ghosh, S., Dutta, T. K., and Ahn, Y. (2016). Current state of knowledge in microbial degradation of polycyclic aromatic hydrocarbons (PAHs): a review. Front. Microbiol. 7:1369. doi: 10.3389/fmicb.2016.01369
Gillbride, K. A., Lee, D. Y., and Beaudette, L. A. (2006). Molecular techniques in wastewater: understanding microbial communities, detecting pathogens, and real-time process control. J. Microbiol. Methods 66, 1–20. doi: 10.1016/j.mimet.2006.02.016
Gilliespie, I. M. M., and Philp, J. C. (2013). Bioremediation, an environmental remediation technology for the bioeconoomy. Trends Biotechnol. 31, 329–332. doi: 10.1016/j.tibtech.2013.01.015
Giri, K., Rawat, A. P., Rawat, M., and Rai, J. (2014). Biodegradation of hexachlorocyclohexane by two species of bacillus isolated from contaminated soil. Chem. Ecol. 30, 97–109. doi: 10.1080/02757540.2013.844795
Godheja, J., Shekhar, S. K., and Modi, D. R. (2014). Advances in molecular biology approaches to guage microbial communities and bioremediation at contaminated sites. Int. J. Environ. Bioremediate. Biodegrade. 2, 167–177. doi: 10.12691/ijebb-2-4-4
Godheja, J., Shekhar, S. K., Siddiqui, S. A., and Modi, D. R. (2016). Xenobiotic compounds present in soil and water: a review on remediation strategies. J. Environ. Anal. Toxicol. 6:2161. doi: 10.4172/2161-0525.1000392
Greene, A. C. (2018). CRISPR-based antibacterials: transforming bacterial defense into offense. Trends Biotechnol. 36, 127–130. doi: 10.1016/j.tibtech.2017.10.021
Griffiths, W. (2007). Metabolomics, Metabonomics and Metabolite Profiling. Cambridge: Royal Society of Chemistry. doi: 10.1039/9781847558107
Grob, C., Taubert, M., Howat, A. M., Burns, O. J., Dixon, J. L., Richnow, H. H., et al. (2015). Combining metagenomics with metaproteomics and stable isotope probing reveales metabolic pathways used by a naturally occurring marine methylotroph. Environ. Microbiol. 17, 4007–4018. doi: 10.1111/1462-2920.12935
Gu, Q., Wu, Q., Zhang, J., Guo, W., Ding, Y., Wang, J., et al. (2018). Isolation and transcriptome analysis of phenol-degrading bacterium from carbon-sand filters in full scale drinking water treatment plant. Front. Microbiol. 9:2162. doi: 10.3389/fmicb.2018.02162
Gursoy, S., and Can, M. (2019). Hypervariable regions in 16S rRNA genes for taxonomic classification. Se. Eur. J. Soft Comput. 8, 23–26. doi: 10.21533/scjournal.v8i1.171
Gutierrez, D. B., Gant-Branum, R. L., Romer, C. E., Farrow, M. A., Allen, J. L., Dahal, N., et al. (2018). An integrated, high-throughput strategy for “multi-omic” system level analysis. J. Proteome Res. 17, 3396–3408. doi: 10.1021/acs.jproteome.8b00302
Gutleben, J., Chaib De Mares, M., van Elsas, J. D., Smidt, H., Overmann, J., and Sipkema, D. (2018). The multi-omics promise in context: from sequence to microbial isolate. Crit. Rev. Microbiol. 44, 212–229. doi: 10.1080/1040841X.2017.1332003
Handelsman, J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68, 669–685. doi: 10.1128/MMBR.68.4.669-685.2004
Hashmi, M. Z., Kumar, V., and Verma, A. (2017). Xenobiotics in the Soil Environment: Monitoring, Toxicity and Management. Cham: Springer International Publishing. doi: 10.1007/978-3-319-47744-2
He, Y., Feng, X., Feng, J., Zhang, Y., and Xiao, X. (2015). Metagenome and metatrancriptome revealed a highly active and intensive sulfur cycle in an oil-immersed hydrothermal chimney in Guaymas Basin. Front. Microbiol. 6:1236. doi: 10.3389/fmicb.2015.01236
He, Z., Deng, Y., van Nostrand, J. D., Xu, M., Hemme, L. H., Tu, Q., et al. (2010). GeoChip 3.0 as a high-throughput tool for analyzing microbial community composition, structure and functional activity. ISME J. 4, 1167–1179. doi: 10.1038/ismej.2010.46
Hegedus, B., Kos, P. B., Bende, G., Bounedjoum, N., Maroti, G., Laczi, K., et al. (2018). Starvation and xenobiotic- related transcriptomic responses of the sulfanilic acid-degrading bacterium Novosphingobium resinvorum SA1. Appl. Microbiol. Biotechnol. 102, 305–318. doi: 10.1007/s00253-017-8553-5
Hettich, R. L., Pan, C., Chourey, K., and Giannone, R. J. (2013). Metaproteomics: Harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal. Chem. 85, 4203–4214. doi: 10.1021/ac303053e
Hidalgo, K. J., Teramoto, E. H., Soriano, A. U., Valoni, E., Baessa, M. P., Richnow, H. H., et al. (2020). Taxonomic and functional diversity of the microbiome in a jet fuel contaminated site as revealed by combined application of in situ microcosms with metagenomic analysis. Sci. Total Environ. 708:135152. doi: 10.1016/j.scitotenv.2019.135152
Hong, Y. H., Deng, M. C., Xu, X. M., Wu, C. F., Xiao, X., and Zhu, Q. (2016). Characterization of the transcriptome of Achromobacter sp. HZ01 with the outstanding hydrocarbon-degrading ability. Gene 584, 185–194. doi: 10.1016/j.gene.2016.02.032
Huang, Y., Lin, Z., Zhang, W., Pang, S., Bhatt, P., Rene, E. R., et al. (2020). New insights into the microbial degradation of D-cyphenothrin in contaminated water/soil environments. Microorganisms 8:473. doi: 10.3390/microorganisms8040473
Huang, Y., Zhang, W., Pang, S., Chen, J., Bhatt, P., Mishra, S., et al. (2021). Insights into the microbial degradation and catalytic mechanisms of chlorpyrifos. Environ. Res. 194:110660. doi: 10.1016/j.envres.2020.110660
Hussain, I., Aleti, G., Naidu, R., Puschenreiter, M., Mahmood, Q., and Rahman, M. M. (2018). Microbe and plant assisted remediation of organic xenobiotics and its enhancement by genetically modified organisms and recombinant technology: a review. Sci. Total Environ. 628, 1582–1599. doi: 10.1016/j.scitotenv.2018.02.037
Iovdijova, A., and Bencko, V. (2010). Potential risk of exposure to selected xenobiotic residues and their fate in the food chain-part I: classification of xenobiotics. Annl. Agric. Environ. Med. 17, 183–192.
Jacob, J., and Cherian, J. (2013). Review of environmental and human exposure to persistent organic pollutant. Asian Social Sci. 9:11. doi: 10.5539/ass.v9n11P107
Jagadeesh, D. S., Kannegundla, U., and Reddy, R. K. (2017). Application of proteomic tools in food quality and safety. Adv. Anim. Vet. Sci. 5, 213–225. doi: 10.17582/journal.aavs/2017/5.5.213.225
Jaiswal, S., and Shukla, P. (2020). Alternative strategies for microbial remediation of pollutants via synthetic biology. Front. Microbiol. 11:808. doi: 10.3389/fmicb.2020.00808
Jaiswal, S., Singh, D. K., and Shukla, P. (2019). Gene editing and systems biology tools for pesticide bioremediation: a review. Front. Microiol. 10:87. doi: 10.3389/fmicb.2019.00087
Jalowiecki, L., Chojniak, J. M., Dorgeloh, E., Hegedusova, B., Ejhed, H., Magner, J., et al. (2016). Microbial community profiles in wastewaters from onsite wastewater treatment system technology. PLoS ONE 11:e0147725. doi: 10.1371/journal.pone.0147725
Janssen, D. B., and Stucki, G. (2020). Perspectives of genetically engineered microbes for groundwater bioremediation. Environ. Sci. Proc. Imp. 22:487. doi: 10.1039/C9EM00601J
Jayaraj, R., Megha, P., and Sreedev, P. (2016). Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip. Toxicol. 9, 90–100. doi: 10.1515/intox-2016-0012
Jeffries, T. C., Rayu, S., Nielsen, U. N., Lai, K., Ijaz, A., Nazaries, L., et al. (2019). Metagenomics function potential predicts functional rates of a model organophosphorus xenobiotic in pesticide-contaminated soil. Front. Microbiol. 9:147. doi: 10.3389/fmicb.2018.00147
Jehmlich, N., Kleinsteuber, S., Vogt, C., Benndorf, D., Harms, H., Schmidt, F., et al. (2010). Phylogenetic and proteomic analysis of an anaerobic toluene-degrading community. J. Appl. Microbiol. 109, 1937–1945. doi: 10.1111/j.1365-2672.2010.04823.x
Jing, R., Fusi, S., and Kjellerup, B. V. (2018). Remediation of polychlorinated bisphenyls (PCBs) in contaminated soils and sediments: state of knowledge and perspectives. Front. Environ. Sci. 6:79. doi: 10.3389/fenvs.2018.00079
Johnson, C. H., Patterson, A. D., Idle, J. R., and Gonzalez, F. J. (2011). Xenobiotic metabolomics: major impact on the metabolome. Annu. Rev. Pharmacol. Toxicol. 52, 37–56. doi: 10.1146/annurev-pharmtox-010611-134748
Johnson, S. J., Spakowicz, D. J., Hong, B.-Y., Petersen, L., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for sepecies and strain-levl microbiome analysis. Nat. Commun. 10:5029. doi: 10.1038/s41467-019-13036-1
Jones, O. A. H., Sdepanian, S., Lofts, S., Svendsen, C., Spurgeon, D. J., Maguire, M. L., et al. (2014). Metabolomic analysis of soil communities can be used for pollution assessment. Environ. Toxicol. Chem. 33, 61–64. doi: 10.1002/etc.2418
Junghare, M., Spiteller, D., and Schink, B. (2019). Anaerobic degradation of xenobiotic isophthalate by the fermenting bacterium Syntrophorhabdus aromaticivorans. ISME J. 13, 1252–1258. doi: 10.1038/s41396-019-0348-5
Kale, N. S., Haug, K., Conesa, P., Jayseelam, K., Moreno, P., Rocca-Serra, P., et al. (2016). MetaboLights: an open-access database repository for metabolomics data. Curr. Protoc. Bioinform. 53:14. doi: 10.1002/0471250953.bi1413s53
Kanehisa, M., Furumichi, M., and Tanabe, M. (2017). KEGG: new perspectives on genome, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kaul, S., Sharma, T., and Dhar, M. K. (2016). “Omics” tools for better understanding the plant endophyte interactions. Front. Plant Sci. 7:955. doi: 10.3389/fpls.2016.00955
Kaur, H., and Kaur, G. (2016). Application of ligninolytic potentials of a white-rot fungus Ganoderma lucidum for degradation of lindane. Environ. Monit. Assess. 188:588. doi: 10.1007/s10661-016-5606-7
Keller, M., and Hettich, R. (2009). Environmental proteomics: a paradigm shift in characterizing microbial activities at the molecular level. Microbiol. Mol. Biol. Rev. 73, 62–70. doi: 10.1128/MMBR.00028-08
Kessner, D., Chambers, M., and Burke, R. (2008). ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536. doi: 10.1093/bioinformatics/btn323
Keum, Y. S., Seo, J. S., Li, Q. X., and Kim, J. H. (2008). Comparative metabolomic analysis of Sinorhizobium sp. C4 during the degradation of phenanthrene. Appl. Microbiol. Biotechnol. 80, 863–872. doi: 10.1007/s00253-008-1581-4
Kim, K. H., Jahan, S. A., Kabir, E., and Brown, R. C. B. (2013). A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effect. Environ. Int. 60, 71–80. doi: 10.1016/j.envint.2013.07.019
Kim, S. I., Song, S. Y., Kim, K. W., Ho, E. M., and Oh, K. H. (2003). Proteomic analysis of the benzoate degradation pathway in Acinetobacter sp. KS-1. Res. Microbiol. 10, 679–703. doi: 10.1016/j.resmic.2003.09.003
Kim, S. J., Jones, R. C., and Cha, C. J. (2004). Identification of proteins induced by polycyclic aromatic hydrocarbon in Mycobacterium vanbaalenii PYR-1 using two-dimensional polyacrylamide gel electrophoresis and denovo sequencing methods. Proteomics 4, 3899–3908. doi: 10.1002/pmic.200400872
Kiyohara, H., Torigoe, S., Kaida, N., Asaki, T., Iida, T., Hayashi, H., et al. (1994). Cloning and characterization of a chromosomal gene cluster, pah, that encodes the upper pathway for phenanthrene and naphthalene utilization by Pseudomonas putida OUS82. J. Bacteriol. 176, 2439–2443. doi: 10.1128/JB.176.8.2439-2443.1994
Knief, C. (2014). Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front. Plant Sci. 5:216. doi: 10.3389/fpls.2014.00216
Krivobok, S., Kuony, S., Meyer, C., and Louwagie, M. (2003). Identification of pyrene-induced proteins in Mycobacterium sp. strain 6PY1: evidence for two-ring hydroxylating dioxygenases. J. Bacteriol. 185, 3828–3841. doi: 10.1128/JB.185.13.3828-3841.2003
Kumar, R., Kumar, D., Pandya, L., Pandit, P. R., Patel, Z., Bhairappanavar, S., et al. (2020). “Gene-targeted metagenomics approach for the degradation of organic pollutants,” in Emerging Technologies in Environmental Bioremediation, eds M. P. Shah, S. Rodriguez-Couto, and S. S. Sengor (Amsterdam: Elsevier), 257–273. doi: 10.1016/B978-0-12-819860-5.00010-9
Kumar, S. S., Shantkriti, S., Muruganandham, T., Murugesh, E., Rane, N., and Govindwar, S. P. (2016). Bioinformatics aided microbial approach for bioremediation of wastewater containing textile dyes. Ecol. Info. 31, 112–121. doi: 10.1016/j.ecoinf.2015.12.001
Kuzyakov, Y., and Blagodatskaya, E. (2015). Microbial hotspots and hot moments in soil, concept and review. Soil Biol. Biochem. 183, 184–199. doi: 10.1016/j.soilbio.2015.01.025
Lallas, P. L. (2001). The Stockholm convention on persistent organic pollutant. Am. J. Int. Law 95, 692–708. doi: 10.2307/2668517
Lam, K. N., Cheng, J., Engel, K., Neufeld, J. D., and Charles, T. C. (2015). Current and future resources for functional metagenomics. Front. Microbiol. 6:1196. doi: 10.3389/fmicb.2015.01196
Lee, S. Y., Kim, G. H., Yun, S. H., Choi, C. W., Yi, Y. S., and Kim, J. (2016a). Proteogenomic characterization of monocyclic aromatic hydrocarbon degrading bacterium Burkholderia sp. K24. PLoS ONE 11:e0154233. doi: 10.1371/journal.pone.0154233
Lee, S. Y., Sekhon, S. S., Ban, Y. H., Ahn, J. Y., Ko, J. H., Lee, L., et al. (2016b). Proteomic analysis of polycyclic aromatic hydrocarbons (PAHs) degradation and detoxification in Sphingobium chungbukense DJ77. J. Microbiol. Biotechnol. 26, 1943–1950. doi: 10.4014/jmb.1606.06005
Li, C., Ma, Y., Mi, Z., Huo, R., Zhou, T., Hai, H., et al. (2018). Screening for Lactobacillus plantarum strains that posses organophosphorus pesticide-degrading activity and metabolomic analysis of phorate degradation. Front. Microbiol. 9:2048. doi: 10.3389/fmicb.2018.02048
Li, J., Wu, C., Chen, S., Lu, Q., Shim, H., Huang, X., et al. (2020). Enriching indigenous microbial consortia as a promising strategy for xenobiotic's cleanup. J. Clean. Prod. 261:121234. doi: 10.1016/j.jclepro.2020.121234
Li, M., Jain, S., Baker, B. J., Taylor, C., and Dick, G. J. (2014). Novel hydrocarbon monooxygenase genes in the metatrancriptome of natural deep-sea hydrocarbon plume. Environ. Microbiol. 16, 60–71. doi: 10.1111/1462-2920.12182
Li, S. H., Yu, X. Y., and Park, D. J. (2015). Rhodococcus soli sp. nov., an actinobacterium isolated from soil using a resuscitative technique. Antonie Van Leeuwenhoek 107, 357–366. doi: 10.1007/s10482-014-0334-x
Li, X., Qu, C., Bian, Y., Gu, C., Jiang, X., and Song, Y. (2019). New insights into responses of soil microorganisms to polycyclic aromatic hydrocarbon stress by combining enzyme activity and sequencing analysis with metabolomics. Environ. Pollut. 255:113312. doi: 10.1016/j.envpol.2019.113312
Lima-Morales, D., Jauregui, R., Camarinha-Silva, A., Geffers, R., Pieper, D. H., and Vilchez-Vergas, R. (2016). Linking microbial community and catabolic gene structures during the adaptation of three contaminated soils under continuous long-term polluted stress. Appl. Environ. Microbiol. 82, 2227–2237. doi: 10.1128/AEM.03482-15
Lin, Z., Pang, S., Zhang, W., Mishra, S., Bhatt, P., and Chen, S. (2020). Degradation of acephate and its intermediate methamidophos: mechanisms and biochemical pathways. Front. Microbiol. 11:2045. doi: 10.3389/fmicb.2020.02045
Lindon, J. C., Nicholson, J. K., and Holmes, E. (2006). The Handbook of Metabonomics and Metabolomics. London: Elsevier Science.
Liu, L., Bilal, M., Duan, X., and Iqbal, H. M. N. (2019). Mitigation of environmental pollution by genetically engineered bacteria-current challenges and future perspectives. Sci. Total. Environ. 667, 444–454. doi: 10.1016/j.scitotenv.2019.02.390
Liu, L., Li, Y., and Li, S. (2012). Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012:251364. doi: 10.1155/2012/251364
Liu, S., Gu, C., Dang, Z., and Liang, X. (2017). Comparitive proteomics reveal the mechanism of Tween 80 enhanced phenantherene biodegradation by Sphingomonas sp. GY2B. Ecotoxicol. Environ. Saf. 137, 256–264. doi: 10.1016/j.ecoenv.2016.12.015
Lourenco, A., Ferreira, A., Veiga, N., Machado, I., Pereira, M. O., and Azevedo, N. F. (2012). BiofOmics: a webplatform for the systematic and standardized collection of high-throughput biofilmdata. PLoS ONE 7:e39960. doi: 10.1371/journal.pone.0039960
Lu, H., Giordano, F., and Ning, Z. (2016). Oxford nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 14, 266–279. doi: 10.1016/j.gpb.2016.05.004
Lu, Z., Gan, J., Cui, X., Moreno, L. D., and Lin, K. (2019). Understanding the bioavailability of pyrethroids in the aquatic environment using chemical approaches. Environ. Int. 129, 194–207. doi: 10.1016/j.envint.2019.05.035
Lueders, T. (2015). “DNA-and RNA based stable isotope probing of hydrocarbon degraders”, in Hydrocarbon and Lipid Microbiology Protocols, eds T. J. McGenity, K. N. Timmis, and B. Nogales (New York, NY: Humana Press), 181–197. doi: 10.1007/8623_2015_74
Luo, C., Tsementzi, D., and Kyrpides, N. (2012). Direct comparison of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 7:e30087. doi: 10.1371/journal.pone.0030087
Machado, L., Leite, D. C. A., Rachid, C. T. C. C., Paes J. E., Martins, E., Peixoto, R., and Rosado, A. (2019). Tracking mangrove oil bioremediation approaches and bacterial diversity at different depths in an in situ mesocosms system. Front. Microbiol. 10:2107. doi: 10.3389/fmicb.2019.02107
Mahmoud, Y. A. G. (2016). Advancement in bioremediation process: a mini review. Int. J. Environ. Tech. Sci. 3, 83–94.
Malik, S., Beer, M., Megharaj, M., and Naidu, R. (2008). The use of molecular techniques to characterize the microbial communities in contaminated soil and water. Environ. Int. 34, 265–276. doi: 10.1016/j.envint.2007.09.001
Malla, M. A., Dubey, A., Yadav, S., Kumar, A., Hashem, A., and Abd-Allah, E. F. (2018). Understanding and designing the strategies for the microbe-mediated remediation of environmental contaminants using omics approaches. Front. Microbiol. 9:1132. doi: 10.3389/fmicb.2018.01132
Mallick, H., Franzosa, E. A., Mclver, L. J., Banerjee, S., Sirota-Madi, A., Kostic, A. D., et al. (2019). Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun. 10:3136. doi: 10.1038/s41467-019-10927-1
Mao, X., Trembley, J., Yu, K., Tringe, S. G., and Alvarez-Cohen, L. (2019). Structural dynamics and transcriptomic analysis of Dehalococcoides mccartyi within a TCE-dechlorinating community in a completely mixed flow. Water Res. 158, 146–156. doi: 10.1016/j.watres.2019.04.038
Marcelino, V. R., Irinyi, L., Eden, J. S., Meyer, W., Holmes, E. C., and Sorrell, T. C. (2019). Metatranscriptomics as a tool to identify fungal species and subspecies in mixed communities- a proof of concept under laboratory conditions. IMA Fungus 10:12. doi: 10.1186/s43008-019-0012-8
Marco, D. E., and Abram, F. (2019). Editorial: using genomics, metagenomics and other “omics” to assess valuable microbial ecosystem services and novel biotechnological applications. Front. Microbiol. 10:151. doi: 10.3389/fmicb.2019.00151
Maurya, P. K. (2016). Bioaccumulation of xenobiotics compound of pesticides in riverine system and its control technique: a critical review. J. Ind. Poll. Control 32, 580–594.
Mazzoli, R., Pessione, E., Giuffrida, M. G., Fattori, P., Barello, C., Giunta, C., et al. (2007). Degradation of aromatic compounds by Acinetobacter radioresistens S13: growth characteristics on single substrates and mixtures. Arch. Microbiol. 188, 55–68. doi: 10.1007/s00203-007-0223-z
McGrath, K. C., Thomas-Hall, S. R., Cheng, C. T., Leo, L., Alexa, A., Schmidt, S., et al. (2008). Isolation and analysis of mRNA from environmental microbial communities. J. Microbiol. Methods 75, 172–176. doi: 10.1016/j.mimet.2008.05.019
Megharaj, M., and Naidu, R. (2010). Soil and brown field bioremediation. Microbial. Biotechnol. 10, 1244–1249. doi: 10.1111/1751-7915.12840
Mishra, S., Zhang, W., Lin, Z., Pang, S., Huang, Y., Bhatt, P., et al. (2020). Carbofuran toxicity and its microbial degradation in contaminated environments. Chemosphere 259:127429. doi: 10.1016/j.chemosphere.2020.127419
Mishra, V. K., Singh, G., and Shukla, R. (2019). Impact of xenobiotics under a changing climatic scenario. Clim. Change Agri. Ecosys. 2, 133–151. doi: 10.1016/B978-0-12-816483-9.00006-2
Misra, B. B., Langefeld, C. D., Olivier, M., and Cox, L.A. (2018). Integrated omics: tools, advances and future approaches. J. Mol. Endocrinol. 62, R21–R45. doi: 10.1530/JME-18-0055
Mocalli, S., and Benedetti, A. (2010). Exploring research frontiers in microbiology: the challenge of metagenomics in soil microbiology. Res. Microbiol. 161, 497–505. doi: 10.1016/j.resmic.2010.04.010
Moniruzzaman, M., Wurch, L. L., Alexander, H., Dyhrman, S. T., Gobler, C. J., and Wilhelm, S. W. (2017). Virus-host relationships of marine single-celled eukaryotes resolved from metatranscriptomics. Nat. Commun. 8:16054. doi: 10.1038/ncomms16054
Moody, J. D., Freeman, J. P., and Cerniglia, C. E. (2005). Degradation of benz[a]anthracene by Microbacterium vanbaalenii strain PYR-1. Biodegradation 16, 513–526. doi: 10.1007/s10532-004-7217-1
Moriya, Y., Shigemizu, D., Hattori, M., Tokimatsu, T., Kotera, M., Goto, S., et al. (2010). Pathpred: an enzyme-catalyzed metabolic pathway prediction server. Nucleic Acid Res. 38, W138–W143. doi: 10.1093/nar/gkq318
Nagata, Y., Kato, H., Ostsubo, Y., and Tsuda, M. (2019). Lessons from the genomes of lindane-degradation sphingomonads. Environ. Microbiol. Rep. 11, 630–644. doi: 10.1111/1758-2229.12762
Nahurira, R., Ren, I., and Song, J. (2017). Degradation of di(2-ethylhexyl) phthalate by a novel Gordonia alkanivorans strain yc-r12. Curr. Microbiol. 74, 1–11. doi: 10.1007/s00284-016-1159-9
Nakano, K., Shiroma, A., Shimoji, M., Tamotsu, H., Ashimine, N., Ohki, S., et al. (2017). Advantages of genome sequencing by long read sequencer using SMRT technology in medical area. Hum. Cell 30, 149–161. doi: 10.1007/s13577-017-0168-8
Nascimento, F. X., Hernandez, G., Glick, B. R., and Rossi, M. J. (2020). Plant growth-promoting activities and genomic analysis of the stress resistant Bacillus megaterium STB1, a bacterium of agriculture and biotechnological interest. Biotenol. Rep. 25:e00406. doi: 10.1016/j.btre.2019.e00406
Negi, G., Gangola, S., Khati, P., Kumar, G., Srivastava, A., and Sharma, A. (2016). Differential expression and characterization of cypermethrin-degrading potential proteins in Bacillus thuringiensis strain SG4. 3 Biotech 6:225. doi: 10.1007/s13205-016-0541-4
Negi, V., and Lal, R. (2017). Metagenomic analysis of a complex community present in pond sediment. J. Genomics 5, 36–47. doi: 10.7150/jgen.16685
Ngara, T. R., and Zhang, H. (2018). Recent advances in function-based metagenomic screening. Genom. Proteom. Bioinform. 6, 405–415. doi: 10.1016/j.gpb.2018.01.002
Niu, J., Rang, Z., and Zhang, C. (2016). The succession pattern of soil microbial communities and its relationship with tobacco bacterial wilt. BMC Microbiol. 16:233. doi: 10.1186/s12866-016-0845-x
Nkongolo, K. K., and Kotha, R. N. (2020). Advances in monitoring soil microbial community dynamic and function. J. Appl. Genet. 61, 249–263. doi: 10.1007/s13353-020-00549-5
Nzila, A. (2013). Update on the catabolism of organic pollutant by bacteria. Environ. Pollut. 178, 474–482. doi: 10.1016/j.envpol.2013.03.042
Nzila, A., Ramirez, C. O., Musa, M. M., Sankara, S., Basheer, C., and Li, Q. X. (2018). Pyrene biodegradation and proteomic analysis in Achromobacter xylosoxidans, PY4 strain. Int. Biodeterior. Biodegrad. 175, 1294–1305. doi: 10.1016/j.ibiod.2018.03.014
Oliver, J. D. (2010). Recent findings on the viable but nonculturable stage in pathogenic bacteria. FEMS Microbiol. Rev. 34, 415–425. doi: 10.1111/j.1574-6976.2009.00200.x
Oliver, J. D., Dagher, M., and Linden, K. (2005). Induction of Escherichia coli and Salmonella typhimuriam into the viable but nonculturable state following chlorination of wastewater. J.Water Health 3, 249–257. doi: 10.2166/wh.2005.040
Ortega-Gonzalez, D. K., Martinez-Gonzalez, G., Flores, C. M., Zaragoza, D., Cancino-Diaz, J. C., Cruz-Maya, J. A., et al. (2015). Amycolatopsis sp. Poz14 isolated from oil-contaminated soil degrades polycyclic aromatic hydrocarbons. Int. Biodeterior. Biodegrad. 99, 165–173. doi: 10.1016/j.ibiod.2015.01.008
Ortiz, H. M. L., Salinas, E. S., Gonzalez, E. D., and Godnez, M. L. C. (2013). “Pesticide biodegradation: mechanisms, genetics and strategies to enhance the process”, in Biodegradation-Life of Science, eds R. Chamy and F. Rosenkranz (Rijeka: InTech Open), 251–287.
Ortiz-Hernandez, M. L., Castrejon-Godinez, M. L., Popoca-Ursino, E. C., and Cervantes-Decasac, F. R. (2018). “Strategies for biodegradation and bioremediation of pesticides in the environment,” in Strategies for Bioremediation of Organic and Inorganic Pollutants, eds M. S. Fuentes, V. L. Colin, and J. M. Saez (Boca Raton, FL: CRC Press), 95–115.
Pandey, A., Tripathi, P. H., Tripathi, A. H., Pandey, S. C., and Gangola, S. (2019). “Omics technology to study bioremediation and respective enzymes,” in Smart Bioremediation Technologies: Microbial Enzymes, ed P. Bhatt (Academic Press), 23–43. doi: 10.1016/B978-0-12-818307-6.00002-0
Pandey, R., Masood, F., Singh, H. P., and Batish, D. R. (2017). Polycyclic aromatic hydrocarbons as environmental pollutants: a review. Int. J. Adv. Res. Sci. Eng. 6, 1361–1369.
Pang, S., Lin, Z., Zhang, W., Mishra, S., Bhatt, P., and Chen, S. (2020). Insights into the microbial degradation and biochemical mechanisms of neonicotinoids. Front. Microbiol. 11, 868. doi: 10.3389/fmicb.2020.00868
Panigrahi, S., Velraj, P., and Rao, T. S. (2019). “Functional microbial diversity in contaminated environment and application in bioremediation,” in Microbial Diversity in the Genomic Era, eds S. Das and H. Dask (Elsevier; Academia Press), 349–385. doi: 10.1016/B978-0-12-814849-5.00021-6
Parisi, V. A., Brubaker, G. R., Zenker, M. J., Prince, R. C., Gieg, L. M., Da Silva, M. L., et al. (2009). Field metabolomics and laboratory assessments of anaerobic intrinsic bioremediation of hydrocarbons at a petroleum-contaminated site. Microbial. Biotechnol. 2, 202–212. doi: 10.1111/j.1751-7915.2009.00077.x
Pathiraja, G., Egodawatta, P., Goonetilleke, A., and Junior Te'o, V. S. (2019). Solubilization and degradation of polychlorinated biphenyls (PCBs) by naturally occurring facultative bacteria. Sci. Total Environ. 651, 2197–2207. doi: 10.1016/j.scitotenv.2018.10.127
Patowary, K., Patowary, R., Kalita, M. C., and Deka, S. (2016). Development of an efficient bacterial consortium for the potential remediation of hydrocarbons from contaminated sites. Front. Microbiol. 7:1092. doi: 10.3389/fmicb.2016.01092
Paul, D., Pandey, G., Pandey, J., and Jain, R. K. (2005). Accessing microbial diversity for bioremediation and environmental pollutant restoration. Trends Biotechnol. 23, 135–142. doi: 10.1016/j.tibtech.2005.01.001
Pazos, F., Guijas, D., Valencia, A., and De Lorenzo, V. (2005). MetaRouter: bioinformatics for bioremediation. Nucleic Acids Res. 45, D588–D592. doi: 10.1093/nar/gki068
Pedersen, J. A., Yeager, M. A., and Suffer, I. H. (2003). Xenobiotic organic compounds in runoff from fields irrigated with treated wastewater. J. Agric. Food Chem. 51, 1360–1372. doi: 10.1021/jf025953q
Perelo, L. W. (2010). In situ and bioremediation of organic pollutants in aquatic sediments. J. Hazard. Mater. 177, 81–89. doi: 10.1016/j.jhazmat.2009.12.090
Perez-Losada, M., Castro-Nallar, E., Bendall, M. L., Freishtat, R. J., and Crandall, K. A. (2015). Dual transcriptomic profiling of host and microbiota during health and disease in pediatric asthma. PLoS ONE 10:e0131819. doi: 10.1371/journal.pone.0131819
Perruchon, C., Vasileiadis, S., Rousidou, C., Papadopoulou, E. S., Tanou, G., Samiotaki, M., et al. (2017). Metabolic and cell adaptation mechanism revealed through genomic, proteomic and transcription analysis of a Sphingomonas haloaromaticamans strain degrading ortho-phenylphenol. Sci. Rep. 7:6449. doi: 10.1038/s41598-017-06727-6
Phale, P. S., Sharma, A., and Gautam, K. (2019). “Microbial degradation of xenobiotics like aromatic pollutants from the terrestrial environments”, in Pharmaceutical and Personal Care Products: Waste Management and Treatment Technology Emerging Contaminants and Micro Pollutants, eds M. N. V. Prasad, M. Vithanage, and A. Kapley (Elsevier), 259–278. doi: 10.1016/B978-0-12-816189-0.00011-1
Pinu, F. R., Beale, D. J., Paten, A. M., Kouremenos, K., Swarup, S., Schirra, H. J., et al. (2019). Systems biology and multi-omics integration: viewpoints from the metabolomics research community. Metabolites 9:76. doi: 10.3390/metabo9040076
Przybylinska, P. A., and Wyszkowski, M. (2016). Environmental contamination with phthalates and its impact on living organisms. Ecol. Chem. Eng. S. 23, 347–356. doi: 10.1515/eces-2016-0024
Puckett, S. P., Samples, R. M., Schloss, P. D., and Balunas, M. J. (2020). “Metabolomics and the microbiome: characterizing molecular diversity in complex microbial communities,” in Comprehensive Natural Products III: Chemistry and Biology, eds H. W. Liu and T. P. Begley (Elsevier), 502–518. doi: 10.1016/B978-0-12-409547-2.14802-4
Rabausch, U., Juergensen, J., Ilmberger, N., Bohnke, S., Fischer, S., Schubach, B., et al. (2013). Functional screening of metagenome and genome libraries for detection of novel flavonoid modifying enzymes. Appl. Environ. Microbiol. 79, 4551–4563. doi: 10.1128/AEM.01077-13
Rahimi, T., Niazi, A., Deihimi, T., Taghavi, S. M., Avatollahi, S., and Ebrahimie, E. (2018). Genome annotation and comparative genomic analysis of Bacillus subtilis MJ01 a new biodegradation strain isolated from oil contaminated soil. Funct. Integr. Genomics 18, 533–543. doi: 10.1007/s10142-018-0604-1
Ram, R. J., Verberkmoes, N. C., and Thelen, M. P. (2005). Community proteomics of a natural microbial biofilms. Science 308, 1915–1920. doi: 10.1126/science.1109070
Rastogi, G., and Sani, R. K. (2011). “Molecular techniques to assess microbial community structure, function and dynamics in the environment,” in Microbes and Microbial Technology, eds I. Ahmed, F. Ahmed, and J. Pichtel (New York, NY: Springer Nature), 29–57. doi: 10.1007/978-1-4419-7931-5_2
Ravindra, A. P., and Haq, S. A. (2019). “Effects of xenobiotics and their biodegradation in marine life,” in Smart Bioremediation Technologies: Microbial Enzymes, ed P. Bhatt (Academic Press), 63–81. doi: 10.1016/B978-0-12-818307-6.00004-4
Reena, R., Majhi, M. C., Arya, A. K., Kumar, R., and Kumar, A. (2012). BioRadBase: a database for bioremediation of radioactive waste. Afr. J. Biotechnol. 11, 8718–8721. doi: 10.5897/AJB12.020
Rodriguez, A., Castrejon-Godinez, M. L., Salazar-Bustamante, E., Gama-Martinez, Y., Sanchez-Salinas, E., Mussali-Galante, P., et al. (2020). Omics approaches to pesticide biodegradation. Curr. Microbiol. 77, 545–563. doi: 10.1007/s00284-020-01916-5
Roh, S. W., Abell, G., Kim, K. H., Nam, Y. D., and Bae, J. W. (2010). Comparing microarray and next generation sequencing technologies for microbial ecology research. Trends Biotechnol. 28, 291–299. doi: 10.1016/j.tibtech.2010.03.001
Rossello-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67. doi: 10.1016/S0168-6445(00)00040-1
Rowdhwal, S. S. S., and Chen, J. (2018). Toxic effect of di-2-ethylhexyl phthalate: an overview. BioMed Res. Int. 2:1750368. doi: 10.1155/2018/1750368
Sajid, M., Basheer, C., Narasimhan, K., Buhmeida, A., Qahtani, M. A., and Al-Ahwal, M. S. (2015). Persistent and endocrine disrupting organic pollutant: advancements and challenges in analysis, health concerns and clinical correlates. Nat. Environ. Poll. Tech. 15, 733–746.
Salam, M., and Verma, A. (2019). Bacterial community structure in soils contaminated with electronic waste pollutants from Delhi NCR, India. Electronic J. Biotechnol. 41, 72–80. doi: 10.1016/j.ejbt.2019.07.003
Salipante, S. J., Kawasahima, T., and Rosenthal, C. (2014). Performance comparison of Illumina and ion torrent next generation sequencing platforms for 16S rRNA-based bacterial community profiling. Appl. Environ. Microbiol. 80, 7583–7591. doi: 10.1128/AEM.02206-14
Sathishkumar, M., Binupriya, A. R., Balk, S., and Yun, S. (2008). Biodegradation of crude oil by individual bacterial strains and mixed bacterial consortium isolated from hydrocarbon contaminated areas. Clean 36, 92–96. doi: 10.1002/clen.200700042
Sato, Y., Hori, T., Koike, H., Navarro, R. R., Ogata, A., and Habe, H. (2019). Transcriptome analysis of activated sludge microbiomes reveals an unexpected role of minority nitrifiers in carbon metabolism. Comm. Biol. 2:179. doi: 10.1038/s42003-019-0418-2
Sayler, G. S., and Ripp, S. (2000). Field application of genetically engineered microorganisms for bioremediation processes. Curr. Opin. Biotechnol. 480, 1–9. doi: 10.1016/S0958-1669(00)00097-5
Schecter, A., Birnbaum, H., Ryan, J. J., and Constable, J. D. (2006). Dioxins: an overview. Environ. Res. 101, 419–428. doi: 10.1016/j.envres.2005.12.003
Scholer, A., Jacquiod, S., and Vestergaard, G. (2017). Analysis of soil microbial communities based on amplicons sequencing of marker genes. Biol. Fertil. Soils 53, 485–489. doi: 10.1007/s00374-017-1205-1
Segata, N., Boernigen, D., Tickle, T. L., Morgan, X. C., Garrett, W. S., and Huttenhower, C. (2013). Computational meta'omics for microbial community studies. Mol. Sys. Biol. 9:666. doi: 10.1038/msb.2013.22
Sengupta, K., Swain, M. T., Livingstone, P. G., Whiteworth, D. E., and Saha, P. (2019). Genome sequencing and comparative transcriptomics provide holistic view of 4-nitrophenol degradation and concurrent fatty acid catabolism by Rhodococcus sp. strain BUPNP1. Front. Microbiol. 9:3209. doi: 10.3389/fmicb.2018.03209
Seo, J., Keum, Y. S., and Li, Q. X. (2013). Metabolomic and proteomic insights into carbaryl catabolism by Burkholderia sp. C3 and degradation of ten N-methylcarbamates. Biodegradation 24, 795–811. doi: 10.1007/s10532-013-9629-2
Seo, J. S., Keum, Y. S., and Li, Q. X. (2009). Bacterial degradation of aromatic compounds. Int. J. Environ. Res. Public Health 6, 278–309. doi: 10.3390/ijerph6010278
Shah, V., Jain, K., Desai, C., and Madamwar, D. (2011). “Metagenomics and integrative-omics technologies in microbial bioremediation: current trends and potential applications”, in Metagenomics: Current Innovations and Future Trends, ed D. Marco (Norfolk: Caister Academic Press), 211–240.
Shakya, M., Lo, C. C., and Chain, P. S. G. (2019). Advances and challenges in metatranscriptomic analysis. Front. Genet. 10:904. doi: 10.3389/fgene.2019.00904
Shanker, J., Abhilash, P. C., Singh, H. B., Singh, R. P., and Singh, D. P. (2011). Genetically engineered bacteria: an emerging tool for environmental remediation and future perspectives. Gene 480, 1–9. doi: 10.1016/j.gene.2011.03.001
Shapiro, R. S., Chavez, A., and Collins, J. J. (2018). CRISPR-based genomic tools for the manipulation ofgenetically intractable microorganism. Nat. Rev. Microbiol. 16, 333–339. doi: 10.1038/s41579-018-0002-7
Sharma, A., Yadav, B., Rohatgi, S., and Yadav, B. (2018). Cypermethrin toxicity: a review. J. Forensic Sci. Criminal Inves. 9:555767. doi: 10.19080/JFSCI.2018.09.555767
Sharma, P. K., Sharma, V., Sharma, A., Bhatia, G., Singh, K., and Sharma, R. (2019). Comparative metatranscriptome analysis revealed broad response of microbial communities in two soil types, agriculture versus organic soil. J. Genet. Eng. Biotechnol. 17:6. doi: 10.1186/s43141-019-0006-3
Shekhar, S. K., Godheja, J., and Modi, D. R. (2020). “Molecular technologies for assessment of bioremediation and characterization of microbial communities of pollutant-contaminated sites” in, Bioremediation of Industrial Waste for Environmental Safety, eds. R. N. Bharagava and G. Saxena (Springer Nature, Singapore), 447–474. doi: 10.1007/978-981-13-3426-9_18
Shi, S., Nuccio, E., Herman, D. J., Rijkers, R., Estera, K., Li, J., et al. (2015). Succesional trajectories of rhizosphere bacterial communities over consecutive seasons. mBio 6, e00746-15. doi: 10.1128/mBio.00746-15
Shokralla, S., Gibson, J. F., and Niknakht, H. (2014). Nest-generation DNA barcoding: using next generation sequencing to enhance and accelerate DNA barcode capture from single specimen. Mol. Ecol. Resour. 14, 892–901. doi: 10.1111/1755-0998.12236
Siles, J. A., and Margesin, R. (2018). Insights into microbial communities mediating the bioremediation of hydrocarbon-contaminated soil from Alpine former military site. Appl. Microbiol. Biotechnol. 102, 4409–4421. doi: 10.1007/s00253-018-8932-6
Singh, A., Chaudhary, S., Dubey, B., and Prasad, V. (2016). “Microbial-mediated management of organic xenobiotic pollutants in agricultural lands”, in Plant Response to Xenobiotics, eds A. Singh, S. M. Prasad, and R. P. Singh (Singapore: Springer), 211–230. doi: 10.1007/978-981-10-2860-1_9
Singh, A. K., Tiwari, M. N., Prakash, O., and Singh, M. P. (2012). A current review of cypermethrin-induced neurotoxicity and nigrostriatal dopaminergic neurodegeneration. Curr. Neuropharmacol. 10, 64–71. doi: 10.2174/157015912799362779
Singh, D. P., Prabha, R., Gupta, V. K., and Verma, M. K. (2018). Metatranscriptomic analysis deciphers multifunctionl genes and enzymes linked with the degradation of aromatic compounds and pesticide in the wheat rhizosphere. Front. Microbiol. 9:1331. doi: 10.3389/fmicb.2018.01331
Singh, O. V. (2006). Proteomics and metabolomics: the molecular make-up of toxic aromatic pollutant bioremediation. Proteomics 6, 5481–5492. doi: 10.1002/pmic.200600200
Singh, O. V., and Nagaraj, N. S. (2006). Transcriptomics, proteomics and interactions: unique approaches to track the insights of bioremediation. Brief. Funct. Genomics Proteomics 4, 355–362. doi: 10.1093/bfgp/eli006
Singh, S., and Li, S. S. L. (2011). Phthalates: toxicological and inferred human diseases. Genomics 97, 148–157. doi: 10.1016/j.ygeno.2010.11.008
Singh, V., Gohil, N., Ramírez García, R., Braddick, D., and Fofié, C. K. (2018). Recent advances in CRISPR-Cas9 genome editing technology for biological and biomedical investigations. J. Cell. Biochem. 119, 81–94. doi: 10.1002/jcb.26165
Song, Y., Li, X., Yao, S., Yang, X., and Jiang, X. (2020). Correlations between soil metabolomics and bacterial community structures in the pepper rhizosphere under plastic greenhouse cultivation. Sci. Total Environ. 728:138439. doi: 10.1016/j.scitotenv.2020.138439
Steele, H. L., Jaeger, K. E., Daniel, R., and Streit, W. R. (2009). Advances in recovery of novel biocatalysts from metagenomes. J. Mol. Microbiol. Biotechnol. 16, 25–37. doi: 10.1159/000142892
Stein, H. P., Navajas-Perez, R., and Aranda, E. (2018). “Potential for CRISPR genetic engineering to increase xenobiotic degradation capacities in model fungi”, in Approaches in Bioremediation, Nanotechonology, eds R. Prasad and E. Aranda (Cham: Springer Nature), 61–78. doi: 10.1007/978-3-030-02369-0_4
Su, X., Chen, X., and Hu, J. (2013). Exploring the potential environmental functions of viable but non-culturable bacteria. World J. Biotechnol. 29, 2213–2218. doi: 10.1007/s11274-013-1390-5
Sun, G., Du, Y., Yin, J., Jiang, Y., Zhang, D., Jiang, B., et al. (2019). Response of microbial communities to different organochlorine pesticide (OCPs) contamination levels in contaminated soil. Chemosphere 215, 461–469. doi: 10.1016/j.chemosphere.2018.09.160
Sunita, V. J., Dolly, P. R., Bateja, S., and Vivek, U. N. (2013). Isolation and screening for hydrocarbon utilizing bacteria (HUB) from petroleum samples. Int. J. Curr. Appl. Sci. 2, 48–60.
Surani, J. J., Akbari, V. G., Purohit, M. K., and Singh, S. P. (2011). Pahbase, a freely available functional database of polycyclic aromatic hydrocarbons (Pahs) degrading bacteria. J. Bioremed. Biodegrad. 2, 116–135. doi: 10.4172/2155-6199.1000116
Szewczyk, R., Kusmierska, A., and Bernat, P. (2017). Ametryn removal by Metarhizium brunneum: biodegradation pathway proposal and metabolic background revealed. Chemosphere 190, 174–183. doi: 10.1016/j.chemosphere.2017.10.011
Szewczyk, R., Sobon, A., Sylwia, R., Dzitko, K., Waidelich, D., and Dlugonski, J. (2014). Intracellukar proteome expression during 4-n-nonylphenol biodegradation by filamentous fungus Metarhizium robertsii. Int. Biodeterior. Biodegrad. 93, 44–53. doi: 10.1016/j.ibiod.2014.04.026
Taupp, M., Mewis, K., and Hallam, S. J. (2011). The art and design of functional metagenomic screens. Curr. Opin. Biotechnol. 22, 465–472. doi: 10.1016/j.copbio.2011.02.010
Temperton, B., and Giovannoni, S. J. (2012). Metagenomics: microbial diversity through a scratched lens. Curr. Opin. Microbiol. 15, 605–612. doi: 10.1016/j.mib.2012.07.001
Theriot, C. M., Koenigsknecht, M. J., Carlson, P. E., Hatton, G. E., Nelson, A. M., Li, B., et al. (2014). Antibiotic-induced shifts in the mouse gut microbiome and metabolome increases susceptibility to Clostridium difficile infection. Nat. Commun. 5:3114. doi: 10.1038/ncomms4114
Tsaboula, A., Papadakis, E. N., Vrvzas, Z., and Kotopoulou, A. (2016). Environmental and human risk hierarchy of pesticide: a monitoring, hazard assessment and environmental fate. Environ. Int. 78–93. doi: 10.1016/j.envint.2016.02.008
Ullah, S., Zuberi, A., Alagawany, M., Farag, M. R., Dadar, M., Karthik, K., et al. (2018). Cypermethrin induced toxicities in fish and adverse health outcomes: its prevention and control measure adaptation. J. Environ. Manage. 206, 863–871. doi: 10.1016/j.jenvman.2017.11.076
Urbance, J. W., Cole, J., and Saxman, P. (2003). BSD: the biodegradative strain database. Nucleic Acids Res. 31, 152–155. doi: 10.1093/nar/gkg032
Vandera, E., Samiotaki, A., Parapouli, M., Panayotou, G., and Koukkou, A. I. (2015). Comparative proteomic analysis of Arthrobacter phenanthrenivorans Sphe3 on phenanthrene, Phthalate and glucose. J. Proteomic 115, 73–89. doi: 10.1016/j.jprot.2014.08.018
Velmurgan, N., Lee, H., Cha, H. J., and Lee, Y. S. (2017). Proteomic analysis of the marine-derived fungus Paecilomyces sp. strain SF-8 in response to polycyclic aromatic hydrocarbons. Botanica Mar. 60:101. doi: 10.1515/bot-2016-0101
Vizcaino, J. A., Csordas, A., del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., et al. (2016). Update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456. doi: 10.1093/nar/gkw880
Wagner, J., Coupland, P., Browne, H. P., Lawley, T. D., Francis, S. C., and Parkhill, J. (2016). Evaluation of PacBio sequencing for full length bacterial 16S rRNA gene classification. BMC Microbiol. 16:274. doi: 10.1186/s12866-016-0891-4
Wang, D. Z., Kong, L. F., Li, Y. Y., and Xie, Z. X. (2016). Environmental microbial community proteomics: status, challenges and perspectives. Int. J. Mol. Sci. 17:1275. doi: 10.3390/ijms17081275
Wang, T., Hu, C., Zhang, R., Sun, A., Li, D., and Shi, X. (2019). Mechanism study of cyfluthrin biodegradation by Photobacterium ganghwense with comparative metabolomics. Appl. Microbiol. Biotechnol. 103, 473–488. doi: 10.1007/s00253-018-9458-7
Wang, W., Wang, L., and Shao, Z. (2018). Polycyclic aromatic hydrocarbon (PAH) degradation pathways of the obligate marine PAH degrader Cycloclasticus sp. strain P1. Appl. Environ. Microbiol. 84:e01261-18. doi: 10.1128/AEM.01261-18
Wang, Y. F., Zhu, H. W., Wang, Y., Zhang, X. L., and Tam, N. F. Y. (2018). Diversity and dynamics of microbial community structure in different mangrove, marine and freshwater sediments during anaerobic debromination of PBDEs. Front. Microiol. 9:952. doi: 10.3389/fmicb.2018.00952
Wei, K., Yin, H., Peng, H., Liu, Z., Lu, G., and Dang, Z. (2017). Characteristics and proteomic analysis of pyrene degradation by Brevibacillus brevis in liquid medium. Chemosphere 178, 80–87. doi: 10.1016/j.chemosphere.2017.03.049
White, R. A., Bottos, E. M., Roy, C. T., Zucker, J. D., Brislawn, C. J., Nicora, C. D., et al. (2016). Moleculo long-read sequencing facilitates assembly and genomic binning from complex soil metagenomes. mSystems 1:e00045-16. doi: 10.1128/mSystems.00045-16
Widada, J., Nojiri, H., and Omori, H. (2002). Recent developments in molecular techniques for identification and monitoring of xenobiotic-degrdaing bacteria and their catabolic genes in bioremediation. Appl. Microbiol. Biotechnol. 60, 45–59. doi: 10.1007/s00253-002-1072-y
Williams, T. J., Wilkins, D., Long, E., Evans, F., DeMaere, M. Z., Raftery, M. J., et al. (2013). The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antartica revealed using metagenomics and metaproteomics. Environ. Microbiol. 15, 1302–1317. doi: 10.1111/1462-2920.12017
Withers, E., Hill, P. W., Chadwick, D. R., and Jones, D. L. (2020). Use of untargeted metabolomics for assessing soil quality and microbial function. Soil Biol. Biochem.143:107758. doi: 10.1016/j.soilbio.2020.107758
Wolf, D. C., Cryder, Z., Khoury, R., Carlan, C., and Gan, J. (2020). Bioremediation of PAH-contaminated shooting range soil using integrated approaches. Sci. Total Environ. 726:13844. doi: 10.1016/j.scitotenv.2020.138440
Wong, D. W. S. (2018). “Gene targeting and genome editing,” in The ABCs of Gene Cloning, ed D. W. S. Wong (Cham: Springer), 187–197. doi: 10.1007/978-3-319-77982-9_20
Wright, R., Bosch, R., Gibson, M. I., and Christie-Oleza, J. (2020). Plasticizer degradation by a marine bacterial isolates: a proteogenomic and metabolomic characterization. Environ. Sci. Technol. 54, 2244–2256. doi: 10.1021/acs.est.9b05228
Wu, X., Yin, Y., Wang, S., and Yu, Y. (2014). Accumulation of chlorothalonil and its metabolite, 4-hydroxychlorothalonil in soil after repeated applications and its effects on soil microbial activities underground greenhouse conditions. Environ. Sci. Pollut. Res. 21, 3452–3459. doi: 10.1007/s11356-013-2318-1
Xie, J., He, Z., Liu, X., Van Nostrand, J. D., and Deng, Y. (2011). GeoChip based analysis of functional gene diversity and metabolic potential of microbial communities in acid mine drainage. Appl. Environ. Microbiol. 77, 991–999. doi: 10.1128/AEM.01798-10
Xiong, J. B., Wu, L. Y., Tu, S. X., Van Nostrand, J. D., He, Z. H., Zhou, J. Z., et al. (2010). Microbial communities and functional genes associated with soil arsenic contamination and the rhizosphere of arsenic-hyperaccumulating plant Pteris vittata L. Appl. Environ. Microbiol. 76, 7277–7284. doi: 10.1128/AEM.00500-10
Xu, Z., Hansen, M. A., Hansen, L. H., Jacquiod, S., and Soresen, S. J. (2014). Bioinformatic approaches reveal metagenomic characterization of soil microbial community. PLoS ONE 9:e93445. doi: 10.1371/journal.pone.0093445
Yan, C., Wang, F., Geng, H., Liu, H., Pu, S., Tian, Z., et al. (2020). Integrating high-throughput sequencing and metagenome analysis to reveal the characteristic and resistance mechanism of microbial community in metal contaminated sediments. Sci. Total Environ. 707:106116. doi: 10.1016/j.scitotenv.2019.136116
Yan, X., Jin, W., Wu, G., Jiang, W., Yang, Z., Ji, J., et al. (2018). Hydrolase CehA and monooxygenase CfdC are responsible for carbofuran degradation in Sphingomonas strain CDS-1. Appl. Environ. Microbiol. 84, 1–15. doi: 10.1128/AEM.00805-18
Yang, J., Feng, Y., Zhan, H., Liu, J., Yang, F., Zhang, K., et al. (2018). Characterization of a pyrethroid-degrading Pseudomonas fulva strain P31 and biochemical degradation pathway of D-phenothrin. Front. Microbiol. 9:1003. doi: 10.3389/fmicb.2018.01003
Yang, T., Ren, L., Jia, Y., fan, S., Wang, J., Nahurira, R., et al. (2018). Biodegradation of di-(2-ethylhexyl) phthalate by Rhodococcus ruber YC-YT1 in contaminated water and soil. Int. J. Environ. Res. Public Health. 15:964. doi: 10.3390/ijerph15050964
Yates, J. R., Ruse, C. L., and Nakorchevsky, A. (2009). Proteomics by mass spectrometry: approaches, advances and applications. Ann. Rev. Biomed. Eng. 11, 49–79. doi: 10.1146/annurev-bioeng-061008-124934
Yu, K., Yi, S., Li, B., Guo, F., Peng, X., Wang, Z., et al. (2019). An integrated meta-omics approach reveals substrates involved in synergistic interactions in a bisphenol A (BPA)-degrading microbial community. Microbiome 7:16. doi: 10.1186/s40168-019-0634-5
Yu, Y., Yin, H., Peng, H., Lu, G., and Dang, Z. (2019). Proteomic mechanism of decabromodiphenyl ether (BDE-209) biodegradation by Microbacterium Y2 and its potential in remediation of BDE-209 contaminated water-sediment system. J. Hazard. Mater. 387:121708. doi: 10.1016/j.jhazmat.2019.121708
Zafra, G., Taylor, T. D., Absalon, A. E., and Cortes-Espinosa, D. V. (2016). Comparative metagenomic analysis of PAH degradation in soil by a mixed microbial consortium. J. Hazard. Mater. 318, 702–710. doi: 10.1016/j.jhazmat.2016.07.060
Zepeda, A. E., de Leon, A. V. P., and Flores, A. S. (2015). The road to metagenomics: from microbiology to DNA sequencing technologies and bioinformatics. Front. Genet. 6:648. doi: 10.3389/fgene.2015.00348
Zhan, H., Huang, Y., Lin, Z., Bhatt, P., and Chen, S. (2020). New insights into the microbial degradation and catalytic mechanism of synthetic pyrethroids. Environ. Res. 182:109138. doi: 10.1016/j.envres.2020.109138
Zhan, H., Wang, H., Liao, L., Feng, Y., Fan, X., Zhang, L., et al. (2018). Kinetics and novel degradation pathway of cypermethrin in Acinetobacter baumannii ZH-14. Front. Microbiol. 9:98. doi: 10.3389/fmicb.2018.00098
Zhang, R., Xu, X., Chen, W., and Huang, Q. (2016). Genetically engineered Pseudomonas putida X3 strain and its potential ability to bioremediate soil microcosms contaminated with methyl parathion and cadmium. Appl. Microbiol. Biotechnol. 100, 1987–1997. doi: 10.1007/s00253-015-7099-7
Zhang, W., Lin, Z., Pang, S., Bhatt, P., and Chen, S. (2020). Insights into the biodegradation of lindane (γ-hexacholocyclohexane) using a microbial system. Front. Microbiol. 11:522. doi: 10.3389/fmicb.2020.00522
Zhang, W., Pang, S., Lin, Z., Mishra, S., Bhatt, P., and Chen, S. (2021). Biotransformation of perfluoroalkyl acid precursors from various environmental systems: advances and perspectives. Environ. Pollut. 268:115908. doi: 10.1016/j.envpol.2020.115908
Zhao, Q., Yue, S., Bilal, M., Hu, H., Wang, W., and Zhang, X. (2017). Comparative genomic analysis of 26 Sphingomonas and Sphingobium strains: dissemination of bioremediation capabilities, biodegradation potential and horizontal gene transfer. Sci. Total Environ. 609, 1238–1247. doi: 10.1016/j.scitotenv.2017.07.249
Zhao, X., Ma, F., Feng, C., Bai, S., Yang, J., and Wang, L. (2017). Complete genome sequence of Arthrobacter sp. ZXY-2 associated with effective atrazine degradation and salt adaptation. J. Biotechnol. 248, 43–47. doi: 10.1016/j.jbiotec.2017.03.010
Zhong, Q., Tian, J., Wang, B., and Wang, L. (2016). PMA based real-time fluoresecent LAMP for the detection of Vibrio parahaemolyticus in viable but nonculturable state. Food Control 63, 230–238. doi: 10.1016/j.foodcont.2015.11.043
Zhou, J., He, Z., and Yang, Y. (2015). High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. mBio 6:e02288-14. doi: 10.1128/mBio.02288-14
Zhu, F., Doyle, E., Zhu, C., Zhou, D., Gu, C., and Gao, J. (2020). Metagenomic analysis exploring microbial assemblages and functional genes potentially involved in di (2-ethylhexyl) phthalate degradation in soil. Sci. Total Environ. 715:137037. doi: 10.1016/j.scitotenv.2020.137037
Zhu, Y., Boye, A., Body-Malape, M., and Herkovits, J. (2017). The toxic effects of xenobiotics on the health of humans and animals. BioMed Res. Int. 17:4627872. doi: 10.1155/2017/4627872
Zhu, Y., Klompe, S. E., Vlot, M., van der Oost, J., and Staals, R. H. (2018). Shooting the messenger: RNA-targetting CRISPR-Cas systems. Biosci. Rep. 38:BSR20170788. doi: 10.1042/BSR20170788
Keywords: bioremediation, microorganisms, xenobiotics, omics, bioinformatics
Citation: Mishra S, Lin Z, Pang S, Zhang W, Bhatt P and Chen S (2021) Recent Advanced Technologies for the Characterization of Xenobiotic-Degrading Microorganisms and Microbial Communities. Front. Bioeng. Biotechnol. 9:632059. doi: 10.3389/fbioe.2021.632059
Received: 22 November 2020; Accepted: 11 January 2021;
Published: 10 February 2021.
Edited by:
Datta Madamwar, Sardar Patel University, IndiaReviewed by:
Masataka Tsuda, Tohoku University, JapanCopyright © 2021 Mishra, Lin, Pang, Zhang, Bhatt and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaohua Chen, c2hjaGVuQHNjYXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.