
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Bioeng. Biotechnol. , 01 December 2020
Sec. Biosafety and Biosecurity
Volume 8 - 2020 | https://doi.org/10.3389/fbioe.2020.591024
This article is part of the Research Topic The Design of Molecular Tools in Relation to Prions and their Biosafety View all 12 articles
 Mohammed Moudjou1Johan Castille2Bruno Passet2Laetitia Herzog1Fabienne Reine1
Mohammed Moudjou1Johan Castille2Bruno Passet2Laetitia Herzog1Fabienne Reine1 Jean-Luc Vilotte2Human Rezaei1
Jean-Luc Vilotte2Human Rezaei1 Vincent Béringue1
Vincent Béringue1 Angélique Igel-Egalon1,3*
Angélique Igel-Egalon1,3*Prions are pathogenic infectious agents responsible for fatal, incurable neurodegenerative diseases in animals and humans. Prions are composed exclusively of an aggregated and misfolded form (PrPSc) of the cellular prion protein (PrPC). During the propagation of the disease, PrPSc recruits and misfolds PrPC into further PrPSc. In human, iatrogenic prion transmission has occurred with incompletely sterilized medical material because of the unusual resistance of prions to inactivation. Most commercial prion disinfectants validated against the historical, well-characterized laboratory strain of 263K hamster prions were recently shown to be ineffective against variant Creutzfeldt-Jakob disease human prions. These observations and previous reports support the view that any inactivation method must be validated against the prions for which they are intended to be used. Strain-specific variations in PrPSc physico-chemical properties and conformation are likely to explain the strain-specific efficacy of inactivation methods. Animal bioassays have long been used as gold standards to validate prion inactivation methods, by measuring reduction of prion infectivity. Cell-free assays such as the real-time quaking-induced conversion (RT-QuIC) assay and the protein misfolding cyclic amplification (PMCA) assay have emerged as attractive alternatives. They exploit the seeding capacities of PrPSc to exponentially amplify minute amounts of prions in biospecimens. European and certain national medicine agencies recently implemented their guidelines for prion inactivation of non-disposable medical material; they encourage or request the use of human prions and cell-free assays to improve the predictive value of the validation methods. In this review, we discuss the methodological and technical issues regarding the choice of (i) the cell-free assay, (ii) the human prion strain type, (iii) the prion-containing biological material. We also introduce a new optimized substrate for high-throughput PMCA amplification of human prions bound on steel wires, as translational model for prion-contaminated instruments.
Transmissible spongiform encephalopathies (TSE) or prion diseases are fatal, uncurable neurodegenerative diseases affecting animals and humans (Collinge, 2001). TSE include scrapie in sheep and goats, bovine spongiform encephalopathy (BSE), chronic wasting disease in cervids and Creutzfeldt-Jakob disease (CJD) in humans. Intra- and inter-species TSE transmission has recurrently occurred in animals and humans via medical and dietary settings. BSE has occurred as an epidemic in cattle and has propagated in human under the form of variant CJD (vCJD). Other dietary exposure in human includes kuru epidemic among Fore people of Papua New Guinea due to funerary cannibalism. Iatrogenic CJD forms are related to the use of contaminated cadaver-extracted human growth hormone and dura mater or to insufficiently sterilized contaminated brain surgery material. The most common forms of CJD are inherited or sporadic.
Transmissible spongiform encephalopathies are caused by prions. Prions are unconventional pathogens exclusively composed of an aggregated and misfolded form (PrPSc) of the cellular prion protein (PrPC). During the disease pathogenesis, PrPSc recruits PrPC and induces its misfolding into further PrPSc. This replicative self-templating process is at the origin of prion infectious nature (Prusiner, 1982). Biochemically, PrPSc and PrPC properties strongly differ. PrPSc is β-sheet rich, contains a protease-resistant core and is prone to aggregation, while PrPC is α-helix rich, protease-sensitive and monomeric (Colby and Prusiner, 2011).
Susceptible mammals, including laboratory species, stably propagate structurally distinct PrPSc assemblies known as prion strains. Prion strains differ biochemically at the level of PrPSc tertiary and quaternary structures. Phenotypically, prion strains encode unique stereotypical biological traits including the time course to disease, neuropathological features and tropism for specific brain regions or lymphoid organs (Bruce, 2003; Collinge and Clarke, 2007; Beringue et al., 2008b; Weissmann et al., 2011). Co-propagation of strains has been observed, notably in CJD (Cassard et al., 2020) and sheep scrapie (Le Dur et al., 2017; Huor et al., 2019). PrPSc structural polymorphism is mostly considered as between strain polymorphism. However, experimental evidence supports the view for further structural diversity and co-propagation of PrPSc assemblies within specific prion populations and strains (Igel-Egalon et al., 2019a).
While human TSE remain relatively rare, they constitute a critical public health concern. First, the disease incubation period is long, asymptomatic, without impact on most biological constants. Early diagnosis of the disease is lacking. Medical, non-disposable instruments may thus be used on individuals incubating silently the disease and potentially be reused. Second, while prions are neurotropic agents, they can replicate extraneurally at significant levels, markedly increasing the range of medical acts that can transmit the disease iatrogenically. Third, prions are highly resistant to common inactivation methods as compared to viruses or bacteria. Since 1999, the World Health Organization guidelines and their national counterparts recommend procedures, which include immersion of non-disposable surgical instruments in 1M sodium hydroxide (NaOH) or 20,000 ppm sodium hypochlorite (2% NaOCl) for 1h, followed by porous autoclaving at 134°C for 18 min (WHO, 1999; DGS, 2011). To circumvent the limits imposed by these methods (e.g., instrument corrosion), the French Medicine Agency (ANSM), for example, recommend since 2011 a list of commercial methods validated for their efficacy to inactivate prions according to a standardized protocol (Ansm, 2011). The validation studies were exclusively based on the inactivation of laboratory prion strains, including as primary model with short incubation time and high infectivity titer hamster-adapted scrapie 263K (or Sc237) prions (Kimberlin and Walker, 1977), as model relevant to BSE/vCJD, mouse-derived BSE prions (Bruce et al., 1994; Lasmezas et al., 1996) or Fukuoka-1 prions derived from the mouse adaptation of human, inherited Gerstmann-Sträussler-Scheinker (GSS) syndrome (Tateishi et al., 1979) as model relevant to human TSE. The inactivation of these prion strains was primarily tested by measuring residual infectivity in hamster or mouse bioassays. However, prion inactivation efficacy is strain-dependent. For example, sporadic CJD prions were shown to be 100,000-fold more resistant than hamster Sc237 prions to acidic SDS treatment (Peretz et al., 2006). The host species in which the strain is passaged can also impact the final efficacy. For example, cattle BSE prions were >1,000-fold more resistant to acidic SDS treatment than BSE-derived mouse 301V prions (Giles et al., 2008). Extended heating or steam sterilization by autoclaving similarly inactivated prions in a strain-dependent manner, BSE and BSE-derived sources being amongst the most resistant strains (Fernie et al., 2012; Marin-Moreno et al., 2019). Worryingly, most ANSM-validated disinfectants which totally inactivated 263K, mouse-BSE or Fukuoka-1 prions were subsequently shown to partially inactivate human vCJD prions (Belondrade et al., 2016; Bélondrade et al., 2020). Collectively, these observations strongly support the view that any inactivation method must be validated against the prions for which they are intended to be used.
The studies by Belondrade et al. (2016); Bélondrade et al. (2020) suggested that cell-free prion amplification assays may replace animal bioassays to quantify prion inactivation efficacy. The European and French Medicine agencies thus implemented their protocols to validate prion inactivation methods. They encourage or request the use of both human prions (or human-relevant prions) and highly sensitive cell-free prion amplification assays (Ansm, 2018; EMA, 2018). These assays measure prion concentration by limiting dilution titration, based on PrPSc seeding activity. Their sensitivity is equivalent or greater than animal bioassays which are measuring prion infectivity. Yet, they are not routinely used in inactivation methods. In this review, we discuss the methodological and technical issues raised by such implementations, with respect to the choice of the cell-free assay, the human prion strain type and the nature of the prion-containing biological material.
Measuring prion concentration in a test sample has for long relied on measuring prion infectivity by bioassay in laboratory animals. Prion infectious titer can be obtained by endpoint dilution titration of the sample in bio-indicator animals or by using incubation time values once prion dose response curves have been established (Prusiner et al., 1980, 1982). Surrogately, cell-free assays estimate prion concentration by measuring prion self-converting activity. Among them, two ultrasensitive amplification assays deserve attention to their effectiveness to detect minute amounts of prions: the real-time quacking-induced conversion (RT-QuIC) assay (Atarashi et al., 2008; Wilham et al., 2010) and the protein misfolding cyclic amplification (PMCA) assay (Saborio et al., 2001; Figure 1). In essence a PrPSc-containing sample is mixed with a substrate containing normally folded, monomeric recombinant PrP (RT-QuIC) or brain PrPC (PMCA). The conversion is favored and accelerated by cycles of shaking (RT-QuIC) or sonication (PMCA) and quiescent incubation. In the RT-QuIC assay, the conversion of recombinant PrP into amyloid aggregates is followed in real-time by incorporation of thioflavin T (ThT), an amyloid-sensitive fluorescent dye. In the PMCA assay, the conversion of PrPC into PrPSc is assessed at the end of the reaction by biochemical purification and immunodetection of PrPSc. Both tests reach similar or greater sensitivities than those of the animal bioassays. Both have a wide range of fundamental and applied applications, including TSE diagnostic. Both have benefits and drawbacks that make them complementary techniques in the TSE field, as summarized in Table 1.
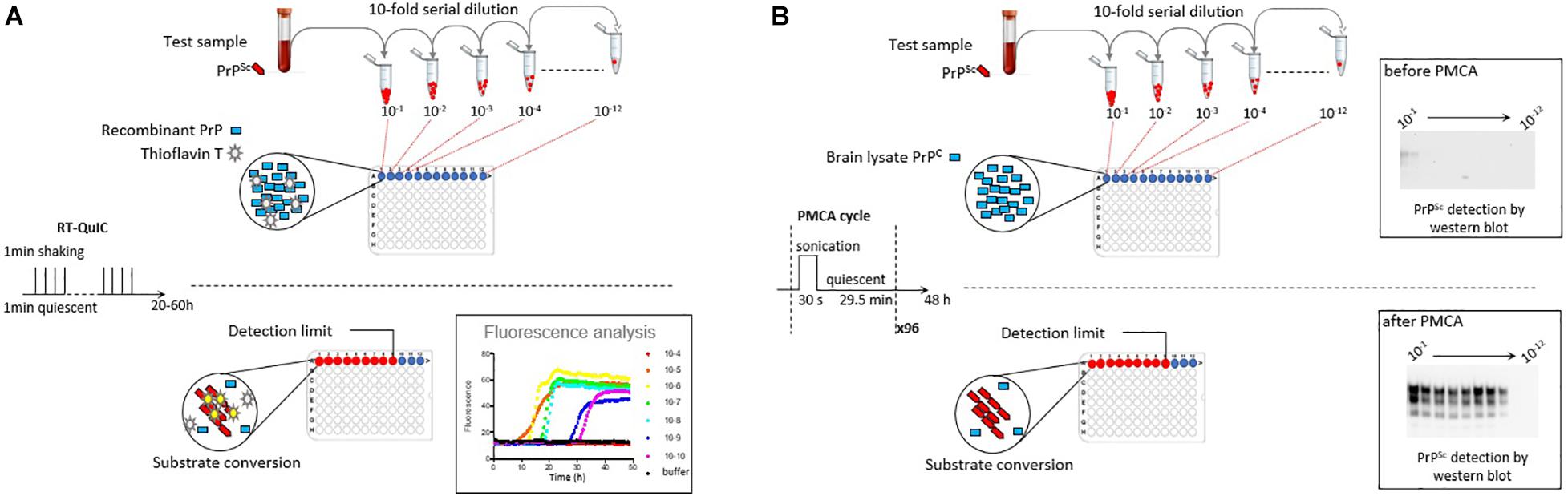
Figure 1. Scheme summarizing the principles of the RT-QuIC and PMCA assays. (A) RT-QuIC is based on the polymerization of recombinant PrP protein by prions in the test sample. During iterative cycles of shaking and quiescent incubation, polymerization is monitored in real-time by the increase in Thioflavin T fluorescence. (B) PMCA is based on the bona fide conversion of brain PrPC by prions in the test sample. Conversion is favored by iterative cycles of quiescent incubation and sonication. At the end of the reaction, the presence of abnormal PrPSc is analyzed by proteinase K digestion and western blot. In both methods, prion seeding activity in the test sample can be quantified by limiting dilution titration.
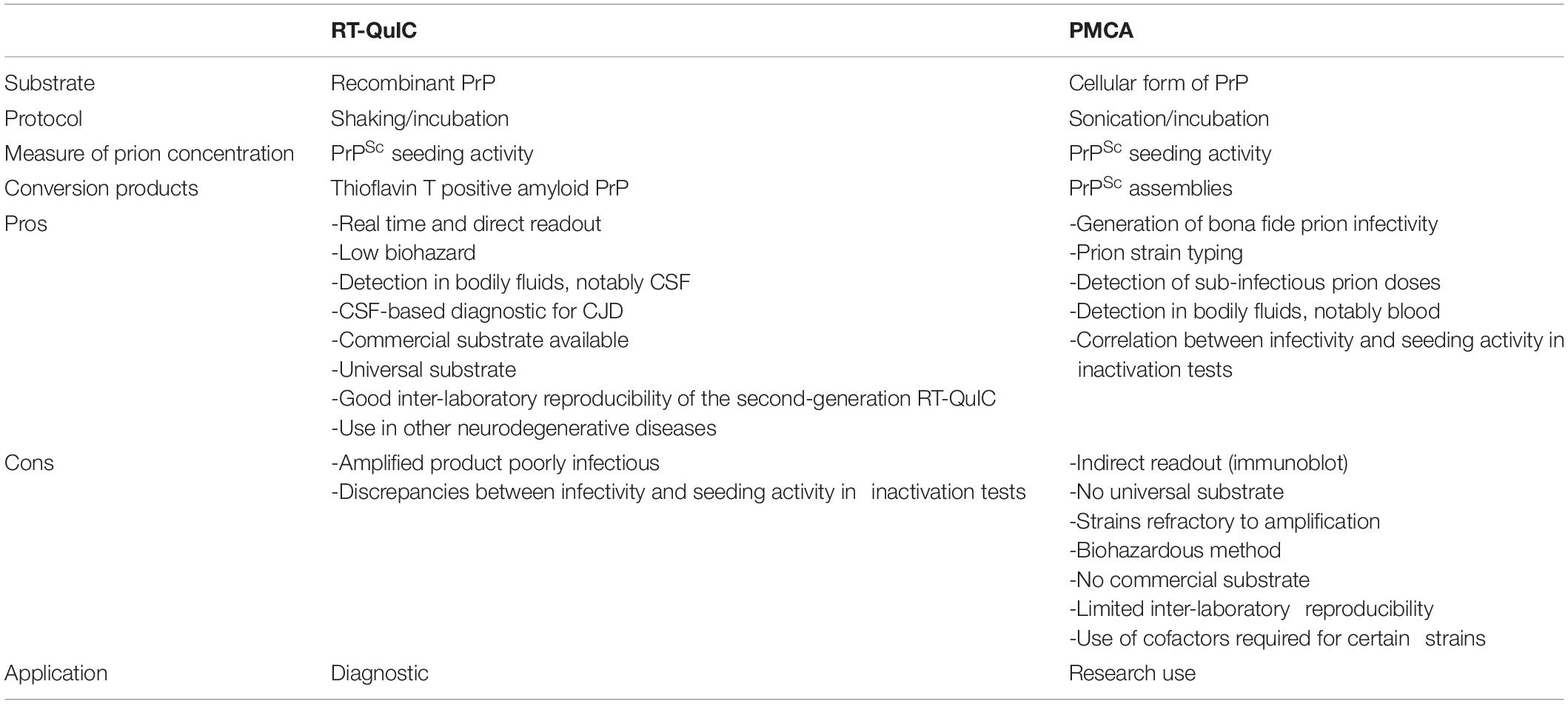
Table 1. Benefits and drawbacks of the RT-QuIC and PMCA assays.
In the RT-QuIC assay, the ThT-positive amyloid assemblies formed by seeded recombinant PrP are poorly infectious (Groveman et al., 2017; Raymond et al., 2020). Manipulation of prion infectivity is thus limited to the seed, limiting biohazards in routine use of the assay. A limiting aspect of RT-QuIC is the possible self-polymerization of recombinant PrP in the absence of any seed. From a diagnostic viewpoint, RT-QuIC is particularly efficient at amplifying prion sub-infectious doses in sample biopsies from the skin (Orru et al., 2017), olfactory mucosa (Orru et al., 2014), urine, saliva (Henderson et al., 2015), blood (Elder et al., 2013), and CSF (McGuire et al., 2012). The second generation of RT-QuIC specially designed for prion detection in the CSF achieved >95% sensitivity and 100% specificity (Orru et al., 2015a), with a good interlaboratory reproducibility, thus opening new avenue for ante-mortem diagnosis of CJD (Rhoads et al., 2020). CSF-based RT-QuIC is now considered as the most powerful versatile technique for diagnosing CJD. It is recommended by the American Center for Disease Control and Prevention (CDC) since 2018. Following this pioneering work, similar CSF-based assays were successfully developed for other prion-like neurodegenerative diseases linked to protein misfolding. These RT-QuIC assays identify alpha-synuclein (Rossi et al., 2020) and Tau (Kraus et al., 2019; Saijo et al., 2020) seeding activity in CSF of patients with Lewy bodies or Parkinson’s disease and Alzheimer’s disease or frontotemporal lobar degeneration, respectively. Proof of concept has been obtained with TDP-43 in amyotrophic lateral sclerosis and frontotemporal lobar degeneration (Scialò et al., 2020).
Given the analytical sensitivity of the RT-QuIC assay and the clear correlation between seeding activity and infectivity in measuring prion concentration (Wilham et al., 2010), its potential to assess prion inactivation methods was assayed. There were discrepancies between the animal bioassay and the RT-QuIC assay. One disinfectant inactivating all measurable prion infectivity by bioassay was not able to eliminate all seeding activity by RT-QuIC (Hughson et al., 2016). In another study, the seeding activity of human CJD prions bound to stainless steel wires was measurable by RT-QuIC and completely removed after a 2 h-treatment with 1M NaOH (Mori et al., 2016), as expected. The reason for such discrepancies is unclear. Prion inactivation treatment may destroy prion infectivity without affecting all PrP seeding activity, given the high sensitivity of the assay. Alternatively, as the RT-QuIC generates amyloid fibers that are off-pathway to prion infectivity, treatment neutralizing these forms may not necessarily affect prion infectivity per se.
As key feature, PMCA mirrors in an accelerated manner the prion replication process (Igel-Egalon et al., 2019b). The reaction products are highly infectious – with infectivity titers equivalent to those found in the brain at the terminal stage of the disease – and contain prions which generally retain the parental seed strain phenotype (Weber et al., 2007; Shikiya and Bartz, 2011; Moudjou et al., 2014). PMCA has been instrumental in identifying co-factors (Deleault et al., 2012a,b; Fernández-Borges et al., 2018), PrP domains (Burke et al., 2020a) or post-translational modifications (Moudjou et al., 2016; Burke et al., 2020b) involved in the prion replication process. It has also been instrumental in studying the diversification of PrPSc assemblies and their dynamic interactions (Igel-Egalon et al., 2019b).
The analytical sensitivities of the PMCA and RT-QuIC assays are generally equivalent. RT-QuIC is the method of choice to quantify the seeding activity of prions responsible for sporadic CJD (notably the most prevalent MM1 subtype), whereas PMCA is more performant at detecting vCJD prions (Moudjou et al., 2014; Camacho et al., 2019). PMCA has been incredibly good at detecting vCJD/BSE prions in blood from asymptomatic carriers, in small and large animal models and in human (Lacroux et al., 2014; Bougard et al., 2016; Concha-Marambio et al., 2016). Yet, the necessity to work with PrPC-containing cell or brain substrate and the absence of real-time detection of the conversion process are major impediments to a diagnostic use. Several laboratories are working at substituting PrPC-containing brain substrate with recombinant PrP (Fernández-Borges et al., 2018; Eraña et al., 2019).
With respect to prion inactivation methods, a strong correlation between animal bioassay and PMCA was found in two compelling studies testing several disinfectants or physical processes on 263K prions or vCJD prions bound on stainless steel wires (Pritzkow et al., 2011; Belondrade et al., 2016; Bélondrade et al., 2020). PMCA thus emerged as a potent tool to evaluate the degree of effectiveness of prion inactivation methods, at least with certain human and laboratory strains.
The choice of the “right” human prion strain to implement the validation of prion inactivation methods is a conundrum:
1. None of the human prion strains have been studied as deeply as hamster 263K with respect to pathogenesis, infectivity titers and response to inactivating treatments by bioassay;
2. The use and relevance of human prions adapted to wild-type laboratory animals must be studied on a case-by-case basis. For example, mouse-adapted Fukuoka-1 prions were not able to propagate on back-passage to transgenic mice expressing human PrP, suggesting loss of parental CJD transmission characteristics (Giles et al., 2017). Oppositely, sporadic CJD prions propagated in bank voles with little or no species barrier, consistent with potential maintain of CJD transmission characteristics (Nonno et al., 2006). Early transmission studies indicated that non-human primates were susceptible to sporadic and vCJD prions (Gibbs et al., 1968; Lasmézas et al., 1996), providing a potential source of human-like prion infectivity. However, macaque and human PrP sequences differ by nine amino acids, which were shown by transgenic modeling to create a substantial species barrier on transmission of sporadic CJD (Espinosa et al., 2019). Whether macaque-passaged prions retain in fine CJD parental properties remain to be determined. Seminal studies by transgenic modeling indicated that amino acid sequence identity between PrPSc and PrPC usually abrogate the barrier to transmission between species (for review Moreno and Telling, 2017). Human prions did not escape the rule. Sporadic and vCJD prions propagate without species barrier in mice transgenic for human PrP, in the absence of mismatch at polymorphic codon 129 (Bishop et al., 2010; Beringue et al., 2012; Chapuis et al., 2016; Jaumain et al., 2016; Cassard et al., 2020). These models, – in the absence of cell models propagating human prions –, allow a relatively rapid, inexpensive, and large production of potentially biologically cloned and well-characterized humanized prions with respect to pathogenesis, strain type and infectivity titers. Oppositely, sporadic and vCJD reference reagents from the World Health Organization contain mixture of strains (Minor et al., 2004; Beringue et al., 2008a; Yull et al., 2009), which may complicate their use in decontamination protocols. It remains to be determined whether humanized prions exhibit similar resistance/sensitivity to inactivation as those that accumulate directly in the human brain. At least, they seem to share similar levels of infectivity (Beringue et al., 2008a; Halliez et al., 2014);
3. The pros and cons of assaying (human or humanized) sporadic or vCJD prions in inactivation trials must be weighed. While the number of clinical cases of vCJD has remained limited (∼235 cases worldwide), this disease is a key issue due to the large number of potential asymptomatic carriers (Gill et al., 2020), the large distribution of infectivity in the body (Douet et al., 2017) and the associated risks of secondary transmission, notably by blood transfusion or surgery (Peden et al., 2004). The disease is due to a unique prion strain type (classical BSE prions) (Diack et al., 2012). When testing heat sterilization methods, BSE/vCJD prions thermostability may increase the predictive value of the assay (Fernie et al., 2012; Marin-Moreno et al., 2019). Sporadic forms of CJD are more prevalent with approximately 1.5 cases per millions per year. The disease is heterogeneous, making the choice of the “right” strain a dilemma; nine sub-types of sporadic CJD are described according to the clinical and neuropathological characteristics of the disease in infected individuals, the polymorphism of the PRNP gene at codon 129 (MM, MV, or VV) and the electrophoretic profile of brain PrPSc (type 1 or type 2) (for review Zerr and Parchi, 2018). At least six distinct strain types are described according to their transmission properties in transgenic mice expressing human PrP. Starting from the most prevalent cases in the population, these strains are classified as MM1/MV1, VV2, MV2, VV1, cortical-MM2, thalamic-MM2 strains (Bishop et al., 2010; Moda et al., 2012; Chapuis et al., 2016; Jaumain et al., 2016; Cassard et al., 2020). Co-propagation of MM1 and VV2 strains has been observed in a significant proportion of sporadic CJD patients (Cassard et al., 2020).
Although direct assay of human prions in inactivation methods may sound highly relevant with respect to iatrogenic risk of prion transmission, it necessitates a thorough examination to ensure that the results obtained are of added value and that extrapolation to the human situation can be made.
Prions and bacterial biofilms are a challenge to proper sterilization of non-disposable medical devices because of their high resistance to inactivation and binding affinity for steel surfaces. To date, six cases of iatrogenic CJD have been reported worldwide by contaminated surgical instruments or depth EEG electrodes (for review Bonda et al., 2016). Prion-contaminated stainless steel wires have become the gold standard to screen and validate prion inactivation methods for medical instruments. This translational model, which is used in most guidelines, came from the seminal work of Charles Weissmann and collaborators (Zobeley et al., 1999; Flechsig et al., 2001). In essence, wires are artificially contaminated in prion-containing brain macerates. To measure prion concentration pre- and post-disinfection, the wires are permanently implanted in the brain of bio-indicator animals and reduction in the disease attack rate is measured. The reduction factor is established by comparing the results obtained with endpoint titration of prions bound on steel wires. A prion-cell endpoint assay (Klohn et al., 2003) was adapted that offered greater sensitivity, practicability and rapidity than the animal bioassay (Edgeworth et al., 2009). However, its use is so far limited to mouse prions. A sensitive assay based on direct immunodetection of surface-bound prions was also reported and validated against human vCJD prions (Edgeworth et al., 2011b).
Key issues in the interpretation and overall validity of the results are the rate of prion adsorption, desorption and/or bio-activity (i.e., ability to initiate infection in the brain). With mouse or hamster prions, a contact of a few minutes between the infected brain homogenate and the wire was sufficient to transmit the disease with 100% attack rate (Flechsig et al., 2001; Giles et al., 2017). A transient insertion of contaminated wires in the brain for 5–30 min was sufficient to transmit the disease to laboratory animals with 100% attack rate, yet with delayed disease tempo (Flechsig et al., 2001; Yan et al., 2004). This suggested relatively rapid rate of adsorption and release/bio-activity. The information on prion fate once surface-adsorbed is relatively limited. Binding to soil fractions (montmorillonite) was reported to potentiate the disease transmission capacity of hamster 263K prions by oral route (Johnson et al., 2007). The opposite was found on binding to silty clay upon intracerebral inoculation (Saunders et al., 2011). In “standard” conditions, wires are contaminated for 1 h and permanently inserted in bio-indicator animals. To further circumvent these uncertainties and address the possibility that the decontamination procedures unbound prions from the wires without inactivating them, an additional study on the inactivation potential of the method on desorbed material may be requested (Ansm, 2011, 2018).
The brain tissue does not represent the more likely source of iatrogenic prion contamination, except during neurosurgical procedures. The use of sensitive animal bioassays with human PrP transgenic mice and/or cell-free assays allowed demonstrating that many tissues outside the brain contain substantial amounts of prions, thus markedly increasing the range of medical acts with non-disposable equipment that may transmit human TSE iatrogenically. This includes dentistry, organ transplant, blood transfusion, and surgery. In individuals infected with vCJD, at the symptomatic or pre-symptomatic stage, prions have been detected in a wider and more unexpected variety of peripheral tissues (Douet et al., 2017) than previously reported (Wadsworth et al., 2001). Those include bone marrow, kidney, salivary glands, skeletal muscle, pancreas, liver or heart in addition to tissue of the lymphoid system. In sporadic CJD, while prion distribution is more intense in the central and peripheral nervous systems, substantial amounts of prions have been found in the bone marrow (Huor et al., 2017), skin (Orru et al., 2017), kidney, lung, liver, adrenal glands (Takatsuki et al., 2016), and muscle (Peden et al., 2006; Rubenstein and Chang, 2013). Prions were also detected in biological fluids, notably blood, and urine of patients with sporadic and variant of CJD, often well before the onset of early clinical signs (Edgeworth et al., 2011a; Lacroux et al., 2014; Moda et al., 2014; Sawyer et al., 2015; Luk et al., 2016). Confirmed cases of iatrogenic transmission of vCJD by blood transfusion, and a probable case in a patient treated with coagulation factors VIII manufactured from plasma indicate that blood constitutes an effective source of iatrogenic contamination (Peden et al., 2004, 2010; Wroe et al., 2006). It was reported that the prion protein PrPC is a major contaminant of the purified urinary-derived gonadotropins used in infertility treatment (Van Dorsselaer et al., 2011). These elements raise specifically the question of prion biosafety of blood, blood-derived products, and urine-derived drug products. Analytical technics for securing drug manufacturing process from blood and urine must be reconsidered and implemented as those securing medical instrumentation, as recently recommended by the European Medicines Agency (EMA, 2018).
Prion presence in several tissues and bodily fluids raises the question of the most physiologically relevant PrPSc aggregates and of the best experimental approach for their inactivation. Most studies use brain macerates or microsomal brain fractions to contaminate steel wires or as spike for studying prion removal. Their high infectious titer compared to other extraneural tissues or fluids obviously increases the analytical sensitivity of the approach. However, PrPSc assemblies in bodily fluids or extraneural tissue may be of smaller size than in the brain. Compelling evidence indicate that different subassemblies with specific structural properties are co-accumulating in the brain at the disease terminal stage. These sub-assemblies harbor different size, different infectivity titer or seeding activity (for review Igel-Egalon et al., 2019a). Their capacity to bind steel wires or their retention properties may vary.
Further, the preparation of the spike or of the dilutions in the ad hoc experimental conditions may modify PrPSc assembly composition. In our previous work, we demonstrated that PrPSc assemblies have two levels of organization (Igel-Egalon et al., 2017). The first one is formed by the packing of oligomeric building blocks (called suPrP) into larger assemblies. In this organization, the cohesion forces are weak, as shown that rapid depolymerization following urea chaotropic treatment. The second level of organization is formed by suPrP itself. suPrP is a very stable oligomer between a dimer or a tetramer that resists >6–8 M urea. This study led us to conclude that PrPSc assemblies and their building blocks are in a highly dynamic equilibrium (Igel-Egalon et al., 2017, 2019a). A simple dilution of purified PrPSc assemblies was able to drive the equilibrium towards their depolymerization into suPrP. A dilution process may thus drive which PrPSc morphotype is submitted to the inactivation/retention process; in return PrPSc morphotype are likely to react to such process in a structural-dependent manner. To conciliate the advantage of using brain extract with the relevance of the model, one alternative could be the use of small PrPSc particles, obtained by fractionation experiments. We previously demonstrated that these assemblies have high specific infectivity values despite their small size (Tixador et al., 2010; Laferriere et al., 2013) and are relatively stable over time out of the conversion process (Igel-Egalon et al., 2019b).
As for the nature of the prion strain, these data collectively support the view that the retention/inactivation methods must be validated against the biological material for which they are intended to be used.
Efficient amplification of human prions by PMCA is strain dependent. It was initially believed that optimal amplification of human prion strains required absence of mismatch at codon 129 between the seed and the substrate. Thus, Jones et al. (2009) classified CJD subtypes in two distinct groups according to their “preference” for the PRNP genotype substrate. The first group, composed of vCJD, MM1, MM2, and MV1 sporadic prions, is preferentially amplified by the PRNP-129MM substrate. The second one, composed of VV1, VV2, and MV2 sporadic CJD prions, is more efficiently amplified by PRNP-129VV substrate. Despite this sequence compatibility, prions were not amplified to a degree of sensitivity sufficient for validating decontamination methods.
Other parameters can be adjusted to improve PMCA efficacy: (1) Obviously, the number of PMCA rounds can be increased. While it was initially suspected to increase the probability of de novo PrPSc formation in the absence of preexisting prions, carefully designed PMCA operating conditions suggested that spontaneous prion formation in PMCA reactions was rather a result of inadvertent cross-contamination (Cosseddu et al., 2011). Three to six rounds of PMCA are routinely used to amplify low amounts of human prions (Lacroux et al., 2014; Bougard et al., 2016; Concha-Marambio et al., 2016; Cassard et al., 2020); (2) The PrPC substrate can be optimized. The higher the PrPC concentration, the higher the sensitivity achieved (Mays et al., 2009; Moudjou et al., 2014, 2016), which lends support for the use of brain from transgenic mice overexpressing PrPC. Further, markedly improved sensitivities, including with human prions, were obtained when PMCA was performed with partially desialylated (Katorcha et al., 2015) or unglycosylated PrPC (Nishina et al., 2006; Camacho et al., 2019) as substrate. The authors attribute this phenomenon to sialic acid electrostatic repulsion forces and the glycan steric hindrance that may interfere with the conversion of PrPC by PrPSc. Whether this would work with all strains given their variable glycoform requirements (Khalili-Shirazi et al., 2005; Nishina et al., 2006) remains to be determined; (3) Conversion enhancers can be added to the PMCA reaction, including polyanions (Fernández-Borges et al., 2018), anionic lipids (Deleault et al., 2007), or dextran sulfate (Moudjou et al., 2016). Their mode of action on the conversion process remains poorly understood. Their impact on ionic strength, osmolarity, water molecule organization, PrPC or assemblies stability may play a role in the replication/templating process; (4) Physical change in the PMCA environment such as the reaction volume or the addition of microbeads in the substrate affected significantly the amplification efficacy with respect to the dilution achieved (Gonzalez-Montalban et al., 2011; Johnson et al., 2012; Moudjou et al., 2014).
The commonly shared view that microbeads addition increases the fragmentation of the newly converted PrPSc assemblies during the sonication steps and therefore multiplies the number of templating interface for conversion was recently challenged. By using sedimentation velocity to explore the quaternary structure of prion assemblies, we found no evidence for fragmentation in PMCA conditions with beads (Igel-Egalon et al., 2019b). Alternative hypotheses such as beads serving as thermoacoustic convertor (Povey et al., 2011) and/or as multidirectional secondary source may be considered.
We propose an alternative model to prion fragmentation to explain prion amplification during PMCA, based on the existence of a detailed-balance between PrPSc assemblies and suPrP (Chyba et al., 2020). A perturbation in prion environment would shift the balance towards PrPSc assemblies depolymerization into suPrP, thus multiplying the number of templating interface. We experimentally investigated this by studying the impact on PrPSc assemblies size of diluting infected brain homogenate in PMCA buffer, out of a replicative context and without sonication. As shown in Figures 2A–C, a >1:3 dilution was sufficient to depolymerize PrPSc assemblies into smaller objects, indicating an equilibrium displacement (Figure 2D). This was observed for three different prion strains, suggesting a possible generic effect. This observation supports the view that the physico-chemical properties of the PMCA buffer are enough to disrupt PrPSc assemblies, suggesting therefore that PrPSc fragmentation does not occur during bead-PMCA reactions.
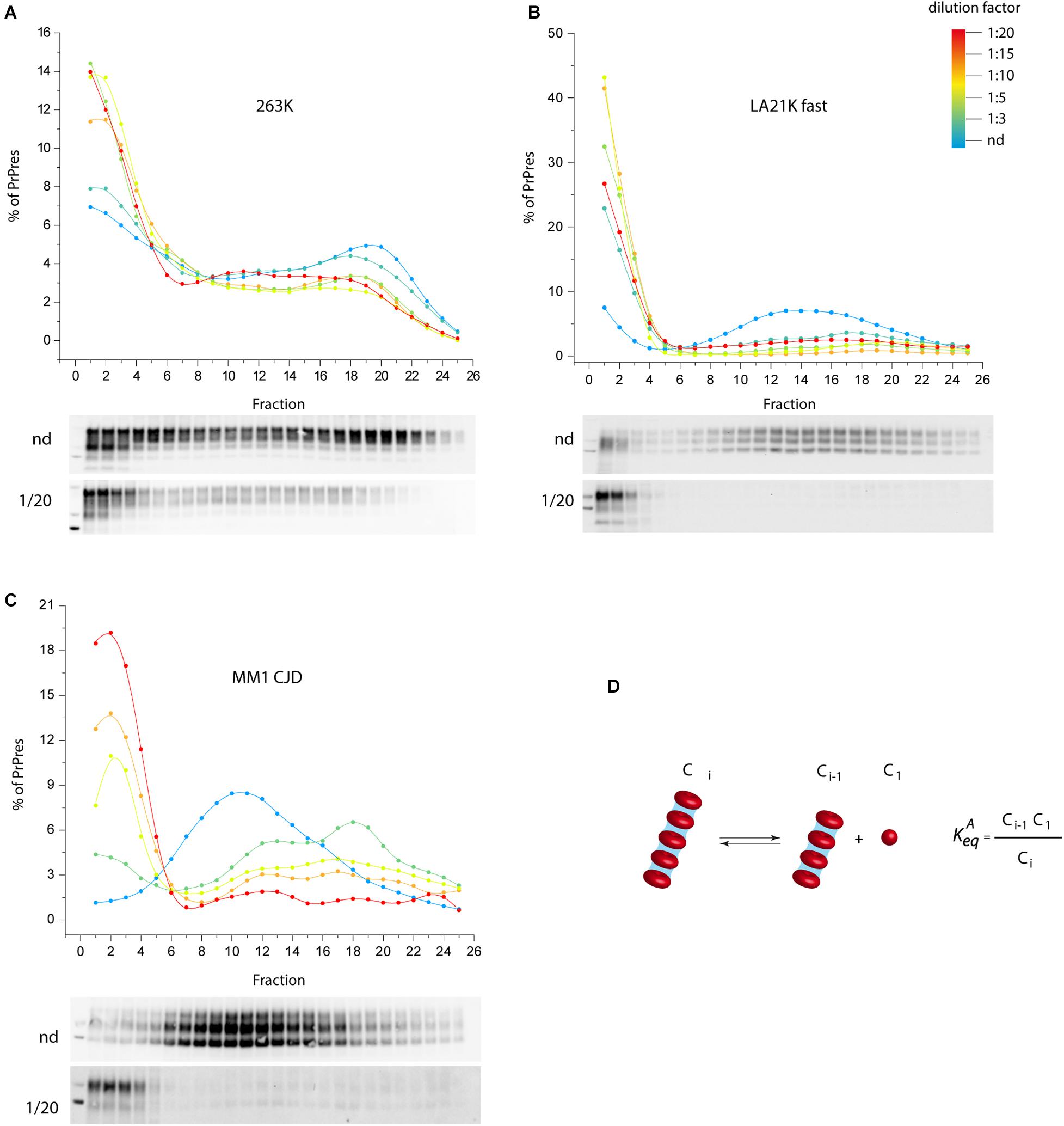
Figure 2. The disassembling effect of PMCA buffer on PrPSc assemblies. (A–C) Twenty percent infected brain homogenate from hamster 263K prions (Kimberlin and Walker, 1977), sheep-derived LA21K fast (Le Dur et al., 2017), and humanized MM1 sporadic CJD prions (Beringue et al., 2008a) were directly diluted [not diluted (nd), 1:3, 1:5, 1:15, 1:20 from blue to red line] in a PrP0/0 lysate made in PMCA buffer (thus in the absence of PrPC substrate). After a 4 h-incubation at 4°C in the absence of sonication, PrPSc assemblies quaternary structure was investigated by sedimentation velocity fractionation (Tixador et al., 2010). The collected fractions (numbered from top to bottom of the gradient) were analyzed for PrPSc content by immunoblot. The graphs show the relative amount of PrPSc per fraction. Representative immunoblots are shown. For the three prion strains, dilution induced a disassembling process generating small PrPSc objects. (D) PrPSc assemblies (Ci) dilution in PMCA buffer tends to displace a detailed-balance toward the formation of smaller objects here termed C1, which are assimilated to suPrP (Igel-Egalon et al., 2017).
Despite these major improvements, certain prion types remain unamplifiable by PMCA or do not achieve degrees of amplification requested by the medicine agencies (6 Log10 of magnitude (Ansm, 2018)). The MM1 subtype remains the most difficult sporadic CJD subtype to amplify. At variance with RT-QuIC (Orru et al., 2015b), there is no universal PMCA substrate for human CJD prions.
With all the potential factors improving PMCA sensitivity in mind, we optimized a substrate to amplify human sporadic CJD prions. First, we designed a new transgenic mouse line homozygous at the locus transgene that overexpressed approximately five- to sixfold the valine allele at codon 129 of human PrPC (BacV line) on a pure FVB/N PrP-knockout background (Passet et al., 2020). The transgene designed for targeting expression of human PrP in these mice is based on a large human BAC insert (the details of the transgenic mice will be published elsewhere). The brain of these mice was used as PrPC substrate in PMCA reactions (see Supplementary Material for the methods section). To improve the sensitivity of the reaction, we added polymers of dextran, one teflon microbeads and worked with a reduced reaction volume, as previously described in our so-called mb-PMCA protocol (Moudjou et al., 2014, 2016). As seed, we used brain homogenates from humanized mice in which we isolated and phenotypically characterized different sporadic CJD subtypes (Jaumain et al., 2016). Those were serially diluted and submitted to one to three rounds of mb-PMCA reaction. Humanized MV2, VV1, VV2 sporadic CJD prions, and vCJD prions were used. Positive reactions were obtained for these humanized strain types up to the 10–7 (MV2 subtype) to 10–10 (VV2 subtype) dilution (Figures 3A,B). Noticeably, mismatch at codon 129 between seed and substrate was not detrimental as the highest levels of amplification were observed with sporadic CJD VV2 prions and vCJD prions serially passaged onto transgenic mice expressing human-PrPC with Met at codon 129. The dynamic of amplification obtained with vCJD prions (limiting dilution at 10–9) or VV2 (10–9 or 10–10 depending on the allele on which the subtype was propagated) offered an analytical sensitivity compatible with the validation of prion inactivation methods. In the case of vCJD prions, it was more sensitive than animal bioassays by at least two orders of magnitude (Douet et al., 2014; Halliez et al., 2014).
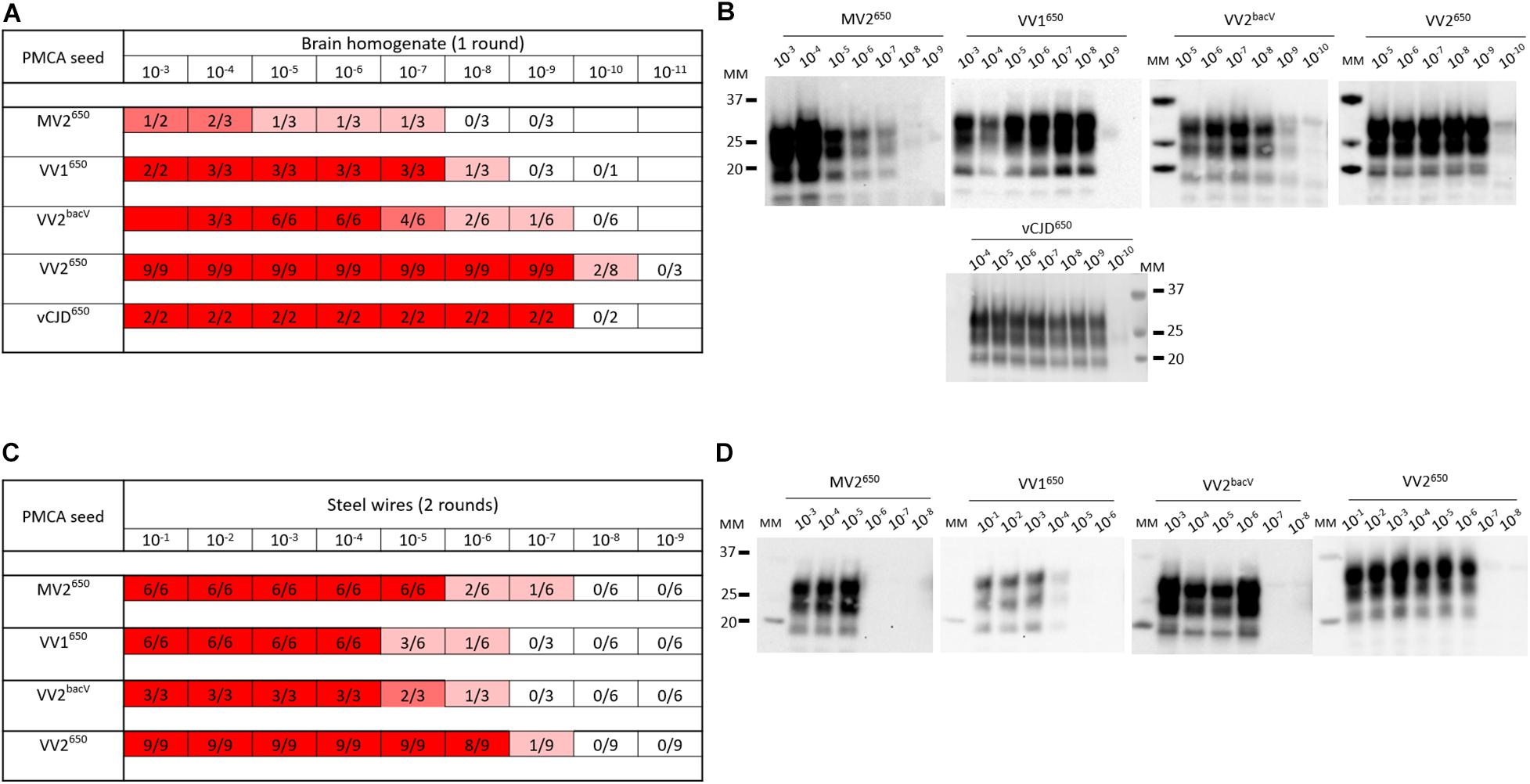
Figure 3. PMCA detection of sporadic and variant CJD prions in suspension or bound to steel wires using BacV-derived human PrP substrate. Serial dilutions of humanized sporadic CJD subtypes and variant CJD (vCJD) prions passaged into human PrP mice [tg650 line (Met129 allele) or BacV line (Val129 allele)] were used in suspension (A,B) or bound to steel wires (C,D). They were mixed with uninfected BacV brain homogenate and submitted to the mb-PMCA for one round (A,B) or two rounds (C,D) of 96 cycles of sonication/incubation for 48 h. The PMCA products were analyzed for PrPSc content by immunoblot. (A,C) Summary of the results obtained. The red boxes indicate the presence of PrPSc in PMCA products, the ratio in each box corresponds to the number of PrPSc-positive amplification products over the number of PMCA replicates performed. (B,D) Representative immunoblots (Sha31 anti-PrP antibody). MM, molecular weight markers.
In the context of the risk evaluation associated with the decontamination of medical instruments, we further evaluated the effectiveness of the BacV-derived mb-PMCA substrate to detect human prions bound onto steel surface. Briefly, fine stainless steel wires were contaminated with serial dilutions of prion-containing brain homogenate (Zobeley et al., 1999; Flechsig et al., 2001) and added directly to the mb-PMCA reaction mix as seed. As summarized in Figures 3C,D, two rounds of PMCA were enough to detect bound prions up to the 10–6–10–7 dilution. Again, these limiting dilution values were in the range required for appraisal of inactivation methods by using steel wire as translational model for medical instruments.
This mini-review addresses the questions raised by the implementations, by the competent authorities, of prionicidal product authorizations. Any validation procedure should be tested against prions for which the inactivant is intended to be used, suggesting that human or humanized prions should be used in fine. However, several pending questions are emerging, including the choice of the prion subtype and of the prion-containing biological matrix. Another operational aspect to consider is the high biohazard level to manipulate these agents as compared to laboratory 263K prions.
Given the correlation between PMCA and animal bioassay in measuring prion concentration bound on steel wires or in suspension, there is a proof of concept that this cell-free assay could complement and even replace animal bioassays. This would provide an economical and ethically sound method. Yet, certain human prion subtypes remain poorly amplifiable, limiting the potential relevance of the assay. Given the high-throughput, rapid format of the assay, the use of several prion subtypes may circumvent this. Animal bioassays using human or humanized prion are still considered as gold standard methods. These models and the PMCA assay are mostly manipulated in academic laboratories and not in Contract Research Organizations (CROs) that routinely perform studies for biocidal products authorizations. This may delay the time to market of these products.
VB, HR, and AI-E designed the study, analyzed the data and drafted the manuscript. J-LV, BP, JC, and VB designed the new mice line. AI-E, MM, LH, and FR performed the experiments. All authors gave their final approval of the submitted version.
This work was funded by the Fondation pour la Recherche Médicale (Equipe FRM DEQ20150331689).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.591024/full#supplementary-material
Atarashi, R., Wilham, J. M., Christensen, L., Hughson, A. G., Moore, R. A., Johnson, L. M., et al. (2008). Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 5, 211–212. doi: 10.1038/nmeth0308-211
Bélondrade, M., Jas-Duval, C., Nicot, S., Bruyére-Ostells, L., Mayran, C., Herzog, L., et al. (2020). Correlation between bioassay and protein misfoldingcyclic amplification for variant Creutzfeldt-Jakob disease decontamination studies. mSphere 5:e00649-19. doi: 10.1128/mSphere.00649-19
Belondrade, M., Nicot, S., Bèringue, V., Coste, J., Lehmann, S., and Bougard, D. (2016). Rapid and highly sensitive detection of variant creutzfeldt - jakob disease abnormal prion protein on steel surfaces by protein misfolding cyclic amplification: application to prion decontamination studies. PLoS One 11:e0146833. doi: 10.1371/journal.pone.0146833
Beringue, V., Herzog, L., Jaumain, E., Reine, F., Sibille, P., Le Dur, A., et al. (2012). Facilitated cross-species transmission of prions in extraneural tissue. Science 335, 472–475. doi: 10.1126/science.1215659
Beringue, V., Le Dur, A., Tixador, P., Reine, F., Lepourry, L., Perret-Liaudet, A., et al. (2008a). Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS One 3:e1419. doi: 10.1371/journal.pone.0001419
Beringue, V., Vilotte, J. L., and Laude, H. (2008b). Prion agent diversity and species barrier. Vet. Res. 39:47. doi: 10.1051/vetres:2008024
Bishop, M. T., Will, R. G., and Manson, J. C. (2010). Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc. Natl. Acad. Sci. U.S.A. 107, 12005–12010. doi: 10.1073/pnas.1004688107
Bonda, D. J., Manjila, S., Mehndiratta, P., Khan, F., Miller, B. R., Onwuzulike, K., et al. (2016). Human prion diseases: surgical lessons learned from iatrogenic prion transmission. Neurosurg. Focus 41:E10.
Bougard, D., Brandel, J. P., Belondrade, M., Beringue, V., Segarra, C., Fleury, H., et al. (2016). Detection of prions in the plasma of presymptomatic and symptomatic patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 8:370ra182. doi: 10.1126/scitranslmed.aag1257
Bruce, M., Chree, A., Mcconnell, I., Foster, J., Pearson, G., and Fraser, H. (1994). Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos. Trans. R. Soc. Lond. B Biol. Sci. 343, 405–411. doi: 10.1098/rstb.1994.0036
Burke, C. M., Mark, K. M. K., Walsh, D. J., Noble, G. P., Steele, A. D., Diack, A. B., et al. (2020a). Identification of a homology-independent linchpin domain controlling mouse and bank vole prion protein conversion. PLoS Pathog. 16:e1008875. doi: 10.1371/journal.ppat.1008875
Burke, C. M., Walsh, D. J., Mark, K. M. K., Deleault, N. R., Nishina, K. A., Agrimi, U., et al. (2020b). Cofactor and glycosylation preferences for in vitro prion conversion are predominantly determined by strain conformation. PLoS Pathog. 16:e1008495. doi: 10.1371/journal.ppat.1008495
Camacho, M. V., Telling, G., Kong, Q., Gambetti, P., and Notari, S. (2019). Role of prion protein glycosylation in replication of human prions by protein misfolding cyclic amplification. Lab. Invest. 99, 1741–1748. doi: 10.1038/s41374-019-0282-1
Cassard, H., Huor, A., Espinosa, J. C., Douet, J. Y., Lugan, S., Aron, N., et al. (2020). Prions from sporadic creutzfeldt-jakob disease patients propagate as strain mixtures. mBio 11:e00393-20. doi: 10.1128/mBio.00393-20
Chapuis, J., Moudjou, M., Reine, F., Herzog, L., Jaumain, E., Chapuis, C., et al. (2016). Emergence of two prion subtypes in ovine PrP transgenic mice infected with human MM2-cortical Creutzfeldt-Jakob disease prions. Acta Neuropathol. Commun. 4, 2–15.
Chyba, M., Kotas, J., Beringue, V., Eblen, C., Igel-Egalon, A., Kravchenko, Y., et al. (2020). An alternative model to prion fragmentation based on the detailed balance between PrP<sup>Sc</sup>and suPrP. bioRxiv[Preprint].
Collinge, J. (2001). Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24, 519–550. doi: 10.1146/annurev.neuro.24.1.519
Collinge, J., and Clarke, A. R. (2007). A general model of prion strains and their pathogenicity. Science 318, 930–936. doi: 10.1126/science.1138718
Concha-Marambio, L., Pritzkow, S., Moda, F., Tagliavini, F., Ironside, J. W., Schulz, P. E., et al. (2016). Detection of prions in blood from patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 8:370ra183.
Cosseddu, G. M., Nonno, R., Vaccari, G., Bucalossi, C., Fernandez-Borges, N., Di Bari, M. A., et al. (2011). Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog. 7:e1002370. doi: 10.1371/journal.ppat.1002370
Deleault, N. R., Harris, B. T., Rees, J. R., and Supattapone, S. (2007). Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746. doi: 10.1073/pnas.0702662104
Deleault, N. R., Piro, J. R., Walsh, D. J., Wang, F., Ma, J., Geoghegan, J. C., et al. (2012a). Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 109, 8546–8551. doi: 10.1073/pnas.1204498109
Deleault, N. R., Walsh, D. J., Piro, J. R., Wang, F., Wang, X., Ma, J., et al. (2012b). Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U.S.A. 109, E1938–E1946.
DGS (2011). DGS/RI3/2011/449 du 1er décembre 2011 relative à l’actualisation des recommandations visant à réduire les risques de transmission d’agents transmissibles non conventionnels lors des actes invasifs. Paris: F.M.O. Health.
Diack, A. B., Ritchie, D., Bishop, M., Pinion, V., Brandel, J. P., Haik, S., et al. (2012). Constant transmission properties of variant Creutzfeldt-Jakob disease in 5 countries. Emerg. Infect. Dis. 18, 1574–1579. doi: 10.3201/eid1810.120792
Douet, J. Y., Lacroux, C., Aron, N., Head, M. W., Lugan, S., Tillier, C., et al. (2017). Distribution and quantitative estimates of variant Creutzfeldt-Jakob disease prions in tissues of clinical and asymptomatic patients. Emerg. Infect. Dis. 23, 946–956. doi: 10.3201/eid2306.161734
Douet, J. Y., Zafar, S., Perret-Liaudet, A., Lacroux, C., Lugan, S., Aron, N., et al. (2014). Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt-Jakob disease. Emerg. Infect. Dis. 20, 114–117. doi: 10.3201/eid2001.130353
Edgeworth, J. A., Farmer, M., Sicilia, A., Tavares, P., Beck, J., Campbell, T., et al. (2011a). Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377, 487–493. doi: 10.1016/s0140-6736(10)62308-2
Edgeworth, J. A., Jackson, G. S., Clarke, A. R., Weissmann, C., and Collinge, J. (2009). Highly sensitive, quantitative cell-based assay for prions adsorbed to solid surfaces. Proc. Natl. Acad. Sci. U.S.A. 106, 3479–3483. doi: 10.1073/pnas.0813342106
Edgeworth, J. A., Sicilia, A., Linehan, J., Brandner, S., Jackson, G. S., and Collinge, J. (2011b). A standardized comparison of commercially available prion decontamination reagents using the Standard Steel-Binding Assay. J. Gen. Virol. 92, 718–726. doi: 10.1099/vir.0.027201-0
Elder, A. M., Henderson, D. M., Nalls, A. V., Wilham, J. M., Caughey, B. W., Hoover, E. A., et al. (2013). In vitro detection of prionemia in TSE-infected cervids and hamsters. PLoS One 8:e80203. doi: 10.1371/journal.pone.0080203
EMA (2018). CHMP position statement on Creutzfeldt-Jakob disease and plasma-derived and urine-derived medicinal products. London: E.M. Agency.
Eraña, H., Charco, J. M., Di Bari, M. A., Díaz-Domínguez, C. M., López-Moreno, R., Vidal, E., et al. (2019). Development of a new largely scalable in vitro prion propagation method for the production of infectious recombinant prions for high resolution structural studies. PLoS Pathog 15:e1008117. doi: 10.1371/journal.ppat.1008117
Espinosa, J. C., Comoy, E. E., Marin-Moreno, A., Aguilar-Calvo, P., Birling, M.-C., Pitarch, J. L., et al. (2019). Transgenic mouse models expressing human and macaque prion protein exhibit similar prion susceptibility on a strain-dependent manner. Sci. Rep. 9:15699.
Fernández-Borges, N., Di Bari, M. A., Eraña, H., Sánchez-Martín, M., Pirisinu, L., Parra, B., et al. (2018). Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol. 135, 179–199. doi: 10.1007/s00401-017-1782-y
Fernie, K., Hamilton, S., and Somerville, R. A. (2012). Limited efficacy of steam sterilization to inactivate vCJD infectivity. J. Hosp. Infect. 80, 46–51. doi: 10.1016/j.jhin.2011.09.004
Flechsig, E., Hegyi, I., Enari, M., Schwarz, P., Collinge, J., and Weissmann, C. (2001). Transmission of scrapie by steel-surface-bound prions. Mol. Med. 7, 679–684. doi: 10.1007/bf03401958
Gibbs, C.J. Jr, Gajdusek, D. C., Asher, D. M., Alpers, M. P., Beck, E., Daniel, P. M., et al. (1968). Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 161, 388–389.
Giles, K., Glidden, D. V., Beckwith, R., Seoanes, R., Peretz, D., Dearmond, S. J., et al. (2008). Resistance of bovine spongiform encephalopathy (BSE) prions to inactivation. PLoS Pathog. 4:e1000206. doi: 10.1371/journal.ppat.1000206
Giles, K., Woerman, A. L., Berry, D. B., and Prusiner, S. B. (2017). Bioassays and Inactivation of Prions. Cold Spring Harb. Perspect. Biol 9:a023499. doi: 10.1101/cshperspect.a023499
Gill, O. N., Spencer, Y., Richard-Loendt, A., Kelly, C., Brown, D., Sinka, K., et al. (2020). Prevalence in Britain of abnormal prion protein in human appendices before and after exposure to the cattle BSE epizootic. Acta Neuropathol. 139, 965–976. doi: 10.1007/s00401-020-02153-7
Gonzalez-Montalban, N., Makarava, N., Ostapchenko, V. G., Savtchenk, R., Alexeeva, I., Rohwer, R. G., et al. (2011). Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 7:e1001277. doi: 10.1371/journal.ppat.1001277
Groveman, B. R., Raymond, G. J., Campbell, K. J., Race, B., Raymond, L. D., Hughson, A. G., et al. (2017). Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Pathog. 13:e1006623. doi: 10.1371/journal.ppat.1006623
Halliez, S., Reine, F., Herzog, L., Jaumain, E., Haik, S., Rezaei, H., et al. (2014). Accelerated, spleen-based titration of variant Creutzfeldt-Jakob disease infectivity in transgenic mice expressing human prion protein with sensitivity comparable to that of survival time bioassay. J. Virol. 88, 8678–8686. doi: 10.1128/jvi.01118-14
Henderson, D. M., Davenport, K. A., Haley, N. J., Denkers, N. D., Mathiason, C. K., and Hoover, E. A. (2015). Quantitative assessment of prion infectivity in tissues and body fluids by real-time quaking-induced conversion. J. Gen. Virol. 96, 210–219. doi: 10.1099/vir.0.069906-0
Hughson, A. G., Race, B., Kraus, A., Sangare, L. R., Robins, L., Groveman, B. R., et al. (2016). Inactivation of prions and amyloid seeds with hypypochlorous acid. PLoS Pathog. 12:e1005914. doi: 10.1371/journal.ppat.1005914
Huor, A., Douet, J. Y., Lacroux, C., Lugan, S., Tillier, C., Aron, N., et al. (2017). Infectivity in bone marrow from sporadic CJD patients. J. Pathol. 243, 273–278. doi: 10.1002/path.4954
Huor, A., Espinosa, J. C., Vidal, E., Cassard, H., Douet, J. Y., Lugan, S., et al. (2019). The emergence of classical BSE from atypical/Nor98 scrapie. Proc. Natl. Acad. Sci. U.S.A. 116, 26853–26862. doi: 10.1073/pnas.1915737116
Igel-Egalon, A., Bohl, J., Moudjou, M., Herzog, L., Reine, F., Rezaei, H., et al. (2019a). Heterogeneity and architecture of pathological prion protein assemblies: time to revisit the molecular basis of the Prion replication process? Viruses 11:429. doi: 10.3390/v11050429
Igel-Egalon, A., Laferriere, F., Moudjou, M., Bohl, J., Mezache, M., Knapple, T., et al. (2019b). Early stage prion assembly involves two subpopulations with different quaternary structures and a secondary templating pathway. Commun. Biol. 2:363.
Igel-Egalon, A., Moudjou, M., Martin, D., Busley, A., Knapple, T., Herzog, L., et al. (2017). Reversible unfolding of infectious prion assemblies reveals the existence of an oligomeric elementary brick. PLoS Pathog. 13:e1006557. doi: 10.1371/journal.ppat.1006557
Jaumain, E., Quadrio, I., Herzog, L., Reine, F., Rezaei, H., Andreoletti, O., et al. (2016). Absence of evidence for a causal link between Bovine Spongiform encephalopathy strain variant L-BSE and known forms of sporadic Creutzfeldt-Jakob disease in human PrP Transgenic Mice. J. Virol. 90, 10867–10874. doi: 10.1128/jvi.01383-16
Johnson, C. J., Aiken, J. M., Mckenzie, D., Samuel, M. D., and Pedersen, J. A. (2012). Highly efficient amplification of chronic wasting disease agent by protein misfolding cyclic amplification with beads (PMCAb). PLoS One 7:e35383. doi: 10.1371/journal.pone.0035383
Johnson, C. J., Pedersen, J. A., Chappell, R. J., Mckenzie, D., and Aiken, J. M. (2007). Oral transmissibility of prion disease is enhanced by binding to soil particles. PLoS Pathog 3:e93. doi: 10.1371/journal.ppat.0030093
Jones, M., Peden, A. H., Yull, H., Wight, D., Bishop, M. T., Prowse, C. V., et al. (2009). Human platelets as a substrate source for the in vitro amplification of the abnormal prion protein (PrP) associated with variant Creutzfeldt-Jakob disease. Transfusion 49, 376–384. doi: 10.1111/j.1537-2995.2008.01954.x
Katorcha, E., Makarava, N., Savtchenko, R., and Baskakov, I. V. (2015). Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci. Rep. 5:16912. doi: 10.1038/srep16912
Khalili-Shirazi, A., Summers, L., Linehan, J., Mallinson, G., Anstee, D., Hawke, S., et al. (2005). PrP glycoforms are associated in a strain-specific ratio in native PrPSc. J. Gen. Virol. 86, 2635–2644. doi: 10.1099/vir.0.80375-0
Kimberlin, R. H., and Walker, C. (1977). Characteristics of a short incubation model of scrapie in the golden hamster. J. Gen. Virol. 34, 295–304. doi: 10.1099/0022-1317-34-2-295
Klohn, P. C., Stoltze, L., Flechsig, E., Enari, M., and Weissmann, C. (2003). A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. U.S.A. 100, 11666–11671. doi: 10.1073/pnas.1834432100
Kraus, A., Saijo, E., Metrick, M. A. II, Newell, K., Sigurdson, C. J., Zanusso, G., et al. (2019). Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. 137, 585–598. doi: 10.1007/s00401-018-1947-3
Lacroux, C., Comoy, E., Moudjou, M., Perret-Liaudet, A., Lugan, S., Litaise, C., et al. (2014). Preclinical Detection of Variant CJD and BSE prions in blood. PLoS Pathog. 10:e1004202. doi: 10.1371/journal.ppat.1004202
Laferriere, F., Tixador, P., Moudjou, M., Chapuis, J., Sibille, P., Herzog, L., et al. (2013). Quaternary structure of pathological prion protein as a determining factor of strain-specific prion replication dynamics. PLoS Pathog. 9:e1003702. doi: 10.1371/journal.ppat.1003702
Lasmezas, C. I., Deslys, J. P., Demaimay, R., Adjou, K. T., Hauw, J. J., and Dormont, D. (1996). Strain specific and common pathogenic events in murine models of scrapie and bovine spongiform encephalopathy. J. Gen. Virol. 77(Pt. 7), 1601–1609. doi: 10.1099/0022-1317-77-7-1601
Lasmézas, C. I., Deslys, J. P., Demaimay, R., Adjou, K. T., Lamoury, F., Dormont, D., et al. (1996). BSE transmission to macaques. Nature 381, 743–744. doi: 10.1038/381743a0
Le Dur, A., Lai, T. L., Stinnakre, M. G., Laisne, A., Chenais, N., Rakotobe, S., et al. (2017). Divergent prion strain evolution driven by PrPC expression level in transgenic mice. Nat. Commun. 8:14170.
Luk, C., Jones, S., Thomas, C., Fox, N. C., Mok, T. H., Mead, S., et al. (2016). Diagnosing sporadic Creutzfeldt-Jakob disease by the detection of abnormal prion protein in patient Urine. JAMA Neurol. 73, 1454–1460. doi: 10.1001/jamaneurol.2016.3733
Marin-Moreno, A., Aguilar-Calvo, P., Moudjou, M., Espinosa, J. C., Beringue, V., and Torres, J. M. (2019). Thermostability as a highly dependent prion strain feature. Sci. Rep. 9:11396.
Mays, C. E., Titlow, W., Seward, T., Telling, G. C., and Ryou, C. (2009). Enhancement of protein misfolding cyclic amplification by using concentrated cellular prion protein source. Biochem. Biophys. Res. Commun. 388, 306–310. doi: 10.1016/j.bbrc.2009.07.163
McGuire, L. I., Peden, A. H., Orru, C. D., Wilham, J. M., Appleford, N. E., Mallinson, G., et al. (2012). Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 72, 278–285. doi: 10.1002/ana.23589
Minor, P., Newham, J., Jones, N., Bergeron, C., Gregori, L., Asher, D., et al. (2004). Standards for the assay of Creutzfeldt-Jakob disease specimens. J. Gen. Virol. 85, 1777–1784.
Moda, F., Gambetti, P., Notari, S., Concha-Marambio, L., Catania, M., Park, K. W., et al. (2014). Prions in the urine of patients with variant Creutzfeldt-Jakob disease. N. Engl. J. Med. 371, 530–539.
Moda, F., Suardi, S., Di Fede, G., Indaco, A., Limido, L., Vimercati, C., et al. (2012). MM2-thalamic Creutzfeldt-Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathol. 22, 662–669. doi: 10.1111/j.1750-3639.2012.00572.x
Moreno, J. A., and Telling, G. C. (2017). Insights into mechanisms of transmission and pathogenesis from transgenic mouse models of Prion diseases. Methods Mol. Biol. 1658, 219–252. doi: 10.1007/978-1-4939-7244-9_16
Mori, T., Atarashi, R., Furukawa, K., Takatsuki, H., Satoh, K., Sano, K., et al. (2016). A direct assessment of human prion adhered to steel wire using real-time quaking-induced conversion. Sci. Rep. 6:24993.
Moudjou, M., Chapuis, J., Mekrouti, M., Reine, F., Herzog, L., Sibille, P., et al. (2016). Glycoform-independent prion conversion by highly efficient, cell-based, protein misfolding cyclic amplification. Sci. Rep. 6:29116.
Moudjou, M., Sibille, P., Fichet, G., Reine, F., Chapuis, J., Herzog, L., et al. (2014). Highly infectious prions generated by a single round of microplate-based protein misfolding cyclic amplification. mBio 5:e00829-13.
Nishina, K. A., Deleault, N. R., Mahal, S. P., Baskakov, I., Luhrs, T., Riek, R., et al. (2006). The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry 45, 14129–14139.
Nonno, R., Di Bari, M. A., Cardone, F., Vaccari, G., Fazzi, P., Dell’omo, G., et al. (2006). Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2:e12. doi: 10.1371/journal.ppat.0020012
Orru, C. D., Bongianni, M., Tonoli, G., Ferrari, S., Hughson, A. G., Groveman, B. R., et al. (2014). A test for Creutzfeldt-Jakob disease using nasal brushings. N. Engl. J. Med. 371, 519–529. doi: 10.1056/nejmoa1315200
Orru, C. D., Groveman, B. R., Hughson, A. G., Zanusso, G., Coulthart, M. B., and Caughey, B. (2015a). Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. mBio 6, e002451-14. doi: 10.1128/mBio.02451-14
Orru, C. D., Groveman, B. R., Raymond, L. D., Hughson, A. G., Nonno, R., Zou, W., et al. (2015b). Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog. 11:e1004983. doi: 10.1371/journal.ppat.1004983
Orru, C. D., Yuan, J., Appleby, B. S., Li, B., Li, Y., Winner, D., et al. (2017). Prion seeding activity and infectivity in skin samples from patients with sporadic Creutzfeldt-Jakob disease. Sci. Transl. Med. 9:eaam7785. doi: 10.1126/scitranslmed.aam7785
Passet, B., Castille, J., Makhzami, S., Truchet, S., Vaiman, A., Floriot, S., et al. (2020). The Prion-like protein Shadoo is involved in mouse embryonic and mammary development and differentiation. Sci. Rep. 10:6765.
Peden, A., Mccardle, L., Head, M. W., Love, S., Ward, H. J., Cousens, S. N., et al. (2010). Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 16, 296–304. doi: 10.1111/j.1365-2516.2009.02181.x
Peden, A. H., Head, M. W., Ritchie, D. L., Bell, J. E., and Ironside, J. W. (2004). Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364, 527–529. doi: 10.1016/s0140-6736(04)16811-6
Peden, A. H., Ritchie, D. L., Head, M. W., and Ironside, J. W. (2006). Detection and localization of PrPSc in the skeletal muscle of patients with variant, iatrogenic, and sporadic forms of Creutzfeldt-Jakob disease. Am. J. Pathol. 168, 927–935. doi: 10.2353/ajpath.2006.050788
Peretz, D., Supattapone, S., Giles, K., Vergara, J., Freyman, Y., Lessard, P., et al. (2006). Inactivation of prions by acidic sodium dodecyl sulfate. J. Virol. 80, 322–331. doi: 10.1128/jvi.80.1.322-331.2006
Povey, M. J. W., Moore, J. D., Braybrook, J., Simons, H., Belchamber, R., Raganathan, M., et al. (2011). Investigation of bovine serum albumin denaturation using ultrasonic spectroscopy. Food Hydrocoll. 25, 1233–1241. doi: 10.1016/j.foodhyd.2010.11.011
Pritzkow, S., Wagenfuhr, K., Daus, M. L., Boerner, S., Lemmer, K., Thomzig, A., et al. (2011). Quantitative detection and biological propagation of scrapie seeding activity in vitro facilitate use of prions as model pathogens for disinfection. PLoS One 6:e20384. doi: 10.1371/journal.pone.0020384
Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. doi: 10.1126/science.6801762
Prusiner, S. B., Cochran, S. P., Groth, D. F., Downey, D. E., Bowman, K. A., and Martinez, H. M. (1982). Measurement of the scrapie agent using an incubation time interval assay. Ann. Neurol. 11, 353–358. doi: 10.1002/ana.410110406
Prusiner, S. B., Groth, D. F., Cochran, S. P., Masiarz, F. R., Mckinley, M. P., and Martinez, H. M. (1980). Molecular properties, partial purification, and assay by incubation period measurements of the hamster scrapie agent. Biochemistry 19, 4883–4891. doi: 10.1021/bi00562a028
Raymond, G. J., Race, B., Orrú, C. D., Raymond, L. D., Bongianni, M., Fiorini, M., et al. (2020). Transmission of CJD from nasal brushings but not spinal fluid or RT-QuIC product. Ann. Clin. Transl. Neurol. 7, 932–944. doi: 10.1002/acn3.51057
Rhoads, D. D., Wrona, A., Foutz, A., Blevins, J., Glisic, K., Person, M., et al. (2020). Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology 95, e1017–e1026.
Rossi, M., Candelise, N., Baiardi, S., Capellari, S., Giannini, G., Orru, C. D., et al. (2020). Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 140, 49–62. doi: 10.1007/s00401-020-02160-8
Rubenstein, R., and Chang, B. (2013). Re-assessment of PrP(Sc) distribution in sporadic and variant CJD. PLoS One 8:e66352. doi: 10.1371/journal.pone.0066352
Saborio, G. P., Permanne, B., and Soto, C. (2001). Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813. doi: 10.1038/35081095
Saijo, E., Metrick, M. A. II, Koga, S., Parchi, P., Litvan, I., Spina, S., et al. (2020). 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 139, 63–77. doi: 10.1007/s00401-019-02080-2
Saunders, S. E., Bartz, J. C., Vercauteren, K. C., and Bartelt-Hunt, S. L. (2011). An enzymatic treatment of soil-bound prions effectively inhibits replication. Appl. Environ. Microbiol. 77, 4313–4317. doi: 10.1128/aem.00421-11
Sawyer, E. B., Edgeworth, J. A., Thomas, C., Collinge, J., and Jackson, G. S. (2015). Preclinical detection of infectivity and disease-specific PrP in blood throughout the incubation period of prion disease. Sci. Rep. 5:17742.
Scialò, C., Tran, T. H., Salzano, G., Novi, G., Caponnetto, C., Chiò, A., et al. (2020). TDP-43 real time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2:fcaa142.
Shikiya, R. A., and Bartz, J. C. (2011). In vitro generation of high-titer prions. J. Virol. 85, 13439–13442. doi: 10.1128/jvi.06134-11
Takatsuki, H., Fuse, T., Nakagaki, T., Mori, T., Mihara, B., Takao, M., et al. (2016). Prion-seeding activity is widely distributed in tissues of sporadic Creutzfeldt-Jakob disease patients. eBioMed. 12, 150–155. doi: 10.1016/j.ebiom.2016.08.033
Tateishi, J., Ohta, M., Koga, M., Sato, Y., and Kuroiwa, Y. (1979). Transmission of chronic spongiform encephalopathy with kuru plaques from humans to small rodents. Ann. Neurol. 5, 581–584. doi: 10.1002/ana.410050616
Tixador, P., Herzog, L., Reine, F., Jaumain, E., Chapuis, J., Le Dur, A., et al. (2010). The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 6:e1000859. doi: 10.1371/journal.ppat.1000859
Van Dorsselaer, A., Carapito, C., Delalande, F., Schaeffer-Reiss, C., Thierse, D., Diemer, H., et al. (2011). Detection of prion protein in urine-derived injectable fertility products by a targeted proteomic approach. PLoS One 6:e17815. doi: 10.1371/journal.pone.0017815
Wadsworth, J. D., Joiner, S., Hill, A. F., Campbell, T. A., Desbruslais, M., Luthert, P. J., et al. (2001). Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358, 171–180. doi: 10.1016/s0140-6736(01)05403-4
Weber, P., Giese, A., Piening, N., Mitteregger, G., Thomzig, A., Beekes, M., et al. (2007). Generation of genuine prion infectivity by serial PMCA. Vet Microbiol 123, 346–357. doi: 10.1016/j.vetmic.2007.04.004
Weissmann, C., Li, J., Mahal, S. P., and Browning, S. (2011). Prions on the move. EMBO Rep 12, 1109–1117. doi: 10.1038/embor.2011.192
WHO (1999). “WHO infection control guidelines for transmissible spongiform encephalopathies,” in World Health Organization Emerging and Other Communicable Diseases, ed. W.H. Organization (Geneva: WHO).
Wilham, J. M., Orru, C. D., Bessen, R. A., Atarashi, R., Sano, K., Race, B., et al. (2010). Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 6:e1001217. doi: 10.1371/journal.ppat.1001217
Wroe, S. J., Pal, S., Siddique, D., Hyare, H., Macfarlane, R., Joiner, S., et al. (2006). Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet 368, 2061–2067. doi: 10.1016/s0140-6736(06)69835-8
Yan, Z. X., Stitz, L., Heeg, P., Pfaff, E., and Roth, K. (2004). Infectivity of prion protein bound to stainless steel wires: a model for testing decontamination procedures for transmissible spongiform encephalopathies. Infect. Control Hosp. Epidemiol. 25, 280–283. doi: 10.1086/502392
Yull, H. M., Ironside, J. W., and Head, M. W. (2009). Further characterisation of the prion protein molecular types detectable in the NIBSC Creutzfeldt-Jakob disease brain reference materials. Biologicals 37, 210–215. doi: 10.1016/j.biologicals.2009.01.009
Zerr, I., and Parchi, P. (2018). Sporadic Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 153, 155–174.
Keywords: prion, sporadic CJD, amplification, PMCA, surface, decontamination
Citation: Moudjou M, Castille J, Passet B, Herzog L, Reine F, Vilotte J-L, Rezaei H, Béringue V and Igel-Egalon A (2020) Improving the Predictive Value of Prion Inactivation Validation Methods to Minimize the Risks of Iatrogenic Transmission With Medical Instruments. Front. Bioeng. Biotechnol. 8:591024. doi: 10.3389/fbioe.2020.591024
Received: 03 August 2020; Accepted: 05 November 2020;
Published: 01 December 2020.
Edited by:
Jesus R. Requena, University of Santiago de Compostela, SpainReviewed by:
Rodrigo Morales, University of Texas Health Science Center at Houston, United StatesCopyright © 2020 Moudjou, Castille, Passet, Herzog, Reine, Vilotte, Rezaei, Béringue and Igel-Egalon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angélique Igel-Egalon, YW5nZWxpcXVlLmVnYWxvbkBpbnJhZS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.