 Wenliang Hao1
Wenliang Hao1 Qiaoqing Chen
Qiaoqing Chen Wenjing Cui
Wenjing Cui Zhemin Zhou
Zhemin Zhou- 1The Key Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi, China
- 2Jiangnan University (Rugao) Food Biotechnology Research Institute, Jiangsu, China
Bacillus subtilis is an important Gram-positive bacterium for industrial biotechnology, which has been widely used to produce diverse high-value added chemicals and industrially and pharmaceutically relevant proteins. Robust and versatile toolkits for genome editing in B. subtilis are highly demanding to design higher version chassis. Although the Streptococcus pyogenes (Sp) CRISPR-Cas9 has been extensively adapted for genome engineering of multiple bacteria, it has many defects, such as higher molecular weight which leads to higher carrier load, low deletion efficiency and complexity of sgRNA construction for multiplex genome editing. Here, we designed a CRISPR-Cpf1-based toolkit employing a type V Cas protein, Cpf1 from Francisella novicida. Using this platform, we precisely deleted single gene and gene cluster in B. subtilis with high editing efficiency, such as sacA, ganA, ligD & ligV, and bac operon. Especially, an extremely large gene cluster of 38 kb in B. subtilis genome was accurately deleted from the genome without introducing any unexpected mutations. Meanwhile, the synthetic platform was further upgraded to a version for multiplex genome editing, upon which two genes sacA and aprE were precisely and efficiently deleted using only one plasmid harboring two targeting sequences. In addition, we successfully inserted foreign genes into the genome of the chassis using the CRISPR-Cpf1 platform. Our work highlighted the availability of CRISPR-Cpf1 to gene manipulation in B. subtilis, including the flexible deletion of a single gene and multiple genes or a gene cluster, and gene knock-in. The designed genome-editing platform was easily and broadly applicable to other microorganisms. The novel platforms we constructed in this study provide a promising tool for efficient genome editing in diverse bacteria.
Introduction
Bacillus subtilis, a well-characterized Gram-positive bacterium, has been regarded to be a “generally recognized as safe” (GRAS) microbe that can naturally secrete numerous extracellular proteins (Correa et al., 2020). B. subtilis is an ideal organism for industrial application, however, the available genetic tools are insufficient compared to other widely used microbial chassis, such as Escherichia coli and Saccharomyces cerevisiae (Keasling, 2010; Dong and Zhang, 2014). Gene-editing is of great utilization to reprogramming and reshaping the genome of synthetic chassis (Bai et al., 2018). In previous studies, several B. subtilis genome editing tools have been developed. Common gene knockout systems in B. subtilis include Cre/loxP recombination (Suzuki et al., 2005; Choi et al., 2015), MazF counter-selectable markers (Westbrook et al., 2016), and synthetic gene circuits (Jeong et al., 2015). Cre/loxP is a recombination system based on resistance selection markers, which can knock out or insert genes by recombining homologous fragments with Cre recombinase. However, this method needs to introduce a foreign resistance gene, which is not in line with the needs of eco-friendly hosts. The counter-selectable method based on MazF knocks out genes by introducing a toxin-antitoxin (TA) system from E. coli (Zhang et al., 2006). Although this method does not introduce resistance markers on chromosomes, its efficiency is very low. Recently, gene knockout methods based on synthetic gene circuits have been constructed in B. subtilis (Jeong et al., 2015). Although it was unnecessary to introduce foreign resistance marker genes, the efficiency of knocking out large gene clusters is very low. Thus, the genome engineering of B. subtilis needs an effective method without antibiotic resistance markers (Suzuki et al., 2005).
Recently, the Class 2 clustered regularly interspaced short palindromic repeat (CRISPR) system has been employed as a powerful tool for genome editing and transcription regulation in many organisms, including bacteria (Jiang et al., 2015; Westbrook et al., 2016; Wu et al., 2018), yeast (Bao et al., 2015), plant (Gao et al., 2017), and mammals (Hwang et al., 2013). CRISPR systems are divided into two categories on the basis of the configuration of their effector molecules (Zetsche et al., 2015). Different from the class 1 CRISPR system, which requires various Cas proteins to coordinate with each other and bind to the crRNA to form a ribonucleoprotein (RNP) complex, class 2 CRISPR system employ a large single-component Cas protein in conjunction with crRNA to mediate genome editing (Zetsche et al., 2015). In type II CRISPR system, Cas9 from Streptococcus pyogenes is widely used because it has been studied very clearly. Zhang et al. (2016) constructed the AIO system in B. subtilis ATCC 6051a by using Cas9 from Streptococcus pyogenes. Using this system, the authors disrupted specific genes (including srfC, spoIIAC, nprE, aprE, and amyE) in B. subtilis ATCC 6051a with 33–53% efficiency (Zhang et al., 2016). The production of β-cyclodextrin glycosyltransferase by modified B. subtilis ATCC 6051a (ΔsrfC, ΔspoIIAC, ΔnprE, ΔaprE, and ΔamyE) is more than 2.5 times that of wild-type B. subtilis ATCC 6051a. Similarly, Westbrook et al. (2016) developed a CRISPR-Cas9-based toolkit that can knockout, knock-in, knockdown and point mutations of target genes in B. subtilis. They employed a strategy of expressing Cas9 and transcribing gRNA on chromosomes, and the authors believe that this method obviates the instability of multicopy plasmid in the host and the pressure of plasmid on the host. CRISPR-Cas9 system requires three essential factors to cleave the genomic DNA: the CRISPR RNA (crRNA), the trans-activating CRISPR RNA (tracrRNA), and the Cas9 nuclease (Chen et al., 2017). In this system, crRNA binds partially to the complementary tracrRNA prior to association with Cas9, allowing to form a gRNA-Cas9 complex. The complex identifies specific target site of genomic DNA based on the PAM sequence and generates blunt-ended double strand breakage (DSB) (Hong et al., 2018). Although CRISPR-Cas9 system has achieved huge success in genome editing in different organisms, it has prominent drawbacks, including severe off-target effects and certain unknown toxicity, resulting in low efficiency in a specific case (Fu et al., 2013; Hsu et al., 2013; Jiang et al., 2017; Hong et al., 2018). Recently, an array of novel Cas proteins has been increasingly developed, such as Cpf1 (Zetsche et al., 2015; Hong et al., 2018), Cas12b (Teng et al., 2018; Strecker et al., 2019), and CasX (Liu et al., 2019) from diverse bacteria. Among these Cas proteins, Cpf1 is a protein from bacterial immune system, which has recently been engineered as a genome editing tool in Clostridium difficile (Hong et al., 2018), Corynebacterium glutamicum (Jiang et al., 2017), and rice (Wang et al., 2017).
Cpf1, derived from a type V CRISPR system, is an effector Cas protein distinct from Cas9 in structure and function (Zetsche et al., 2015). The prominent advantage of Cpf1 over Cas9 is that the maturity of CRISPR arrays does not require additional tracrRNA, so that Cpf1 is able to process pre-crRNA to mature crRNA (Zetsche et al., 2015, 2017). This feature resolves the drawback in the construction of multiple or large expression constructs using Cas9, upon which the procedure of multiplexed-gene editing is possible to be simplified (Zetsche et al., 2015, 2017; Fonfara et al., 2016). Moreover, CRISPR-Cpf1 complexes efficiently cleave the target DNA utilizing a T-rich PAM sequence rather than the G-rich PAM sequence in CRISPR-Cas9 system (Zetsche et al., 2015), which is probably more efficient in B. subtilis. In addition, Cpf1 leaves a staggered end with a 5′ overhang after cleaving DNA, which facilitates repairing the nicked DNA by non-homologous end joining (NHEJ) or homology-directed repair (HDR) after cutting (Zetsche et al., 2015; Jiang et al., 2017).
Previous studies have shown that Cpf1 is greatly superior to Cas9 in genome editing (Kim et al., 2016). Compared with Cas9, Cpf1 is a small protein that contains a single well–identified nuclease domain rather than two nuclease domains for Cas9 (Zetsche et al., 2015). For instance, Cpf1 has only 1300 amino acids (Zetsche et al., 2015), which is more suitable to deliver Cas–sgRNA complex. Importantly, Cpf1 is lower in potential toxicity to the host compared to Cas9 (such as SpCas9) (Jiang et al., 2017). Therefore, it is a more suitable candidate Cas protein for genome editing (Zetsche et al., 2017). In previous studies, CRISPR–Cpf1 system has been engineered as a powerful genome–editing tool and applied to different organisms, including rice (Wang et al., 2017), soybean (Kim et al., 2017), mouse (Hur et al., 2016), zebrafish (Hwang et al., 2013), human cell (Kim et al., 2016), Mycobacterium smegmatis (Yan et al., 2017), yeast (Buchmuller et al., 2019), and C. difficile (Hong et al., 2018). Recently, Wu et al. (2020) constructed a gene editing system based on CRISPR–Cpf1 in B. subtilis, and used the system to perform gene knock–out, knock–in, and regulation of target gene expression. The authors employed a two–plasmid model and improved the efficiency of gene editing (including double gene knockout, knock–in, and point mutation) by overexpressing mutated NgAgo∗ on plasmids. However, it is unclear whether the efficiency of gene knock-out by CRISPR-Cpf1 system in B. subtilis will be varied with the size of the target gene fragment. And it is still to be determined whether CRISPR-Cpf1 system can mediate deletion of large gene cluster in B. subtilis.
In this study, we broadened genome editing toolkit based on CRISPR-Cpf1 system employing different strategies in B. subtilis and successfully applied the CRISPR-Cpf1 system to the deletion of single gene of various sizes as well as multiplex-gene editing in high efficiency (Figure 1). Importantly, we also established an efficient chromosome-integration genome editing (CIGE) platform to achieve precise insertion of gene of interest. These results exhibit that the CRISPR-Cpf1-based tools we have designed and built in this study have highly flexible property that is not only used to deletion of gene of diverse sizes but also serve as a proficient platform to precisely insert heterologous genes into chromosome. This toolbox is of great importance to develop high version of chassis by precisely editing the genome B. subtilis, which is great potential to extend the synthetic biology of B. subtilis.
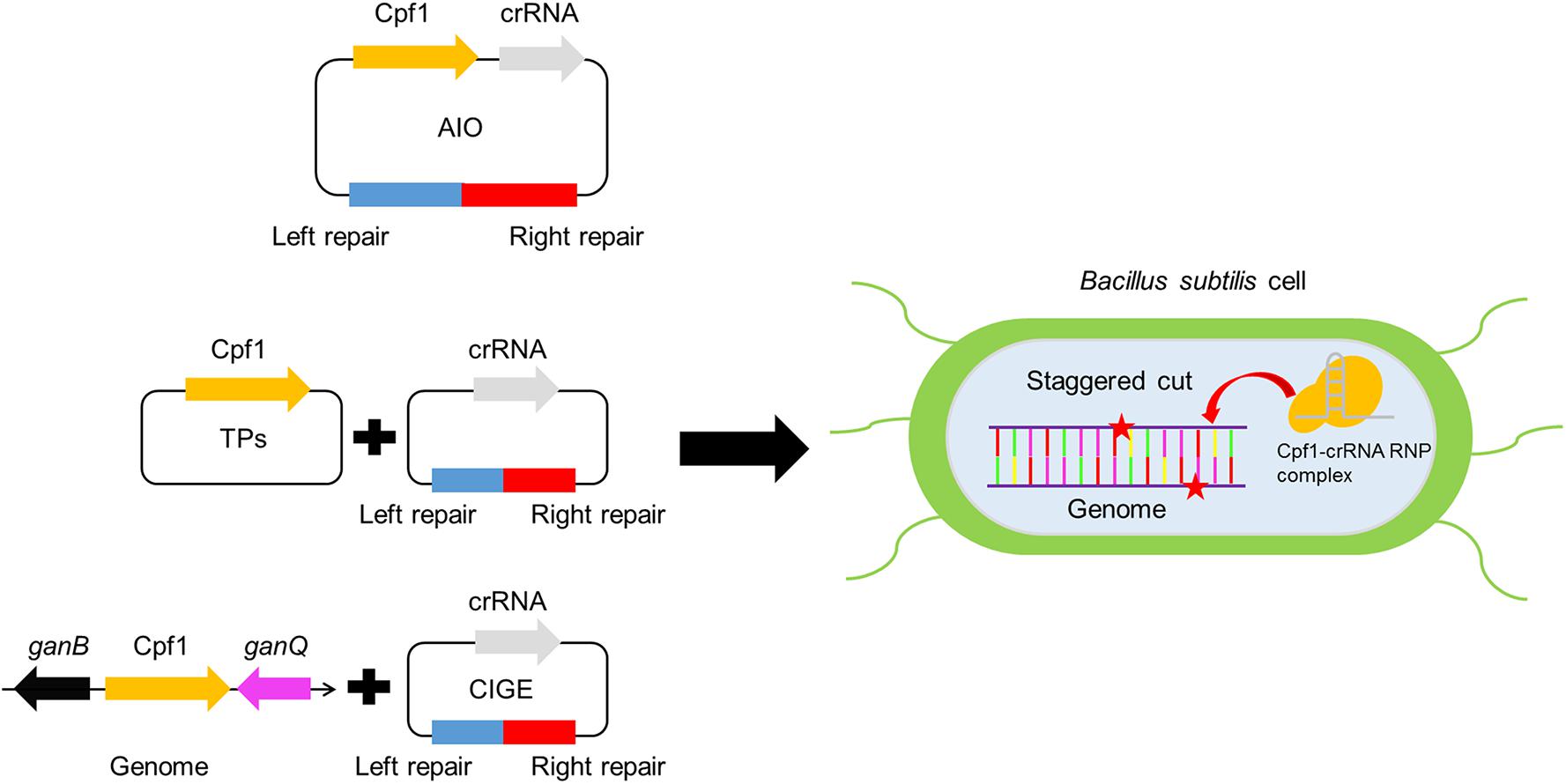
Figure 1. Development of genome editing toolkit based on CRISPR-Cpf1 in Bacillus subtilis.
Materials and Methods
Bacterial Strains and Growth Conditions
All the E. coli and B. subtilis strains used in this study are listed in Supplementary Table S1. The JM109 clone E. coli strains (General Biosystems, China) was used as the general host for plasmid construction and gene cloning. The transformation of the JM109 E. coli strains was conducted through chemical transformation using Competent cells from General Biosystems. E. coli strains were grown in Luria-Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl, pH 7.0) supplement with ampicillin (100 μg/mL) or spectinomycin (100 μg/mL) when necessary. Transformation of B. subtilis cells was carried out by the two-step transformation procedure (Anagnostopoulos and Spizizen, 1961). B. subtilis strains were cultivated in Luria-Bertani (LB) medium supplement with spectinomycin (100 μg/mL), chloramphenicol (6 μg/mL) or kanamycin (50 μg/mL) and LB solid medium supplemented with 1% glucose.
Plasmids Construction
All the plasmids used in this study were listed in Supplementary Table S1. All the DNA primers used in this study were listed in Supplementary Table S2. All the crRNA used in this study were listed in Supplementary Table S3. The nucleotide and amino acid sequences of FnCpf1 and SpCas9 nucleases were shown in the complementary sequences. The crRNA and sgRNA sequences used in this paper were also listed in the Supplementary Sequences.
Construction of All-in-One System
The pHTsacA plasmid was derived from pHT01, an expression plasmid for B. subtilis (MoBiTec, Göttingen, Germany). P43 promoter and RBS were amplified from pBSG03 using the primers pHT-P43-F and pHT-P43-R while the backbone of the plasmid, harboring lacI gene, was amplified using the primer pHT-P43-b-F and pHT-P43-b-R. Then, P43 promoter and RBS were inserted into the shuttle vectors using Gibson assembly (Gibson et al., 2009), yielding pHT-P43-RBS. Accordingly, Cpf1 gene was cloned into pHT-P43-RBS using the primers P43-FnCpf1-F/R and P43-FnCpf1-b-F/R, yielding pHT-P43-RBS-FnCpf1. The sacA-targeting crRNA expression cassette under the control of a strong promoter Pveg was synthesized by GENEWIZ Inc., Ltd. (Wuxi, China) and cloned into pHT-P43-RBS-FnCpf1 using the primers pHT-pVeg-sacAcrRNA-F/R and pHT-sacAcr-b-F/R, producing pHT-FnCpf1-sacAcrRNA. Primer pair sacA-HA-F/R and pHT-HA-b-F/R was used to amplify the 1.2-kb donor DNA template and its bone, respectively. Finally, sacA homologous arm was cloned into pHT-FnCpf1-sacAcrRNA, yielding pHTsacA (for the detailed construction method of pHTganA-Cpf1,pHTganA-Cas9, and pHTDV, refer to the Supplementary Method in the Supplementary Material).
The plasmid pHTsfGFPKiT was used to delete sacA from chromosome of B. subtilis. To construct this plasmid, the sfGFP DNA fragment was amplified from pBPylbp-sfGFP-Ter plasmid using primers sfGFPKi-F/R. The backbone of the plasmid pHTsacA was amplified at the same time using primers pHT-all-sfGFPKi-b-F/R. Then, sfGFP was cloned between the homologous arm of the plasmid pHTsacA, yielding pHTsfGFPKi.
Construction of Two Plasmids System
To construct gene deletion tool of two-plasmid format, gene Cpf1 was firstly cloned into pHT01 vector using the primer pHT-Pgrac-Cpf1-F/R and pHT-Pgrac-Cpf1-b-F/R by the Gibson assembly, generating pHT01-Cpf1 activated by Isopropyl-β-D-thiogalactopyranoside (IPTG). To generate the vector for expression of the sacA-targeting crRNA, we cloned sacA-targeting crRNA expression cassette to pAD123 vector, harboring coding sequences (CDSs) of gfpmut3a and rep60 from pAT1060 origin using the primers pAD123-pVeg-sacAcrT-F/R and pAD123-sacAcrT-b-F/R, generating pAD-pVeg-sacAcrRNA. The sacA homologous arm was cloned into pAD-pVeg-sacAcrRNA using the primer pAD-sacAH-F/R and pAD-sacAH-b-F/R, yielding pADsacA.
Construction of Chromosomally Integrated Genome Editing System (CIGE)
To construct a CIGE system that had Cpf1 integrated in the chromosome in B. subtilis, pAX01-Cpf1 under the control of PxylA was constructed to this end using the primer pAX01-Cpf1-F/R and pAX01-Cpf1-b-F/R. Then, rrnBT1 terminator and rrnBT2 terminator was fused downstream of Cpf1 expression cassette. Chloramphenicol-resistant gene (cat), lox66-71 site and the homologous arm of ganA gene was amplified using the primer lacA-Cpf1-F/R, the PCR fragment was integrated specifically into ganA site of genome, yielding the strain BS-ganA’-Cpf1. To construct plasmid harboring sacA-targeting crRNA, we cloned sacAcrRNA expression cassette and sacA homologous arm from pHTsacA using the primer pB-pVeg-sacAHA-F/R and pB-pVeg-sacAHA-b-F/R into pBSG03 vector, producing the plasmid pBsacA (for the detailed construction method of pBbac and pBpps, refer to the Supplementary Method in the Supplementary Material).
pBsfGFPKi was constructed to insert super folder green fluorescence protein (sfGFP) into the sacA locus. The sacAcrRNA expression cassette, the homologous arms of sacA, and the sequence of sfGFP were amplified from pHTsfGFPKi using the primers pB-sacAHA-sfGFP-F/R. Meanwhile, the backbone of the plasmid pHTsfGFPKi was amplified using the primers pB-sacAHA-sfGFP-b-F/R. Accordingly, the two PCR products were fused by Gibson assembly, generating pBsfGFPKi.
The plasmid pB-sacAKo-aprEKi was constructed by one-pot Golden Gate assembly reaction (Engler et al., 2009). Specifically, pB-sacAcrRNA-HA used primers AmpM-BsaI-F/R and RepM-BsaI-F/R to mutate ampicillin-resistance gene and repB replication gene to eliminate BsaI recognition sites. The purpose to do this was to prevent the BsaI restriction enzyme from non-specifically cutting other positions in the reaction of Golden Gate assembly. Then, we amplified the 500-bp homologous arms upstream and downstream of aprE from the plasmid pB-aprEHA using the primers pB-aprEHA-F/R. The amplicon was further fused to the plasmid pBsacA using primers pB-aprEHA-b-F/R, producing pBsacA-aprEHA. The gene of mCherry was amplified from the plasmid pBP43-GFP-mCherry using primers pB-mCherry-F/R prior to fusing with the linearized plasmid pBsacA-aprEHA (amplified by primers pB-mCherry-b-F/R) by Gibson assembly, generating pBsacA-aprEHA-mCh. As Golden gate assembly requires two BsaI restriction site and orthogonal overhangs to ensure the target fragment correctly being inserted into the recipient vector, we used pBsacA-aprEHA-mCh as a template and perform two rounds of rPCR to obtain two BsaI restriction sites using primers BsaI-1-F/R and BsaI-2-F/R, so as to enable the resulting plasmid being digested by BsaI. The crRNA sequence targeting aprE was amplified from pHTaprE using primers aprEcrRNA-BsaI-F/R. To construct pB-sacAKo-aprEKi, a restriction-ligation was performed in a mixture containing crRNA expression cassette targeting aprE, the recipient vector pBsacA-aprEHA-mCh, BsaI enzyme and T4 DNA ligase, generating pB-sacAKo-aprEKi. For double deletion of sacA and aprE, a single crRNA array was synthesized from GENEWIZ, Inc. (Suzhou, China), which contained PvegM promoter, crRNA targeting sacA and aprE, as well as BT5 terminator (screened by our laboratory). The array was amplified from pUC57 plasmid using primers SA-BT5-F/R, the resulting PCR product was inserted into pB-sacAHA-aprEHA plasmid (containing the homologous arms of sacA and aprE) with primers SA-BT5-b-F/R by Gibson Assembly, yielding the recombinant plasmid pB-PvegM-SAKo.
Plasmid Curing
To cure mutant strains of the pBSG so as to enable their use in a second round of genome editing, the mutant strains were inoculated into LB medium with a final concentration of 0.0005% SDS without antibiotics (Trevors, 1986), and then incubated at 37°C, 200 rpm for 20 h. Accordingly, the culture was diluted and spread on LB plates without antibiotics. Colonies were carefully picked up and dotted at the same positions on two LB plates with and without antibiotics, respectively. Antibiotics-sensitive colonies were picked and propagated in 5-mL LB medium. Then, plasmid-free mutants were further confirmed through PCR. For elimination of pHT01 and pAD123 plasmid, we referred to previous research and achieved it (Yamashiro et al., 2011; So et al., 2017).
Results and Discussion
Design of a CRISPR-Cpf1 in All-in-One (AIO) System for Deletion of Large Genes in B. subtilis
To achieve the goal for highly efficient editing the genome in B. subtilis, we employed a type V Cas protein, Cpf1 from Francisella novicida to design an all-in-one system (AIO). In this system, crRNA and Cpf1 were expressed constitutively by Pveg and P43 promoters, respectively. The expressed crRNA carried Cpf1 to a specific site of the target gene, where the cleavage of Cpf1 to the target gene would cause the precise recombination of homologous segments, leading to the deletion of the target gene (Figure 2A). To compare the performance of gene editing between Cpf1 and Cas9, we constructed a CRISPR-Cas9-based AIO system. To verify the functionality of two AIO systems, we selected the ganA gene (gene of medium size) to identify the efficiency of deletion mediated by the two AIO systems. The ganA, which was of 2064 bp in length, encodes β-galactosidase, which participates in the degradation of galactose. Disruption or complete deletion of ganA does not affect the growth of host. Therefore, we chose ganA gene as the target to explore the functionality of the two designed CRISPR systems (CRISPR-Cpf1 and CRISPR-Cas9) for deletion of large genes. The two editing plasmids, pHTganA-Cpf1 (refer to addgene #158647) and pHTganA-Cas9, were separately introduced into B. subtilis 168. The transformants were separately picked up and then inoculated into newly prepared LB medium to grow. Then the culture was diluted to appropriate density and spread onto LB agar plates. Twenty clones were randomly picked for cPCR to screen ganA-disrupted mutants from pHTganA-Cpf1 and pHTganA-Cas9 plates, respectively. The results showed that 20 clones of ganA-disrupted mutants had smaller cPCR product than that of wild-type strain (ck), indicating that ganA has been successfully deleted on pHTganA-Cpf1 plate (efficiency was 100%, Figure 2B). However, we only screened 15 ganA-deletion mutants on the pHTganA-Cas9 plate (efficiency was 75%, Figure 2B). Furthermore, the precision of deletion sites for these engineered strains were validated by sequencing the cPCR products (Figure 2B). These data displayed that the efficiency of CRISPR-Cpf1 and CRISPR-Cas9 systems was sufficient for general gene editing. However, CRISPR-Cpf1 system would be more suitable for larger fragments to be knocked out. Therefore, our following experiments all aimed at the engineering design of CRISPR-Cpf1 system. To further verify the functionality of the AIO system based on CRISPR-Cpf1, we selected sacA to identify the efficiency of deletion mediated by the system. The sacA (NC_000964:3902858), which was of 2400 bp in length, encodes sucrose-6-phosphate hydrolase, which is not an essential gene for B. subtilis (Zhu and Stulke, 2018). In most integrated systems for B. subtilis, sacA site was widely used to an integration site (Radeck et al., 2013). Here, we selected PAM sequence 5′-TTTG-3′ (Zetsche et al., 2015) in sacA gene, and then constructed PHTsacA plasmid harboring Cpf1 gene and target-sacA crRNA that under the control of constitutive promoters P43 and Pveg, respectively (Figure 2A). The 1200-bp homologous arm was inserted downstream of the CRISPR-Cpf1 expression cassette. Based on the deletion design, the fragment should be 240 bp after deletion. Plasmid pHTsacA was then introduced into B. subtilis 168. The transformations were picked, inoculated into 5 mL of LB medium, and incubated aerobically at 37°C overnight. Then the culture was spread onto LB plates. Twelve colonies were randomly collected to perform colony PCR (cPCR) to verify the deletion efficiency. The PCR product should be of 1655 bp if sacA-deletion was unsuccessful using designed primers, in contrast, it should be of 240 bp smaller than that of the wild type B. subtilis 168. The data showed that introduction of pHTsacA into B. subtilis 168 resulted in 100% deletion (Figure 2C). These results were further confirmed by sequencing of one successfully deleted mutant (Figure 2C), indicating that the designed synthetic CRISPR-Cpf1 tool has relatively high editing efficiency targeting genes with short lengths.
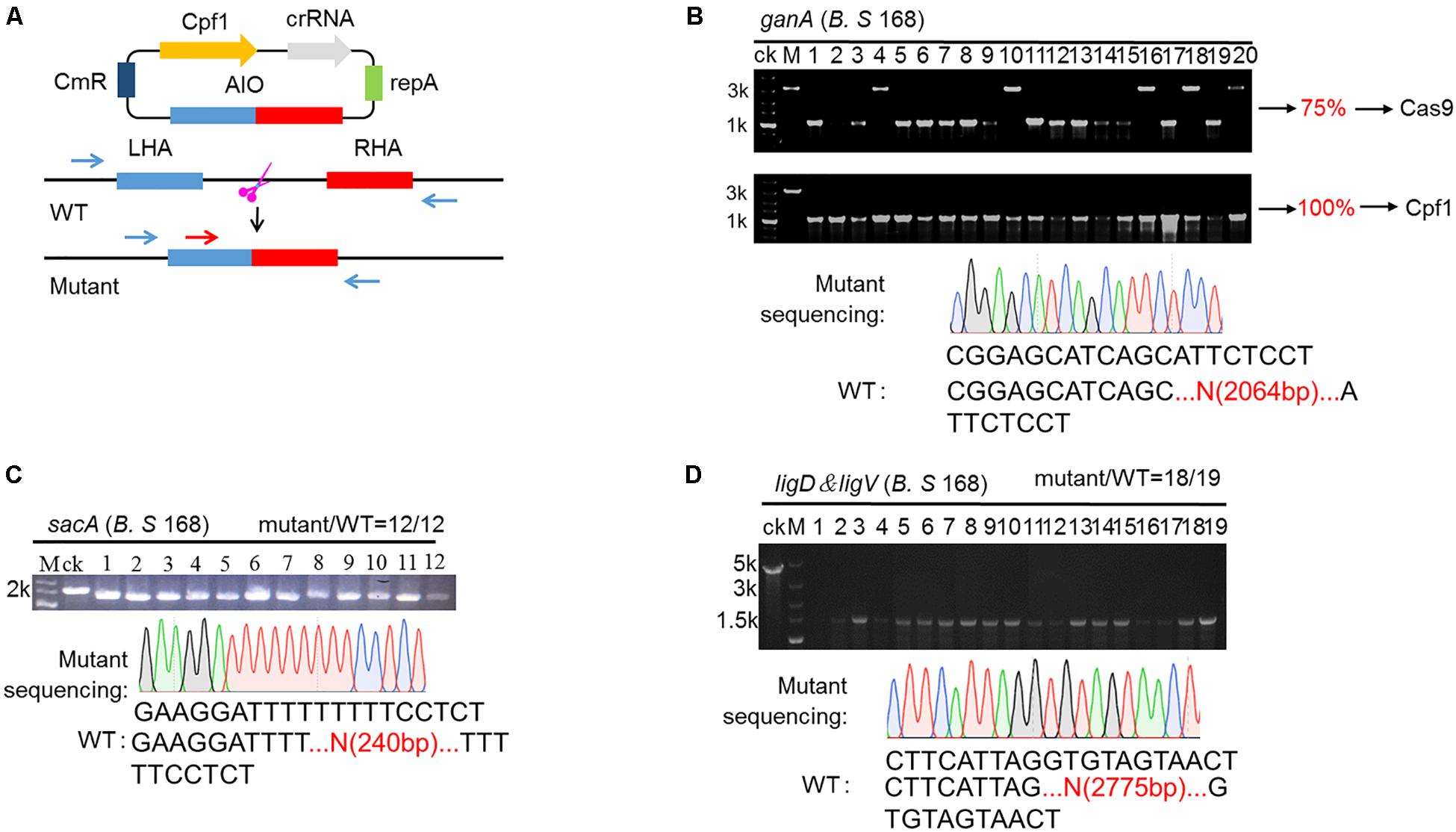
Figure 2. CRISPR-Cpf1-mediated genome editing in the Bacillus subtilis via using AIO system. (A) Schematic illustration of the editing procedures. The blue arrows are the primers utilized for PCR validation of the editing efficiency. The red arrow is the primer used for sequencing. (B) Deletion of ganA gene mediated by AIO system based on CRISPR-Cpf1 and CRISPR-Cas9 in B. subtilis 168. The efficiency of deletion of ganA by AIO system based on CRISPR-Cpf1 was 20/20 (100%). The efficiency of deletion of ganA gene by AIO system based on CRISPR-Cas9 was 15/20 (75%). Lane M, the 5-k DNA marker from Takara with number on the left representing the band size in kb. The lane labeled “ck” is the PCR product from the wild-type strain as a control. (C) AIO-mediated deletion of the sacA gene in the B. subtilis 168 strain. The editing efficiency was 12/12. (D) AIO-mediated deletion of the ligD & ligV genes in the B. subtilis 168 strain. The editing efficiency was 18/19.
Furthermore, to further identify the available range of AIO-based CRISPR-Cpf1 system, we employed the system to delete larger gene cluster. The ligD & ligV gene cluster, which is of 2775 bp in length, was selected as the target. The gene cluster ligD & ligV is involving in the non-homologous end-joining (NHEJ) process (Pitcher et al., 2007). Although the gene cluster is important in maintaining chromosomal stability in bacteria, it is non-fatal to B. subtilis when ligD & ligV cluster is deficient. Therefore, we chose ligD & ligV gene cluster as the target to test the functionality of deletion of large gene cluster using designed CRISPR-Cpf1 system. The AIO system was selected to construct the editing system (Figure 2A). The pHTDV plasmid harboring ligD & ligV-targeting crRNA and the homologous arms was constructed in the similar procedure as that of pHTganA but had longer homologous arms (600 bp). The editing plasmid, pHTDV, was introduced into B. subtilis 168. The transformants were picked and then inoculated into newly prepared LB medium to grow. Then the culture was diluted to appropriate density and spread onto LB agar plates. Nineteen colonies were randomly picked up to screen ligD & ligV-disrupted mutants by cPCR. The results showed that 18 of the 19 clones were confirmed to be the ligD & ligV-deficient strains, suggesting an editing efficiency of 94.7% (Figure 2D). Furthermore, the precision of deletion sites for these engineered strains were validated by sequencing the cPCR products. The data displayed that all these ganA, sacA and ligD & ligV mutants had accurate deletion sites as we had designed (Figures 2B–D).
In this work, we authenticated that the AIO system was capable of being applied to delete single gene with high efficiency in B. subtilis. AIO-based gene editing employing CRISPR-Cas9 was also constructed in B. subtilis ATCC 6051a (Zhang et al., 2016). The prominent feature of AIO was that Cas protein, sgRNA and homologous arms were all concentrated on one plasmid. When the gene editing was completed, the plasmid will be eliminated and no foreign genes will be introduced into the genome. However, the plasmids of AIO were often large, which led to low transformation efficiency and instability of plasmids (Supplementary Table S5). To improve the stability of plasmid replication, we use pHT01 vector as the skeleton of AIO, which carries a repA replicon. The vector pHT01 belongs to a medium copy plasmid. Because repA protein in the way of θ replication to multiply, the pHT01 vector is relatively stable. If we want to further improve the stability of the plasmid, we can integrate Cpf1 into the host genome, which can reduce the replication pressure of plasmid. Therefore, to break through the limitations of AIO system and provide flexible tools to facilitate gene editing in complex genetic context, more robustness, and efficiency of CRISPR-Cpf1 system should be built to allow reliable genome editing.
Construction and Validation of Two-Plasmid (TP)-Based, Cpf1-Mediated Gene-Deleting System in B. subtilis
To broaden an alternative form of CRISPR-Cpf1 system, the two plasmids (TP) system, and verify whether the system was able to efficiently delete genes in B. subtilis. In this system, we also chose sacA as the target. First, we constructed plasmid pHT01-Cpf1 (refer to addgene #158648), harboring Cpf1 under the control of the inducible promoter Pgrac (Figure 3A). Because pHT01 and pAD123 contained the same chloramphenicol resistance gene, we substituted the chloramphenicol-resistant gene from pHT01 vector with spectinomycin-resistant gene, allowing to screen the transformants with different antibiotics (Figure 3A). We constructed plasmid pADsacA (refer to addgene #158649), derived from the backbone of pAD123, to constitutively express the crRNA targeting sacA mediated by Pveg promoter (Figure 3A). Then, we sought to identify the efficiency of disruption of sacA by TPs system. We sequentially transformed plasmids pHT01-Cpf1 and pAD-sacA into B. subtilis. The two types of transformants were picked up, and inoculated into LB medium to propagation. When OD600 reached approximately 0.5, Isopropyl-β-D-thiogalactopyranoside (IPTG) with final concentration of 1 mM was added to induce the expression of Cpf1. Then the culture was spread onto LB agar plates. After incubated overnight, 23 colonies were randomly picked to perform cPCR to verify the deletion efficiency. The results exhibit that all the colonies (23/23) were the successfully deleted mutant harboring a 240 bp-deleted sequence of sacA (Figure 3B). To further validate the accuracy of gene deletion, we confirmed the sequence of disrupted sacA by sequencing. The data validated the successful deletion of 240 bp of sacA, suggesting that the deletion efficiency mediated by TPs strategy is as high as 100% (Figure 3B). To further compare the performance CRISPR-Cpf1 and CRISPR-Cas9 using two-plasmid strategy to knock out the same gene in B. subtilis 168, we selected ganA (2064 bp) gene as our target. The results showed that the knockout efficiency of CRISPR-Cpf1 system was much higher than that of CRISPR-Cas9 system (10/11 vs. 6/11, Supplementary Figure S2). These data suggest that the function of CRISPR-Cpf1 in knockout of single gene is higher than that of CRISPR-Cas9 with the same strategy (two-plasmid system).
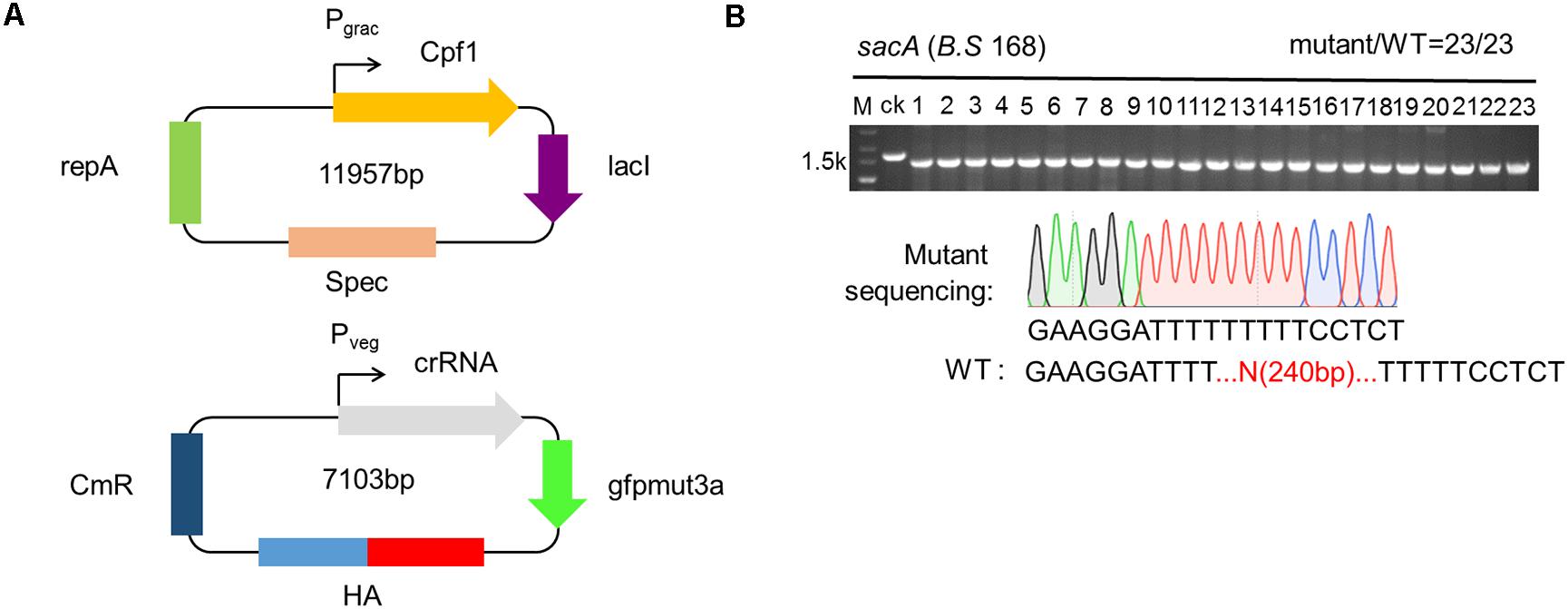
Figure 3. Construction of the two plasmids (TPs) for genome engineering system. (A) Schematic illustration of pHTCpf1 plasmid and pADsacA plasmid containing sacA crRNA transcription module as well as donor DNA template. (B) TPs-mediated disruption of the sacA gene in the Bacillus subtilis 168 strain. The editing efficiency was 20/20. The lane labeled “ck” is the PCR product from the wild-type strain as a control.
CRISPR-Cpf1-Based CIGE (CCB-CIGE) Platform Is Superior to Highly Efficient Gene Insertion in B. subtilis
Gene insertion is another critical issue to genome editing. Therefore, robust and efficient insertion system is valuable tool for precisely editing the genome of B. subtilis. To evaluate the availability of the CRISPR-Cpf1-based AIO system for insertion of heterologous gene into the genome of B. subtilis, we firstly constructed an AIO plasmid pHTsfGFPKi, of which the expression of Cpf1 and sacA-targeting crRNA was controlled by P43 and Pveg, respectively. The coding region of sfGFP was flanked by upstream and downstream homologous arms of sacA (Figure 4A). We introduced pHTsfGFPKi into B. subtilis 168 to implement insertion function. We randomly chose 23 colonies from the plate and performed cPCR. By evaluating the size of cPCR product for each selected mutant, we found that two clones contained larger size of cPCR product than that of the “ck,” whereas the others had the same size as that of the ck, indicating that sfGFP was successfully inserted into the genome of the two clones among the 23 selected colonies. The efficiency of insertion was calculated to be 9% (2/23) (Figure 4B). We infer that because the expression of Cpf1 or crRNA might be unstable from the plasmid that is too large in size.
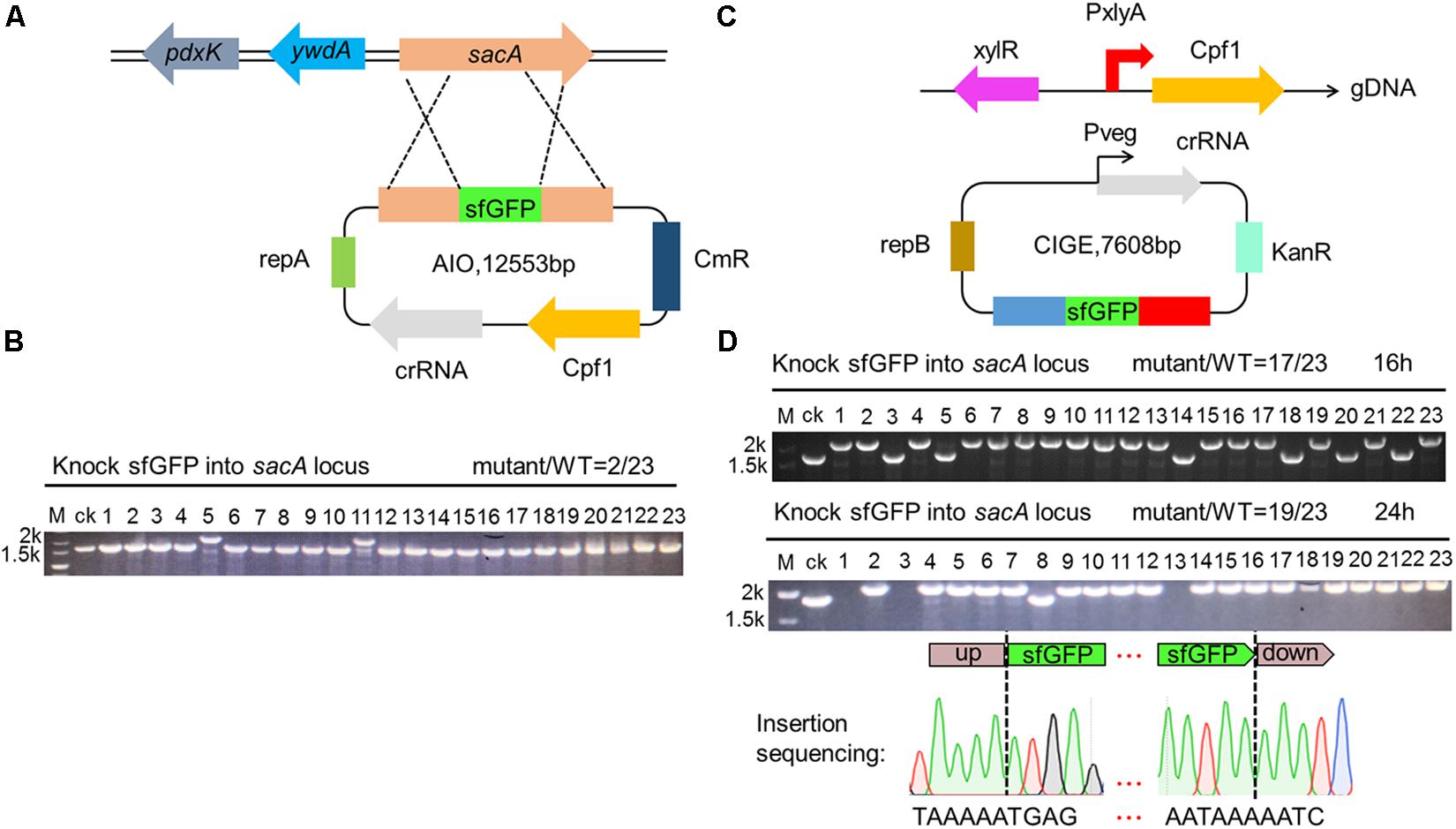
Figure 4. Construction of superfolder green fluorescence protein (sfGFP) insertion in sacA. (A) Scheme showing the procedures for gene insertion into Bacillus subtilis 168 by AIO system. (B) cPCR results shown that 9% (lane 5 and lane 11) of colonies had the sfGFP insertion mutant. (C) Scheme showing the procedures for gene insertion into B. subtilis 168 by CCB-CIGE system. (D) CIGE enables highly efficient sfGFP insertion mutation in the B. subtilis 168 strain. The sacA gene was replaced by the sfgfp gene. The efficiency for sfgfp gene insertion was 17/23 in the B. subtilis 168 strain. Prolonged incubation time under selective pressure increased the mutation efficiency to 82%.
To resolve the problem of low insertion efficiency by AIO system, we designed CRISPR-Cpf1-based CIGE (CCB-CIGE) platform to improve the expression of the components to elevate efficiency of gene insertion (Figure 4C). In this strategy, we chose pBSG as the skeleton and constructed a plasmid, pBsfGFPKi, harboring a sacA-targeted crRNA sequence controlled by Pveg. The coding region of sfGFP was flanked by upstream and downstream homologous arms. Because pBSG vector belongs to high copy plasmid, we can improve the expression of each component by using it. However, the replication protein of repB carried by the plasmid will multiply in the form of rolling circle replication, which may lead to instability of the plasmid. The plasmid was then introduced into the host BS-ganA’-Cpf1 (the Cpf1 gene under the control of PxylA was integrated in the chromosome of B. subtilis via substitution for ganA). After 16 h induction by 1% xylose, 23 colonies were randomly chosen to identify the insertion results by cPCR. The data showed that the cPCR products from 17 out of 23 clones were larger compared to that of ck, indicating that the sfGFP had been successfully inserted into the chromosome of these clones (Figure 4D). The insertion efficiency was confirmed to be 74% (Figure 4D). According to the previous research, efficiency of gene knock-in in B. subtilis can be significantly improved by iterative genome engineering (So et al., 2017). Therefore, we prolonged the incubation time to 24 h, and the insertion efficiency increased to 82% (Figure 4D). To further validate that insertion occurred precisely at the sacA site, we sequenced the region from the upstream to the downstream homologous arm of No. 1 mutant among the successfully-inserted mutants. The map of sequencing revealed that sfGFP has been accurately inserted into specific site of the sacA gene using an optimized CRISPR-Cpf1-mediated gene knock-in strategy, without any unintended mutations (Figure 4D). These results manifest that our optimized CRISPR-Cpf1 system accurately insert target genes into preset positions in the chromosome.
Gene knock-in based on CRISPR system is an important technology for metabolic engineering and genomic function research. A potential application is that when we construct metabolic engineering strains, we often need to overexpress some genes to improve the titer of metabolites. However, overexpression of some genes on plasmids often brings great pressure to the host. Therefore, integrating the target gene into the genome for overexpression has become a preferred strategy. Another advantage is to study the function of some genes in the genome. When we study a gene in the genome, we often take the surrounding genetic environment into account. At this time, in situ expression of gene is often much more important than overexpression on plasmid. Although we have constructed a gene knock-in system based on CRISPR-Cpf1 in this paper, we only verified it using sfGFP. In the current research, large gene cluster knock-in is an indispensable technology for the study of synthetic biology. Therefore, the strategy of gene knock in based on CRISPR-Cpf1 constructed in this work needs to be further optimized and the ability of large gene cluster knock-in needs to be improved. As the CCB-CIGE strategy was more efficient than AIO, we used CCB-CIGE in the following experiments.
Design of CRISPR-Cpf1 for Precise Deletion of Large Gene Cluster
Previous studies have shown that the deletion of large genome in Bacillus sp. plays a crucial role in heterologous expression of proteins, genome reduction (Westers et al., 2003), strain improvement (Thwaite et al., 2002) and overproduction of antibiotics (Zobel et al., 2015). There are three large gene clusters in B. subtilis encoding polyketide synthase (pks), plipastatin synthetase (pps), and surfactin (srf), which account for 7.7% of the total genome (So et al., 2017). There are also gene clusters in B. subtilis that synthesize peptide antibiotics, such as bac operon (Ozcengiz and Ogulur, 2015). The deletion of these gene clusters that synthesize secondary metabolites has minor effect on the growth of B. subtilis. Moreover, deletion of these non-essential regions in B. subtilis is essential to construction of the minimal genome in microbial chassis. Although a counter-selectable marker system based on synthetic gene circuits has been developed to delete pps operon, the knockout efficiency was only 6.4% (Jeong et al., 2015). Recently, a new editing system based on CRISPR-Cas9 employing a single sgRNA was developed to delete pps operon (So et al., 2017). However, the pps operon was not completely deleted even though the efficiency of deletion was further improved by optimizing. We infer that the DSB site and the repair site of homologous arm are too far to initiate an efficient double cross-over.
In view of those results that CIGE-based CRISPR-Cpf1 system was highly functioned to insertion of gene in chromosome, we considered that whether the system was also suitable to deletion of large gene clusters in B. subtilis. To verify the deduction, we first evaluated the function of deletion of sacA (Figure 5A). Plasmid pBsacA (refer to addgene #158650), containing sacA-targeted crRNA and the corresponding homologous arms identical to that of AIO and TPs (refer to pHTsacA), was constructed to implement the function. Then, pBsacA was introduced into BS-ganA’-Cpf1. We randomly selected 20 colonies from the agar plate and perform cPCR to identify the deletion efficiency. The results demonstrated that all the colonies harbored the disrupted sacA smaller than that of the wild-type sacA (ck), suggesting that the genome editing efficiencies mediated by CIGE system was 100% (20/20) (Figure 5B, upper panel). Sequencing results also authenticated that sacA has been disrupted by 240 bp in the CIGE edited hosts (Figure 5B, lower panel).
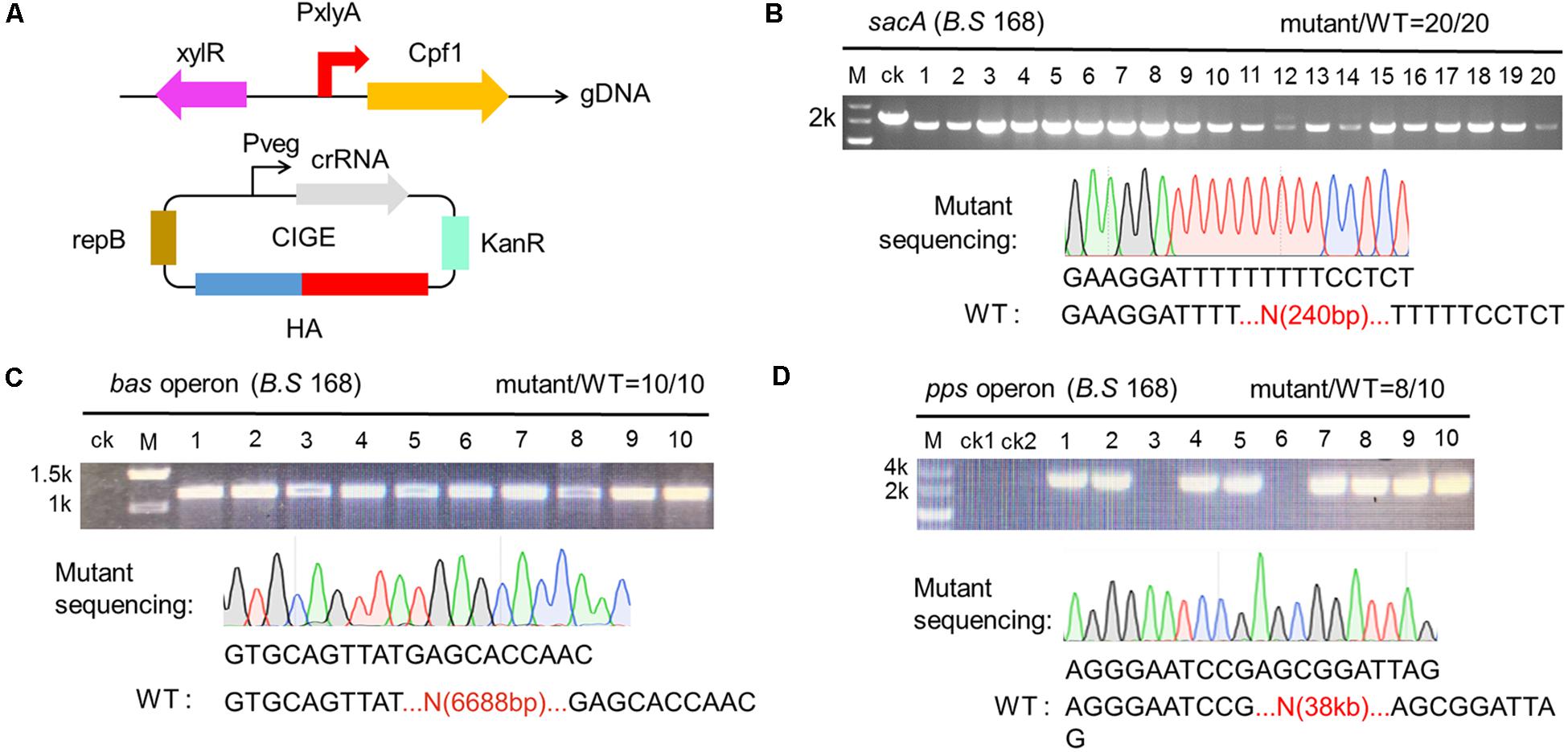
Figure 5. Deletion of larger gene clusters mediated by CCB-CIGE system derived from CRISPR-Cpf1. (A) Scheme showing the composition of the CIGE system for the deletion of larger gene clusters. (B) CIGE-mediated disruption of the sacA gene in the Bacillus subtilis 168 strain. The editing efficiency was 20/20. (C) Identification of bac operon deletion mutants. bac operon is composed of seven genes (bacABCDEFG), about 6688 bp. Ten transformants were selected and verified by colony PCR. M and ck represent the 2-kb DNA ladder and wild-type B. subtilis 168, respectively. (D) Confirmation of pps operon deletion using colony PCR. Lane M, the 5-kb DNA marker from Takara. Lane ck1 and ck2, PCR amplification with the B. subtilis 168 and B. subtilis comK genomic DNA (gDNA) using external primers, respectively. Lane 1-10, PCR amplification with the mutant gDNA using external primer. The editing efficiency was 8/10.
After validating that sacA was deleted by the engineering CRISPR-Cpf1 system, we sought to further explore whether the system can be used to delete larger gene clusters. The bac operon was selected as the deletion target. Firstly, we constructed the targeting plasmid, pBbac, containing expression cassette composed of a pair of 500-bp homologous arms and a bacD-targeted crRNA under the control of Pveg. After transformation in BS-ganA’-Cpf1, the transformants harboring pBbac was cultured and screened in line with the procedure above. Since the designed primers for verification flanked the homologous arms, the product of cPCR should be approximate of 1320 bp rather than 8008 bp (the full-length of bac operon) if bac operon was successfully deleted. We randomly selected 10 colonies from the agar plate and performed cPCR to identify the deletion efficiency. Interestingly, the cPCR product from the 10 colonies had expected gene size while the control (ck) hadn’t, indicating that bacD was successfully deleted from the operon (Figure 5C). In parallel, we sequenced the cPCR product from one of the successfully deleted mutants and confirmed the deletion (Figure 5C).
To testify whether the tool functioned to a broad range of targets, we selected another target, the pps operon, to evaluate the deletion efficiency. We firstly verified that the pps operon exists in the B. subtilis 168 prior to performing the deletion of the pps operon (Supplementary Figure S1). Then, we replaced bacD-targeting crRNA with ppsC-targeting crRNA on plasmid pBbac, and extended the length of homologous arms to 800 bp to ensure the deletion efficiency. Plasmid used to delete pps was termed pBpps. Then, pBpps was transformed into BS-ganA’-Cpf1. The screening and verification procedure were identical to that of bacD deletion. The cPCR product from ppsC-deleted mutant should be of 1772 bp by the designed primers. The results of cPCR showed that 8 out of 10 colonies had the products of expected size, suggesting that ppsC is deleted in these 8 mutants (Figure 5D). These results were further confirmed by sequencing (Figure 5D). The deletion efficiency for bac and pps was of 100% and 80%, respectively, based on the CRISPR-Cpf1-dependent CIGE platform (Figures 5C,D).
These results manifest that our designed CRISPR-Cpf1 system (CIGE) is more reliable and portable than the CRISPR-Cas9 system, especially for deletion of large gene clusters. To our best knowledge, this is the most efficient system to delete a pps operon with a single crRNA (Figure 5D). Deletion of large gene fragments is very important in synthetic biology, especially in deleting non-essential genes to construct minimal genomic microorganisms. Therefore, our customized CRISPR-Cpf1 system has great potential to implement this.
CCB-CIGE Platform Is Upgraded to a Higher Version to Exert Multiplex-Gene Editing
Wild-type bacteria usually are unable to exert programmed functions preset in cell factories. Therefore, engineering natural bacteria is a practical strategy to design a higher version of microbial chassis for synthetic biology. Precise and portable multiplex gene editing is an absolutely indispensable approach to do this. However, some commonly used methods, Cre/loxP (Suzuki et al., 2005; Choi et al., 2015) and Red recombination system (Datsenko and Wanner, 2000), were difficult to efficiently edit at genome-scale because of some technical hurdles. First of all, Cre/loxP system is not a scarless knockout technology, which leaves loxPLR sites at the edited position (Suzuki et al., 2007). These scars may affect the growth of host cells, and the cycle of this technique is longer. Secondly, the efficiency of this gene deletion method (Cre/loxP) is not very efficient because two rounds of homologous recombination are required and mutant selection after the second recombination is time-consuming (Cleto et al., 2016). Thirdly, it is difficult to achieve simultaneous editing of multiple targets in the genome editing system based on Cre/loxP. For genome editing in prokaryotes, phage-derived lambda red recombinases have been employed in recombineering, which facilitates homology-dependent integration/replacement of a donor DNA or oligonucleotide. However, these systems require the genetic background of the target strains such as deficiency of methyl-directed mismatch repair or RecA that involves in the recombinational DNA repair system (Wang et al., 2009). Multiplex genome editing systems based on CRISPR-Cas9 have been applied to different organisms, including E. coli (Zerbini et al., 2017), B. subtilis (Westbrook et al., 2016), Streptomyces species (Cobb et al., 2015), Rhodosporidium toruloides (Otoupal et al., 2019), and mammalian (Cong et al., 2013).
To fully exploit the function of CRISPR-Cpf1 in multiplex genome editing, we employed two genes used above, sacA and aprE, to design and build a multiple gene editing system. Firstly, we incorporated sacA- and aprE- crRNA cassettes into pBSG, by which the two crRNAs can simultaneously target sacA and aprE. In this form, we found when the crRNAs were transformed into BS-ganA’-Cpf1, only one gene was deleted in the same transformant (data not shown). Previous study reported that B. subtilis has more complex recombination systems and diverse plasmid replication modes. Therefore, we inferred that the failure might be due to the exchange of some fragments of the plasmid in the process of replication. After sequencing the transformants with single gene deletion, we confirmed that the sacA crRNA expression cassette was lost from plasmid, resulting in failure of deleting sacA (data not shown).
According to previous studies, Cpf1 intrinsically processes pre-crRNA into mature crRNA by cleaving specific site (Fonfara et al., 2016; Zetsche et al., 2017). Thus, we inferred that it might be feasible to implement multiplex genome editing by integrating multiple crRNAs to single expression cassette. Two crRNAs targeting sacA and aprE were genetically fused and insulated them by synthetic two repeats of “Direct Repeat (DR)-Spacer” units. The homologous arms of sacA and aprE tandem were sequentially cloned downstream of these crRNAs, generating a complete targeting sequence. Pveg was equipped to trigger the expression cassette. The expression cassette was then cloned into pBSG, yielding pB-PvegW-SAKo (Figure 6A). pB-PvegW-SAKo was transformed into BS-ganA’-Cpf1. Through the cultivation and screening as described above, cPCR results showed that 27.2% (6/22) of colonies were sacA- and aprE-deficient (Figure 6B). Although we successfully achieved double-gene deletion by CCB-CIGE platform, the efficiency still needs to be improved so as to elevate the work performance.
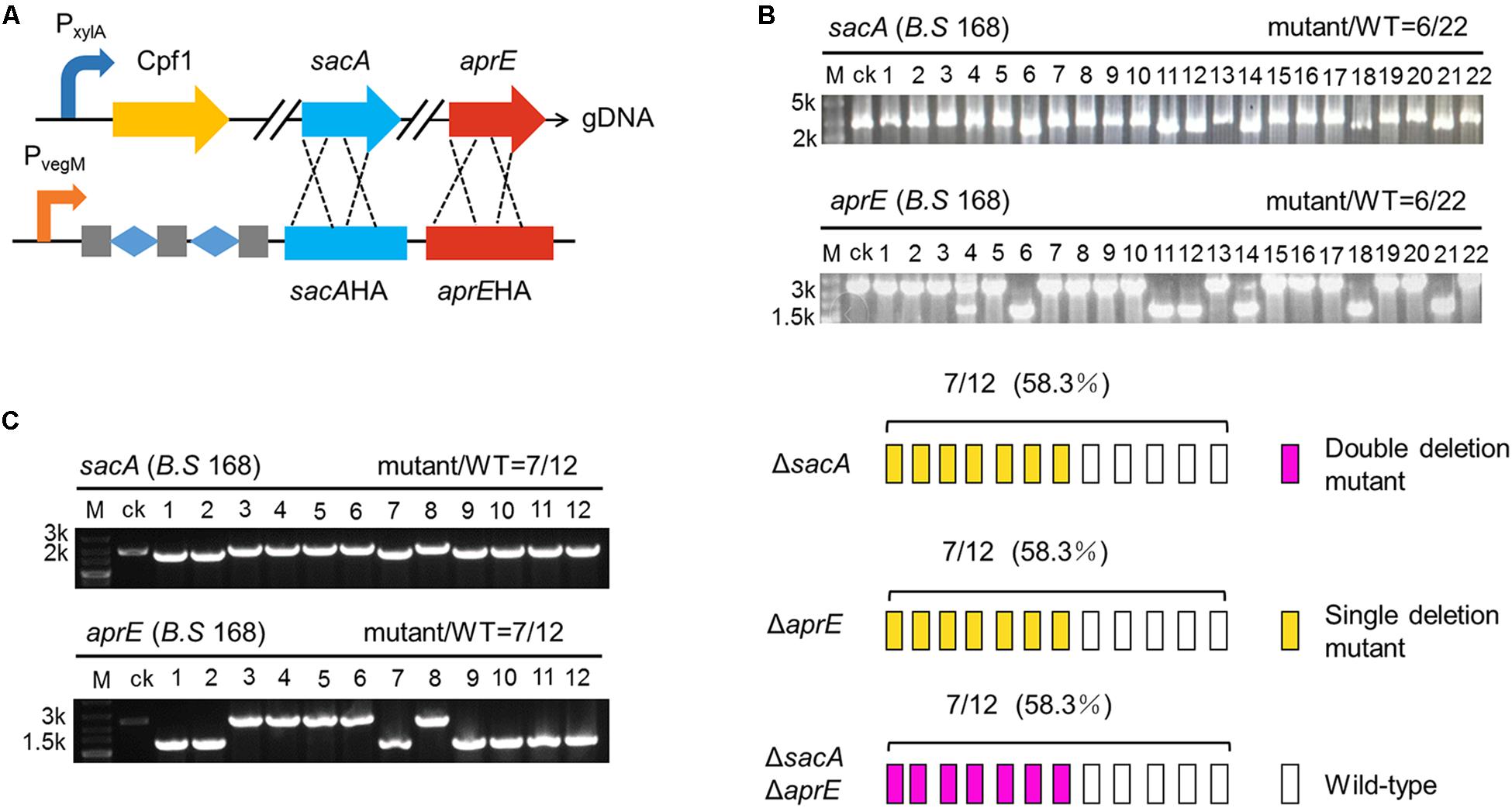
Figure 6. CRISPR-Cpf1-assisted simultaneous deletion of double genes (ΔsacAΔaprE). (A) The construction of the Cpf1 multiplex gene editing system contains a Cpf1 expression cassette and a multi-crRNA expression cassette. Cpf1 was inserted ganA site of genome DNA (gDNA) of Bacillus subtilis 168. The multi-crRNA expression cassette contains three DR guide units, and each unit includes one mature DR and 23 bp of guide sequence. (B) Multi-crRNA expression cassette is controlled by the Pveg promoter. Twenty-two colonies were picked and screened for mutations. The efficiency of simultaneous deletion of double genes is 6/22. (C) Multi-crRNA expression cassette is controlled by the Pveg promoter variant PvegM. The colonies of 7/12 is identified as a double deletion mutant strain (ΔsacAΔaprE). The open rectangle, orange rectangle, and dark pink rectangle represent the WT strain, the single deletion mutant of ΔsacA or ΔaprE, and the ΔsacAΔaprE double deletion mutant, respectively.
Accordingly, the next thrust is to engineer the components on this platform. Because the wild type Pveg promoter is a super strong promoter, we deduced that excessive activity of Pveg in the cell might induce growth burden in B. subtilis, rendering low editing efficiency through disturbing expression of crRNAs. Therefore, to rebalance the expression, we mutated the -10 region of the wild-type Pveg promoter to decrease its activity, variant was termed PvegM (Figure 6A). By substitution for the wild-type Pveg, pB-PvegM-SAKo was constructed, and then transformed into BS-ganA’-Cpf1. We randomly chose 12 colonies to verify the deletion by cPCR. The results showed that 7 selected clones among the 12 clones harbored the deleted sacA and aprE genome as the bands were smaller than that of the control “ck” (Figure 6C). The double-deficient mutants accounted for 58.3% of total tested mutants (Figure 6C), which was higher than that of previously reported by two folds (Figures 6B,C). These results suggest that the optimized CRISPR-Cpf1 system has great potential to achieve multiplex-gene editing at genome-scale.
Up to date, CRISPR-Cas9 system has been widely used in different organisms for genome editing. However, the function relies on a complex composed of crRNA and tracrRNA (or a chimera gRNA), which guides Cas9 to the target in genome. In contrast, one single crRNA is sufficient to guide CRISPR-Cpf1 RNP to a gene target. Practically, CRISPR-Cas9 system mediates multiplex genome editing by expression of multiple gRNAs. Nevertheless, the functional construct is often relatively large and complex, which would be rather difficult to construct and transform plasmid. Compared with Cas9 nucleases, the most significant feature of Cpf1 nucleases is that they not only have DNase activity but also RNase activity, which gives them the ability to process their own crRNA from a long precursor. This feature greatly facilitates their application for multiplex genome editing, transcriptional regulation and imaging, which tasks typically need to locate multiple loci in the genome for efficient operation. By taking advantage of this feature, we successfully constructed the multiplex genome editing system by using a single crRNA array in one vector (Figure 6A). The potential advantage of this system is that it can delete those genes that are difficult to delete individually. Another consideration is that the transformation efficiency of multiplex-gene-targeting plasmid is significantly lower than that of single-gene-targeting plasmid. It might also be due to the strong cleavage effect of crRNA by the constitutive transcription of the Pveg promoter, which makes it difficult for bacteria to repair in time. Therefore, inducible promoters can be used to regulate the expression of crRNA to enrich biomass in later studies.
Up to now, we have constructed a set of CRISPR-Cpf1-based toolkit (including AIO, TP, and CCB-CIGE) in B. subtilis. Zhang et al., Westbrook et al., and Wu et al., have developed genome editing tools based on CRISPR-Cas9 and CRISPR-Cpf1 in previous studies. However, compared with their systems, our newly constructed system has some advantages.
Firstly, although Zhang et al., constructed a genome editing system based on CRISPR-Cas9 in B. subtilis ATCC 6051a, they only verified that CRISPR-Cas9 system can disrupt gene in B. subtilis ATCC 6051a, and did not testify that the system can accurately delete and insert gene in the frame. In this work, we constructed a genome editing system based on CRISPR-Cpf1 with the same strategy (AIO). In this system, we used strong promoters P43 and Pveg from B. subtilis to express Cpf1 and crRNA, respectively. And we also verified that our engineered AIO system is capable of knocking out fragments of different sizes (240 bp, 2064 bp and 2775 bp) with efficiency over 95% (Figures 2B–D). Simultaneously, we construct two AIO systems (based on CRISPR-Cpf1 and CRISPR-Cas9) to prove that CRISPR-Cpf1 system is superior to CRISPR-Cas9 system in B. subtilis (take gene ganA as an example, 100% of Cpf1 vs. 75% of Cas9, Figure 2B). However, the AIO system we constructed in this study is relatively larger, which can make the construction and transformation of plasmid a little more difficult.
Secondly, in the study of Westbrook et al. (2016) they constructed a tool kit based on CRISPR-Cas9 in B. subtilis. In their whole research, they all adopt chromosome integration strategy to achieve different functions (including knockout, knock-in and transcription interference) based on CRISPR-Cas9. In this way, foreign genes (for example, Cas9, gRNA, and resistance marker) will inevitably be introduced into the genome of B. subtilis. However, as an important platform for metabolic engineering and synthetic biology research, the effect of exogenous gene introduction on the expected function of B. subtilis is unknown. Concurrently, insertion of antibiotic resistance marker will disqualify the use of engineered Bacillus strains as an eco-friendly host for food-grade applications, industrial fermentation and bioremediation. Westbrook et al. (2016) have also constructed a multi-gRNA delivery vector for multiplex genome editing. However, it is relatively difficult and time-consuming to insert multiple repeat sequences into one vector at the same time. In this study, we constructed a toolkit based on CRISPR-Cpf1, which can be used in different scenarios. AIO and TP systems can be used for traceless knockout, and are suitable for use without introducing foreign gene (for example, fermentation of food-grade enzymes). CCB-CIGE system can be used for engineering modification of B. subtilis to produce industrial enzyme. Recently, Wu et al., developed a toolkit (including knockout, knock-in, point mutation and transcription interference and activation) based on CRISPR-Cpf1 in B. subtilis. However, they did not explore the editing efficiency of a single gene of CRISPR-Cpf1 system in B. subtilis. It is unclear whether the editing efficiency varied in deletion of different size of the fragment. In our study, the AIO system we developed can knock-out small and medium-sized fragment, and the knock-out efficiency is almost not affected (efficiency is between 95% and 100%, Figures 2B–D). And in previous studies, it has been shown that the knockout of non-essential large gene clusters is indispensable for the construction of the minimal genomic chassis microorganism. However, in the study of Wu et al., deletion of large gene clusters was not validated. Here, we developed an upgraded CRISPR-Cpf1 system, which can knock-out bac (efficiency is 100%) and pps operons (efficiency is 80%, Figures 5C,D). And as far as we know, this is the first example to knock out 38-kb gene cluster with CRISPR-Cpf1 system in B. subtilis. Next, we also try to use CCB-CIGE platform to delete extremely large cluster pks operon (about 78 kb). However, we only obtain lower deletion efficiency (about 16.7%, data not show). Although, we still believe that the optimized CCB-CIGE platform can continue to improve the knock-out efficiency of pks operon. For double gene editing, Wu et al., used a double plasmid expression system, and enhanced the efficiency of homologous recombination by overexpression of mutated NgAgo∗. However, they need to add two inducers to achieve the goal of knocking out two genes, which is more laborious. Moreover, the transformation efficiency of the double plasmid system is significantly lower than that of the CCB-CIGE system in our study. Similarly, in the research of Jiang et al., they also overexpressed the recombinant factor recT to improve the efficiency of recombination. However, in our study, Cpf1 is integrated into the chromosome, the decrease of Cpf1 expression may weaken the knockout efficiency. And we did not introduce the factor NgAgo∗ and recT, to enhance homologous recombination, which will also reduce the editing efficiency. It is necessary to continue to improve the efficiency of homologous recombination and multi-target editing of our system.
Overall, we identified that engineering CRISPR-Cpf1 system significantly facilitates gene manipulation in B. subtilis. The development of this system has accelerated the construction of high-version microbial chassis. We believe that the optimized CRISPR-Cpf1 system can also be applied to other Gram-positive bacteria, such as Bacillus thuringiensis and Bacillus licheniformis.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
WH and WC conceived the project and designed the experiments. FS performed the molecular cloning experiments. QL and QC performed the colony PCR. WH, WC, and ZZ analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by a project funded by the International S&T Innovation Cooperation Key Project (2017YFE0129600), the National Natural Science Foundation of China (21878125), the Natural Science Foundation of Jiangsu Province (BK20181206), the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 111 Project (No. 111-2-06), the Jiangsu Province “Collaborative Innovation Center for Advanced Industrial Fermentation” Industry Development Program, and First-Class Discipline Program of Light Industry Technology and Engineering (LITE2018-04).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.524676/full#supplementary-material
References
Anagnostopoulos, C., and Spizizen, J. (1961). Requirements for transformation in bacillus subtilis. J. Bacteriol. 81, 741–746.
Bai, H., Deng, A., Liu, S., Cui, D., Qiu, Q., Wang, L., et al. (2018). A novel tool for microbial genome editing using the restriction-modification system. ACS Synth. Biol. 7, 98–106. doi: 10.1021/acssynbio.7b00254
Bao, Z., Xiao, H., Liang, J., Zhang, L., Xiong, X., Sun, N., et al. (2015). Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth. Biol. 4, 585–594. doi: 10.1021/sb500255k
Buchmuller, B. C., Herbst, K., Meurer, M., Kirrmaier, D., Sass, E., Levy, E. D., et al. (2019). Pooled clone collections by multiplexed CRISPR-Cas12a-assisted gene tagging in yeast. Nat. Commun. 10:2960. doi: 10.1038/s41467-019-10816-7
Chen, W., Zhang, Y., Yeo, W. S., Bae, T., and Ji, Q. (2017). Rapid and efficient genome editing in Staphylococcus aureus by using an engineered CRISPR/Cas9 system. J. Am. Chem. Soc. 139, 3790–3795. doi: 10.1021/jacs.6b13317
Choi, J. W., Yim, S. S., Kim, M. J., and Jeong, K. J. (2015). Enhanced production of recombinant proteins with Corynebacterium glutamicum by deletion of insertion sequences (IS elements). Microb. Cell Fact. 14:207. doi: 10.1186/s12934-015-0401-7
Cleto, S., Jensen, J. V., Wendisch, V. F., and Lu, T. (2016). Corynebacterium glutamicum metabolic engineering with CRISPR Interference (CRISPRi). ACS Synth. Biol. 5, 375–385. doi: 10.1021/acssynbio.5b00216
Cobb, R. E., Wang, Y., and Zhao, H. (2015). High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth. Biol. 4, 723–728. doi: 10.1021/sb500351f
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Correa, G. G., Rachel da Costa Ribeiro Lins, M., Silva, B. F., Barbosa, de Paiva, G., Bertolazzi Zocca, V. F., et al. (2020). A modular autoinduction device for control of gene expression in Bacillus subtilis. Metab. Eng. 61, 326–334. doi: 10.1016/j.ymben.2020.03.012
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Dong, H., and Zhang, D. (2014). Current development in genetic engineering strategies of Bacillus species. Microb. Cell Fact. 13, 63. doi: 10.1186/1475-2859-13-63
Engler, C., Gruetzner, R., Kandzia, R., and Marillonnet, S. (2009). Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One 4:e5553. doi: 10.1371/journal.pone.0005553
Fonfara, I., Richter, H., Bratovic, M., Le Rhun, A., and Charpentier, E. (2016). The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521. doi: 10.1038/nature17945
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826. doi: 10.1038/nbt.2623
Gao, W., Long, L., Tian, X., Xu, F., Liu, J., Singh, P. K., et al. (2017). Genome editing in cotton with the CRISPR/Cas9 system. Front. Plant Sci. 8:1364. doi: 10.3389/fpls.2017.01364
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A. III, and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. doi: 10.1038/nmeth.1318
Hong, W., Zhang, J., Cui, G., Wang, L., and Wang, Y. (2018). Multiplexed CRISPR-Cpf1-Mediated Genome Editing in Clostridium difficile toward the Understanding of Pathogenesis of C. difficile Infection. ACS Synth. Biol. 7, 1588–1600. doi: 10.1021/acssynbio.8b00087
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. doi: 10.1038/nbt.2647
Hur, J. K., Kim, K., Been, K. W., Baek, G., Ye, S., Hur, J. W., et al. (2016). Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat. Biotechnol. 34:807. doi: 10.1038/nbt.3596
Hwang, W. Y., Fu, Y., Reyon, D., Maeder, M. L., Tsai, S. Q., Sander, J. D., et al. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229. doi: 10.1038/nbt.2501
Jeong, D. E., Park, S. H., Pan, J. G., Kim, E. J., and Choi, S. K. (2015). Genome engineering using a synthetic gene circuit in Bacillus subtilis. Nucleic Acids Res. 43:e42. doi: 10.1093/nar/gku1380
Jiang, Y., Chen, B., Duan, C., Sun, B., Yang, J., and Yang, S. (2015). Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 81, 2506–2514. doi: 10.1128/aem.04023-14
Jiang, Y., Qian, F., Yang, J., Liu, Y., Dong, F., Xu, C., et al. (2017). CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat. Commun. 8:15179. doi: 10.1038/ncomms15179
Keasling, J. D. (2010). Manufacturing molecules through metabolic engineering. Science 330, 1355–1358. doi: 10.1126/science.1193990
Kim, D., Kim, J., Hur, J. K., Been, K. W., Yoon, S. H., and Kim, J. S. (2016). Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 34, 863–868. doi: 10.1038/nbt.3609
Kim, H., Kim, S. T., Ryu, J., Kang, B. C., Kim, J. S., and Kim, S. G. (2017). CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat. Commun. 8:4406. doi: 10.1038/ncomms14406
Liu, J. J., Orlova, N., Oakes, B. L., Ma, E., Spinner, H. B., Baney, K. L. M., et al. (2019). CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 566, 218–223. doi: 10.1038/s41586-019-0908-x
Otoupal, P. B., Ito, M., Arkin, A. P., Magnuson, J. K., Gladden, J. M., and Skerker, J. M. (2019). Multiplexed CRISPR-Cas9-Based genome editing of Rhodosporidium toruloides. mSphere 4:e00099-19. doi: 10.1128/mSphere.00099-19
Ozcengiz, G., and Ogulur, I. (2015). Biochemistry, genetics and regulation of bacilysin biosynthesis and its significance more than an antibiotic. N. Biotechnol. 32, 612–619. doi: 10.1016/j.nbt.2015.01.006
Pitcher, R. S., Brissett, N. C., and Doherty, A. J. (2007). Nonhomologous end-joining in bacteria: a microbial perspective. Annu. Rev. Microbiol. 61, 259–282. doi: 10.1146/annurev.micro.61.080706.093354
Radeck, J., Kraft, K., Bartels, J., Cikovic, T., Durr, F., Emenegger, J., et al. (2013). The Bacillus BioBrick Box: generation and evaluation of essential genetic building blocks for standardized work with Bacillus subtilis. J. Biol. Eng. 7:29. doi: 10.1186/1754-1611-7-29
So, Y., Park, S. Y., Park, E. H., Park, S. H., Kim, E. J., Pan, J. G., et al. (2017). A highly efficient CRISPR-Cas9-mediated large genomic deletion in Bacillus subtilis. Front. Microbiol. 8:1167. doi: 10.3389/fmicb.2017.01167
Strecker, J., Jones, S., Koopal, B., Schmid-Burgk, J., Zetsche, B., Gao, L., et al. (2019). Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 10:212. doi: 10.1038/s41467-018-08224-4
Suzuki, N., Inui, M., and Yukawa, H. (2007). Site-directed integration system using a combination of mutant lox sites for Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 77, 871–878. doi: 10.1007/s00253-007-1215-2
Suzuki, N., Tsuge, Y., Inui, M., and Yukawa, H. (2005). Cre/loxP-mediated deletion system for large genome rearrangements in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 67, 225–233. doi: 10.1007/s00253-004-1772-6
Teng, F., Cui, T., Feng, G., Guo, L., Xu, K., Gao, Q., et al. (2018). Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov. 4:63. doi: 10.1038/s41421-018-0069-3
Thwaite, J. E., Baillie, L. W., Carter, N. M., Stephenson, K., Rees, M., Harwood, C. R., et al. (2002). Optimization of the cell wall microenvironment allows increased production of recombinant Bacillus anthracis protective antigen from B. subtilis. Appl. Environ. Microbiol. 68, 227–234. doi: 10.1128/aem.68.1.227-234.2002
Trevors, J. T. (1986). Plasmid curing in bacteria. FEMS Microbiol. Rev. 32, 149–157. doi: 10.1111/j.1574-6968.1986.tb01189.x
Wang, H. H., Isaacs, F. J., Carr, P. A., Sun, Z. Z., and Xu, G. (2009). Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898. doi: 10.1038/nature08187
Wang, M., Mao, Y., Lu, Y., Tao, X., and Zhu, J. K. (2017). Multiplex gene editing in rice using the CRISPR-Cpf1 system. Mol. Plant 10, 1011–1013. doi: 10.1016/j.molp.2017.03.001
Westbrook, A. W., Moo-Young, M., and Chou, C. P. (2016). Development of a CRISPR-Cas9 tool kit for comprehensive engineering of Bacillus subtilis. Appl. Environ. Microbiol. 82, 4876–4895. doi: 10.1128/aem.01159-16
Westers, H., Dorenbos, R., van Dijl, J. M., Kabel, J., Flanagan, T., Devine, K. M., et al. (2003). Genome engineering reveals large dispensable regions in Bacillus subtilis. Mol. Biol. Evol. 20, 2076–2090. doi: 10.1093/molbev/msg219
Wu, Y., Chen, T., Liu, Y., Lv, X., Li, J., Du, G., et al. (2018). CRISPRi allows optimal temporal control of N-acetylglucosamine bioproduction by a dynamic coordination of glucose and xylose metabolism in Bacillus subtilis. Metab. Eng. 49, 232–241. doi: 10.1016/j.ymben.2018.08.012
Wu, Y., Liu, Y., Lv, X., Li, J., Du, G., and Liu, L. (2020). CAMERS-B: CRISPR/Cpf1 assisted multiple-genes editing and regulation system for Bacillus subtilis. Biotechnol. Bioeng. 117, 1817–1825. doi: 10.1002/bit.27322
Yamashiro, D., Minouchi, Y., and Ashiuchi, M. (2011). Moonlighting role of a poly-gamma-glutamate synthetase component from Bacillus subtilis: insight into novel extrachromosomal DNA maintenance. Appl. Environ. Microbiol. 77, 2796–2798. doi: 10.1128/aem.02649-10
Yan, M. Y., Yan, H. Q., Ren, G. X., Zhao, J. P., Guo, X. P., and Sun, Y. C. (2017). CRISPR-Cas12a-assisted recombineering in bacteria. Appl. Environ. Microbiol. 83:e00947-17. doi: 10.1128/aem.00947-17
Zerbini, F., Zanella, I., Fraccascia, D., Konig, E., Irene, C., Frattini, L. F., et al. (2017). Large scale validation of an efficient CRISPR/Cas-based multi gene editing protocol in Escherichia coli. Microb. Cell Fact. 16:68. doi: 10.1186/s12934-017-0681-1
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. doi: 10.1016/j.cell.2015.09.038
Zetsche, B., Heidenreich, M., Mohanraju, P., Fedorova, I., Kneppers, J., DeGennaro, E. M., et al. (2017). Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 35, 31–34. doi: 10.1038/nbt.3737
Zhang, K., Duan, X., and Wu, J. (2016). Multigene disruption in undomesticated Bacillus subtilis ATCC 6051a using the CRISPR/Cas9 system. Sci. Rep. 6:27943. doi: 10.1038/srep27943
Zhang, X. Z., Yan, X., Cui, Z. L., Hong, Q., and Li, S. P. (2006). mazF, a novel counter-selectable marker for unmarked chromosomal manipulation in Bacillus subtilis. Nucleic Acids Res. 34:e71. doi: 10.1093/nar/gkl358
Zhu, B., and Stulke, J. (2018). SubtiWiki in 2018: from genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res. 46, D743–D748. doi: 10.1093/nar/gkx908
Keywords: CRISPR-Cpf1, Bacillus subtilis, multiplex genome editing, large fragment deletion, gene insertion, chassis microorganisms
Citation: Hao W, Suo F, Lin Q, Chen Q, Zhou L, Liu Z, Cui W and Zhou Z (2020) Design and Construction of Portable CRISPR-Cpf1-Mediated Genome Editing in Bacillus subtilis 168 Oriented Toward Multiple Utilities. Front. Bioeng. Biotechnol. 8:524676. doi: 10.3389/fbioe.2020.524676
Received: 06 January 2020; Accepted: 12 August 2020;
Published: 02 September 2020.
Edited by:
Yi Wang, Auburn University, United StatesReviewed by:
Zengyi Shao, Iowa State University, United StatesJunjie Yang, Center for Excellence in Molecular Plant Sciences (CAS), China
Copyright © 2020 Hao, Suo, Lin, Chen, Zhou, Liu, Cui and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenjing Cui, d2pjdWlAamlhbmduYW4uZWR1LmNu; Zhemin Zhou, emhtemhvdUBqaWFuZ25hbi5lZHUuY24=