 Xingyue Gu
Xingyue Gu Zhihua Chen1*
Zhihua Chen1*- 1Institute of Computing Science and Technology, Guangzhou University, Guangzhou, China
- 2Department of General Surgery, Heilongjiang Province Land Reclamation Headquarters General Hospital, Harbin, China
The G Protein-Coupled Receptor (GPCR) family consists of more than 800 different members. In this article, we attempt to use the physicochemical properties of Composition, Transition, Distribution (CTD) to represent GPCRs. The dimensionality reduction method of MRMD2.0 filters the physicochemical properties of GPCR redundancy. Matplotlib plots the coordinates to distinguish GPCRs from other protein sequences. The chart data show a clear distinction effect, and there is a well-defined boundary between the two. The experimental results show that our method can predict GPCRs.
Introduction
G protein-coupled receptors (GPCRs) are the largest receptor superfamily. According to their sequence similarity, they are divided into 6 subfamilies (AF), of which the Rhodopsin or rhodopsin-like family is the largest and most widely studied family (Fredriksson et al., 2003; Liu and Zhu, 2019; Ru et al., 2020). Class A has approximately 284 members in humans, and Class B subfamilies can be further divided into two unused families: Class B1, named secretin, secrete protein-like receptors, and Class B2 (adhesion) adhere to GPCRs. Class B1 and Class B2 contain 15 members and 33 members in humans, respectively. The adhesive G protein-coupled receptor (ADGR) family is one of the oldest GPCR families. It exists in primitive animals, and even in several basic fungi, and is the ancestor of the B1 subfamily of GPCRs (Nordstrm et al., 2009; Krishnan et al., 2012). Finally, the class C glutamate family is composed of peptide receptors. The class F frizzled protein family has appsroximately 11 members in humans.
Protein classification is one of the key issues in bioinformatics and plays an important role in the identification and study of gene markers (Tibshirani, 1996; Cheng and Hu, 2018; Feng, 2019; Guo et al., 2019). With the development of machine learning, protein classification and prediction have entered a new era. Machine learning can use previous experience and data to automatically improve the performance of algorithms, build appropriate models, and discriminate new protein sequences. Islam et al. (2017) applied a natural language processing N-Gram model to classify proteins. The above machine learning methods have achieved certain effects in protein classification. This article uses feature extraction and dimension reduction of GPCR proteins to distinguish between the properties of the extracted proteins. Finally, Matplotlab is used to distinguish GPCRs from non-GPCRs. In the article Prediction of G Protein-Coupled Receptors (Liao et al., 2016), the 188D method is used to extract the protein features, and then cross validation and random forest are used to accurately divide the GPCR and non-gpcr protein sequences. In this paper, the CTD mode (Zou et al., 2013) is used, where C represents the content of each hydrophobic amino acid, T represents the frequency of the divalent peptide, and D represents the amino acid distribution at the five positions of the sequence. After using CTDC feature extraction method, the innovative feature of this experiment is that the redundant features are well-extracted using dimensionality reduction. Finally, the machine learning method and Matplotlib are used to draw a graph that distinguishes GPCRs from non-GPCRs.
Materials and Methods
Datasets
1. The original 5027 G protein-coupled receptors (GPCRs) were obtained in fasta format from the database (http://www.UniProt.org/); 2. The initial sequence was pre-processed using the protein clustering programme CDHIT (http://cd-hit.org/) to improve the analysis performance and reduce the homology of the predicted sequence (Zou et al., 2020). The critical value of sequence identity was located at 0.8. Finally, 2,495 GPCR sequences were obtained from the positive data set. 3. The positive sequences of all the protein sequences were removed, and 10,386 non-GPCR protein sequences were produced as the positive dataset (Liao et al., 2016).
Feature Extraction Methods
Principle
CTD represents the composition, transition, and distribution, respectively. Its principle is to replace the amino acid sequence with mathematical symbols representing physical and chemical properties (Cheng et al., 2018a). Because the protein sequence information is of different lengths, CTD is used to obtain fixed-length information from proteins as input to machine learning. In protein or peptide sequences, CTD represents physicochemical properties or amino acid distribution patterns of specific structures (Dubchak et al., 1995, 1999; Cai et al., 2003; Zhang et al., 2011; Ding et al., 2017). These features are very important for protein sequence analysis (Wei et al., 2018; Liu et al., 2019; Liu et al., 2019a; Yan et al., 2019; Chen et al., 2020). According to the main amino acid indicators of Tomii and Kanehisa (Kentaro and Minoru, 1996), amino acids are divided into three groups according to seven physical and chemical properties, as shown in Table 1.
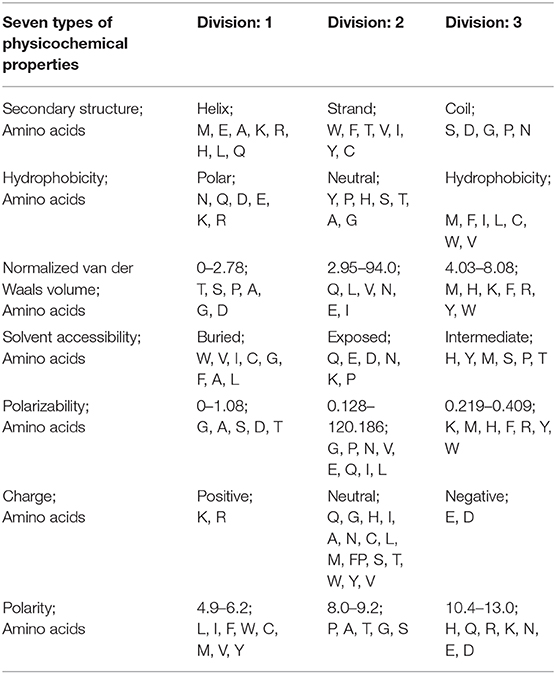
Table 1. Seven types of physicochemical properties and the division of amino acids.
CTD (Dubchak et al., 1999) is very helpful for enzyme prediction. Composition (Cai et al., 2003; Han et al., 2004; Chen W. et al., 2019; Liu, 2019) refers to the number of specific amino acids in a protein sequence divided by the total length N of the amino acid in the protein sequence:
where ne represents the sum of the number of e, a particular amino acid, in the sequence. e could be 1, 2, or 3, which represents the type of amino acid.
Assuming two specific amino acids are a and b, transition (T) means the number of ab and ba divided by the length of the protein sequence N-1:
The distribution is the position of a specific amino acid in the protein/the total length of the protein sequence, which represents the chain length at which the first, 25, 50, 100% amino acids of this particular amino acid are located.
For example, take the following protein sequence: DEKRADGSTAGPSTDGNPS. According to Table 1, DE is the amino acid sequence of classification 2 under Charge, KR is the amino acid sequence of category 3 under Charge, and ADGST is the amino acid sequence of classification 1 under Polarizability. AGPST is an amino acid sequence of Polarity 2, and DGNPS is the amino acid sequence of classification 1 under the Secondary Structure. Thus, our protein sequence is converted by CTD to 2233111112222211111. The following shows how the protein sequence Composition, Transition, Distribution is calculated (see Figure 1).
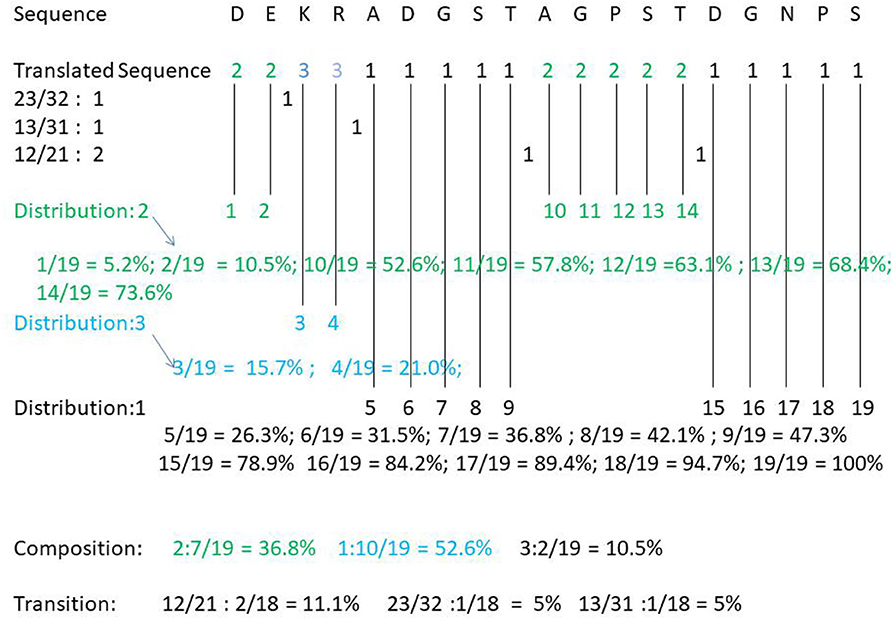
Figure 1. Computational flow of CTD eigenvectors in protein sequences.
Composition of category 2: 7/(7 + 2 + 10 = 19)= 36.8%; Composition of category 3: 2/19 = 10.5%; Composition of category 1: 10/19 = 52.6%. Transition (23, 32) = 1/18 = 5.5%; Transition (12, 21) = 2/18 = 11.1%; Transition (13, 31) = 1/18 = 5.5%. Distribution (1) = 5/19, 6/19, 7/19, 8/19, 15/19, 16/19, 17/19, 18/19, 19/19; Distribution (2) =1/19, 2/19, 10/19, 11/19, 12/19, 13/19, 14/19; Distribution 3 is equal to 3/19, 4/19. The final CTD results of DEKRADGSTAGPSTDGNPS are as follows: Composition (2): 36.8%, Composition (3): 10.5%, Composition (1): 52.6%. T (23, 32): 5.5%, T (12, 21): 11.1%, T (13, 31): 5.5%; D (1): 26.3, 31.5, 36.8, 42.1, 78.9, 84.2, 89.4, 94.7, 100%; D (2): 5.2, 10.5, 52.6, 57.8, 63.1, 68.4, 73.6%; D (3): 15.7, 21.0%.
Dimensionality Reduction
The MRMD2.0 (Wei et al., 2015; Zou et al., 2016a,b) algorithm is used to reduce the dimensions of the files after using CTDC to extract features. The specific process of dimensionality reduction is:
1. Attribute selection: Using analysis of variance to test the significance of the difference between the mean values of two or more samples; maximum correlation and maximum distance MRMD feature classification and accuracy and stability of prediction tasks; MIC is based on a non-parametric information-based maximum parameter exploration for measuring the linear or non-linear strength of two variables X and Y; the minimum absolute contraction and selection operator (LASSO) (Tibshirani, 1996; Guo et al., 2019) uses an L1 regularized linear regression method; Minimal Redundancy-Maximum Correlation (mRMR) method expands the representativeness of a feature set by requiring features to be maximally different from each other; chi-square test is a widely used hypothesis test based on the chi-square distribution for common hypothesis testing; Recursive Feature Elimination (RFE) classifies data according to the size of the correlation coefficients or importance of feature attributes. Through recursive elimination of functions in each cycle, RFE attempts to eliminate possible dependencies and collinearity in the model.
2. Function ranking PageRank algorithm: In the attribute selection method used above, point a to b because feature b is more important than feature a. Finally, the result of each function selection method forms a link list. Using the PageRank algorithm to rank these links, a directed graph is formed, and each feature receives a score. A ranking is then obtained according to the level of the feature, a, b, c, d, e .
3. Finally, choose the best outcome of the sequence. Since the first feature “a” in the new sequence has the highest score, random forest (Pang et al., 2006; Ding et al., 2016; Cheng et al., 2018b; Liu et al., 2019b; Su et al., 2019; Wei et al., 2019; Xu et al., 2019c; Lv et al., 2020) is used for 5-fold cross-validation starting from the first feature. The highest standard score is made by comparing the three sequences: “a,” “a,b;” “a,b,c,d,e.” Finally, five data indicators were used: f-score, precision, recall, MCC and AUC (Xu et al., 2018a; Cheng, 2019; Cheng L. et al., 2019; Ding et al., 2019; Zeng et al., 2019a, 2020; Zhang et al., 2019; Liu and Chen, 2020; Wang et al., 2020), and the sequence with the highest index and the highest score for dimension reduction was found. The specific dimension reduction process is shown in Figure 2.
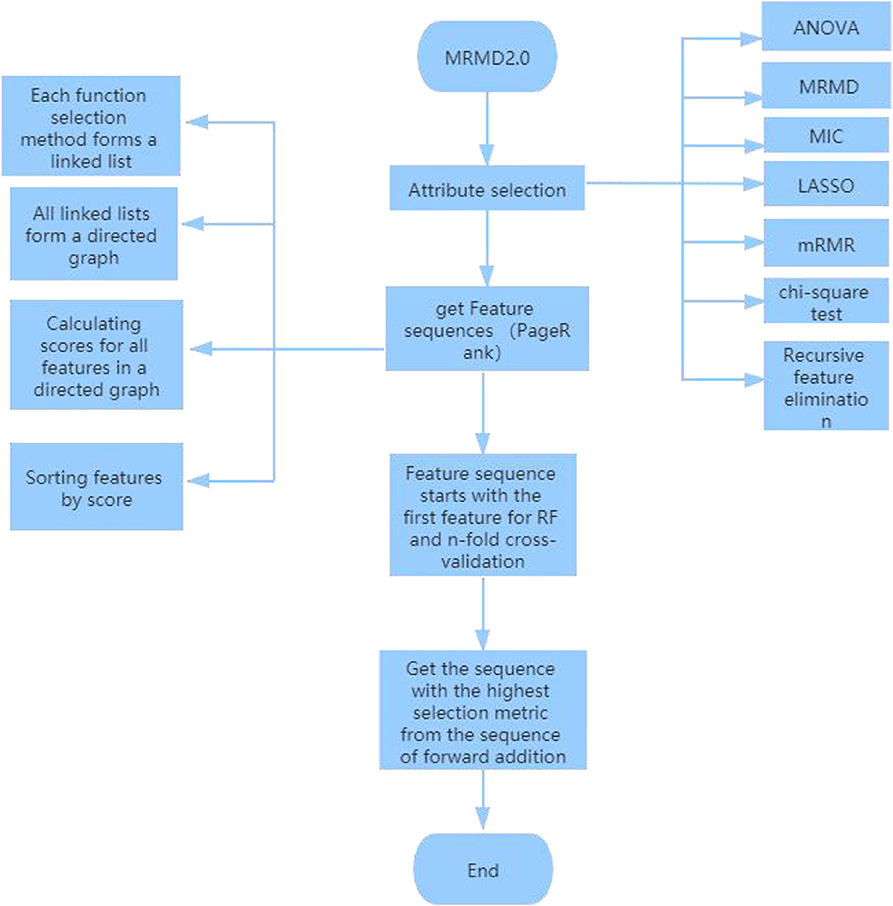
Figure 2. The specific dimension reduction process.
Algorithm Steps
GPCR sequence protein features are extracted using specific protein extraction methods. Any two attributes in the extracted features are divided into GPCRs and non-GPCRs. Finally, Matplotlib is used to divide any two attributes in the extracted features into GPCRs and non-GPCRs (the experimental flow chart is shown in Figure 3):
(1) Using all the different positive protein samples, extract the corresponding Pfam protein sequence from the “family and domain” of the UniProt website and delete the redundant and identical Pfam number. Then, the unique Pfam number obtained for the positive data set (Liao et al., 2016).
(2) All the protein sequences are integrated into the Pfam number file, and the protein sequences with the same Pfam sequence are then merged into the same file named after the Pfam number.
(3) Delete the files with a positive Pfam number. In the remaining Pfam number files, the negative data set (Liao et al., 2016) is extracted from the longest sequence of each Pfam.
(4) Use the CTDC method command to extract specific features in fasta files to generate GPCRs and non-GPCRs .csv files; positive GPCRs sample are marked as 0, negative sample are marked as −1, and the GPCRs and non-GPCRs .csv files are combined into one file.
(5) The combined .csv file was reduced by MRMD2.0, and the reduced CTDC-mRMD2.0.csv file was obtained.
(6) Select any two attributes of the 39 attributes in the CTDC sequence. GPCRs are purple and marked 0, and non-GPCRs are green and marked 1; Using Matplotlib, plot the picture of GPCRs and non-GPCRs.
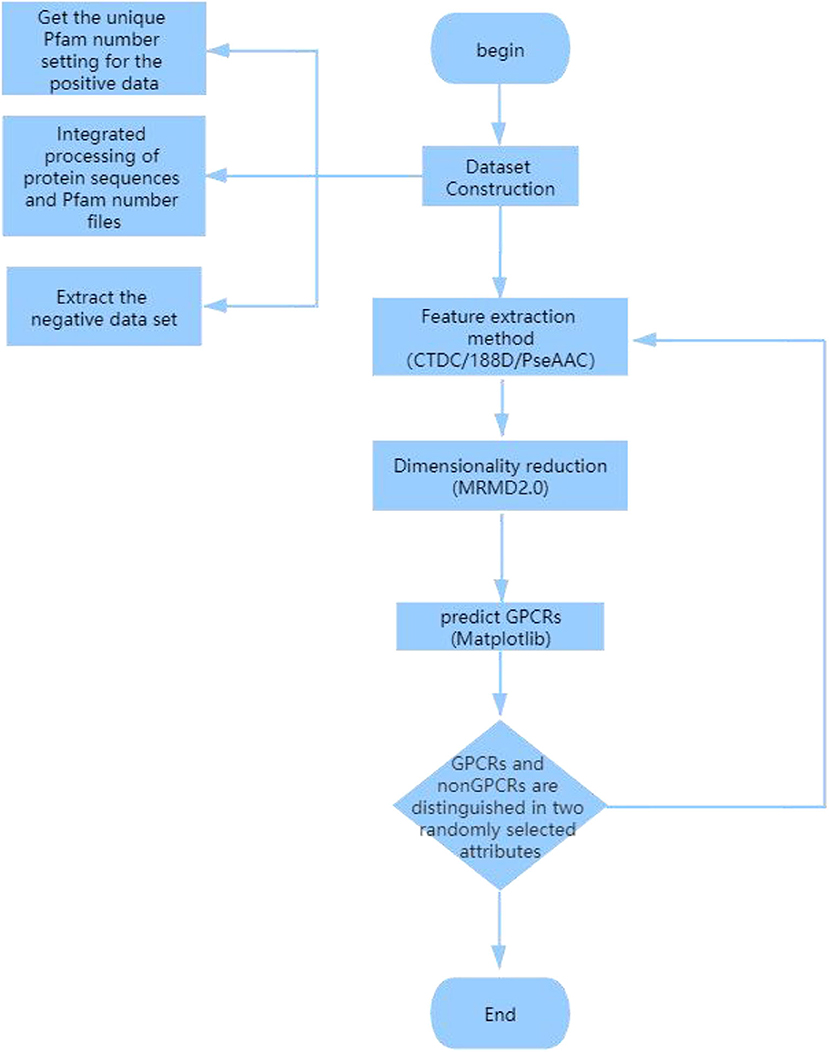
Figure 3. Experimental flow chart for prediction of G protein-coupled receptors.
Results
Comparison of Effects of Different Features
CTDC was used to extract the characteristics of the GPCR protein feature sequences sample, including 39 properties. Previous studies showed that feature extraction is very important for constructing the computational predictors (Wei et al., 2017a,b; Xu et al., 2018b; Liang et al., 2019; Liu and Li, 2019; Patil and Chouhan, 2019; Shen et al., 2019; Zhang and Liu, 2019; Junwei et al., 2020; Liu et al., 2020; Wen et al., 2020). Any two of the 39 attributes were selected and plotted using Matplotlab to obtain the sample differentiation graph of GPCRs and non-GPCRs, as shown in Figure 4. Among them, the abscissa and the ordinate in the chart represent two of the 39 attributes. The x-coordinate of Figure 4 on the left is the first of the 39 properties, “hydrophobicity_PRAM900101,” named “RKEDQN,” which is hydrophilic. The y-coordinate is the 14th property, “hydrophobicity_PRAM900101,” named “GASTPHY,” which is neutral. In the right diagram of Figure 4, the X coordinate is the fourth attribute in the CTDC feature extraction method, normwaalsvolume: NVEQIL. The Y coordinate is the 25th attribute in CTDC, hydrophobicity_ENGD860101: CVLIMF. As seen from the chart, GPCRs and non-GRCRs are represented by blue and green, respectively, in which GPCRs and non-GPCRs can be clearly distinguished.
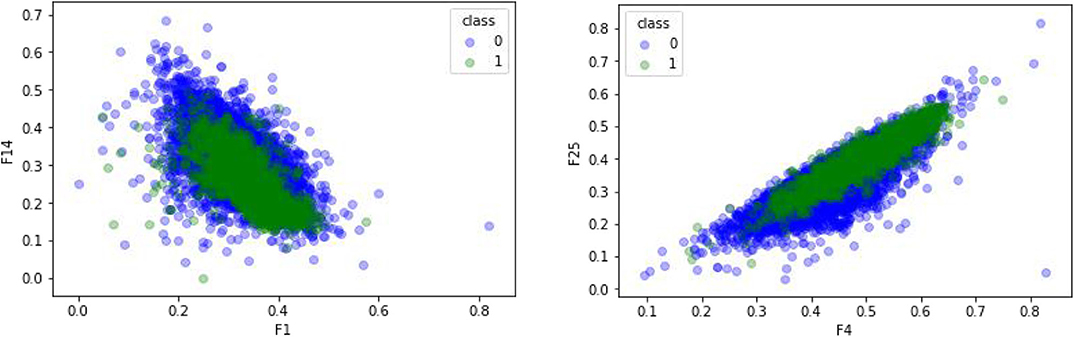
Figure 4. Comparison of effects of different features.
Comparison of Different Feature Extraction Methods
A comparative experiment was conducted, and the GPCR protein feature sequences are extracted by the 188D feature extraction method. The experimental effect is shown in Figure 5. In Figure 5, 120 and 100 dimensions of 188D are used. Non-GPCRs and GPCRs are marked as −1 and 1, respectively. It can be seen from the chart that the differentiation effect of GPCRs and non-GPCRs is very poor, but the differentiation effect of Figure 4 is very good. Thus, whether GPCRs and non-GPCRs can be distinguished well is related to the selected feature extraction method.
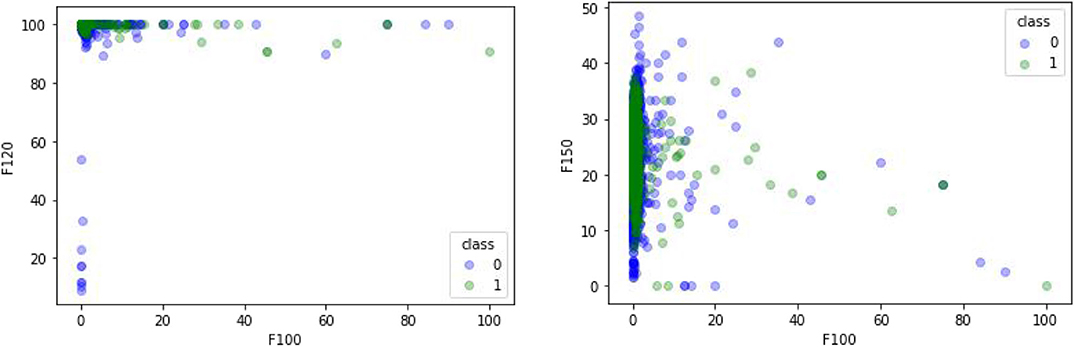
Figure 5. Comparison of different feature extraction methods.
Comparison of Results of Different Dimensionality Reduction Methods
The feature sequences of GPCR protein are extracted by the mRMR (Ding and Peng, 2005; Peng et al., 2005; Wang et al., 2018) dimensionality reduction method. 0 represents negative sample non-GPCRs, and 1 represents positive sample GPCRs. The experimental results are shown in Figure 6. In comparison with Figure 4, the two figures adopt the same feature extraction method of CTDC, the same attribute features and different dimension reduction methods. As seen from the figure, the difference between GPCRs and non-GPCRs was also very high after the dimension reduction method was used, and positive and negative samples are clearly distinguished.
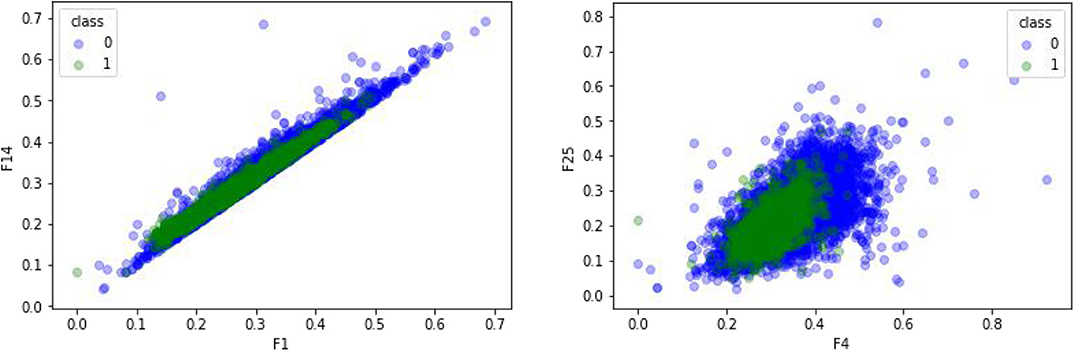
Figure 6. Comparison of results of different dimensionality reduction methods.
Comparison With Others
In the study of Prediction of G Protein-Coupled Receptors with SVM-Prot Features and Random Forest (Liao et al., 2016), the researchers adopted a method different from the method in this paper to predict GPCRs and non-GPCRs. The experimental steps they adopted were as follows: 1. Extract GPCR and non-GPCR sample characteristics with 188D (Balfanz et al., 2013) 2. The sample sequences were divided into five parts, four of which were for the training set and the remaining one for the test set. In these four parts, positive and negative samples were treated with a strike balance 3. Random Forest was applied to the training samples, and the accuracy of the test samples was measured 4. Finally, Sn, Sp, Acc, MCC, and AUC standards were adopted to measure the accuracy. The correct classification rate of the five independent test sets was 90.64, 90.37, 88.04, 93.28, and 95.73, with an average rate of 91.61 ± 2.96%.
Conclusion
With the feature extraction method of CTDC, GPCRs and non-GPCRs can be well-distinguished from the two randomly selected dimensions. The same CTDC feature extraction method was used, but another dimension reduction method, mRMR, was selected. Compared with mRMD2.0, the differentiation effect was similar, and GPCRs and non-GPCRs could be significantly predicted. Using different feature extraction methods (188D) and the same dimensionality reduction method (mRMD2.0), GPCRs and non-GPCRs had no clear dividing line. In conclusion, different methods of feature extraction and the same method of dimensionality reduction have different effects on GPCRs and non-GPCRs. Therefore, the feature extraction method is the direct factor for distinguishing GPCRs from non-GPCRs.
However, a similar work was done in the Prediction of G protein-coupled sensor (Nordstrm et al., 2009) study. Compared with our study, the defects were as follows: 1. The 188D feature extraction method with more dimensions was adopted, the 188D feature extraction method had more feature dimensions, and the feature information of proteins was more complete and more comprehensive. The dimension information extracted by the CTDC method in this experiment has only 39 attribute characteristics, and there are less data. In addition, there is less redundant information after dimension reduction. 2. Five independent test sets and training sets were divided in the Prediction of G protein-coupled sensor study, and the positive and negative samples in the training set tended to be balanced by the use of strike. However, defects in the strike method lead to inaccuracy of the data. In this paper, on the basis of original data collection, feature extraction and dimensionality reduction were directly carried out to distinguish GPCRs sample from non-GPCRs sample to obtain more accurate prediction results. Compared with this paper, the advantages are as follows: 1. The accuracy of the Prediction of G Protein by Coupled sensor study is approximately 90%; while the GPCRs and non-GPCRs differentiation diagram in this paper is shown by Matplotlab, and the accuracy was not calculated correctly. 2. The universality of this experiment is relatively low. The CTDC method and MRMD2.0 dimension reduction method may only be applicable to GPCRs protein sequence but not to other protein sequence. In the study of Prediction of G protein-coupled sensor, cross validation and Random Forest can be used on other protein sequences (Lai et al., 2018; Tang et al., 2018), especially the proposed framework can be applied to protein fold recognition (Wei et al., 2016; Liu et al., 2017), protein remote homology (Liu et al., 2020), protein subcellular localization (Lv et al., 2019), etc.
Discussion
Like other macromolecules, proteins are important parts of the living body, the material basis of life, and they participate in almost every activity in the cell. Proteins perform many functions in the body. Through the study of proteins, the mechanism of diseases can be studied, and the design of new drugs can also be promoted. With the advent of machine learning, the function prediction of proteins has also flourished. Obtaining high-performance classification models, accurately and efficiently extracting protein sequences, and converting them into equal-length amino acid sequences have become research directions of many scientists.
Compared with the traditional experimental method, a set of experimental schemes in this paper replaces the redundant experimental steps. Using the CTDC method and dimensionality reduction in CTD, the redundant attributes in the protein sequence features are successfully removed, and they are drawn intuitively using Matplotlib. The division map between GPCRs and non-GPCRs is then drawn. In the division map, there can be a clear distinction between GPCRs and non-GPCRs. This experiment has achieved a certain degree of accuracy.
There are still many aspects that need to be further studied. The Matplotlib coordinate chart used to classify GPCRs and non-GPCRs can only distinguish the relatively large positive and negative samples after being divided by attributes, extracting several solutions: 1. The use of a single Matplotlib coordinate diagram is simple to operate and has many limitations; thus, it cannot reach high accuracy. In the later stage, more comprehensive computational intelligence method such as neural networks (Song et al., 2018a; Zhou et al., 2018; Bao et al., 2019; Hong et al., 2019; Sun et al., 2020), network methods (Sun et al., 2014; Zhou et al., 2015, 2016; Song et al., 2018b; Zeng et al., 2018) and evolutionary strategies (Xu et al., 2019a,b; Zeng et al., 2019b) can be adopted to take the extracted protein features as input. Thus, the positive and negative samples can be divided more accurately, and accuracy can be obtained. 2. In terms of high extraction accuracy, a more comprehensive protein feature extraction method combined with the dimension reduction method (Yang et al., 2019; Zhu et al., 2019) for GPCRs pruning was attempted to screen out features with higher differentiation between GPCRs and non-GPCRs.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author Contributions
ZC made the design of the subject and the whole idea of the whole experiment in the early stage. XG did comparative experiments and experimental data analysis. DW analyzed the results of the comparative experiment. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Chinese National Natural Science Foundation under Grant 61876047.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Balfanz, S., Jordan, N., Langenstück, T., Breuer, J., Bergmeier, V., and Baumann, A. (2013). Molecular, pharmacological, and signaling properties of octopamine receptors from honeybee (Apis mellifera) brain. J. Neurochem. 129, 284–296. doi: 10.1111/jnc.12619
Bao, S., Zhao, H., Yuan, J., Fan, D., Zhang, Z., Su, J., et al. (2019). Computational identification of mutator-derived lncRNA signatures of genome instability for improving the clinical outcome of cancers: a case study in breast cancer. Brief. Bioinform. bbz118. doi: 10.1093/bib/bbz118
Cai, C. Z., Han, L. Y., Ji, Z. L., Chen, X., and Chen, Y. Z. (2003). SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucl. Acids Res. 31, 3692–3697. doi: 10.1093/nar/gkg600
Chen, W., Feng, P., Liu, T., and Jin, D. (2019). Recent advances in machine learning methods for predicting heat shock proteins. Curr Drug Metab 20, 224–228. doi: 10.2174/1389200219666181031105916
Chen, W., Nie, F., and Ding, H. (2020). Recent advances of computational methods for identifying bacteriophage virion proteins. Protein Pept. Lett. 27, 259–264. doi: 10.2174/0929866526666190410124642
Cheng, J. H., Yang, H., Liu, M. L., Su, W., Feng, P. M., Ding, H., et al. (2018a). Prediction of bacteriophage proteins located in the host cell using hybrid features. Chemometr. Intell. Lab. Syst. 180, 64–69. doi: 10.1016/j.chemolab.2018.07.006
Cheng, L. (2019). Computational and biological methods for gene therapy. Curr. Gene Ther. 19:210. doi: 10.2174/156652321904191022113307
Cheng, L., and Hu, Y. (2018). Human disease system biology. Curr Gene Ther. 18, 255–256. doi: 10.2174/1566523218666181010101114
Cheng, L., Jiang, Y., Ju, H., Sun, J., Peng, J., Zhou, M., et al. (2018b). InfAcrOnt: calculating cross-ontology term similarities using information flow by a random walk. BMC Genom. 19:919. doi: 10.1186/s12864-017-4338-6
Cheng, L., Zhao, H., Wang, P., Zhou, W., Luo, M., Li, T., et al. (2019). Computational methods for identifying similar diseases. Molecular therapy. Nucl. Acids 18, 590–604. doi: 10.1016/j.omtn.2019.09.019
Ding, C., and Peng, H. (2005). Minimum redundancy feature selection from microarray gene expression data. J. Bioinf. Comput. Biol. 3, 185–205. doi: 10.1142/S0219720005001004
Ding, Y., Tang, J., and Guo, F. (2016). Predicting protein-protein interactions via multivariate mutual information of protein sequences. Bmc Bioinformatics 17:398. doi: 10.1186/s12859-016-1253-9
Ding, Y., Tang, J., and Guo, F. (2017). Identification of drug-target interactions via multiple information integration. Inf. Sci. 418, 546–560. doi: 10.1016/j.ins.2017.08.045
Ding, Y., Tang, J., and Guo, F. (2019). Identification of drug-side effect association via multiple information integration with centered kernel alignment. Neurocomputing 325, 211–224. doi: 10.1016/j.neucom.2018.10.028
Dubchak, I., Muchnik, I., Holbrook, S. R., and Kim, S. H. (1995). Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. U.S.A. 92, 8700–8704. doi: 10.1073/pnas.92.19.8700
Dubchak, I., Muchnik, I., Mayor, C., and Dralyuk, I. (1999). Recognition of a protein fold in the context of the SCOP classification. Proteins Struct. Funct. Bioinform. 35, 401–407. doi: 10.1002/(SICI)1097-0134(19990601)35:4<401::AID-PROT3>3.0.CO;2-K
Feng, Y. M. (2019). Gene therapy on the road. Curr. Gene Ther. 19:6. doi: 10.2174/1566523219999190426144513
Fredriksson, R., Lagerström, M. C., Lundin, L. G., and Schiöth, H. B. (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 63, 1256–1272. doi: 10.1124/mol.63.6.1256
Guo, Y., Wu, C., Guo, M., Zou, Q., Liu, X., and Keinan, A. (2019). Combining sparse group lasso and linear mixed model improves power to detect genetic variants underlying quantitative traits. Front. Genet. 10:271. doi: 10.3389/fgene.2019.00271
Han, L. Y., Cai, C. Z., Ji, Z. L., Cao, Z. W., Cui, J., and Chen, Y. Z. (2004). Predicting functional family of novel enzymes irrespective of sequence similarity: a statistical learning approach. Nucl. Acids Res. 32, 6437–6444. doi: 10.1093/nar/gkh984
Hong, Z., Zeng, X., Wei, L., and Liu, X. J. B. (2019). Identifying enhancer-promoter interactions with neural network based on pre-trained DNA vectors and attention mechanism. Bioinformatics 36, 1037–1043. doi: 10.1093/bioinformatics/btz694
Islam, S. M. A., Heil, B. J., Kearney, C. M., and Baker, E. J. (2017). Protein classification using modified n-grams and skip-grams. Bioinformatics. 34, 1481–1487. doi: 10.1093/bioinformatics/btx823
Junwei, H., Xudong, H., Qingfei, K., and Liang, C. (2020). psSubpathway: a software package for flexible identification of phenotype-specific subpathways in cancer progression. Bioinformatics. 36, 2303–2305. doi: 10.1093/bioinformatics/btz894
Kentaro, T., and Minoru, K. (1996). Analysis of amino acid indices and mutation matrices for sequence comparison and structure prediction of proteins. Protein Eng. 9, 27–36.
Krishnan, A., Sällman, M., Robert, A., Helgi, F., and Schiöth, H. B. (2012). The origin of GPCRs: identification of mammalian like rhodopsin, adhesion, glutamate and frizzled GPCRs in fungi. PLoS ONE 7:e29817. doi: 10.1371/journal.pone.0029817
Lai, H. Y., Feng, C. Q., Zhang, Z. Y., Tang, H., Chen, W., and Lin, H. (2018). A brief survey of machine learning application in cancerlectin identification. Curr. Gene Ther. 18, 257–267. doi: 10.2174/1566523218666180913112751
Liang, C., Changlu, Q., He, Z., Tongze, F., and Xue, Z. (2019). gutMDisorder: a comprehensive database for dysbiosis of the gut microbiota in disorders and interventions. Nucl. Acids Res. 48, D554–D560. doi: 10.1093/nar/gkz843
Liao, Z., Ying, J., and Quan, Z. (2016). Prediction of G protein-coupled receptors with SVM-prot features and random forest. Scientifica 2016:8309253. doi: 10.1155/2016/8309253
Liu, B. (2019). BioSeq-analysis: a platform for DNA, RNA, and protein sequence analysis based on machine learning approaches. Brief. Bioinf. 20, 1280–1294. doi: 10.1093/bib/bbx165
Liu, B., Chen, S., Yan, K., and Weng, F. (2019b). iRO-PsekGCC: identify DNA replication origins based on Pseudo k-tuple GC composition. Front. Genet. 10:842. doi: 10.3389/fgene.2019.00842
Liu, B., Gao, X., and Zhang, H. (2019a). BioSeq-analysis2.0: an updated platform for analyzing DNA, RNA, and protein sequences at sequence level and residue level based on machine learning approaches. Nucl. Acids Res. 47:e127. doi: 10.1093/nar/gkz740
Liu, B., Jiang, S., and Zou, Q. (2020). HITS-PR-HHblits: protein remote homology detection by combining PageRank and hyperlink-induced topic search. Brief. Bioinf. 21, 298–308. doi: 10.1093/bib/bby104
Liu, B., Li, C., and Yan, K. (2019). DeepSVM-fold: protein fold recognition by combining Support Vector Machines and pairwise sequence similarity scores generated by deep learning networks. Brief. Bioinf . doi: 10.1093/bib/bbz098
Liu, B., and Li, K. (2019). iPromoter-2L2.0: identifying promoters and their types by combining Smoothing Cutting Window algorithm and sequence-based features. Mol. Ther. Nucl. Acids 18, 80–87. doi: 10.1016/j.omtn.2019.08.008
Liu, B., Luo, Z., and He, J. (2020). sgRNA-PSM: predict sgRNAs on-target activity based on position specific mismatch. Mol. Ther. Nucl. Acids. 20, 323–330. doi: 10.1016/j.omtn.2020.01.029
Liu, B., and Zhu, Y. (2019). ProtDec-LTR3.0: protein remote homology detection by incorporating profile-based features into Learning to Rank. IEEE Access 7, 102499–102507. doi: 10.1109/ACCESS.2019.2929363
Liu, B., Zhu, Y., and Yan, K. (2017). Fold-LTR-TCP: protein fold recognition based on triadic closure principle. Brief. Bioinf. 25. doi: 10.1093/bib/bbz139
Liu, K., and Chen, W. (2020). iMRM:a platform for simultaneously identifying multiple kinds of RNA modifications. Bioinformatics. 36, 3336–3342. doi: 10.1093/bioinformatics/btaa155
Lv, H., Dao, F. Y., Zhang, D., Guan, Z. X., Yang, H., Su, W., et al. (2020). iDNA-MS: an integrated computational tool for detecting DNA modification sites in multiple genomes. iScience 23:100991. doi: 10.1016/j.isci.2020.100991
Lv, Z., Jin, S., Ding, H., and Zou, Q. (2019). A random forest sub-Golgi protein classifier optimized via dipeptide and amino acid composition features. Front. Bioeng. Biotechnol. 7:215. doi: 10.3389/fbioe.2019.00215
Nordstrm, K. J. V., Linn, M. C. L., Wallér, M. J., Fredriksson, R., and Schith, H. B. (2009). The secretin GPCRs descended from the family of adhesion GPCRs. Mol. Biol. Evol. 26, 71–84. doi: 10.1093/molbev/msn228
Pang, H., Lin, A., Holford, M., Enerson, B., Lu, B., Lawton, M., et al. (2006). Pathway analysis using random forests classification and regression. Bioinformatics 22, 2028–2036. doi: 10.1093/bioinformatics/btl344
Patil, K., and Chouhan, U. (2019). Relevance of machine learning techniques and various protein features in protein fold classification: a review. Curr. Bioinf. 14, 688–697. doi: 10.2174/1574893614666190204154038
Peng, H., Long, F., and Ding, C. (2005). Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 27, 1226–1238. doi: 10.1109/TPAMI.2005.159
Ru, X., Wang, L., Li, L., Ding, H., Ye, X., and Zou, Q. (2020). Exploration of the correlation between GPCRs and drugs based on a learning to rank algorithm. Comput. Biol. Med. 119:103660. doi: 10.1016/j.compbiomed.2020.103660
Shen, C., Jiang, L., Ding, Y., Tang, J., and Guo, F. (2019). LPI-KTASLP: prediction of lncRNA-protein interaction by semi-supervised link learning with multivariate information. IEEE Access 7, 13486–13496. doi: 10.1109/ACCESS.2019.2894225
Song, T., Rodríguez-Patón, A., Zheng, P., and Zeng, C. (2018a). Spiking neural P systems with colored spikes. IEEE Trans. Cogn. Dev. Syst. 10, 1106–1115. doi: 10.1109/TCDS.2017.2785332
Song, T., Zeng, X., Zheng, P., Jiang, M., and Rodríguez-Patón, A. (2018b). A parallel workflow pattern modelling using spiking neural p systems with colored spikes. IEEE Trans. Nanobiosci. 17, 474–484. doi: 10.1109/TNB.2018.2873221
Su, R., Wu, H., Xu, B., Liu, X., and Wei, L. (2019). Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data. IEEE ACM Trans. Comput. Biol. Bioinf. 16, 1231–1239. doi: 10.1109/TCBB.2018.2858756
Sun, J., Shi, H., Wang, Z., Zhang, C., Liu, L., Wang, L., et al. (2014). Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 10, 2074–2081. doi: 10.1039/C3MB70608G
Sun, J., Zhang, Z., Bao, S., Yan, C., Hou, P., Wu, N., et al. (2020). Identification of tumor immune infiltration-associated lncRNAs for improving prognosis and immunotherapy response of patients with non-small cell lung cancer. J. Immunother. Cancer 8:e000110. doi: 10.1136/jitc-2019-000110
Tang, H., Zhao, Y. W., Zou, P., Zhang, C. M., Chen, R., Huang, P., et al. (2018). HBPred: a tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 14, 957–964. doi: 10.7150/ijbs.24174
Tibshirani, R. (1996). Regression shrinkage and selection via the lasso. J. R. Stat. Soc. 58, 267–288. doi: 10.1111/j.2517-6161.1996.tb02080.x
Wang, H., Ding, Y., Tang, J., and Guo, F. (2020). Identification of membrane protein types via multivariate information fusion with Hilbert-Schmidt Independence Criterion. Neurocomputing 383, 257–269. doi: 10.1016/j.neucom.2019.11.103
Wang, S. P., Zhang, Q., Lu, J., and Cai, Y. D. (2018). Analysis and prediction of nitrated tyrosine sites with the mRMR method and support vector machine algorithm. Curr. Bioinform. 13, 3–13. doi: 10.2174/1574893611666160608075753
Wei, L., Ding, Y., Su, R., Tang, J., and Zou, Q. (2018). Prediction of human protein subcellular localization using deep learning. J. Parallel Distrib. Comput. 117, 212–217. doi: 10.1016/j.jpdc.2017.08.009
Wei, L., Liao, M., Gao, X., and Zou, Q. (2015). An improved protein structural prediction method by incorporating both sequence and structure information. IEEE Trans. Nanobiosci. 14, 339–349. doi: 10.1109/TNB.2014.2352454
Wei, L., Wan, S., Guo, J., and Wong, K. K. (2017b). A novel hierarchical selective ensemble classifier with bioinformatics application. Artif. Intell. Med. 83, 82–90. doi: 10.1016/j.artmed.2017.02.005
Wei, L., Xing, P., Shi, G., Ji, Z., and Zou, Q. (2019). Fast prediction of methylation sites using sequence-based feature selection technique. IEEE ACM Trans. Comput. Biol. Bioinf. 16, 1264–1273. doi: 10.1109/TCBB.2017.2670558
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017a). Improved prediction of protein–protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Wei, L., Zou, Q., Liao, M., Lu, H., and Zhao, Y. (2016). A novel machine learning method for cytokine-receptor interaction prediction. Combinatorial Chem. High Throughput Screen. 19, 144–152. doi: 10.2174/1386207319666151110122621
Wen, S., Dong, M., Yang, Y., Zhou, P., Huang, T., and Chen, Y. (2020). End-to-end detection-segmentation network for face labeling. IEEE Trans. Emerg. Top. Comput. Intell. 1–11. doi: 10.1109/TETCI.2019.2947319
Xu, H., Zeng, W., Zeng, X., and Yen, G. G. (2019b). An evolutionary algorithm based on minkowski distance for many-objective optimization. IEEE Trans. Cybernet. 49, 3968–3979. doi: 10.1109/TCYB.2018.2856208
Xu, H., Zeng, W., Zhang, D., and Zeng, C. (2019a). MOEA/HD: a multiobjective evolutionary algorithm based on hierarchical decomposition. IEEE Trans. Cybernet. 49, 517–526. doi: 10.1109/TCYB.2017.2779450
Xu, L., Liang, G., Liao, C., Chen, G.-D., and Chang, C.-C. (2019c). k-skip-n-gram-RF: a random forest based method for Alzheimer's disease protein identification. Front. Genet. 10:33. doi: 10.3389/fgene.2019.00033
Xu, L., Liang, G., Shi, S., and Liao, C. (2018b). SeqSVM: a sequence-based support vector machine method for identifying antioxidant proteins. Int. J. Mol. Sci. 19:1773. doi: 10.3390/ijms19061773
Xu, L., Liang, G., Wang, L., and Liao, C. (2018a). A novel hybrid sequence-based model for identifying anticancer peptides. Genes 9:158. doi: 10.3390/genes9030158
Yan, K., Fang, X., Xu, Y., and Liu, B. (2019). Protein fold recognition based on multi-view modeling. Bioinformatics 35, 2982–2990. doi: 10.1093/bioinformatics/btz040
Yang, W., Zhu, X. J., Huang, J., Ding, H., and Lin, H. (2019). A brief survey of machine learning methods in protein sub-Golgi localization. Curr. Bioinf. 14, 234–240. doi: 10.2174/1574893613666181113131415
Zeng, X., Wang, W., Chen, C., and Yen, C. (2019b). A consensus community-based particle swarm optimization for dynamic community detection. IEEE Trans. Cybernet. 1–12. doi: 10.1109/TCYB.2019.2938895
Zeng, X., Zhu, S., Liu, X., Zhou, Y., Nussinov, R., and Cheng, F. (2019a). deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics 35, 5191–5198. doi: 10.1093/bioinformatics/btz418
Zeng, X., Zhu, S., Lu, W., Liu, Z., Huang, J., Zhou, Y., et al. (2020). Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 11, 1775–1797. doi: 10.1039/C9SC04336E
Zeng, X. X., Liu, L., Lu, L. Y., and Zou, Q. (2018). Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 34, 2425–2432. doi: 10.1093/bioinformatics/bty112
Zhang, J., and Liu, B. (2019). A review on the recent developments of sequence-based protein feature extraction methods. Curr. Bioinf. 14, 190–199. doi: 10.2174/1574893614666181212102749
Zhang, X., Zou, Q., Rodriguez-Paton, A., and Zeng, X. (2019). and Bioinformatics, Meta-path methods for prioritizing candidate disease miRNAs. IEEE ACM Trans. Comput. Biol. Bioinf. 16, 283–291. doi: 10.1109/TCBB.2017.2776280
Zhang, Y., Liu, B., Dong, Q., and Jin, V. X. (2011). An improved profile-level domain linker propensity index for protein domain boundary prediction. Protein Peptide Lett. 18, 7–16. doi: 10.2174/092986611794328717
Zhou, M., Hu, L., Zhang, Z., Wu, N., Sun, J., and Su, J. (2018). Recurrence-associated long non-coding RNA signature for determining the risk of recurrence in patients with colon cancer. Mol. Ther. Nucl. Acids 12, 518–529. doi: 10.1016/j.omtn.2018.06.007
Zhou, M., Wang, X., Li, J., Hao, D., Wang, Z., Shi, H., et al. (2015). Prioritizing candidate disease-related long non-coding RNAs by walking on the heterogeneous lncRNA and disease network. Mol. Biosyst. 11, 760–769. doi: 10.1039/C4MB00511B
Zhou, M., Wang, X., Shi, H., Cheng, L., Wang, Z., Zhao, H., et al. (2016). Characterization of long non-coding RNA-associated ceRNA network to reveal potential prognostic lncRNA biomarkers in human ovarian cancer. Oncotarget. 7, 12598–12611. doi: 10.18632/oncotarget.7181
Zhu, X. J., Feng, C. Q., Lai, H. Y., Chen, W., and Lin, H. (2019). Predicting protein structural classes for low-similarity sequences by evaluating different features. Knowl. Based Syst. 163, 787–793. doi: 10.1016/j.knosys.2018.10.007
Zou, Q., Lin, G., Jiang, X., Liu, X., and Zeng, X. (2020). Sequence clustering in bioinformatics: an empirical study. Brief. Bioinform. 21, 1–10. doi: 10.1093/bib/bby090
Zou, Q., Wan, S., Ju, Y., Tang, J., and Zeng, X. (2016a). Pretata: predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 10:114. doi: 10.1186/s12918-016-0353-5
Zou, Q., Wang, Z., Guan, X., Liu, B., Wu, Y., and Lin, Z. (2013). An approach for identifying cytokines based on a novel ensemble classifier. BioMed Res. Int. 2013:686090. doi: 10.1155/2013/686090
Keywords: feature extraction, CTD, MRMD2.0, Matplotlib, predict GPCRs
Citation: Gu X, Chen Z and Wang D (2020) Prediction of G Protein-Coupled Receptors With CTDC Extraction and MRMD2.0 Dimension-Reduction Methods. Front. Bioeng. Biotechnol. 8:635. doi: 10.3389/fbioe.2020.00635
Received: 21 April 2020; Accepted: 26 May 2020;
Published: 25 June 2020.
Edited by:
Zhibin Lv, University of Electronic Science and Technology of China, ChinaCopyright © 2020 Gu, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihua Chen, Y3poZ2QmI3gwMDA0MDtnemh1LmVkdS5jbg==; Donghua Wang, d2FuZ2RvbmdodWE3ODg1JiN4MDAwNDA7MTYzLmNvbQ==