 Xizi Luo1†
Xizi Luo1† Qingyu Meng
Qingyu Meng Guoqing Wang
Guoqing Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 30 June 2020
Sec. Computational Genomics
Volume 8 - 2020 | https://doi.org/10.3389/fbioe.2020.00607
This article is part of the Research Topic Explainable Intelligent Processing of Biological Resources Integrating Data, Information, Knowledge, and Wisdom View all 21 articles
Background: Mycobacterium tuberculosis is one of the deadliest pathogens in humans. Co-infection of M. tuberculosis with HIV and the emergence of multi-drug-resistant tuberculosis (TB) constitute a serious global threat. However, no effective anti-TB drugs are available, with the exception of first-line drugs such as isoniazid. The cell wall of M. tuberculosis, which is primarily responsible for the lack of effective anti-TB drugs and the escape of the bacteria from host immunity, is an important drug target. The core components of the cell wall of M. tuberculosis are peptidoglycan, arabinogalactan, and mycotic acid. However, the functional genome and metabolic regulation pathways for the M. tuberculosis cell wall are still unknown. In this study, we used the biclustering algorithm integrated into cMonkey, sequence alignment, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and other bioinformatics methods to scan the whole genome of M. tuberculosis as well as to identify and statistically analyze the genes related to the synthesis of the M. tuberculosis cell wall.
Method: We performed high-throughput genome-wide screening for M. tuberculosis using Biocarta, KEGG, National Cancer Institute Pathway Interaction Database (NCI-PID), HumanCyc, and Reactome. We then used the Database of Origin and Registration (DOOR) established in our laboratory to classify the collection of operons for M. tuberculosis cell wall synthetic genes. We used the cMonkey double clustering algorithm to perform clustering analysis on the gene expression profile of M. tuberculosis for cell wall synthesis. Finally, we visualized the results using Cytoscape.
Result and Conclusion: Through bioinformatics and statistical analyses, we identified 893 M. tuberculosis H37Rv cell wall synthesis genes, distributed in 20 pathways, involved in 46 different functions related to cell wall synthesis, and clustered in 386 modules. We identified important pivotal genes and proteins in the cell wall synthesis pathway such as murA, a class of operons containing genes involved in cell wall synthesis such as ID6951, and a class of operons indispensable for the survival of the bacteria. In addition, we found 41 co-regulatory modules for cell wall synthesis and five co-expression networks of molecular complexes involved in peptidoglycan biosynthesis, membrane transporter synthesis, and other cell wall processes.
Mycobacterium tuberculosis is considered one of the world’s most successful pathogens. The disease caused by it has been a major global health challenge (Sher et al., 2020). Since the 1950s, the discovery of first-line anti-tuberculosis (TB) drugs such as isoniazid, rifampicin, and ethambutol has effectively improved the cure rate and survival rate of TB patients. However, the emergence of multiple forms of drug-resistant strains, including a single isoniazid-resistant strain, a multi-drug-resistant strain, and a widely drug-resistant strain, has again made M. tuberculosis one of the leading causes of death worldwide, with a mortality of 1.5 million people in 2018 (Merker et al., 2020). Co-infection of HIV and M. tuberculosis increases the burden of curing TB; therefore, the development of new and effective anti-TB drugs is critical (Turner et al., 2020).
The cell wall structure of M. tuberculosis is unique and is extremely important for the invasion, survival, and reproduction of the bacterium in a host. The main reason for the difficulty in developing drugs for M. tuberculosis is that the bacterium has a hard cell wall and very low permeability. The development of M. tuberculosis resistance is also associated with the cell wall. Howard et al. (2018) found that M. tuberculosis carrying a rifampicin-resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Maitra et al. (2019) described M. tuberculosis cell wall peptidoglycan as its fatal weakness. Thus, the cell wall of M. tuberculosis is an important target for the development of new anti-TB drugs.
In this study, we performed high-throughput screening of M. tuberculosis cell wall synthesis genes and screened key genes using bioinformatics and statistical methods to obtain new key targets for the development of anti-TB drugs.
The relevant data for M. tuberculosis cell wall synthesis genes used in this study were obtained from the screening and integration of the following databases: TubercuList (Lew et al., 2011), TBDB (Galagan et al., 2010), PATRIC (Gillespie et al., 2011), MycoDB (Chaudhuri, 2009), GenoMycDB (Catanho et al., 2006), MyBASE (Zhu et al., 2009), MabsBase (Heydari et al., 2013), and MGDD (Vishnoi et al., 2008).
We used online software1 to compare the amino acid sequence of M. tuberculosis H37Rv with the amino acid sequence of Mycobacterium smegmatis, Mycobacterium leprae, Mycobacterium bovis, and M. tuberculosis H37Ra. Genes with homology greater than 60% were selected (Kuroda et al., 2001; Hellweger et al., 2014).
The whole genome information for M. tuberculosis H37Rv was obtained from the National Center for Biotechnology Information (NCBI) and annotated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database with KEGG Orthology (KO) in accordance with the “binary relationships” provided by the KEGG Brite database. The types and functions of cell wall synthesis genes were determined using Clusters of Orthologous Groups with KO (KO COG) and the P-Score and E-Score for each KO were calculated. The E-Score was calculated with KO using the same path annotation and the P-Score was determined from the e-score. The P-Score-KEGG and P-Score-COG were also calculated based on the KEGG and COG annotations (Kong et al., 2019). These two values were in the range of 0 to 1, with 0 indicating a lack of necessity and 1 indicating necessity.
We applied the operon Database of Origin and Registration (DOOR) (Cao et al., 2019) established in our laboratory to classify the operon collection of cell wall genes. The DOOR database uses two prediction procedures. For operon genomes with a large number of experimental verifications, we used a non-linear classifier to train the known operon subsets based on the general characteristics of the genome and the characteristics of specific genomes. For genomes without experimental data, we used linear classification to predict operons for the general characteristics of the genome.
We selected all M. tuberculosis H37Rv gene chips in NCBI after filtering out irrelevant chip data and performed min-max normalization on each chip. We used the cMonkey double clustering algorithm to establish seed clusters (Waltman et al., 2010). We calculated the P-values of three such model components based on the amount of co-expressed genes, upstream sequences, and association networks. We optimized seed clusters by adding or removing related genes and proceeded to build new clusters. We used the Monte Carlo procedure to calculate the probability of each gene or condition sampled as a dual cluster gene with the conditional probability at each stage. Through these procedures, the genomic co-regulation network was identified.
We performed a Gene Ontology (GO) analysis of the target genes using the comprehensive database Davide2 for enrichment analysis, annotation, and visualization. We used the Biocarta, KEGG, National Cancer Institute Pathway Interaction Database (NCI-PID), HumanCyc, and Reactome pathway databases for pathway enrichment of the target genes. P < 0.05 was considered statistically significant when the threshold was ≥ two genes. We used R software and the Perl language to visualize the enrichment results. We also installed “Rcpp,” “ggplot2,” and other related software packages (Postma and Goedhart, 2019).
The protein–protein interaction (PPI) network was constructed using a gene interaction search tool database (STRING) and Cytoscape 3.6.1 was used for visualization. The Minimal Common Oncology Data Elements (MCODE), a Cytoscape network analysis plug-in for molecular complex detection, was used to deeply mine the existing modules in the network structure to find the core gene clustering modules with the highest levels of interaction.
Through database annotation and sequence alignment, we screened the cell wall synthesis genes for mycobacteria. As shown in Table 1, there were 892 cell wall synthesis genes for M. tuberculosis H37Rv, 888 for M. tuberculosis H37Ra, 780 for M. bovis, 508 for M. smegmatis, and 454 for M. leprae.
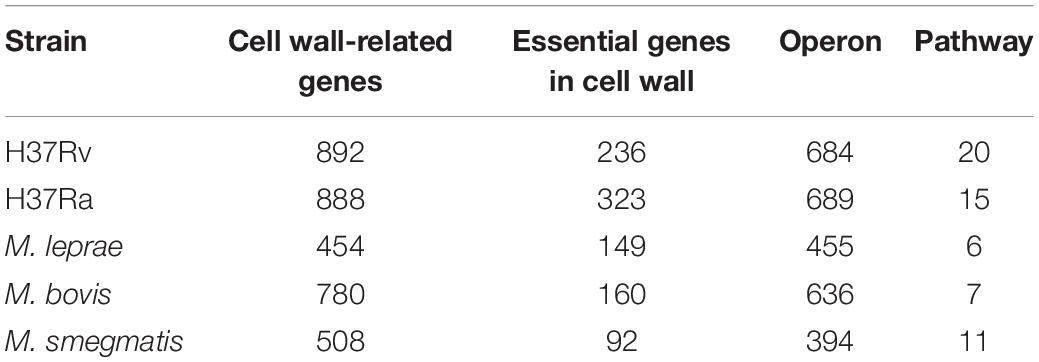
Table 1. Cell wall synthesis network module in mycobacteria.
We used the operon database DOOR to assess the module distribution of cell wall synthesis genes. In M. tuberculosis H37Rv, 893 genes related to cell wall synthesis were located in 684 operons and 37 operons contained three or more cell wall-related genes. Multiple genes located in an operon are usually regulated by the same control region and constitute a transcription unit. The 149 genes contained in these 37 operons may be key genes that play an important role in the synthesis of the M. tuberculosis cell wall. There are four sets of operons, which contain more than seven genes related to the cell wall, including operons with ID numbers 7375, 7760, 6927, and 7590 displayed in the DOOR database. The ID number of the operon with the largest number of genes is 7558, up to 9. The genes yrbE1A and yrbE1B encode cell wall membrane proteins (Pasricha et al., 2011). The proteins encoded by mce3A and mce3B are not only present in the cell wall, but are also important for the virulence of M. tuberculosis during host invasion (Ahmad et al., 2005); 37 pairs of operons in this pathway and their details are shown in Supplementary Table S1.
The main cause of infection of the host with M. tuberculosis is the virulence factor. We obtained all coding genes related to virulence of TB from the VFDB database, of which 115 genes are cell wall synthesis genes. The cell wall genes that belong to virulence included the mmpl family which encoded cell wall lipid transporters, the cell wall mycolic acid synthase mmA4, and Rv2224c with little research and unknown specific function. Genes related to cell walls and virulence factors are shown in Supplementary Table S2.
Essential genes are often critical for sustaining the activities of living organisms. As shown in Table 1, there are 236 essential genes related to cell wall synthesis in the whole genome of M. tuberculosis H37Rv. These genes are located in 161 operons, among which there are 10 operons containing more than three essential genes and five operons with more than four essential genes. Three or more operons have five or more essential genes. The six genes controlled by the operon ID6951 are all required genes that play key roles in cell wall synthesis. The six required genes include eccA3-E3, a member of the ESAT6 secretory system (ESX), and membrane-anchored mycosin mycP3. ESX secretion systems mediate various functions, participate in the metabolism of zinc and iron, and play an important role in cell wall integrity (Gaur et al., 2017).
We used KEGG, BioCyc, and Reactome pathway data to analyze cell wall synthetic genes (Figures 1A,B). The 892 cell wall synthesis genes in the H37Rv strain were distributed in 39 signaling pathways. The essential gene murA participates in the metabolic pathway (KEGG mtu01100), peptidoglycan biosynthesis (KEGG mtu00550), and UDP-N-acetylmuramoyl-pentapeptide biosynthesis I (BioCyc pwy6387). AftB is involved in the super pathways of mycolyl-arabinogalactan-peptidoglycan complex biosynthesis (BioCyc pwy6404) and Lipoarabinomannan biosynthesis (KEGG mtu00571). MurA and aftB are thought to be key node genes in the cell wall biosynthesis pathway. In addition, we identified some genes whose functions are currently unknown but which are located in important pathways such as the mycolyl-arabinogalactan-peptidoglycan complex biosynthesis (BioCyc PWY-6397) pathway.
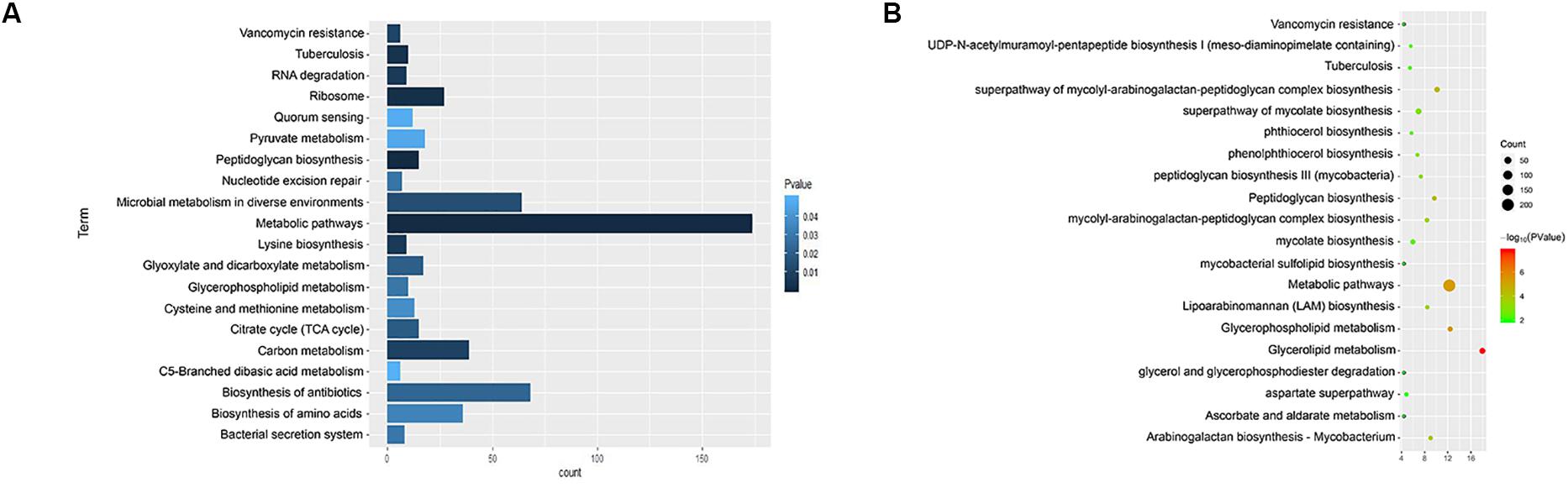
Figure 1. Enrichment of cell wall-related gene pathways in the standard strain of M. tuberculosis H37Rv. (A) The top 20 pathways with the lowest p-values (<0.05) for KEGG were selected and a histogram was created. (B) The top 20 pathways with the lowest p-values (<0.05) for Biocarta, KEGG, NCI-PID, HumanCyc, and Reactome are shown in a bubble chart.
We annotated the gene functions using GO and identified 46 GO items in the 892 cell wall synthesis genes. As shown in Figure 2, there were 19 items related to biological processes (BPs), nine items related to cell components (CCs), and 18 items related to molecular function (MF). The most significant BP terms were related to cell wall organization (GO:0071555), regulation of cell shape (GO:0008360), and peptidoglycan biosynthetic process (GO:0009252), as shown in Figure 2A.
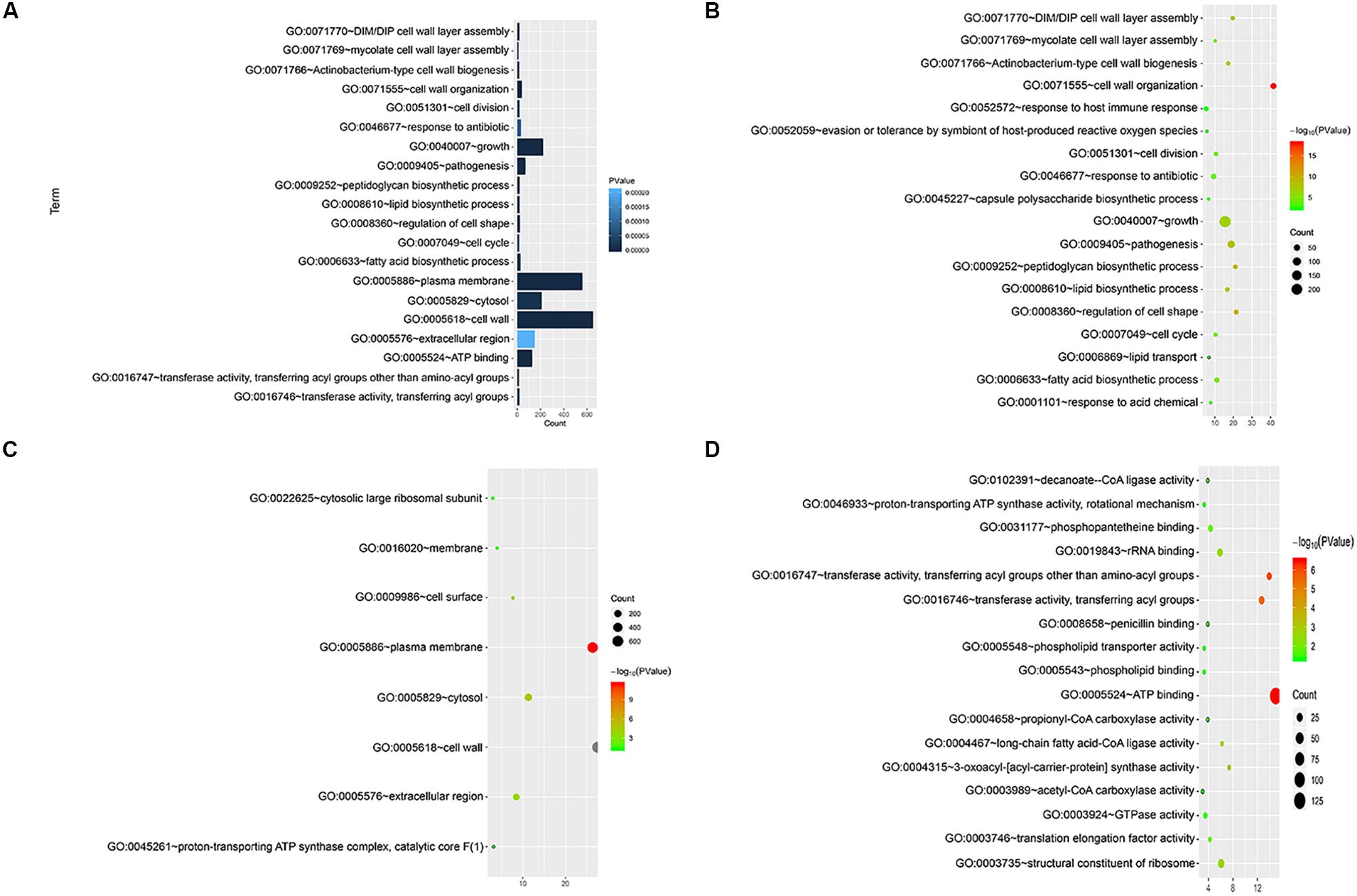
Figure 2. Functional significance analysis of cell wall-related genes in the standard strain of M. tuberculosis H37Rv. (A) Histogram showing the top 20 pathways identified in function analysis of cell wall-related genes. (B–D) Bubble charts showing the top 20 pathways in BP, CC, and MF.
We also visualized and clustered the enriched GO and KEGG terms using the cluego in Cytoscape (Figure 3). We found that most genes are enriched in important cell wall-related pathways, such as lipid biosynthetic process, peptidoglycan-base cell wall synthesis, lipid synthesis, and 3-oxoacyl-acyl-carrier-protein sythase activity. In addition, it is closely related to the pathogenicity of the host, symbiosis of the host, secretion, and pathogenesis.
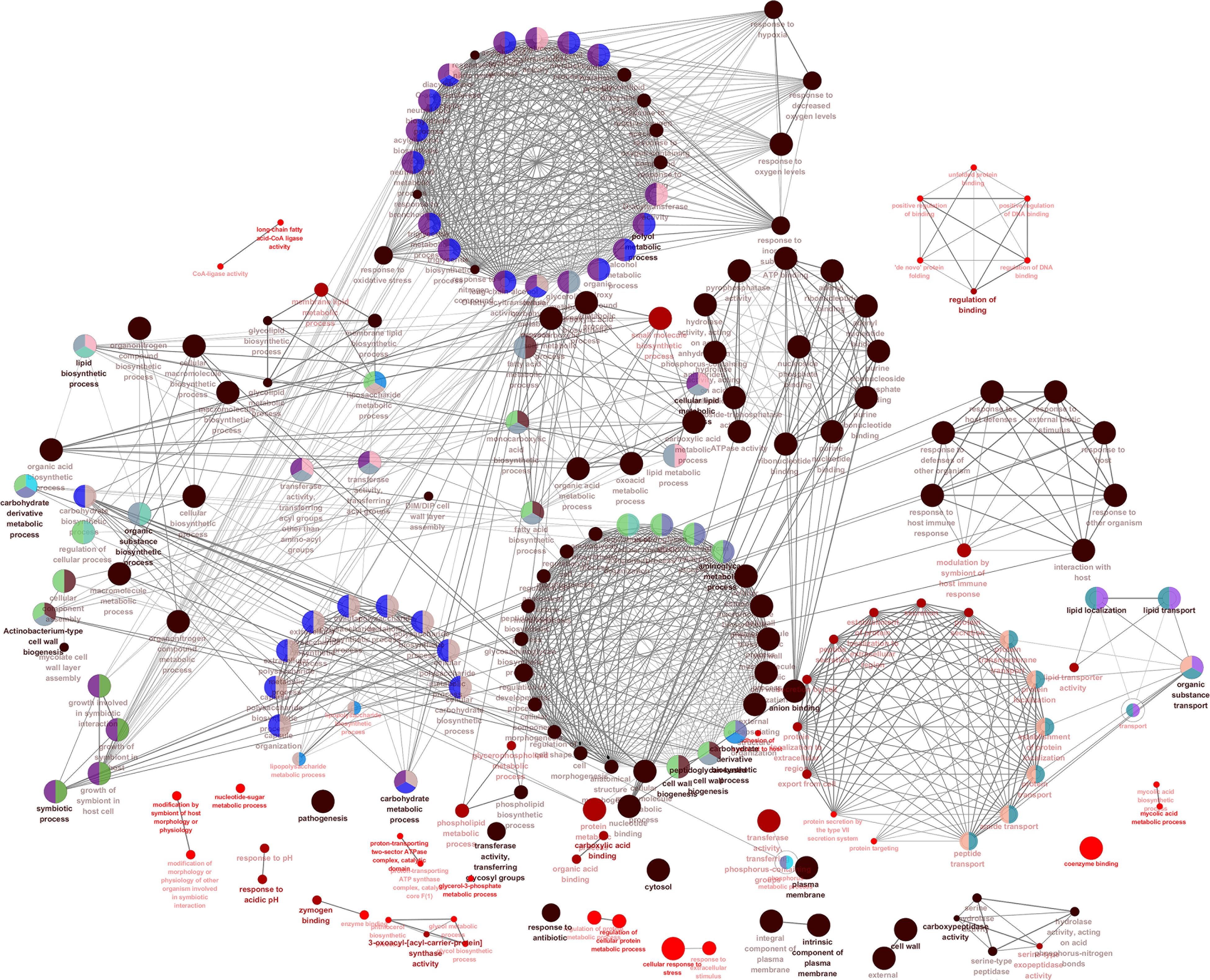
Figure 3. The enrichment map of GO annotation and KEGG pathway. Node size represents the number of cell wall genes expressed in specific terms. The edge thickness represents the number of genes shared by the two items connected by the edge.
By screening the M. tuberculosis cell wall modules using gene chips and the cMonkey double clustering algorithm, we found that the total number of M. tuberculosis modules was 600, among which 386 contained the target genes for cell wall synthesis.
Among the modules containing the target genes, 41 modules contained more than four target genes. Among these 41 modules, 16 were related to the synthesis of sugar in the cell wall, such as bicluster_0098 for the mannosyl transfer process and bicluster_0329 for the peptidoglycosyl transfer process. Fifteen modules were related to the synthesis of lipids. The modules bicluster_0068 and bicluster_0012 were related to the synthesis of mycobacterial acid (Saelens et al., 2018). There were 10 modules related to cell wall surface proteins and virulence. Among them, bicluster_0384 contained the largest number of target genes for cell wall synthesis in a single module. The nine genes contained in this module are all involved in the biosynthetic process for arabinose. For example, the Rv0129c coding protein plays a role in the addition of mycosyl residues in the cell wall arabinose (Jiang et al., 2020) and Rv3806c plays a role in the synthesis of decenyl phosphate D-arabinose (Safi et al., 2013).
In the process of gene transcription, transcription factors complete the binding of proteins to DNA by identifying specific sequences of the double helix structure (motif). The motif is short and conservative, consisting of about 20 base pairs. Many key regulatory pathways in the cell are usually recruited by a motif (Ivarsson and Jemth, 2019). Genes located in a module are regulated by a transcription factor and have the same motif. We mapped the motif base distribution for the four modules with the largest number of cell wall genes, as shown in Figures 4A–D.
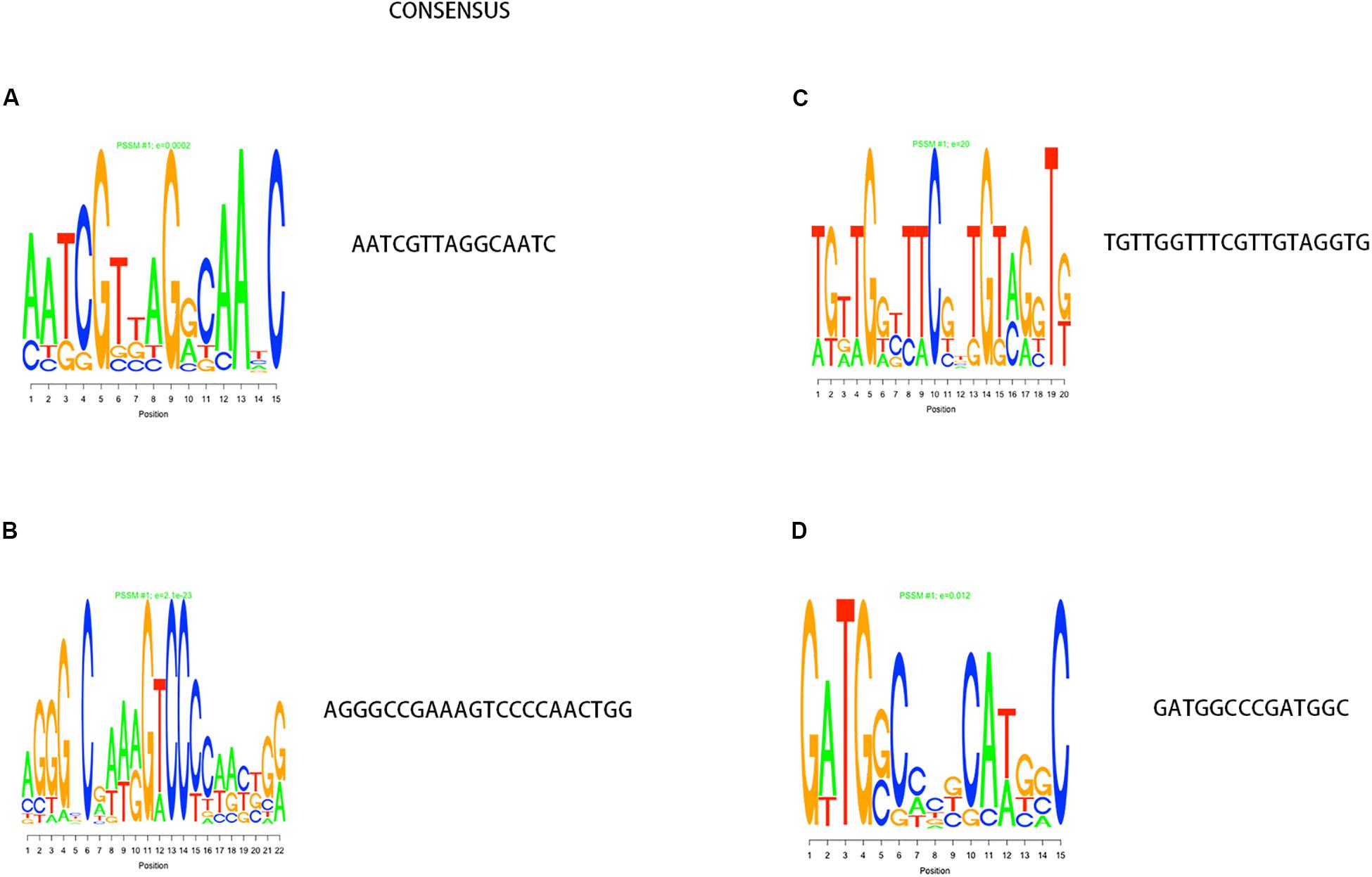
Figure 4. Motif analysis results with the largest number of cell wall genes in a single module cluster. (A–D) The motif base distribution of the four modules with the largest number of cell wall genes. In each BP base distribution in the motif, the size of the base is proportional to the corresponding frequency.
We enriched the function of cell wall synthesis gene and constructed the network between cell wall synthesis gene and gene function. As shown in Figure 5, the cell wall genes screened are mainly related to 18 functions, including fatty acid biosynthesis process, DIM cell wall layer assembly, and plasma membrane.
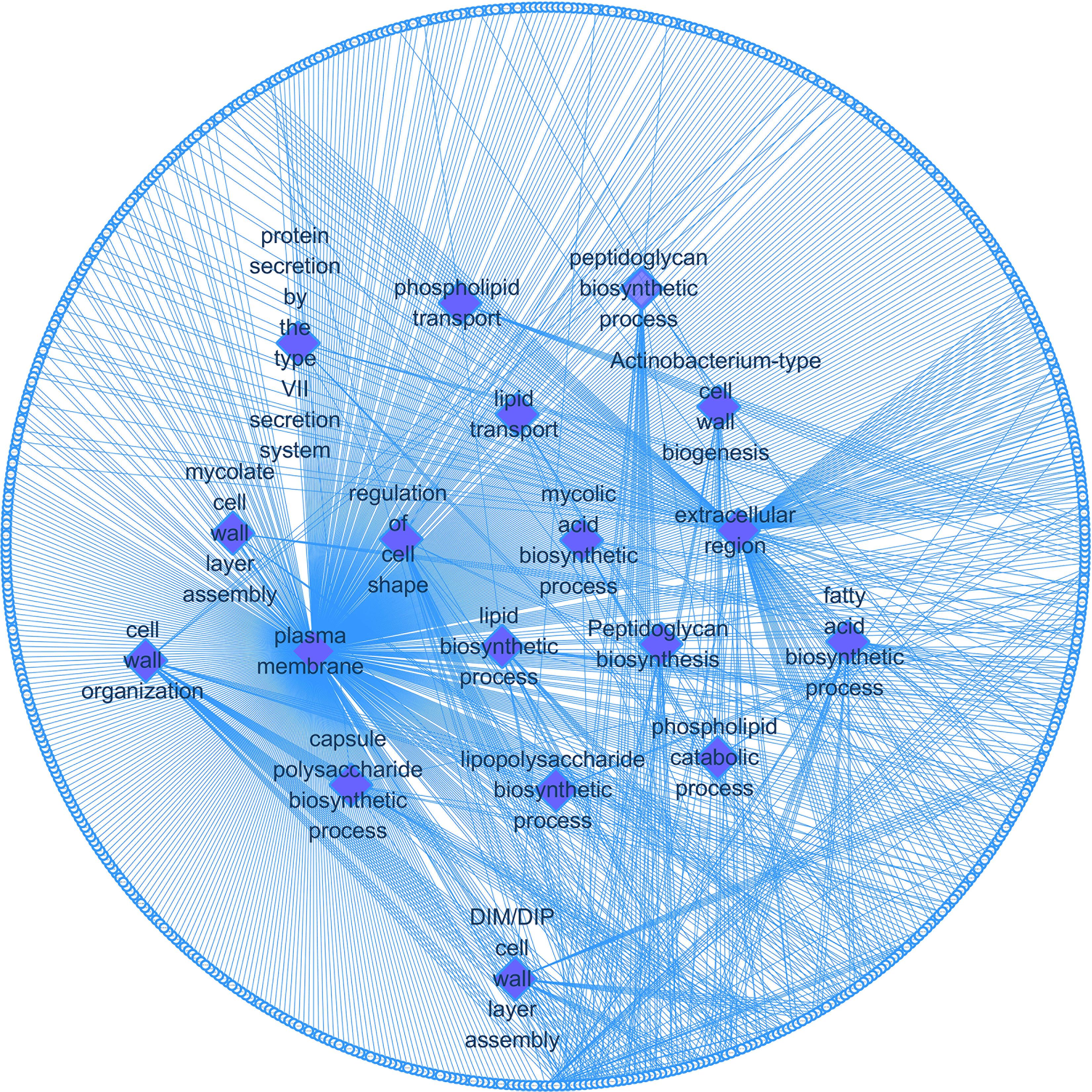
Figure 5. Cell wall synthesis gene and gene function regulatory network. Diamond-shaped nodes and rectangular nodes, respectively, represent gene functions and genes related to cell wall synthesis.
Using the STRING database, we analyzed the interaction relationships between the cell wall synthesis genes of M. tuberculosis and constructed a PPI network of cell wall-related genes after deleting unconnected nodes. As shown in Figure 6A, in order to identify the key genes in the network diagram, we used MCODE to screen out five important subnets and several related genes under the condition of k-score = 2.
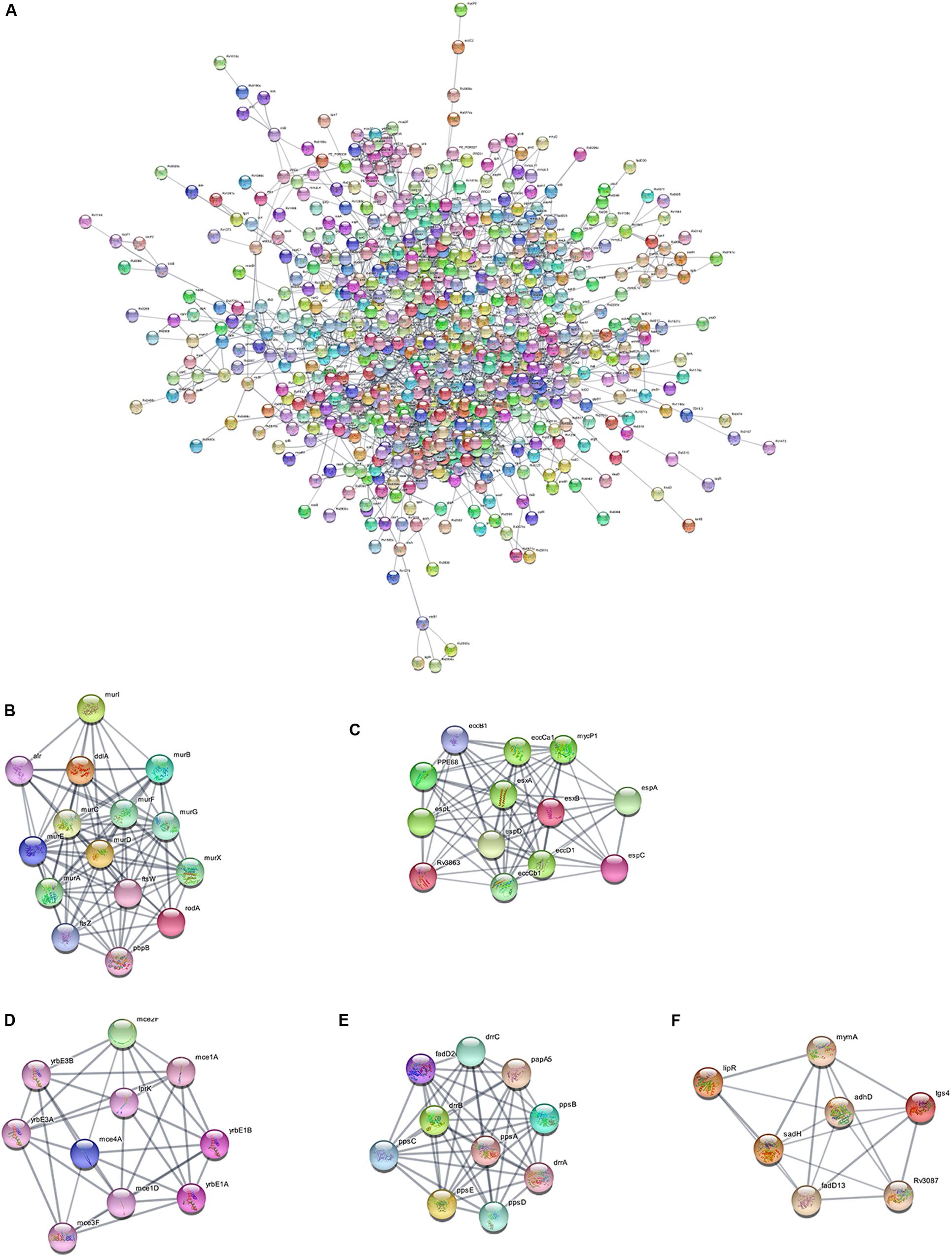
Figure 6. PPI network of cell wall-related genes of M. tuberculosis. (A) Protein interaction networks visualized with Cytoscape. (B) Molecular complex detection (MCODE) with deep excavation of the core subnet. A modular gene involved in peptidoglycan synthesis in Subnet 1. (C) The gene cluster for the ESX-1 secretory system. (D) Gene clusters encoding membrane lipid transporters. (E) Key gene clusters for cell wall resistance. (F) Fatty acids modify gene clusters.
As shown in Figure 6B, Subnet 1 contains 14 key genes in the cell wall peptidoglycan synthesis process. Alr, the ddla coding protein, plays a role in the synthesis of alanine peptidoglycan (Bhat et al., 2017; Meng et al., 2019). Ftsw, ftsz, and pbp3-encoded proteins can form a ternary complex to potentially regulate peptidoglycan biogenesis. Roda glycosyltransferase is also involved in peptidoglycan synthesis (Wu et al., 2016). Figure 6C shows that Subnet 2 contains 13 ESX-1 secretory system-related genes. The ESX-1 secretory system is not only an important determinant of M. tuberculosis virulence, but is also closely related to cell wall synthesis (Wong, 2017). After elimination of the espa gene encoding the ESX-1 substrate, M. tuberculosis bacteria lose the ability to synthesize a complete cell wall structure (Chen et al., 2013). ESX-A is an early secreted antigen target that promotes the synthesis of the ESX-1 substrate and interacts with the cell membrane and cell wall of bacteria. Subnet 3 (Figure 6D) contains membrane lipid transporters. In Subnet 4 (Figure 6E), ddrA-C is not only the key gene in cell wall synthesis, but also the key gene for drug resistance in M. tuberculosis bacteria (Selvam et al., 2013). The other eight genes are related to the synthesis of lipid phthiocerol dimycocerosates (PDIM) in the cell wall. Among them, ppsA-E encodes the PDIM catecholic dipolyoleate (Gopal et al., 2016). All seven genes in Subnet 5 (Figure 6F) are regulated by the mymA operon and play a role in cell wall fatty acid modification (Singh et al., 2005).
Through PPI, we identified some node genes that are crucial in cell wall biosynthesis of important sugars and lipids. In Figure 7, the petal diagram shows the genes contained in the top five annotations in BP, MF, and CC and the genes contained in the first five paths in KEGG BioCyc and Reactome. We selected the top five groups with the lowest P-values in all enrichments. This is to intuitively demonstrate the functional enrichment pathway for the co-regulation of key genes. The key genes for this pathway are shown in Supplementary Table S3.
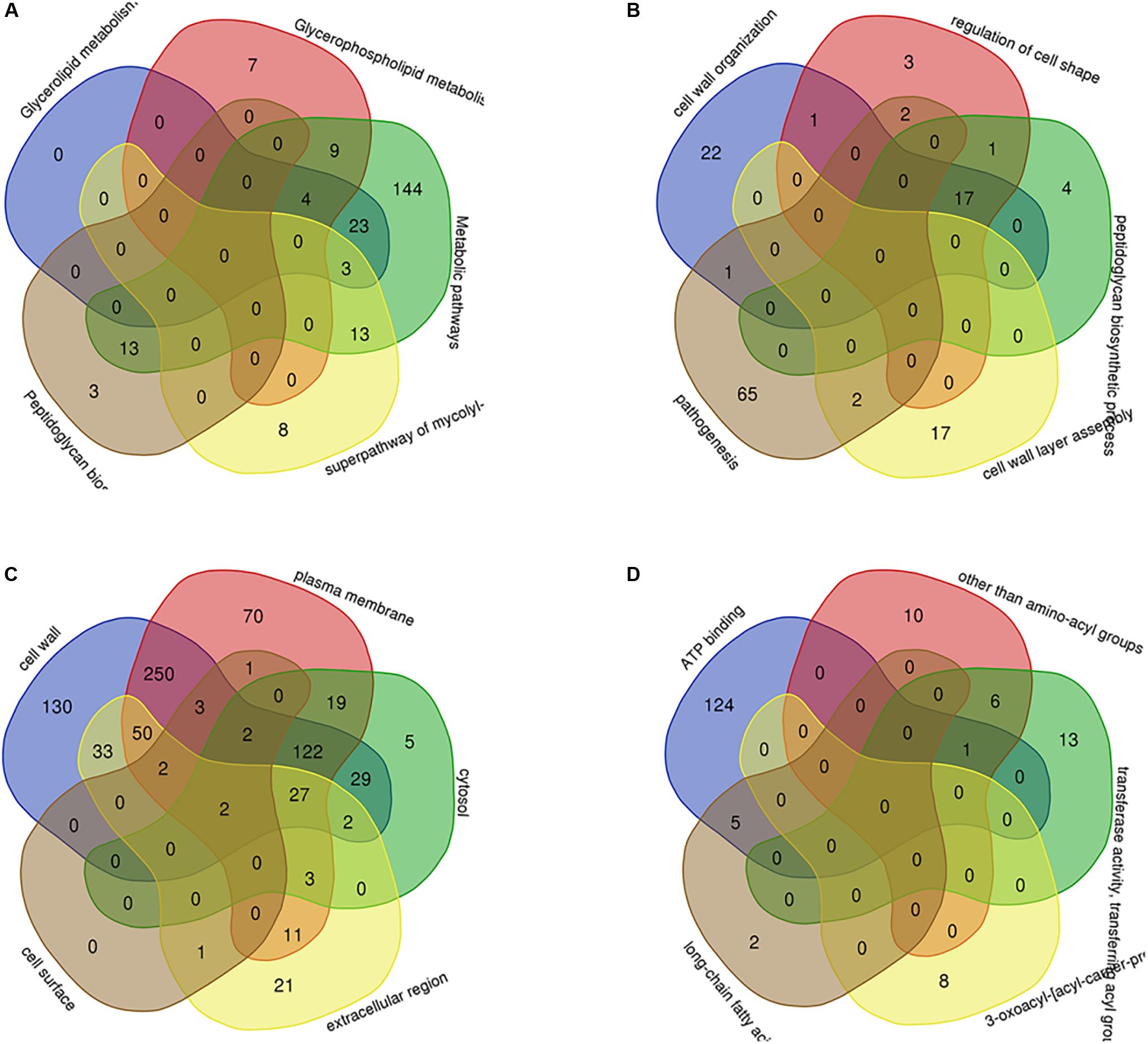
Figure 7. The key genes with multifunctional correlation intermingle through multiple pathways screened for the cell wall-related genes of M. tuberculosis. Intersecting genes in the five pathways with the most significant differences in KEGG (A), BP (B), CC (C), and MF (D) are displayed in a petal diagram.
As an important target for the development of new anti-TB drugs, the M. tuberculosis cell wall has attracted increasing attention. Maan and Kaur (2019) discovered Rv2223c in the cell wall of M. tuberculosis, which is a carboxyl transferase. Bothra et al. knocked out mmpl11 and the resulting mutant strain exhibited a change in the biological activity related to mycolate wax and long-chain triacylglycerol. The knockout strain was also damaged compared to the wild strain in vitro granuloma model, thus demonstrating the important role of mmpl11 in cell wall and biofilm syntheses (Bothra et al., 2018). Quigley et al. (2017) found that the expression of lipid PDIM in the cell wall of M. tuberculosis was negatively regulated by a novel transcription repressor, Rv3167c. Although extensive M. tuberculosis cell wall-related research has been conducted, there is still no comprehensive summary of the key genes involved in the process of cell wall synthesis.
In this study, we first screened the genes related to cell wall anabolism using multiple M. tuberculosis gene annotation databases. Next, we screened the essential genes for cell wall synthesis by GO functional annotation. We then evaluated the distribution of cell wall synthesis genes in the whole genome using the DOOR database established in our laboratory. Using the above methods, we obtained a lot of valuable information. For example, we identified the entire operon containing genes involved in cell wall synthesis, which is necessary for the survival of the bacterium. We employed module analysis and the cMonkey double clustering algorithm to cluster the cell wall synthesis genes. We also identified key genes by screening co-regulatory clustering modules. Through functional analysis of cell wall synthesis genes by GO and KEGG, we screened the key genes for the synthesis of important components of the cell wall, such as mycotic acid and peptidoglycan, and the key hub genes involved in multi-pathway synthesis. Finally, we created a PPI network and identified five important subnets through MCODE analysis. The intrinsic relationship between proteins in the network was used to deeply explore the genes. Molecular complexes containing key genes were extracted based on closely related regions in the PPI. Finally, we obtained the five most valuable subnets. Using Subnet 3 as an example, all genes contained in this subnet are part of the mammalian cell entry (MCE) operon (Gioffre et al., 2005). The MCE operon is present in all genera of mycobacteria and actinomycetes. However, the number of MCE operons in different strains varies, with MCE 4 in M. tuberculosis, MCE 3 in M. smegmatis, and MCE 1, 2, and 4 in M. bovis. It is unknown why the MCE 3 operon is absent from M. bovis (Kumar et al., 2005). The MCE operons help M. tuberculosis ingest cholesterol in the host to keep the bacteria alive. Lack of the MCE operon causes a serious imbalance of lipid content in the M. tuberculosis cell wall. Sally et al. reported free mycolic acid accumulation in the cell wall of the MCE 1 operon mutant strain of M. tuberculosis (Singh et al., 2018). However, the genes contained in Subnet 4, such as ppsa and ppsb, were significantly altered in drug-resistant bacteria (Cantrell et al., 2013). We believe that ppsa changes the expression of PDIM in the cell wall by changing the approach of the multi-subunit non-iterated polyketide synthase system (Vergnolle et al., 2015). This makes the bacterial cell wall thicker and causes bacterial drug efflux. We used bioinformatics and statistical methods to comprehensively scan all the genes synthesized in the M. tuberculosis cell wall and to screen out new targets that can be used as new anti-M. tuberculous cell wall targeting drugs.
All datasets presented in this study are included in the article/Supplementary Material.
GW designed this study. XL and JP wrote the manuscript. XL analyzed the data. QM, WW, JH, and NZ contributed in the data collection. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (#81871699), Foundation of Jilin Province Science and Technology Department (#172408GH010234983), and the epidemiology, early warning, and response techniques of major infectious diseases in the Belt and Road Initiative (#2018ZX10101002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Medjaden Bioscience Limited (Hong Kong, China) for editing and proofreading this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00607/full#supplementary-material
Ahmad, S., El-Shazly, S., Mustafa, A. S., and Al-Attiyah, R. (2005). The six mammalian cell entry proteins (Mce3A-F) encoded by the mce3 operon are expressed during in vitro growth of Mycobacterium tuberculosis. Scand. J. Immunol. 62, 16–24. doi: 10.1111/j.1365-3083.2005.01639.x
Bhat, A. H., Pathak, D., and Rao, A. (2017). The alr-groEL1 operon in Mycobacterium tuberculosis: an interplay of multiple regulatory elements. Sci. Rep. 7:43772. doi: 10.1038/srep43772
Bothra, A., Arumugam, P., Panchal, V., Menon, D., Srivastava, S., Shankaran, D., et al. (2018). Phospholipid homeostasis, membrane tenacity and survival of Mtb in lipid rich conditions is determined by MmpL11 function. Sci. Rep. 8:8317. doi: 10.1038/s41598-018-26710-z
Cantrell, S. A., Leavell, M. D., Marjanovic, O., Iavarone, A. T., Leary, J. A., and Riley, L. W. (2013). Free mycolic acid accumulation in the cell wall of the mce1 operon mutant strain of Mycobacterium tuberculosis. J. Microbiol. 51, 619–626. doi: 10.1007/s12275-013-3092-y
Cao, H., Ma, Q., Chen, X., and Xu, Y. (2019). DOOR: a prokaryotic operon database for genome analyses and functional inference. Brief. Bioinform. 20, 1568–1577. doi: 10.1093/bib/bbx088
Catanho, M., Mascarenhas, D., Degrave, W., and Miranda, A. B. (2006). GenoMycDB: a database for comparative analysis of mycobacterial genes and genomes. Genet. Mol. Res. 5, 115–126.
Chaudhuri, R. R. (2009). MycoDB: an online database for comparative genomics of the mycobacteria and related organisms. Methods Mol. Biol. 465, 419–431. doi: 10.1007/978-1-59745-207-6_27
Chen, J. M., Zhang, M., Rybniker, J., Basterra, L., Dhar, N., Tischler, A. D., et al. (2013). Phenotypic profiling of Mycobacterium tuberculosis EspA point mutants reveals that blockage of ESAT-6 and CFP-10 secretion in vitro does not always correlate with attenuation of virulence. J. Bacteriol. 195, 5421–5430. doi: 10.1128/JB.00967-13
Galagan, J. E., Sisk, P., Stolte, C., Weiner, B., Koehrsen, M., Wymore, F., et al. (2010). TB database 2010: overview and update. Tuberculosis 90, 225–235. doi: 10.1016/j.tube.2010.03.010
Gaur, A., Sharma, V. K., Shree, S., Rai, N., and Ramachandran, R. (2017). Characterization of EccA3, a CbbX family ATPase from the ESX-3 secretion pathway of M. tuberculosis. Biochim. Biophys. Acta Proteins Proteom. 1865, 715–724. doi: 10.1016/j.bbapap.2017.04.001
Gillespie, J. J., Wattam, A. R., Cammer, S. A., Gabbard, J. L., Shukla, M. P., Dalay, O., et al. (2011). PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect. Immun. 79, 4286–4298. doi: 10.1128/IAI.00207-11
Gioffre, A., Infante, E., Aguilar, D., Santangelo, M. P., Klepp, L., Amadio, A., et al. (2005). Mutation in mce operons attenuates Mycobacterium tuberculosis virulence. Microbes Infect. 7, 325–334. doi: 10.1016/j.micinf.2004.11.007
Gopal, P., Yee, M., Sarathy, J., Low, J. L., Sarathy, J. P., Kaya, F., et al. (2016). Pyrazinamide resistance is caused by two distinct mechanisms: prevention of coenzyme a depletion and loss of virulence factor synthesis. ACS Infect. Dis. 2, 616–626. doi: 10.1021/acsinfecdis.6b00070
Hellweger, F. L., van Sebille, E., and Fredrick, N. D. (2014). Biogeographic patterns in ocean microbes emerge in a neutral agent-based model. Science 345, 1346–1349. doi: 10.1126/science.1254421
Heydari, H., Wee, W. Y., Lokanathan, N., Hari, R., Mohamed Yusoff, A., Beh, C. Y., et al. (2013). MabsBase: a Mycobacterium abscessus genome and annotation database. PLoS One 8:e62443. doi: 10.1371/journal.pone.0062443
Howard, N. C., Marin, N. D., Ahmed, M., Rosa, B. A., Martin, J., Bambouskova, M., et al. (2018). Mycobacterium tuberculosis carrying a rifampicin drug resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Nat. Microbiol. 3, 1099–1108. doi: 10.1038/s41564-018-0245-0
Ivarsson, Y., and Jemth, P. (2019). Affinity and specificity of motif-based protein-protein interactions. Curr. Opin. Struct. Biol. 54, 26–33. doi: 10.1016/j.sbi.2018.09.009
Jiang, M. J., Liu, S. J., Su, L., Zhang, X., Li, Y. Y., Tang, T., et al. (2020). Intranasal vaccination with Listeria ivanovii as vector of Mycobacterium tuberculosis antigens promotes specific lung-localized cellular and humoral immune responses. Sci. Rep. 10:302. doi: 10.1038/s41598-019-57245-6
Kong, X., Zhu, B., Stone, V. N., Ge, X., El-Rami, F. E., Donghai, H., et al. (2019). ePath: an online database towards comprehensive essential gene annotation for prokaryotes. Sci. Rep. 9:12949. doi: 10.1038/s41598-019-49098-w
Kumar, A., Chandolia, A., Chaudhry, U., Brahmachari, V., and Bose, M. (2005). Comparison of mammalian cell entry operons of mycobacteria: in silico analysis and expression profiling. FEMS Immunol. Med. Microbiol. 43, 185–195. doi: 10.1016/j.femsim.2004.08.013
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., et al. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240. doi: 10.1016/s0140-6736(00)04403-2
Lew, J. M., Kapopoulou, A., Jones, L. M., and Cole, S. T. (2011). TubercuList–10 years after. Tuberculosis 91, 1–7. doi: 10.1016/j.tube.2010.09.008
Maan, P., and Kaur, J. (2019). Rv2223c, an acid inducible carboxyl-esterase of Mycobacterium tuberculosis enhanced the growth and survival of Mycobacterium smegmatis. Future Microbiol. 14, 1397–1415. doi: 10.2217/fmb-2019-0162
Maitra, A., Munshi, T., Healy, J., Martin, L. T., Vollmer, W., Keep, N. H., et al. (2019). Cell wall peptidoglycan in Mycobacterium tuberculosis: an Achilles’ heel for the TB-causing pathogen. FEMS Microbiol. Rev. 43, 548–575. doi: 10.1093/femsre/fuz016
Meng, J., Gao, P., Wang, X., Guan, Y., Liu, Y., and Xiao, C. (2019). Digging deeper to save the old anti-tuberculosis target: D-Alanine-D-Alanine Ligase With a Novel Inhibitor, IMB-0283. Front. Microbiol. 10:3017. doi: 10.3389/fmicb.2019.03017
Merker, M., Kohl, T. A., Barilar, I., Andres, S., Fowler, P. W., Chryssanthou, E., et al. (2020). Phylogenetically informative mutations in genes implicated in antibiotic resistance in Mycobacterium tuberculosis complex. Genome Med. 12:27. doi: 10.1186/s13073-020-00726-5
Pasricha, R., Chandolia, A., Ponnan, P., Saini, N. K., Sharma, S., Chopra, M., et al. (2011). Single nucleotide polymorphism in the genes of mce1 and mce4 operons of Mycobacterium tuberculosis: analysis of clinical isolates and standard reference strains. BMC Microbiol. 11:41. doi: 10.1186/1471-2180-11-41
Postma, M., and Goedhart, J. (2019). PlotsOfData-A web app for visualizing data together with their summaries. PLoS Biol. 17:e3000202. doi: 10.1371/journal.pbio.3000202
Quigley, J., Hughitt, V. K., Velikovsky, C. A., Mariuzza, R. A., El-Sayed, N. M., and Briken, V. (2017). The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. mBio 8:e00148-17. doi: 10.1128/mBio.00148-17
Saelens, W., Cannoodt, R., and Saeys, Y. (2018). A comprehensive evaluation of module detection methods for gene expression data. Nat. Commun. 9:1090. doi: 10.1038/s41467-018-03424-4
Safi, H., Lingaraju, S., Amin, A., Kim, S., Jones, M., Holmes, M., et al. (2013). Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-beta-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 45, 1190–1197. doi: 10.1038/ng.2743
Selvam, K., Duncan, J. R., Tanaka, M., and Battista, J. R. (2013). DdrA, DdrD, and PprA: components of UV and mitomycin C resistance in Deinococcus radiodurans R1. PLoS One 8:e69007. doi: 10.1371/journal.pone.0069007
Sher, J. W., Lim, H. C., and Bernhardt, T. G. (2020). Global phenotypic profiling identifies a conserved actinobacterial cofactor for a bifunctional PBP-type cell wall synthase. eLife 9:54761. doi: 10.7554/eLife.54761
Singh, A., Gupta, R., Vishwakarma, R. A., Narayanan, P. R., Paramasivan, C. N., Ramanathan, V. D., et al. (2005). Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J. Bacteriol. 187, 4173–4186. doi: 10.1128/JB.187.12.4173-4186.2005
Singh, P., Sinha, R., Tyagi, G., Sharma, N. K., Saini, N. K., Chandolia, A., et al. (2018). PDIM and SL1 accumulation in Mycobacterium tuberculosis is associated with mce4A expression. Gene 642, 178–187. doi: 10.1016/j.gene.2017.09.062
Turner, C. T., Gupta, R. K., Tsaliki, E., Roe, J. K., Mondal, P., Nyawo, G. R., et al. (2020). Blood transcriptional biomarkers for active pulmonary tuberculosis in a high-burden setting: a prospective, observational, diagnostic accuracy study. Lancet Respir. Med. 8, 407–419. doi: 10.1016/S2213-2600(19)30469-2
Vergnolle, O., Chavadi, S. S., Edupuganti, U. R., Mohandas, P., Chan, C., Zeng, J., et al. (2015). Biosynthesis of cell envelope-associated phenolic glycolipids in Mycobacterium marinum. J. Bacteriol. 197, 1040–1050. doi: 10.1128/JB.02546-14
Vishnoi, A., Srivastava, A., Roy, R., and Bhattacharya, A. (2008). MGDD: Mycobacterium tuberculosis genome divergence database. BMC Genom. 9:373. doi: 10.1186/1471-2164-9-373
Waltman, P., Kacmarczyk, T., Bate, A. R., Kearns, D. B., Reiss, D. J., Eichenberger, P., et al. (2010). Multi-species integrative biclustering. Genome Biol. 11:R96. doi: 10.1186/gb-2010-11-9-r96
Wong, K. W. (2017). The Role of ESX-1 in Mycobacterium tuberculosis pathogenesis. Microbiol. Spectr. 5, 627–634. doi: 10.1128/microbiolspec.TBTB2-0001-2015
Wu, M. L., Gengenbacher, M., Chung, J. C., Chen, S. L., Mollenkopf, H. J., Kaufmann, S. H., et al. (2016). Developmental transcriptome of resting cell formation in Mycobacterium smegmatis. BMC Genomics 17:837. doi: 10.1186/s12864-016-3190-4
Keywords: Mycobacterium tuberculosis, cell wall, module, regulatory networks, enrichment analysis
Citation: Luo X, Pan J, Meng Q, Huang J, Wang W, Zhang N and Wang G (2020) High-Throughput Screen for Cell Wall Synthesis Network Module in Mycobacterium tuberculosis Based on Integrated Bioinformatics Strategy. Front. Bioeng. Biotechnol. 8:607. doi: 10.3389/fbioe.2020.00607
Received: 06 April 2020; Accepted: 18 May 2020;
Published: 30 June 2020.
Edited by:
Yungang Xu, University of Texas Health Science Center at Houston, United StatesCopyright © 2020 Luo, Pan, Meng, Huang, Wang, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoqing Wang, cWluZ0BqbHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.