 Mingyue Li
Mingyue Li Jiuzhou Chen
Jiuzhou Chen Yu Wang
Yu Wang Jiao Liu2
Jiao Liu2 Jibin Sun
Jibin Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Bioeng. Biotechnol. , 24 April 2020
Sec. Industrial Biotechnology
Volume 8 - 2020 | https://doi.org/10.3389/fbioe.2020.00357
Corynebacterium glutamicum is an important workhorse for industrial production of diversiform bioproducts. Multiplex control of metabolic pathway genes is crucial for maximizing biosynthesis of desired products. However, few tools for simultaneously regulating multiple genes in C. glutamicum have been reported. Here, a CRISPR-dCpf1-based multiplex gene repression system was developed for C. glutamicum. This system successfully repressed two fluorescent reporter genes simultaneously by expressing a dCpf1 (E1006A, D917A) and a designed single crRNA array. To demonstrate applications of this CRISPR-dCpf1 system in metabolic engineering, we applied this system to repress four genes involved in lysine biosynthesis (gltA, pck, pgi, and hom) with a single array, which increased the lysine titer and yield for over 4.0-fold. Quantitative PCR demonstrated that transcription of all the four endogenous target genes were repressed by over 90%. Thus, the CRISPR-dCpf1 system is a simple and effective technique for multiplex gene repression in C. glutamicum and holds promise for metabolic engineering of C. glutamicum to produce valuable chemicals and fuels.
Corynebacterium glutamicum, a non-pathogenic Gram-positive bacterium, is an important platform strain and has been widely used for industrial production of various amino acids, biochemicals, and biofuels (Becker and Wittmann, 2012; Yokota and Ikeda, 2017). To maximize biosynthesis of desired product, expression of multiple metabolic pathway genes needs to be balanced (Lee et al., 2019). However, identification of the optimum expression level for each target gene is time-consuming. Considering the underlying intricacies and interrelationships of metabolic pathways, optimizing the expression of target genes one by one usually cannot lead to desired outcomes (Schafer et al., 1994; Ikeda and Nakagawa, 2003; Okibe et al., 2011). Therefore, development of efficient multiplex gene regulation techniques is in urgent demand.
Recently, synthetic small regulatory RNA (sRNA) system and class 2 type II-A clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas) system (Cas9) have been repurposed as RNA interference (RNAi) and CRISPR interference (CRISPRi) tools, respectively, for gene repression in C. glutamicum (Cleto et al., 2016; Park et al., 2019; Sun et al., 2019). However, for both sRNA-mediated RNAi and deactivated Cas9 (dCas9)-mediated CRISPRi, a sRNA or CRISPR RNA (crRNA) will be required for each target gene repression because these systems are incapable of processing RNA arrays into individual functional RNAs. By recruiting native RNase III and expressing a dCas9:crRNA:transactivating crRNA (tracrRNA) complex, a crRNA array processing system was developed for dCas9-mediated multiplex transcriptional repression in Escherichia coli (Cress et al., 2015). However, such system has not been established in C. glutamicum due to limited knowledge of functional RNase. As a result, multiple RNA expression cassettes with high sequence and architecture similarity need to be constructed for multiplex gene regulation in C. glutamicum, which is difficult to assemble in vitro and also unstable in vivo (Reis et al., 2019).
Unlike Cas9, class 2 type V-A CRISPR effector Cpf1 (also known as Cas12a) possesses both DNase and RNase activities, which can process crRNA arrays into mature crRNAs and offer an alternative tool for genetic modifications (Luo et al., 2016). Furthermore, the direct repeat sequence of Cpf1 crRNA (∼20 nt) is much shorter than the handle sequence of Cas9 crRNA (∼60 nt) (Fonfara et al., 2016; Liu et al., 2017), which makes synthesis and assembly of Cpf1 crRNA array cheaper and easier. These properties provide distinct advantages to perform multiplex gene editing and perturbation (Zetsche et al., 2015). Over the last 2 years, CRISPR-dCpf1 (DNase-deactivated Cpf1) systems have been employed for gene repression in several microorganisms, such as E. coli, Streptomyces, and Yarrowia lipolytica (Kim et al., 2017; Zhang et al., 2017; Li et al., 2018; Zhang et al., 2018). Until very recently, Liu and colleagues reported the first attempt to use catalytically active Cpf1 with truncated crRNAs for gene repression in C. glutamicum (Liu et al., 2019). However, the low repression efficiency (up to 60%) and high risk of introducing double-stranded DNA breaks (DSBs) would limit its application. CRISPR-dCpf1 system that is capable of processing crRNA arrays but will not generate DSBs has not been systematically explored in C. glutamicum so far.
In this study, CRISPR-dCpf1 from Francisella novicida was employed for gene repression and metabolic engineering of C. glutamicum. By optimizing dCpf1 expression and testing different dCpf1 variants, an efficient CRISPRi system was successfully established in C. glutamicum, which simultaneously repressed expression of multiple target genes with high efficiencies over 90%. A Golden Gate assembly-based method was also developed for simple and rapid assembly of crRNA array. To demonstrate an application of this CRISPR-dCpf1 system in metabolic engineering of C. glutamicum, combinational repression of four potential target genes was conducted to maximize lysine production. The CRISPR-dCpf1-mediated multiplex gene repression technique developed here will enable the rapid development of high-performance C. glutamicum strains.
Bacterial strains used in this study are listed in Supplementary Table S1. E. coli DH5α and DB.3.1 were used for the plasmid construction and cultivated in Luria-Bertani (LB) medium (5 g/L yeast extract, 10 g/L tryptone, 10 g/L NaCl) at 37°C. Kanamycin (50 μg/mL) or chloramphenicol (20 μg/mL) was added to LB medium as required. C. glutamicum strains were cultivated at 30°C in LB medium supplement with 5 g/L glucose (LBG medium). Kanamycin (25 μg/mL), chloramphenicol (5 μg/mL), or isopropyl-β-D-thiogalactopyranoside (IPTG) (1 mM) was added when necessary.
All the plasmids and primers used in this study are listed in Supplementary Tables S1, S2, respectively. E. coli–C. glutamicum shuttle expression vector pXMJ19 was used to express dCpf1. The E1006A mutation of Cpf1 from F. novicida was first introduced by PCR using primer pair pY003-E1006A-F/R. Then the dCpf1 gene was amplified using the primer pair pXM-01-F/R and cloned into the HindIII and BamHI site of pXMJ19, generating plasmid pXM-01. To optimize the expression of dCpf1, the start codon (ATG) of dCpf1 was first replaced by GTG with primer pair pXM-02-GTG-F/R, resulting in plasmid pXM-02. Then the original ribosome binding site (RBS) of dCpf1 in pXM-01 and pXM-02 (RBS1) was replaced with RBS2 and RBS3 (Supplementary Table S3) chosen from previously constructed libraries (Zhang et al., 2015) by PCR, resulting in plasmids pXM-03, pXM-04, pXM-05, and pXM-06, respectively. To replace the E1006A mutation in dCpf1 with D917A mutation, D917A mutation was first introduced into pXM-04 by PCR using primer pair pXM-07-F/R, generating plasmid pXM-07. Then the E1006A mutation of dCpf1 in pXM-07 was reversed by PCR using primer pair pXM-08-F/R, generating plasmid pXM-08.
E. coli–C. glutamicum shuttle expression vector pEC-XK99E was used to express crRNA. To construct a basic plasmid, the initial Ptrc promoter was replaced with a constitutive promoter (P11F) by PCR using primer pair pEC-01-11F-F/R. Then the backbone of pEC-01 was amplified with primer pair and pEcrRNA-F/R. A ccdB cassette was amplified from pgRNA-ccdB (Wang et al., 2018b) with primer pair pEC-02-ccdB-F/R. These two PCR fragments were purified and ligated to generate plasmid pEC-02. Protospacers possessing the requisite 5′ PAM sequence (BTTV) were identified near the start codon of the coding region, and 23 nucleotides downstream of the PAM were selected as the spacer. Two oligonucleotides were annealed and assembled into BbsI-digested pEC-02 backbone using a Golden Gate assembly method. All single-stranded DNA (ssDNA) oligonucleotides utilized for construction of crRNAs were listed in Supplementary Table S4.
To integrate a gfp expression cassette downstream lysA gene of the ATCC 13032::rfp chromosome and construct a dual-fluorescence reporter strain, plasmid pK18mobsacB-gfp was first constructed. To this end, gfp gene was amplified from plasmid pXM-gfp (Sun et al., 2019) using primer pair gfp-F/R. Then, the plasmid backbone of pK18mobsacB (Schafer et al., 1994), upstream and downstream recombination arms (about 1,000 bp) targeting the lysA locus were amplified with primer pairs pK18mobsacB-F/R, gfp-up-F/R and gfp-down-F/R, respectively. Finally, these PCR fragments were ligated using the CloneExpress MultiS One Step Cloning Kit (Vazyme Biotech, Nanjing, China). pK18mobsacB-gfp was transformed into strain ATCC 13032::rfp and markerless gfp expression cassette insertion was performed as described previously (Xu et al., 2014).
The overnight cultures of C. glutamicum were transferred to fresh LBG medium supplemented with 1 mM IPTG to induce dCpf1 expression. After cultivated at 30°C and with shaking at 220 rpm for 24 h, cells were harvested by centrifugation at 5,000 × g for 10 min, washed once, and re-suspended in phosphate buffer (pH 7.4). Red fluorescent protein (RFP) and green fluorescent protein (GFP) fluorescence intensities were determined using a microplate reader (SpectraMax M5, Molecular Devices, RFP: λ excitation = 560 nm, λ emission = 607 nm; GFP: λ excitation = 488 nm, λ emission = 520 nm). The fluorescence intensities were normalized with OD600.
For RNA extraction, cells were collected after 24 h cultivation. Total RNA extraction, reverse transcription and quantitative PCR (qPCR) were performed according to protocols described previously (Wang et al., 2018a). Briefly, RNA was extracted from the cell pellet using an RNAprep Pure Cell/Bacteria kit (Tiangen Biotech, Beijing, China). Then, cDNA was synthesized using random primers and a FastQuant RT kit (Tiangen Biotech, Beijing, China). The resultant cDNA was used as a template for qPCR analysis. The total RNA sample was used as a template for amplifying the target gene by PCR and no product could be detected by electrophoresis, suggesting that genomic DNA contamination during RNA extraction was minimal. Specific primers for qPCR were designed with Beacon Designer software v7.7 (PREMIER Biosoft International, United States). qPCR was performed by using a SuperReal Premix SYBR green kit (Tiangen Biotech, Beijing, China) and the Applied Biosystems 7,500 real-time PCR system (Thermo Fisher Scientific, United States) according to the manufacturer’s instructions. The gene encoding 16s rRNA was used as a reference for signal normalization. Data analysis was performed according to procedures described previously (Schmittgen and Livak, 2008).
C. glutamicum SCgL30 with a feedback deregulated aspartokinase (T311I) was used for lysine production. Strain SCgL30 and its derivatives were cultivated in LBG medium at 30°C and with shaking at 220 rpm. The overnight cultures were transferred into 24-well plates with 600 μL fermentation medium (80 g/L glucose, 8 g/L yeast extract, 9 g/L urea, 1.5 g/L K2HPO4⋅3H2O, 0.01 g/L MnSO4, 0.6 g/L MgSO4⋅7H2O, 0.01 g/L FeSO4⋅7H2O, 42 g/L MOPS) to an optical density at 600 nm (OD600) of 0.5. Then, cells were cultivated for 24 h at 30°C and with shaking at 800 rpm in INFORS Microtron (INFORS HT Multitron Pro, Switzerland). Samples were taken periodically and glucose and lysine concentrations were quantified using an SBA-40D biosensor analyzer (Institute of Biology of Shandong Province Academy of Sciences, Shandong, China). Cell biomass was determined as OD600 with a UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan) after proper dilution with distilled water.
To construct a DNase-deactivated Cpf1 variant, E1006A mutation was introduced to Cpf1 of F. novicida as described previously (Zetsche et al., 2015). The dCpf1 gene was cloned to pXMJ19 plasmid under control of an IPTG-inducible promoter (Ptrc), producing pXM-01. However, transformation of pXM-01 into C. glutamicum failed. Previous studies reported that excess expression of dCas9 in E. coli inhibited cell growth and down-regulation of its expression level relieved this adverse effect (Cho et al., 2018; Cui et al., 2018). To decrease the leaky expression of dCpf1 (E1006A) and avoid potential toxicity to host cells, two RBSs with lower translation initiation efficiency were selected to replace the original strong RBS (Zhang et al., 2015). In addition, the start codon ATG of dCpf1 was also replaced by GTG. All of the five new modified plasmids were successfully transformed into C. glutamicum. To construct an expression plasmid for crRNA array, a cassette consisting of a constitutive promoter (P11F), two direct repeats (DRs), a ccdB flanked by two BbsI sites, and a terminator was first assembled and cloned to pEC-XK99E, resulting the basic plasmid pEC-02. Bacterial toxin gene ccdB was used as a counter-selectable marker for negative selection in plasmid construction (Wang et al., 2014). By using Golden Gate assembly and annealed ssDNA oligodeoxynucleotides, multiple spacers and DRs could be efficiently assembled in a single reaction (Figure 1). To rapidly determine repression efficiency of aforementioned dCpf1 (E1006A) expression systems, RFP was used as a reporter. A spacer sequence targeting the template strand of rfp with a 5′-BTTV-3′ PAM sequence preferred by F. novicida Cpf1 (Leenay et al., 2016) was selected and inserted into plasmid pEC-02 (Figure 1). The potential off-target sites of rfp-targeting crRNA were examined using Cas-OFFinder (Bae et al., 2014). The resulting plasmid pEC-03 and different dCpf1 (E1006A) plasmids were co-transformed into RFP expressing C. glutamicum strain ATCC 13032::rfp (Figure 2A). The RFP fluorescence intensities were reduced in all the strains expressing both rfp-targeting crRNA and dCpf1 (E1006A). The highest repression efficiency (63%) was achieved when RBS2 (AAAGGTGGTTCAT) and start codon of GTG were used for dCpf1 (E1006A) expression. No significant changes in cell growth were observed when dCpf1 was expressed with different start codons or RBSs (Figure 2B). Furthermore, individual expression of rfp-targeting crRNA or dCpf1 (E1006A) did not reduce RFP fluorescence intensity (Supplementary Figure S1), suggesting the observed gene repression was contributed by the CRISPR-dCpf1 system.
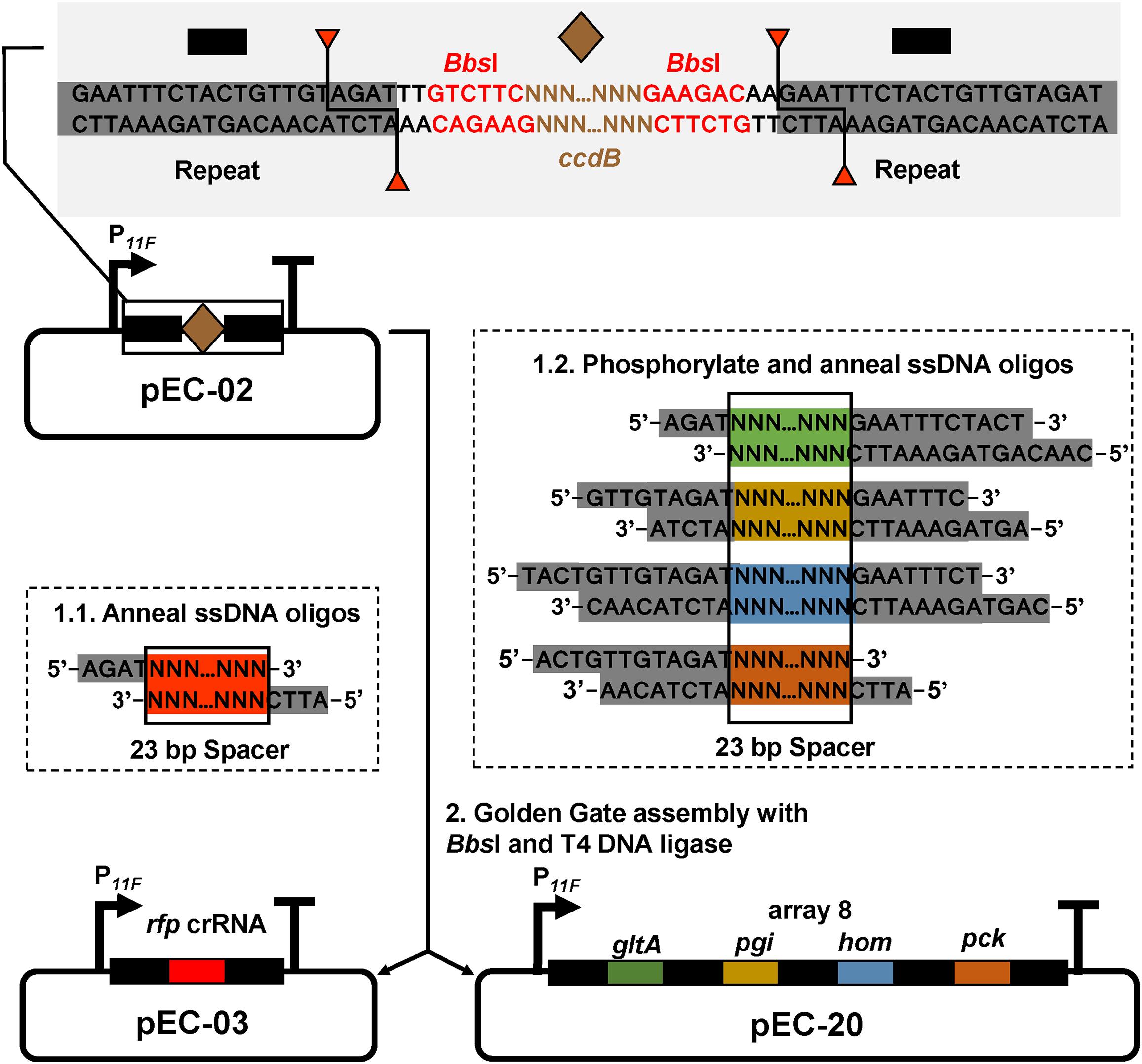
Figure 1. crRNA assembly strategy. pEC-02 harbors a constitutive promoter (P11F), two DRs (black rectangle), and a ccdB cassette (brown diamond) flanked by two BbsI sites (red font). Each 43 bp spacer-DR brick is assembled by 5′ phosphorylation and annealing of two offset complementary ssDNA oligonucleotides. For crRNA with a single targeting spacer, the ends of ssDNA oligonucleotides were designed as sequences complementary to the sticky ends of BbsI in plasmid pEC-02. For crRNA arrays, the ends of ssDNA oligonucleotides were designed as sequences complementary to the sticky ends of BbsI in plasmid pEC-02 or different sites inside DRs.
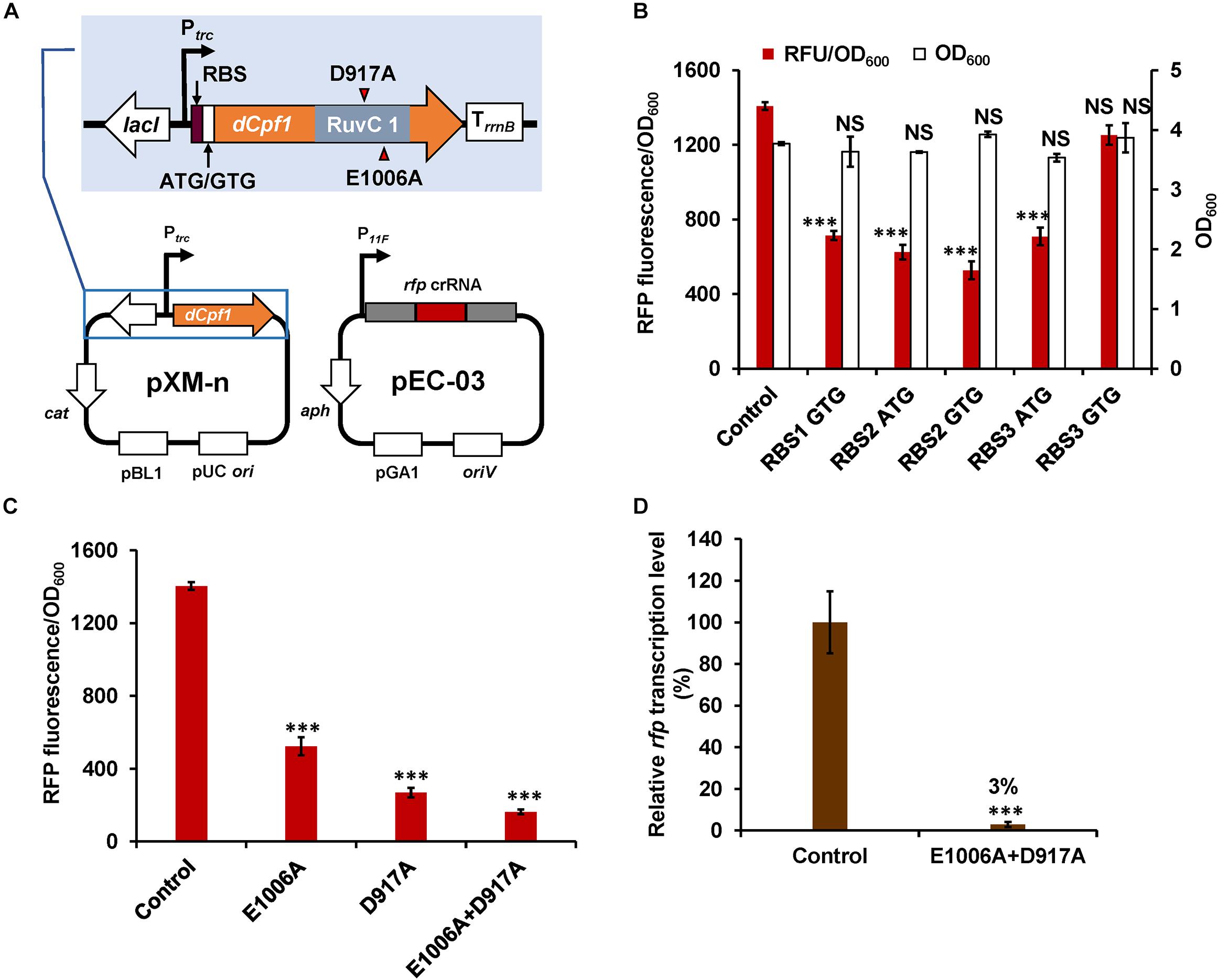
Figure 2. Development of CRISPR-dCpf1-mediated gene repression system in C. glutamicum. (A) Schematic diagram of the CRISPR-dCpf1 tool plasmids for rfp repression. (B) Effects of different RBSs and start codons of dCpf1 gene on rfp repression efficiency and cell growth. (C) Effects of different dCpf1 variants on rfp repression efficiency. (D) Relative mRNA levels of rfp in C. glutamicum ATCC 13032::rfp strains with or without the CRISPR-dCpf1 system. The strain expressing rfp-targeting crRNA but no dCpf1 was used as a control. IPTG (1 mM) was added for inducing dCpf1 expression. Error bars indicate standard deviations from three parallel experiments. All t-tests compare the fluorescence, OD600 or transcription level obtained by strains expressing rfp-targeting crRNA and dCpf1 against control strain expressing rfp-targeting crRNA but no dCpf1 (∗∗∗P < 0.001; NS, non-significant).
Either D917A or E1006A mutation in the RuvC domain of Cpf1 could completely deactivate its DNA cleavage activity (Zetsche et al., 2015). However, different mutations may affect DNA binding abilities of dCpf1 and result in different repression activities. A recent study reported that dCpf1 (D917A) showed higher repression activity than dCpf1 (E1006A) in E. coli (Miao et al., 2019). Therefore, another two dCpf1 variants, dCpf1 (D917A) and dCpf1 (E1006A, D917A), were constructed and evaluated for their performances in gene repression. Employment of dCpf1 (D917A) and dCpf1 (E1006A, D917A) led to 81 and 89% repression of RFP expression, respectively (Figure 2C), which is significantly higher than that obtained by dCpf1 (E1006A) (63%). qPCR further confirmed that dCpf1-based CRISPRi system significantly decreased the mRNA level of rfp by 97% relative to the control strain (Figure 2D). It was noticed that the repression efficiency at protein level (89%) was lower than that at mRNA level (97%). We speculate that the translation process might contribute to this difference. Similar phenomena have been observed in previous studies reporting CRISPRi (Park et al., 2018; Zhang et al., 2018). For example, Park and colleagues applied CRISPR-dCas9 to knock down gltA in C. glutamicum DM1919. The efficiency of mRNA knockdown reached 96% while the enzyme acidity was reduced by 70–80% (Park et al., 2018). Taken together, an efficient gene repression technique based on CRISPR-dCpf1 was successfully developed for C. glutamicum by optimizing dCpf1 expression and screening the most suitable dCpf1 variants. dCpf1 (E1006A, D917A) was used in subsequent experiments due to its higher repression activity.
Next, we tested the application of CRISPR-dCpf1 system in multiple gene repression in C. glutamicum. A dual-fluorescence reporter system was constructed with RFP and GFP, resulting in strain ATCC 13032::rfp::gfp (Supplementary Figure S2A). RFP and GFP fluorescence can be determined in the recombinant strain without interfering with each other, which can be used for double gene repression test (Supplementary Figure S2B). A crRNA array harboring rfp- and gfp-targeting spacers (array 1) and two crRNAs with individual rfp- or gfp-targeting spacers were assembled (Figure 3A). The crRNA expression plasmid was introduced into strain 13032::rfp::gfp with plasmid pXM-07 and RFP and GFP fluorescence intensities were detected. Expression of crRNA array 1 decreased fluorescence of RFP and GFP by 86 and 83%, respectively (Figure 3B). Transcription of rfp and gfp was also decreased by 98 and 92%, respectively (Figure 3C). The efficiencies of simultaneous repression of rfp and gfp obtained with crRNA array 1 were similar to those obtained with crRNAs harboring individual rfp- or gfp-targeting spacers (∼90% at protein level and ∼96% at mRNA level) (Figures 3B,C). Furthermore, the expression of rfp (or gfp) was not significantly affected by the gfp-targeting (or rfp-targeting) crRNA (Figure 3B). The results demonstrate the high efficiency and specificity of CRISPR-dCpf1-mediated multiplex gene repression.
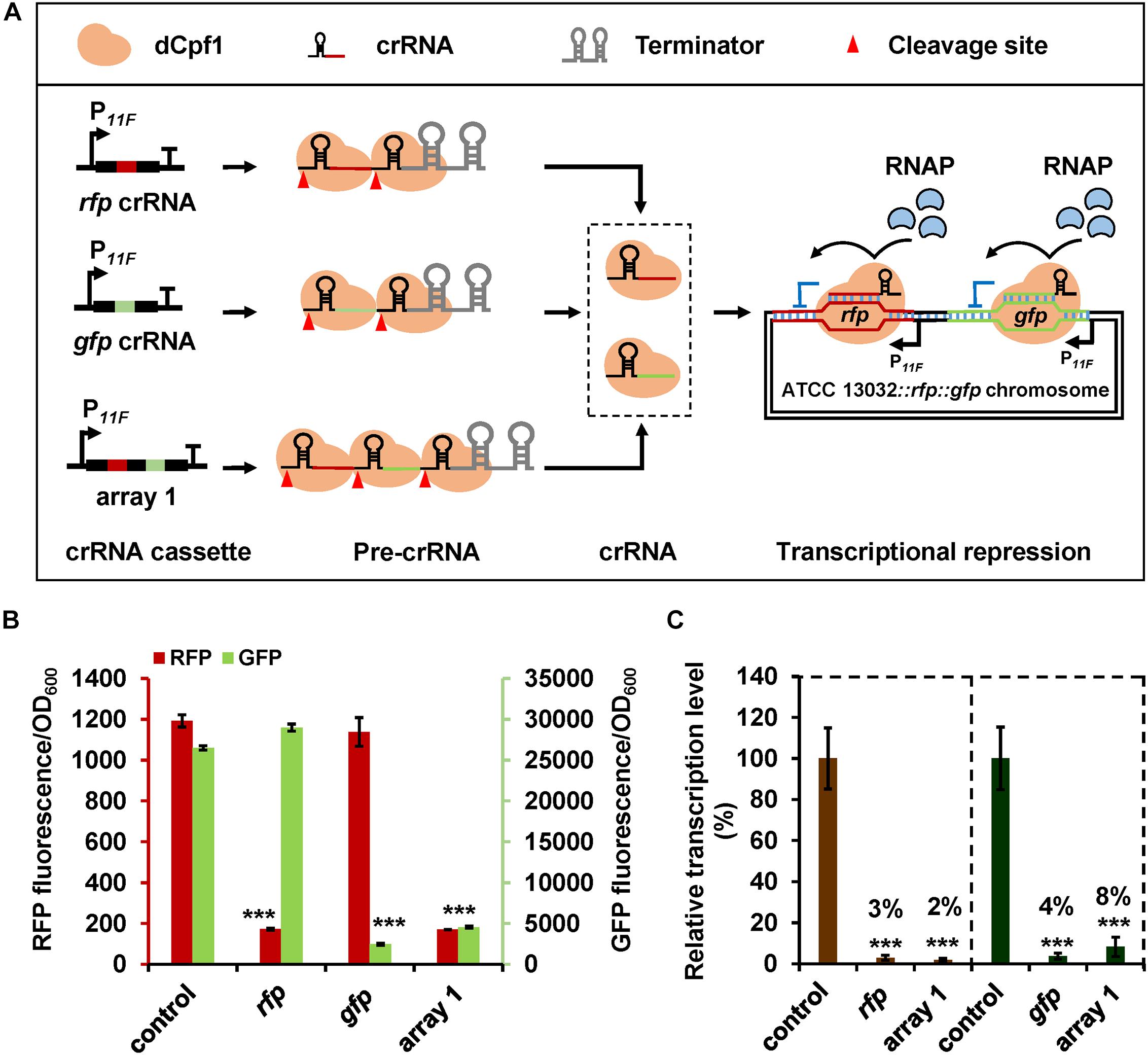
Figure 3. CRISPR-dCpf1-mediated multiplex gene repression in C. glutamicum. (A) Schematic diagram of application of CRISPR-dCpf1 for multiplex gene repression. Chromosomal expressed rfp and gfp were employed as reporter genes. (B) Fluorescence intensities of RFP and GFP controlled by the CRISPR-dCpf1 system with rfp-targeting crRNA, gfp-targeting crRNA, and crRNA array 1 harboring both rfp- and gfp-targeting spacers. The strain expressing dCpf1 but no crRNA was used as the control. (C) Relative transcription levels of rfp and gfp in C. glutamicum ATCC 13032::rfp::gfp strains expressing dCpf1 and different crRNAs. The strain expressing dCpf1 but no crRNA was used as the control. Error bars indicate standard deviations from three parallel experiments. All t-tests compare the fluorescence or transcription level obtained by strains expressing dCpf1 and crRNA against control strain expressing dCpf1 but no crRNA (***P < 0.001).
To explore the potential of CRISPR-dCpf1 system for pathway engineering via endogenous gene regulation, four genes (gltA, pck, pgi, and hom) were selected as targets for enhancing lysine production in C. glutamicum (Figure 4A). Repression of gltA (encoding citrate synthase) and pck (encoding phosphoenolpyruvate carboxykinase) is expected to increase availability of oxaloacetate, which is the precursor for lysine biosynthesis (van Ooyen et al., 2012; Eggeling and Bott, 2015; Zhou and Zeng, 2015; Park et al., 2018). The disruption of pgi (encoding glucose-6-phosphate isomerase) would benefit lysine production by improving NADPH supply via enhancing pentose-phosphate pathway flux (Marx et al., 2003). Weakening threonine biosynthesis pathway branch by repressing homoserine dehydrogenase (encoded by hom) activity would also result in lysine accumulation due to an enhanced flux to lysine synthesis (Eggeling and Bott, 2015).
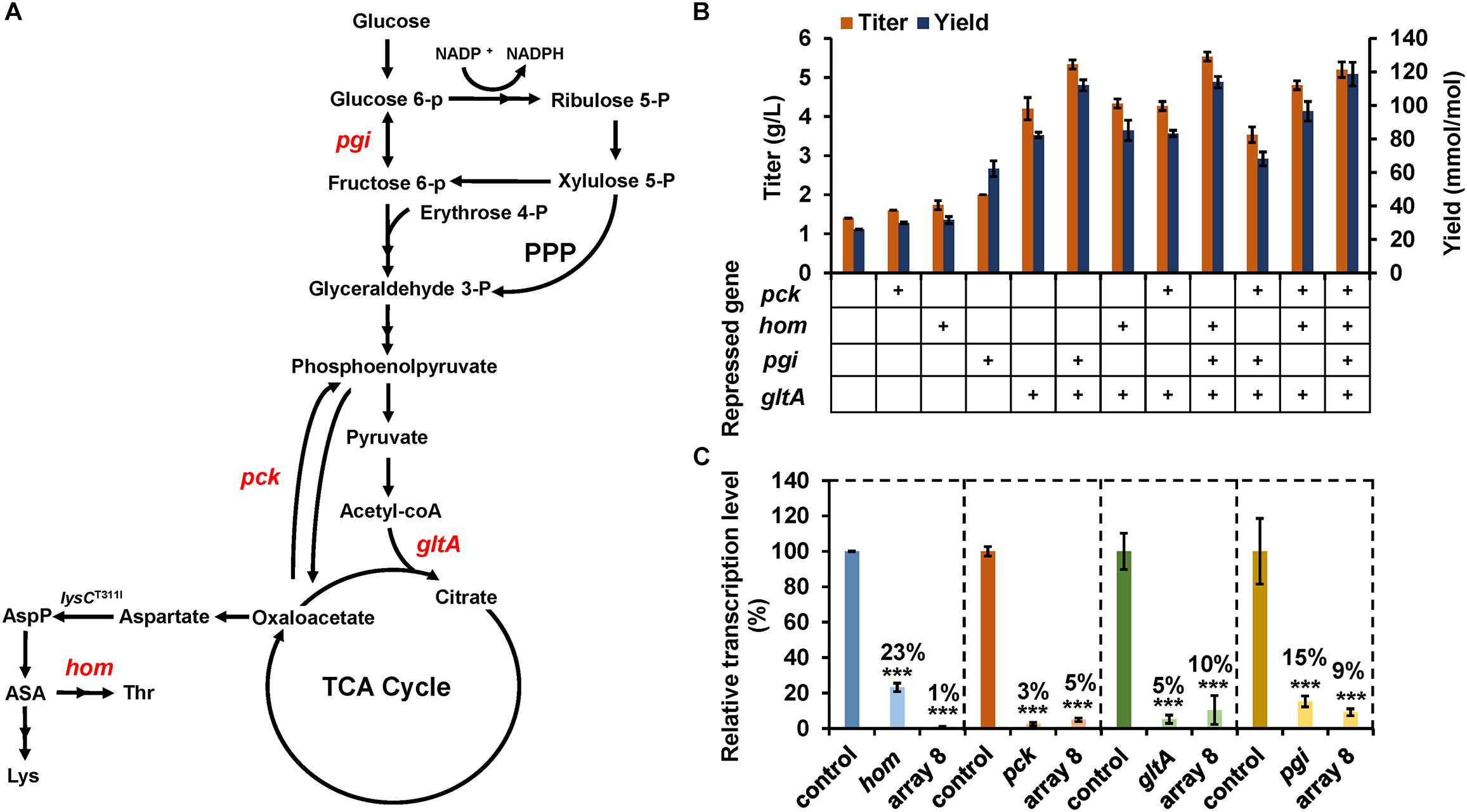
Figure 4. Enhancing lysine production by CRISPR-dCpf1-mediated multiplex gene repression. (A) Lysine biosynthesis pathway of C. glutamicum. Target genes repressed by CRISPR-dCpf1 are colored. AspP, aspartyl phosphate; ASA, aspartate semialdehyde; Thr, threonine; Lys, lysine. (B) Effects of CRISPR-dCpf1-mediated single and multiple genes repression on lysine production. SCgL30 strains co-expressing dCpf1 with an individual crRNA or a crRNA array were cultivated in fermentation medium for 24 h. An engineered strain expressing dCpf1 but no crRNA was used as the control. (C) Relative transcription levels of target genes. Four target genes (gltA, pgi, hom, and pck) were simultaneously repressed by dCpf1 and crRNA array 8. SCgL30 strain expressing dCpf1 but no crRNA was used as the control. Error bars indicate standard deviations from three parallel experiments. All t-tests compare the transcription level obtained by strains expressing dCpf1 and crRNA against control strain expressing dCpf1 but no crRNA (***P < 0.001).
Firstly, single gene repression for these endogenous genes was studied. Four crRNAs were designed to target the 5′-end of the coding regions of target genes and cloned to pEC-02, respectively. The resultant crRNA expression plasmid was transformed into lysine-producing strain SCgL30 with plasmid pXM-07. Fermentation in 24-well plates was performed to evaluate lysine production. Compared with the control strain, repression of pck, hom, pgi, and gltA enhanced extracellular lysine concentrations by 14.3, 23.8, 42.9, and 200%, respectively (Figure 4B). The yields of glucose to lysine conversion were also increased from 25.9 to 29.8, 31.5, 62.2, and 82.3 mmol/mol by repression of pck, hom, pgi, and gltA, respectively (Figure 4B). Subsequently, mRNA levels of the target genes were analyzed. As expected, transcription of these genes was significantly down-regulated by 77–97% in the recombinant strains (Figure 4C). These results indicated that CRISPR-dCpf1 system could be employed to efficiently repress endogenous genes and used for metabolic engineering in C. glutamicum. The gltA gene was also selected as a target for improving lysine production via CRISPR-dCas9 and lysine riboswitch in previous studies and 40–60% improvements in lysine yield were obtained (Zhou and Zeng, 2015; Park et al., 2018). The differences in specific values may be mainly ascribed to the different repression efficiencies and mechanisms of these regulatory tools.
Next, combinational repression of multiple target genes was performed to investigate the best combination for lysine production. Considering the superior effect of gltA repression on lysine production, all the combinations contained gltA repression. The highest lysine titer (5.5 g/L), which was 4.0-fold higher compared with the control strain, was obtained by simultaneously repressing gltA, pgi, and hom. Quadruple repression of gltA, pgi, hom, and pck resulted in a slight decrease in lysine titer (5.2 g/L) but increase in lysine yield (119.0 mmol/mol) (Figure 4B), compared with the triple gene repression. Further qPCR analysis revealed that the mRNA levels of gltA, pgi, hom, and pck were simultaneously reduced by 90, 91, 99, and 95%, respectively (Figure 4C). Unexpectedly, quadruple repression of gltA, pgi, hom, and pck led to an increased repression efficiency of hom (99%), compared to individual repression of hom (77%). To investigate the possible cause, we performed a blast between the spacers of gltA-, pgi-, and pck-targeting crRNAs and the hom transcript. Two potential off-target sites with functional 5′-BTTV-3′ PAM sequences were identified for gltA- and pgi-targeting crRNA in hom transcript with nine mismatches (Supplementary Figure S3). It is uncertain whether the extra repression of hom in quadruple repression experiment was caused by such weak off-target effects or other unknown mechanism. In summary, these results indicate that the CRISPR-dCpf1 system could repress four genes simultaneously with high efficiencies and could be employed for quickly identifying the optimum gene regulation strategy for metabolic engineering in C. glutamicum.
Due to the complexity of cell metabolism, multiple metabolic engineering targets need to be manipulated to balance metabolic flux and maximize biosynthesis of desired products (Jones et al., 2015; Lee and Kim, 2015; Nielsen and Keasling, 2016). For example, genes involved in byproduct formation and intermediate competition are usually knocked out to develop efficient microbial cell factories. However, consecutive deletion of multiple genes is time consuming and laborious (Schafer et al., 1994). Furthermore, for those genes which are essential for cell growth and metabolism, gene knockout is probably not the best choice. As an alternative, multiplex gene repression can be used for optimizing metabolic pathway and is preferred for balancing cell growth and hyper-production of molecules of interests (Solomon et al., 2012; Na et al., 2013). CRISPR-dCas9- and sRNA-mediated gene repression systems have been developed and used for the metabolic engineering of C. glutamicum. By introducing multiple crRNA or sRNA expression cassettes, up to three genes were down-regulated simultaneously in C. glutamicum (Cleto et al., 2016; Zhang et al., 2016; Park et al., 2018, 2019; Gauttam et al., 2019; Sun et al., 2019). However, the complexity of assembling multiple expression cassettes and genetic instability of plasmid with repeated sequences make simultaneous repression of more target genes difficult. In this study, dCpf1-based CRISPRi system was successfully established for multiplex gene repression in C. glutamicum. Due to its inherent function of maturing crRNA, a single crRNA array is sufficient for repressing multiple target genes by CRISPR-dCpf1, which can largely simplify plasmid construction. We tested repression of four genes by the CRISPR-dCpf1 system with a crRNA array, which resulted in over 90% repression efficiencies for all targets. We envision that the CRISPR-dCpf1 system can facilitate simultaneous repression of more target genes in C. glutamicum.
CRISPR-dCpf1 has been applied in E. coli and Streptomyces to modulate multiple genes expression. In E. coli and our C. glutamicum cases, quadruple genes repression showed similar efficiencies with individual gene repression (Zhang et al., 2017), while increasing the number of target genes in Streptomyces decreased repression efficiency (∼70% for triple genes vs. 82.1–95.2% for single gene) (Li et al., 2018). Another interesting phenomenon is dCpf1 (D917A) outperformed dCpf1 (E1006A) and dCpf1 (E1006A, D917A) in E. coli (Miao et al., 2019). In C. glutamicum, however, dCpf1 (E1006A, D917A) showed the highest repression activity compared to the rest two variants with a single mutation. These results suggest that the repression activity of dCpf1 might be host-dependent. For practical applications, target genes may need to be repressed at various levels. To this end, several strategies are applicable. It has been reported that the repression efficiency of CRISPR-dCpf1 system can be adjusted by using truncated or mutated spacers, engineered DRs, and difference PAM patterns (Liu et al., 2019; Miao et al., 2019; Wang et al., 2019). Tuning the expression levels of CRISPRi components (dCpf1 and crRNA) with various constitutive or inducible promoters can also facilitate a quantitative control of the repression efficiency (Fontana et al., 2018). Similar strategies can be investigated in C. glutamicum to achieve gradient knockdown of multiple targets in further studies.
All data generated or analyzed during this study are included in this published article and Supplementary Material. The authors are willing to provide any additional data and materials related to this research that may be requested for research purposes.
ML, JC, PZ, and JS conceptualized the project and designed the study. ML, JC, and JH conducted the experiments and collected data. ML, JC, YW, and JL performed data analysis. ML, JC, YW, PZ, and JS composed the manuscript. PZ, JS, and NC provided critical feedback on the manuscript and provided resources. All authors read and approved the manuscript.
This work was supported by the National Key R&D Program of China (2018YFA0903600 and 2018YFA0900300) and the National Natural Science Foundation of China (31700044 and 31870044).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully thank Dr. Changhao Bi from Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences for providing the FnCpf1 gene.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00357/full#supplementary-material
Bae, S., Park, J., and Kim, J. S. (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. doi: 10.1093/bioinformatics/btu048
Becker, J., and Wittmann, C. (2012). Bio-based production of chemicals, materials and fuels-Corynebacterium glutamicum as versatile cell factory. Curr. Opin. Biotechnol. 23, 631–640. doi: 10.1016/j.copbio.2011.11.012
Cho, S., Choe, D., Lee, E., Kim, S. C., Palsson, B., and Cho, B. K. (2018). High-level dCas9 expression induces abnormal cell morphology in Escherichia coli. ACS Synth. Biol. 7, 1085–1094. doi: 10.1021/acssynbio.7b00462
Cleto, S., Jensen, J. V., Wendisch, V. F., and Lu, T. K. (2016). Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi). ACS Synth. Biol. 5, 375–385. doi: 10.1021/acssynbio.5b00216
Cress, B. F., Toparlak, O. D., Guleria, S., Lebovich, M., Stieglitz, J. T., Englaender, J. A., et al. (2015). CRISPathBrick: modular combinatorial assembly of Type II-A CRISPR arrays for dCas9-mediated multiplex transcriptional repression in E. coli. ACS Synth. Biol. 4, 987–1000. doi: 10.1021/acssynbio.5b00012
Cui, L., Vigouroux, A., Rousset, F., Varet, H., Khanna, V., and Bikard, D. (2018). A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat. Commun. 9:1912. doi: 10.1038/s41467-018-04209-5
Eggeling, L., and Bott, M. (2015). A giant market and a powerful metabolism: L-lysine provided by Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 99, 3387–3394. doi: 10.1007/s00253-015-6508-2
Fonfara, I., Richter, H., Bratovic, M., Le Rhun, A., and Charpentier, E. (2016). The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521. doi: 10.1038/nature17945
Fontana, J., Dong, C., Ham, J. Y., Zalatan, J. G., and Carothers, J. M. (2018). Regulated expression of sgRNAs tunes CRISPRi in E. coli. Biotechnol. J. 13:1800069. doi: 10.1002/biot.201800069
Gauttam, R., Seibold, G. M., Mueller, P., Weil, T., Weiss, T., Handrick, R., et al. (2019). A simple dual-inducible CRISPR interference system for multiple gene targeting in Corynebacterium glutamicum. Plasmid 103, 25–35. doi: 10.1016/j.plasmid.2019.04.001
Ikeda, M., and Nakagawa, S. (2003). The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 62, 99–109. doi: 10.1007/s00253-003-1328-1
Jones, J. A., Toparlak, O. D., and Koffas, M. A. (2015). Metabolic pathway balancing and its role in the production of biofuels and chemicals. Curr. Opin. Biotechnol. 33, 52–59. doi: 10.1016/j.copbio.2014.11.013
Kim, S. K., Kim, H., Ahn, W. C., Park, K. H., Woo, E. J., Lee, D. H., et al. (2017). Efficient transcriptional gene repression by Type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth. Biol. 6, 1273–1282. doi: 10.1021/acssynbio.6b00368
Lee, S. Y., and Kim, H. U. (2015). Systems strategies for developing industrial microbial strains. Nat. Biotechnol. 33, 1061–1072. doi: 10.1038/nbt.3365
Lee, S. Y., Kim, H. U., Chae, T. U., Cho, J. S., Kim, J. W., Shin, J. H., et al. (2019). A comprehensive metabolic map for production of bio-based chemicals. Nat. Catal. 2, 18–33. doi: 10.1038/s41929-018-0212-4
Leenay, R. T., Maksimchuk, K. R., Slotkowski, R. A., Agrawal, R. N., Gomaa, A. A., Briner, A. E., et al. (2016). Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol. Cell 62, 137–147. doi: 10.1016/j.molcel.2016.02.031
Li, L., Wei, K., Zheng, G., Liu, X., Chen, S., Jiang, W., et al. (2018). CRISPR-Cpf1-assisted multiplex genome editing and transcriptional repression in Streptomyces. Appl. Environ. Microbiol. 84:e0827-18. doi: 10.1128/aem.00827-18
Liu, J., Wang, Y., Lu, Y. J., Zheng, P., Sun, J. B., and Ma, Y. H. (2017). Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb. Cell Fact. 16:205. doi: 10.1186/s12934-017-0815-5
Liu, W., Tang, D. D., Wang, H. J., Lian, J. Z., Huang, L., and Xu, Z. N. (2019). Combined genome editing and transcriptional repression for metabolic pathway engineering in Corynebacterium glutamicum using a catalytically active Cas12a. Appl. Microbiol. Biotechnol. 103, 8911–8922. doi: 10.1007/s00253-019-10118-4
Luo, M. L., Leenay, R. T., and Beisel, C. L. (2016). Current and future prospects for CRISPR-based tools in bacteria. Biotechnol. Bioeng. 113, 930–943. doi: 10.1002/bit.25851
Marx, A., Hans, S., Mockel, B., Bathe, B., and de Graaf, A. A. (2003). Metabolic phenotype of phosphoglucose isomerase mutants of Corynebacterium glutamicum. J. Biotechnol. 104, 185–197. doi: 10.1016/S0168-1656(03)00153-6
Miao, C. S., Zhao, H. W., Qian, L., and Lou, C. B. (2019). Systematically investigating the key features of the DNase deactivated Cpf1 for tunable transcription regulation in prokaryotic cells. Synth. Syst. Biotechnol. 4, 1–9. doi: 10.1016/j.synbio.2018.11.002
Na, D., Yoo, S. M., Chung, H., Park, H., Park, J. H., and Lee, S. Y. (2013). Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 31, 170–174. doi: 10.1038/nbt.2461
Nielsen, J., and Keasling, J. D. (2016). Engineering cellular metabolism. Cell 164, 1185–1197. doi: 10.1016/j.cell.2016.02.004
Okibe, N., Suzuki, N., Inui, M., and Yukawa, H. (2011). Efficient markerless gene replacement in Corynebacterium glutamicum using a new temperature-sensitive plasmid. J. Microbiol. Methods 85, 155–163. doi: 10.1016/j.mimet.2011.02.012
Park, J., Shin, H., Lee, S. M., Um, Y., and Woo, H. M. (2018). RNA-guided single/double gene repressions in Corynebacterium glutamicum using an efficient CRISPR interference and its application to industrial strain. Microb. Cell Fact. 17:4. doi: 10.1186/s12934-017-0843-1
Park, J., Yu, B. J., Cho, J. I., and Woo, H. M. (2019). Heterologous production of squalene from glucose in engineered Corynebacterium glutamicum using multiplex CRISPR interference and high-throughput fermentation. J. Agric. Food. Chem. 67, 308–319. doi: 10.1021/acs.jafc.8b05818
Reis, A. C., Halper, S. M., Vezeau, G. E., Cetnar, D. P., Hossain, A., Clauer, P. R., et al. (2019). Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays. Nat. Biotechnol. 37, 1294–1301. doi: 10.1038/s41587-019-0286-9
Schafer, A., Tauch, A., Jager, W., Kalinowski, J., Thierbach, G., and Puhler, A. (1994). Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145, 69–73. doi: 10.1016/0378-1119(94)90324-7
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Solomon, K. V., Sanders, T. M., and Prather, K. L. (2012). A dynamic metabolite valve for the control of central carbon metabolism. Metab. Eng. 14, 661–671. doi: 10.1016/j.ymben.2012.08.006
Sun, D. H., Chen, J. Z., Wang, Y., Li, M. Y., Rao, D. M., Guo, Y. M., et al. (2019). Metabolic engineering of Corynebacterium glutamicum by synthetic small regulatory RNAs. J. Ind. Microbiol. Biotechnol. 46, 203–208. doi: 10.1007/s10295-018-02128-4
van Ooyen, J., Noack, S., Bott, M., Reth, A., and Eggeling, L. (2012). Improved L-lysine production with Corynebacterium glutamicum and systemic insight into citrate synthase flux and activity. Biotechnol. Bioeng. 109, 2070–2081. doi: 10.1002/bit.24486
Wang, H. L., Bian, X. Y., Xia, L. Q., Ding, X. Z., Muller, R., Zhang, Y. M., et al. (2014). Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res. 42:e37. doi: 10.1093/nar/gkt1339
Wang, J., Lu, A., Bei, J., Zhao, G., and Wang, J. (2019). CRISPR/ddCas12a-based programmable and accurate gene regulation. Cell Discov. 5:15. doi: 10.1038/s41421-019-0085-y
Wang, Y., Cao, G., Xu, D., Fan, L., Wu, X., Ni, X., et al. (2018a). A novel Corynebacterium glutamicum L-glutamate exporter. Appl. Environ. Microbiol. 84:e2691-17. doi: 10.1128/aem.02691-17
Wang, Y., Liu, Y., Liu, J., Guo, Y. M., Fan, L. W., Ni, X. M., et al. (2018b). MACBETH: multiplex automated Corynebacterium glutamicum base editing method. Metab. Eng. 47, 200–210. doi: 10.1016/j.ymben.2018.02.016
Xu, J. Z., Xia, X. H., Zhang, J. L., Guo, Y. F., Qian, H., and Zhang, W. G. (2014). A method for gene amplification and simultaneous deletion in Corynebacterium glutamicum genome without any genetic markers. Plasmid 72, 9–17. doi: 10.1016/j.plasmid.2014.02.001
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system. Cell 163, 759–771. doi: 10.1016/j.cell.2015.09.038
Zhang, B., Liu, Z., Liu, C., and Zheng, Y. (2016). Application of CRISPRi in Corynebacterium glutamicum for shikimic acid production. Biotechnol. Lett. 38, 2153–2161. doi: 10.1007/s10529-016-2207-z
Zhang, B., Zhou, N., Liu, Y. M., Liu, C., Lou, C. B., Jiang, C. Y., et al. (2015). Ribosome binding site libraries and pathway modules for shikimic acid synthesis with Corynebacterium glutamicum. Microb. Cell Fact. 14:71. doi: 10.1186/s12934-015-0254-0
Zhang, J. L., Peng, Y. Z., Liu, D., Liu, H., Cao, Y. X., Li, B. Z., et al. (2018). Gene repression via multiplex gRNA strategy in Y. lipolytica. Microb. Cell Fact. 17:62. doi: 10.1186/s12934-018-0909-8
Zhang, X., Wang, J., Cheng, Q., Zheng, X., Zhao, G., and Wang, J. (2017). Multiplex gene regulation by CRISPR-ddCpf1. Cell Discov. 3:17018. doi: 10.1038/celldisc.2017.18
Keywords: Corynebacterium glutamicum, CRISPR-dCpf1, multiplex gene repression, metabolic engineering, lysine
Citation: Li M, Chen J, Wang Y, Liu J, Huang J, Chen N, Zheng P and Sun J (2020) Efficient Multiplex Gene Repression by CRISPR-dCpf1 in Corynebacterium glutamicum. Front. Bioeng. Biotechnol. 8:357. doi: 10.3389/fbioe.2020.00357
Received: 21 January 2020; Accepted: 31 March 2020;
Published: 24 April 2020.
Edited by:
Evangelos Topakas, National Technical University of Athens, GreeceReviewed by:
Lothar Eggeling, Julich Research Centre, Helmholtz Association of German Research Centers (HZ), GermanyCopyright © 2020 Li, Chen, Wang, Liu, Huang, Chen, Zheng and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Zheng, emhlbmdfcEB0aWIuY2FzLmNu; Jibin Sun, c3VuX2piQHRpYi5jYXMuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.