
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 07 April 2020
Sec. Synthetic Biology
Volume 8 - 2020 | https://doi.org/10.3389/fbioe.2020.00235
This article is part of the Research Topic Synthetic Nucleic Acids for Expanding Genetic Codes and Probing Living Cells View all 9 articles
 Han-Kai Jiang1,2,3
Han-Kai Jiang1,2,3 Man-Nee Lee1
Man-Nee Lee1 Jo-Chu Tsou1Kuan-Wen Chang1Hsueh-Wei Tseng1Kuang-Po Chen4Yaw-Kuen Li4
Jo-Chu Tsou1Kuan-Wen Chang1Hsueh-Wei Tseng1Kuang-Po Chen4Yaw-Kuen Li4 Yane-Shih Wang1,2,5*
Yane-Shih Wang1,2,5*The Methanosarcina mazei pyrrolysyl-tRNA synthetase (PylRS)⋅tRNAPyl pair can be used to incorporate non-canonical amino acids (ncAAs) into proteins at installed amber stop codons. Although engineering of the PylRS active site generates diverse binding pockets, the substrate ranges are found similar in charging lysine and phenylalanine analogs. To expand the diversity of the ncAA side chains that can be incorporated via the PylRS⋅tRNAPyl pair, exploring remote interactions beyond the active site is an emerging approach in expanding the genetic code research. In this work, remote interactions between tRNAPyl, the tRNA binding domain of PylRS, and/or an introduced non-structured linker between the N- and C-terminus of PylRS were studied. The substrate range of the PylRS⋅tRNAPyl pair was visualized by producing sfGFP-UAG gene products, which also indicated amber suppression efficiencies and substrate specificity. The unstructured loop linking the N-terminal and C-terminal domains (CTDs) of PylRS has been suggested to regulate the interaction between PylRS and tRNAPyl. In exploring the detailed role of the loop region, different lengths of the linker were inserted into the junction between the N-terminal and the C-terminal domains of PylRS to unearth the impact on remote effects. Our findings suggest that the insertion of a moderate-length linker tunes the interface between PylRS and tRNAPyl and subsequently leads to improved suppression efficiencies. The suppression activity and the substrate specificity of PylRS were altered by introducing three mutations at or near the N-terminal domain of PylRS (N-PylRS). Using a N-PylRS⋅tRNAPyl pair, three ncAA substrates, two S-benzyl cysteine and a histidine analog, were incorporated into the protein site specifically.
Expanding the genetic code is a pragmatic approach to incorporate over 200 different kinds of non-canonical amino acids (ncAAs) into proteins in vivo (Vargas-Rodriguez et al., 2018). This technology employs a bioorthogonal aminoacyl-tRNA synthetase (AARS)⋅tRNA pair to decode nonsense or rare codons in living systems genetically (O’Donoghue et al., 2012). One of most used AARS⋅tRNA systems for genetic code expansion is the pyrrolysyl-tRNA synthetase (PylRS)⋅tRNAPyl derived from the archaea Methanosarcina barkeri (Mb) or Methanosarcina mazei (Mm) or the eubacteria Desulfitobacterium hafniense (Dh) (Herring et al., 2007; Neumann et al., 2008; Yanagisawa et al., 2008). The PylRS⋅tRNAPyl pair has a wide range of bio-orthogonality in different species from prokaryotes to eukaryotes (Mukai et al., 2008; Neumann et al., 2008; Han et al., 2017). The MmPylRS⋅MmtRNAPyl (PylRS⋅tRNAPyl) pair was naturally evolved for amber TAG codon recoding with slow enzyme kinetic properties (Guo et al., 2014). Uniquely, PylRS harbors a sophisticated and dynamic active site for recognizing pyrrolysine, a lysine analog modified with a 4-methylpyrroline-ring through an Nε-amide bond linkage (Hao et al., 2002). Various evolved PylRS⋅tRNAPyl pairs for incorporating lysine and phenylalanine analogs have been reported through directed-evolution and rational design approaches (Mukai et al., 2008; Neumann et al., 2008; Wang Y.S. et al., 2012; Wang et al., 2010; Guo et al., 2014). The diverse substrate scope and co-crystal structures of ncAA substrate-PylRS mutants reveal that the substrate can have multiple binding modes and a slower feature in enzymatic kinetic studies (Kavran et al., 2007; Nozawa et al., 2009). The directed evolution of full-length PylRS has been studied. However, a systematic investigation of interactions between the C-terminal domain (CTD) and the N-terminal domain (NTD) of the protein and the cognate tRNA, as well as the impact of those interactions of the dynamic substrate range, remains unexplored.
The co-crystal structure of MmPylRS CTD with pyrrolysine revealed a distinct binding mode for the amino acid, which is deeply embedded into the hydrophobic pocket of the catalytic domain. The CTD of PylRS contains the binding domain that harbors tRNAPyl and ATP (Nozawa et al., 2009). The interplay between tRNAPyl and PylRS plays a key role in affecting enzymatic activity and substrate scope. The G1:C72 base pair and discriminator G73, which are located on the acceptor arm of tRNAPyl, are thought to be required for PylRS recognition (Herring et al., 2007). Mutations in the D loop, T loop, or variable loop of tRNAPyl spatially perturb its shape, which in turn diminishes the activity of PylRS. The suppression efficiency is slightly influenced by a change in the anticodon loop compared to the D or the T loop, suggesting the inessential element for PylRS recognition (Ambrogelly et al., 2007).
The anticodon loop and acceptor arm of the tRNA are fundamental and essential elements for endogenous AARSs recognition (Schimmel et al., 1993; O’Donoghue and Luthey-Schulten, 2003). As opposed to this binding mode, PylRS recognizes its cognate tRNAPyl distinctly. This characteristic helps PylRS to selectively distinguish tRNAPyl in the translational system. The T loop and the variable loop of tRNAPyl have hydrophilic interactions with the PylRS-NTD. The tRNAPyl is oppositely embraced by the NTD and the CTD of PylRS, which is in connection with the postulated flexible loops (Suzuki et al., 2017; Figure 1A). This compatible binding mode dynamically controls the interaction between PylRS and tRNAPyl.
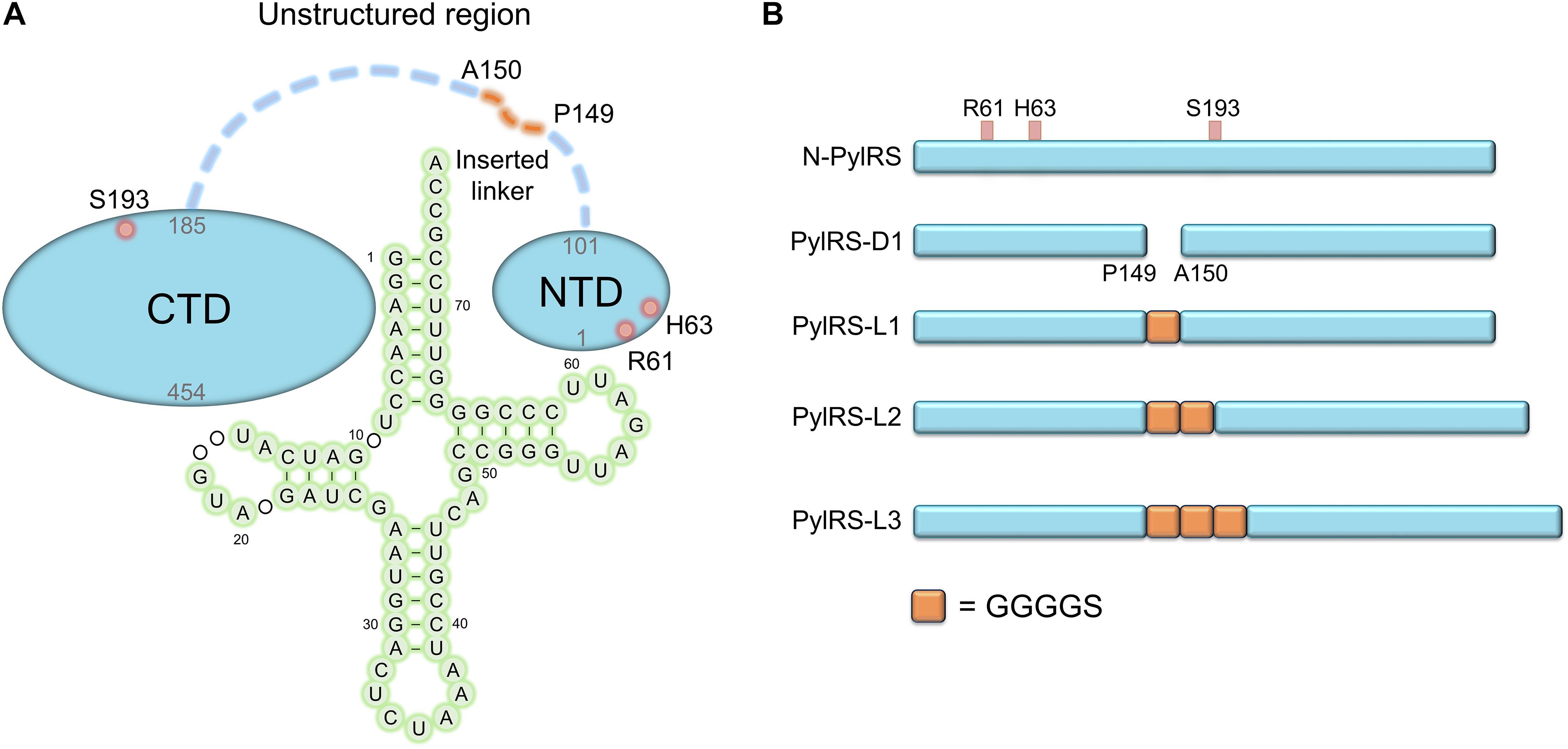
Figure 1. Protein designs of evolved MmPylRS and their linker engineering. (A) Illustration of the interaction between MmPylRS and tRNAPyl. R61K/H63Y/S193R mutations (labeled in open red circles) are distributed on the tRNA binding domain of MmPylRS NTD or CTD (labeled as filled blue circle). The detailed interactions from X-ray crystal structures in PylRS-NTD/tRNA (Suzuki et al., 2017) and PylRS-CTD/tRNA (Nozawa et al., 2009) are indicated in Figures 2, 3. tRNA 2D topology and sequence are labeled in green filled circles. The orange dashed line denotes the inserted linker between P149 and A150. (B) Engineered MmPylRS constructs used in this work. N-PylRS represents MmPylRS with R61K/H63Y/S193R mutations; PylRS-D1 represents MmPylRS with insertion of the spacer containing the stop and the start codons between P149 and A150; PylRS-L1 indicates the insertion of SGGGGS linker between P149 and A150; PylRS-L2 for S(GGGGS)2 linker insertion; PylRS-L3 for S(GGGGS)3 linker insertion.
Moreover, the truncation of PylRS NTD significantly precludes the binding specificity of PylRS by making it incapable to charge tRNAPyl (Jiang and Krzycki, 2012). The mutations on NTD of the evolved chimeric PylRS (chPylRS) have played a crucial role in tuning the binding of tRNAPyl (Suzuki et al., 2017). The previous study of class II AARS seryl-tRNA synthetase (SerRS) showed that the R76 and the R94 of the NTD of MbSerRS were curial for tRNA recognition, and the alanine mutations of both had abolished the amber suppression efficiency (Jaric et al., 2009). N-terminal Add-1 domain from an exceptional class I AARS arginyl-tRNA synthetase (ArgRS) of Saccharomyces cerevisiae was found to bind the D loop of tRNAArg, which is a rare spatial arrangement among class I AARSs but similar to MmPylRS and MbSerRS (Cavarelli et al., 1998). The length of NTD extensively affects the orientation of tRNAArg by the positional change of ArgRS (Shimada et al., 2001). Using directed evolution, certain mutations generated on PylRS NTD accompanied by variations surrounding the catalytic site also led to increased suppression efficiencies as well as substrate specificity (Mukai et al., 2015). Evolved PylRS harboring three mutations on NTD was found to enhance the suppression efficiency in charging BocK (1, Scheme 1) (Sharma et al., 2018). The above studies support the idea that the mutations on the NTD might influence the binding mode and the activity of PylRS.

Scheme 1. Chemical structures of the non-canonical amino acids described in this study. ncAA 1: Nε-(tert-butoxycarbonyl)-L-lysine (BocK); 2: Nε- (alloyloxycarbonyl)-L-lysine (AlloK); 3: S-(4-methylbenzyl)-L-cysteine (MbzlC); 4: S-(4-methoxylbenzyl)-L-cysteine (MeObzlC); 5: N-π-methyl-L-histidine (3MeH); 6: Nε-(carbobenzyloxy)-L-lysine (CbzK); 7: Nε-(2-chlorocarbobenzyloxy)-L-lysine (ClCbzK); 8: Nε-(carbobenzyloxy)-D-lysine (DCbzK); 9: Nε-(2-chlorocarbobenzyloxy)- D-lysine (DClCbzK); 10: (R)-2-amino-3-(2-benzyloxycarbonylaminoethylselanyl)propanoic acid (SeCbzK); 11: (S)-6-(benzyloxycarbonylamino)-2-hydroxygexanoic acid (CbzKOH).
Here we report the generation of PylRS variants with mutations beyond the catalytic site, which has allowed us to further decipher the interaction between PylRS and tRNAPyl. Fast screening of a ncAAs library using an amber-codon-installed superfolder green fluorescent protein (sfGFP) gene allowed us to determine the substrate range and suppression efficiency and, in turn, fine-tuning of the interaction between PylRS and tRNAPyl. We found that extending the length of a linker between the NTD and the CTD of PylRS further probed the management of flexible loop and their effectiveness in ncAA incorporation yield.
Natural amino acids and ncAAs were purchased from Chem Impex Inc (Wood Dale, IL, United States). The ncAA SeCbzK (10) (Wang Z.U. et al., 2012) and CbzKOH (11) (Li et al., 2012; Wang, 2012) (Scheme 1) were synthesized by the reported methods. PCR was performed using the KOD hot start polymerase kit (Merck). Oligonucleotide synthesis and DNA sequencing were done by Genomics Inc. (Taipei, Taiwan). The primer sequences are listed in the Supplementary Materials. Construction of the pET-pylT-sfGFP-TAG2 and the pET-pylT-sfGFP-TAG27 plasmids followed general cloning protocols. The sfGFP-TAG2 gene was amplified by PCR from pET-pylT-sfGFP (Guo et al., 2014) with the primers sfGFP-TAG2-NdeI-F and sfGFP-SacI-R2. The sfGFP-TAG27 was generated by overlap extension PCR with two fragments (fragment 1: primer sfGFP-NdeI-F1 and sfGFP-TAG27-R; fragment 2: sfGFP-TAG27-F and sfGFP-SacI-R2). The overlapped sfGFP gene products and pET-pylT plasmid were then double-digested with restriction enzyme NdeI and SacI, and then T4 DNA ligase was used for DNA ligation.
In the construction of the plasmid pCDF-N-PylRS variants, the PylRS-R61K/H63Y/S193R (N-PylRS) gene was generated by overlap extension PCR with three fragments using the template pCDF-PylRS: fragment 1: PylRS-NcoI-F and PylRS-R61K/H63Y-R; fragment 2: PylRS-R61K/H63Y-F and PylRS-S193R-R; fragment 3: PylRS-S193R-F and PylRS-BamHI-R. In preparing pCDF-PylRS-R61K, pCDF-PylRS-H63Y, and pCDF-PylRS-S193R, the same procedures were performed with the following primers to produce PylRS-R61K, PylRS- H63Y, and PylRS-S193R genes with pCDF-PylRS template and following the overlap extension PCR; PylRS-R61K gene: fragment 1, PylRS-NcoI-F and PylRS-R61K-R; fragment 2, PylRS-R61K-F and PylRS-BamHI-R; PylRS-H63Y gene: fragment 1, PylRS-NcoI-F and PylRS-H63Y-R; fragment 2, PylRS-H63Y-F and PylRS-BamHI-R; PylRS-S193R gene: fragment 1, PylRS-NcoI-F and PylRS-S193R-R; fragment 2, PylRS-S193R-F and PylRS-BamHI-R. In preparing pCDF-PylRS-ND, the PylRS-ND gene fragment was generated by PCR with two primers, PylRS-NcoI-F and PylRS-P149-BamHI-R, and pCDF-PylRS was used as a template. In the preparation of pCDF-PylRS-D1, the spacer sequence (TGAAAAAAGCGATG), including the TGA stop codon and the ATG start codon, was installed between positions P149 and A150 of the PylRS gene. The PylRS-D1 gene fragment was generated by overlap extension PCR with two fragments using pCDF-PylRS as a template. Fragment 1 was generated by the primers PylRS-NcoI-F and PylRS-P149SSS-R1; fragment 2 was generated by the primers PylRS-P149SSS-F2 and PylRS-BamHI-R. Different lengths of linker were installed in PylRS variants between sposition 149 and 150. In preparing pCDF-PylRS-L1, pCDF-PylRS-L2, and pCDF-PylRS-L3 (Figure 1B), the same procedures were performed by the following primers to produce PylRS-L1, PylRS-L2, and PylRS-L3 genes with pCDF-PylRS template and following overlap extension PCR: PylRS-L1 gene, PylRS-NcoI-F, and Linker-1XG4S-R1, Linker-1XG4S-F2, and PylRS-BamHI-R; PylRS-L2 gene: fragment 1, PylRS-NcoI-F and Linker-2XG4S-R1; fragment 2, Linker-2XG4S-F2 and Linker-2XG4S-R2; fragment 3, Linker-2XG4S-F3 and PylRS-BamHI-R; PylRS-L3 gene: fragments 1 and 3 were from the PylRS-L2 gene; fragment 2, Linker-3XG4S-F2 and Linker-3XG4S-R2.
The pCDF-N-PylRS plasmids with the inserted linker, pCDF-N-PylRS-D1, pCDF-N-PylRS-L1, pCDF-N-PylRS-L2, and pCDF-N-PylRS-L3 (Figure 1B), were then constructed using the procedures mentioned above. pCDF-ZRS was derived from pEVOL-mKRS1-pylT (Wang Z.U. et al., 2012). The N-PylRS mutations were transplanted to ZRS by the methods mentioned earlier to construct pCDF-N-ZRS. The linkers were inserted into ZRS to generate pCDF-N-ZRS-D1, pCDF-ZRS-L1, pCDF-ZRS-L2, and pCDF-ZRS-L3 plasmids. All the ligated products were transformed to E. coli DH5α, and the colonies were selected for DNA sequencing, respectively.
To produce ncAA-encoded sfGFP proteins, the pET-pylT-sfGFP-TAG2 or pET-pylT-sfGFP-TAG27 plasmid was co-transformed with different pCDF-PylRS variants into E. coli BL21 (DE3) individually. After an hour of recovery, the bacteria were spread on a plate containing ampicillin (Amp) (100 μg/ml) and streptomycin (Sm) (100 μg/ml). A single colony was chosen from the plate and cultured in 1 ml LB medium overnight. The cultured bacteria were then transferred to 50 ml fresh LB medium and incubated at 37°C until the OD595 reached 0.6–0.8. Protein expression was induced with the supplement of 1 mM IPTG and ncAA (except for sfGFP-3 and sfGFP-4 protein production, where the medium was changed to GMML medium supplemented with 2 mM ncAA) and incubated at 37°C for 12 h. The bacteria were then harvested and resuspended in lysis buffer [1X phosphate-buffered saline (PBS), pH 7.4] and sonicated. After centrifugation (60 min, 20,000 × g, 4°C), the supernatant was collected and incubated with 0.5 ml Ni2+-NTA resin (Roche) for protein purification. A total of 5 ml lysis buffer and 2.5 ml washing buffer (1X PBS, 5 mM imidazole, pH 7.4) were used to remove proteins bound non-specifically to the resin. The target protein was eluted from the resin with 2.5 ml elution buffer (1X PBS, 200 mM imidazole, pH 7.4). The buffer of the eluted fractions was changed to 1X PBS with Amicon Ultra-15 Centrifugal Filter Units (MWCO 10 kDa). Purified sfGFP was analyzed by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) with instant blue staining.
Whole cells were collected and lysed at 100°C with SDS loading dye for 15 min and then subjected to 12% SDS-PAGE analysis. The gels were stained with InstantBlueTM Stain to visualize the target proteins with the expected molecular weight of around 28 kDa. The suppression efficiency of the amber codon in the sfGFP proteins with a C-terminal His tag was observed by western blot with an anti-6X His tag antibody. Western blots were performed using a Trans-Blot Turbo System (Bio-Rad) and an RTA transfer kit. Anti-His (SignalChem, H99-61M-100) and horseradish peroxidase (HRP)-conjugated secondary antibody (Cell Signaling Technology, 7076P2) were used for immunoblotting. After SDS-PAGE analysis, the gel was immersed in the transfer buffer and then blotted with a polyvinylidene fluoride (PVDF) membrane (25 V/1.3 A, 10 min). After the transfer process was finished, the PVDF membrane was washed thrice with phosphate-buffered saline with Tween® (PBST) buffer for 5 min. Next, the membrane was blocked with 5% skimmed milk for 1 h at room temperature. Then, the membrane was washed thrice with PBST buffer for 5 min (washing step). The primary antibody (1:1,000 dilution) was added and incubated with the membrane for 1 h at room temperature following the washing step. Subsequently, the HRP-conjugated secondary antibody (1:5,000 dilution) was added and incubated with the membrane for 1 h at room temperature. The membrane was then treated by a washing step. Finally, the WesternBright ECL HRP substrate (Advansta, K-12045-D50) was mixed and spread onto the membrane to visualize the band signals using the ChemiDoc Imaging Systems (Bio-Rad) in bioluminescence detection mode.
The pure protein was diluted with 50% acetonitrile and 1% formic acid. An aliquot corresponding to 1 pmol of the pure protein was injected into an ESI source (Waters LockSpray Exact Mass Ionization Source) with a syringe pump (Harvard Apparatus, MA, United States) and a flow rate of 5 μl/min was held throughout the analysis. The mass of the intact proteins was determined using Waters Synapt G2 HDMS mass spectrometer (Waters, Milford, MA, United States). The acquired spectra were deconvoluted to single-charge state using the MaxEnt1 algorithm of the MassLynx 4.1 software (Waters).
Details of in-gel digestion are as follows: after the staining procedure, the gel bands were excised and cut into small pieces. The gel pieces were sequentially washed with 25 mM NH4HCO3, 40% methanol solution, and 100% acetonitrile before being treated with DTT and then iodoacetamide. Washing of the gel pieces with 25 mM NH4HCO3 and 50% acetonitrile and then drying in a vacuum centrifuge provided the materials for trypsin digestion. A solution of 65 to 100 ng of sequencing-grade modified trypsin (Promega) in 25 mM NH4HCO3 and 10% acetonitrile (25–30 μl) was added and incubated with the gel pieces for 12–16 hrs at 37°C. The reaction was stopped by adding 1–2 μl of 5% formic acid. Details of the MS and data analysis are as follows: the digested samples (0.5 μl) were carefully mixed with the matrix solution (0.5 μl of 5 mg/ml DHB in 0.1% TFA/30% acetonitrile) and 0.5 μl of the mixture was deposited onto a MTP 600/384 AnchorChip (Bruker Daltonics). All mass spectrometry experiments were done using a Bruker Autoflex III MALDI TOF/TOF mass spectrometer (Bremen, Germany) equipped with a 200-Hz SmartBean Laser in positive ion mode with delayed extraction in the reflectron mode. Data acquisition was done manually with FlexControl 3.4, and data processing was performed with Flex-Analysis 3.4 (both Bruker Daltonik). Protein database searches through Mascot, using combined PMF and tandem mass spectrometry MS/MS datasets, were performed via Biotools 3.2 (Bruker).
To understand the substrate range of PylRS variants, screening of a 359 ncAAs library was performed. The plasmid pET-pylT-sfGFP-TAG2 or pET-pylT-sfGFP-TAG27 and pCDF-PylRS variants were co-transformed into E. coli BL21 (DE3) individually. The bacteria were spread on a plate supplemented with Amp (100 μg/ml) and Sm (100 μg/ml). The plate was incubated at 37°C overnight. Ten colonies were then inoculated and cultured in 3 ml LB medium at 37°C overnight before 500 μl of each cultured bacteria was transferred to 25 ml of fresh LB medium and incubated at 37°C until the OD595 reached 0.6–0.8. The cells were harvested and washed twice with M9 salts and suspended in M9 medium (M9 salts, 1% glycerol, 2 mM MgSO4, and 0.1 mM CaCl2) containing 1 mM IPTG. Aliquots (50 μl) of the suspended cells were loaded into a 384-well plate containing a different ncAAs (1 mM) in 359 wells (Supplementary Table S2). Cells were incubated in a plate reader (BioTek) at 37°C for 12 h, with continuous monitoring of the fluorescence intensity (excitation 535 nm and emission 595 nm) as well as OD595. Twelve wells were used as controls to measure the background signals (six wells without ncAAs and IPTG; six wells without ncAAs but containing IPTG). The fluorescence intensity of sfGFP was divided by the OD595 following the subtraction of the control signals (containing IPTG but no ncAAs) to generate the relative fluorescence intensity.
To explore novel substrate ranges, this work aimed to generate novel PylRS variants without active site mutations for studying remote effects in altering the interaction between the tRNA and the PylRS NTD or CTD. In a previous study, the evolved PylRS, HarRS, had R61K, H63Y, S193R, N203T, L305H, L309W, N346D, C348S, L367M, Y384F, K429M, K431M, D433G, and G444E mutations. These mutations of HarRS have been found to enhance the activity and the selectivity in charging homoarginine (Mukai et al., 2015). To understand how the mutations on NTD affect the suppression efficiency of PylRS, the first three mutation sites in HarRS are transplanted to PylRS to generate N-PylRS (R61K/H63Y/S193R) (Figure 1). The R62K and H63Y mutations are in the NTD and at the interface between the PylRS-NTD and the tRNA T loop region (Figure 2). The S193R mutation is in the CTD and is located at the interface with the tRNA D-loop region (Figure 3) based on an overlapped model of the DhPylRS CTD/tRNAPyl co-crystal and the MmPylRS CTD crystal structures. In phage-assisted non-continuous evolution (PANCE) approach for evolving chPylRS, some mutants were found to be active in charging BocK with two separated genes by inserted TGA stop codon and following ATG start codon between NTD and CTD (Suzuki et al., 2017). The chPylRS was generated by fusing MbPylRS NTD (1–149 residues) with the MmPylRS C-terminus (185–454 residues). In this work, wild-type MmPylRS (wt-PylRS) was used to translate the inserted mutation between P149 and A150 to form the PylRS-D1 construct (Figure 1B) in generating MmPylRS NTD (1–149 residues) and MmPylRS CTD (150–454 residues) proteins. The truncated MmPylRS NTD (1–149 residues), namely, PylRS-ND, was also generated for comparison.
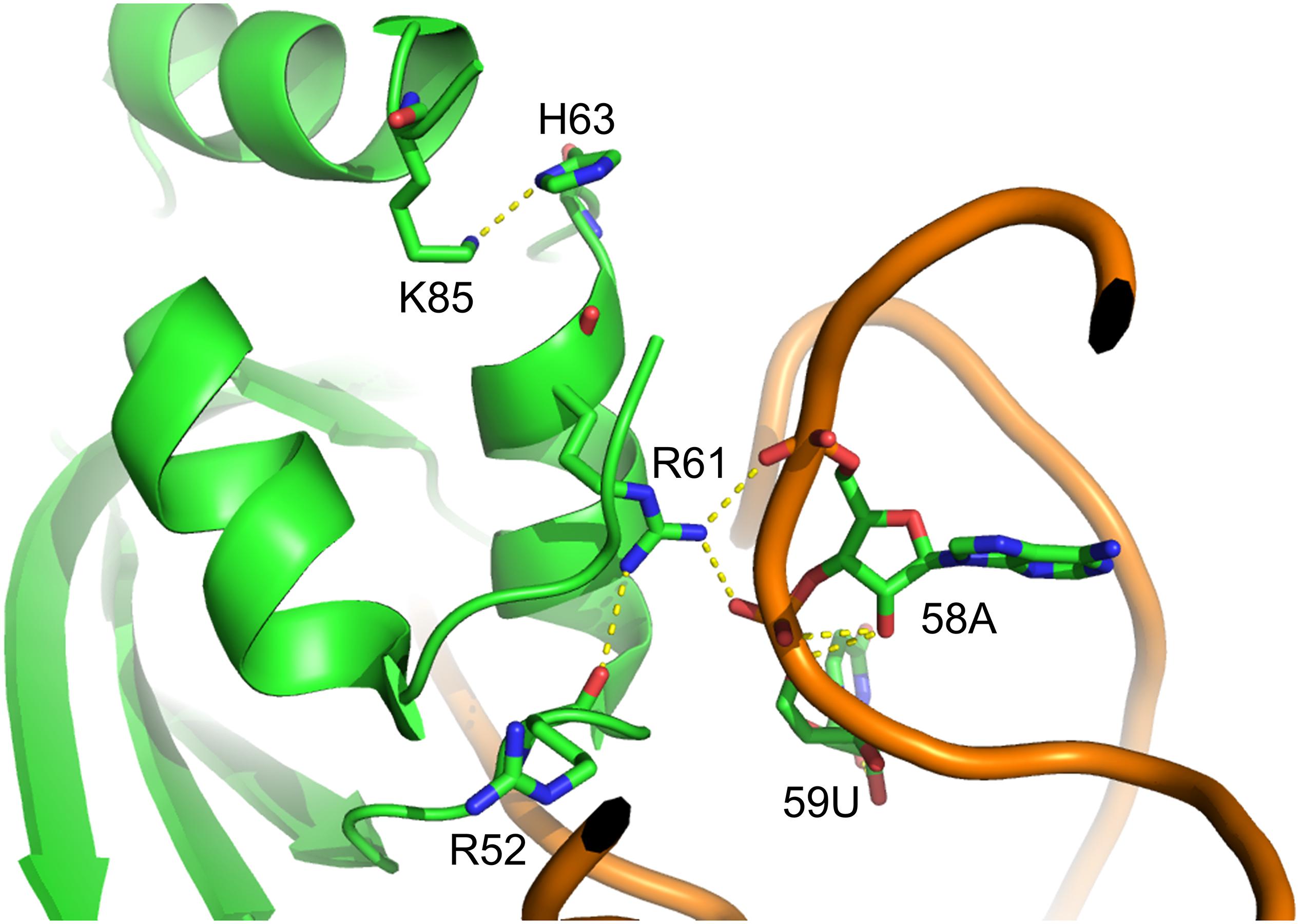
Figure 2. Structure of MmPylRS NTD complex with tRNAPyl. R61 and H63 located in the PylRS NTD; the yellow dashed lines represent potential hydrogen-bonding interactions. Three such interactions are illustrated between the side chain of R61 and R52 within PylRS NTD and the phosphodiester backbone of 58A and 59U in tRNAPyl. One hydrogen-bonding interaction was found between the side chain of H63 and K85 within the PylRS NTD. The structure is based on the PDB entry 5UD5.
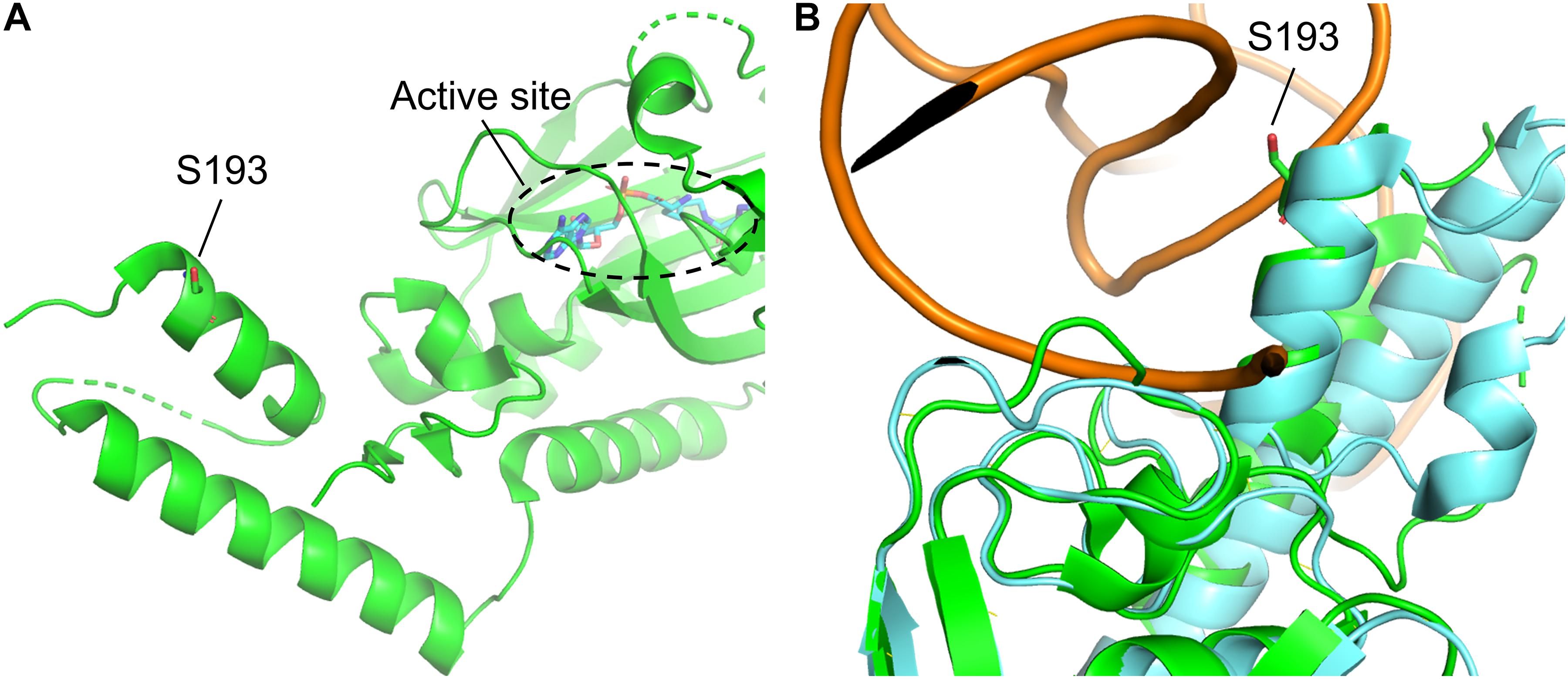
Figure 3. Structure of MmPylRS CTD and its superimposition with the DhPylRS CTD/tRNAPyl complex. (A) S193 is located in the tRNA binding domain of MmPylRS CTD (green). (B) The superimposed structure of MmPylRS CTD (green) and DhPylRS CTD⋅DhtRNAPyl complex (cyan). The structures are based on the PDB entry 2Q7H and 2ZNI.
To probe the crosstalk between the PylRS NTD and the CTD in charging amino acid substrates, three flexible loops of different lengths were inserted between these domains in PylRS-D1. The linkers were the hexapeptide SGGGGS (PylRS-L1), the tridecapeptide S(GGGGS)2 (PylRS-L2), and the non-adecapeptide S(GGGGS)3 (PylRS-L3) (Figure 1B). In addition to these three mutants, we also compared wt-PylRS and PylRS-D1; these five PylRS variants were subjected to test substrate range with the supplement of co-transformed E. coli carrying MmPylRS/tRNAPyl gene cassettes and reporter gene sfGFP-TAG2 or sfGFP-TAG27, respectively. The fluorescence intensities of the sfGFP-UAG2 and sfGFP-UAG27 gene products indicated the read-through of the amber codon in response to the ncAA. Generally, the sfGFP-UAG2 suppression test has 4.7 to 1.6 times higher signal than sfGFP-UAG27 suppression test in charging ncAA 1 and 2 (Scheme 1), but the reverse results are seen with ncAA 3–5. After producing the sfGFP-UAG2 and sfGFP-UAG27 gene products, 359 ncAAs (see Supplementary Table S1 for structures) were tested as substrates. The screening results (Supplementary Figures S15–S24) show various intensities of signals in charging BocK (1) and AlloK (2) (Scheme 1), which are considered as good substrates for wt-PylRS recognition (Yanagisawa et al., 2008). In sfGFP-UAG2 gene production, PylRS-D1 preserved 34% of activity in charging AlloK (2) compared to wt-PylRS. The PylRS variants with linkers generate better activities (Figure 4A). All three PylRS variants (PylRS-L1, PylRS-L2, and PylRS-L3) were rescued by inserted linkers and showed enhanced activity at 120–230% compared to the activity of wt-PylRS. In sfGFP-UAG27 gene production (Figure 4B), however, the activity of all five PylRS variants maintained a similar pattern with less charging of ncAA 1 and 2. Small signals in charging 3MeH (5) (Scheme 1) were found in wt-PylRS, PylRS-L1, PylRS-L2, and PylRS-3 in this amber suppression test.
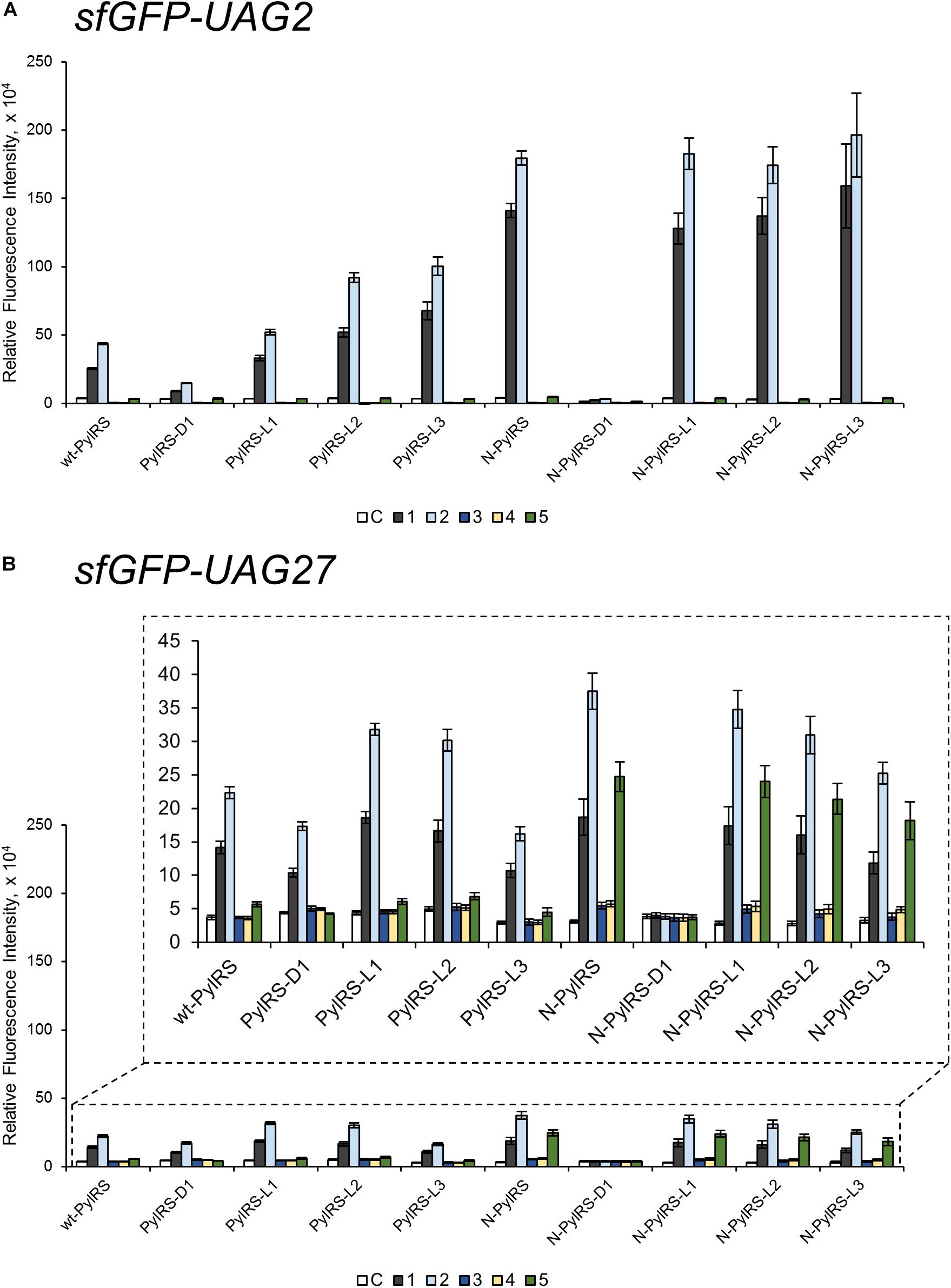
Figure 4. The sfGFP-UAG2 and sfGFP-UAG27 gene suppression efficiencies of PylRS enzyme variants. Incorporation efficiencies of PylRS variants (Figure 1B) as measured by fluorescence intensities of sfGFP with an amber mutation at position 2 (A) and 27 (B). The proteins were expressed in 1 mM ncAA and IPTG in GMML medium at 37°C for 12 h. The cells were excited at 485 nm and the fluorescence intensities were detected at 535 nm. The cell density was monitored by absorbance at 595 nm. “C” indicates the Control experiments of cells with the supplement of 1 mM IPTG; 1–5 denote the supplement of 1 mM IPTG and ncAA 1–5 (Scheme 1). The background signals from cells without adding IPTG were subtracted from each group. The error bars represent the standard deviation of sfGFP production from four repeated experiments.
The effects of PylRS-R61K, PylRS-H63Y, and PylRS-S193R variants were determined and found to improve the suppression efficiency against ncAA 1–2 in sfGFP-UAG2 production compared to wt-PylRS, whereas no fluorescence signals were observed in PylRS-ND (Supplementary Figure S47). Combining the three beneficial mutations, N-PylRS harboring R61K/H63Y/S193R mutations was evaluated in its substrate range by sfGFP-UAG2 and sfGFP-UAG27 gene production (Supplementary Figures S25, S26). Thus, we decided to investigate N-PylRS and its four variants, N-PylRS-D1 and N-PylRS-L1–L3 (Supplementary Figures S27–S34). Noticeably, N-PylRS showed nearly 5.6 and 4.1 times higher fluorescent signals in charging BocK (1) and AlloK (2) than wt-PylRS in the sfGFP-UAG2 gene suppression yield (Figure 4A). In the sfGFP-UAG27 gene suppression study, N-PylRS was found to recognize ncAA 1–5. While AlloK (2) is still the best substrate, 3MeH (5) has a higher signal than BocK (1) (Figure 4B). In addition, N-PylRS was capable of incorporating S-benzyl cysteine analogs MbzlC (3) and MeObzlC (4) in low suppression efficiencies in sfGFP-UAG27 production. In contrast to PylRS-D1, the substrate specificity profiles of N-PylRS-D1 have revealed abolished fluorescence intensities in charging ncAA 1–5 in sfGFP-UAG2 and sfGFP-UAG27 suppression tests. The N-PylRS-L1–L3 variants showed reinstalled signals in sfGFP-UAG2 suppression with similar activities in charging BocK (1) and AlloK (2). The sfGFP-UAG27 gene suppression test in N-PylRS-L1–L3 had a gradual decrease in activity along with increasing linker length in charging ncAA 1–5.
ZRS was evolved from MmPylRS with Y306M/L309A/C348T/T364K/Y384F mutations at the active site (Wang et al., 2010). To validate the influence on the activity of ZRS by the R61K/H63Y/S193R mutations, as well as the introduction of the linker, these variations in the wt-PylRS study were transplanted and tested on ZRS, which was initially found to incorporate CbzK (6) and CbzK analogs (7, 10–11) (Scheme 1) (Wang Z.U. et al., 2012). Thus, ZRS variants, N-ZRS, ZRS-D1, ZRS-L1, ZRS-L2, and ZRS-L3, were constructed for evaluating their effects with active site mutation. The screening results of the substrate range in sfGFP-UAG2 and sfGFP-UAG27 suppression for the ZRS and the five variants are shown in Supplementary Figures S35–S46. NcAA 3–11 (Scheme 1) had a positive response and are illustrated in Figure 5.
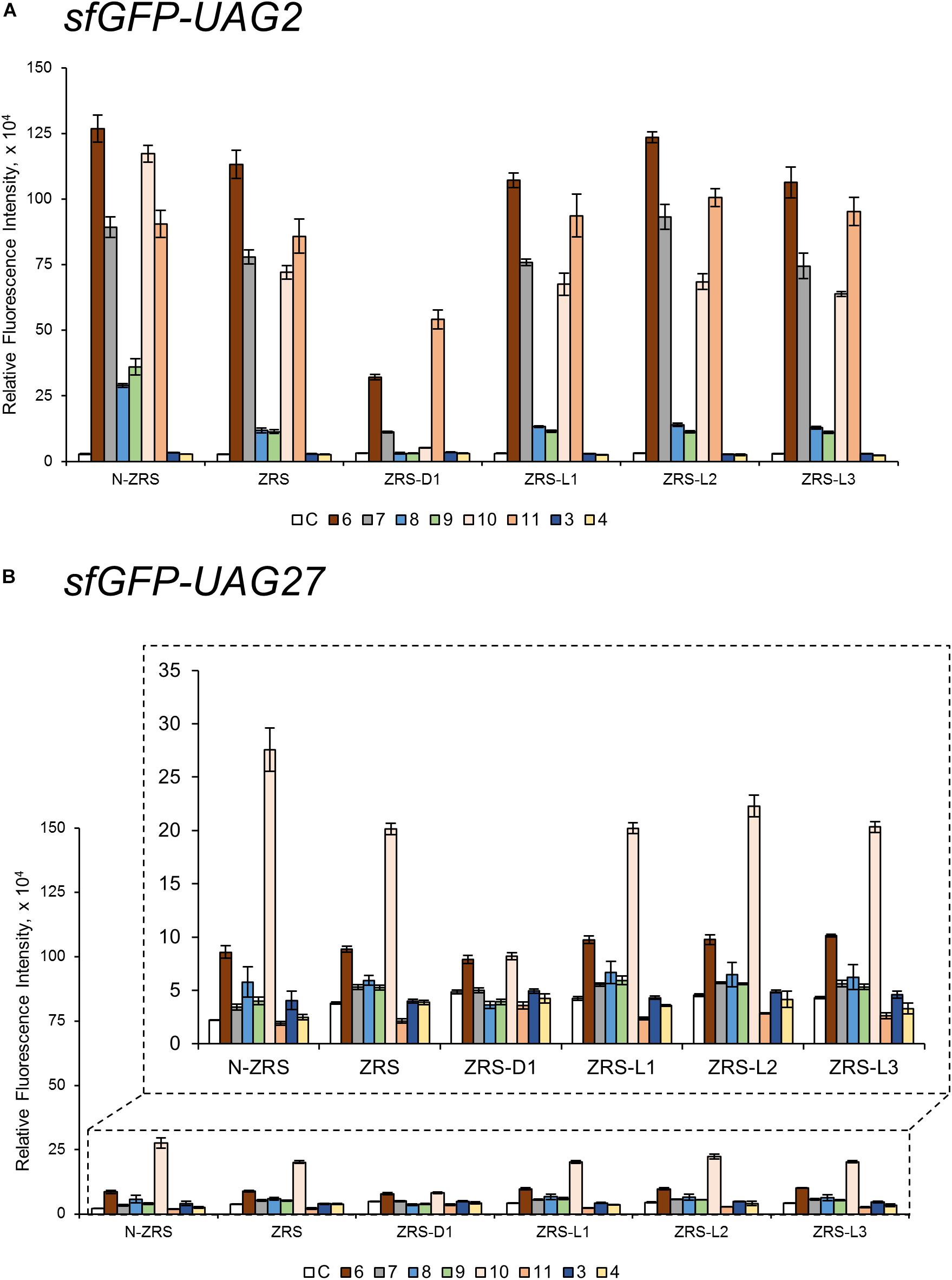
Figure 5. The sfGFP-UAG2 and sfGFP-UAG27 gene suppression efficiencies of ZRS enzyme variants. Incorporation efficiencies of ZRS variants (Figure 1B) as measured by the fluorescence intensities of sfGFP with amber mutation at position 2 (A) and 27 (B). The proteins were expressed in 1 mM ncAA and IPTG in GMML medium at 37°C for 12 h. The cells were excited at 485 nm and the fluorescence intensities were detected at 535 nm. The cell density was monitored by absorbance at 595 nm. “C” indicate the Control experiments of cells with the supplement of 1 mM IPTG; 3, 4, 6–11 denote the supplement of 1 mM IPTG and ncAA 3, 4, 6–11 (Scheme 1). Error bars represent the standard deviation of sfGFP production from four repeated experiments.
CbzK (6) was efficiently incorporated into sfGFP in response to the amber codon as reported for ZRS. The CbzK analogs, ClCbzK (7), SeCbzK (10), and CbzKOH (11) (Scheme 1), also showed similar intensity in the sfGFP-UAG2 suppression study. Two D-form CbzK analogs, DCbzK (8) and DClCbzK (9), were positive but had lower signals (Figure 5A). sfGFP-UAG27 suppression in ncAAs library screening of ZRS showed 12.8 times lower signal in charging CbzK compared to the sfGFP-UAG2 suppression study but was only 1.6 times lower in charging SeCbzK. The substrate range of N-ZRS remained the same as that of ZRS; nevertheless, the substrate specificity profiles of N-ZRS in the sfGFP-UAG2 suppression study showed a higher fluorescence intensity as compared to ZRS, especially in charging D-ncAA 8 and 9. Unexpectedly, S-benzyl cysteine analogs ncAA 3 and 4 were also incorporated into sfGFP with low efficiencies by N-ZRS in the sfGFP-UAG27 suppression study. The ZRS-D1 screening results showed a significant decrease in activity in the sfGFP-UAG2 and sfGFP-UAG27 suppression studies compared to ZRS and other ZRS variants. The ZRS-D1 activity with CbzKOH (11), however, was the best in the sfGFP-UAG2 screening and had diminished activity in the sfGFP-UAG27 screening (Figure 5). The addition of a linker did not raise the suppression efficiencies of ZRS albeit a slight increase in the fluorescence intensities of ZRS-L2 can be observed with ncAA 6 and 10 in response to sfGFP-UAG2 and sfGFP-UAG27 (Figure 5).
Some amber screening results show reverse or different intensity patterns, and western blotting analysis was used to confirm sfGFP protein yield to support the sfGFP fluorescence intensity results. N-ZRS and ZRS-D1 were chosen for analyzing sfGFP-UAG27 and sfGFP-UAG2 protein productions (Figure 6A). An anti-His tag antibody was used to detect the C-terminal-His tag present in full-length sfGFP, which indicates the amount of read-through at the amber stop codon. Western blotting analysis of N-ZRS/sfGFP-UAG27 indicates strong amber read-through for ncAA 6–8 and 10 and weaker response for ncAA 9 and 11 in SDS-PAGE and anti-His channel. Clearly, sfGFP-11 showed an additional band at lower molecular weight, near the 25-kDa protein marker in SDS-PAGE, which was also detected by western blotting (Figure 6A). These results are in partial agreement with the sfGFP fluorescent screening. They match for ncAA 3, 4, 9, and 10, not for ncAA 6–8 and 11 (Figure 5B). A ZRS-D1/sfGFP-UAG2 analysis (Figure 6A), however, matches well with sfGFP fluorescent screening results (Figure 5A), confirming that ZRS-D1 charges CbzKOH to generate acylated tRNA with high activity. The purified sfGFP-11∗ protein produced by the ZRS-D1⋅tRNAPyl pair generated two additional major mass peaks because of ester bond cleavage and Cbz group deprotection: 27,773 Da (without the N-terminal Met) and 27,639 Da (without the N-terminal Met and Cbz group) (Table 1 and Supplementary Figure S7). Purified sfGFP-6, sfGFP-7, and sfGFP-11, which were produced by N-ZRS⋅tRNAPyl pair, were analyzed by electrospray ionization-mass spectrometry (ESI-MS) (Table 1 and Figure 6B, Supplementary Figures S4–S6). The experimental mass of sfGFP-6 (28,086 and 27,955 Da) and sfGFP-7 (28,120, and 27,988 Da) matched well to the calculated molecular weight of sfGFP-6 (28,085 and 27,955 Da) and sfGFP-7 (28,120 and 27,989 Da). A matrix-assisted laser desorption/ionization-time of flight-tandem mass spectrometry (MALDI-TOF-MS/MS) analysis of peptide 27XSV30R fragments which encoded ncAA 6 and 7 further provides the correct molecular mass and sufficient b and y fragments to indicate ncAA purity at position 27 of the sfGFP (Table 1 and Supplementary Figures S10, S11). In characterizing the mass of sfGFP-11, the replacement of the amino group with a hydroxyl group in the main chain results in additional mass peaks. Three mass peaks, at 27,955, 27,820, and 25,036 Da, were observed in the sfGFP-11 mass spectrum. The calculated mass of full-length and truncated sfGFP-11 is 27,955 (-Met) and 25,320 Da, respectively. The full-length sfGFP-11 mass agrees with the calculated mass, and the peak at 27,820 Da matches the expected mass of sfGFP-11 with Cbz group deprotection. The mass of sfGFP truncated at 28 position, 25,036 Da, does not fully match the calculated mass 25,057 Da and instead shows a loss of 21 Da, which indicates CbzKOH deletion at 27 position of the truncated sfGFP-11 (Table 1 and Figure 6B, Supplementary Figure S6).
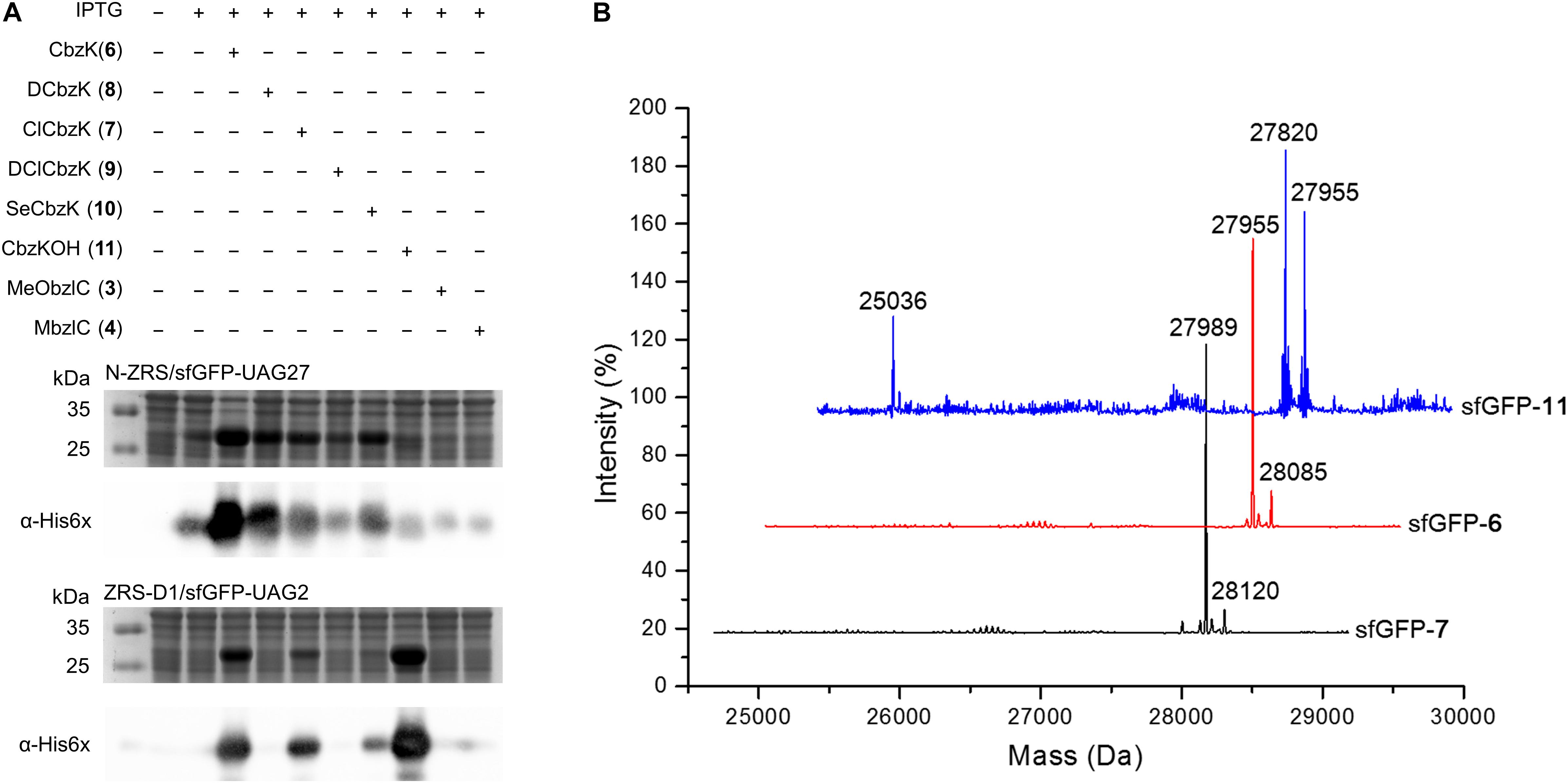
Figure 6. sfGFP production by ZRS variants and mass characterization. (A) Amber suppression of the sfGFP-UAG27 gene (N-ZRS⋅tRNAPyl pair) and the sfGFP-UAG2 gene (ZRS-D1⋅tRNAPyl pair) product with ncAA 3–4 and 6–11. The sfGFP proteins were produced in E. coli BL21 (DE3) coding N-ZRS⋅tRNAPyl or ZRS-D1⋅tRNAPyl pair with the supplement of 1 mM IPTG and ncAAs in GMML medium at 37°C for 12 h. The whole-cell lysate was analyzed by SDS-PAGE and western blotting by anti-His tag antibody indicated as α-His6X. (B) ESI-MS determination of sfGFP-UAG27 proteins with ncAA 6, 7, and 11. Full-length sfGFP-6 and sfGFP-7 proteins were produced by N-ZRS⋅tRNAPyl pair in E. coli BL21 (DE3) with the supplement of 1 mM IPTG and ncAA 6 or 7 in LB medium at 37°C for 12 h. Full-length sfGFP-11 proteins were produced with the same condition but with 1 mM ncAA 11 and in GMML minimal medium. The calculated molecular masses of sfGFP-7 are 28,121 and 27,989 Da (–Met); the observed molecular masses are 28,120 and 27,989 Da (–Met). The calculated molecular masses of sfGFP-6 are 28,085 and 27,954 Da (–Met); the observed molecular masses are 28,085 and 27,955 Da (–Met). The calculated molecular masses of sfGFP-11 are 28,086, 27,955 (–Met), and 25,320 Da (truncated sfGFP at 27 position); the observed molecular masses are 27,955 (–Met) and 27,820 Da (without Cbz group at 27 position and N-terminal Met residues) and 25,036 Da. The detailed electrospray and deconvoluted mass spectra are shown in Supplementary Figures S4–S6. ESI-MS determination of sfGFP-UAG2 protein. sfGFP-11* with ncAA 11 (ZRS-D1⋅tRNAPyl pair) is shown in Supplementary Figure S7.
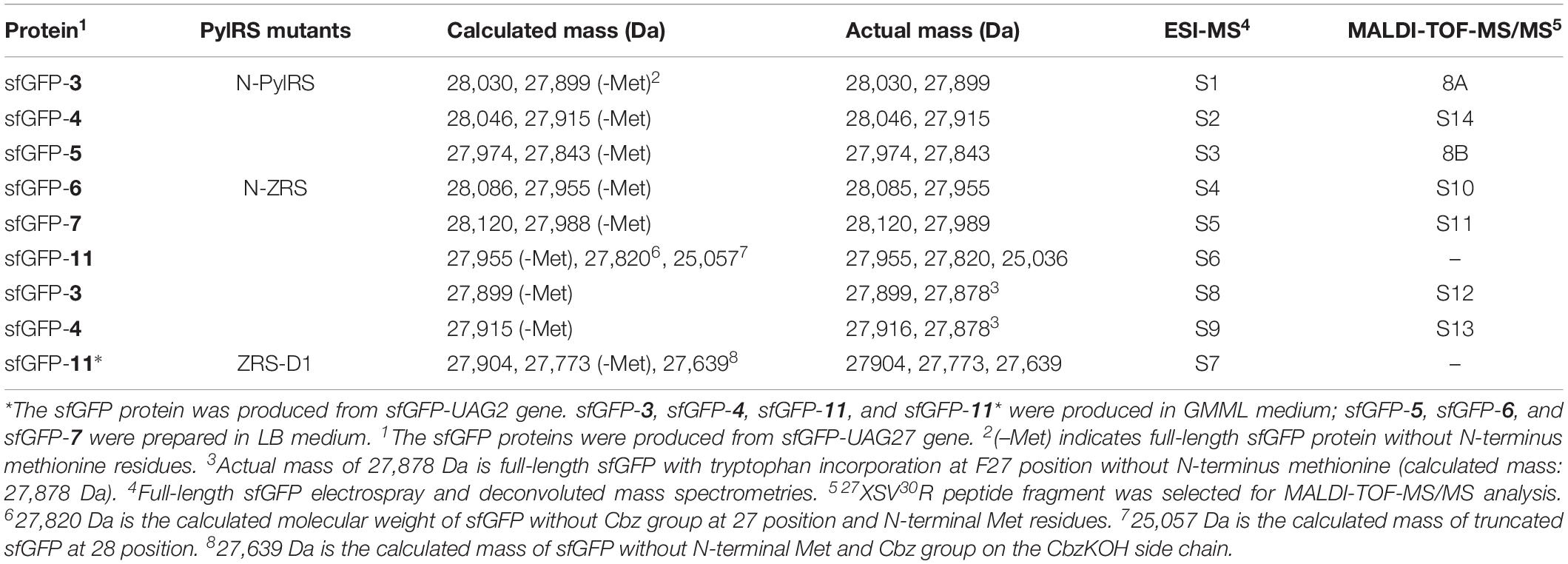
Table 1. Electrospray ionization-mass spectrometry (ESI-MS) analysis of sfGFP proteins.
The purified sfGFP-3 and sfGFP-4 proteins with ncAA 3 and 4 encoded at 27 position that were produced by the N-ZRS⋅tRNAPyl pair in E. coli were characterized, and they matched the calculated mass (Table 1 and Supplementary Figures S8, S9). The mass spectra of sfGFP-3 and sfGFP-4 show a molecular peak indicating tryptophan incorporation, 27,878 Da. A MALDI-TOF-MS/MS analysis of sfGFP-3 and sfGFP-4 also confirmed the presence of ncAA 3 and 4 at position 27 of sfGFP. To confirm the ncAAs screening results mentioned in the N-PylRS⋅tRNAPyl pair study, sfGFP-3, sfGFP-4, and sfGFP-5 were expressed and purified, respectively, and then subjected to ESI-MS analysis (Table 1 and Supplementary Figures S1–S3). The calculated molecular weight of sfGFP-3, sfGFP-4, and sfGFP-5, generated by N-PylRS, matched the observed mass in the full-length sfGFP (Figure 7). The incorporation of ncAA 3, 4, and 5 into sfGFP at position 27 was shown by MALDI-TOF-MS/MS (Figure 8 and Supplementary Figures S12–S14). This multiple evidence indicates that MbzlC (3), MeObzlC (4), and 3MeH (5) are incorporated into proteins by non-active, mutated N-PylRS⋅tRNAPyl pair site-specifically.
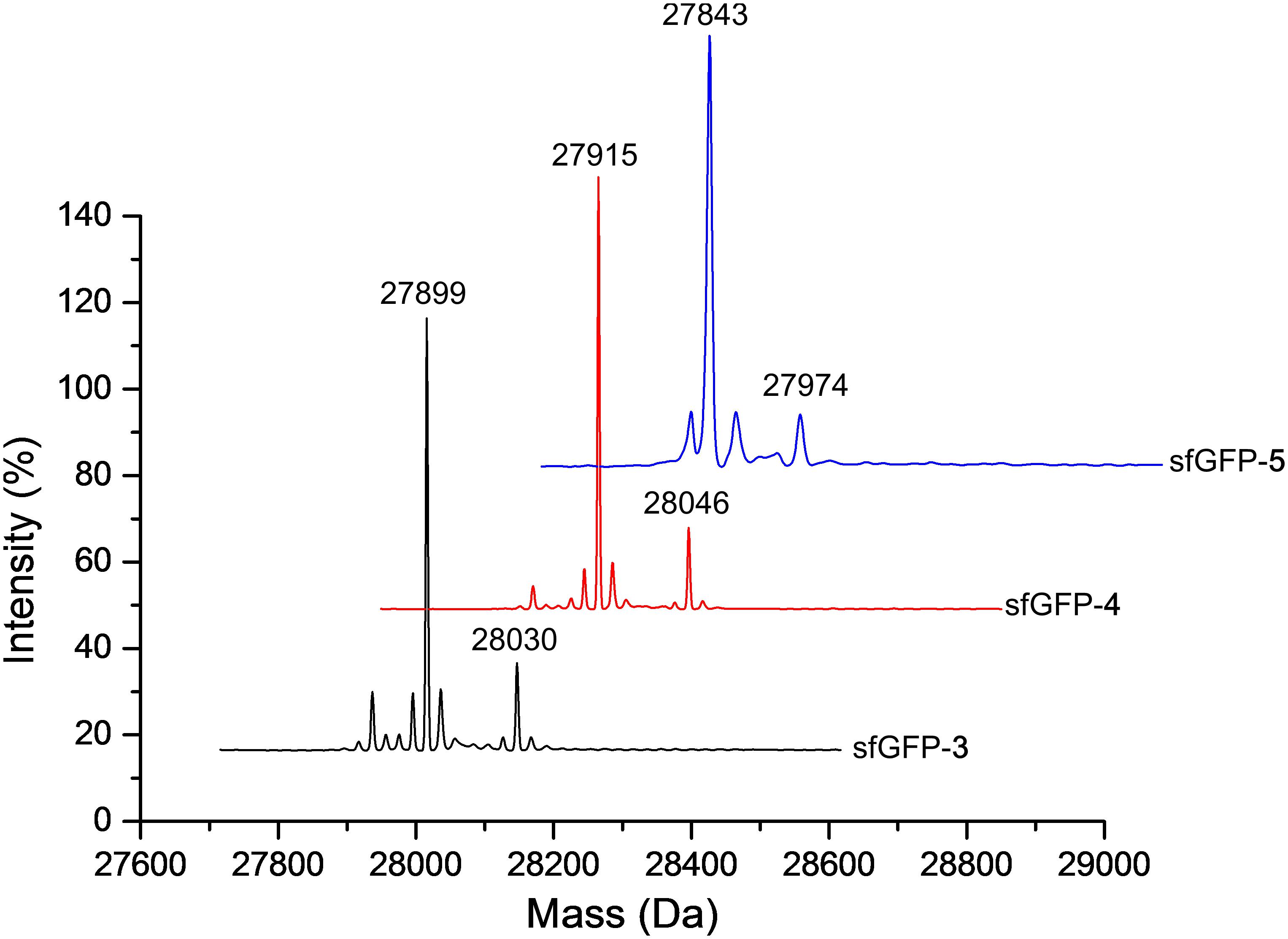
Figure 7. Deconvoluted ESI-MS spectra of sfGFP containing ncAAs at position 27. Full-length sfGFP-3, sfGFP-4, and sfGFP-5 were expressed using the N-PylRS⋅tRNAPyl pair in the presence of ncAA 3, 4 (2 mM) or 5 (1 mM), respectively, in GMML medium (sfGFP-3 and sfGFP-4) or LB medium (sfGFP-5). The purified protein samples were subjected to ESI-MS for identification of the incorporation of defined ncAA. Deconvoluted ESI-MS of ncAA 3–5 incorporated sfGFP at position 27 are shown above. sfGFP-X in which X represents one of the ncAA 3–5 was incorporated into sfGFP at position 27. The calculated molecular masses of sfGFP-3 are 28,030 and 27,899 Da (-Met); the observed molecular masses are 28,030 and 27,899 Da (-Met). The calculated molecular masses of sfGFP-4 are 28,046 and 27,915 Da (-Met); the observed molecular masses are 28,046 and 27,915 Da (-Met). The calculated molecular masses of sfGFP-5 are 27,974 and 27,843 Da (-Met); the observed molecular masses are 27,974 and 27,843 Da (-Met). The detailed electrospray and deconvoluted mass spectra are shown in Supplementary Figures S1–S3.
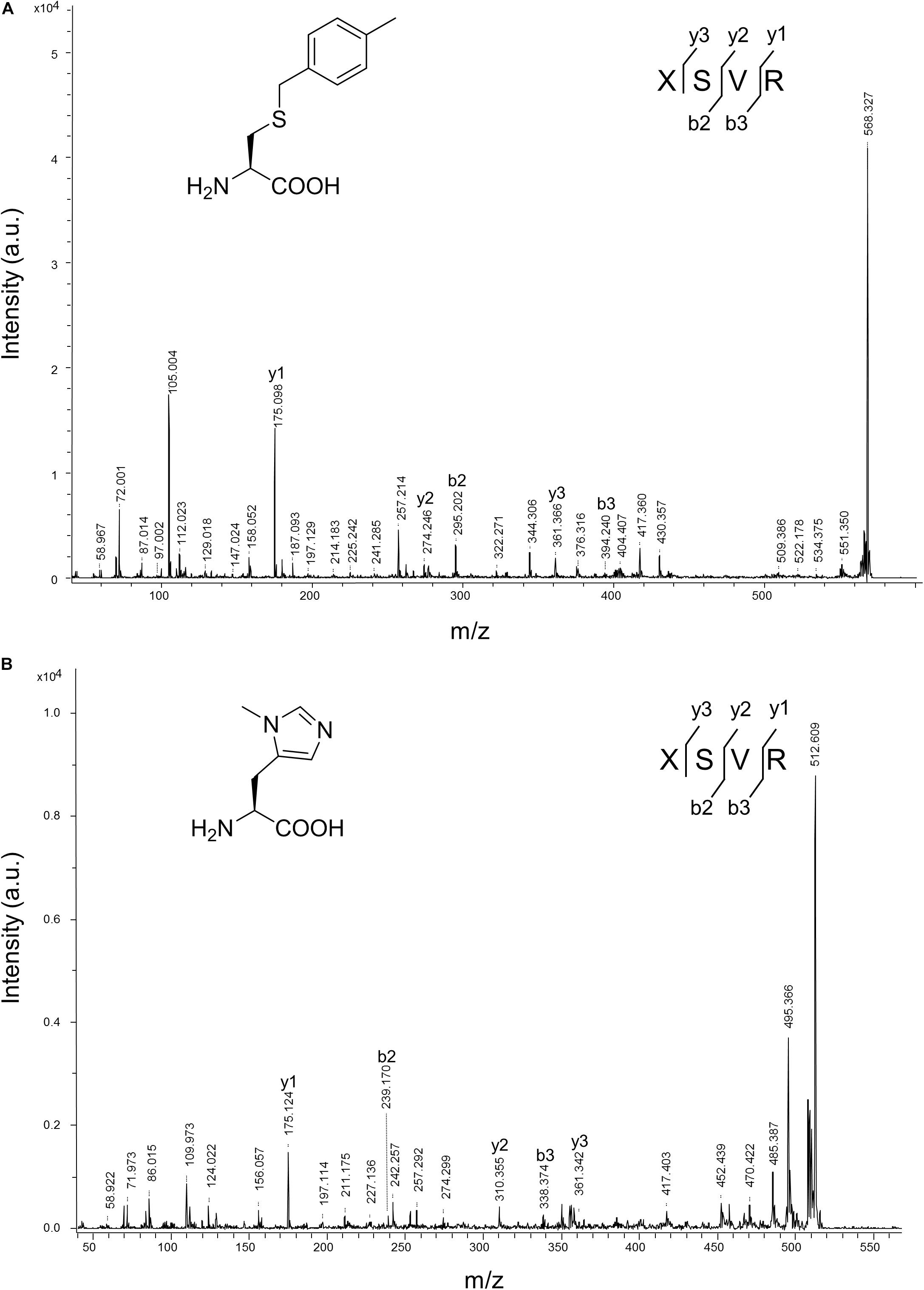
Figure 8. MALDI-TOF-MS/MS analysis of sfGFP-3 and sfGFP-5. The sfGFP-3 and sfGFP-5 proteins were in-gel digested with trypsin and subjected to MALDI-TOF-MS/MS analysis. (A) The tandem mass spectrum of the X(3)SVR; X denotes a fragment from sfGFP-3, with ncAA 3 incorporated at position 27. The calculated molecular mass of the X(3)SVR peptide fragment is 567.284 Da and the actual molecular mass is 568.327 Da. Full-length sfGFP-3 was expressed using N-PylRS in the presence of 2 mM ncAA 3 in GMML medium. (B) The tandem mass spectrum of the X(5)SVR; X denotes a fragment from sfGFP-5 with ncAA 5 incorporated at position 27. The calculated molecular mass of the X(5)SVR peptide fragment is 511.287 Da and the actual molecular mass is 512.609 Da. Full-length sfGFP-5 was produced using N-PylRS⋅tRNAPyl pair in the presence of 1 mM ncAA 5 in LB medium.
In this study, we showed that the R61K/H63Y/S193R mutations, which are located beyond the catalytic site, increase the suppression efficiencies of PylRS and ZRS toward their preferred substrates. The substrate range of N-PylRS allowed the incorporation of ncAA 3–5, two S-benzyl cysteine analogs and a histidine analog, with high efficiency as compared to wt-PylRS. This suggests that the remote mutations tune the interaction between PylRS and tRNAPyl, which leads to the successful incorporation of ncAA 3–5 despite the active site engineering of PylRS (Xiao et al., 2014; Sharma et al., 2016). A similar tuning effect has been observed in PylRS N-terminal mutations by global-directed evolution and PANCE, which results in enhancement of enzyme efficiencies (Sharma et al., 2018). We chose R61 and H63 of PylRS as they are clustered on the loop adjacent to the extremity of the α-helix. Specifically, the guanidinium side-chain of R61 has a polar interaction with the phosphodiester backbone near 58A of tRNAPyl as well as the R52 residue within the NTD. In addition, the imidazole side-chain of H63 hydrogen bonds with the amino group on the side chain of K85 (Figure 2). The R61K and H63Y mutations may disrupt these interactions. The S193 is located on the tRNA binding domain of the PylRS CTD. Although S193 is relatively far from the NTD, it does not interact with residues alongside the catalytic site. We envision that the S193R mutation moderately tunes the binding between PylRS and tRNAPyl (Figure 3). By placing the R61K/H63Y/S193R mutations on wt-PylRS, the substrate range was shifted to introduce ncAA 3–5. We postulate that the catalytic core of wt-PylRS expands locally to harbor ncAA with shorter bulky side-chains owing to the sophisticated regulation with the binding of tRNAPyl. The ZRS shows its best incorporation efficiencies toward ncAA 6, and it was also found to incorporate ncAA 7, 3, and 4 into sfGFP based on the screening results. Unanticipatedly, the substrate range of N-ZRS remained the same as that of ZRS. The fine-tuning effect of the mutations seen with wt-PylRS is not seen with ZRS, which might result from its tightly regulated substrate range of the evolved catalytic pocket. This result also implies the current directed-evolution approach at the PylRS active site which leads to a stabilized local arrangement that prevents it from the tuning effects of N-terminal mutations. This also explains no significant activity enhancement in introducing a linker to N-PylRS and ZRS. Although no significant improvement of an introduced linker was found, the repeated peptide sequence, (GGGGS)1–3, could be evolved to explore additional interaction with tRNAPyl for altering substrate range and activity enhancement. Importantly, the D-form ncAA 8 and 9 gave small but positive signals in ZRS and a yield enhancement in N-ZRS based on fluorescence screening and western blotting analysis. ZRS-D1 with separated NTD and CTD seems tightening up the chiral specificity by releasing the structural remote effects, which results in a losing activity in charging the D-form ncAAs 8 and 9 (Figures 5, 6A).
Two different sfGFP proteins, namely, sfGFP-UAG2 and sfGFP-UAG27, were used in this study to compare the incorporation efficiencies of the PylRS variants. Our studies have proven that the former fluorescence intensity of lysine derivatives was considerably stronger than the latter one. This indicates that the serine 2 of sfGFP located on the flexible loop might be suitable for the long and polar side-chains of ncAAs. On the contrary, the latter substrate range of amino acids with bulky and aromatic side-chain derivatives were easier to be observed than the former one. However, western blotting analysis of N-ZRS in producing the sfGFP-UAG27 proteins by installing ncAA 6–9 (Figure 6A) shows reverse intensity in fluorescent screening. This suggests that the sfGFP variants containing these ncAAs have lost their folded structure, resulting in fluorescent quenching by penetrating water molecules or protein precipitation. Surprisingly, sfGFP-10 has a higher fluorescent signal than would be expected due to the western-blotting results. This may be due to a smaller bond angle at the selenium atom that causes a bended side-chain and stabilizes the sfGFP β-barrel structure. The characterization of sfGFP-11 and sfGFP-11∗, which are produced by N-ZRS and ZRS-D1, also indicate ester bond hydrolysis and partial Cbz group deprotection.
Various PylRS have been discovered in nature to catalyze the acylation reactions between pyrrolysine and tRNAPyl. Previous studies have shown that PylRS can be expressed in the form of either a single polypeptide (MmPylRS or MbPylRS) or separated polypeptides (DhPylRS) (Jiang and Krzycki, 2012). ΔNPylRS is a group of PylRS which lacks the NTD but which shows decent suppression efficiencies in vivo (Willis and Chin, 2018). Our findings demonstrate that the separated polypeptide of MmPylRS, PylRS-D1, still retains the lower catalytic activity, but this was not the case for N-PylRS-D1. Noticeably, the substrate specificity profiles of N-PylRS-D1 showed a decrease in activity as compared to PylRS-D1. The abolished activity of N-PylRS-D1 implies that the R61K/H63Y/S193R mutations need a linkage between the NTD and the CTD to transfer the remote effect in improving enzyme activity. In addition, our results point out the indispensability of the unstructured flexible loop connecting the NTD and the CTD of PylRS. The separately expressed NTD and CTD of MmPylRS are believed to be functionally reassembled in cells according to the substrate specificity profiles of PylRS-D1, showing the reasonable decrease in activity toward the same substrates in contrast to wt-PylRS. Recombining the linker between the junctions has conspicuously restored the activity of N-PylRS-D1. This suggests that the tuning effect of R61K/H63Y/S193R between PylRS and tRNAPyl could also mutually regulate the interplay between the NTD and the CTD and that the truncation of the flexible loop might perturb their binding mode, leading to a reduction in activity. Increasing the length of a linker leads to an upward trend in the suppression efficiencies of wt-PylRS, whereas this effect is not observed with ZRS. These findings lead us to believe that the identity of the residues in the linker affects PylRS activity via tuning flexibility interactions involving the NTD and the CTD or providing beneficial interactions between linker residues and tRNA.
Introduction of R61K/H63Y/S193R mutations to wt-PylRS has altered its substrate range for the incorporation of ncAA 3–5, which, we propose, arises from the refinement of the dynamics between PylRS and tRNAPyl. The addition of a linker provides extra flexibility to potentially regulate the interaction between NTD and CTD of PylRS and changes the interface between PylRS and tRNAPyl, in turn enhancing the suppression efficiencies. Our findings not only indicate the sophisticated interaction between NTD and CTD of PylRS and tRNAPyl but also reveal the importance of N-terminus engineering in exploring the novel substrate range and improving the suppression efficiency.
All datasets generated for this study are included in the article/Supplementary Material.
Y-SW: conceptualization, resources, supervision, project administration, and funding acquisition. H-KJ, J-CT, K-WC, H-WT, K-PC, Y-KL, and Y-SW: methodology. Y-SW and H-KJ: validation. H-KJ and M-NL: formal analysis. H-KJ, M-NL, and Y-SW: data curation. H-KJ: writing – original draft preparation. Y-SW, Y-KL, and H-KJ: writing – review and editing.
This research was funded by the Taiwan Protein Project (AS-KPQ-105-TPP) and the Ministry of Science and Technology (MOST), Taiwan (MOST 107-2113-M-001-025-MY3). Y-SW was supported by the Award for Supporting Talent Junior Faculty, MOST and Academia Sinica.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Shu-Yu Lin for the assistance in full-length protein mass spectrometry analysis and data processing that were acquired at the Academia Sinica Common Mass Spectrometry Facilities for the proteomics and protein modification analysis located at the Institute of Biological Chemistry, Academia Sinica, supported by Academia Sinica Core Facility and Innovative Instrument Project (AS-CFII-108-107). We also thank Cheng-Hsilin Hsieh for the technical assistance in MALDI-TOF/TOF mass spectrometry from Genomics Core at the Institute of Molecular Biology, Academia Sinica. We are grateful to Prof. Todd L. Lowary (Institute of Biological Chemistry, Academia Sinica) for critical discussion and helpful suggestions. We thank AS-TIGP for supporting H-KJ with AS-TIGP Research Performance Fellowship Award.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00235/full#supplementary-material
Ambrogelly, A., Gundllapalli, S., Herring, S., Polycarpo, C., Frauer, C., and Söll, D. (2007). Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc. Natl. Acad. Sci. U.S.A. 104, 3141–3146. doi: 10.1073/pnas.0611634104
Cavarelli, J., Delagoutte, B., Eriani, G., Gangloff, J., and Moras, D. (1998). L-arginine recognition by yeast arginyl-tRNA synthetase. EMBO J. 17, 5438–5448. doi: 10.1093/emboj/17.18.5438
Guo, L. T., Wang, Y. S., Nakamura, A., Eiler, D., Kavran, J. M., Wong, M., et al. (2014). Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc. Natl. Acad. Sci. U. S. A. 111, 16724–16729. doi: 10.1073/pnas.1419737111
Han, S., Yang, A., Lee, S., Lee, H.-W., Park, C. B., and Park, H.-S. (2017). Expanding the genetic code of Mus musculus. Nat. Commun. 8:14568. doi: 10.1038/ncomms14568
Hao, B., Gong, W., Ferguson, T. K., James, C. M., Krzycki, J. A., and Chan, M. K. (2002). A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science 296, 1462–1466. doi: 10.1126/science.1069556
Herring, S., Ambrogelly, A., Polycarpo, C. R., and Soll, D. (2007). Recognition of pyrrolysine tRNA by the Desulfitobacterium hafniense pyrrolysyl-tRNA synthetase. Nucleic Acids Res. 35, 1270–1278. doi: 10.1093/nar/gkl1151
Jaric, J., Bilokapic, S., Lesjak, S., Crnkovic, A., Ban, N., and Weygand-Durasevic, I. (2009). Identification of amino acids in the N-terminal domain of atypical methanogenic-type seryl-tRNA synthetase critical for tRNA recognition. J. Biol. Chem. 284, 30643–30651. doi: 10.1074/jbc.M109.044099
Jiang, R. S., and Krzycki, J. A. (2012). PylSn and the homologous N-terminal domain of pyrrolysyl-tRNA synthetase bind the tRNA that is essential for the genetic encoding of pyrrolysine. J. Biol. Chem. 287, 32738–32746. doi: 10.1074/jbc.M112.396754
Kavran, J. M., Gundllapalli, S., O’Donoghue, P., Englert, M., Soll, D., and Steitz, T. A. (2007). Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. U.S.A. 104, 11268–11273. doi: 10.1073/pnas.0704769104
Li, Y.-M., Yang, M.-Y., Huang, Y.-C., Li, Y.-T., Chen, P. R., and Liu, L. (2012). Ligation of expressed protein α-hydrazides via genetic incorporation of an α-hydroxy acid. ACS Chem. Biol. 7, 1015–1022. doi: 10.1021/cb300020s
Mukai, T., Kobayashi, T., Hino, N., Yanagisawa, T., Sakamoto, K., and Yokoyama, S. (2008). Adding l-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochem. Biophys. Res. Commun. 371, 818–822. doi: 10.1016/j.bbrc.2008.04.164
Mukai, T., Yamaguchi, A., Ohtake, K., Takahashi, M., Hayashi, A., Iraha, F., et al. (2015). Reassignment of a rare sense codon to a non-canonical amino acid in Escherichia coli. Nucleic Acids Res. 43, 8111–8122. doi: 10.1093/nar/gkv787
Neumann, H., Peak-Chew, S. Y., and Chin, J. W. (2008). Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 4, 232–234. doi: 10.1038/nchembio.73
Nozawa, K., O’Donoghue, P., Gundllapalli, S., Araiso, Y., Ishitani, R., Umehara, T., et al. (2009). Pyrrolysyl-tRNA synthetase-tRNA(Pyl) structure reveals the molecular basis of orthogonality. Nature 457, 1163–1167. doi: 10.1038/nature07611
O’Donoghue, P., and Luthey-Schulten, Z. (2003). On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol. Mol. Biol. Rev. 67, 550–573. doi: 10.1128/mmbr.67.4.550-573.2003
O’Donoghue, P., Prat, L., Heinemann, I. U., Ling, J., Odoi, K., Liu, W. R., et al. (2012). Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 586, 3931–3937. doi: 10.1016/j.febslet.2012.09.033
Schimmel, P., Giege, R., Moras, D., and Yokoyama, S. (1993). An operational RNA code for amino acids and possible relationship to genetic code. Proc. Natl. Acad. Sci. U.S.A. 90, 8763–8768. doi: 10.1073/pnas.90.19.8763
Sharma, V., Wang, Y.-S., and Liu, W. R. (2016). Probing the catalytic charge-relay system in alanine racemase with genetically encoded histidine mimetics. ACS Chem. Biol. 11, 3305–3309. doi: 10.1021/acschembio.6b00940
Sharma, V., Zeng, Y., Wang, W. W., Qiao, Y., Kurra, Y., and Liu, W. R. (2018). Evolving the N-terminal domain of Pyrrolysyl-tRNA synthetase for improved incorporation of noncanonical amino acids. ChemBioChem 19, 26–30. doi: 10.1002/cbic.201700268
Shimada, A., Nureki, O., Goto, M., Takahashi, S., and Yokoyama, S. (2001). Structural and mutational studies of the recognition of the arginine tRNA-specific major identity element, A20, by arginyl-tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 98, 13537–13542. doi: 10.1073/pnas.231267998
Suzuki, T., Miller, C., Guo, L. T., Ho, J. M. L., Bryson, D. I., Wang, Y. S., et al. (2017). Crystal structures reveal an elusive functional domain of pyrrolysyl-tRNA synthetase. Nat. Chem. Biol. 13, 1261–1266. doi: 10.1038/nchembio.2497
Vargas-Rodriguez, O., Sevostyanova, A., Soll, D., and Crnkovic, A. (2018). Upgrading aminoacyl-tRNA synthetases for genetic code expansion. Curr. Opin. Chem. Biol. 46, 115–122. doi: 10.1016/j.cbpa.2018.07.014
Wang, Y.-S. (2012). Expanding Genetic Code for Protein Lysine and Phenylalanine Modifications. Ph.D. thesis, Texas A&M University, College Station, TX.
Wang, Y. S., Fang, X., Wallace, A. L., Wu, B., and Liu, W. R. (2012). A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. J. Am. Chem. Soc. 134, 2950–2953. doi: 10.1021/ja211972x
Wang, Z. U., Wang, Y. S., Pai, P. J., Russell, W. K., Russell, D. H., and Liu, W. R. (2012). A facile method to synthesize histones with posttranslational modification mimics. Biochemistry 51, 5232–5234. doi: 10.1021/bi300535a
Wang, Y.-S., Wu, B., Wang, Z., Huang, Y., Wan, W., Russell, W. K., et al. (2010). A genetically encoded photocaged Nε-methyl-l-lysine. Mol. Biosyst. 6, 1557–1560. doi: 10.1039/C002155E
Willis, J. C. W., and Chin, J. W. (2018). Mutually orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs. Nat. Chem. 10, 831–837. doi: 10.1038/s41557-018-0052-5
Xiao, H., Peters, F. B., Yang, P. Y., Reed, S., Chittuluru, J. R., and Schultz, P. G. (2014). Genetic incorporation of histidine derivatives using an engineered pyrrolysyl-tRNA synthetase. ACS Chem. Biol. 9, 1092–1096. doi: 10.1021/cb500032c
Yanagisawa, T., Ishii, R., Fukunaga, R., Kobayashi, T., Sakamoto, K., and Yokoyama, S. (2008). Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(epsilon)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 15, 1187–1197. doi: 10.1016/j.chembiol.2008.10.004
Keywords: non-canonical amino acids, pyrrolysyl-tRNA synthetase, linker engineering, amber codon suppression, tRNA binding domain
Citation: Jiang H-K, Lee M-N, Tsou J-C, Chang K-W, Tseng H-W, Chen K-P, Li Y-K and Wang Y-S (2020) Linker and N-Terminal Domain Engineering of Pyrrolysyl-tRNA Synthetase for Substrate Range Shifting and Activity Enhancement. Front. Bioeng. Biotechnol. 8:235. doi: 10.3389/fbioe.2020.00235
Received: 14 January 2020; Accepted: 06 March 2020;
Published: 07 April 2020.
Edited by:
Chenguang Fan, University of Arkansas, United StatesReviewed by:
Wenshe R. Liu, Texas A&M University, United StatesCopyright © 2020 Jiang, Lee, Tsou, Chang, Tseng, Chen, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yane-Shih Wang, eWFuZXNoaWh3YW5nQGdhdGUuc2luaWNhLmVkdS50dw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.