 Priscilla S. Briquez1*†
Priscilla S. Briquez1*† Sylvie Hauert1†
Sylvie Hauert1† Alexandre de Titta2†
Alexandre de Titta2† Laura T. Gray1
Laura T. Gray1 Aaron T. Alpar1Melody A. Swartz1,3,4
Aaron T. Alpar1Melody A. Swartz1,3,4 Jeffrey A. Hubbell1,4*
Jeffrey A. Hubbell1,4*- 1Pritzker School of Molecular Engineering, The University of Chicago, Chicago, IL, United States
- 2Agap2 Zürich, Zurich, Switzerland
- 3Ben May Department of Cancer Research, The University of Chicago, Chicago, IL, United States
- 4Committee on Immunology, The University of Chicago, Chicago, IL, United States
Therapeutic cancer vaccines constitute a valuable tool to educate the immune system to fight tumors and prevent cancer relapse. Nevertheless, the number of cancer vaccines in the clinic remains very limited to date, highlighting the need for further technology development. Recently, cancer vaccines have been improved by the use of materials, which can strongly enhance their intrinsic properties and biodistribution profile. Moreover, vaccine efficacy and safety can be substantially modulated through selection of the site at which they are delivered, which fosters the engineering of materials capable of targeting cancer vaccines to specific relevant sites, such as within the tumor or within lymphoid organs, to further optimize their immunotherapeutic effects. In this review, we aim to give the reader an overview of principles and current strategies to engineer therapeutic cancer vaccines, with a particular focus on the use of site-specific targeting materials. We will first recall the goal of therapeutic cancer vaccination and the type of immune responses sought upon vaccination, before detailing key components of cancer vaccines. We will then present how materials can be engineered to enhance the vaccine’s pharmacokinetic and pharmacodynamic properties. Finally, we will discuss the rationale for site-specific targeting of cancer vaccines and provide examples of current targeting technologies.
Introduction
Cancer ranks as the second leading cause of global deaths, according to the World Health Organization, with nearly 15% of people dying from it (World Health Organization, 2018). More alarmingly, the rate of cancer incidence is increasing and is expected to reach more than 20 million newly diagnosed cases per year and 13 million cancer-related deaths in 2030 (American Cancer Society, 2018; World Health Organization, 2018). Among the various types of cancer, the most prevalent are lung, liver, colorectal, stomach and breast cancers. While cancer can affect any part of the body and is very heterogeneous between patients, most malignant tumors share biological similarities – defined as the “hallmarks of cancer” (Hanahan and Weinberg, 2011) – which help researchers break down the disease complexity and subsequently guide them toward the development of effective cancer therapies.
Currently, surgery, radiotherapy and chemotherapy remain the first-lines of cancer treatments that are prescribed as a single therapy or in combination, in a patient-tailored fashion that depends on the tumor characteristics (e.g., type, stage, aggressiveness and accessibility), as well as the patient’s symptoms and health conditions (National Institutes of Health of the USA, 2019). In recent years, immunotherapies have emerged as highly promising treatments to educate the patient’s immune system to efficiently fight its own cancer cells. The most successful clinical cancer immunotherapies that have received United States FDA approval to date include adoptive T cells therapies, immunomodulatory therapies, targeted cancer therapies, oncolytic virus therapies and cancer vaccines, as detailed in Table 1. Some of these treatments are currently being established as first-line treatments in cases of advanced cancers (Peters et al., 2019), highlighting the strong clinical potential of such immunotherapies. Furthermore, a multitude of novel immunotherapeutic compounds are currently being tested in clinical trials, foreseeing a fast evolution of the cancer therapy landscape in upcoming years.
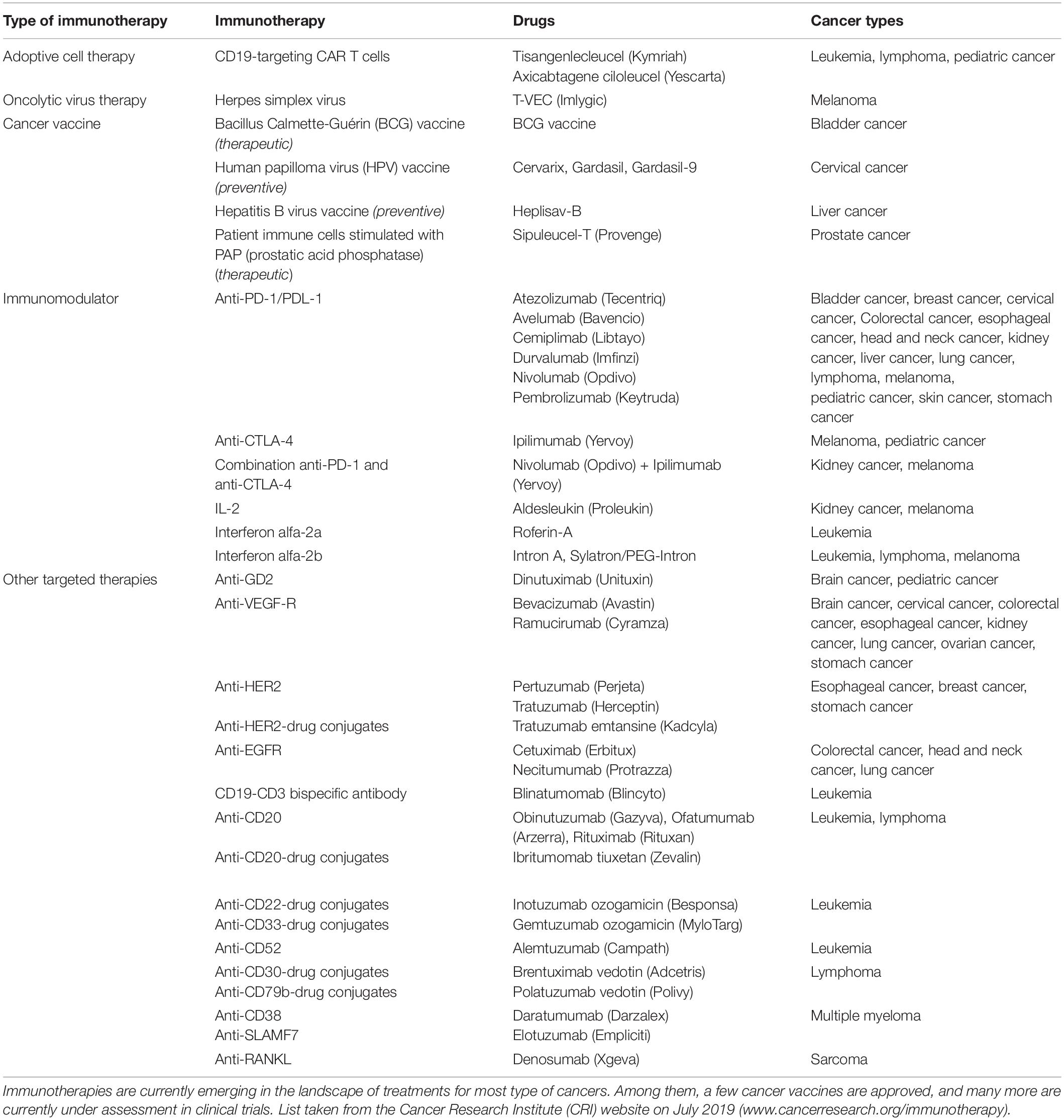
Table 1. Current approved United States FDA immunotherapies.
Among these immunotherapies, vaccines aim at reducing cancer occurrence by preventing cancer-causing infections, in the case of prophylactic vaccines, or at developing strong host immune reactions and subsequent immune memory to efficiently eradicate primary tumor cells and metastasis, in the case of therapeutic vaccines. Therapeutic vaccines hold great promises for long-term remission in patients, in that they can install immunological memory directed against the tumor. Unfortunately, despite extensive research, only four cancer vaccines have made it into the clinic to date: two prophylactic ones, the human papilloma virus vaccine and the hepatitis B virus vaccine for prevention of cervical and liver cancer respectively, and two therapeutic ones, namely the Bacillus Calmette-Guérin (BCG) vaccine, which reduces relapse and metastasis of early stage bladder cancers, and Sipuleucel-T, a cell-based vaccine for advanced prostate cancer (DeMaria and Bilusic, 2019). Therefore, important additional efforts are required to achieve the high expectations of especially therapeutic cancer vaccines at the bedside.
Indeed, the next generation of therapeutic cancer vaccines will necessitate improvements both in terms of vaccine compositions and delivery strategies. In this review, we will discuss how such improvements could be achieved via materials engineering. Furthermore, while vaccination can lead to toxicity concerns due to strong systemic activation of the patient’s immune system, we will present how engineering of targeting materials can enhance the safety profile of vaccines by localizing their effects to specific sites. Therefore, here we aim at providing the reader with design considerations, current challenges and lines of thoughts for the development of potent and safe site-specific targeting therapeutic cancer vaccines. Because very few of these vaccines have been developed to date, this review will take selected examples of targeting cancer immunotherapies – not only vaccines – to highlight possible engineering strategies that can be further applied to vaccination.
Which Type of Immune Reactions Should Therapeutic Cancer Vaccines Induce?
Engineering an optimal therapeutic cancer vaccine requires a good understanding of the type of immune reactions needed to eradicate tumors. Ideally, the vaccine should elicit potent immune responses that specifically recognize and eliminate all tumor cells present in the body, including those in the primary tumor, in circulation and in metastatic lesions. These immune responses should therefore be cancer cell antigen-specific to limit unwanted systemic side effects and prevent adverse autoimmune reactions. In addition, the vaccine should induce a strong immune memory against the cancer cells, able to efficiently reactivate anti-tumor immunity upon detection of cancer relapse, which is necessary to achieve long-term disease remission. In fact, cancer mortality has been largely imputed to relapses rather than to the primary tumor (Mehlen and Puisieux, 2006). As specificity and memory are hallmarks of the adaptive immune system, therapeutic cancer vaccines aim at activating endogenous cellular or humoral mechanisms of adaptive immunity against tumors.
The most common strategy exploited for the development of therapeutic cancer vaccines relies on the generation of endogenous cancer-specific cytotoxic CD8+ T cells (cytotoxic T lymphocytes, CTLs), due to their unique ability to kill cancer cells upon specific recognition (Farhood et al., 2018). This recognition is mediated by the T cell receptor (TCR) of CTLs that can bind to cancer antigen epitopes mounted on the major histocompatibility complex class I (MHCI) displayed at the cancer cell surface. TCR-signaled CTLs can then induce cancer cell death via multiple pathways, including by degranulation, which releases perforin (PRF)/granzyme B (GZMB), or by upregulation of FasL or TRAIL that signal cancer cell apoptotic pathways (Figure 1A).
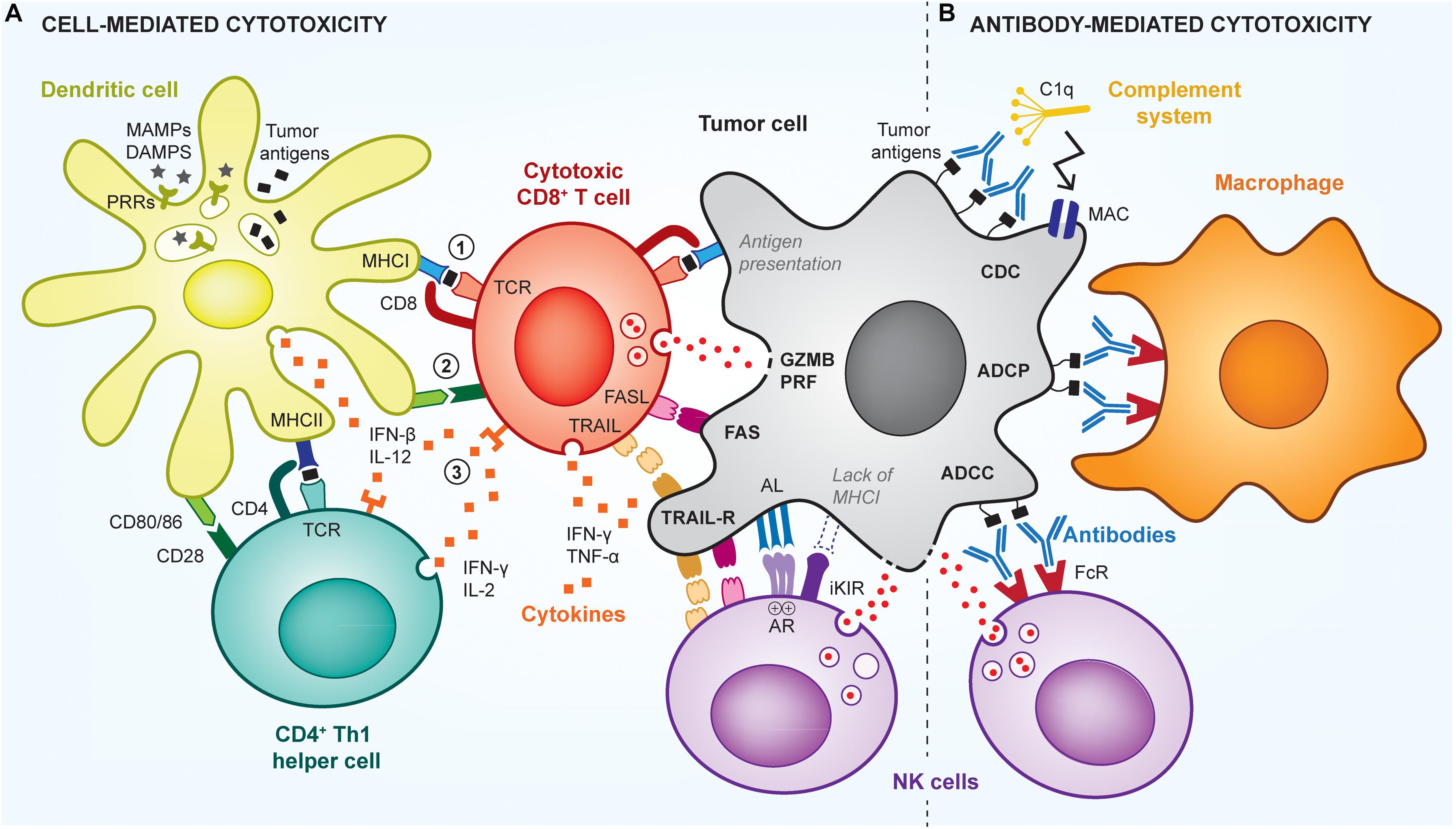
Figure 1. Cell- and antibody-mediated cytotoxic mechanisms of anti-tumor immunity. The immune system uses multiple mechanisms to efficiently kill tumor cells, via cytotoxic CD8+ T cells (CTLs), NK cells, or antibody-mediated mechanisms. (A) To be activated, T cells need 3 signals from antigen-presenting cells (APCs; e.g., dendritic cell): signal (1) is the presentation of cancer antigens (black squares) via MHC complexes; signal (2) is the signaling induced by co-stimulatory receptors (e.g., CD80/86), which are expressed by the APCs in the presence of adjuvants (e.g., MAMPs/DAMPs); and signal (3) is the stimulation by pro-inflammatory cytokines. Such cytokines are produced by APCs as well as by CD4+ Th1 helper cells that further enhance activation of CTLs. Upon activation and recognition of tumor cells, CTLs can induce their death via various pathways, including perforin (PRF)/granzyme B (GZMB), FasL-Fas, TRAIL or inflammatory cytokines. Using similar pathways, NK cells can kill cancer cells that have downregulated their MHCI expression and fail to signal through the inhibitory killer immunoglobulin-like receptor (iKIR), or that overstimulate NK activating receptors (AR). (B) Tumors cells can also be targeted by antibodies that induce direct killing via the activation of the complement cascade, through a mechanism called complement-dependent cytotoxicity (CDC), which leads to the formation of membrane attack complexes (MAC) perforating the tumor cell membrane. In addition, antibodies can signal via Fc receptors (FcR) on innate immune cells, to induce antibody-dependent cell phagocytosis (ADCP) of tumor cells by macrophages, or antibody-dependent cell cytotoxicity (ADCC) by NK cells or neutrophils.
To become effective, CTLs need to be educated by antigen-presenting cells (APCs) prior to cancer cell recognition, the most professional APCs being dendritic cells (DCs) and especially CD103+ migratory DCs. APCs provide CTLs with three required activation signals, namely (1) the cancer antigen epitope mounted on MHCI, (2) co-stimulatory molecules such as CD80/86 and CD40, and (3) pro-inflammatory cytokines such as IL-12, IFNβ and TNFα (Locy et al., 2018). In addition, activation and survival of CTLs is further supported by activated CD4+ T helper (Th) cells via the secretion of additional cytokines, as well as via a process known as “DC licensing,” by which a CD4+ Th cell activates a DC through interaction with CD40, and subsequently supports CTLs that come in contact with the same DC (Ridge et al., 1998; Laidlaw et al., 2016). CD4+ Th cells are activated by APCs similarly to CD8+ T cells, with the exception that the cancer antigen epitope is presented on MHCII instead of MHCI.
Upon activation, CTLs and CD4+ Th cells acquire particular phenotypes, which strongly determine the subsequent efficacy of CTL cytotoxic responses (Thorsson et al., 2018). Commonly, phenotypes of CTLs are defined by the cocktail of cytokines they produce, as well as by the cytotoxic pathway they use to induce cell death. Many studies have shown that the production of IFNγ and TNFα by CTLs correlates with better control of tumor burden and enhanced patient survival (Matsushita et al., 2015; Bhat et al., 2017). In parallel, other studies have demonstrated an increased patient survival when CD4+ Th cells acquire a Th1 phenotype, characterized by the secretion of IFNγ, TNFα and IL-2. Although more controversial (Chen and Gao, 2019), it has been shown that combination of the Th1 response with a Th17 orientation, characterized by IL-17 production, can be further beneficial (Punt et al., 2015; Thorsson et al., 2018). Noteworthily, not only the type of secreted cytokine matters, but also their variety and amount. Indeed, T cells that secrete multiple cytokines are known to be more efficient than those expressing a single one (Seder et al., 2008). In addition, because each T cell has a unique TCR able to recognize only a single antigenic epitope, immune responses that generate a broad clonality of anti-tumor T cells (i.e., multiple T cells clones) are more robust (Thorsson et al., 2018). All that being said, the ideal type of immune response sought upon vaccination might vary between cancers.
In addition, antibody-mediated cytotoxic mechanisms can also efficiently control tumor growth (Zahavi et al., 2018) and be harnessed by cancer vaccination (Figure 1B). Particularly, antibodies that specifically bind to cancer cells can trigger their elimination by antibody-mediated cellular cytotoxicity (ADCC), antibody-mediated cellular phagocytosis (ADCP) or complement-dependent cytotoxicity (CDC) (Almagro et al., 2018). To date, such mechanisms have been mostly exploited in passive cancer immunotherapies via the infusion of therapeutic antibodies in patients, rather than in the context of humoral-based cancer vaccines, which aim to activate endogenous host anti-cancer antibody responses (Huijbers and Griffioen, 2017). In both cases, antibodies recognize short conformational or linear epitopes exposed on the cancer cell surface (Bayrami et al., 2016; Tarek et al., 2018). Then, innate immune cells, mostly natural killer (NK) cells, macrophages and neutrophils, can detect bound antibodies via their Fc receptors and subsequently induce cell lysis, in case of ADCC, or phagocytosis, in case of ADCP. In contrast, CDC is independent of immune cells and can directly activate the complement pathway to form cytolytic pores in cancer cell membranes, inducing their death. Such antibody-mediated anti-tumor responses are antigen-specific and can provide immune memory if the tumor-specific antibodies are produced endogenously.
Finally, the anti-cancer adaptive immune response sought by cancer vaccination can be further supported by co-activation of other mechanisms of innate immunity. For example, innate lymphoid cells (ILCs), such as NK cells or invariant NK T cells (iNKT), have the ability to control cancer cells in complementary ways to CTLs (Nair and Dhodapkar, 2017; Souza-Fonseca-Guimaraes et al., 2019). For example, NK cells possess cytotoxic capabilities and can lyse cancer cells that downregulate MHCI to avoid T cell recognition or that overstimulate activating receptors on NK cells (e.g., NKG2D, 4-1BB) (Souza-Fonseca-Guimaraes et al., 2019). On the other hand, activation of iNKT cells can result in secretion of Th1 or Th2 cytokines in the microenvironment and increase their CD40L expression. Therefore, iNKT cells can strongly stimulate DC and B cell maturation and indirectly promote T cell responses, thus illustrating a pivotal role in modulating adaptive immune responses (Cerundolo et al., 2009). Nevertheless, because NK or iNKT cells are not antigen-specific and do not establish immune memory, they are often not the primary targets of cancer vaccines.
What Should Therapeutic Cancer Vaccines Be Composed Of?
The composition of therapeutic cancer vaccines directly relates to their anticipated biological outcomes. Commonly, cancer vaccines are made of antigens that define what is recognized on or within cancer cells, as well as adjuvants that determine the type of immune response that will be induced.
Cancer Antigens
Including cancer antigens in vaccines is essential to induce targeted cancer cell death as well as to avoid toxic, non-specific immune reactions. Determining the appropriate immunogenic antigen to incorporate in therapeutic cancer vaccines, however, remains extremely challenging. Indeed, the self-origin of cancer cells makes them hard to discriminate from healthy cells, as they carry most of the host proteome, which is naturally immune tolerized to prevent autoimmune reactions. Nevertheless, different types of cancer antigens have been successfully used in cancer vaccines, as detailed below (Vigneron, 2015; Finn, 2017).
First, cancer vaccines often use tumor-associated antigens (TAAs) as targets, which are molecules largely overexpressed in tumor cells (e.g., 10–1000 fold increase in some cases) as compared to healthy cells (Vigneron, 2015). Notably, TAAs can be proteins involved in tumor cell survival and proliferation, such as HER2, EGFR, p53, telomerase, survivin, and Ras, in tumor metabolism, such as folate-related proteins and glucose receptors (e.g., GLUT1), or other proteins such as MUC-1 and mesothelin (Vigneron, 2015). However, the main drawbacks of TAAs reside in the difficulty of inducing strong immunity against them, which must break endogenous immune tolerance mechanisms, while preventing autoimmune reactions against healthy cells.
As an alternative, vaccines can use tumor-specific antigens, which are absent in healthy cells or expressed in limited areas. Particularly, cancer cells can re-express MAGE antigens, NY-ESO-1, 5T4 or the carcinoembryonic antigen (CEA), which are only present during the developmental stage or in very specific tissues (e.g., placenta, testis) in the body (Vigneron, 2015). The restricted expression of such tumor-specific antigens provides the advantage of limiting off-target reactions. Interestingly, it has been shown that some of these antigens are naturally immunogenic and able to raise specific T cell responses in spite of their endogenous origin. Additionally, some types of cancer exhibit specific mutations that can be conserved across patients, such as the BRAF mutation in melanoma, which can constitute good antigenic targets as they are exclusively present in cancerous tissues (Mandalà and Voit, 2013).
Similarly, most tumors contain specific mutated antigens, called neoantigens, due to the higher mutational rate of cancer cells as compared to healthy ones (Schumacher and Schreiber, 2015). These antigens constitute a subclass of tumor-specific antigens, and can be similarly targeted by cancer vaccines. However, the use of neoantigens implies more personalized therapies, as they are different between patients, tumors or even tumor cell subsets. Nevertheless, recent technological advances and ease in genome sequencing allow the fast emergence of such therapies. Neoantigens have been proven to be immunogenic, and their restricted expression in cancer cells highly limits the risk of T cell reactions against healthy self cells. Neoantigens are being widely addressed, with many researchers focusing on understanding how to select, design and deliver the most relevant neoepitopes to incorporate into vaccines (Schumacher et al., 2019).
Furthermore, other strategies use whole cancer cells or cancer cell-derived materials as antigenic components in vaccines, instead of selecting single or combinations of defined antigens (Vermaelen, 2019). Such approaches are particularly interesting as they allow vaccination against multiple cancer antigens, while bypassing the need of identifying them. For example, lethally irradiated cancer cells derived from the primary tumor have been used to induce effective polyantigenic anti-tumor immune responses (Vermaelen, 2019). In addition, tumor cells can be prepared as lysates (González et al., 2014). In such cases, it is expected that the immunogenic responses will be mainly directed against cancer-specific antigens rather than against co-delivered endogenous proteins, since immune tolerance mechanisms would dampen responses to self-antigens. Lastly, cancer antigens have been shown to be present on tumor-derived extracellular vesicles, such as exosomes, as well as on tumor apoptotic debris, which can then also serve as antigenic materials (André et al., 2002). Importantly, targeting multiple antigens provides the advantage of reducing the risk of tumor immune escape, a mechanism by which cancer cells downregulate targeted antigens, mutate them or limit their presentation on MHC to avoid recognition and killing by CTLs.
Finally, cancer vaccines could also be rationally designed to target the tumor in vivo and use it as an in situ source of cancer antigens, as further discussed in the section “Rationale for Site-Specific Targeting of Therapeutic Cancer Vaccines”. Because these tumor-targeting vaccines can be composed of only adjuvants (i.e., without added antigens), whether it is classified as a therapeutic vaccine or as another type of immunotherapy is arguable.
Immune Adjuvants
The delivery of antigens alone may induce immune tolerance rather than activation. As a consequence, vaccines need to combine antigens with adjuvants, which are immunostimulatory molecules able to skew immune cells toward the desired type of immune response. Adjuvants can be derived from microbes, so called microbial-associated molecular patterns (MAMPs) or pathogen-associated molecular patterns (PAMPs), from endogenous danger signals released upon cell damage or immunogenic cell death, known as damage-associated molecular patterns (DAMPs), or can simply be cytokines that are naturally secreted to support endogenous immune responses (Tovey and Lallemand, 2010; Tang et al., 2012).
Both MAMPs and DAMPs are able to generate Th1 and CTL immune responses, as mostly intended in cancer vaccines, via the activation of pattern-recognizing receptors (PRRs) on APCs (Tang et al., 2012). Among these PRRs, Toll-Like receptors (TLRs) have been the most studied, with 6 gathering a significant interest in cancer vaccines, namely TLR-2, -3, -4, -7/-8, and -9 (Gay and Gangloff, 2007). These receptors are located in the endosomal compartment of APCs, except for TLR-2 and -4 which are on the cell surface. Consistent with their subcellular location, TLR-3, -7/-8, and -9 primarily recognize nucleic acid ligands from viruses or bacteria, double-stranded RNA, single-stranded RNA and unmethylated CpG oligodinucleotides (ODN), respectively, whereas TLR-2 recognizes bacterial lipoproteins (Lpp) upon dimerization with TLR-1 or -6, and TLR-4 recognizes lipopolysaccharides (LPS) from bacterial outer membranes. Examples of well-known TLR ligands that have been assessed in cancer vaccines are Pam3CSK4 (Zom et al., 2018) and Pam2Cys (Zhou et al., 2019) for TLR-2/1 and -2/6 respectively, poly(I:C) for TLR-3 (Ammi et al., 2015), LPS and monophosphoryl lipid A (MPLA) for TLR-4 (Cluff, 2010), imiquimod and other imidazoquinolines for TLR-7/-8 (Dowling, 2018), and CpG-B for TLR-9 (Shirota et al., 2015). Although these TLR agonists are very potent in activating immune responses, they can be associated with toxicity, which affects their clinical translation. Interestingly, some endogenous extracellular proteins have also been identified as TLR agonists and might be potentially safer considering their endogenous origin. For instance, the extra domain A (EDA) of fibronectin, a matrix protein, can bind to TLR-4 upon proteolytic cleavage and has showed some promises as adjuvant in cancer vaccines in pre-clinical models (Lasarte et al., 2007; Julier et al., 2015).
In addition to TLRs, other PRRs can be targeted by cancer vaccines. For example, the cytosolic DNA sensor cGAS detects aberrant concentrations of DNA in the cytosol and triggers the simulator of interferon genes (STING) pathway (Li et al., 2019). Another example is the cytosolic RNA sensor RIG-I that detects particular viral dsRNA (Tang et al., 2012; Elion and Cook, 2018). Stimulators of these cytosolic nucleic-acid sensor pathways are currently being explored as adjuvants for cancer immunotherapies.
Upon PRR signaling, APCs undergo maturation, which results in increased antigen presentation, expression of co-stimulatory receptors and secretion of cytokines, thus providing the three signals necessary for T cell activation, as previously detailed. Additionally, the nature of the co-stimulatory receptors and cytokine expression by APCs depends on the type of delivered adjuvants. Interestingly, it has been shown that secretion of IFNα and IFNβ by APCs upon maturation can induce direct inhibitory effects on tumor cell proliferation and activate their apoptotic pathways, inducing cancer cell death (Apelbaum et al., 2013).
Since cytokines themselves can strongly support immune responses, they have also been considered as adjuvants in cancer vaccines. Particularly, cytokines can be delivered to promote activation of immune cells, recruit them at specific sites, or induce their proliferation. For instance, IL-2, IL-12, IFNα, and IFNβ have been used to increase survival and activation of T cells, NK cells and APCs. Despite being very effective in boosting anti-tumor immune responses, these cytokines suffer from toxicity-related issues, similarly to TLR agonists, and require further development of appropriate delivery systems to harness their potential in the clinic. On the other hand, chemokines – a subset of cytokines – have been used to attract APCs at the vaccine site, thus enhancing overall antigen presentation and subsequent immune cell activation. While some chemokines induce the recruitment of multiple types of APC (e.g., DCs, macrophages), such as CCL3 and CCL4 (Nguyen-Hoai et al., 2016; Allen et al., 2018), some others recruit specific APC subsets. For example, the delivery of XCL1 specifically attracts the CD103+ DCs (Russell et al., 2007; Sánchez-Paulete et al., 2018), known to express the cognate receptor XCR1 and be highly efficient in generating CTLs. Moreover, chemokines can also be used to recruit T cells, rather than APCs. Notably, CXCL10 and CXCL11 have been delivered to increase infiltration of activated T cells in tumors (Groom and Luster, 2011). Lastly, as an alternative to recruitment, in situ proliferation of immune cells can be promoted by the delivery of growth factors. Particularly, granulocyte-macrophage colony-stimulating factor (GM-CSF) has been used to expand DC populations in a therapy called GVAX (Simons and Sacks, 2006).
Interestingly, the secretion of multiple cytokines by activated iNKT cells can also be exploited as an adjuvant in cancer immunotherapies, including vaccines (Wolf et al., 2018; Fujii and Shimizu, 2019). Upon activation by CD1d-bound α-Galactosylceramide (α-GalCer; KRN7000) on APCs, iNKT cells secrete large amounts of IFNγ and IL-4 that enhance DC maturation and subsequent antigen-specific T cell responses.
Finally, in addition to exogenous adjuvant delivery, another important strategy in cancer vaccines is to exploit the release of endogenous DAMPs by the tumor itself to self-adjuvant vaccines (Hernandez et al., 2016). Indeed, induction of immunogenic cancer cell death by current therapies, such as chemotherapy, radiotherapy, and/or immunotherapy, can substantially increase the release of DAMPs from dying tumor cells, such as heat-shock proteins (HSPs), adenosine-triphosphate (ATP) or the high mobility group protein B1 (HMGB1) (Tang et al., 2012). Along with endogenous DAMPs, immunogenic cancer cell death co-releases cancer antigens, together promoting antigen spreading, a complex mechanism by which immune reactions are mounted against antigens that were not originally targeted by a therapy (Gulley et al., 2017). As a consequence, any method capable of killing cancer cells in an immunogenic way can potentially boost the effects of cancer vaccines and broaden the anti-cancer immune response to multiple antigens.
How to Engineer Targeting Materials for Therapeutic Cancer Vaccine Delivery?
Once the cancer vaccine components have been defined, the way they are delivered will significantly impact overall efficacy and safety. As a consequence, the vaccine delivery needs to be rationally designed from the entry into the patient to its terminal effect; this includes the administration route into the patient, the targeting to correct tissues, cell types, subcellular locations and specific receptors, and ultimately the onset of appropriate immune responses. All together, these steps constitute the pharmacokinetic and pharmacodynamic profiles of the vaccine and can be fine-tuned by the use of materials. Here, we will discuss how materials can be engineered to optimally deliver its components and target them into relevant sites.
Where to Target Therapeutic Cancer Vaccines?
Possible Delivery Routes for Therapeutic Cancer Vaccines
From a clinical point of view, cancer vaccines can be conveniently administered to patients via intradermal, subcutaneous, intramuscular, intravenous or intratumoral routes, if the tumor is easily accessible at the body surface, as in skin cancers. However, intratumoral administration can become challenging depending on the tumor size, location, the number of tumors to inject, as well as on the intrinsic heterogeneity of tumor structures that can lead to non-homogenous drug distribution (Marabelle et al., 2018). Similarly, intralymphatic, intranodal and intrasplenic delivery routes are relatively complex, although they may be relevant from a biological point of view, as discussed below. Other routes, such as topical, oral or intranasal, are often less utilized yet might be appropriate in specific types of cancer.
The choice of the vaccine delivery route should be based on the anticipated biological mechanisms of action, since its efficacy depends on its bioavailability at the targeted sites. In the case of therapeutic cancer vaccines, tumor tissues and lymphoid organs are generally considered as the most interesting sites to target, and many direct or indirect strategies have been explored to deliver drugs at these locations.
Rationale for Site-Specific Targeting of Therapeutic Cancer Vaccines
Tumors are the primary location where therapeutic efficacy is sought, when they cannot be fully removed by surgery. Accordingly, targeting cancer vaccines into tumors is an appealing strategy to induce direct in situ cytotoxic effects and promote potent antigen-specific adaptive immune responses (Marabelle et al., 2014). Of particular importance, the tumor is the main source of cancer antigens, and thus can be used in place of or in combination with antigens from the vaccine (Figure 2A). Because the tumor gathers high concentrations of all cancer antigens at the same location, it theoretically constitutes an ideal target to promote broad polyantigenic immune responses. Interestingly, targeting the vaccine into tumors may also allow induction of immune reactions against antigens expressed only by small subpopulations of cancer cells, such as cancer stem cells, which are particularly important to eradicate (Saygin et al., 2019). Another advantage of targeting tumors in vivo is provided by the local release of antigens and DAMPs upon intratumoral cytotoxicity, which can enhance antigen spreading and thus the vaccine’s effects, as discussed in the section “Immune Adjuvants” (Hernandez et al., 2016; Gulley et al., 2017). Finally, some inflamed tumors are the battlefield of pre-existing anti-tumor immune reactions, which can be further supported or re-activated in situ by tumor-targeting vaccines.
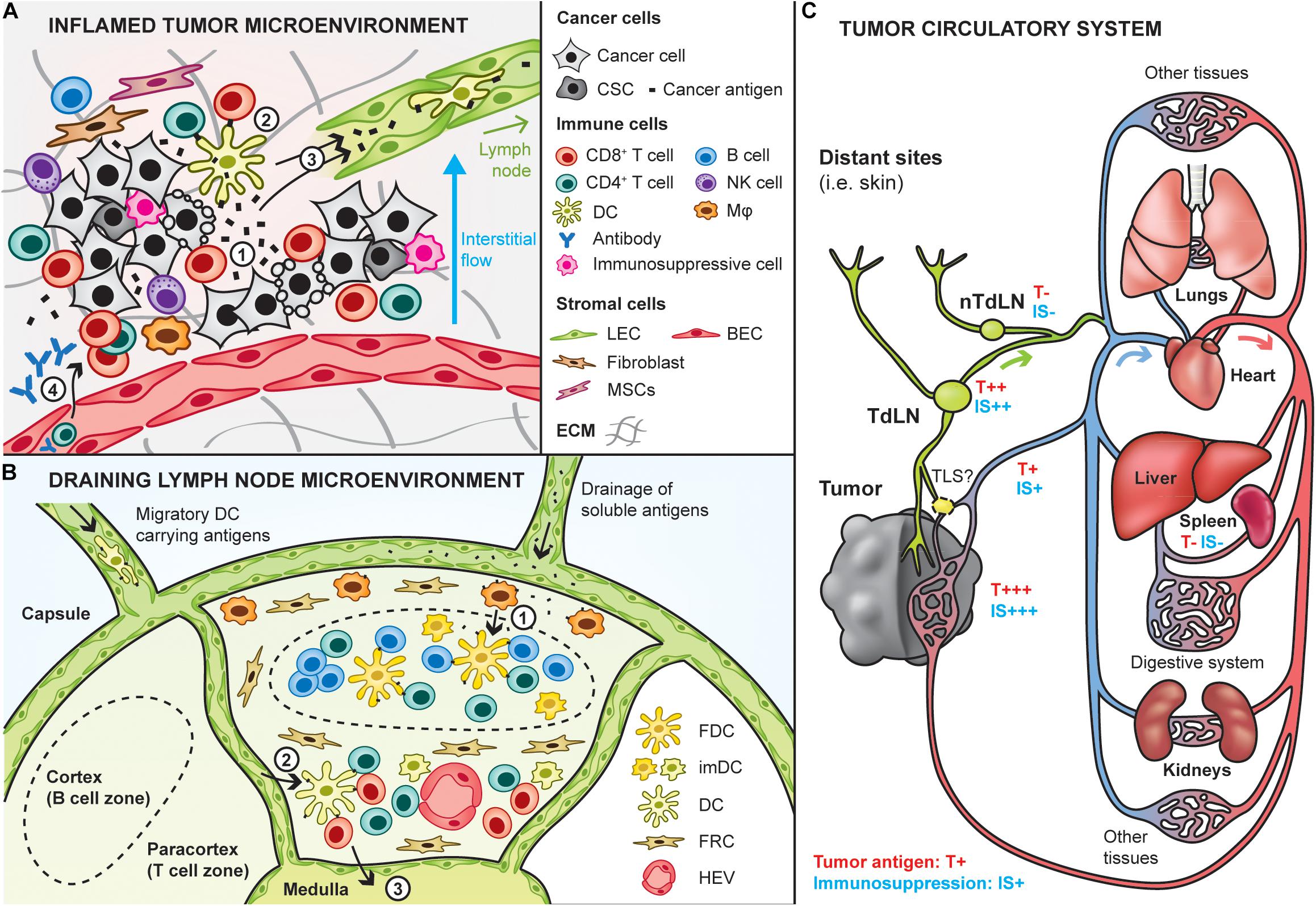
Figure 2. Site-specific targeting of therapeutic cancer vaccines. Cancer vaccines can be designed to specifically target sites that are potent for inducing anti-tumor immunity, including the tumor and lymphoid tissues. (A) Interactions between the tumor and immune infiltrates in the inflamed tumor microenvironment. Upon cancer cell death (e.g., T cell-mediated), cancer antigens are released in the local environment (1) and can be taken up by dendritic cells (DCs) to induce in situ activation of cancer-specific T cells (2). In addition, cancer antigens are either passively drained by lymphatic vessels or actively transported by immune cells trafficking to the draining lymph node (3), where potent immune responses can be induced. Then, T cells activated in the lymph nodes can home into the tumor to kill tumor cells (4). In addition, humoral immune responses may be triggered in the lymph node and can lead to the production of cancer-targeting antibodies that enter the tumor bed to induce antibody-mediated cytotoxic mechanisms (4). It is important to note here that the depicted tumor-immune interactions do not take place in tumors that are known as immune deserts (i.e., non-inflamed). CSC: cancer stem cells, Mφ: macrophage, LEC/BEC: lymphatic/blood endothelial cell, MSC: mesenchymal stem cells, ECM extracellular matrix. (B) Lymph nodes are relevant to target by therapeutic cancer vaccines as they are naturally optimized to induce strong immune responses, due to their high content in immature DCs, and naive T and B cells. Naive lymphocytes enter the lymph node via the high endothelial venules (HEV). Antigens and adjuvants can enter lymph nodes in soluble form and be further transported by subcapsular macrophages to follicular DCs to induce B cell responses (1). In addition, migratory DCs loaded with cancer antigens can go into the paracortical zone (2), where they activate antigen-specific CD4+ and CD8+ T cells. Activated T cells exit the lymph node via the medulla (3). FDC: follicular dendritic cell, imDC: immature dendritic cell, FRC: fibroblastic reticular cells. (C) Overview of the tumor circulatory system and tumor antigen biodistribution. Tumors are connected to the blood circulatory system by blood vessels (arterial system in red, venous system in blue) and lymphatic routes (in green). The tumor is a relevant site to target by cancer vaccines as it has the highest concentration of tumor antigens (high exposure: T+++), although its high immunosuppression (IS+++) might impair vaccine efficacy. Alternative sites to target can be the tumor-draining lymph node (TdLN) also relatively highly exposed to tumor antigens. As they are less immunosuppressed, targeting non-tumor-draining lymph nodes (nTdLN) and the spleen might lead to better immune activation upon vaccination, yet their poor exposure to tumor antigens might require the use of exogenous cancer antigens.
On the other hand, tumor-targeting strategies also present some important limitations for vaccination. First, although intratumoral activation of immune cells has been demonstrated, tumors are not physiologically optimized to mount strong immune responses, as opposed to lymphoid tissues (Thompson et al., 2010). Secondly, the high heterogeneity of tumor compositions may strongly affect the efficacy of the vaccines, possibly requiring tailoring per tumor characteristics (Binnewies et al., 2018). In particular, vaccines targeted to tumors that are known as immune deserts (i.e., lack of immune infiltrates) are likely to be poorly effective as compared to targeting into inflamed tumors. Thirdly, the tumor microenvironment is known to be strongly immunosuppressive, which would undeniably prevent potent anti-tumor immune reactions to be mounted upon vaccination.
To overcome these limitations, other sites might be interesting to target by cancer vaccines, notably lymphoid organs, including lymph nodes and the spleen, which are physiologically optimized to build potent immune responses and might be exposed to tumor antigens (Thomas et al., 2014; Rotman et al., 2019; Figure 2B). Indeed, tumor interstitial fluid and debris are drained from the tumor to the lymph nodes through lymphatic vessels, and then to the blood systemic circulation. In addition to lymphatic routes, some debris can directly enter the blood circulation via tumor venous drainage. Eventually, they are filtered by the spleen, liver and kidneys (Figure 2C). Recently, it has been highlighted that tertiary lymphoid structures (TLS) can form in proximity of tumors, which might constitute another relevant site to target, although our current knowledge on these structures remain limited to date (Sautès-Fridman et al., 2019).
Targeting the tumor-draining lymph nodes (TdLNs) might therefore constitute a good alternative to direct tumor-targeting, considering their high exposure to cancer antigens, optimal content and organization of immune cell populations and conserved structures, which would permit the development of more generic (i.e., less tumor-specific) cancer vaccines (Jeanbart et al., 2014). Nevertheless, TdLNs can also be affected by tumor-derived immunosuppression, as they drain immunosuppressive factors from the tumor. In addition, tumor-draining lymph nodes are sometimes surgically removed for diagnosis purposes, to establish the metastatic and aggressiveness profile of cancers.
Should this be the case, targeting cancer vaccines to the spleen or to non-tumor draining lymph nodes (nTdLNs) remain other relevant options (Jeanbart et al., 2014). Since those are located downstream in the circulatory system, they are less exposed to the tumor immunosuppression, although also less supplied with tumor antigens. As a consequence, adding exogenous tumor antigens in vaccines targeting these sites might be necessary to achieve proper efficacy.
Practically, tumors can be directly targeted via intratumoral delivery or indirectly by the use of tumor-targeting technologies. On the other hand, lymph nodes can be indirectly targeted by delivering the vaccine in the tissues they drain, via topical, intradermal, subcutaneous or intramuscular routes, for example, which might be more convenient than via intranodal or intralymphatic injections. Lastly, the spleen can be efficiently targeted via intravenous perfusion, or less commonly via an intrasplenic route.
Passive Targeting Using Material Engineering
Establishing the vaccine delivery route and strategy will inform on the intrinsic properties required for the vaccine to be efficient, notably providing criteria on the components’ half-life, stability, solubility, toxicity or biodistribution. Modulation of these parameters has been widely achieved by the use of materials. In addition, some materials can act themselves as immunostimulants (Sun et al., 2017), as targeting tools (Weissleder and Pittet, 2008), or have direct cytotoxic effects on cancer cells (Zou et al., 2016), further enhancing the vaccine outcomes.
Choice of Material Physicochemical Properties
When developing a new material for cancer vaccines, or using an already existing one, the choice of material primarily depends on its physicochemical properties, such as its size, shape, charge, solubility and elasticity. These parameters will affect the vaccine by modifying its biodistribution, cell internalization and activation capabilities, as well as its half-life and release kinetics. Thus, the material needs to be chosen according to both the delivery route into the patient and its ability to target and get metabolized by the correct cell types.
Size
First, the size of a material, or particle, can range from a few nanometers up to several microns and will influence its drainage, biodistribution, which cells internalize it as well as its retention time. It has been demonstrated that intradermal injection of nanoparticles ranging from 20 to 200 nm can enter the lymphatic system and drain to the lymph node, with a preference for particles ranging around 40 nm, whereas larger ones will be retained at the injection site and internalized by APCs before being transported to the lymph node via cellular trafficking (Swartz, 2001; Reddy et al., 2006; Irvine et al., 2013). On the other hand, if the formulation is injected intravenously, carriers smaller than 5 nm will not only be filtered by the kidney in less than 5 min but will also escape the vessels to diffuse in the neighboring tissues, whereas larger particles have a longer half-life in the blood (Choi et al., 2011; Hoshyar et al., 2016). Interestingly, compared to the tight junction of the endothelium of blood vessels in healthy tissues (5–10 nm), fast growing cancer vessels have looser junctions with pores ranging from 200 to 1200 nm, allowing particle extravasation within that range (Chauhan et al., 2012). Upon extravasation, particles can additionally be retained in the tumor for extended time, as a result of impaired tissue drainage. This is a process known as the enhanced permeability and retention (EPR) effect that has been widely exploited in cancer animal models for the development of tumor targeting nanosystems, but that remains controversial for use in humans (Danhier, 2016; Golombek et al., 2018). Unfortunately, although larger particles have several advantages when injected directly in the bloodstream, their penetration efficiency in the tumor is reduced compared to smaller particles (Hauert and Bhatia, 2014).
At a cellular level, the size of the carrier influences endocytosis by specific cell types. Indeed, it has been demonstrated that particles of 20–600 nm are preferentially taken up by DCs, whereas particles from 0.5 to 5 μm are rather taken up by macrophages (Xiang et al., 2006; Kanchan and Panda, 2007). Finally, the material fate upon intracellular trafficking will directly affect the efficacy of the vaccine itself since the payload has to reach the correct subcellular compartment to activate the cognate receptors or signaling cascade. Briefly, particles ranging from 250 nm up to 3 μm are preferentially taken up by phagocytosis whereas smaller particles enter the cell through pino- or macro-pinocytosis (Rivolta et al., 2012). Several uptake studies on cancer cells have demonstrated that the highest uptake was observed for particles around 50 nm (Chithrani et al., 2006).
Shape
Second, the shape of the material also affects its systemic biodistribution, circulation in blood, cellular uptake and interactions. For example, it has been shown that for some materials, such as gold nanoparticles, rod-shape structures tend to accumulate more in the spleen and less in the liver than their spherical counterparts (Arnida et al., 2011; Black et al., 2014); although not a general rule, this exemplifies how material shape can influence biodistribution. In addition, non-spherical particles in the bloodstream tend to marginate more and escape the blood flow (Toy et al., 2014). Microscopically, it has been demonstrated that the rate of cellular internalization of non-spherical particles depends on their angular orientation relative to the cell membrane (Sharma et al., 2010; Behzadi et al., 2017). Furthermore, spherical particles are favorably internalized by monocytes/macrophages compared to particles with a high aspect ratio, which will marginate and target the endothelial cell and evade macrophage uptake (Peiris et al., 2012). Finally, an interesting study has highlighted that T cell activation is enhanced when using ellipsoidal synthetic APCs rather than spherical ones, due to an increased contact interactions with the immune cell membrane (Meyer et al., 2015).
Elasticity
Thirdly, it is hypothesized that the elasticity of particles influences cellular uptake and tumor accumulation properties. Generally, quantum dots, gold or magnetic particles are considered hard particles, whereas hydrogels, liposomes or polymersomes are described as soft particles. Overall, harder particles are better internalized than soft materials (Beningo and Wang, 2002; Anselmo et al., 2015). Anselmo et al. (2015) also showed that soft particles circulate at a higher concentration in the blood at early times after intravenous delivery and were slower and less endocytosed compared to hard particles. In addition, it was demonstrated that soft nanolipogels accumulated more in tumors compared to hard ones (Guo et al., 2018).
Charge, hydrophobicity and other chemical properties
Lastly, compared to the parameters discussed above, the chemical properties are based on intrinsic characteristics of the material, such as its charge, hydrophobicity and functional groups. Indeed, the material charge – cationic, anionic or neutral – influences cell internalization, immune activation and blood half-life. Since cell membranes are negatively charged, they will take up positively charged molecules much faster due to electrostatic interactions compared to other particles (Foged et al., 2005). However, the uptake of positively charged particles can disrupt cell membranes, leading to increased cell toxicity (Fröhlich, 2012). Furthermore, several studies have demonstrated that changing the charge of a material from negative to positive can induce a higher immune response (Wen et al., 2016). Finally it has been demonstrated that neutral particles have a slower internalization rate than charged ones (Owens and Peppas, 2006).
With regard to hydrophobic materials, a shorter half-life in the bloodstream is observed compared to their hydrophilic counterparts due to the reticulo-endothelial system recognizing them as foreign and removing them in the liver or the spleen (Owens and Peppas, 2006). In addition, a positive correlation has been demonstrated between hydrophobicity and immune activation (Moyano et al., 2012). However, hydrophobic materials can be “masked” to prevent removal and reduce intrinsic immune activation by coating them with a hydrophilic material such as polyethylene glycol (PEG), for example. Such a strategy is useful to improve the delivery of vaccines with hydrophobic compounds in the particle core (Maiti et al., 2019). Nevertheless, surface modification of particles additionally modulates their half-life and distribution profile in the body.
The interplay of all these parameters and how they affect treatment outcomes shows the importance of thoroughly characterizing new materials and their intrinsic properties in vitro and in vivo. Beyond that, core material properties can be further tuned by modifying the material with particular molecules, ligands or polymers, to fulfill specific criteria and needs.
Selected Examples of Different Types of Material
The extensive research on material engineering for drug delivery has provided a tremendous amount of available materials and technologies that could be used for the development of cancer vaccines. Here, we present a few examples of different types of materials to illustrate possible designs and structures (Figure 3).
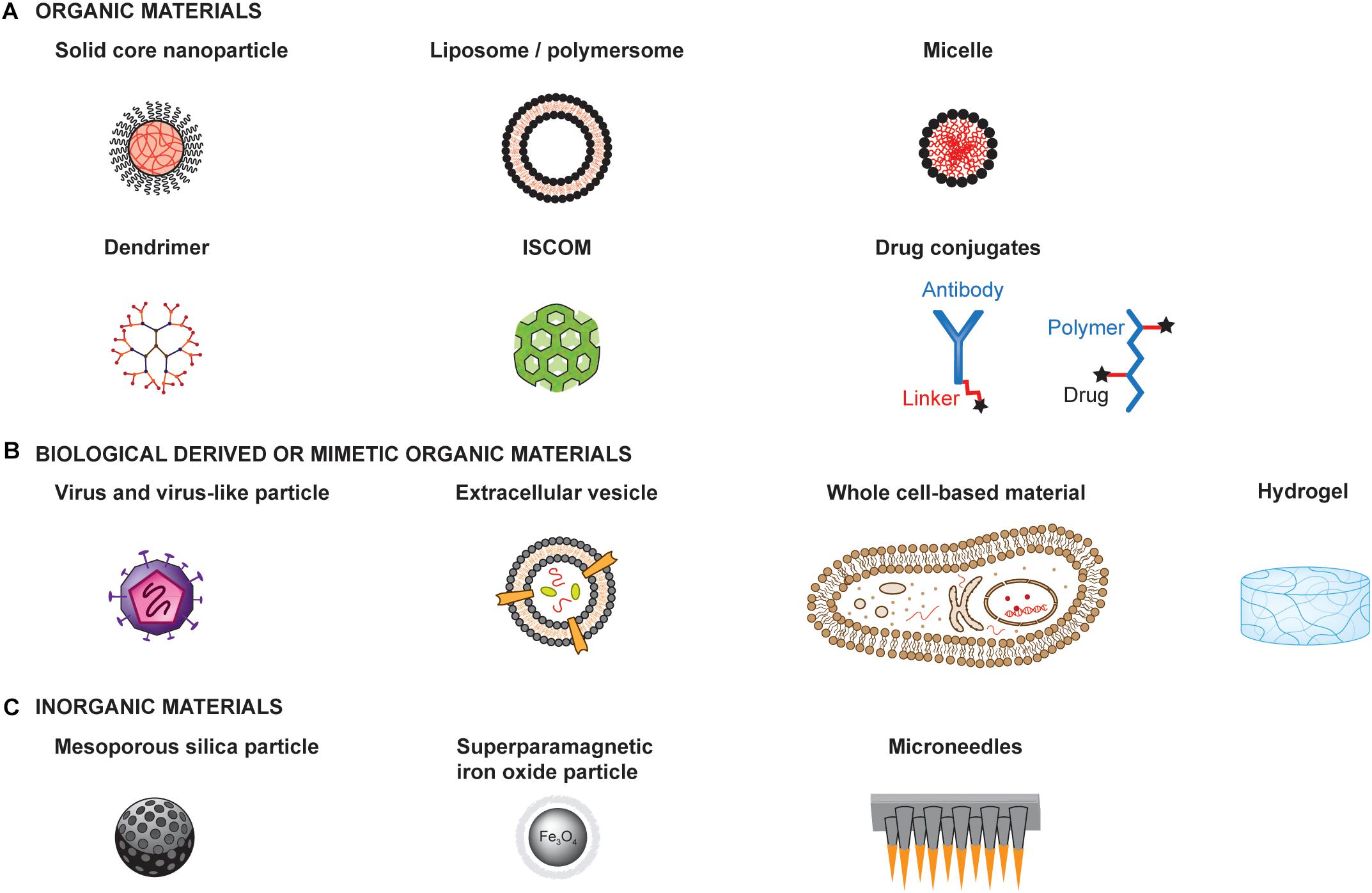
Figure 3. Examples of materials for the development of therapeutic cancer vaccines. Materials can be engineered to enhance the therapeutic efficacy and safety profiles of cancer vaccines. Materials with very different structures and physicochemical properties can be used as a basis for engineering the delivery of adjuvants and antigens to optimize immune activation. Such materials can be organic materials (A), including those derived from or mimicking biological materials (B), or inorganic materials (C) ISCOM: Immunostimulatory complex.
Materials can be organic or inorganic, each having specific properties, advantages and limitations that should guide the choice of a material. Furthermore, organic material can either be synthetic, such as poly(D,L-lactide-co-glycolide) (PLGA), poly(γ-carboxyglutamic acid) (γ-PGA), and PEG, or natural, such as dextran, alginate, lipids and chitosan (Figure 3A). Similarly, they can be synthetically produced or derived from biological origin (Figure 3B). They can form a broad range of structures, including solid core particles, vesicles, micelles, emulsions, dendrimers or hydrogels. On the other hand, inorganic particles have the advantage of having rigid structure, controllable synthesis, with a size range of 2 to 150 nm, as well as low toxicity, although most are not biodegradable. Examples of inorganic particles include silica-based and magnetic particles (Figure 3C).
Solid core nanoparticles (NPs)
Nanoparticles are spherical particles with solid cores in the nanoscale size and have been extensively used for drug delivery over the past decades (de Titta et al., 2013; Tran et al., 2017; Ankita et al., 2019). Apart from their spherical shape, most of their parameters and characteristics can be tuned, such as their charges, hydrophobicity or surface properties (i.e., by conjugation of specific moieties), to give a few examples (van der Vlies et al., 2010).
Liposomes and polymersomes
Liposomes and polymersomes are 50–500 nm often spherical bilayered vesicles composed of phospholipids or block copolymers, respectively. They can incorporate hydrophobic or viral envelope glycoproteins on their bilayer as well as encapsulate hydrophilic molecule in their core (Senapati et al., 2018). Similarly, to NPs, these vesicles are highly versatile since it is possible to modify most of their parameters, by modifying their surface charge (Mo et al., 2012) or conjugating targeting ligands (Noble et al., 2014), for instance. Interestingly, they can be designed to release their payload in specific subcellular compartments (Jiang et al., 2012).
Micelles
Micelles are self-assembled spherical materials composed of amphiphilic block copolymers with a hydrophobic core and a hydrophilic corona (Hanafy et al., 2018). These colloids will spontaneously form at a specific concentration, called the critical micelle concentration (CMC), and temperature. Hydrophobic molecules can be encapsulated into micelles through physical, chemical or electrostatic interactions (Park et al., 2008).
Dendrimers
Dendrimers are spherical macromolecules composed of many branches originating from a central point forming a star-like structure. The advantages of these particles are their highly tunable properties since their molecular weight, size, flexibility, branching density, and solubility can be modulated (Tran et al., 2017; Sherje et al., 2018). Interestingly, it is possible to both conjugate dendrimers with several different drugs using different chemistry and “encapsulate” poorly water-soluble molecules into them (Wang et al., 2011). Furthermore, if the polymer used in the dendrimer is positively charged, DNA or RNA can be complexed to it for delivery into cells (Shan et al., 2012). The main drawback of this material is its potential toxicity, and bio-incompatibility, depending on its surface physico-chemical properties (Palmerston Mendes et al., 2017).
Immunostimulating complex (ISCOM)
Immunostimulating complexes are cage like particles of 40 nm composed of phospholipids, cholesterol, saponin adjuvant Quil A and protein antigens (Homhuan et al., 2004). Usually the antigen is not directly conjugated to the particle but rather interacts by hydrophobicity (Peek et al., 2008). In addition, they naturally induce an immune response, thus acting as immunostimulant materials.
Hydrogels
Hydrogels are a three-dimensional network of hydrophilic polymers cross-linked together. They have the capacity to retain large quantities of fluids and can be chemically modified to insert enzymatic, hydrolytic or stimuli-responsive components to ensure their biodegradability (Peppas et al., 2000). The main advantage of hydrogels is their high water-content similar to biological tissues, thus reducing surface tension induced by the material. In addition, the drug loading and release rate can be tightly controlled by modifying the quantity of gel cross-linking (Lin and Metters, 2006). An interesting feature of hydrogels is the possibility to induce their gelation in situ with a specific stimuli such as pH, temperature or light (Van Tomme et al., 2008).
Drug-conjugates
These materials simply consist of a drug conjugated to a polymer, or a protein via a linker, which can be cleavable or not. This delivery system has the advantage of reducing treatment toxicity and adverse side effects, solubilizing the drug as well as an easy synthesis (Dan et al., 2018). Cleavable linkers are either acid-sensitive, glutathione-sensitive, lysosomal protease-sensitive or β-glucuronide-sensitive, whereas non-cleavable linkers usually have thioether bonds, which do not have the risk of releasing the drug at the wrong time (Dan et al., 2018).
Viruses and virus-like particles
Viruses and virus-like particles both have the advantage of naturally inducing a strong immune response due to their envelope (Zhang et al., 2000). In addition, they can be used as a delivery system for genes, antigens or drugs into tumor cells (Chulpanova et al., 2018). The choice of virus for a treatment will depend on the virus tropism, size and longevity of the desired gene that has to be delivered, as well as on its safety profile. The most common viruses currently tested in clinical trials as oncolytic viruses for cancer therapies are adenoviruses, herpes viruses, measles viruses, retroviruses, vaccinia viruses, and vesicular stomatitis viruses. Despite their relative success with inducing tumor regression, a major drawback of viruses is that they are strongly neutralized by host antibody responses upon re-injection. In addition, the immune response can be diverted from tumor antigens to viral antigens (Cawood et al., 2012). Another option is to use only highly immunogenic virus-like particles (VLP) to induce a strong immune response against tumor antigens without having the issue of self-replication and safety concerns caused by viruses (Cubas et al., 2011; Li et al., 2013; Palladini et al., 2018; Thong et al., 2019). These particles can usually range between 20 and 800 nm (Pushko et al., 2013).
Extracellular vesicles (EVs)
Extracellular vesicles are biological materials naturally secreted by cells and delimited by a lipid bilayer, commonly found with a size of 20–500 nm, although some can reach several microns (van Niel et al., 2018). They can be derived from the cell plasma membrane in case of microvesicles or from endosomal origin in case of exosomes. As important mediators of intercellular communication, extracellular vesicles can carry proteins, nucleic acid, metabolites and lipids from one cell to another. As such, they have raised interest for possible use as drug delivery systems (Vader et al., 2016). Interestingly, it has been shown that the composition of extracellular vesicles can be modified by engineering either the producing cells or the vesicles after isolation. Although EVs are considered poorly immunogenic carriers (Saleh et al., 2019), they play a role in mediating immunostimulating or immunosuppressive responses (Robbins and Morelli, 2014).
Whole cell-based materials
Mammalian cells are living materials also delimited by a lipid bilayer, with a typical size of 10–50 μm of various shapes, that can be used as carriers to deliver drugs or as therapeutic agents per se when administered into patients (Cheng et al., 2019; Gong et al., 2019). To prevent cell rejection upon delivery, cells for clinical use are often derived from autologous sources, processed ex vivo and re-administered into the patient. One key advantage of using living cells as delivery materials is their ability to actively migrate to specific sites and to dynamically interact with endogenous cells and tissues (Leibacher and Henschler, 2016). Nevertheless, controlling the fate of living materials upon delivery can be challenging due to their high complexity.
Silica-based nanoparticles
Silica-based nanoparticles (SiNPs), especially porous SiNPs such as mesoporous silica nanoparticles (MSN), are used for drug delivery due to their high loading capacity and the possibility to control the release and encapsulation of different molecular weight drugs (Lai et al., 2003). In addition, MSN can be functionalized with targeting ligands, antibodies, peptides and even magnetic particles (Mamaeva et al., 2009).
Superparamagnetic iron oxide nanoparticles
Superparamagnetic iron oxide nanoparticles (SPIONs) are receiving increasing attention due to their broad applications in chemotherapy, hypothermia, magnetic resonance imaging (MRI), cell and tissue targeting, to mention a few (Quinto et al., 2015; Senapati et al., 2018). They are composed of an inner magnetic core and a hydrophilic coating polymer, such as PEG, polysaccharide and poly(vinyl alcohol), which can be used to deliver drugs or conjugate targeting ligand (Laurent et al., 2014). Due to their magnetic properties, studies showed the possibility to use an external magnetic field to localize them in the correct tissue and/or heat them to kill cancer cells.
Microneedles
Microneedles are sharp protrusions measuring from 100 μm to less than 1 mm, and are used as topical materials for local drug delivery. They are minimally painful for the patient and can be self-administered. The needle tips can be coated with protein, viruses, drugs or immunotherapy and will release the payload in a controlled slow manner (Ingrole and Gill, 2019). In the context of melanoma, for instance, transdermal delivery of immunotherapies with microneedles has demonstrated promising efficacy (Ye et al., 2017).
Engineering Tumor-Targeting Materials
In addition to their intrinsic physicochemical properties, materials can be further engineered to preferentially or specifically target tumors. Until recently, tumor-targeting materials have been primarily developed to deliver immunotherapeutic or chemotherapeutic drugs, or for diagnostic purposes, rather than for cancer vaccination. Therefore, we here focus on the different targeting technologies used by cancer immunotherapies in a broader scope, considering that they could inspire the design of future cancer vaccines. Particularly, we detail how tumors can be targeted at different levels, including macroscopic targeting of the tumor environment and microscopic targeting of cancer cells, tumor-associated stromal cells and the tumor extracellular matrix (ECM).
Targeting the Tumor Biochemical Environment
Due to unusual metabolism, the tumor environment has unique biochemical properties that differ from those of healthy tissues, and that can be used to activate or release drugs in a stimuli-responsive fashion, for example based on pH, oxygenation, protease contents, and chemokine secretion. Using stimuli-responsive materials, it may be possible to improve drug safety by limiting activity in off-target sites.
pH-responsive materials
Due to a high metabolism, the tumor environment is at a pH of 6.5 compared to the physiological one at 7.4 (Tian and Bae, 2012). This decrease in pH is caused by an increase in lactate and hydrogen ions produced to permit the substantial and rapid tumor growth. This pH difference has been exploited to develop materials capable of shrinking, aggregating or even enhancing cellular uptake upon tumor microenvironment entry (Wu et al., 2018). For example, particles coated with a zwitterionic monolayer change charge on their surface from negative to positive upon entering the tumor thus enhancing cell uptake and aggregation (Mizuhara et al., 2015). Another option to induce aggregation or release would be to have acid-labile amide bond breakage (Wu et al., 2018). Such strategies have been used to deliver chemotherapy (Yang et al., 2017), thermal therapy (Liu et al., 2017) or for tumor imaging (Hoffmann et al., 2012).
Hypoxia-responsive materials
A well-known characteristic of tumors is their low content of oxygen (Shannon et al., 2003; Bennewith and Dedhar, 2011). Based on this property, engineers have developed materials that incorporate bioreductive linkers, such as nitroimidazole analogs, thiol groups, and azobenzene moieties to deliver drugs upon entry into tumors (Guise et al., 2014; Ahmad et al., 2016; Kulkarni et al., 2016). For example, hypoxia-responsive nanoparticles have been developed to release doxorubicin in squamous carcinomas (Thambi et al., 2014). Similarly, prodrugs have been engineered to be activated in low-oxygen environments (Hunter et al., 2016). However, as hypoxia increases with tumor growth, hypoxia-sensitive drugs may have limited efficacy for early stage tumor targeting.
Protease-responsive materials
Many tumors exhibit abnormal enzymatic activity (Anderson and Cui, 2017; Yao et al., 2018), including the overexpression of matrix-metalloproteinases (MMPs) (Gialeli et al., 2010), caspases (Nejadnik et al., 2015), urokinase-type plasminogen activators (uPA) and cathepsins (Joyce et al., 2004). Therefore, including protease substrate sequences in materials and prodrugs has been exploited to specifically release drugs into tumors and limit their side-effects. Furthermore, some materials can change size and shape upon protease exposure, for example forming nanostructures (Hu et al., 2014; Anderson and Cui, 2017). As an example, Tanaka et al. designed a gelator precursor that self-assembles into nanofibers upon exposure to MMP-7 in tumor cells, inducing their death (Tanaka et al., 2015). Another study used caspase-sensitive gold NPs (AuNPs) as an apoptosis-inducing imaging probe (Sun et al., 2010).
Chemotaxis-based cellular tumor targeting
Tumor inflammation induces the secretion of chemokines, such as CXCL12, which are able to recruit specific cell types. Based on this mechanism, active tumor targeting can be achieved by the delivery of cells capable of sensing these chemokine gradients and actively migrating into tumor-inflamed regions (Cheng et al., 2019), such as myeloid cells, T cells, neural stem cells and mesenchymal stem cells (MSCs) for example (Leibacher and Henschler, 2016; Combes et al., 2018). Furthermore, these cells can be engineered to deliver anti-cancer drugs; for example, MSCs have been genetically modified to overexpress IFNβ or to carry paclitaxel into tumors (Ling et al., 2010; Sadhukha et al., 2014).
Targeting Tumor Cells
As cancer cells are the ones to eradicate, they constitute the ultimate target of cancer immunotherapies, including vaccines. Currently, many clinical treatments use targeted therapies to directly kill tumor cells. Coupling such targeting strategies with immune adjuvants would be valuable to turn them into therapeutic cancer vaccines, thus colocalizing tumor cell antigens and immunostimulatory molecules. Cancer cells can be targeted at multiple levels, including cell surface, intracellular or genomic levels, or by other approaches that use infectious materials (Figure 4).
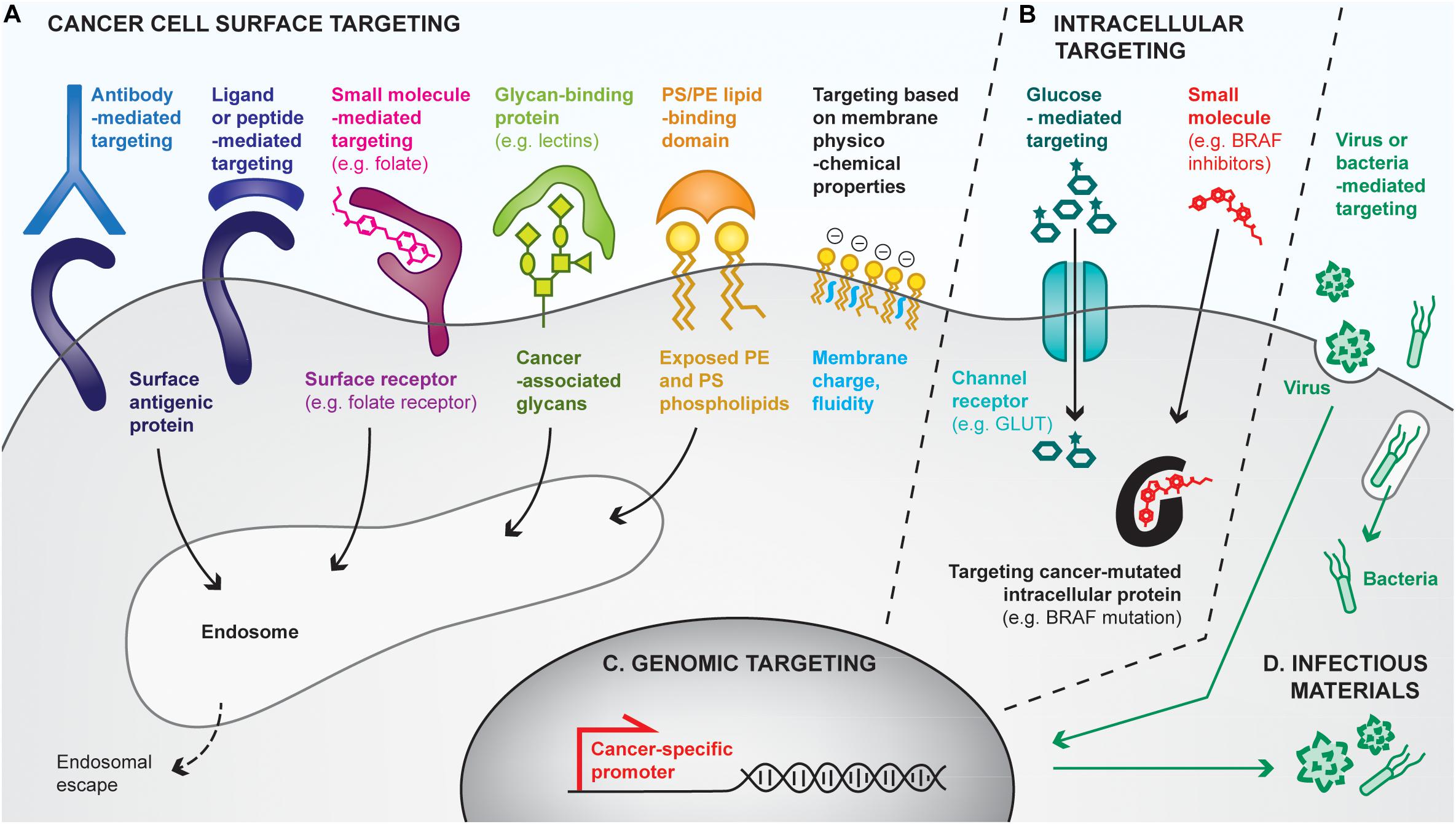
Figure 4. Overview of strategies for cancer cell targeting. Biomolecular engineering can be used to preferentially target the cancer cell at multiple levels, using differences between cancer cells and healthy ones to discriminate between them. (A) Cancer cells can be targeted with cell surface-binding moieties, based on specific affinities with cell-surface antigens, receptors, glycans, lipids or based on physicochemical properties (e.g., membrane charges). Most cell surface targeting strategies will lead to endocytosis of the targeting moiety. (B) The cancer cell cytoplasm can be directly targeted by using channel receptors that transport small molecules, or by using small molecules capable of crossing cell membranes. (C) Cancer cells reactivate specific promoters that are silenced in healthy cells, allowing cancer cell targeting by the delivery of genes placed under cancer specific promoters. (D) Finally, some pathogens (e.g., oncolytic viruses, bacteria) favorably infect and replicate in cancer cells, often leading to their death, thus providing additional means for preferential cancer cell targeting.
Targeting the cancer cell surface (Figure 4A)
One of the most common approaches to target cancer cells relies on affinity-based interactions of surface tumor-associated antigens with antibodies or antibody derivatives (e.g., Fab, scFv). As a clinical example, HER2-positive tumor cells can be targeted by intravenous or subcutaneous injection of anti-HER2 antibodies, which accumulate at the cancer cell surface due to their high specific affinity for the receptor, in both the primary tumor and metastases. Such antibodies can display intrinsic activities to affect tumor cell growth, notably by blocking the surface protein functions and by triggering antibody-mediated cytotoxicity. They can additionally be modified with anti-cancer drugs or adjuvants for the development of cancer vaccines (Hong et al., 2011). For example, Sharma et al. (2008) have chemically conjugated anti-HER2 to CpG, which has led to tumor eradication and induction of protective memory when combined with anti-GITR immunotherapy. In addition, tumor-targeting antibodies have been conjugated to material surfaces, such as nanoparticles (Kubota et al., 2018) or liposomes (Espelin et al., 2016), to confer them the ability to target tumors. Interestingly, biological cell-based materials can be similarly engineered; for instance, in the context of chimeric antigen receptor (CAR)-T cell therapies, patient-derived T lymphocytes are transduced with a modified TCR that comprises a scFv fragment recognizing a specific cancer antigen (Jackson et al., 2016). Although not a cancer vaccine, CAR-T cell therapies strongly mimic their purpose, by both delivering tumor-specific cytotoxic T cells and having the potential to establish anti-tumor memory (McLellan and Ali Hosseini Rad, 2019).
In addition to antibodies, cell surface receptors can be targeted using receptor ligands, peptides, or small molecules. Indeed, ligand/receptor interactions have been exploited to target the epidermal growth factor receptor (EGFR) on urinary bladder cancer, by delivering an EGF-diphteria toxin fusion protein, for example (Yang et al., 2013). Alternatively, cancer-targeting peptides can be found by phage display screening (Zhang et al., 2001), and have the advantages of being smaller and potentially easier to synthesize than antibodies. Lastly, small molecules have been used to functionalize materials to target cancer cell receptors. Particularly, materials conjugated to folate can successfully bind to cancer cell folate receptors with very high affinity (Xia and Low, 2010; Tao et al., 2015).
Not only proteins can be targeted at the cancer cell surface, but also glycans or lipids. Particularly, cancer cells express specific glycans or overexpress others as compared to healthy cells (Dube and Bertozzi, 2005), which can be targeted using glycan-binding proteins, notably lectins. For example, the conjugation of the specific rBC2LC-N lectin to bacterial exotoxin has shown successful targeting and therapeutic effects in pancreatic cancer (Shimomura et al., 2018). Interestingly, lectins themselves can induce autophagy or apoptosis of cancer cells (Yau et al., 2015). Similarly, cancer cells lack the ability to maintain the natural lipid asymmetry in cell membranes, thus exposing phosphatidylserine (PS) and phosphatydilethanolamine (PE) on the outer leaflet of their membranes, has encouraged the development PS/PE-targeting drug delivery systems (De et al., 2018).
Finally, differences have been found in the physicochemical properties of cancer cell membranes, which are more negative and more fluid, as compared to healthy cells (Bernardes and Fialho, 2018). Approaches targeting such differences have been attempted (Chen et al., 2016a), but are likely to be less efficient than those relying on specific interactions for drug delivery purposes.
Targeting the cancer cell cytosol (Figure 4B)
Some strategies have been developed to target the cancer cell cytoplasm, for example by using molecular transport via specific channel receptors. Particularly, many cancer cells overexpress the GLUT1 glucose channel receptor to increase their glucose metabolism. Conjugation of glucose to small molecules enables their transport through GLUT1 into the cytoplasm, as exemplified by Glucosfamid or glucose-conjugated paclitaxel (Liu et al., 2007; Calvaresi and Hergenrother, 2013). Interestingly, conjugation of glucose to larger moieties, such as nanoparticles, has allowed an increase of their uptake by cancer cells, yet via clathrin-dependent endocytosis (Dreifuss et al., 2018).
Another approach that can be considered as cytoplasmic targeting is the delivery of small molecules that passively diffuse through cell membranes, but that mostly display activity in cancer cells upon binding to their cytoplasmic target. For example, BRAF inhibitors selectively target the BRAFV600E mutated protein present in melanoma cells but not in healthy ones (Sharma et al., 2012; Karoulia et al., 2017). Similarly, prodrugs can be engineered to be activated in the cytosol of cancer cells specifically (Zhang et al., 2017b).
Targeting the cancer cell genome (Figure 4C)
Cancer cells additionally upregulate some specific promoters that can be targeted for gene delivery. For example, the telomerase hTERT promoter has been shown to be re-activated in 90% of human cancers while being silenced in healthy cells (Jafri et al., 2016). Delivering genes under the control of such promoters allows restriction of their expression to cancer cells (Zarogoulidis et al., 2013). Such approaches have mostly been used to deliver cytotoxic genes, such as suicide genes inducing cancer cell death (Xu and Goldkorn, 2016). Interestingly, multiple cancer-specific promoters can be used in combination to further enhance cancer-targeting specificity (Li et al., 2005).
Targeting the cancer cell with infectious materials (Figure 4D)
It has been demonstrated that cancer cells are more prone to infection than healthy cells, and that some pathogens tend to favorably infect cancer cells (Marelli et al., 2018; Zhou et al., 2018). As a consequence, strategies targeting cancer cells with infectious materials have been developed. As an example of choice, one of the two approved therapeutic cancer vaccines uses the BCG bacteria as a tumor-killing agent. Currently, most of the infectious-based cancer therapies under development focus on the use of oncolytic viruses, which naturally infect, replicate inside and lyse cancer cells, thus releasing additional viruses in the tumor. Viruses can be engineered at multiple levels to improve their specificity to cancer cells. First, their capsid or envelope can be modified to enhance tropism to cancer cell surfaces (Büning and Srivastava, 2019). For instance, Münch et al. (2013) have modified the capsid of an adeno-associated virus to specifically target HER2-positive tumors upon systemic delivery. Secondly, the virus genome can be manipulated to incorporate cancer-specific promoters, as described above. Similarly, bacteria have been engineered for enhanced cancer-targeting. For instance, Salmonella typhimurium decorated with anti-CEA scFv have efficiently targeted CEA-expressing MC38 colon cancer models (Bereta et al., 2007). In cancer vaccines, the use of pathogens as delivery tools is particularly interesting as they naturally contain multiple MAMPs that act as adjuvants.
Targeting Tumor-Associated Stromal Cells
Tumors are not only composed of cancer cells, but also of tumor-associated stromal cells (TASCs), including endothelial cells and fibroblasts. Since stromal cells are less heterogenous populations than cancer cells, their markers are more conserved across cancers and patients, possibly enabling the development of less personalized targeting therapies. To date, most strategies that target TASCs focus on the use of antibodies, peptides and small molecules, although gene therapy remains a feasible option. Since TASCs often support tumor development, progression and dissemination, most therapies aim at blocking or killing them to stop tumor nurturing.
Targeting tumor-associated blood endothelial cells (BECs)
Because tumors get nutrients and oxygen from blood vessels, many strategies have been developed to prevent tumor angiogenesis and subsequently starve the tumor. Clinically, tumor angiogenesis has been mostly inhibited by blocking the vascular endothelial growth factor-A (VEGF-A), using either blocking antibodies against it or against its receptor VEGF-R2, or tyrosine kinase inhibitors that block VEGF-R2 downstream signaling (Zirlik and Duyster, 2018). Furthermore, tumoral BECs express some surface proteins that are not commonly present in healthy vasculature, such as VEGF-R3 or endoglin, which can be used to preferentially target them (Laakkonen et al., 2007; Dallas et al., 2008). Importantly, it has been shown that modulating tumor vasculature strongly impacts the outcomes of immunotherapies, since blood vessels directly control immune cell trafficking into tumors. Indeed, some anti-angiogenic therapies have been shown to enhance intratumoral trafficking of lymphocytes in some cancers, notably by increasing the expression of adhesion molecules involved in T cell homing and by increased chemokine expression (Tartour et al., 2011; Wu et al., 2016). Alternatively, several strategies have been explored to normalize tumor vasculature rather than blocking it to relieve hypoxia, which has been shown to impair T cell cytotoxic activities (Huang et al., 2013; Uldry et al., 2017).
Targeting tumor-associated lymphatic endothelial cells (LECs)
The presence of lymphatic vessels in tumors has been associated with increased metastasis and overall poor diagnosis. Nevertheless, when combined with immunotherapies, lymphatic vessels can instead promote anti-tumor responses by increasing immune infiltrates into tumors, as it has been demonstrated in mouse melanoma models and positively correlated in human melanoma (Fankhauser et al., 2017). By draining tumor interstitial fluid and debris, lymphatic capillaries of LECs are strongly exposed to tumor antigens, which they scavenge and present on their MHCs to further modulate immune responses (Hirosue et al., 2014; Kimura et al., 2015). Due to their endocytic capability, targeting LECs has been achieved passively by injecting drugs upstream to their draining route, using liposomes for instance (Oussoren et al., 1997), although such passive approaches can side-target other phagocytic cells. In contrast, preferential targeting of LECs has been achieved by using anti-VEGF-R3 antibodies (Saif et al., 2016). More recently, a dual targeting approach using anti-Lyve-1 combined with anti-Podoplanin antibodies coated on magnetic nanoparticles has been engineered, although the specificity of this last approach remains to be demonstrated in vivo (Wu et al., 2019). Noteworthily, LECs can not only be targeted in the tumor and lymphatic vessels, but also in the lymph nodes where they strongly interact with immune cells.
Targeting tumor-associated fibroblasts (TAFs)
Tumor-associated fibroblasts, which constitute a preponderant population in solid tumors, have been shown to promote cancer progression, mediate tumor immunosuppression and affect T cell infiltration (Shiga et al., 2015; Liu et al., 2019), and have therefore been chosen as targets for direct killing. For example, TAFs have been preferentially targeted using lipid-protamine-DNA nanoparticles displaying aminoethylanisamide (AEAA) ligands at their surface, which can bind to their cognate sigma receptors highly expressed by TAFs (Liu et al., 2019). Another interesting approach for the therapeutic use of TAFs in cancer immunotherapy has been proposed by Müller et al. (2008) who have engineered a bispecific antibody-derived fusion protein that targets on one side the Fibroblast Activation Protein (FAP) receptors on TAFs, and on the other side display the extracellular portion of 4-1BBL, a co-stimulatory molecule that promotes T cell activation. This approach of rendering TAFs immunostimulatory could be highly relevant in the context of therapeutic vaccines, to increase intratumoral immunogenicity and endogenous T cell stimulation.
Targeting other stromal cells
Other stromal cells present in the tumor microenvironment could be used as potential targets to localize vaccines in the tumor, for example, stromal stem cells such as MSCs (Poggi et al., 2018). Interestingly, MSCs can actively migrate from the systemic circulation into the tumor microenvironment via chemokine gradient sensing, as seen before in the section “Targeting the Tumor Biochemical Environment.”
Targeting Tumor Extracellular Matrix (ECM)
In addition to cell targeting, the tumoral ECM has been exploited for the delivery of anti-cancer or immunomodulatory drugs. During tumorigenesis, the ECM is remodeled and dysregulated, leading to changes in composition as compared to healthy ECMs. Such differences in tumor ECM composition make it amenable to targeting of cancer therapeutics. Targeting the tumoral ECM is particularly relevant for the delivery of cytokines or similar signaling molecules, considering the important physiological roles of the ECM in regulating cytokine spatiotemporal release and activity in vivo (Frantz et al., 2010; Martino et al., 2014; Briquez et al., 2016). Various components of the ECM have been targeted within tumors, particularly glycoproteins, fibrous scaffold proteins (e.g., collagen), and glycosaminoglycans.
Glycoprotein targeting
Some glycoproteins are differently spliced or overexpressed in tumors; for example, the EDA and EDB domains of fibronectin are present in tumors and wounded tissues, but absent in normal matrices. Both of these domains have served as targets for the delivery of cytokines and small molecules into tumoral ECMs (Kaspar et al., 2005; Hutmacher and Neri, 2019). Particularly, the anti-EDB antibody fused to IL-2 and TNF have shown promising results in enhancing anti-tumor immunity and are currently being tested in clinical trials (Hutmacher and Neri, 2019). Other glycoproteins can be similarly targeted, such as tenascin-C or the G45 domain of laminin-332 that are respectively, absent and degraded in physiological ECMs, but often expressed in tumoral matrices. Interestingly, functionalization of drug-loaded nanoliposomes with tenascin-C-binding peptides or sulfatide – a tenascin-C-binding glycosphingolipid – showed successful tumor targeting and reduction of drug side-effects (Lin et al., 2014; Chen et al., 2016b).
Collagen targeting
Collagen is another ECM component that can be targeted, as it is present at higher levels in many types of tumors compared to normal tissue (Provenzano et al., 2008; Zhou et al., 2017). Consequently, engineering drugs for collagen targeting can increase in situ retention within the tumor. Indeed, intratumoral delivery of IL-2 and IL-12 fused with lumican, a collagen VI-binding protein, has led to increased sustainability, efficacy and safety of these cytokines (Momin et al., 2019). Additionally, collagen is not naturally exposed to the blood stream in healthy tissues, but is accessible upon increase of vessel permeability during inflammation and in cancer. As such, drugs conjugated to appropriate collagen-binding moieties can accumulate in the tumor microenvironment upon systemic delivery (e.g., intravenous). As an example, the fusion of an anti-EGFR Fab to a collagen-binding peptide exhibited localization to A431 xenografts and enhanced retention time compared to untargeted anti-EGFR Fab when injected intraperitoneally (Liang et al., 2016). In addition, a recent study by Ishihara et al. used a collagen I and III-binding domain derived from von Willebrand factor to target checkpoint blockade antibodies and IL-2 to various tumors, which resulted in tumor growth suppression and lowered drug toxicity (Ishihara et al., 2019).
Proteoglycan and glycosaminoglycan (GAG) targeting
Similarly to other ECM components, GAGs are also dysregulated in cancers, and many of them, such as hyaluronan (HA) or chondroitin sulfate (CS) are overexpressed by tumor cells (Toole, 2004; Vallen et al., 2014). Accordingly, GAG-binding peptides have been used to target materials into tumors. For instance, silver nanoparticles functionalized with the IP3 HA-binding peptide have successfully localized into peritoneal tumors (Ikemoto et al., 2017). In addition to being overexpressed, some tumoral GAGs also display differences in sulfation patterns, which can be further exploited to enhance targeting specificity (Vallen et al., 2014). As an example, Salanti et al. targeted a specific sulfated form of CS, called CSA, in melanoma and prostate tumors by using a peptide derived from the Plasmodium falciparum VAR2CSA protein (Salanti et al., 2015). Other approaches to target CS have used antibody fragments (van der Steen et al., 2017) or liposomes containing a cationic lipid TRX-20 (Lee et al., 2002). Lastly, other GAGs can be used as tumoral ECM targets, notably heparan sulfates and aggrecans (Raavé et al., 2018).
Promiscuous ECM Targeting
Finally, a more versatile approach for cancer ECM targeting is to target multiple ECM components at the same time rather than just a single component at a time. For example, this strategy has been exploited by conjugating materials to the heparin-binding domain (HBD) of placenta growth factor-2 (PlGF-2123–144), which has been shown to display a super-affinity for multiple ECM components (Martino et al., 2014). In one study, PlGF-2123–144 successfully allowed the retention of checkpoint blockade antibodies within the tumor environment, improving efficacy and safety of these antibodies (Ishihara et al., 2017). However, such an approach is limited to local delivery as it also targets fibrinogen in the blood. Another multi-targeting strategy used a bispecific peptide (PL1) that binds to both fibronectin EDB and tenascin-C. Lingasamy et al. used PL1 to target iron oxide nanoworms loaded with proapoptotic peptides into glioblastoma and prostate carcinoma tumors (Lingasamy et al., 2019).
Engineering Immune Cell-Targeting Materials
Immune cells are the main actors to mediate tumor cell killing and establish anti-cancer memory. As such, the main challenge of cancer vaccines is to leverage their potential by targeting and stimulating the correct set of immune cells with the appropriate signals. Immune cells can be targeted at various places including in the tumor (depending on the presence of immune infiltrates), in lymphoid organs or in distant tissues, from where they can migrate to the lymphoid organs. As immune cells are dynamically migrating between the different sites, we here classified targeting strategies per cell type rather than by site. We focus on strategies targeting DCs, T cells, B cells, and NK cells as being the most studied targeted cell types for cancer vaccines. However, all immune cells and mechanisms able to mediate or enhance cytotoxicity and memory could play a significant role in the development of future strategies. In particular, macrophages and neutrophils, capable of ADCP, or the complement system are of emerging interests in cancer immunotherapies (Gul and van Egmond, 2015; Reis et al., 2017). Whether their potential could be harnessed to enhance cancer vaccination, however, remains unclear.
Targeting Dendritic Cells (DCs)
Dendritic Cells are considered as the most potent APCs and play a pivotal role in triggering adaptive immune responses, due to their ability to promote T and B cell maturation. Consequently, targeting antigens and adjuvants to DCs is an important strategy pursued in cancer vaccines, and has been primarily explored ex vivo and more recently in vivo.
Ex vivo DC vaccines
The development of DC-targeting therapies has been made possible by the technical advances in DC isolation and in vitro culture. Modifying DCs ex vivo has the unique advantage of avoiding the influence of the tumor immunosuppression, which has been shown to impair DCs functions in vivo (Pinzon-Charry et al., 2005). Conventional DCs can be isolated from the patient peripheral blood and targeted in vitro with both cancer-associated antigens and adjuvants to stimulate their maturation. Delivery of tumor-associated antigens to DCs has been tested in the form of mRNA transfection (Borch et al., 2016), tumor lysates (Yu et al., 2004), co-culture with tumor cells or even fusion of tumor cells with DCs (Yu et al., 2004). Similarly, many adjuvants have been used to mature the DCs, notably exposure to TLR-3, -4, -7/-8, and -9 ligands and co-stimulatory receptor ligands such as CD40L or cytokines (Saxena and Bhardwaj, 2018). Following in vitro antigen loading and maturation, DCs are re-administered to the patient via intradermal, intravenous, intranodal or intralymphatic, or intratumoral injection (Shang et al., 2017). Noteworthily, the currently approved therapeutic cancer vaccine Sipuleucel-T for prostate cancer treatment is an ex vivo DC-based vaccine.
In vivo DC-targeting vaccines
While delivered nanomaterials are taken up by DCs in vivo, more specific targeting has been achieved by the use of antibodies against DC-specific receptors, such as anti-DEC205, anti-CLEC9A or anti-DC-SIGN (Bonifaz et al., 2004; Hesse et al., 2013; Tullett et al., 2016). Interestingly, it has been shown that targeting different regions on the DC-SIGN receptor can modulate the internalization pathway and influence the extent of antigen presentation on MHCI by DCs (Tacken et al., 2011). In this study, Tacken et al. additionally highlighted that co-targeting of the antigen and adjuvants at the same time using PLGA nanoparticles enhances DC maturation (Tacken et al., 2011; Zitvogel and Palucka, 2011). In addition to antibodies, DCs have been targeted by materials conjugated to mannose or TLR-ligands, the latter being used as targeting tools in addition to being adjuvants (Thomann et al., 2011; Wilson et al., 2019). Upon in vivo targeting, DCs migrate to lymph nodes wherein they can efficiently educate T cells. Interestingly, it has been shown that pre-conditioning the vaccine site with pro-inflammatory cytokines enhances DC migration to lymphoid organs (Mitchell et al., 2015).
Targeting T Lymphocytes
T lymphocytes are the main actors in anti-tumor cell-mediated cytotoxicity, and their presence in the tumor correlates with good prognosis (Gooden et al., 2011). Currently, the most widely used T cell-targeting strategy relies on antibodies that bind to T cell surface receptors, such as PD1, CTLA4 or LAG3, and has been developed in the context of checkpoint blockade immunotherapies (Grywalska et al., 2018; Havel et al., 2019). In cancer vaccination, however, T cells have been mainly targeted by materials that mimic the role of APCs, aiming at promoting endogenous cancer-specific T cell responses.
DC-derived exosomes
One major challenge of DC vaccine remains the control of DC fates upon delivery, which has fostered the development of cell-free alternative strategies. In particular, DCs secrete exosomes that carry antigen-MHC complexes, both MHCI and MHCII, as well as co-stimulatory receptors, which make them capable of activating T cell responses (Zitvogel et al., 1998). DC-derived exosomes have been tested in clinical trials with encouraging outcomes (Morse et al., 2005; Tian and Li, 2017).
Artificial APCs (aAPCs)
Similar to the use of exosomes, synthetic materials have been developed to bypass APCs and directly activate T cells ex vivo and in vivo. Indeed, aAPCs are composed of a biomaterial (lipid-, polymeric- or inorganic-based) with all three signals required for T-cell activation, including MHC-antigen complex, co-stimulatory molecules and cytokines. Generally the co-stimulatory moieties are the antibodies anti-CD3 or anti-CD28 and the cytokines can be any T cell stimulating cytokines, such as IL-2, IL-7, IL-15, or IL-23 (Steenblock et al., 2011; Eggermont et al., 2014; Wang et al., 2017). Interestingly, it has been demonstrated that microparticles are more suited for the design of aAPCs than smaller sized ones as they provide increased interaction with T cells, due to their lower curvature. Particularly, ellipsoidal nanoworm particles induce higher activation efficacy compared to spherical ones (Mandal et al., 2013; Sunshine et al., 2014). In this context, it has been additionally shown that more sustained release of cytokines elicits a stronger immune response. Lastly, these aAPC platforms have also been used to deliver immunosuppressive blocking antibodies and showed promising results in delayed tumor growth (Eggermont et al., 2014; Zhang et al., 2017a).
Targeting B Cells
B cells have the dual role of serving as APCs and mediating humoral immune responses. Although the first role has taken more attention in the development of therapeutic cancer vaccines, new strategies are being explored to induce potent anti-tumor immunity relying on humoral responses.
B cells as APCs in cancer vaccines
B cells can be turned into potent APCs upon stimulation of their CD40 receptors, using soluble CD40L recombinant proteins, CD40 agonist antibodies or co-culturing them with CD40L-expressing feeder cells (Wennhold et al., 2019). CD40-activated B cells have been then shown to present antigens on both MHCI and MHCII to trigger CD8+ and CD4+ T cell responses respectively, upon adoptive transfer in humans (Schultze et al., 1997; Lapointe et al., 2003; Wennhold et al., 2019). As a consequence, B cells are being considered as an alternative source of APCs to DCs in cancer vaccines, as they are easier to isolate in sufficient number from patient peripheral blood, even from cancer patients, and are less sensitive to tumor immunosuppression, among other advantages (von Bergwelt-Baildon et al., 2002; Shimabukuro-Vornhagen et al., 2012). In addition, B cells can be targeted in vivo for antigen presentation, notably using anti-CD19 antibody as a targeting tool, as exemplified by Ding et al. (2008).
Humoral-based cancer vaccines
Another interesting approach relying on B cells is the vaccination against B-cell cancer epitopes, which generates large amounts of endogenous antibodies able to target the cancer cell surface and induce their death via antibody-mediated cytotoxicity (Kaumaya, 2015). As an example, HER2-Vaxx (developed by Imugene) induces polyclonal antibody responses against a specific epitope of HER2 and is being tested in patients with HER2-positive gastric, esophageal, and breast cancers. In the current vaccine, the HER2 peptide is formulated using the carrier protein CRM197, a non-toxic mutant of diphtheria toxin, combined with an adjuvant, whereas the previous formulation used virosome-based materials. This therapy has successfully passed a Phase I safety trial and is under evaluation in a Phase II trial. In addition to direct cytotoxicity, such a B-cell epitope peptide vaccine could provide immune protection against cancer relapse, via the presence of long-term circulating antibodies and memory B cells.
Targeting NK Cells
Although NK cells are not the primary targeted cell type in cancer vaccines, they are generating substantial interest due to their cytotoxic ability as well as their strong cooperation with T and B cells. The role of NK cells in cancer immunotherapy is particularly important as they can detect cancer cells that avoid T cell recognition by downregulating their MHC and can mediate antigen-specific ADCC via recognition of cancer cell membrane-bound antibodies (Collins et al., 2011).
Because NK cells are innate immune cells, they do not have, according to our traditional understanding, the ability to mediate direct antigen-specific recognition and immunologic memory, which are central criteria for vaccines. Nevertheless, new roles for NK cells have been unveiled recently, notably in the context of viral infections, suggesting some features of antigen specificity and long-term memory in mouse and primate NK cells (Paust et al., 2010; Reeves et al., 2015; Pahl et al., 2018). Particularly in mice, it has been shown that NK cells can develop specific memory against antigens from murine cytomegalovirus (MCMV), influenza, and vesicular stomatitis virus (VSV) and human immunodeficient virus-1 (HIV), which can be effectively recalled upon antigen re-encounter (Sun et al., 2009; Paust et al., 2010). Currently, researchers are exploring whether such NK memory cells can be harnessed for vaccination purposes in cancer (Sun and Lanier, 2018; Capuano et al., 2019). For example, a study by Romee et al. (2016) has shown that adoptive transfer of cytokine-induced memory-like NK cells reduce leukemia burden in humans.
Post-targeting Fate of Materials
Targeting the material to the appropriate cells is not sufficient to ensure the therapeutic effect of a cancer vaccine. Indeed, upon targeting, the drugs – antigens and/or adjuvants – remain to be delivered in the appropriate subcellular compartments (i.e., where their targets or receptors are located), which can be the endosomes, cytoplasm, nucleus or other cell organelles. Interestingly, most cell-surface targeting strategies will result in drug internalization in endosomes, yet via multiple pathways (Elkin et al., 2016; Owens et al., 2016). Particularly, from the endosomes, drugs can travel to late endosomes and lysosomes, escape into the cytosol, be recycled at the cell surface or be transported to other organelles such as in the endoplasmic reticulum (Cullen and Steinberg, 2018). Upon escape in the cytosol, drugs can further be directed to the nucleus or to other specific sites, for example targeting mitochondria (Battogtokh et al., 2018). Importantly, the material itself (e.g., size, shapes, ligands, etc.) can influence which internalization pathway will be favored (Gratton et al., 2008).
As a consequence, the type of drug to deliver should be rationally chosen; for example, antigens can be in the form of protein, peptides, RNA or DNA. Importantly, proteinaceous antigens delivered from the extracellular space can be presented on MHCII but less so on MHCI, which is generally restricted to presentation of intracellular antigens. As a consequence, extracellularly-delivered proteins should undergo endosomal escape and reach the cytoplasm to trigger CD8+ T cell responses. Accordingly, materials can be engineered to promote endosomal escape (Selby et al., 2017), for example by being pH-sensitive, redox-sensitive, or osmotic change-sensitive to burst the endosomes (Phillips and Gibson, 2014; Kongkatigumjorn et al., 2018; Rangasamy et al., 2018). In addition, some antigenic peptides can be designed to bind to MHCI from the extracellular space, thus bypassing this challenge (Ilca et al., 2018). Interestingly, some APCs subsets (e.g., CD103+ DCs) (Joffre et al., 2012) are known to be naturally capable of mounting extracellular antigens onto MHCI, through a mechanism called antigen cross-presentation, and have therefore raised particular attention in vaccination (Fehres et al., 2014). As an alternative to protein forms, antigens can be delivered as DNA or RNA, which are directly translated into the cytosol. This particular advantage has encouraged the development of DNA vaccines to enhance CTL responses (Tiptiri-Kourpeti et al., 2016).
As to adjuvants, their receptors can be similarly located at the cell surface, in the endosomes or intracellularly. Therefore, materials that aim at co-delivering antigens and adjuvants need to be carefully designed in an integrated way.
Combination of Therapeutic Cancer Vaccines With Other Cancer Therapies
While potent cancer vaccines will strongly activate the immune system to recognize and fight tumors, the activated immune cells still have to infiltrate the tumor and be locally effective to achieve therapeutic effects. These final steps constitute additional challenges, which could be partly overcome by the combination of the vaccine with other cancer therapies.
First, immune cells need to be in contact with the tumor cells to induce their death. The migration of immune cells into tumors is highly dependent on the tumor structure, and can be further dampened through impaired chemokine expression and downregulation of adhesion molecules on endothelial cells (Lanitis et al., 2015; Harjunpää et al., 2019). Consequently, combining cancer vaccines with therapies that ameliorate intratumoral immune infiltrations, such as those modulating local angiogenesis (Wu et al., 2016; Calcinotto et al., 2012) and lymphangiogenesis (Fankhauser et al., 2017), might be highly beneficial.
Secondly, upon reaching the tumor, the immune cells will face an immunosuppressive environment hampering their cytotoxic activity. Indeed, multiple mechanisms are at play in the tumor to prevent tumor cell killing by immune cells, via myeloid-derived suppressive cells, regulatory T cells or the secretion of immunosuppressive factors (e.g., IL-10, TGFβ, IDO) (Binnewies et al., 2018). Nevertheless, the reduction of tumor immunosuppression can potentially be achieved using immune checkpoint blockade, pro-inflammatory cytokines or IDO inhibitors, to mention a few examples.
Lastly, in the tumor, the immune cells still need to detect their targeted antigens in sufficient amount on cancer cells. Particularly, cancer cells that downregulate expression of the antigens or of MHCs will avoid immune recognition and subsequent killing, leading to cancer relapse. Therefore, diversifying antigen targets and immune-mediated cytotoxic mechanisms is essential to reduce the risk of tumor escape. To do so, cancer vaccines can be combined with other therapies that enhance cancer antigen spreading and local DAMPs release from the tumor, such as chemo-, radio- or other immunotherapies (Wang et al., 2018; Joshi and Durden, 2019).
Conclusion and Perspectives
In conclusion, cancer vaccines hold the promises of eradicating tumors and preventing relapse by inducing strong antigen-specific immune responses and long-term memory. Nevertheless, despite the extensive efforts invested in their development over the last decades, very few have thus far been approved in the clinic. However, lessons from successes, developments-in-progress and failures have increased our knowledge on the design of cancer vaccines, providing some rules to rationally engineer materials that enhance their therapeutic outcomes. Overall, we here proposed that the development of potent vaccines requires careful considerations of their (1) intrinsic composition, i.e., antigens and adjuvants, (2) formulation with materials, (3) delivery route and subsequent targeting to specific relevant sites and cell types, (4) subcellular targeting of their receptors and downstream biological pathways, and (5) combination with other cancer treatments. Although not discussed in this review, the optimal dosing and delivery regimen of the vaccine would also need to be precisely determined, which includes the number of doses to be administered and the delay between repeated delivery.
Defining all these parameters constitutes a major challenge as their combined effects remain poorly predictable, thus requiring thorough investigations in vitro and in vivo. Therefore, improvement of cancer models for better translatability would be beneficial to select the most relevant formulations and strategies to move forward in the clinic. In addition, establishing which tumor types/subtypes and subset of patients would be the most responsive to a cancer vaccine remains difficult to date, despite advances in diagnostic tools and immunological tests. In that perspective, clinical data collection, standardization and availability are essential to allow meta-analyses that help researchers and clinicians to draw criteria for the vaccine efficacy. Overall, improving the predictability of vaccines’ outcomes would permit to reduce their cost and the time required for their development.
Finally, the vaccine would also need to be manufactured at scale and to comply with the regulatory authorities. The multitude of available materials and engineering strategies could lead to highly complex formulations, optimized for biological efficacy yet challenging to produce and become approved therapies. Therefore, a good compromise between efficacy and feasibility is essential to accelerate clinical translation of therapeutic cancer vaccines in the near future.
Author Contributions
All authors have contributed in writing of the manuscript. MS and JH have supervised the work and contributed in writing and corrections.
Conflict of Interest
AT was employed by the company Agap2 Zürich, Switzerland.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Renata Mezyk-Kopec for her advice and corrections on the manuscript.
References
Ahmad, Z., Lv, S., Tang, Z., Shah, A., and Chen, X. (2016). Methoxy poly (ethylene glycol)-block-poly (glutamic acid)-graft-6-(2-nitroimidazole) hexyl amine nanoparticles for potential hypoxia-responsive delivery of doxorubicin. J. Biomater. Sci. Polym. Ed. 27, 40–54. doi: 10.1080/09205063.2015.1107707
Allen, F., Bobanga, I. D., Rauhe, P., Barkauskas, D., Teich, N., Tong, C., et al. (2018). CCL3 augments tumor rejection and enhances CD8+ T cell infiltration through NK and CD103+ dendritic cell recruitment via IFNγ. OncoImmunology 7:e1393598. doi: 10.1080/2162402X.2017.1393598
Almagro, J. C., Daniels-Wells, T. R., Perez-Tapia, S. M., and Penichet, M. L. (2018). Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front. Immunol. 8, 495–19. doi: 10.3389/fimmu.2017.01751
American Cancer Society (2018). Cancer Facts and Figures 2018. Atlanta, GA: American Cancer Society.
Ammi, R., De Waele, J., Willemen, Y., Van Brussel, I., Schrijvers, D. M., Lion, E., et al. (2015). Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol. Ther. 146, 120–131. doi: 10.1016/j.pharmthera.2014.09.010
Anderson, C. F., and Cui, H. (2017). Protease-sensitive nanomaterials for cancer therapeutics and imaging. Ind. Eng. Chem. Res. 56, 5761–5777. doi: 10.1021/acs.iecr.7b00990
André, F., Schartz, N. E. C., Chaput, N., Flament, C., Raposo, G., Amigorena, S., et al. (2002). Tumor-derived exosomes: a new source of tumor rejection antigens. Vaccine 20(Suppl 4), A28–A31.
Ankita, D., Ashish, B., and Raj, K. N. (2019). Nanoparticles as carriers for drug delivery in cancer. Artif. Cells Nanomed. Biotechnol. 46, S295–S305. doi: 10.1080/21691401.2018.1457039
Anselmo, A. C., Zhang, M., Kumar, S., Vogus, D. R., Menegatti, S., Helgeson, M. E., et al. (2015). Elasticity of nanoparticles influences their blood circulation, phagocytosis, endocytosis, and targeting. ACS Nano 9, 3169–3177. doi: 10.1021/acsnano.5b00147.
Apelbaum, A., Yarden, G., Warszawski, S., Harari, D., and Schreiber, G. (2013). Type I interferons induce apoptosis by balancing cFLIP and caspase-8 independent of death ligands. Mol. Cell. Biol. 33, 800–814. doi: 10.1128/MCB.01430-1412
Arnida Janát-Amsbury, M. M., Ray, A., Peterson, C. M., and Ghandehari, H. (2011). Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur. J. Pharm. Biopharm. 77, 417–423. doi: 10.1016/j.ejpb.2010.11.010
Battogtokh, G., Cho, Y. Y., Lee, J. Y., Lee, H. S., and Kang, H. C. (2018). Mitochondrial-targeting anticancer agent conjugates and nanocarrier systems for cancer treatment. Front. Pharmacol. 9, 17450–20. doi: 10.3389/fphar.2018.00922.
Bayrami, V., Keyhanfar, M., Mohabatkar, H., Mahdavi, M., and Moreau, V. (2016). In silico prediction of B cell epitopes of the extracellular domain of insulin-like growth factor-1 receptor. Mol. Biol. Res. Commun. 5, 201–214.
Behzadi, S., Serpooshan, V., Tao, W., Hamaly, M. A., Alkawareek, M. Y., Dreaden, E. C., et al. (2017). Cellular uptake of nanoparticles: journey inside the cell. Chem. Soc. Rev. 46, 4218–4244. doi: 10.1039/C6CS00636A
Beningo, K. A., and Wang, Y. L. (2002). Fc-receptor-mediated phagocytosis is regulated by mechanical properties of the target. J. Cell Sci. 115, 849–856.
Bennewith, K. L., and Dedhar, S. (2011). Targeting hypoxic tumour cells to overcome metastasis. BMC Cancer 11:504. doi: 10.1186/1471-2407-11-504
Bereta, M., Hayhurst, A., Gajda, M., Chorobik, P., Targosz, M., Marcinkiewicz, J., et al. (2007). Improving tumor targeting and therapeutic potential of Salmonella VNP20009 by displaying cell surface CEA-specific antibodies. Vaccine. 25, 4183–4192. doi: 10.1016/j.vaccine.2007.03.008
Bernardes, N., and Fialho, A. (2018). Perturbing the dynamics and organization of cell membrane components: a new paradigm for cancer-targeted therapies. IJMS 19, 3871–19. doi: 10.3390/ijms19123871
Bhat, P., Leggatt, G., Waterhouse, N., and Frazer, I. H. (2017). Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 8:e2836. doi: 10.1038/cddis.2017.67.
Binnewies, M., Roberts, E. W., Kersten, K., Chan, V., Fearon, D. F., Merad, M., et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550. doi: 10.1038/s41591-018-0014-x
Black, K. C. L., Wang, Y., Luehmann, H. P., Cai, X., Xing, W., Pang, B., et al. (2014). Radioactive 198Au-doped nanostructures with different shapes for in vivo analyses of their biodistribution, tumor uptake, and intratumoral distribution. ACS Nano 8, 4385–4394. doi: 10.1021/nn406258m
Bonifaz, L. C., Bonnyay, D. P., Charalambous, A., Darguste, D. I., Fujii, S.-I., Soares, H., et al. (2004). In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 199, 815–824. doi: 10.1084/jem.20032220
Borch, T. H., Engell-Noerregaard, L., Iversen, T. Z., Ellebaek, E., Met, Ö., Hansen, M., et al. (2016). mRNA-transfected dendritic cell vaccine in combination with metronomic cyclophosphamide as treatment for patients with advanced malignant melanoma. OncoImmunology 5, 1–12. doi: 10.1080/2162402X.2016.1207842
Briquez, P. S., Clegg, L. E., Martino, M. M., Gabhann, F. M., and Hubbell, J. A. (2016). Design principles for therapeutic angiogenic materials. Nat. Rev. Mater. 1, 15006–15. doi: 10.1038/natrevmats.2015.6
Büning, H., and Srivastava, A. (2019). Capsid modifications for targeting and improving the efficacy of AAV vectors. Mol. Ther. Methods Clin. Dev. 12, 248–265. doi: 10.1016/j.omtm.2019.01.008
Calcinotto, A., Grioni, M., Jachetti, E., Curnis, F., Mondino, A., Parmiani, G., et al. (2012). Targeting TNF-α to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J. Immunol. 188, 2687–2694. doi: 10.4049/jimmunol.1101877
Calvaresi, E. C., and Hergenrother, P. J. (2013). Glucose conjugation for the specific targeting and treatment of Cancer. Chem. Sci. 4, 2319–30. doi: 10.1039/c3sc22205e
Capuano, C., Pighi, C., Battella, S., Santoni, A., Palmieri, G., and Galandrini, R. (2019). Memory NK cell features exploitable in anticancer immunotherapy. J. Immunol. Res. 2019, 1–8. doi: 10.1155/2019/8795673
Cawood, R., Hills, T., Wong, S. L., Alamoudi, A. A., Beadle, S., Fisher, K. D., et al. (2012). Recombinant viral vaccines for cancer. Trends Mol. Med. 18, 564–574. doi: 10.1016/j.molmed.2012.07.007
Cerundolo, V., Silk, J. D., Masri, S. H., and Salio, M. (2009). Harnessing invariant NKT cells in vaccination strategies. Nat. Rev. Immunol. 9, 28–38. doi: 10.1038/nri2451.
Chauhan, V. P., Stylianopoulos, T., Martin, J. D., Popoviæ, Z., Chen, O., Kamoun, W. S., et al. (2012). Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 7, 383–388. doi: 10.1038/nnano.2012.45
Chen, B., Le, W., Wang, Y., Li, Z., Wang, D., Lin, L., et al. (2016a). Targeting negative surface charges of cancer cells by multifunctional nanoprobes. Theranostics 6, 1887–1898. doi: 10.7150/thno.16358
Chen, B., Wang, Z., Sun, J., Song, Q., He, B., Zhang, H., et al. (2016b). A tenascin C targeted nanoliposome with navitoclax for specifically eradicating of cancer-associated fibroblasts. Nanomedicine 12, 131–141. doi: 10.1016/j.nano.2015.10.001
Chen, C., and Gao, F. -H. (2019). Th17 Cells paradoxical roles in melanoma and potential application in immunotherapy. Front. Immunol. 10:187. doi: 10.3389/fimmu.2019.00187.
Cheng, S., Nethi, S. K., Rathi, S., Layek, B., and Prabha, S. (2019). Engineered mesenchymal stem cells for targeting solid tumors: therapeutic potential beyond regenerative therapy. J. Pharmacol. Exp. Ther. 370, 231–241. doi: 10.1124/jpet.119.259796
Chithrani, B. D., Ghazani, A. A., and Chan, W. C. W. (2006). Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 6, 662–668. doi: 10.1021/nl052396o
Choi, C. H. J., Zuckerman, J. E., Webster, P., and Davis, M. E. (2011). Targeting kidney mesangium by nanoparticles of defined size. Proc. Natl. Acad. Sci. U.S.A. 108, 6656–6661. doi: 10.1073/pnas.1103573108
Chulpanova, D., Solovyeva, V., Kitaeva, K., Dunham, S., Khaiboullina, S., and Rizvanov, A. (2018). Recombinant viruses for cancer therapy. Biomedicines 6, 94–14. doi: 10.3390/biomedicines6040094.
Cluff, C. W. (2010). Monophosphoryl lipid A (MPL) as an adjuvant for anti-cancer vaccines: clinical results. Adv. Exp. Med. Biol. 667, 111–123. doi: 10.1007/978-1-4419-1603-7-10
Collins, D. M., O’Donovan, N., McGowan, P. M., O’Sullivan, F., Duffy, M. J., and Crown, J. (2011). Trastuzumab induces antibody-dependent cell-mediated cytotoxicity (ADCC) in HER-2-non-amplified breast cancer cell lines. Ann. Oncol. 23, 1788–1795. doi: 10.1093/annonc/mdr484
Combes, F., Cafferty, S. M., Meyer, E., and Sanders, N. N. (2018). Off-target and tumor-specific accumulation of monocytes, macrophages and myeloid-derived suppressor cells after systemic injection. Neoplasia 20, 848–856. doi: 10.1016/j.neo.2018.06.005.
Cubas, R., Zhang, S., Li, M., Chen, C., and Yao, Q. (2011). Chimeric Trop2 virus-like particles: a potential immunotherapeutic approach against pancreatic cancer. J. Immunother. 34, 251–263. doi: 10.1097/CJI.0b013e318209ee72
Cullen, P. J., and Steinberg, F. (2018). To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 19, 679–696. doi: 10.1038/s41580-018-0053-57
Dallas, N. A., Samuel, S., Xia, L., Fan, F., Gray, M. J., Lim, S. J., et al. (2008). Endoglin (CD105): a marker of tumor vasculature and potential target for therapy. Clin. Cancer Res. 14, 1931–1937. doi: 10.1158/1078-0432.CCR-07-4478
Dan, N., Setua, S., Kashyap, V., Khan, S., Jaggi, M., Yallapu, M., et al. (2018). Antibody-drug conjugates for cancer therapy: chemistry to clinical implications. Pharmaceuticals 11:32. doi: 10.3390/ph11020032
Danhier, F. (2016). To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 244, 108–121. doi: 10.1016/j.jconrel.2016.11.015
de Titta, A., Ballester, M., Julier, Z., Nembrini, C., Jeanbart, L., van der Vlies, A. J., et al. (2013). Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc. Natl. Acad. Sci. U.S.A. 110, 19902–19907. doi: 10.1073/pnas.1313152110
De, M., Ghosh, S., Sen, T., Shadab, M., Banerjee, I., Basu, S., et al. (2018). A novel therapeutic strategy for cancer using phosphatidylserine targeting stearylamine-bearing cationic liposomes. Mol. Ther. Nucleic Acid 10, 9–27. doi: 10.1016/j.omtn.2017.10.019
DeMaria, P. J., and Bilusic, M. (2019). Cancer vaccines. Hematol. Oncol. Clin. North Am. 33, 199–214. doi: 10.1016/j.hoc.2018.12.001
Ding, C., Wang, L., Marroquin, J., and Yan, J. (2008). Targeting of antigens to B cells augments antigen-specific T-cell responses and breaks immune tolerance to tumor-associated antigen MUC1. Blood 112, 2817–2825. doi: 10.1182/blood-2008-05-157396
Dowling, D. J. (2018). Recent advances in the discovery and delivery of tlr7/8 agonists as vaccine adjuvants. Immunohorizons 2, 185–197. doi: 10.4049/immunohorizons.1700063
Dreifuss, T., Ben-Gal, T. -S., Shamalov, K., Weiss, A., Jacob, A., Sadan, T., et al. (2018). Uptake mechanism of metabolic-targeted gold nanoparticles. Nanomedicine 13, 1535–1549. doi: 10.2217/nnm-2018-2022
Dube, D. H., and Bertozzi, C. R. (2005). Glycans in cancer and inflammation — potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 4, 477–488. doi: 10.1038/nrd1751
Eggermont, L. J., Paulis, L. E., Tel, J., and Figdor, C. G. (2014). Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. 32, 456–465. doi: 10.1016/j.tibtech.2014.06.007
Elion, D. L., and Cook, R. S. (2018). Harnessing RIG-I and intrinsic immunity in the tumor microenvironment for therapeutic cancer treatment. Oncotarget 9, 29007–29017. doi: 10.18632/oncotarget.25626
Elkin, S. R., Lakoduk, A. M., and Schmid, S. L. (2016). Endocytic pathways and endosomal trafficking: a primer. Wien. Med. Wochenschr. 166, 196–204. doi: 10.1007/s10354-016-0432-437
Espelin, C. W., Leonard, S. C., Geretti, E., Wickham, T. J., and Hendriks, B. S. (2016). Dual HER2 targeting with trastuzumab and liposomal-encapsulated doxorubicin (MM-302) demonstrates synergistic antitumor activity in breast and gastric cancer. Cancer Res. 76, 1517–1527. doi: 10.1158/0008-5472.CAN-15-1518
Fankhauser, M., Broggi, M. A. S., Potin, L., Bordry, N., Jeanbart, L., Lund, A. W., et al. (2017). Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci. Transl. Med. 9:eaal4712. doi: 10.1126/scitranslmed.aal4712
Farhood, B., Najafi, M., and Mortezaee, K. (2018). CD8 +cytotoxic T lymphocytes in cancer immunotherapy: a review. J. Cell. Physiol. 234, 8509–8521. doi: 10.1002/jcp.27782
Fehres, C. M., Unger, W. W. J., Garcia-Vallejo, J. J., and van Kooyk, Y. (2014). Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front. Immunol. 5:149. doi: 10.3389/fimmu.2014.00149.
Finn, O. J. (2017). Human tumor antigens yesterday, today, and tomorrow. Cancer Immunol. Res. 5, 347–354. doi: 10.1158/2326-6066.CIR-17-0112
Foged, C., Brodin, B., Frokjaer, S., and Sundblad, A. (2005). Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 298, 315–322. doi: 10.1016/j.ijpharm.2005.03.035
Frantz, C., Stewart, K. M., and Weaver, V. M. (2010). The extracellular matrix at a glance. J. Cell Sci. 123, 4195–4200. doi: 10.1242/jcs.023820.
Fröhlich, E. (2012). The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 19, 5577–15. doi: 10.2147/IJN.S36111
Fujii, S. -I., and Shimizu, K. (2019). Immune networks and therapeutic targeting of inkt cells in cancer. Trends Immunol. 40, 984–997. doi: 10.1016/j.it.2019.09.008
Gay, N. J., and Gangloff, M. (2007). Structure and function of toll receptors and their ligands. Annu. Rev. Biochem. 76, 141–165. doi: 10.1146/annurev.biochem.76.060305.151318
Gialeli, C., Theocharis, A. D., and Karamanos, N. K. (2010). Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 278, 16–27. doi: 10.1111/j.1742-4658.2010.07919.x
Golombek, S. K., May, J. -N., Theek, B., Appold, L., Drude, N., Kiessling, F., et al. (2018). Tumor targeting via EPR: strategies to enhance patient responses. Adv. Drug Del. Rev. 130, 17–38. doi: 10.1016/j.addr.2018.07.007
Gong, X., Li, J., Tan, T., Wang, Z., Wang, H., Wang, Y., et al. (2019). Emerging approaches of cell-based nanosystems to target cancer metastasis. Adv. Funct. Mater. 29, 1903441–36. doi: 10.1002/adfm.201903441
González, F. E., Gleisner, A., Falcón-Beas, F., Osorio, F., López, M. N., and Salazar-Onfray, F. (2014). Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum. Vaccin. Immunother. 10, 3261–3269. doi: 10.4161/21645515.2014.982996
Gooden, M. J. M., de Bock, G. H., Leffers, N., Daemen, T., and Nijman, H. W. (2011). The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br. J. Cancer 105, 93–103. doi: 10.1038/bjc.2011.189
Gratton, S. E. A., Ropp, P. A., Pohlhaus, P. D., Luft, J. C., Madden, V. J., Napier, M. E., et al. (2008). The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. U.S.A. 105, 11613–11618. doi: 10.1073/pnas.0801763105
Groom, J. R., and Luster, A. D. (2011). CXCR3 in T cell function. Exp. Cell Res. 317, 620–631. doi: 10.1016/j.yexcr.2010.12.017
Grywalska, E., Pasiarski, M., Góźdź, S., and Roliński, J. (2018). Immune-checkpoint inhibitors for combating T-cell dysfunction in cancer. OTT Vol. 11, 6505–6524. doi: 10.2147/OTT.S150817
Guise, C. P., Mowday, A. M., Ashoorzadeh, A., Yuan, R., Lin, W. -H., Wu, D. -H., et al. (2014). Bioreductive prodrugs as cancer therapeutics: targeting tumor hypoxia. Chin. J. Cancer 33, 80–86. doi: 10.5732/cjc.012.10285
Gul, N., and van Egmond, M. (2015). Antibody-dependent phagocytosis of tumor cells by macrophages: a potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Res. 75, 5008–5013. doi: 10.1158/0008-5472.CAN-15-1330
Gulley, J. L., Madan, R. A., Pachynski, R., Mulders, P., Sheikh, N. A., Trager, J., et al. (2017). Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. J. Natl. Cancer Inst. 109:djw261 doi: 10.1093/jnci/djw261.
Guo, P., Liu, D., Subramanyam, K., Wang, B., Yang, J., Huang, J., et al. (2018). Nanoparticle elasticity directs tumor uptake. Nat. Commun. 9:130. doi: 10.1038/s41467-017-02588-2589
Hanafy, N., El-Kemary, M., and Leporatti, S. (2018). Micelles structure development as a strategy to improve smart cancer therapy. Cancers 10, 238–14. doi: 10.3390/cancers10070238
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Harjunpää, H., Llort Asens, M., Guenther, C., and Fagerholm, S. C. (2019). Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 10, 883–24. doi: 10.3389/fimmu.2019.01078
Hauert, S., and Bhatia, S. N. (2014). Mechanisms of cooperation in cancer nanomedicine: towards systems nanotechnology. Trends Biotechnol. 32, 448–455. doi: 10.1016/j.tibtech.2014.06.010
Havel, J. J., Chowell, D., and Chan, T. A. (2019). The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 19, 133–150. doi: 10.1038/s41568-019-0116-x
Hernandez, C., Huebener, P., and Schwabe, R. F. (2016). Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35, 5931–5941. doi: 10.1038/onc.2016.104
Hesse, C., Ginter, W., Förg, T., Mayer, C. T., Baru, A. M., Arnold-Schrauf, C., et al. (2013). In vivo targeting of human DC-SIGN drastically enhances CD8 +T-cell-mediated protective immunity. Eur. J. Immunol. 43, 2543–2553. doi: 10.1002/eji.201343429
Hirosue, S., Vokali, E., Raghavan, V. R., Rincon-Restrepo, M., Lund, A. W., Corthésy-Henrioud, P., et al. (2014). Steady-state antigen scavenging, cross-presentation, and CD8+ T cell priming: a new role for lymphatic endothelial cells. J. Immunol. 192, 5002–5011. doi: 10.4049/jimmunol.1302492
Hoffmann, S., Vystrèilová, L., Ulbrich, K., Etrych, T., Caysa, H., Mueller, T., et al. (2012). Dual fluorescent HPMA copolymers for passive tumor targeting with pH-sensitive drug release: synthesis and characterization of distribution and tumor accumulation in mice by noninvasive multispectral optical imaging. Biomacromolecules 13, 652–663. doi: 10.1021/bm2015027
Homhuan, A., Prakongpan, S., Poomvises, P., Maas, R. A., Crommelin, D. J. A., Kersten, G. F. A., et al. (2004). Virosome and ISCOM vaccines against Newcastle disease: preparation, characterization and immunogenicity. Eur. J. Pharm. Sci. 22, 459–468. doi: 10.1016/j.ejps.2004.05.005
Hong, L. P. T., Scoble, J. A., Doughty, L., Coia, G., and Williams, C. C. (2011). Cancer-targeting antibody–drug conjugates: site-specific conjugation of doxorubicin to anti-EGFR 528 fab’ through a polyethylene glycol linker. Aust. J. Chem. 64, 779–11. doi: 10.1071/CH11071
Hoshyar, N., Gray, S., Han, H., and Bao, G. (2016). The effect of nanoparticle size on in vivopharmacokinetics and cellular interaction. Nanomedicine 11, 673–692. doi: 10.2217/nnm.16.5
Hu, Q., Katti, P. S., and Gu, Z. (2014). Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale 6, 12273–12286. doi: 10.1039/C4NR04249B.
Huang, Y., Goel, S., Duda, D. G., Fukumura, D., and Jain, R. K. (2013). Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 73, 2943–2948. doi: 10.1158/0008-5472.CAN-12-4354
Huijbers, E. J. M., and Griffioen, A. W. (2017). The revival of cancer vaccines - the eminent need to activate humoral immunity. Hum. Vaccin. Immunother. 13, 1112–1114. doi: 10.1080/21645515.2016.1276140
Hunter, F. W., Wouters, B. G., and Wilson, W. R. (2016). Hypoxia-activated prodrugs: paths forward in the era of personalised medicine. Br. J. Cancer 114, 1071–1077. doi: 10.1038/bjc.2016.79
Hutmacher, C., and Neri, D. (2019). Antibody-cytokine fusion proteins: biopharmaceuticals with immunomodulatory properties for cancer therapy. Adv. Drug Del. Rev. 141, 67–91. doi: 10.1016/j.addr.2018.09.002
Ikemoto, H., Lingasamy, P., Anton Willmore, A. -M., Hunt, H., Kurm, K., Tammik, O., et al. (2017). Hyaluronan-binding peptide for targeting peritoneal carcinomatosis. Tumour Biol. 39:1010428317701628. doi: 10.1177/1010428317701628
Ilca, F. T., Neerincx, A., Wills, M. R., de la Roche, M., and Boyle, L. H. (2018). Utilizing TAPBPR to promote exogenous peptide loading onto cell surface MHC I molecules. Proc. Natl. Acad. Sci. U.S.A. 115, E9353–E9361. doi: 10.1073/pnas.1809465115
Ingrole, R. S. J., and Gill, H. S. (2019). Microneedle coating methods: a review with a Perspective. J. Pharmacol. Exp. Ther. 370, 555–569. doi: 10.1124/jpet.119.258707.
Irvine, D. J., Swartz, M. A., and Szeto, G. L. (2013). Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 12, 978–990. doi: 10.1038/nmat3775
Ishihara, J., Fukunaga, K., Ishihara, A., Larsson, H. M., Potin, L., Hosseinchi, P., et al. (2017). Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med. 9:eaan0401. doi: 10.1126/scitranslmed.aan0401
Ishihara, J., Ishihara, A., Sasaki, K., Lee, S. S.-Y., Williford, J.-M., Yasui, M., et al. (2019). Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl. Med. 11:eaau3259. doi: 10.1126/scitranslmed.aau3259
Jackson, H. J., Rafiq, S., and Brentjens, R. J. (2016). Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 13, 370–383. doi: 10.1038/nrclinonc.2016.36
Jafri, M. A., Ansari, S. A., Alqahtani, M. H., and Shay, J. W. (2016). Roles of telomeres and telomerase in cancer, and advances in telomerase- targeted therapies. Genome Med. 8:69. doi: 10.1186/s13073-016-0324-x
Jeanbart, L., Ballester, M., de Titta, A., Corthésy, P., Romero, P., Hubbell, J. A., et al. (2014). Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol. Res. 2, 436–447. doi: 10.1158/2326-6066.CIR-14-0019-T
Jiang, T., Zhang, Z., Zhang, Y., Lv, H., Zhou, J., Li, C., et al. (2012). Dual-functional liposomes based on pH-responsive cell-penetrating peptide and hyaluronic acid for tumor-targeted anticancer drug delivery. Biomaterials 33, 9246–9258. doi: 10.1016/j.biomaterials.2012.09.027
Joffre, O. P., Segura, E., Savina, A., and Amigorena, S. (2012). Cross-presentation by dendritic cells. Nat Rev Immunol. 12, 557–69. doi: 10.1038/nri3254
Joshi, S., and Durden, D. L. (2019). Combinatorial approach to improve cancer immunotherapy: rational drug design strategy to simultaneously hit multiple targets to kill tumor cells and to activate the immune system. J. Oncol. 2019, 1–18. doi: 10.1155/2019/5245034
Joyce, J. A., Baruch, A., Chehade, K., Meyer-Morse, N., Giraudo, E., Tsai, F. -Y., et al. (2004). Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 5, 443–453. doi: 10.1038/nrc1396
Julier, Z., Martino, M. M., de Titta, A., Jeanbart, L., and Hubbell, J. A. (2015). The TLR4 agonist fibronectin extra domain a is circulation, phagocytosis, endocytosis, and targeting. Sci. Rep. 5:8569. doi: 10.1038/srep08569
Kanchan, V., and Panda, A. K. (2007). Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response. Biomaterials 28, 5344–5357. doi: 10.1016/j.biomaterials.2007.08.015
Karoulia, Z., Gavathiotis, E., and Poulikakos, P. I. (2017). New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 17, 676–691. doi: 10.1038/nrc.2017.79
Kaspar, M., Zardi, L., and Neri, D. (2005). Fibronectin as target for tumor therapy. Int. J. Cancer 118, 1331–1339. doi: 10.1002/ijc.21677
Kaumaya, P. T. (2015). A paradigm shift: cancer therapy with peptide-based B-cell epitopes and peptide immunotherapeutics targeting multiple solid tumor types: emerging concepts and validation of combination immunotherapy. Hum. Vaccin. Immunother. 11, 1368–1386. doi: 10.1080/21645515.2015.1026495
Kimura, T., Sugaya, M., Oka, T., Blauvelt, A., Okochi, H., and Sato, S. (2015). Lymphatic dysfunction attenuates tumor immunity through impaired antigen presentation. Oncotarget 6, 18081–18093. doi: 10.18632/oncotarget.4018
Kongkatigumjorn, N., Smith, S. A., Chen, M., Fang, K., Yang, S., Gillies, E. R., et al. (2018). Controlling endosomal escape using pH-responsive nanoparticles with tunable disassembly. ACS Appl. Nano Mater. 1, 3164–3173. doi: 10.1021/acsanm.8b00338
Kubota, T., Kuroda, S., Kanaya, N., Morihiro, T., Aoyama, K., Kakiuchi, Y., et al. (2018). HER2-targeted gold nanoparticles potentially overcome resistance to trastuzumab in gastric cancer. Nanomedicine 14, 1919–1929. doi: 10.1016/j.nano.2018.05.019
Kulkarni, P., Haldar, M. K., You, S., Choi, Y., and Mallik, S. (2016). Hypoxia-responsive polymersomes for drug delivery to hypoxic pancreatic cancer cells. Biomacromolecules 17, 2507–2513. doi: 10.1021/acs.biomac.6b00350
Laakkonen, P., Waltari, M., Holopainen, T., Takahashi, T., Pytowski, B., Steiner, P., et al. (2007). Vascular endothelial growth factor receptor 3 is involved in tumor angiogenesis and growth. Cancer Res. 67, 593–599. doi: 10.1158/0008-5472.CAN-06-3567
Lai, C.-Y., Trewyn, B. G., Jeftinija, D. M., Jeftinija, K., Xu, S., Jeftinija, S., et al. (2003). A mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules. J. Am. Chem. Soc. 125, 4451–4459. doi: 10.1021/ja028650l
Laidlaw, B. J., Craft, J. E., and Kaech, S. M. (2016). The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 16, 102–111. doi: 10.1038/nri.2015.10
Lanitis, E., Irving, M., and Coukos, G. (2015). Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 33, 55–63. doi: 10.1016/j.coi.2015.01.011
Lapointe, R., Bellemare-Pelletier, A., Housseau, F., Thibodeau, J., and Hwu, P. (2003). CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 63, 2836–2843.
Lasarte, J. J., Casares, N., Gorraiz, M., Hervás-Stubbs, S., Arribillaga, L., Mansilla, C., et al. (2007). The extra domain A from fibronectin targets antigens to TLR4-expressing cells and induces cytotoxic T cell responses in vivo. J. Immunol. 178, 748–756. doi: 10.4049/jimmunol.178.2.748
Laurent, S., Saei, A. A., Behzadi, S., Panahifar, A., and Mahmoudi, M. (2014). Superparamagnetic iron oxide nanoparticles for delivery of therapeutic agents: opportunities and challenges. Expert. Opin. Drug Deliv. 11, 1449–1470. doi: 10.1517/17425247.2014.924501
Lee, C. M., Tanaka, T., Murai, T., Kondo, M., Kimura, J., Su, W., et al. (2002). Novel chondroitin sulfate-binding cationic liposomes loaded with cisplatin efficiently suppress the local growth and liver metastasis of tumor cells in vivo. Cancer Res. 62, 4282–4288.
Leibacher, J., and Henschler, R. (2016). Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res. Ther. 7:7. doi: 10.1186/s13287-015-0271-272
Li, A., Yi, M., Qin, S., Song, Y., Chu, Q., and Wu, K. (2019). Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol. 12:35. doi: 10.1186/s13045-019-0721-x
Li, K., Peers-Adams, A., Win, S. J., Scullion, S., Wilson, M., Young, V. L., et al. (2013). Antigen incorporated in virus-like particles is delivered to specific dendritic cell subsets that induce an effective antitumor immune response in vivo. J. Immunother. 36, 11–19. doi: 10.1097/CJI.0b013e3182787f5e
Li, Y., Idamakanti, N., Arroyo, T., Thorne, S., Reid, T., Nichols, S., et al. (2005). Dual promoter-controlled oncolytic adenovirus CG5757 has strong tumor selectivity and significant antitumor efficacy in preclinical models. Clin. Cancer Res. 11, 8845–8855. doi: 10.1158/1078-0432.CCR-05-1757
Liang, H., Li, X., Bin, W., Chen, B., Zhao, Y., Sun, J., et al. (2016). A collagen-binding EGFR antibody fragment targeting tumors with a collagen-rich extracellular matrix. Sci. Rep. 6:18205. doi: 10.1038/srep18205
Lin, C.-C., and Metters, A. T. (2006). Hydrogels in controlled release formulations: Network design and mathematical modeling. Adv. Drug Deliv. Rev. 58, 1379–1408. doi: 10.1016/j.addr.2006.09.004
Lin, J., Shigdar, S., Fang, D. Z., Xiang, D., Wei, M. Q., Danks, A., et al. (2014). Improved efficacy and reduced toxicity of doxorubicin encapsulated in sulfatide-containing nanoliposome in a glioma model. PLoS One 9:e103736. doi: 10.1371/journal.pone.0103736
Ling, X., Marini, F., Konopleva, M., Schober, W., Shi, Y., Burks, J., et al. (2010). Mesenchymal stem cells overexpressing IFN-β inhibit breast cancer growth and metastases through stat3 signaling in a syngeneic tumor model. Cancer Microenviron. 3, 83–95. doi: 10.1007/s12307-010-0041-48
Lingasamy, P., Tobi, A., Haugas, M., Hunt, H., Paiste, P., Asser, T., et al. (2019). Bi-specific tenascin-C and fibronectin targeted peptide for solid tumor delivery. Biomaterials 219:119373. doi: 10.1016/j.biomaterials.2019.119373
Liu, D.-Z., Sinchaikul, S., Reddy, P. V. G., Chang, M.-Y., and Chen, S.-T. (2007). Synthesis of 2’-paclitaxel methyl 2-glucopyranosyl succinate for specific targeted delivery to cancer cells. Bioorg. Med. Chem. Lett. 17, 617–620. doi: 10.1016/j.bmcl.2006.11.008
Liu, M., Song, W., and Huang, L. (2019). Drug delivery systems targeting tumor-associated fibroblasts for cancer immunotherapy. Cancer Lett. 448, 31–39. doi: 10.1016/j.canlet.2019.01.032
Liu, Y., Zhi, X., Yang, M., Zhang, J., Lin, L., Zhao, X., et al. (2017). Tumor-triggered drug release from calcium carbonate-encapsulated gold nanostars for near-infrared photodynamic/photothermal combination antitumor therapy. Theranostics 7, 1650–1662. doi: 10.7150/thno.17602
Locy, H., Melhaoui, S., Maenhout, S. K., and Thielemans, K. (2018). “Dendritic cells: the tools for cancer treatment,” in Dendritic Cells, ed. S. P. Chapoval, (London: InTech), 1–29. doi: 10.5772/intechopen.79273
Maiti, S., Manna, S., Shen, J., Esser-Kahn, A. P., and Du, W. (2019). Mitigation of hydrophobicity-induced immunotoxicity by sugar poly(orthoesters). J. Am. Chem. Soc. 141, 4510–4514. doi: 10.1021/jacs.8b12205
Mamaeva, V., Rosenholm, J. M., Bate-Eya, L. T., Bergman, L., Peuhu, E., Duchanoy, A., et al. (2009). Mesoporous silica nanoparticles as drug delivery systems for targeted inhibition of notch signaling in cancer. Mol. Ther. 19, 1538–1546. doi: 10.1038/mt.2011.105
Mandal, S., Eksteen-Akeroyd, Z. H., Jacobs, M. J., Hammink, R., Koepf, M., Lambeck, A. J. A., et al. (2013). Therapeutic nanoworms: towards novel synthetic dendritic cells for immunotherapy. Chem. Sci. 4:4168. doi: 10.1039/c3sc51399h
Mandalà, M., and Voit, C. (2013). Targeting BRAF in melanoma: biological and clinical challenges. Crit. Rev. Oncol. 87, 239–255. doi: 10.1016/j.critrevonc.2013.01.003
Marabelle, A., Andtbacka, R., Harrington, K., Melero, I., Leidner, R., de Baere, T., et al. (2018). Starting the fight in the tumor: expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann. Oncol. 29, 2163–2174. doi: 10.1093/annonc/mdy423
Marabelle, A., Kohrt, H., Caux, C., and Levy, R. (2014). Intratumoral immunization: a new paradigm for cancer therapy. Clin. Cancer Res. 20, 1747–1756. doi: 10.1158/1078-0432.CCR-13-2116
Marelli, G., Howells, A., Lemoine, N. R., and Wang, Y. (2018). Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 9:866. doi: 10.3389/fimmu.2018.00866
Martino, M. M., Briquez, P. S., Guc, E., Tortelli, F., Kilarski, W. W., Metzger, S., et al. (2014). Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science 343, 885–888. doi: 10.1126/science.1247663
Matsushita, H., Hosoi, A., Ueha, S., Abe, J., Fujieda, N., Tomura, M., et al. (2015). Cytotoxic T lymphocytes block tumor growth both by lytic activity and IFNγ-dependent cell-cycle arrest. Cancer Immunol. Res. 3, 26–36. doi: 10.1158/2326-6066.CIR-14-0098
McLellan, A. D., and Ali Hosseini Rad, S. M. (2019). Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 129, 664–674. doi: 10.1111/imcb.12254
Mehlen, P., and Puisieux, A. (2006). Metastasis: a question of life or death. Nat. Rev. Cancer 6, 449–458. doi: 10.1038/nrc1886
Meyer, R. A., Sunshine, J. C., Perica, K., Kosmides, A. K., Aje, K., Schneck, J. P., et al. (2015). Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-Cell activation. Small 11, 1519–1525. doi: 10.1002/smll.201402369
Mitchell, D. A., Batich, K. A., Gunn, M. D., Huang, M.-N., Sanchez-Perez, L., Nair, S. K., et al. (2015). Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519, 366–369. doi: 10.1038/nature14320
Mizuhara, T., Saha, K., Moyano, D. F., Kim, C. S., Yan, B., Kim, Y.-K., et al. (2015). Acylsulfonamide-functionalized zwitterionic gold nanoparticles for enhanced cellular uptake at tumor pH. Angew. Chem. Int. Ed. 54, 6567–6570. doi: 10.1002/anie.201411615
Mo, R., Sun, Q., Xue, J., Li, N., Li, W., Zhang, C., et al. (2012). Multistage pH-Responsive liposomes for mitochondrial-targeted anticancer drug delivery. Adv. Mater. 24, 3659–3665. doi: 10.1002/adma.201201498
Momin, N., Mehta, N. K., Bennett, N. R., Ma, L., Palmeri, J. R., Chinn, M. M., et al. (2019). Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci. Transl. Med. 11:eaaw2614. doi: 10.1126/scitranslmed.aaw2614
Morse, M. A., Garst, J., Osada, T., Khan, S., Hobeika, A., Clay, T. M., et al. (2005). A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 3:9. doi: 10.1186/1479-5876-3-9
Moyano, D. F., Goldsmith, M., Solfiell, D. J., Landesman-Milo, D., Miranda, O. R., Peer, D., et al. (2012). Nanoparticle hydrophobicity dictates immune response. J. Am. Chem. Soc. 134, 3965–3967. doi: 10.1021/ja2108905
Müller, D., Frey, K., and Kontermann, R. E. (2008). A novel antibody-4-1BBL fusion protein for targeted costimulation in cancer immunotherapy. J. Immunother. 31, 714–722. doi: 10.1097/CJI.0b013e31818353e9
Münch, R. C., Janicki, H., Völker, I., Rasbach, A., Hallek, M., Büning, H., et al. (2013). Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 21, 109–118. doi: 10.1038/mt.2012.186
Nair, S., and Dhodapkar, M. V. (2017). Natural killer T cells in cancer immunotherapy. Front. Immunol. 8:1178. doi: 10.3389/fimmu.2017.01178
National Institutes of Health of the USA (2019). Type of Cancer Treatments. Available at: www.cancer.gov/about-cancer/treatment/types (accessed 2019).
Nejadnik, H., Ye, D., Lenkov, O. D., Donig, J. S., Martin, J. E., Castillo, R., et al. (2015). Magnetic resonance imaging of stem cell apoptosis in arthritic joints with a caspase activatable contrast agent. ACS Nano 9, 1150–1160. doi: 10.1021/nn504494c
Nguyen-Hoai, T., Pham-Duc, M., Gries, M., Dörken, B., Pezzutto, A., and Westermann, J. (2016). CCL4 as an adjuvant for DNA vaccination in a Her2/neu mouse tumor model. Cancer Gene Ther. 23, 162–167. doi: 10.1038/cgt.2016.9
Noble, G. T., Stefanick, J. F., Ashley, J. D., Kiziltepe, T., and Bilgicer, B. (2014). Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol. 32, 32–45. doi: 10.1016/j.tibtech.2013.09.007
Oussoren, C., Zuidema, J., Crommelin, D. J., and Storm, G. (1997). Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid compostion and lipid dose. Biochim. Biophys. Acta 1328, 261–272. doi: 10.1016/s0005-2736(97)00122-123
Owens, D. E., and Peppas, N. A. (2006). Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 307, 93–102. doi: 10.1016/j.ijpharm.2005.10.010
Owens, G. J., Singh, R. K., Foroutan, F., Alqaysi, M., Han, C.-M., Mahapatra, C., et al. (2016). Sol-gel based materials for biomedical applications. Prog. Mater. Sci. 77, 1–79. doi: 10.1016/j.pmatsci.2015.12.001
Pahl, J. H. W., Cerwenka, A., and Ni, J. (2018). Memory-like NK cells: remembering a previous activation by cytokines and NK cell receptors. Front. Immunol. 9:2796. doi: 10.3389/fimmu.2018.02796
Palladini, A., Thrane, S., Janitzek, C. M., Pihl, J., Clemmensen, S. B., de Jongh, W. A., et al. (2018). Virus-like particle display of HER2 induces potent anti-cancer responses. Oncoimmunology 7:e1408749. doi: 10.1080/2162402X.2017.1408749
Palmerston Mendes, L., Pan, J., and Torchilin, V. (2017). Dendrimers as nanocarriers for nucleic acid and drug delivery in cancer therapy. Molecules 22, 1401–1421. doi: 10.3390/molecules22091401
Park, J. H., Lee, S., Kim, J.-H., Park, K., Kim, K., and Kwon, I. C. (2008). Polymeric nanomedicine for cancer therapy. Prog. Polym. Sci. 33, 113–137. doi: 10.1016/j.progpolymsci.2007.09.003
Paust, S., Gill, H. S., Wang, B.-Z., Flynn, M. P., Moseman, E. A., Senman, B., et al. (2010). Critical role for the chemokine receptor CXCR6 in NK cell–mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 11, 1127–1135. doi: 10.1038/ni.1953
Peek, L. J., Middaugh, C. R., and Berkland, C. (2008). Nanotechnology in vaccine delivery. Adv. Drug Deliv. Rev. 60, 915–928. doi: 10.1016/j.addr.2007.05.017
Peiris, P. M., Bauer, L., Toy, R., Tran, E., Pansky, J., Doolittle, E., et al. (2012). Enhanced delivery of chemotherapy to tumors using a multicomponent nanochain with radio-frequency-tunable drug release. ACS Nano 6, 4157–4168. doi: 10.1021/nn300652p
Peppas, N. A., Bures, P., Leobandung, W., and Ichikawa, H. (2000). Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 50, 27–46.
Peters, S., Reck, M., Smit, E. F., Mok, T., and Hellmann, M. D. (2019). How to make the best use of immunotherapy as first-line treatment of advanced/metastatic non-small-cell lung cancer. Ann. Oncol. 30, 884–896. doi: 10.1093/annonc/mdz109
Phillips, D. J., and Gibson, M. I. (2014). Redox-sensitive materials for drug delivery: targeting the correct intracellular environment, tuning release rates, and appropriate predictive systems. Antioxid. Redox Signal. 21, 786–803. doi: 10.1089/ars.2013.5728
Pinzon-Charry, A., Maxwell, T., and López, J. A. (2005). Dendritic cell dysfunction in cancer: a mechanism for immunosuppression. Immunol. Cell Biol. 83, 451–461. doi: 10.1111/j.1440-1711.2005.01371.x
Poggi, A., Varesano, S., and Zocchi, M. R. (2018). How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. Front. Immunol. 9:262. doi: 10.3389/fimmu.2018.00262
Provenzano, P. P., Inman, D. R., Eliceiri, K. W., Knittel, J. G., Yan, L., Rueden, C. T., et al. (2008). Collagen density promotes mammary tumor initiation and progression. BMC Med. 6:11. doi: 10.1186/1741-7015-6-11
Punt, S., Langenhoff, J. M., Putter, H., Fleuren, G. J., Gorter, A., and Jordanova, E. S. (2015). The correlations between IL-17 vs. Th17 cells and cancer patient survival: a systematic review. OncoImmunology 4:e984547. doi: 10.4161/2162402X.2014.984547
Pushko, P., Pumpens, P., and Grens, E. (2013). Development of virus-like particle technology from small highly symmetric to large complex virus-like particle structures. Intervirology 56, 141–165. doi: 10.1159/000346773
Quinto, C. A., Mohindra, P., Tong, S., and Bao, G. (2015). Multifunctional superparamagnetic iron oxide nanoparticles for combined chemotherapy and hyperthermia cancer treatment. Nanoscale 7, 12728–12736. doi: 10.1039/C5NR02718G
Raavé, R., van Kuppevelt, T. H., and Daamen, W. F. (2018). Chemotherapeutic drug delivery by tumoral extracellular matrix targeting. J. Control. Rel. 274, 1–8. doi: 10.1016/j.jconrel.2018.01.029
Rangasamy, L., Chelvam, V., Kanduluru, A. K., Srinivasarao, M., Bandara, N. A., You, F., et al. (2018). New mechanism for release of endosomal contents: osmotic lysis via Nigericin-Mediated K+/H+ exchange. Bioconjugate Chem. 29, 1047–1059. doi: 10.1021/acs.bioconjchem.7b00714
Reddy, S. T., Rehor, A., Schmoekel, H. G., Hubbell, J. A., and Swartz, M. A. (2006). In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Rel. 112, 26–34. doi: 10.1016/j.jconrel.2006.01.006
Reeves, R. K., Li, H., Jost, S., Blass, E., Li, H., Schafer, J. L., et al. (2015). Antigen-specific NK cell memory in rhesus macaques. Nat. Immunol. 16, 927–932. doi: 10.1038/ni.3227
Reis, E. S., Mastellos, D. C., Ricklin, D., Mantovani, A., and Lambris, J. D. (2017). Complement in cancer: untangling an intricate relationship. Nat. Publ. Group 18, 5–18. doi: 10.1038/nri.2017.97
Ridge, J. P., Di Rosa, F., and Matzinger, P. (1998). A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393, 474–478. doi: 10.1038/30989
Rivolta, I., Panariti, A., and Miserocchi, G. (2012). The effect of nanoparticle uptake on cellular behavior: disrupting or enabling functions? NSA 5:87. doi: 10.2147/NSA.S25515
Robbins, P. D., and Morelli, A. E. (2014). Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208. doi: 10.1038/nri3622
Romee, R., Rosario, M., Berrien-Elliott, M. M., Wagner, J. A., Jewell, B. A., Schappe, T., et al. (2016). Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
Rotman, J., Koster, B. D., Jordanova, E. S., Heeren, A. M., and de Gruijl, T. D. (2019). Unlocking the therapeutic potential of primary tumor-draining lymph nodes. Cancer Immunol. Immunother. 68, 1681–1688. doi: 10.1007/s00262-019-02330-y
Russell, H. V., Strother, D., Mei, Z., Rill, D., Popek, E., Biagi, E., et al. (2007). Phase I trial of vaccination with autologous neuroblastoma tumor cells genetically modified to secrete IL-2 and lymphotactin. J. Immunother. 30, 227–233. doi: 10.1097/01.cji.0000211335.14385.57
Sadhukha, T., O’Brien, T. D., and Prabha, S. (2014). Nano-engineered mesenchymal stem cells as targeted therapeutic carriers. J. Control. Rel. 196, 243–251. doi: 10.1016/j.jconrel.2014.10.015
Saif, M. W., Knost, J. A., Chiorean, E. G., Kambhampati, S. R. P., Yu, D., Pytowski, B., et al. (2016). Phase 1 study of the anti-vascular endothelial growth factor receptor 3 monoclonal antibody LY3022856/IMC-3C5 in patients with advanced and refractory solid tumors and advanced colorectal cancer. Cancer Chemother. Pharmacol. 78, 815–824. doi: 10.1007/s00280-016-3134-3133
Salanti, A., Clausen, T. M., Agerbæk, M. Ø., Nakouzi, A. N. I, Dahlbäck, M., Oo, H. Z., et al. (2015). Targeting human cancer by a Glycosaminoglycan binding malaria protein. Cancer Cell 28, 500–514. doi: 10.1016/j.ccell.2015.09.003
Saleh, A. F., Lázaro-Ibáñez, E., Forsgard, M. A. M., Shatnyeva, O., Osteikoetxea, X., Karlsson, F., et al. (2019). Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale 11, 6990–7001. doi: 10.1039/C8NR08720B
Sánchez-Paulete, A. R., Teijeira, Á., Quetglas, J. I, Rodríguez-Ruiz, M. E., Sánchez-Arráez, Á., Labiano, S., et al. (2018). Intratumoral immunotherapy with XCL1 and sFlt3L Encoded in recombinant semliki forest virus-derived vectors fosters dendritic cell-mediated t-cell cross-priming. Cancer Res. 78, 6643–6654. doi: 10.1158/0008-5472.CAN-18-0933
Sautès-Fridman, C., Petitprez, F., Calderaro, J., and Fridman, W. H. (2019). Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325. doi: 10.1038/s41568-019-0144-146
Saxena, M., and Bhardwaj, N. (2018). Re-emergence of dendritic cell vaccines for cancer treatment. Trends Cancer 4, 119–137. doi: 10.1016/j.trecan.2017.12.007
Saygin, C., Matei, D., Majeti, R., Reizes, O., and Lathia, J. D. (2019). Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell 24, 25–40. doi: 10.1016/j.stem.2018.11.017
Schultze, J. L., Michalak, S., Seamon, M. J., Dranoff, G., Jung, K., Daley, J., et al. (1997). CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J. Clin. Invest. 100, 2757–2765. doi: 10.1172/JCI119822
Schumacher, T. N., and Schreiber, R. D. (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. doi: 10.1126/science.aaa4971
Schumacher, T. N., Scheper, W., and Kvistborg, P. (2019). Cancer neoantigens. Annu. Rev. Immunol. 37, 173–200. doi: 10.1146/annurev-immunol-042617-053402
Seder, R. A., Darrah, P. A., and Roederer, M. (2008). T-cell quality in memory and protection: implications for vaccine design. Nat. Rev. Immunol. 8, 247–258. doi: 10.1038/nri2274
Selby, L. I., Cortez-Jugo, C. M., Such, G. K., and Johnston, A. P. R. (2017). Nanoescapology: progress toward understanding the endosomal escape of polymeric nanoparticles. WIREs Nanomed. Nanobiotechno.l 9:e1452. doi: 10.1002/wnan.1452
Senapati, S., Mahanta, A. K., Kumar, S., and Maiti, P. (2018). Controlled drug delivery vehicles for cancer treatment and their performance. Signal. Transduct. Target Ther. 3, 7–19. doi: 10.1038/s41392-017-0004-3
Shan, Y., Luo, T., Peng, C., Sheng, R., Cao, A., Cao, X., et al. (2012). Gene delivery using dendrimer-entrapped gold nanoparticles as nonviral vectors. Biomaterials 33, 3025–3035. doi: 10.1016/j.biomaterials.2011.12.045
Shang, N., Figini, M., Shangguan, J., Wang, B., Sun, C., Pan, L., et al. (2017). Dendritic cells based immunotherapy. Am. J. Cancer Res. 7, 2091–2102.
Shannon, A. M., Bouchier-Hayes, D. J., Condron, C. M., and Toomey, D. (2003). Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treatment Rev. 29, 297–307. doi: 10.1016/S0305-7372(03)00003-3
Sharma, A., Shah, S. R., Illum, H., and Dowell, J. (2012). Vemurafenib: targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs 72, 2207–2222. doi: 10.2165/11640870-000000000-00000
Sharma, G., Valenta, D. T., Altman, Y., Harvey, S., Xie, H., Mitragotri, S., et al. (2010). Polymer particle shape independently influences binding and internalization by macrophages. J. Control. Rel. 147, 408–412. doi: 10.1016/j.jconrel.2010.07.116
Sharma, S., Dominguez, A. L., Manrique, S. Z., Cavallo, F., Sakaguchi, S., and Lustgarten, J. (2008). Systemic targeting of CpG-ODN to the tumor microenvironment with anti-neu-CpG hybrid molecule and T regulatory cell depletion induces memory responses in BALB-neuT tolerant mice. Cancer Res. 68, 7530–7540. doi: 10.1158/0008-5472.CAN-08-1635
Sherje, A. P., Jadhav, M., Dravyakar, B. R., and Kadam, D. (2018). Dendrimers_ A versatile nanocarrier for drug delivery and targeting. Int. J. Pharm. 548, 707–720. doi: 10.1016/j.ijpharm.2018.07.030
Shiga, K., Hara, M., Nagasaki, T., Sato, T., Takahashi, H., and Takeyama, H. (2015). Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers 7, 2443–2458. doi: 10.3390/cancers7040902
Shimabukuro-Vornhagen, A., Draube, A., Liebig, T. M., Rothe, A., Kochanek, M., and Bergwelt-Baildon, et al. (2012). The immunosuppressive factors IL-10, TGF-β, and VEGF do not affect the antigen-presenting function of CD40-activated B cells. J. Exp. Clin. Cancer Res. 31, 47–47. doi: 10.1186/1756-9966-31-47
Shimomura, O., Oda, T., Tateno, H., Ozawa, Y., Kimura, S., Sakashita, S., et al. (2018). A Novel therapeutic strategy for pancreatic cancer: targeting cell surface glycan using rBC2LC-N Lectin-Drug Conjugate (LDC). Mol. Cancer Ther. 17, 183–195. doi: 10.1158/1535-7163.MCT-17-0232
Shirota, H., Tross, D., and Klinman, D. (2015). CpG oligonucleotides as cancer vaccine adjuvants. Vaccines 3, 390–407. doi: 10.3390/vaccines3020390
Simons, J. W., and Sacks, N. (2006). Granulocyte-macrophage colony-stimulating factor-transduced allogeneic cancer cellular immunotherapy: the GVAX vaccine for prostate cancer. Urol. Oncol. 24, 419–424. doi: 10.1016/j.urolonc.2005.08.021
Souza-Fonseca-Guimaraes, F., Cursons, J., and Huntington, N. D. (2019). The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. 40, 142–158. doi: 10.1016/j.it.2018.12.003
Steenblock, E. R., Fadel, T., Labowsky, M., Pober, J. S., and Fahmy, T. M. (2011). An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. J. Biol. Chem. 286, 34883–34892. doi: 10.1074/jbc.M111.276329
Sun, I.-C., Lee, S., Koo, H., Kwon, I. C., Choi, K., Ahn, C.-H., et al. (2010). Caspase sensitive gold nanoparticle for apoptosis imaging in live cells. Bioconjugate Chem. 21, 1939–1942. doi: 10.1021/bc1003026
Sun, J. C., and Lanier, L. L. (2018). Is there natural killer cell memory and can it be harnessed by vaccination? Cold Spring Harb. Perspect. Biol. 10, a29538–a29539. doi: 10.1101/cshperspect.a029538
Sun, J. C., Beilke, J. N., and Lanier, L. L. (2009). Adaptive immune features of natural killer cells. Nature 457, 557–561. doi: 10.1038/nature07665
Sun, Z.-Y., Chen, P.-G., Liu, Y.-F., Shi, L., Zhang, B.-D., Wu, J.-J., et al. (2017). Self-assembled nano-immunostimulant for synergistic immune activation. ChemBioChem 18, 1721–1729. doi: 10.1002/cbic.201700246
Sunshine, J. C., Perica, K., Schneck, J. P., and Green, J. J. (2014). Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells. Biomaterials 35, 269–277. doi: 10.1016/j.biomaterials.2013.09.050
Tacken, P. J., Ginter, W., Berod, L., Cruz, L. J., Joosten, B., Sparwasser, T., et al. (2011). Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood 118, 4111–4119. doi: 10.1182/blood-2011-04-346957
Tanaka, A., Fukuoka, Y., Morimoto, Y., Honjo, T., Koda, D., Goto, M., et al. (2015). Cancer cell death induced by the intracellular self-assembly of an enzyme-responsive supramolecular gelator. J. Am. Chem. Soc. 137, 770–775. doi: 10.1021/ja510156v
Tang, D., Kang, R., Coyne, C. B., Zeh, H. J., and Lotze, M. T. (2012). PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev. 249, 158–175. doi: 10.1111/j.1600-065X.2012.01146.x
Tao, W., Zhang, J., Zeng, X., Liu, D., Liu, G., Zhu, X., et al. (2015). Blended nanoparticle system based on miscible structurally similar polymers: a safe, simple, targeted, and surprisingly high efficiency vehicle for cancer therapy. Adv. Healthc. Mater. 4, 1203–1214. doi: 10.1002/adhm.201400751
Tarek, M. M., Shafei, A. E., Ali, M. A., and Mansour, M. M. (2018). Computational prediction of vaccine potential epitopes and 3-dimensional structure of XAGE-1b for non-small cell lung cancer immunotherapy. Biomed. J. 41, 118–128. doi: 10.1016/j.bj.2018.04.002
Tartour, E., Pere, H., Maillere, B., Terme, M., Merillon, N., Taieb, J., et al. (2011). Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis. Rev. 30, 83–95. doi: 10.1007/s10555-011-9281-9284
Thambi, T., Deepagan, V. G., Yoon, H. Y., Han, H. S., Kim, S.-H., Son, S., et al. (2014). Hypoxia-responsive polymeric nanoparticles for tumor-targeted drug delivery. Biomaterials 35, 1735–1743. doi: 10.1016/j.biomaterials.2013.11.022
Thomann, J.-S., Heurtault, B., Weidner, S., Brayé, M., Beyrath, J., Fournel, S., et al. (2011). Antitumor activity of liposomal ErbB2/HER2 epitope peptide-based vaccine constructs incorporating TLR agonists and mannose receptor targeting. Biomaterials 32, 4574–4583. doi: 10.1016/j.biomaterials.2011.03.015
Thomas, S. N., Vokali, E., Lund, A. W., Hubbell, J. A., and Swartz, M. A. (2014). Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 35, 814–824. doi: 10.1016/j.biomaterials.2013.10.003
Thompson, E. D., Enriquez, H. L., Fu, Y.-X., and Engelhard, V. H. (2010). Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J. Exp. Med. 207, 1791–1804. doi: 10.1084/jem.20092454
Thong, Q. X., Biabanikhankahdani, R., Ho, K. L., Alitheen, N. B., and Tan, W. S. (2019). Thermally-responsive virus-like particle for targeted delivery of cancer drug. Sci. Rep. 9:3945. doi: 10.1038/s41598-019-40388-x
Thorsson, V., Gibbs, D. L., Brown, S. D., Wolf, D., Bortone, D. S., Ou Yang, T.-H., et al. (2018). The immune landscape of cancer. Immunity 48, 812.e–830.e. doi: 10.1016/j.immuni.2018.03.023
Tian, H., and Li, W. (2017). Dendritic cell-derived exosomes for cancer immunotherapy: hope and challenges. Ann. Transl. Med. 5, 221–221. doi: 10.21037/atm.2017.02.23
Tian, L., and Bae, Y. H. (2012). Cancer nanomedicines targeting tumor extracellular pH. Colloids Surf. B 99, 116–126. doi: 10.1016/j.colsurfb.2011.10.039
Tiptiri-Kourpeti, A., Spyridopoulou, K., Pappa, A., and Chlichlia, K. (2016). DNA vaccines to attack cancer: Strategies for improving immunogenicity and efficacy. Pharmacol. Ther. 165, 32–49. doi: 10.1016/j.pharmthera.2016.05.004
Toole, B. P. (2004). Hyaluronan: from extracellular glue to pericellular cue. Nat. Rev. Cancer 4, 528–539. doi: 10.1038/nrc1391
Tovey, M. G., and Lallemand, C. (2010). Adjuvant activity of cytokines. Methods Mol. Biol. 626, 287–309. doi: 10.1007/978-1-60761-585-9_19
Toy, R., Peiris, P. M., Ghaghada, K. B., and Karathanasis, E. (2014). Shaping cancer nanomedicine: the effect of particle shape on the in vivojourney of nanoparticles. Nanomedicine 9, 121–134. doi: 10.2217/nnm.13.191
Tran, S., DeGiovanni, P.-J., Piel, B., and Rai, P. (2017). Cancer nanomedicine: a review of recent success in drug delivery. Clin. Transl. Med. 6:44. doi: 10.1186/s40169-017-0175-170
Tullett, K. M., Leal Rojas, I. M., Minoda, Y., Tan, P. S., Zhang, J.-G., Smith, C., et al. (2016). Targeting CLEC9A delivers antigen to human CD141+ DC for CD4+ and CD8+T cell recognition. JCI Insight 1:e87102. doi: 10.1172/jci.insight.87102
Uldry, E., Faes, S., Demartines, N., and Dormond, O. (2017). Fine-tuning tumor endothelial cells to selectively kill cancer. IJMS 18, 1401–1416. doi: 10.3390/ijms18071401
Vader, P., Mol, E. A., Pasterkamp, G., and Schiffelers, R. M. (2016). Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 106, 148–156. doi: 10.1016/j.addr.2016.02.006
Vallen, M. J. E., van der Steen, S. C. H. A., van Tilborg, A. A. G., Massuger, L. F. A. G., and van Kuppevelt, T. H. (2014). Sulfated sugars in the extracellular matrix orchestrate ovarian cancer development: “When sweet turns sour. Gynecol. Oncol. 135, 371–381. doi: 10.1016/j.ygyno.2014.08.023
van der Steen, S. C. H. A., Raavé, R., Langerak, S., van Houdt, L., van Duijnhoven, S. M. J., van Lith, S. A. M., et al. (2017). Targeting the extracellular matrix of ovarian cancer using functionalized, drug loaded lyophilisomes. Eur. J. Pharm. Biopharm. 113, 229–239. doi: 10.1016/j.ejpb.2016.12.010
van der Vlies, A. J., O’Neil, C. P., Hasegawa, U., Hammond, N., and Hubbell, J. A. (2010). Synthesis of Pyridyl disulfide-functionalized nanoparticles for conjugating thiol-containing small molecules. Peptides, and Proteins. Bioconjugate Chem. 21, 653–662. doi: 10.1021/bc9004443
van Niel, G., D’Angelo, G., and Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nat. Publ. Group 19, 213–228. doi: 10.1038/nrm.2017.125
Van Tomme, S. R., Storm, G., and Hennink, W. E. (2008). In situ gelling hydrogels for pharmaceutical and biomedical applications. Int. J. Pharm. 355, 1–18. doi: 10.1016/j.ijpharm.2008.01.057
Vermaelen, K. (2019). Vaccine strategies to improve anti-cancer cellular immune responses. Front. Immunol. 10:8. doi: 10.3389/fimmu.2019.00008
Vigneron, N. (2015). Human tumor antigens and cancer immunotherapy. BioMed. Res. Int. 2015:948501. doi: 10.1155/2015/948501
von Bergwelt-Baildon, M. S., Vonderheide, R. H., Maecker, B., Hirano, N., Anderson, K. S., Butler, M. O., et al. (2002). Human primary and memory cytotoxic T lymphocyte responses are efficiently induced by means of CD40-activated B cells as antigen-presenting cells: potential for clinical application. Blood 99, 3319–3325. doi: 10.1182/blood.v99.9.3319.
Wang, C., Sun, W., Ye, Y., Bomba, H. N., and Gu, Z. (2017). Bioengineering of artificial antigen presenting cells and lymphoid organs. Theranostics 7, 3504–3516. doi: 10.7150/thno.19017
Wang, Y., Deng, W., Li, N., Neri, S., Sharma, A., Jiang, W., et al. (2018). Combining immunotherapy and radiotherapy for cancer treatment: current challenges and future directions. Front. Pharmacol. 9:185. doi: 10.3389/fphar.2018.00185
Wang, Y., Guo, R., Cao, X., Shen, M., and Shi, X. (2011). Encapsulation of 2-methoxyestradiol within multifunctional poly(amidoamine) dendrimers for targeted cancer therapy. Biomaterials 32, 3322–3329. doi: 10.1016/j.biomaterials.2010.12.060
Weissleder, R., and Pittet, M. J. (2008). Imaging in the era of molecular oncology. Nature 452, 580–589. doi: 10.1038/nature06917
Wen, Y., Waltman, A., Han, H., and Collier, J. H. (2016). Switching the immunogenicity of peptide assemblies using surface properties. ACS Nano 10, 9274–9286. doi: 10.1021/acsnano.6b03409
Wennhold, K., Shimabukuro-Vornhagen, A., and von Bergwelt-Baildon, M. (2019). B Cell-based cancer immunotherapy. Transfus. Med. Hemother. 46, 36–46. doi: 10.1159/000496166
Wilson, D. S., Hirosue, S., Raczy, M. M., Bonilla-Ramirez, L., Jeanbart, L., Wang, R., et al. (2019). Antigens reversibly conjugated to a polymeric glyco-adjuvant induce protective humoral and cellular immunity. Nat. Mater. 18, 175–185. doi: 10.1038/s41563-018-0256-255
Wolf, B. J., Choi, J. E., and Exley, M. A. (2018). Novel approaches to exploiting invariant NKT cells in cancer immunotherapy. Front. Immunol. 9:384. doi: 10.3389/fimmu.2018.00384
World Health Organization, (2018). Cancer: Key Facts. Available at: www.who.int/en/news-room/fact-sheets/detail/cancer (accessed 2019).
Wu, S., Liu, X., He, J., Wang, H., Luo, Y., Gong, W., et al. (2019). A dual targeting magnetic nanoparticle for human cancer detection. Nanosc. Res. Lett. 14:228. doi: 10.1186/s11671-019-3049-3040
Wu, W., Luo, L., Wang, Y., Wu, Q., Dai, H.-B., Li, J.-S., et al. (2018). Endogenous pH-responsive nanoparticles with programmable size changes for targeted tumor therapy and imaging applications. Theranostics 8, 3038–3058. doi: 10.7150/thno.23459
Wu, X., Giobbie-Hurder, A., Liao, X., Lawrence, D., McDermott, D., Zhou, J., et al. (2016). VEGF neutralization plus CTLA-4 blockade alters soluble and cellular factors associated with enhancing lymphocyte infiltration and humoral recognition in melanoma. Cancer Immunol. Res. 4, 858–868. doi: 10.1158/2326-6066.CIR-16-0084
Xia, W., and Low, P. S. (2010). Folate-targeted therapies for cancer. J. Med. Chem. 53, 6811–6824. doi: 10.1021/jm100509v
Xiang, S. D., Scholzen, A., Minigo, G., David, C., Apostolopoulos, V., Mottram, P. L., et al. (2006). Pathogen recognition and development of particulate vaccines: does size matter? Methods 40, 1–9. doi: 10.1016/j.ymeth.2006.05.016
Xu, Y., and Goldkorn, A. (2016). Telomere and telomerase therapeutics in cancer. Genes 7:22. doi: 10.3390/genes7060022
Yang, G., Wang, X., Fu, S., Tang, R., and Wang, J. (2017). pH-triggered chitosan nanogels via an ortho ester-based linkage for efficient chemotherapy. Acta Biomaterialia 60, 232–243. doi: 10.1016/j.actbio.2017.05.003
Yang, X., Kessler, E., Su, L.-J., Thorburn, A., Frankel, A. E., Li, Y., et al. (2013). Diphtheria toxin-epidermal growth factor fusion protein DAB389EGF for the treatment of bladder cancer. Clin. Cancer Res. 19, 148–157. doi: 10.1158/1078-0432.CCR-12-1258
Yao, Q., Kou, L., Tu, Y., and Zhu, L. (2018). MMP-responsive “Smart” drug delivery and tumor targeting. Trends Pharmacol. Sci. 39, 766–781. doi: 10.1016/j.tips.2018.06.003
Yau, T., Dan, X., Ng, C., and Ng, T. (2015). Lectins with potential for anti-cancer therapy. Molecules 20, 3791–3810. doi: 10.3390/molecules20033791
Ye, Y., Wang, C., Zhang, X., Hu, Q., Zhang, Y., Liu, Q., et al. (2017). A melanin-mediated cancer immunotherapy patch. Sci. Immunol. 2:eaan5692. doi: 10.1126/sciimmunol.aan5692
Yu, J. S., Liu, G., Ying, H., Yong, W. H., Black, K. L., and Wheeler, C. J. (2004). Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 64, 4973–4979. doi: 10.1158/0008-5472.CAN-03-3505
Zahavi, D., AlDeghaither, D., O’Connell, A., and Weiner, L. M. (2018). Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antibody Therapeutics 1, 7–12. doi: 10.1093/abt/tby002
Zarogoulidis, P., Darwiche, K., Sakkas, A., Yarmus, L., Huang, H., Li, Q., et al. (2013). Suicide gene therapy for cancer - current strategies. J. Genet. Syndr. Gene Ther. 4:16849. doi: 10.4172/2157-7412.1000139
Zhang, J., Spring, H., and Schwab, M. (2001). Neuroblastoma tumor cell-binding peptides identified through random peptide phage display. Cancer Lett. 171, 153–164. doi: 10.1016/s0304-3835(01)00575-574
Zhang, L. F., Zhou, J., Chen, S., Cai, L. L., Bao, Q. Y., Zheng, F. Y., et al. (2000). HPV6b virus like particles are potent immunogens without adjuvant in man. Vaccine 18, 1051–1058. doi: 10.1016/s0264-410x(99)00351-355
Zhang, L., Wang, L., Shahzad, K. A., Xu, T., Wan, X., Pei, W., et al. (2017a). Paracrine release of IL-2 and anti-CTLA-4 enhances the ability of artificial polymer antigen-presenting cells to expand antigen-specific T cells and inhibit tumor growth in a mouse model. Cancer Immunol. Immunother. 66, 1229–1241. doi: 10.1007/s00262-017-2016-2019
Zhang, X., Li, X., You, Q., and Zhang, X. (2017b). Prodrug strategy for cancer cell-specific targeting: A recent overview. Eur. J. Med. Chem. 139, 542–563. doi: 10.1016/j.ejmech.2017.08.010
Zhou, S., Gravekamp, C., Bermudes, D., and Liu, K. (2018). Tumour-targeting bacteria engineered to fight cancer. Nat. Rev. Cancer 18, 727–743. doi: 10.1038/s41568-018-0070-z
Zhou, Y., Banday, A. H., Hruby, V. J., and Cai, M. (2019). Development of N-Acetylated dipalmitoyl-s-glyceryl cysteine analogs as efficient TLR2/TLR6 agonists. Molecules 24:3512. doi: 10.3390/molecules24193512
Zhou, Z.-H., Ji, C.-D., Xiao, H.-L., Zhao, H.-B., Cui, Y.-H., and Bian, X.-W. (2017). Reorganized collagen in the tumor microenvironment of gastric cancer and its association with prognosis. J. Cancer 8, 1466–1476. doi: 10.7150/jca.18466
Zirlik, K., and Duyster, J. (2018). Anti-angiogenics: current situation and future perspectives. Oncol. Res. Treat. 41, 166–171. doi: 10.1159/000488087
Zitvogel, L., and Palucka, K. (2011). Antibody co-targeting of DCs. Blood 118, 6726–6727. doi: 10.1182/blood-2011-10-384552
Zitvogel, L., Regnault, A., Lozier, A., Wolfers, J., Flament, C., Tenza, D., et al. (1998). Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med. 4, 594–600.
Zom, G. G., Willems, M. M. J. H. P., Khan, S., van der Sluis, T. C., Kleinovink, J. W., Camps, M. G. M., et al. (2018). Novel TLR2-binding adjuvant induces enhanced T cell responses and tumor eradication. J. Immunol. Ther. Cancer 6:146. doi: 10.1186/s40425-018-0455-452
Keywords: cancer, vaccines, material engineering, targeting strategies, immunoengineering, immunotherapy
Citation: Briquez PS, Hauert S, de Titta A, Gray LT, Alpar AT, Swartz MA and Hubbell JA (2020) Engineering Targeting Materials for Therapeutic Cancer Vaccines. Front. Bioeng. Biotechnol. 8:19. doi: 10.3389/fbioe.2020.00019
Received: 10 October 2019; Accepted: 10 January 2020;
Published: 11 February 2020.
Edited by:
Filippo Rossi, Politecnico di Milano, ItalyReviewed by:
Benoit Frisch, UMR 7199 Laboratoire de Conception et Application de Molécules Bioactives, FrancePradipta Ranjan Rauta, University of Texas MD Anderson Cancer Center, United States
Copyright © 2020 Briquez, Hauert, de Titta, Gray, Alpar, Swartz and Hubbell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Priscilla S. Briquez, cGJyaXF1ZXpAdWNoaWNhZ28uZWR1; Jeffrey A. Hubbell, amh1YmJlbGxAdWNoaWNhZ28uZWR1
†These authors have contributed equally to this work and share first authorship