 James L. Galman
James L. Galman Deepankar Gahloth
Deepankar Gahloth Fabio Parmeggiani
Fabio Parmeggiani
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 21 December 2018
Sec. Bioprocess Engineering
Volume 6 - 2018 | https://doi.org/10.3389/fbioe.2018.00205
This article is part of the Research Topic PLP-Dependent Enzymes: Extraordinary Versatile Catalysts and Ideal Biotechnological Tools for the Production of Unnatural Amino Acids and Related Compounds View all 7 articles
The reductive amination of prochiral ketones using biocatalysts has been of great interest to the pharmaceutical industry in the last decade for integrating novel strategies in the production of chiral building blocks with the intent of minimizing impact on the environment. Amongst the enzymes able to catalyze the direct amination of prochiral ketones, pyridoxal 5′-phosphate (PLP) dependent ω-transaminases have shown great promise as versatile industrial biocatalysts with high selectivity, regioselectivity, and broad substrate scope. Herein the biochemical characterization of a putrescine transaminase from Pseudomonas putida (Pp-SpuC) was performed, which showed an optimum pH and temperature of 8.0 and 60°C, respectively. To gain further structural insight of this enzyme, we crystallized the protein in the apo form and determined the structure to 2.1 Å resolution which revealed a dimer that adopts a class I transaminase fold comparable to other class III transaminases. Furthermore we exploited its dual substrate recognition for biogenic diamines (i.e., cadaverine) and readily available monoamines (i.e., isopropylamine) for the synthesis of benzylamine derivatives with excellent product conversions and extremely broad substrate tolerance.
Substituted benzylamines are essential building blocks for the synthesis of a diverse range of agrochemicals, pharmaceuticals, chiral ligands, and polymers (Roughley and Jordan, 2011; Froidevaux et al., 2016). Furthermore, they are key components with potent bioactivities in natural products and synthetic drugs, such as the topical antifungal treatment butenafine (Krauss et al., 2017), glucocorticoid receptor modulators with potent anti-inflammatory and immunosuppressive activity (Link et al., 2005), epoxide hydrolase inhibitors employed in the treatment of type II diabetes (Shen and Hammock, 2012), and most recently anti-tumor drugs (Mojena et al., 2018). The classical direct amination route, which has often been used on an industrial scale, is based on the reaction of alcohols with ammonia using transition metal supported catalysts (Figure 1A); however, this heterogeneous process requires high temperatures and pressures resulting sometimes in low yields of desired product (Hayes, 2001). For reasons of availability, a cost effective alternative would use a one-pot conversion of a carbonyl compound into an amine via the formation of an iminium ion intermediate which is subsequently reduced in situ by a reducing agent. Homogenous catalysis for the amination of carbonyl groups using rhodium, iridium, and ruthenium transition metal catalysts with ammonia under hydrogen pressure (Figure 1B) demonstrate this effective and clean procedure (Gross et al., 2002; Tan et al., 2018). Apart from transition metal catalysis, a well-known two-step approach involving the reaction of hydroxylamine with carbonyl groups to give oxime ethers and subsequent reduction with borane to yield amine products has also been applied (Figure 1C), but this cumbersome approach requires isolation and purification steps of the stable reaction intermediates (Feuer and Braunstein, 1969; Mirjafary et al., 2015).
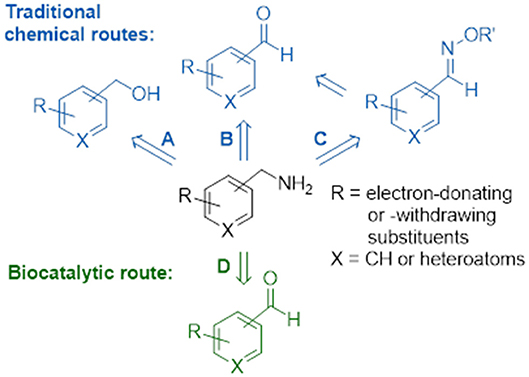
Figure 1. Chemical and biocatalytic routes to substituted benzylamines. (A) Direct amination of alcohols. (B) Reductive amination of aldehydes. (C) Reduction of oxime ethers. (D) Biocatalytic transamination.
Biocatalysis provides numerous advantages to the traditional chemical synthetic methods, allowing for biosustainable processes that exhibit excellent stereo- and regioselectivities, mild reaction conditions and no requirement for expensive and toxic heavy metals or protection-deprotection strategies. The expanding toolbox of enzymes enables scalable asymmetric amine synthesis to access chiral amine derivatives. Several biocatalytic strategies that have been developed include amine dehydrogenases (Ye et al., 2015), ammonia lyases (Parmeggiani et al., 2018), reductive aminases (Aleku et al., 2017) or via kinetic resolutions using monoamine oxidases (Ghislieri et al., 2013; Batista et al., 2018), and/or hydrolases (Reetz and Schimossek, 1996). Of these the reductive amination of prochiral ketones has been listed in the ACS Green Chemistry Roundtable discussion of implementing an atom efficient and environmentally benign method to produce chiral amines (Constable et al., 2007). The direct amination of prochiral ketones via the transfer of ammonia from an amine donor using ω-transaminases (Figure 1D) has an attractive advantage over dehydrogenases as the latter require an external nicotinamide cofactor regeneration system whilst ω-transaminases utilize pyridoxal 5′-phosphate (PLP) as a tightly bound prosthetic group (Slabu et al., 2017). However, ω-transaminase suffers from thermodynamic equilibrium limitations (Tufvesson et al., 2014) and substrate inhibition (Shin and Kim, 1997). Studies have shown this can be circumvented using inexpensive amine donors in large excess such as isopropylamine (which in the formation of volatile acetone that drives the equilibrium to the desired product) or using sacrificial amine cosubstrates which displaces equilibria via ring aromatization or cyclisation (Gomm et al., 2016; Martínez-Montero et al., 2016; Galman et al., 2017). To date there are many examples that have demonstrated the use of ω-transaminases toward the production of industrially relevant amines, alkaloids, and other bioactive natural products (Guo and Berglund, 2017; Costa et al., 2018; Galman et al., 2018).
We earlier identified a putrescine transaminase spuC gene in Pseudomonas putida with preference to aliphatic diamines other than putrescine such as cadaverine and spermidine (Galman et al., 2017). A basic local alignment search tool (BLAST) analysis (Altschul et al., 1990) revealed a 76% protein sequence identity to a spuC gene from Pseudomonas aeruginosa, that was previously characterized to be part of a polyamine uptake and utilization pathway converting putrescine to 4-aminobutyraldehyde (Lu et al., 2002). Further investigations supported the physiological role of SpuC by maintaining alanine homeostasis via the transamination of pyruvate (Han et al., 2008). However, the promiscuous role of P. putida Pp-SpuC may also contribute to a complementary pathway in L-lysine catabolism via the cadaverine route (Revelles et al., 2005) to produce sought after polyamide building blocks 5-aminovalerate and glutaric acid (Adkins et al., 2013). In this study, we report on the detailed characterization (structural and biochemical) of the putrescine transaminase Pp-SpuC, and its application in the transamination of a comprehensive range of ketones/aldehydes for the production of a variety of benzylamine derivatives.
Putrescine transaminase Pp-SpuC was cloned into a pET28b vector containing an N-terminal His6-tag and recombinantly expressed in E. coli BL21 (DE3) as previously reported (Galman et al., 2017). The initial thermoactivity of purified Pp-SpuC was investigated by incubating the enzyme with (S)-α-methylbenzylamine and sodium pyruvate at 25–90°C in Tris-HCl buffer pH 8.0 (Figure 2). The activity was measured according to the UV absorbance change due to the formation of acetophenone. The activity of the enzyme significantly improved as the temperature increased to its suboptimum activity at 50°C, and reached 100% optimum relative activity at 60°C. At higher temperature, it showed gradual denaturation but maintaining significant residual activity (77%) up to 70°C. Interestingly, an activity above 50% of the maximum was observed over a large temperature range (45–75°C).
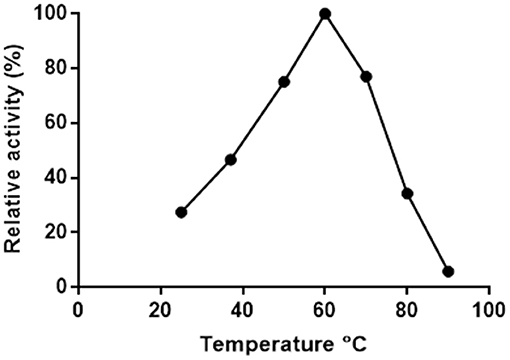
Figure 2. Effect of temperature on the activity of Pp-SpuC. The activity was determined spectrophotometrically based the formation of acetophenone.
The thermostability of Pp-SpuC was assayed by incubating the enzyme (5 mg mL−1) at optimum and sub-optimum temperatures for different times, then assaying the residual activity at 30°C as described above (Figure 3). Each result was an average of triplicate measurements from the initial rates upon the formation of acetophenone and was linearly plotted as ln(residual activity %) vs. time. The deactivation rate constants kd were calculated at 4.65 and 0.156 h−1 for incubated enzyme temperatures at 60 and 50°C, respectively. The half-life times (t1/2 = ln2/kd) were calculated as 4.4 h at 50°C which is more stable than the optimal relative activity conditions at 60°C with a half-life time of 8.9 min.
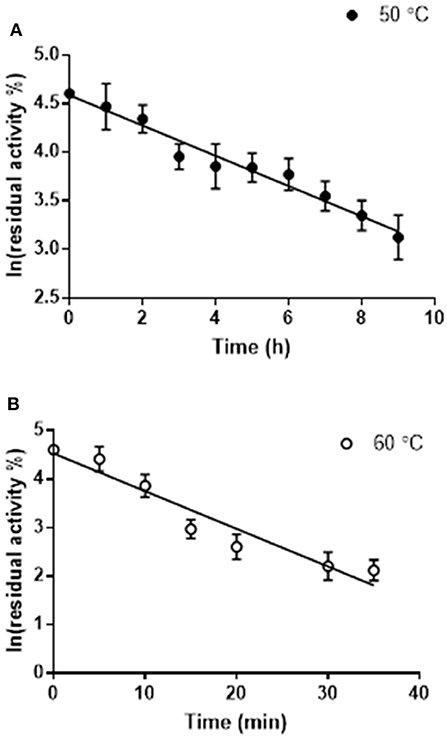
Figure 3. Residual activity of purified Pp-SpuC after incubation in 50 mM Tris-HCl, 0.1 mM PLP at different temperatures. (A) Incubation at 50°C (●) with a half life t1/2 = 7.7 min. (B) Incubation at 60°C (○) with a half life t1/2 = 4.7 h. All reactions were performed in triplicate and the standard deviations <9%.
The pH-stability profile of Pp-SpuC was determined by incubating at 30°C for 24 h the purified enzyme in different buffer solutions (50 mM), then measuring residual activity as described above (Figure 4). The pH profile reveals the effect on transaminase activity in defined buffers shown as relative activity using 2-fluoroacetophenone as the acceptor substrate and isopropylamine (50 equiv.) as the amine donor. Pp-SpuC was found inactive at acidic pH values pH < 6.0 and active at pH 7.0–9.0 with optimum activity at pH 8.0, which is typical of a majority of (S)-selective transaminases such as those from Halomonas elongata He-ωTA (Cerioli et al., 2015), Vibrio fluvialis Vf-ωTA (Shin et al., 2003), and Chromobacterium violaceum Cv-ωTA (Schell et al., 2009). Tris-HCl buffer was found to stabilize the protein, with the highest activity at pH 7.0 (compared to phosphate buffer which showed a 40% decrease in activity at the same pH). The decrease in activity is likely to be due to the displacement of the cofactor pyridoxamine-5′-phosphate (PMP), the aminated form of PLP, via competing phosphate ions, as previously reported within Class III transaminases (Schell et al., 2009; Mathew et al., 2015). Enzyme stability decreases by about 55% when the pH was increased to 9.0 and only negligible activity could be seen at pH > 10.0.
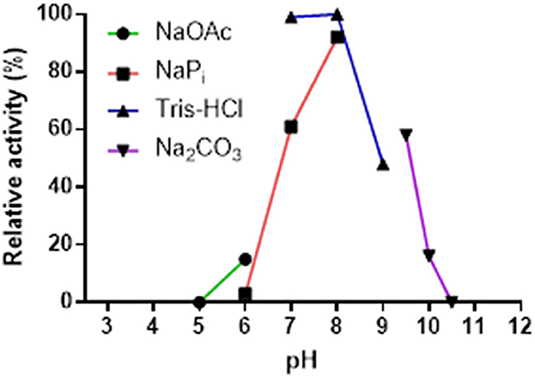
Figure 4. Effect of pH on the activity of Pp-SpuC. The activity was determined by HPLC based on the formation of the corresponding amine. The buffers used were acetate (pH 5.0–6.0), phosphate (pH 6.0–8.0), Tris-HCl (pH 7.0–9.0), and carbonate (pH 9.5–10.5) at a concentration of 50 mM.
Transaminase catalyzed reactions often rely on the use of organic co-solvents, primarily to increase the solubility of hydrophobic substrates. Biotransformation reactions with Pp-SpuC transaminase were previously conducted using 1% ν/ν DMSO and gave high product conversions (Galman et al., 2017). However, a detailed study of the influence of various organic co-solvents was not performed (Figure 5). In order to investigate the effects, Pp-SpuC reactions were performed on a 5 mM scale using 3-fluorobenzaldehyde 1a as the substrate acceptor with 50 equivalents of IPA in the presence of 10–30% ν/ν of several common co-solvents. Pp-SpuC was shown to tolerate up to 10% ν/ν with all the organic co-solvents tested. Increasing the co-solvents to 20% ν/ν revealed a 45–55% decrease in activity with MeCN, MeOH, and THF. Interestingly, the activity of Pp-SpuC remained unaffected with DMSO at 30% ν/ν, therefore higher concentrations were tested, revealing almost identical activity at up to 40% ν/ν DMSO. The high DMSO stability of Pp-SpuC is comparable to a recently published wild type transaminase (pQR2189, 52% protein sequence identity to Pp-SpuC) isolated from a household drain metagenome, displaying activity up to 50% ν/ν DMSO (Leipold et al., in press).
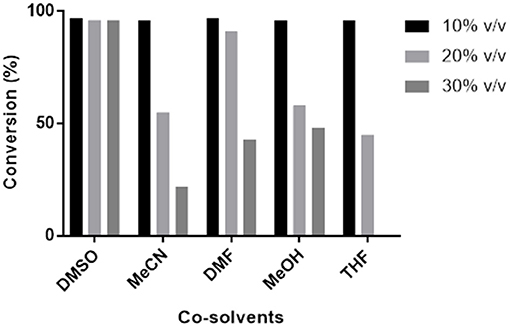
Figure 5. Effect of co-solvents on the activity of Pp-SpuC against 1a. The activity was determined by LC-MS method based on the formation of the corresponding amine 1b.
In an attempt to rationalize the substrate scope and gain better insight into the catalytic mechanism, the crystal structure of Pp-SpuC was solved as follows. The His-tagged Pp-SpuC purified by nickel affinity chromatography was further purified by ion-exchange before crystallization. A commercially available crystallogenesis screening set was employed, obtaining crystals under a number of different conditions after overnight incubation at 20°C. The best diffracting crystals were obtained in 0.2 M MgCl2, 0.1 M Bis-Tris pH 6.5, 25% PEG3350, then cryoprotected before freezing. X-ray diffraction data was collected from single crystal at IO4 beamline at the Diamond Light Source Syncrotron (UK). The structure was solved by the molecular replacement method using as a search model the aspartate aminotransferase from Pseudomonas sp. (Pse-ωTA) (PDB id: 5TI8) (Wilding et al., 2017) which shows 77% identity with Pp-SpuC. The final refined structure of Pp-SpuC has been deposited in the PDB database (PDB id: 6HX9).
Lastly, to demonstrate the applicability of Pp-SpuC as an efficient biocatalyst, the transamination of a broad range of aromatic carbonyl compounds (a representative panel of 30 aldehyde/ketone substrates substituted with halogens, electron-donating groups, and electron-withdrawing groups, was tested. Reactions were carried out with recombinant Pp-SpuC (2 mg mL−1) in Tris-HCl buffer (50 mM) pH 9.0, containing PLP (1.0 mM), at 30°C for 18 h. For comparison, two equilibrium-shifting approaches were tested: either a large excess isopropylamine (50 equiv.) as a low-cost amine donor, or bio-derived cadaverine (5 equiv.) as a cleaner, “green” alternative sacrificial co-substrate, which drives the reaction by spontaneous cyclisation. Conversions were monitored by LC-MS and the identity of the products was verified by MS and/or by comparison with authentic standards. In most cases, almost quantitative conversions could be observed (see Discussion section), proving the broad substrate tolerance of this enzyme.
In order to demonstrate the feasibility of the reaction with reduced catalyst loadings, biotransformations of the model substrate 1a were performed with successive ten-fold dilutions, under the same conditions specified above. With a catalyst concentration of 0.2 mg mL−1 full conversion could still be achieved, while reducing the enzyme loading to 0.02 mg mL−1 only a modest conversion (19%) was observed. From the latter data, with the substrate only partially consumed, a total turnover number (TTN) could be estimated to ~2400.
The overall structure of Pp-SpuC (PDB id: 6HX9) is similar to Pseudomonas sp. Pse-ωTA (PDB id: 5TI8) (Wilding et al., 2017) and C. violaceum Cv-ωTA (PDB id: 4A6T) (Humble et al., 2012; Sayer et al., 2013) and typical to the class I transaminase fold. There are two monomers in the asymmetric unit and the enzyme forms a tight dimer with predicted surface area of about 28710 Å2, with buried surface area of 3980 Å Each monomer can be divided into two major domains (Figure 6). The large domain comprises residues 93–313 and folds into a sandwich of α/β/α comprising a core of seven stranded β-sheets with only β10 being antiparallel. The large domain is flanked by a small domain composed of two subdomains joined by a parallel β interaction between strand β3 and β14. The N-terminal subdomain is composed of three antiparallel β strands. In the case of holo Cv-ωTA (PDB id: 4A6U) these anti parallel β strands capped by two most N-terminal α-helices, which were absent in our structure, similarly to the apo structures of CV-ωTA (PDB id: 4A6U) and Pse-ωTA (PDB id: 5TI8). The N-terminal subdomain forms extensive interactions with monomer 2 of the dimer which is important for active site formation, similar to what was observed with Cv-ωTA and Pse-ωTA. The C-terminal subdomain is formed of four antiparallel β-sheets.
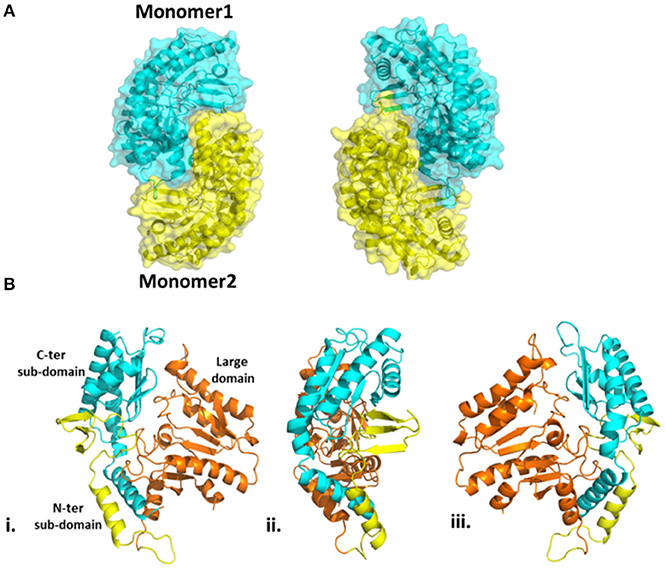
Figure 6. (A) Homodimeric structure of Pp-SpuC. Monomer 1 shown in cyan and monomer 2 shown in yellow. (B) Domain organization of the Pp-SpuC monomer, divided into one large domain (residue 93–313, orange) and two sub-domains, N-terminal (residues 1–92, yellow) and C-terminal (residues 314–352, cyan). Views (ii) and (iii) are obtained by 90° rotation of (i).
In the active site of aminotransferases, the PLP cofactor binds to the enzyme to form an aldimine intermediate in which the C4′ of PLP forms a Schiff base with an active site lysine during the catalytic interaction (Eliot and Kirsch, 2004). Unfortunately, all our attempts to obtain the structure of the complex of Pp-SpuC with PLP bound were unsuccessful. The superposition of the Pp-SpuC structure with the holo structure of Cv-ωTA (PDB id: 4A6T) reveals the position of the active site. In the case of SpuC, the catalytic residue Lys289 is located on the loop between β4 and β10. The Pp-Spuc structure is very similar to the Pse-ωTA (PDB id: 5TI8) (Wilding et al., 2017) and the apo form of Cv-ωTA (PDB id: 4A6U) (Humble et al., 2012) as compared to holo structure of Cv-ωTA (PDB id: 4A6T). The first N-terminal 33 residues are missing from the pp-SpuC structure which forms the corresponding two α helices in the holo Cv-ωTA (PDB id: 4A6T). Another disordered region of Pp-SpuC are amino acid residues 154–179 and 313–324. The homologous region 154–179 forms a “roof” above the cofactor binding site in the holo structure of Cv-ωTA, indicating its importance in PLP binding (Figure 7). This region is also absent in case of apo-Cv-ωTA and Pse-ωTA. The additional missing loop from 313–324 which should connect two alpha helices and comes close to the phosphate moiety of the PLP cofactor in holo-Cv-ωTA (PDB id: 4A6T).
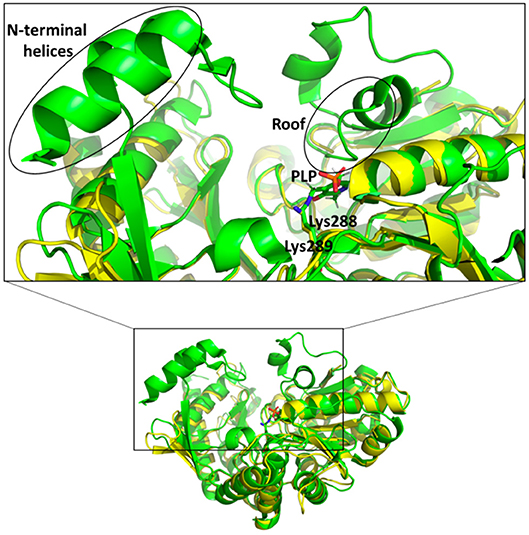
Figure 7. Superposition of holo Cv-ωTA (PDB id: 4A6T, green) structure on Pp-SpuC crystal structure (PDB id: 6HX9, yellow). The Pp-SpuC catalytic lysine (Lys289) residue superimposes well with the catalytic Lys288 of holo Cv-ωTA. In the case of Pp-SpuC the N-terminal helices and the residues corresponding to the roof of the PLP binding site (highlighted in the box) remain disordered.
Two structurally homologous genes Vf-ωTA (Shin and Kim, 2002; Midelfort et al., 2013) and Cv-ωTA with a sequence identity of 36 and 55% respectively have been extensively studied with crystals isolated in the apo and holo form co-crystallized with PLP or bound to a suicide inhibitor such as gabaculine (Sayer et al., 2013). Superposition of Pp-SpuC structure with the Cv-ωTA apo monomer (PDB id: 4A6U) (Humble et al., 2012), in which the authors also experienced difficulties obtaining crystals with PLP bound, gave an root mean square deviation (RMSD) value of 1.5 Å between 376 Cα atoms of aligned residues (out of 389) with the same “roof” residues missing in the apo structure of Cv-ωTA. The disordered region may indicate why no electron density is observed for the PLP cofactor at the interface of the two enzyme subunits. A structure-based sequence alignment of closely related ω-TAs was constructed with Pp-SpuC as a template, revealing several important characteristics for these industrially relevant enzymes for the production of chiral amines (Figure S1, Supplementary Information). Similar to other Class III transaminases of fold type I, the active site of ω-TAs contain two binding pockets for binding of the substrates (Figure 8). The active key amino acid residues which constitute the small binding pocket and allow accommodation of substituents less bulky than an ethyl group are identified in Pp-SpuC as Phe23, Phe89′ (from the other subunit), Tyr154 and His155 (absent in the Pp-SpuC apo structure). A clear observation of these residues acting as a steric barrier narrowing the entrance to the active site cavity (Malik et al., 2012) is the decrease in amino donor specificities from (S)-methylbenzylamine to (S)-ethylbenzylamine which was also observed in Vf-ωTA (Nobili et al., 2015) and Ochrobactrum anthropi Oa-ωTA (Han et al., 2015). Several previous studies have mutated the corresponding Trp61 in homologous ωTA sequences resulting in improved activities toward aromatic and aliphatic amines without compromising enantioselectivity (Cho et al., 2008; Cassimjee et al., 2012). On the other hand, the highly conserved amino acid residues which surround the large hydrophobic binding pocket were identified as Leu60, Trp61, Ala232, Ile263, and Arg415 which has dual recognition for hydrophobic substituents and a carboxylate group (Hirotsu et al., 2005). This was shown to be in general agreement with previous studies of ω-TAs with high affinity for acetophenone derivatives as well as aliphatic methyl ketones and aldehydes (Kaulmann et al., 2007; Cerioli et al., 2015).
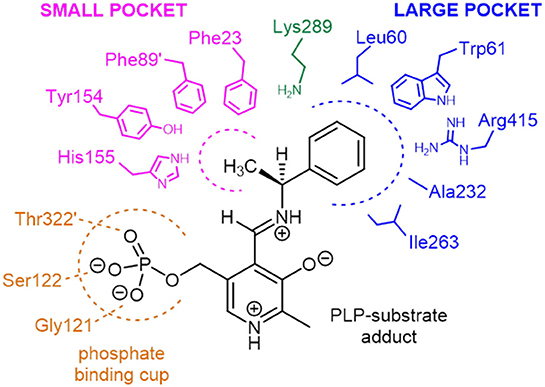
Figure 8. Schematic representation of the amino acid residues forming the small and large binding pockets of Pp-SpuC around the external aldimine intermediate (PLP-substrate adduct). The model is based on the multiple sequence alignment analysis and on the active site structure of Vf-ωTA (PDB id: 4E3Q).
A plethora of ω-transaminases have been reported to aminate acetophenone derivatives and bulkier ketones. There have been recent reports on the use cinnamaldehyde derivatives as amine acceptor molecules using ω-transaminases from Cv-ωTA, Vf-ωTA, and isolated transaminase genes from metagenomic samples furnishing modest yields of the allylic amines (Baud et al., 2017). To our surprise, only a few have been described on the acceptance of benzylic aldehydes (Cerioli et al., 2015; Guidi et al., 2018). In one of the reports, the benzaldehydes were generated by biocatalytic oxidation of the corresponding benzylic alcohols, followed by transamination to the corresponding benzylamines (Fuchs et al., 2012). Interestingly, within the panel of ω-transaminases investigated, significant differences in the amination of benzaldehyde and analogs could be observed, transamination biocatalysts with a broad substrate tolerance toward this class of compounds are rare. Moreover, an additional cofactor recycling system in the presence of a sacrificial cosubstrate was essential to drive the equilibrium to completion.
Since Pp-SpuC was already demonstrated to have high activity with a variety of prochiral ketones (Slabu et al., 2018), we envisaged to investigate the amination of a range of benzaldehyde analogs bearing halogen substituents (1a-6a), electron-donating groups (7a-12a), electron-withdrawing groups (13a-18a) and heteroatoms in the ring (19a-24a). Considering the importance of non-natural amino acids, we also investigated aromatic aldehydes and ketones that lead to pharmaceutically relevant examples of this class of compounds (25a-30a). All compounds were screened using either a large excess of isopropylamine as a donor (Figure 9A) or only a moderate excess of cadaverine, a biogenic amine donor that favors reaction equilibria by cyclising spontaneously to 1-piperideine after transamination (Figure 9B).
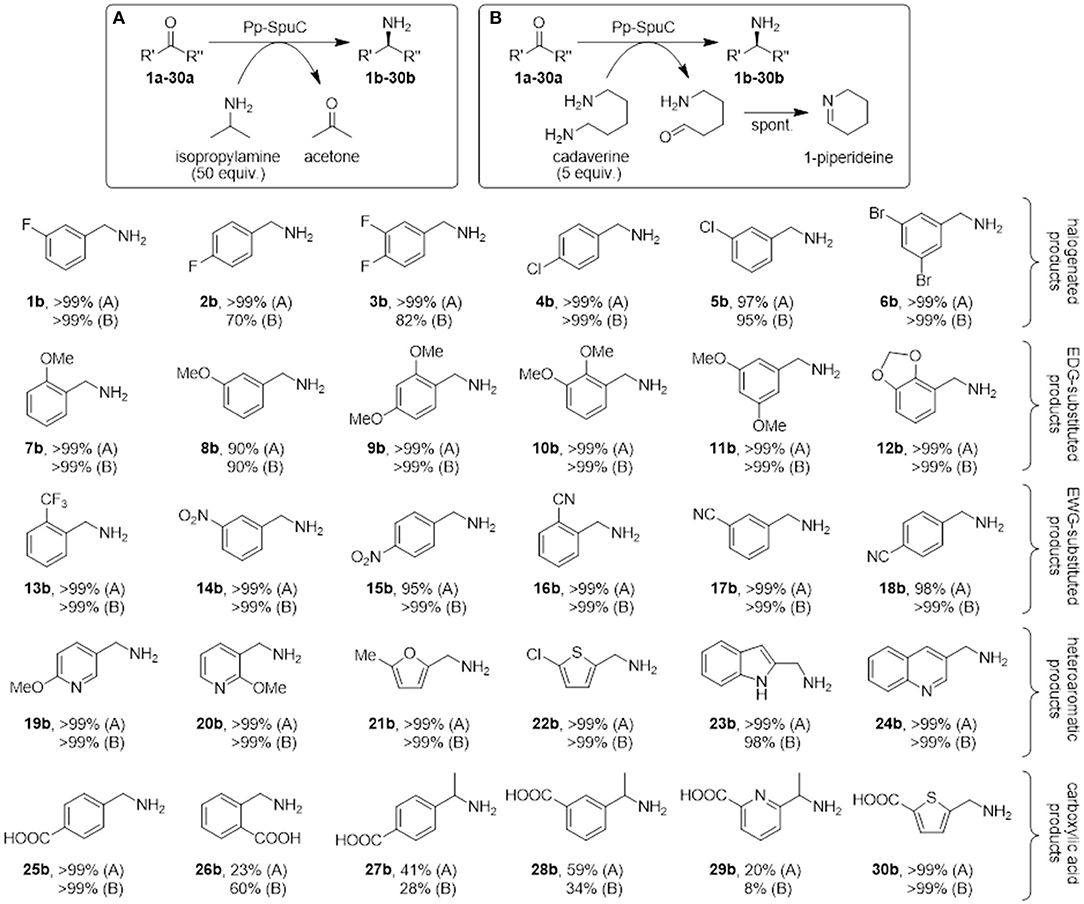
Figure 9. Biocatalytic synthesis of a range of substituted benzylamines mediated by Pp-SpuC. Reaction conditions: 5 mM substrate, 2 mg mL−1 Pp-SpuC, 1 mM PLP, 1% v/v DMSO, 250 mM isopropylamine (A) or 25 mM cadaverine (B), in 50 mM Tris-HCl, pH 9.0, 30 °C, 18 h. Conversions were measured by LC-MS (Supplementary Information).
Mono- and dihalogenated benzaldehydes 1a-6a afforded excellent conversions to the halogenated benzylamines 1b-6b using isopropylamine as the amine donor within 18 h (in the preliminary screening, substrate 1a was converted quantitatively in < 30 min with both amine donors). Interestingly, a decrease in conversions was observed using para-fluorinated substrates 2a-3a with cadaverine as the amine donor. We previously observed a similar low conversion with a para-substituted fluoroacetophenone derivative (Galman et al., 2017) which is presumably ascribed to the interaction between the piperideine monomer from the transamination of cadaverine and the activated para-fluoro substrates. Encouraged by these results, other benzylic derivatives were tested to assess the stereoelectronic effects of electron-donating (methoxy, methylenedioxy), or electron-withdrawing (nitro, cyano, trifluoromethyl) substituents. All functional groups were well-tolerated, giving almost quantitative conversion with both amine donors (except for m-anisaldehyde 8a that afforded amine 8b in 90% conversion). No overall detrimental effects were observed due to the higher or lower electrophilicity of the carbonyl group due to the substituents, or due to the steric hindrance due to multiple substitutions at ortho-, meta-, and para-positions. Similarly, heteroaromatic aldehydes proved to be accepted very well by Pp-SpuC, affording complete conversions with pyridines (19a-20a), furan (21a), thiophene (22a), indoline (23a), and quinoline (24a) moieties.
In addition, we attempted to carry out the biocatalytic synthesis of benzylamines carrying the carboxylic acid functionality from the transamination of formylbenzoic acid derivatives (25a-26a). The para-substituted compound was readily accepted to produce the corresponding amine 25b with excellent conversion, while the ortho-derivative gave considerably lower conversions to 26b (23 and 60%, respectively), indicating the presence of a carboxylic acid substituent at the ortho-position can have a detrimental effect on the reactivity. This facile approach to generate 25b, which is used as an antifibrinolytic agent, provides a sustainable alternative to other reductive amination protocols requiring toxic metal catalysts and harsh reaction conditions (Yraola et al., 2006). Prochiral acetylbenzoic acids 27a and 28a are considerably more challenging due to the presence of the methylketone moiety. Indeed only moderate conversions to 27b (41%) and 28b (59%) could be achieved. This can be rationalized by looking at the active site shape in the crystal structure of Pp-SpuC, which provides an insight into the substrate scope of the enzyme: the hydrophobic environment of the large pocket can accommodate large aromatic groups, presumably by stabilization from π-π stacking interactions, while the smaller pocket imposes a steric constraint on the other substituent of the carbonyl group and might hinder access to larger substituents. This explains why almost any aromatic aldehyde derivative is accepted easily, while the range of methyl (and higher) ketones is very limited, limiting the biocatalytic synthesis of chiral amines and is configured to accept aromatic aldehydes readily. Lastly, we screened heteroaryl oxoacid derivatives 29a and 30a. Unsurprisingly, 29a afforded lower conversions (20 and 8%, respectively), presumably for the same reasons discussed above, while 30a was converted completely, indicating that the presence of a carboxylic acid substituent does not have a detrimental effect on the reactivity of these heteroaromatic substrates.
In summary, we have performed the biochemical characterization of putrescine transaminase Pp-SpuC which reveals a high relative activity at 60°C and excellent stability in 50 mM Tris-HCl at pH 8.0. The enzyme structure was obtained, showing a high degree of similarity to the apo form of Cv-ωTA (PDB id: 4A6U) which can accommodate large aromatic substrates for catalysis. We recognized the important dual substrate recognition of Pp-SpuC, accepting cadaverine or isopropylamine as the equilibrium shifting amine donor, and having excellent activity against a broad range of aromatic aldehyde derivatives bearing various electron-donating and electron-withdrawing substituents, as well as different heteroatoms in the ring, with excellent conversions. This method also eliminates the need for an expensive cofactor recycling system making this approach to important amine scaffolds greener and more economically feasible.
All commercially available reagents, analytical standards, and solvents were purchased from Sigma-Aldrich, Alfa Aesar, or Fluorochem and used without further purification. Escherichia coli DH5α and BL21 (DE3) cells were purchased from New England Biolabs (Ipswich, MA, USA). Expression vector pET-28b was purchased from Novagen (Darmstadt, Germany) and was used for gene expression. P. putida NBRC 14161 (Asc no. 9494) was purchased from the NCIMB culture collection (Aberdeen, UK). The putrescine transaminase spuC from P. putida was cloned in a pET-28b vector containing an N-terminal His6-tag and was recombinantly overexpressed in E. coli BL21 (DE3) cells according to a previously published procedure (Galman et al., 2017) with slight modifications.
Plasmid pET28b-SpuC was transformed into E. coli BL21 (DE3). A fresh colony was used to inoculate LB medium (4 mL) containing kanamycin (50 μg mL−1). This freshly prepared overnight culture was grown at 220 rpm at 37°C, and was used to inoculate 500 mL of LB medium supplemented with kanamycin (50 μg mL−1) in a 2 L Erlenmeyer flask at 220 rpm at 37°C. The recombinant protein expression was induced by adding isopropyl-β-D-1-thiogalactopyranoside (IPTG) (0.2 mM, final) when OD600 reached 0.6–0.8. The cell cultures were then incubated at 18°C for 16 h. The cells were harvested by centrifugation at 4°C (3,250 g, 20 min) and were resuspended (1g in 10 mL) in lysis buffer (50 mM Tris-HCl, 5 mM imidazole, pH 7.0) and lysed in an iced bath by ultra-sonication by Soniprep 150 (20 s on, 20 s off, for 20 cycles, at 30% amplitude). After centrifugation (4°C, 16,000 g, 20 min) the clarified lysate was used for protein purification via a Ni-NTA agarose column. The bound enzyme was washed with 20 mL wash buffer (50 mM Tris-HCl, 30 mM imidazole pH 7.0), and eluted with 50 mM Tris-HCl, 250 mM imidazole at pH 7.0. The collected fractions were concentrated in VivaspinTM filter spin membrane columns (10,000 MWCO). The purified enzyme was buffer exchanged using PD-10 desalting columns using 50 mM Tris-HCl containing 1 mM PLP at pH 9.0. The purity was analyzed by SDS/PAGE and the protein was more than 95% pure and the protein stock was determined by the Bradford assay using bovine serum albumin as standard.
Unless otherwise specified, all biotransformation were performed in 2 mL Eppendorf tubes, at 30°C in biotransformation buffer: Tris-HCl (50 mM) containing PLP (1 mM), pH 9.0. To the addition of amine donor, cadaverine (25 mM), or isopropylamine (250 mM) in biotransformation buffer adjusted to pH 9.0 and carbonyl substrate (5 mM, from a 500 mM stock solution in DMSO), was added purified Pp-SpuC (2 mg mL−1, purified as described above) in a final volume of 0.5 mL. After 18 h, the reaction mixture was quenched by adding aqueous NaOH solution (100 μL, 2 M).
The residual activities of the enzyme after incubating at 50 and 60°C at the allotted time was determined by the generation of acetophenone from (S)-methylbenzylamine with pyruvate as the amine acceptor substrate molecule (Schätzle et al., 2009). The absorbance was measured at 300 nm using a UV transparent 96-well plate (Tecan infinite M200 spectrophotometer, Austria). One unit of enzyme activity (U) was defined as the amount of enzyme required to produce 1 μmole of acetophenone produced per min. The deactivation rate constant kd was determined from the slope and the half-lives were calculated as t1/2 = ln2/kd.
The effect of pH on enzyme stability was carried at various pH buffers from pH 4.0–10.5. The enzyme (2 mg mL−1) was incubated for 24 h at 4°C in different buffers containing 1 mM PLP: sodium acetate (pH 4.0–6.0), sodium phosphate (pH 6.0–8.0), Tris-HCl (pH 7.0–9.0), and sodium carbonate/bicarbonate (pH 9.2–10.0). The residual activity was assayed from the biotransformations as described above.
The effect of solvent stability was performed using a variety of different co-solvents (DMSO, MeOH, MeCN, DMF, and THF) at different solvent concentrations % (v/v) using 5 mM of 1a, and 2 mg mL−1 of purified Pp-SpuC, 1 mM PLP, and 250 mM isopropylamine at pH 9.0.
Conversions were calculated by HPLC analysis on an Agilent 1,100 Series system equipped with a quaternary pump, a HiChrom ACE 5 C18-AR column (15cm × 4.6 mm), a diode UV array detector and an Agilent LC/MSD SL LCMS System. Separation conditions: 1 mL min−1 flowrate, mobile phases: H2O + 0.1% v/v TFA, MeOH + 0.1% v/v TFA. Gradient: 95:5 for 4 min, to 0:100 over 25 min. Temperature: 30°C. Detection wavelength: 210 nm. Injection volume: 10 μL. Compounds were ionized using API-electrospray technique and detected in positive mode on the LCMS System. Drying gas temperature 250°C at 12 L min−1, and nebulizer pressure at 25 psig. All products, unless otherwise specified, were identified by their [M+H]+ signal and confirmed via chemical standards.
C-terminal His-tagged Pp-SpuC purified by affinity chromatography as described above was exchanged to 20 mM HEPES buffer pH 7.5 and submitted to an additional step of ion-exchange purification on a ResourceQ column (6 mL, GE healthcare), to further purify the protein for crystallogenesis. Pp-SpuC was loaded on the column in 20 mM HEPES pH 7.5 buffer and elution was performed with a linear gradient (25 column volumes) of NaCl in buffer. All the fractions were analyzed by SDS-PAGE. Fractions containing Pp-SpuC were pooled and concentrated in a Vivaspin™ filter spin membrane columns (10,000 MWCO).
Apo and PLP (equimolar) mixed protein was screened against commercially available crystallogenesis screens using a mosquito nanodispenser robot (LLP Biotech). Overnight crystals appeared in a number of conditions at 20°C. The best diffracting crystals were obtained in 0.2 M MgCl2, 0.1 M Bis-Tris pH 6.5, 25% PEG3350. SpuC crystals cryoprotected with 10–20% PEG200 in mother liquor before freezing. Data was collected from single crystal at IO4 beamline at the Diamond Light Source Syncrotron (UK). Data was indexed and integrated with XDS (Kabsch, 2010) and scaled with AIMLESS (Evans and Murshudov, 2013). Data processing and refinement statistics are summarized in Table 1.

Table 1. Data collection and refinement statistics.
The structure of Pp- SpuC was solved using molecular replacement method by Phaser (Eliot and Kirsch, 2004). Crystal structure of Pse-ωTA (PDB id: 5TI8, sequence identity 77%) was used as a phasing model. Iterative cycles of manual building in COOT (Emsley et al., 2010) and refinement in Phenix.refine (Murshudov et al., 1997; Eliot and Kirsch, 2004) were carried out to complete the model. Validation of structure was done with Molprobity (Chen et al., 2010) and PDB-redo (Joosten et al., 2014) were integrated into the iterative building and refinement procedure. The final refined structure of Pp-SpuC has been deposited in the PDB database (PDB id: 6HX9).
JG, FP, and NT conceived the project. JG performed molecular biology procedures associated with gene cloning and construct preparations. IS worked on enzyme preparations and biochemical characterisations. FP performed the LC-MS analytics. DG, and DL undertook all structural analyses.
JG acknowledges the support of the BIOINTENSE project, financed through the European Union 7th Framework Programme (grant agreement no. 312148). FP was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) and Glaxo-SmithKline (GSK) under the Strategic Longer and Larger (sLoLa) grant initiative ref BB/K00199X/1. IS acknowledges a CASE award from BBSRC and Dr. Reddy's (Grant code BB/K013076/1). NT acknowledges the ERC for the award of an Advanced Grant (grant number 742987).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2018.00205/full#supplementary-material
Adkins, J., Jordan, J., and Nielsen, D. R. (2013). Engineering Escherichia coli for renewable production of the 5-carbon polyamide building-blocks 5-aminovalerate and glutarate. Biotechnol. Bioeng. 110, 1726–1734. doi: 10.1002/bit.24828
Aleku, G. A., France, S. P., Man, H., Mangas-Sanchez, J., Montgomery, S. L., Sharma, M., et al. (2017). A reductive aminase from Aspergillus oryzae. Nat. Chem. 9, 961–969. doi: 10.1038/nchem.2782
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Batista, V. F., Galman, J. L., Pinto, D. C. G. A., Silva, A. M. S., and Turner, N. J. (2018). Monoamine oxidase: tunable activity for amine resolution and functionalization. ACS Catal. 8, 11889–11907. doi: 10.1021/acscatal.8b03525
Baud, D., Jeffries, J. W. E., Moody, T. S., Ward, J. M., and Hailes, H. C. (2017). A metagenomics approach for new biocatalyst discovery: application to transaminases and the synthesis of allylic amines. Green Chem. 19, 1134–1143. doi: 10.1039/C6GC02769E
Cassimjee, K. E., Humble, M. S., Land, H., Abedi, V., and Berglund, P. (2012). Chromobacterium violaceum ω-transaminase variant Trp60Cys shows increased specificity for (S)-1-phenylethylamine and 4′-substituted acetophenones, and follows Swain-Lupton parameterisation. Org. Biomol. Chem. 10, 5466–5470. doi: 10.1039/c2ob25893e
Cerioli, L., Planchestainer, M., Cassidy, J., Tessaro, D., and Paradisi, F. (2015). Journal of molecular catalysis b : enzymatic characterization of a novel amine transaminase from halomonas elongata. J. Mol. Catal. B. Enzym. 120, 141–150. doi: 10.1016/j.molcatb.2015.07.009
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 12–21. doi: 10.1107/S0907444909042073
Cho, B.-K., Park, H.-Y., Seo, J.-H., Kim, J., Kang, T.-J., Lee, B.-S., et al. (2008). Redesigning the substrate specificity of ω-aminotransferase for the kinetic resolution of aliphatic chiral amines. Biotechnol. Bioeng. 99, 275–284. doi: 10.1002/bit.21591
Constable, D. J. C., Dunn, P. J., Hayler, J. D., Humphrey, G. R., Leazer, J. L., Linderman, R. J., et al. (2007). Key green chemistry research areas - A perspective from pharmaceutical manufacturers. Green Chem. 9, 411–420. doi: 10.1039/B703488C
Costa, B. Z., Galman, J. L., Slabu, I., France, S. P., Marsaioli, A. J., and Turner, N. J. (2018). Synthesis of 2,5-disubstituted pyrrolidine alkaloids via A one-pot cascade using transaminase and reductive aminase biocatalysts. ChemCatChem 10, 4733–4738. doi: 10.1002/cctc.201801166
Eliot, A. C., and Kirsch, J. F. (2004). Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415. doi: 10.1146/annurev.biochem.73.011303.074021
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501. doi: 10.1107/S0907444910007493
Evans, P. R., and Murshudov, G. N. (2013). How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 1204–1214. doi: 10.1107/S0907444913000061
Feuer, H., and Braunstein, D. M. (1969). The Reduction of oximes, oxime ethers, and oxime esters with diborane. A novel synthesis of amines. J. Org. Chem. 34, 1817–1821. doi: 10.1021/jo01258a062
Froidevaux, V., Negrell, C., Caillol, S., Pascault, J. P., and Boutevin, B. (2016). Biobased amines: from synthesis to polymers; present and future. Chem. Rev. 116, 14181–14224. doi: 10.1021/acs.chemrev.6b00486
Fuchs, M., Tauber, K., Sattler, J., Lechner, H., Pfeffer, J., and Faber, K. (2012). RSC advances amination of benzylic and cinnamic alcohols via a biocatalytic, aerobic, oxidation – transamination cascade. 2, 6262–6265. doi: 10.1039/C2RA20800H
Galman, J. L., Slabu, I., Parmeggiani, F., and Turner, N. J. (2018). Biomimetic synthesis of 2-substituted N-heterocycle alkaloids by one-pot hydrolysis, transamination and decarboxylative Mannich reaction. Chem. Commun. 54, 11316–11319. doi: 10.1039/C8CC06759G
Galman, J. L., Slabu, I., Weise, N. J., Iglesias, C., Parmeggiani, F., Lloyd, R. C., et al. (2017). Biocatalytic transamination with near-stoichiometric inexpensive amine donors mediated by bifunctional mono- and di-amine transaminases. Green Chem. 19, 361–366. doi: 10.1039/C6GC02102F
Ghislieri, D., Green, A. P., Pontini, M., Willies, S. C., Rowles, I., Frank, A., et al. (2013). Engineering an enantioselective amine oxidase for the synthesis of pharmaceutical building blocks and alkaloid natural products. J. Am. Chem. Soc. 135, 10863–10869. doi: 10.1021/ja4051235
Gomm, A., Lewis, W., Green, A. P., and O'Reilly, E. (2016). A new generation of smart amine donors for transaminase-mediated biotransformations. Chemistry 22, 12692–12695. doi: 10.1002/chem.201603188
Gross, T., Seayad, A. M., Ahmad, M., and Beller, M. (2002). Synthesis of primary amines: first homogeneously catalyzed reductive amination with ammonia. Org. Lett. 4, 2055–2058. doi: 10.1021/ol0200605
Guidi, B., Planchestainer, M., Contente, M. L., Laurenzi, T., Eberini, I., Gourlay, L. J., et al. (2018). Strategic single point mutation yields a solvent- and salt-stable transaminase from Virgibacillus sp. in soluble form. Sci. Rep. 8:16441. doi: 10.1038/s41598-018-34434-3
Guo, F., and Berglund, P. (2017). Transaminase biocatalysis: optimization and application. Green Chem. 19, 333–360. doi: 10.1039/C6GC02328B
Han, S. W., Park, E. S., Dong, J. Y., and Shin, J. S. (2015). Active-site engineering of ω-transaminase for production of unnatural amino acids carrying a side chain bulkier than an ethyl substituent. Appl. Environ. Microbiol. 81, 6994–7002. doi: 10.1128/AEM.01533-15
Han, T. C., Kwon, D. H., Hegazy, M., and Lu, C. D. (2008). Transcriptome analysis of agmatine and putrescine catabolism in Pseudomonas aeruginosa PAO1. J. Bacteriol. 190, 1966–1975. doi: 10.1128/JB.01804-07
Hayes, K. S. (2001). Industrial processes for manufacturing amines. Appl. Catal. A Gen. 221, 187–195. doi: 10.1016/S0926-860X(01)00813-4
Hirotsu, K., Goto, M., Okamoto, A., and Miyahara, I. (2005). Dual substrate recognition of aminotransferases. Chem. Rec. 5, 160–172. doi: 10.1002/tcr.20042
Humble, M. S., Cassimjee, K. E., Hakansson, M., Kimbung, Y. R., Walse, B., Abedi, V., et al. (2012). Crystal structures of the Chromobacterium violaceum ω-transaminase reveal major structural rearrangements upon binding of coenzyme PLP. FEBS J. 279, 779–792. doi: 10.1111/j.1742-4658.2012.08468.x
Joosten, R. P., Long, F., Murshudov, G. N., and Perrakis, A. (2014). The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220. doi: 10.1107/S2052252514009324
Kabsch, W. (2010). Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132. doi: 10.1107/S0907444909047337
Kaulmann, U., Smithies, K., Smith, M. E. B., Hailes, H. C., and Ward, J. M. (2007). Substrate spectrum of ω-transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzyme Microb. Technol. 41, 628–637. doi: 10.1016/j.enzmictec.2007.05.011
Krauss, J., Stadler, M., and Bracher, F. (2017). Synthesis and structure–activity relationships of novel benzylamine-type antifungals as butenafine-related antimycotics. Arch. Pharm (Weinheim). 350, 1–20. doi: 10.1002/ardp.201600342
Leipold, L., Dobrijevic, D., Jeffries, J. W. E., Bawn, M., Moody, T. S., Ward, J. M., et al. (in press). The identification use of robust transaminases from a domestic drain metagenome. Green Chem. doi: 10.1039/C8GC02986E
Link, J. T., Sorensen, B., Patel, J., Grynfarb, M., Goos-Nilsson, A., Wang, J., et al. (2005). Antidiabetic activity of passive non-steroidal glucocorticoid receptor modulators. J. Med. Chem. 48, 5295–5304. doi: 10.1021/jm050205o
Lu, C.-D., Itoh, Y., Nakada, Y., and Jiang, Y. (2002). Functional analysis and regulation of the divergent spuABCDEFGH-spuI operons for polyamine uptake and utilization in Pseudomonas aeruginosa PAO1. J. Bacteriol. 184, 3765–3773. doi: 10.1128/JB.184.14.3765-3773.2002
Malik, M. S., Park, E.-S., and Shin, J.-S. (2012). Features and technical applications of ω-transaminases. Appl. Microbiol. Biotechnol. 94, 1163–1171. doi: 10.1007/s00253-012-4103-3
Martínez-Montero, L., Gotor, V., Gotor-Fernández, V., and Lavandera, I. (2016). But-2-ene-1,4-diamine and But-2-ene-1,4-diol as donors for thermodynamically favored transaminase- and alcohol dehydrogenase-catalyzed processes. Adv. Synth. Catal. 358, 1618–1624. doi: 10.1002/adsc.201501066
Mathew, S., Bea, H., Nadarajan, S. P., Chung, T., and Yun, H. (2015). Production of chiral β-amino acids using ω-transaminase from Burkholderia graminis. J. Biotechnol. 196–197, 1–8. doi: 10.1016/j.jbiotec.2015.01.011
Midelfort, K. S., Kumar, R., Han, S., Karmilowicz, M. J., McConnell, K., Gehlhaar, D. K., et al. (2013). Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialis aminotransferase for the synthesis of imagabalin. Protein Eng. Des. Sel. 26, 25–33. doi: 10.1093/protein/gzs065
Mirjafary, Z., Abdoli, M., Saeidian, H., Boroon, S., and Kakanejadifard, A. (2015). Oxime ethers as versatile precursors in organic synthesis: a review. RSC Adv. 5, 79361–79384. doi: 10.1039/C5RA15299B
Mojena, M., Povo-Retana, A., González-Ramos, S., Fernández-García, V., Regadera, J., Zazpe, A., et al. (2018). Benzylamine and thenylamine derived drugs induce apoptosis and reduce proliferation, migration and metastasis formation in melanoma cells. Front. Oncol. 8:328. doi: 10.3389/fonc.2018.00328
Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 240–255. doi: 10.1107/S0907444996012255
Nobili, A., Steffen-munsberg, F., Kohls, H., and Trentin, I. (2015). Engineering the active site of the amine transaminase from vibrio fluvialis for the asymmetric synthesis of Aryl – Alkyl amines and amino alcohols. ChemCatChem 7, 757–760. doi: 10.1002/cctc.201403010
Parmeggiani, F., Weise, N. J., Ahmed, S. T., and Turner, N. J. (2018). Synthetic and therapeutic applications of ammonia-lyases and aminomutases. Chem. Rev. 118, 73–118. doi: 10.1021/acs.chemrev.6b00824
Reetz, M. T., and Schimossek, K. (1996). Lipase-catalyzed dynamic kinetic resolution of chiral amines: use of palladium as the racemization catalyst. Chimia (Aarau) 50, 668–669.
Revelles, O., Espinosa-urgel, M., Fuhrer, T., Sauer, U., and Ramos, J. L. (2005). Multiple and interconnected pathways for L-lysine catabolism in Pseudomonas putida KT2440. J Bacteriol.187, 7500–7510. doi: 10.1128/JB.187.21.7500-7510.2005
Roughley, S. D., and Jordan, A. M. (2011). The medicinal chemist's toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 54, 3451–3479. doi: 10.1021/jm200187y
Sayer, C., Isupov, M. N., Westlake, A., and Littlechild, J. A. (2013). Structural studies of Pseudomonas and Chromobacterium ω- aminotransferases provide insights into their differing substrate specificity. Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 564–576. doi: 10.1107/S0907444912051670
Schätzle, S., Höhne, M., Redestad, E., Robins, K., and Bornscheuer, U. T. (2009). Rapid and sensitive kinetic assay for characterization of ω-transaminases. Anal. Chem. 81, 8244–8248. doi: 10.1021/ac901640q
Schell, U., Wohlgemuth, R., and Ward, J. M. (2009). Synthesis of pyridoxamine 5′-phosphate using an MBA:pyruvate transaminase as biocatalyst. J. Mol. Catal. B Enzym. 59, 279–285. doi: 10.1016/j.molcatb.2008.10.005
Shen, H. C., and Hammock, B. D. (2012). Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J. Med. Chem. 55, 1789–1808. doi: 10.1021/jm201468j
Shin, J., and Kim, B. (2002). Exploring the active site of amine:pyruvate aminotransferase on the basis of the substrate structure-reactivity relationship: how the enzyme controls substrate specificity and stereoselectivity. J. Org. Chem. 67, 2848–2853. doi: 10.1021/jo016115i
Shin, J.-S., Yun, H., Jang, J.-W., Park, I., and Kim, B.-G. (2003). Purification, characterization, and molecular cloning of a novel amine:pyruvate transaminase from Vibrio fluvialis JS17. Appl. Microbiol. Biotechnol. 61, 463–471. doi: 10.1007/s00253-003-1250-6
Shin, J. S., and Kim, B. G. (1997). Kinetic resolution of alpha-methylbenzylamine with omicron-transaminase screened from soil microorganisms: application of a biphasic system to overcome product inhibition. Biotechnol. Bioeng. 55, 348–358. doi: 10.1002/(SICI)1097-0290(19970720)55:2<348::AID-BIT12>3.0.CO;2-D
Slabu, I., Galman, J. L., Iglesias, C., Weise, N. J., Lloyd, R. C., and Turner, N. J. (2018). n -Butylamine as an alternative amine donor for the stereoselective biocatalytic transamination of ketones. Catal. Today 306, 96–101. doi: 10.1016/j.cattod.2017.01.025
Slabu, I., Galman, J. L., Lloyd, R. C., and Turner, N. J. (2017). Discovery, engineering, and synthetic application of transaminase biocatalysts. ACS Catal. 7, 8263–8284. doi: 10.1021/acscatal.7b02686
Tan, X., Gao, S., Zeng, W., Xin, S., Yin, Q., and Zhang, X. (2018). Asymmetric synthesis of chiral primary amines by ruthenium-catalyzed direct reductive amination of alkyl aryl ketones with ammonium salts and molecular H2. J. Am. Chem. Soc. 140, 2024–2027. doi: 10.1021/jacs.7b12898
Tufvesson, P., Bach, C., and Woodley, J. M. (2014). A model to assess the feasibility of shifting reaction equilibrium by acetone removal in the transamination of ketones using 2-propylamine. Biotechnol. Bioeng. 111, 309–319. doi: 10.1002/bit.25017
Wilding, M., Scott, C., Peat, T. S., and Newman, J. (2017). X-ray crystal structure of a malonate-semialdehyde dehydrogenase from Pseudomonas sp. strain AAC. Acta Crystallogr. Sect. F Struct. Biol. Commun. 73, 24–28. doi: 10.1107/S2053230X16020008
Ye, L. J., Toh, H. H., Yang, Y., Adams, J. P., Snajdrova, R., and Li, Z. (2015). Engineering of amine dehydrogenase for asymmetric reductive amination of ketone by evolving Rhodococcus phenylalanine dehydrogenase. ACS Catal. 5, 1119–1122. doi: 10.1021/cs501906r
Keywords: biocatalysis, transaminase, green chemistry, benzylamines, protein structure
Citation: Galman JL, Gahloth D, Parmeggiani F, Slabu I, Leys D and Turner NJ (2018) Characterization of a Putrescine Transaminase From Pseudomonas putida and its Application to the Synthesis of Benzylamine Derivatives. Front. Bioeng. Biotechnol. 6:205. doi: 10.3389/fbioe.2018.00205
Received: 17 October 2018; Accepted: 12 December 2018;
Published: 21 December 2018.
Edited by:
Kateryna Fesko, Graz University of Technology, AustriaReviewed by:
Volker F. Wendisch, Bielefeld University, GermanyCopyright © 2018 Galman, Gahloth, Parmeggiani, Slabu, Leys and Turner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas J. Turner, bmljaG9sYXMudHVybmVyQG1hbmNoZXN0ZXIuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.