 Sayaka Mino
Sayaka Mino Naoki Yoneyama1
Naoki Yoneyama1 Satoshi Nakagawa
Satoshi Nakagawa Ken Takai
Ken Takai Tomoo Sawabe
Tomoo Sawabe
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 28 November 2018
Sec. Bioprocess Engineering
Volume 6 - 2018 | https://doi.org/10.3389/fbioe.2018.00184
This article is part of the Research Topic Bioprospecting and Biotechnology of Extremophiles View all 11 articles
Nitrous oxide (N2O) is a greenhouse gas and also leads to stratospheric ozone depletion. In natural environments, only a single N2O sink process is the microbial reduction of N2O to N2, which is mediated by nitrous oxide reductase (NosZ) encoded by nosZ gene. The nosZ phylogeny has two distinct clades, clade I and formerly overlooked clade II. In deep-sea hydrothermal environments, several members of the class Campylobacteria are shown to harbor clade II nosZ gene and perform the complete denitrification of nitrate to N2; however, little is known about their ability to grow on exogenous N2O as the sole electron acceptor. Here, we obtained an enrichment culture from a deep-sea hydrothermal vent in the Southern Mariana Trough, which showed a respiratory N2O reduction with H2 as an electron donor. The single amplicon sequence variant (ASV) presenting 90% similarity to Hydrogenimonas species within the class Campylobacteria was predominant throughout the cultivation period. Metagenomic analyses using a combination of short-read and long-read sequence data succeeded in reconstructing a complete genome of the dominant ASV, which encoded clade II nosZ gene. This study represents the first cultivation analysis that shows the occurrence of N2O-respiring microorganisms in a deep-sea hydrothermal vent and provides the opportunity to assess their capability to reduce N2O emission from the environments.
Nitrous oxide (N2O) is a stable greenhouse gas and a major stratospheric ozone layer-depleting substance of the 21th century (Ravishankara et al., 2009). Its 100-years global warming potential is around 300 times higher than CO2 (IPPC., 2007). Since atmospheric N2O concentration has steadily increased at a rate of 0.2–0.3% per year and this increase is thought be due to anthropogenic emissions (IPCC., 2006), mitigation of N2O release is an international challenge in the context of controlling global nitrogen cycle.
Nitrous oxide (N2O) can be reduced at the final step of the microbial denitrification pathway, which consists of the sequential reduction of to , NO, N2O, and N2. N2O reductase (NosZ), encoded by nosZ gene, is the only known enzyme that converts N2O to N2 gas. NosZ is phylogenetically classified into two clades; clade I (typical NosZ) and clade II (atypical NosZ). Newly described clade II NosZ (Sanford et al., 2012; Jones et al., 2013) is often identified within non-denitrifying N2O-reducing microorganisms that lack other denitrification genes or perform dissimilatory nitrate reduction to ammonium. The abundance of microorganisms possessing the clade II NosZ outnumbers that of microorganisms possessing the clade I NosZ in the diverse environments (Jones et al., 2013, 2014), and clade II organisms have been reported with the higher affinity to N2O than clade I organisms (Yoon et al., 2016), suggesting that microorganisms possessing the clade II NosZ play a crucial role in attenuating N2O emission in the various natural environments.
Deep-sea hydrothermal vent fields are representative environments where the ecosystem is fueled by chemosynthetic microorganisms that are taxonomically and metabolically diverse (Takai and Nakamura, 2011; Waite et al., 2017; Mino and Nakagawa, 2018). Members of the class Campylobacteria are known as one of the predominant bacterial groups there (Muto et al., 2017). Nitrate is a primary electron acceptor for chemosynthetic Campylobacteria (Sievert and Vetriani, 2012), and several campylobacterial isolates mediate complete denitrification of to N2 (Nakagawa et al., 2005; Takai et al., 2006), indicating their ability to reduce N2O to N2. Clade II nosZ gene has been detected in the genomes of several Campylobacteria isolated from deep-sea hydrothermal environments (Inagaki et al., 2003, 2004; Nakagawa et al., 2005; Giovannelli et al., 2016). In addition to metabolic and genetic characteristics of the isolates, multiple omics analyses have provided the insights into the in situ N2O-reducing metabolic potential of Campylobacteria (Fortunato and Huber, 2016; Pjevac et al., 2018). The reduction of exogeneous N2O, however, has never been characterized for Campylobacteria living in deep-sea hydrothermal vents. Here, we, for the first time, report on the direct N2O reduction of deep-sea vent chemolithoautotrophs and on its ability of N2O consumption.
The chimney structure was taken with R/V Yokosuka and DSV Shinkai 6500 from the Baltan chimney at the Urashima site (12°55.3014'N, 143°38.8946'E) in the South Mariana Trough in 2010 during the JAMSTEC cruise YK10-10. After retrieval on board, the sample was anaerobically processed as described previously (Mino et al., 2013). Samples were stored at 4°C until use. Serial dilution cultures were performed using HNN medium. HNN medium contained 0.1% (w/v) NaHCO3 per liter of modified MJ synthetic seawater (Sako et al., 1996). Modified MJ synthetic seawater is composed of 25 g NaCl, 4.2 g MgCl2 6H2O, 3.4 g MgSO4 7H2O, 0.5g KCl, 0.25 g NH4Cl, 0.14 g K2HPO4, 0.7 g CaCl2 2H2O, and 10 ml trace mineral solution per liter of distilled water. To prepare HNN medium, concentrated solution of NaHCO3 was added before gas purging of 100% N2O. The tubes were then tightly sealed with butyl rubber stopper, autoclaved, and pressurized the headspace to 300 kPa with 80% H2 + 20% CO2. H2 is a sole energy source of the medium. The enrichment was performed with HNN medium under 33°C. The N2 production under N2O-reducing condition was confirmed using a Shimazu GC-2014 gas chromatograph (Shimadzu, Kyoto, Japan).
The microbial community was precultured in HNN medium for 24 h at 38°C. Then, 2 mL of preculture was inoculated to 50 mL glass vials each containing 20 mL medium (33% N2O + 54% H2 + 13% CO2, 300 kPa). 5 vials were cultivated at once at 38°C, with mixing using a magnetic stirrer (MS-51M; AS ONE, Osaka, Japan) at 850 rpm. The concentration of headspace N2O of each cultivation vial at each time point (t = 0, 12, 18, 24, 36, 42, and 48 h) was measured using a gas chromatograph (GC-2014; Shimadzu, Kyoto, Japan) with the SHINCARBON ST 50/80 (2 m × 3 mmϕ) column (Shinwa Chemical Industries, Kyoto, Japan). The cultivation was continued until decrease of N2O concentration reaches a plateau. At each time point (t = 12, 18, 24, 36, and 48 h), one of the replicate vials was used and 20 mL of culture medium was transferred into 50 ml plastic centrifugation tubes. After centrifugation at 10,000 × g for 20 min, the supernatant was removed and cell pellet was stored at −30°C until DNA extraction. Cultivation experiment was carried out in triplicate.
DNA extraction from the pelleted cell culture samples were performed with NucleoSpin Soil DNA kit (Macherey-Nagel, Düren, Germany) according to the protocol provided by the manufacturer. The DNA samples were stored at −30°C until sequencing analysis. A total of 15 DNA samples was used for 16S rRNA gene-based microbial community analysis. Four DNA samples from two time points (t = 12 and 36 h, n = 2) were also used for metagenomic analysis.
Each amplicon library was constructed by amplifying V1-V2 paired-end libraries using 27Fmod (5′-AGRGTTTGATYMTGGCTCAG-3′) and 338R (5′-TGCTGCCTCCCGTAGGAGT-3') according to Nextera library preparation methods. Equal amounts of each PCR amplicons were pooled, followed by amplification and 2 × 300 bp paired-end sequencing on the MiSeq platform according to the manufacturer's instructions.
A total of 519,290 raw reads across 15 samples, with an average 34,619 reads per sample, was further processed by QIIME 2 software package (version 2018.4). Quality control was performed using DADA2 (Callahan et al., 2016) by removing chimeras and residual PhiX reads, and low-quality regions of sequences. Forward and reverse reads were, respectively, truncated to 230 and 209 bp for DADA2 analysis based on the average quality scores determined. After quality filtering, the dataset contained 421,406 reads, with an average of 28,093 sequences per samples. Dereplication was then performed by DADA2, which combines identical reads into amplicon sequence variants (ASVs), which is analog to the traditional Operational Taxonomic Unit (OTU). Representative sequences were classified into taxonomic groups using the SILVA 132 database. Mitochondrial and chloroplast sequences were then removed from the feature table, and bacterial and archaeal sequences were used further analysis. We used BLASTN search against the online nucleotide collection (nr/nt) database on NCBI to find the closest GenBank neighbor for these representative sequences.
For two replicates from two time points (t = 12 and 36 h), paired-end libraries for metagenome sequencing were generated using Nextera library preparation methods. Genome sequencing was then performed on an Illumina MiSeq platform (2 × 300 bp paired-end). Read data from Illumina sequencing were trimmed with Platanus_trim (Kajitani et al., 2014).
Nanopore sequencing was conducted with a DNA sample from a time point 36 h (n = 1). Sequencing library preparation was carried out using the Rapid Barcoding Sequence kit (SQK-RBK001) (Oxford Nanopore Technologies, Oxford, UK) according to the standard protocol provided by the manufacturer. The constructed library was loaded into the FlowCell (FLO-MIN106) on a MinION device and performed 24 h sequencing run with MinKNOW1.7.14 software. The fast5 read file was basecalled with ONT Albacore command line tool (v2.0.2). The adapter sequences were trimmed from the fastq read file using Porechop v0.2.1 (https://github.com/rrwick/Porechop).
For assembly, Canu version 1.6 (Koren et al., 2017) was run using Nanopore reads with following options: genome size 2.1 Mb, and correctedErrorRate = 0.120. The Illumina short reads were mapped to the Canu's assembly by BWA version 0.7.12 (Li, 2013). Pilon version 1.22 (Walker et al., 2014) was run on the alignments to polish the assembly sequences. Circlator version 1.5.1 (Hunt et al., 2015) program was used for circularizing the genome assembly. Two contigs were again polished by Pilon using Illumina reads. Metagenome assembled genome (MAG) sequences were assessed using CheckM v1.0.8 (Parks et al., 2015) for the completion estimate, and then larger MAG was annotated with the Rapid Annotation using Subsystem Technology (RAST) server v2.0 (Aziz et al., 2008). rRNA genes were predicted using RNAmmer (Lagesen et al., 2007). 16S rRNA gene sequences of the MAG were compared to a representative sequence of a dominant ASV obtained by 16S rRNA amplicon analysis. MAG was functionally annotated using KEGG database with the BlastKOALA pipeline (Kanehisa et al., 2016) and KO numbers were mapped using KEGG mapper to compare the metabolic pathway with a closely related species.
Taxonomic placement of the MAG was assessed based on both 16S rRNA and genomic approaches. 16S rRNA gene sequence of the MAG was aligned to closely related species using SILVA Incremental Aligner v1.2.11 (Pruesse et al., 2012). A phylogenetic tree was constructed by Maximum Likelihood (ML) method using MEGA 7.0.26 package (Kumar et al., 2016) with GTR+I+G substitution model. Phylogenomic tree construction was performed using PhyloPhlAn (Segata et al., 2013), which extracts up to 400 conserved proteins coded in the genomes of closely related species and generates multiple alignments of each protein. Multiple alignments of each universal protein were then concatenated into a single amino acid sequence. The optimal model for phylogenomic analysis was determined by Modelgenerator (Keane et al., 2006), and then ML tree was constructed using RAxML version 8.2.11 (Stamatakis, 2014) with BLOSUM62+G+F model. Average nucleotide identity (ANI), average amino acid identity (AAI) and in-silico DNA-DNA hybridization (DDH) values were calculated by ANI calculator (http://enve-omics.ce.gatech.edu/ani/index), AAI calculator (http://enve-omics.ce.gatech.edu/aai/) and Genome-to-Genome Distance Calculator (GGDC 2.1) (Meier-Kolthoff et al., 2013), respectively.
The genomes of chemosynthetic Campylobacteria were used to reconstruct the nos gene clusters. Sequences of functionally characterized NosZ protein and its accessory genes from Wolinella succinogenes were used to search the genes adjacent to inferred nosZ on Hydrogenimonas sp. MAG. Multiple amino acid sequence alignment was generated from representatives of clade I and clade II nosZ sequences using MUSCLE version 3.8.31 (Edgar, 2004). ML tree based on nosZ amino acid sequences was constructed by MEGA with WAG+G model based upon 500 bootstrapped replicates. Trees were visualized using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
In attempt to evaluate the nitrate-respiring ability of the enrichment, each of the potential electron donors, such as H2, elemental sulfur (1%, w/v), or thiosulfate (0.1%, w/v) was tested with nitrate (0.1%, w/v) as an electron acceptor under 40°C. The ability to use H2 was examined with 80% H2 + 20% CO2 (300 kPa) in the headspace. For testing growth on elemental sulfur and thiosulfate as an electron donor, 80% N2 + 20 % CO2 was used as the gas phase (300 kPa).
Approximately 90% of initial headspace N2O was consumed after 42 h of cultivation (Figure 1). The maximum rate of N2O consumption was 3.3 μmol h−1 per ml of culture, higher than other clade I and II reducers (Sanford et al., 2012; Yoon et al., 2016), though a higher initial N2O concentration in this study was provided than in the previous studies. The aqueous-phase N2O concentration was calculated from the Ostwald coefficient (0.4190) at 1 atm partial pressure and 40°C (Wilhelm et al., 1977). 2.75 mmol of vessel N2O in 20 ml of medium corresponded to an initial aqueous-phase N2O concentration 56.5 mM. This concentration is more than million-fold higher than the N2O level observed in deep-sea hydrothermal environments, where N2O is mainly supplied by microbial nitrification and denitrification processes (Lilley et al., 1982; Kawagucci et al., 2010).
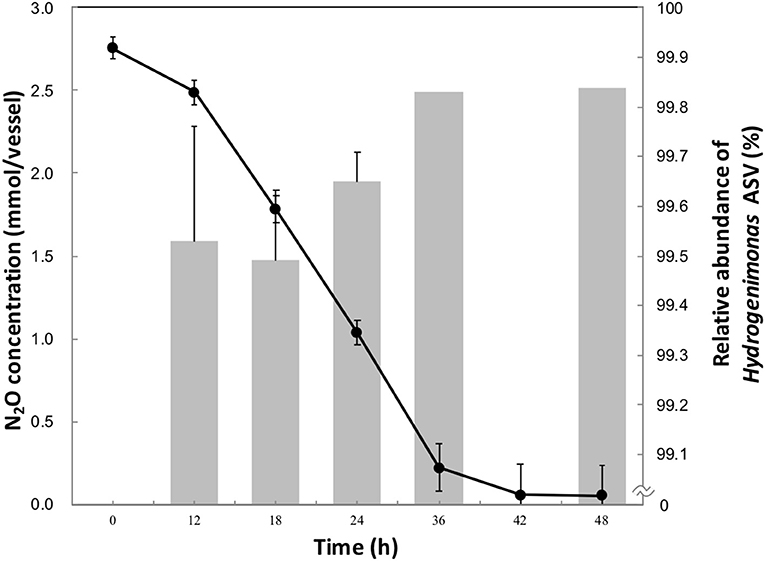
Figure 1. N2O consumption (•) and relative abundance of Hydrogenimonas ASV in the enrichment culture (bar chart) during the 48 h-cultivation. The data points and error bars represent the means and standard errors, respectively.
To identify the N2O-reducing organisms in the enrichment culture, we analyzed the time-series community structure during the cultivation under the HNN medium. Dereplicate 421,406 Illumina reads resulted in 11 bacterial ASVs. The genus Hydrogenimonas consisting of a single ASV was the most predominant bacterial group through the cultivation period and its relative abundance was above 98% (Figure 1 and Figure S1). In order to further identify this dominant ASV, a representative sequence was extracted from the sequence data and analyzed by BLAST search. The best match represented Hydrogenimonas thermophila EP1-55-1%T (= JCM 11971T = ATCC BAA-737T) with 90% identity, suggesting the host organism of the ASV would be a new species of Hydrogenimonas or even a new genus of the class Campylobacteria. Strain EP1-55-1%, isolated from a deep-sea hydrothermal vent field in the Indian Ocean, is able to utilize nitrate as an electron acceptor and shows the optimum growth at 55°C (Takai et al., 2004). These characteristics are different from the N2O-reducing enrichment culture in this study, which shows an optimum growth at 40°C and no growth under nitrate-respiring condition. The genus Thalassospira consisting of a single ASV accounted for about 0.7% of reads at 12 h, and its abundance gradually decreased to 0.1% at 36 h of cultivation. The representative sequence of the ASV assigned to Thalassospira showed 100% similarity to T. xianhensis P-4T (= CGMCC 1.6849T = JCM 14850T) isolated from an oil-polluted saline soil (Zhao et al., 2010). T. xianhensis engages denitrification, possibly having N2O-reducing ability, but it cannot grow under autotrophic condition. Along with the increase of relative abundance of Hydrogenimonas, headspace N2O decreased and reached 0.05 mmol/vessel at 48 h, showing that members belonging to Hydrogenimonas significantly contribute to N2O consumption of the enrichment culture.
In order to understand the genomic characteristics of most dominant ASV species, we employed the MAG reconstruction strategy, because cells formed pellicle-like structures that was unable to be purified to a single pure strain by the dilution-to-extinction technique. High-quality 14,951,908 reads (4.6 Gb) and 236,546 reads (1.1 Gb) of metagenomic data were obtained by Illumina and Nanopore sequencing, respectively, and assembled into two contigs. After circularization, a larger contig resulted in a complete circular, which is 2,193,435 bp in length with an average G + C content of 50.2% and >99% genome completeness (Figure S2). In total, 2,185 coding sequence (CDS) regions and 54 RNAs were annotated. Three copies of 16S rRNA gene were annotated by RNAmmer, and its V1-V2 regions were identical with each other and with the represent sequence of the dominant ASV. With full length of 16S rRNA gene sequences as a query in BLAST search, the highest similarity was estimated with H. thermophila EP1-55-1%T (94%). The size of the other contig is 49,484 bp with 0% completeness level estimated by CheckM.
Both 16S rRNA gene-based and genome-based phylogenetic trees revealed that the MAG was most closely related to H. thermophila (Figures 2,3). In-silico DDH and ANI values between the strain BAL40 MAG and H. thermophila genome sequences were 17.4% and 74.4%, respectively, well below species cutoff (Richter and Rosselló-Móra, 2009). AAI value with H. thermophila was 71.4%, which is higher than the genus criterion level (Konstantinidis and Tiedje, 2005; Konstantinidis et al., 2017). Taken together, the strain BAL40 MAG described here represents a novel species within the genus Hydrogenimonas.
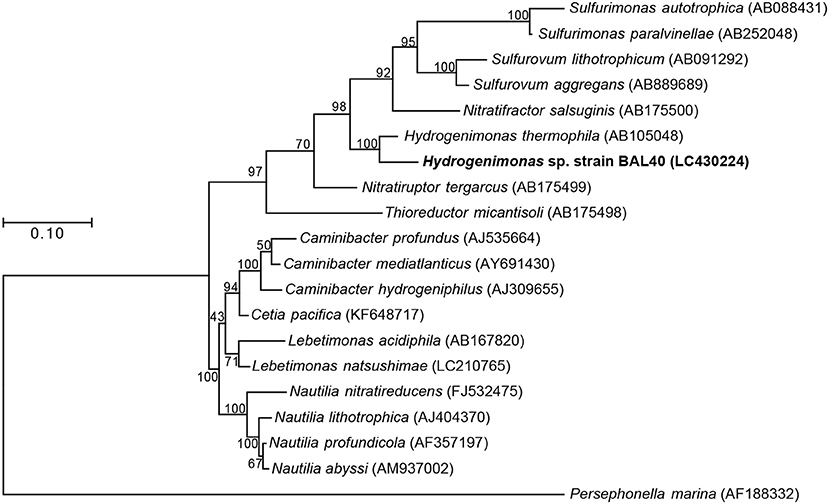
Figure 2. ML Phylogenetic tree based on 1,196 nucleotide position of 16S rRNA gene sequences. Bootstrap values based on 500 resampling replicates are shown as percentages at branch nodes.

Figure 3. ML tree based on genome sequences and schematic comparison of the nos gene clusters in different Campylobacteria. Bootstrap values based on 500 resampling replicates are shown as percentages at branch nodes. Clade II nosZ and its accessory genes (BDFGHLY) are labeled and colored according to homology across the different species.
Reconstruction of denitrification pathway using KEGG database revealed that the MAG possessed whole set of denitrification genes (nap, nir, nor, and nos) (Figure S3), suggesting the ability to utilize nitrate as an electron acceptor. However, the enrichment culture did not show any growth under the nitrate-respiring condition in this study. This discrepancy between genetic and physiological characteristics implies the existence of heretofore overlooked N2O-reducing microorganisms, which are genetically denitrifiers sensu stricto but are physiologically non-denitrifiers. However, the influence of in situ physicochemical conditions on their denitrification metabolisms could not be examined here. Considering our results, we speculate that together diverse microbial species drive different steps to the full denitrification pathway of nitrate to N2 in deep-sea hydrothermal environments. It should be noted that because most environmental studies investigating abundance, diversity and function of denitrifiers have been performed based solely on the PCR-based marker gene analysis (Braker and Tiedje, 2003; Henry et al., 2006; Smith et al., 2007; Sanford et al., 2012; Jones et al., 2013), it is virtually impossible to identify which organism is actually responsible for specific steps of the denitrification process. Further culture-dependent studies could demonstrate the actual function of both denitrifying and non-denitrifying microorganisms. In addition, cultivation under N2O-respiring condition might decrease the number of yet-to-be cultured microorganisms and unveil the N2O mitigation potential hidden in deep-sea hydrothermal environments.
We reconstructed nos gene clusters from the genomes of MAG and related Campylobacteria including Wolinella succinogenes, which has been used as a campylobacterial model organism (Figure 3). Similar to W. succinogenes, Hydrogenimonas sp. MAG possessed a clade II nos gene cluster that contains nosZ, -B, -D, -G, -H, -C1, -C2, -H, -Y, -L genes, and the presence and organization of these genes were well-conserved in the related Campylobacteria members, as previously described (Torres et al., 2016). A phylogenetic tree based on amino acid sequences of nosZ gene indicated that the sequence of Hydrogenimonas sp. MAG is closely related to Nitratifractor salsuginis E9I37-1T, which is known as a nitrate-reducing mesophile (Nakagawa et al., 2005) (Figure S4). Multiple alignment of NosZ amino acid sequences that contained both clade I and clade II sequences showed that NosZ of Campylobacteria including Hydrogenimonas sp. MAG conserved ligands of two cooper sites; CuA electron transfer center and CuZ catalytic center (Zumft and Kroneck, 2007) (Figure S5). It is therefore deduced that N2O-respiring metabolism supports more diverse Campylobacteria in deep-sea hydrothermal environments.
A variety of microorganisms contributing to the denitrification process have been reported in a wide range of environments such as sediments, paddy soils, hydrothermal vents, and geothermal streams (Shao et al., 2010; Masuda et al., 2017; Kato et al., 2018). N2O emissions by denitrification are the net result of the balance between its production and reduction to N2. In deep-sea hydrothermal environments, diverse microorganisms including Campylobacteria involved in denitrification processes (Bourbonnais et al., 2012; Bowles et al., 2012; Fortunato and Huber, 2016; Pjevac et al., 2018). The occurrence of microorganisms with the high ability to reduce exogenous N2O could explain the previous physicochemical results that in situ N2O level is quite low (Lilley et al., 1982; Kawagucci et al., 2010), and its flux to the deep waters is negligible (Bange and Andreae, 1999). Our findings imply the capacity of deep-sea hydrothermal environments to act as a sink for N2O is greater than its capacity to emit N2O.
We conducted, to the best of our knowledge, the first culture-dependent study of deep-sea hydrothermal vent N2O-reducing microorganisms. Evaluation of N2O consumption ability and genome reconstruction from metagenomic data demonstrated that the notable N2O consumption rate of the novel member within genus Hydrogenimonas. Our findings will open door for new ideas to implement biotechnological applications of NosZ from thermophiles, such as nitrogen removal from wastewater. Future studies, such as comparative cultivation analysis of N2O-reducing Campylobacteria, transcriptional analysis of nos genes, and measurement of N2O consumption kinetics, may provide the insights into the mechanisms allowing to their high N2O consumption ability.
This project has been deposited at DDBJ/EMBL/GenBank under the BioProject PRJDB7296. Sequences for MAG and 16S rRNA gene of Hydrogenimonas sp. strain BAL40 are available with DDBJ/EMBL/GenBank AP019005 and LC430224, respectively.
SM, SN, and TS designed research. SM, NY performed the experiments and analyzed the data. SN and KT contributed the sample collection. SM and TS contributed reagents, materials, and analysis tools. SM wrote the paper with inputs from SN, KT, and TS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the captain and crew of R/V Yokosuka and DSV Shinkai 6500 operation team on cruise JAMSTEC YK10-10. We are also grateful to Ms. Mami Tanaka (Hokkaido University, Hakodate, Japan) for invaluable help with Nanopore sequencing, and to Prof. Masahira Hattori and Dr. Wataru Suda (the University of Tokyo, Kashiwa, Japan) for assistance with sequencing of metagenome by Illumina. We would like to thank Muneyuki Fukushi (Hokkaido University, Hakodate, Japan) for kindly assistance with cultivation experiments. This work was partially supported by the JSPS KAKENHI (No. 12J03037 and No. 15H05991).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2018.00184/full#supplementary-material
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bange, H. W., and Andreae, M. O. (1999). Nitrous oxide in the deep waters of the world's oceans. Glob. Biogeochem. Cycles 13, 1127–1135. doi: 10.1029/1999GB900082
Bourbonnais, A., Juniper, S. K., Butterfield, D. A., Devol, A. H., Kuypers, M. M. M., Lavik, G., et al. (2012). Activity and abundance of denitrifying bacteria in the subsurface biosphere of diffuse hydrothermal vents of the Juan de Fuca Ridge. Biogeosciences 9, 4661–4678. doi: 10.5194/bg-9-4661-2012
Bowles, M. W., Nigro, L. M., Teske, A. P., and Joye, S. B. (2012). Denitrification and environmental factors influencing nitrate removal in Guaymas Basin hydrothermally altered sediments. Front. Microbiol. 3:377. doi: 10.3389/fmicb.2012.00377
Braker, G., and Tiedje, J. M. (2003). Nitric oxide reductase (norB) genes from pure cultures and environmental samples. Appl. Environ. Microbiol. 69, 3476–3483. doi: 10.1128/AEM.69.6.3476-3483.2003
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fortunato, C. S., and Huber, J. A. (2016). Coupled RNA-SIP and metatranscriptomics of active chemolithoautotrophic communities at a deep-sea hydrothermal vent. ISME J. 10, 1925–1938. doi: 10.1038/ismej.2015.258
Giovannelli, D., Chung, M., Staley, J., Starovoytov, V., Le Bris, N., and Vetriani, C. (2016). Sulfurovum riftiae sp. nov., a mesophilic, thiosulfate-oxidizing, nitrate-reducing chemolithoautotrophic epsilonproteobacterium isolated from the tube of the deep-sea hydrothermal vent polychaete Riftia pachyptila. Int. J. Syst. Evol. Microbiol. 66, 2697–2701. doi: 10.1099/ijsem.0.001106
Henry, S., Bru, D., Stres, B., Hallet, S., and Philippot, L. (2006). Quantitative detection of the nosZ gene, encoding nitrous oxide reductase, and comparison of the abundances of 16S rRNA, narG, nirK, and nosZ genes in soils. Appl. Environ. Microbiol. 72, 5181–5189. doi: 10.1128/AEM.00231-06
Hunt, M., Silva, N. D., Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16:294. doi: 10.1186/s13059-015-0849-0
Inagaki, F., Takai, K., Kobayashi, H., Nealson, K. H., and Horikoshi, K. (2003). Sulfurimonas autotrophica gen. nov., sp. nov., a novel sulfur-oxidizing ε-proteobacterium isolated from hydrothermal sediments in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 53, 1801–1805. doi: 10.1099/ijs.0.02682-0
Inagaki, F., Takai, K., Nealson, K. H., and Horikoshi, K. (2004). Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the ε-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 54, 1477–1482. doi: 10.1099/ijs.0.03042-0
IPPC. (2007). Climate Change 2007: Mitigation of Climate Change; Contribution of Working Group III to the the 4th Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge.
Jones, C. M., Graf, D. R., Bru, D., Philippot, L., and Hallin, S. (2013). The unaccounted yet abundant nitrous oxide-reducing microbial community: a potential nitrous oxide sink. ISME J. 7, 417–426. doi: 10.1038/ismej.2012.125
Jones, C. M., Spor, A., Brennan, F. P., Breuil, M.-C., Bru, D., Lemanceau, P., et al. (2014). Recently identified microbial guild mediates soil N2O sink capacity. Nat. Clim. Chang. 4, 801–805. doi: 10.1038/nclimate2301
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kato, S., Sakai, S., Hirai, M., Tasumi, E., Nishizawa, M., et al. (2018). Long-term cultivation and metagenomics reveal ecophysiology of previously uncultivated thermophiles involved in biogeochemical nitrogen cycle. Microbes Environ. 33, 107–110. doi: 10.1264/jsme2.ME17165
Kawagucci, S., Shirai, K., Lan, T. F., Takahata, N., Tsunogai, U., Sano, Y., et al. (2010). Gas geochemical characteristics of hydrothermal plumes at the HAKUREI and JADE vent sites, the Izena Cauldron, Okinawa Trough. Geochem. J. 44, 507–518. doi: 10.2343/geochemj.1.0100
Keane, T. M., Creevey, C. J., Pentony, M. M., Naughton, T. J., and Mclnerney, J. O. (2006). Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 6:29. doi: 10.1186/1471-2148-6-29
Konstantinidis, K. T., Rosselló-Móra, R., and Amann, R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. doi: 10.1038/ismej.2017.113
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic. Acids. Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. arXiv:1303.3997. Available online at: http://arxiv.org/abs/1303.3997
Lilley, M. D., de Angelis, M. A., and Gordon, L. I. (1982). CH4, H2, CO and N2O in submarine hydrothermal vent waters. Nature 300, 48–50. doi: 10.1038/300048a0
Masuda, Y., Itoh, H., Shiratori, Y., Isobe, K., Otsuka, S., and Senoo, K. (2017). Predominant but previously-overlooked prokaryotic drivers of reductive nitrogen transformation in paddy soils, revealed by metatranscriptomics. Microbes Environ. 32, 180–183. doi: 10.1264/jsme2.ME16179
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Mino, S., Makita, H., Toki, T., Miyazaki, J., Kato, S., Watanabe, H., et al. (2013). Biogeography of Persephonella in deep-sea hydrothermal vents of the Western Pacific. Front. Microbiol. 4:107. doi: 10.3389/fmicb.2013.00107
Mino, S., and Nakagawa, S. (2018). “Deep-sea ven extrimophiles: cultivation, physiological characteristics, and ecological significance,” in Extremophiles: From biology to biotechnology, eds R. V. Durvasula and D. V. S. Rao (New York, NY: CRC Press), 165–183.
Muto, H., Takaki, Y., Hirai, M., Mino, S., Sawayama, S., Takai, K., et al. (2017). A simple and efficient RNA extraction method from deep-sea hydrothermal vent chimney structures. Microbes Environ. 32, 330–335. doi: 10.1264/jsme2.ME17048
Nakagawa, S., Takai, K., Inagaki, F., Horikoshi, K., and Sako, Y. (2005). Nitratiruptor tergarcus gen. nov., sp. nov. and Nitratifractor salsuginis gen. nov., sp. nov., nitrate-reducing chemolithoautotrophs of the ε-Proteobacteria isolated from a deep-sea hydrothermal system in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 55, 925–933. doi: 10.1099/ijs.0.63480-0
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015), CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pjevac, P., Meier, D. V., Markert, S., Hentschker, C., Schweder, T., Becher, D., et al. (2018). Metaproteogenomic profiling of microbial communities colonizing actively venting hydrothermal chimneys. Front. Microbiol. 9:680. doi: 10.3389/fmicb.2018.00680
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Ravishankara, A. R., Daniel, J. S., and Portmann, R. W. (2009). Nitrous oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century. Science 326, 123–125. doi: 10.1126/science.1176985
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Sako, Y., Takai, K., Ishida, Y., Uchida, A., and Katayama, Y. (1996). Rhodothermus obamensis sp. nov., a modern lineage of extremely thermophilic marine bacteria. Int. J. Syst. Bacteriol. 46, 1099–1104. doi: 10.1099/00207713-46-4-1099
Sanford, R. A., Wagner, D. D., Wu, Q., Chee-Sanford, J. C., Thomas, S. H., Cruz-García, C., et al. (2012). Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc. Natl. Acad. Sci. USA. 109, 19709–19714. doi: 10.1073/pnas.1211238109
Segata, N., Börnigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4:2304. doi: 10.1038/ncomms3304
Shao, M. F., Zhang, T., and Fang, H. H. (2010). Sulfur-driven autotrophic denitrification: diversity, biochemistry, and engineering applications. Appl. Microbiol. Biotechnol. 88, 1027–1042. doi: 10.1007/s00253-010-2847-1
Sievert, S. M., and Vetriani, C. (2012). Chemoautotrophy at deep-sea vents: past, present, and future. Oceanography 25, 218–233. doi: 10.5670/oceanog.2012.21
Smith, C. J., Nedwell, D. B., Dong, L. F., and Osborn, A. M. (2007). Diversity and abundance of nitrate reductase genes (narG and napA), nitrite reductase genes (nirS and nrfA), and their transcripts in estuarine sediments. Appl. Environ. Microbiol. 73, 3612–3622. doi: 10.1128/AEM.02894-06
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Takai, K., and Nakamura, K. (2011). Archaeal diversity and community development in deep-sea hydrothermal vents. Curr. Opin. Microbiol. 14, 282–291. doi: 10.1016/j.mib.2011.04.013
Takai, K., Nealson, K. H., and Horikoshi, K. (2004). Hydrogenimonas thermophila gen. nov., sp. nov., a novel thermophilic, hydrogen-oxidizing chemolithoautotroph within the ε-Proteobacteria, isolated from a black smoker in a Central Indian Ridge hydrothermal field. Int. J. Syst. Evol. Microbiol. 54, 25–32. doi: 10.1099/ijs.0.02787-0
Takai, K., Suzuki, M., Nakagawa, S., Miyazaki, M., Suzuki, Y., Inagaki, F., et al. (2006). Sulfurimonas paralvinellae sp. nov., a novel mesophilic, hydrogen- and sulfur-oxidizing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent polychaete nest, reclassification of Thiomicrospira denitrificans as Sulfurimonas denitrificans comb. nov. and emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 56, 1725–1733. doi: 10.1099/ijs.0.64255-0
Torres, M. J., Simon, J., Rowley, G., Bedmar, E. J., Richardson, D. J., Gates, A. J., et al. (2016). Nitrous oxide metabolism in nitrate-reducing bacteria: physiology and regulatory mechanisms. Adv. Microb. Physiol. 68, 353–432. doi: 10.1016/bs.ampbs.2016.02.007
Waite, D. W., Vanwonterghem, I., Rinke, C., Parks, D. H., Zhang, Y., Takai, K., et al. (2017). Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 8:682. doi: 10.3389/fmicb.2017.00682
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Wilhelm, E., Battino, R., and Wilcock, R. J. (1977). Low-pressure solubility of gases in liquid water. Chem. Rev. 77, 219–262. doi: 10.1021/cr60306a003
Yoon, S., Nissen, S., Park, D., Sanford, R. A., and Löffler, F. E. (2016). Nitrous oxide reduction kinetics distinguish bacteria harboring clade I NosZ from those harboring clade II NosZ. Appl. Environ. Microbiol. 82, 3793–3800. doi: 10.1128/AEM.00409-16
Zhao, B., Wang, H., Li, R., and Mao, X. (2010). Thalassospira xianhensis sp. nov., a polycyclic aromatic hydrocarbon-degrading marine bacterium. Int. J. Syst. Evol. Microbiol. 60, 1125–1129. doi: 10.1099/ijs.0.013201-0
Keywords: N2O-reducing bacterium, deep-sea hydrothermal field, Campylobacteria, Epsilonproteobacteria, nosZ, nitrous oxide
Citation: Mino S, Yoneyama N, Nakagawa S, Takai K and Sawabe T (2018) Enrichment and Genomic Characterization of a N2O-Reducing Chemolithoautotroph From a Deep-Sea Hydrothermal Vent. Front. Bioeng. Biotechnol. 6:184. doi: 10.3389/fbioe.2018.00184
Received: 16 August 2018; Accepted: 13 November 2018;
Published: 28 November 2018.
Edited by:
Milko Alberto Jorquera, Universidad de La Frontera, ChileReviewed by:
Zongze Shao, Third Institute of Oceanography, ChinaCopyright © 2018 Mino, Yoneyama, Nakagawa, Takai and Sawabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sayaka Mino, c2F5YWthLm1pbm9AZmlzaC5ob2t1ZGFpLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.