 Dario Veneziano
Dario Veneziano Giovanni Nigita
Giovanni Nigita Alfredo Ferro
Alfredo Ferro
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Bioeng. Biotechnol. , 03 June 2015
Sec. Computational Genomics
Volume 3 - 2015 | https://doi.org/10.3389/fbioe.2015.00077
This article is part of the Research Topic Bioinformatics of Non-Coding RNAs with Applications to Biomedicine: Recent Advances and Open Challenges View all 11 articles
The majority of the human transcriptome is defined as non-coding RNA (ncRNA), since only a small fraction of human DNA encodes for proteins, as reported by the ENCODE project. Several distinct classes of ncRNAs, such as transfer RNA, microRNA, and long non-coding RNA, have been classified, each with its own three-dimensional folding and specific function. As ncRNAs are highly abundant in living organisms and have been discovered to play important roles in many biological processes, there has been an ever increasing need to investigate the entire ncRNAome in further unbiased detail. Recently, the advent of next-generation sequencing (NGS) technologies has substantially increased the throughput of transcriptome studies, allowing an unprecedented investigation of ncRNAs, as regulatory pathways and novel functions involving ncRNAs are now also emerging. The huge amount of transcript data produced by NGS has progressively required the development and implementation of suitable bioinformatics workflows, complemented by knowledge-based approaches, to identify, classify, and evaluate the expression of hundreds of ncRNAs in normal and pathological conditions, such as cancer. In this mini-review, we present and discuss current bioinformatics advances in the development of such computational approaches to analyze and classify the ncRNA component of human transcriptome sequence data obtained from NGS technologies.
For over five decades, the central dogma of molecular biology has represented the basis of genetics (Crick, 1970), essentially describing the genetic information flow of life in which DNA and protein, as respectively repository and functional incarnation of that information, have been viewed as the two main actors in the life of the cell, confining RNA simply to the role of template for protein synthesis. Nevertheless, this view of the biological role of RNA, initially apparently exhaustive, has been over time subjected to challenges, as firstly suggested by Gilbert in 1986 (Gilbert, 1986).
As interest on the hypothesized “RNA world” grew, subsequent studies allowed to explore the potential of such new vision (Lee et al., 1993; Fire et al., 1998), eventually leading to one of the most significant biological discoveries of the past decade: the existence of several types of RNAs, each with their specific functions in eukaryotic cells (Eddy, 2001; Todd and Karbstein, 2007). As the ENCODE project has confirmed, most of the human genome is in fact transcribed, but only a very small fraction of it encodes for proteins (Birney et al., 2007; Elgar and Vavouri, 2008). Indeed, the larger remaining portion of the transcribed genomic output is represented by a diverse family of untranslated transcripts that play crucial roles in many biochemical cellular processes (Mattick, 2001).
These non-coding RNAs (ncRNAs) are divided into two major categories most commonly according to their nucleotide sequence length: small (<200 bp) and long (200 bp or more). Within each category, there are several distinct classes, each one with its own three-dimensional folding and specific function.
From the more popular classes of small structural ncRNAs, such as transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), focus has shifted in the last 10 years to a set of small RNA classes involved in post-transcriptional regulation: microRNAs (miRNAs) whose precursors (pre-miRNAs) form a peculiar hairpin structure; small interfering RNAs (siRNAs); piwi-interacting RNAs (piRNAs).
Growing interest has more recently emerged also toward long ncRNAs (lncRNAs) which constitute the majority of the non-protein-coding transcripts (Ponting et al., 2009). Having length >200 nt, lncRNAs, already thought of potentially regulating transcription via chromatin modulation, may be also involved in post-transcriptional regulation, organization of protein complexes, and cell–cell signaling (Meldrum et al., 2011).
Finally, an additional class of ncRNAs is represented by circular RNAs (circRNAs) which have been proven to be untranslated, very stable, abundant, and conserved RNA molecules in animals (Jeck et al., 2013).
Yet, despite having been more than a decade since the human genome was sequenced, most transcribed regions are still of unknown molecular function and biological significance.
A potential approach to solving this problem is provided by the ever increasing application of high throughput sequencing technology (HTS), also known as next-generation sequencing (NGS). In fact, numerous transcriptomic sequencing projects are accumulating with increasing rapidity, generating data which are enabling the identification of different types of ncRNAs, and the quantification of their expression levels in different tissues, conditions, and developmental stages.
Deep sequencing provides a very promising tool. NGS can produce millions of sequences at lower cost in shorter time than before (Meldrum et al., 2011) delivering greater sensitivity and accuracy than previous technologies. Its sensitivity and specificity are above microarray techniques (‘t Hoen et al., 2008; Wang et al., 2009); it does not rely on target probe hybridization, permitting the sequencing of the exact transcript on a single nucleotide resolution (Zhou et al., 2011), thus allowing the identification of variations in length or composition (Jung et al., 2010); it requires no previous transcript information (Isakov et al., 2012), utilizing any relevant database to compare and characterize the sequence population (Ronen et al., 2010); it provides high depth of coverage for any library of nucleic acids and it can be modified to study specific properties, e.g., small RNA-seq (sRNA-seq) (Landgraf et al., 2007); it can be used on species for which a full-genome sequence is not yet available; RNA editing events can be detected, and knowledge of polymorphisms can provide direct measurement of allele-specific expression (Malone and Oliver, 2011).
Several HTS platforms are commercially available, each characterized by specific data throughput, read length, error rate, and price (Zhou et al., 2011), providing a wide choice of options.
Earlier attempts at whole genome identification of ncRNAs generally had already focused on distinct expression patterns and novel RNA structural families to better characterize the properties of ncRNAs. An example of this is the incRNA pipeline employed by Lu et al. (2011) who have developed a comprehensive machine-learned model integrating sequence, structure, and large-scale expression data, both deep sequencing and array. This proves how the complementary nature of combined features can clearly separate ncRNAs from other genomic elements and potentially differentiate between distinct ncRNA types, representing an important advantage of integrative approaches.
Such characterization studies have provided methods that can be adapted to different organisms to identify novel ncRNAs from unannotated genomic regions, paving the way for the development of integrated tools.
Moreover, the large amount of data generated by HTS experiments has made it absolutely necessary to dispose of bioinformatics methods in order to properly store, analyze, and visualize such data.
Generally, a ncRNA bioinformatics analysis system can be comprised of three essential components: a post-sequencing data analysis pipeline for ncRNA detection, classification and expression analysis representing the core of the system; a data module to provide annotation information and storage for the analysis results; a visualization/query system for viewing and functionally analyzing raw data and elaborated results.
As proven by Cordero et al. (2012), statistical detection of differential expression of NGS data gives efficient results when computational strategies employ statistical models based on NB distribution [i.e., baySeq (Hardcastle and Kelly, 2010)] or on variance [i.e., DESeq (Anders and Huber, 2010), DESeq2 (Love et al., 2014)], as opposed to non-parametric methods which are frequently used for microarray-generated data but are very sensitive to background composition when applied to NGS data.
In order to satisfy the urgent demand for intuitive and efficient data exploration and relieve the growing pressure on handling massive quantities of short-read sequences, several NGS-based RNA transcriptome bioinformatics analysis tools/pipelines have been developed (Tables 1 and 2), and below we give an overview of the current most popular ones.

Table 1. Small non-coding RNA Tool comparison.
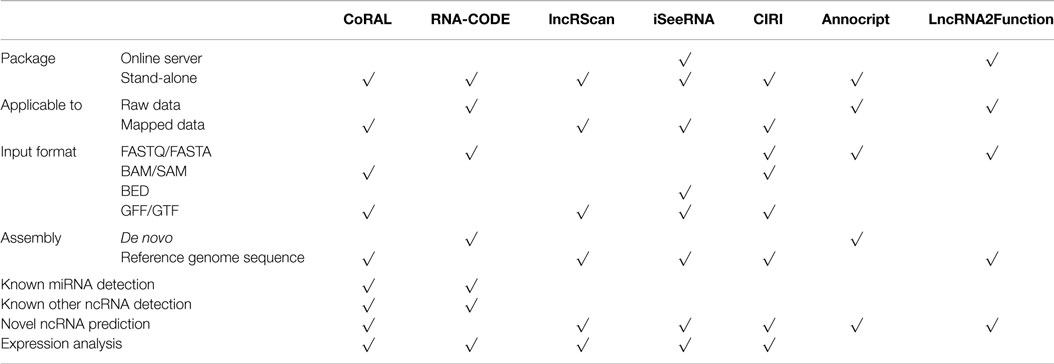
Table 2. Long non-coding RNA Tool comparison.
Throughout the last decade, the study of the small RNA transcriptome has been gradually recognized to be essential to fully comprehend the complex scenario of transcriptional regulation. For this reason, most currently available tools/pipelines for transcriptome investigation through NGS concentrate on detection/prediction/expression quantification of small RNAs, especially miRNAs.
miRDeep (Friedländer et al., 2008) is believed to be the first stand-alone tool used to analyze large-scale sRNA-seq data in order to detect both known and novel miRNAs. miRDeep employs Bayesian probability controls along the steps of miRNA biogenesis to estimate the false-positive rate and the sensitivity of predictions. The algorithm assumes that if a read is truly related to a pre-miRNA, then it must be a portion either of the loop sequence or of one of the potential two mature sequences in the hairpin. Thus, given the higher abundance of the dominant mature sequence in the cell compared to any other sequence of a pre-miRNA, the higher number of reads in the data will likely correspond to mature sequences, while less frequent reads may map to other parts of the hairpins. Algorithms for mapping and evaluation of free energy, previously under user control, are carried out by Bowtie and Randfold in miRDeep2 (Bonnet et al., 2004; Langmead et al., 2009; Friedländer et al., 2012) in which species conservation has been a key addition as well (Mackowiak, 2011).
Modeled off miRDeep, mirDeep* (An et al., 2013) employs a miRNA precursor prediction strategy which the authors have proved to outperform both versions of miRDeep as it adopts a different strategy to excise the potential precursor locus range, resulting in a lower number of false negatives. Users can also apply the original miRDeep prediction algorithm, as well as the TargetScan (Lewis et al., 2005) algorithm in order to predict targets for identified known and novel miRNAs.
Great innovation in terms of portability and the elaboration of miRNA processing information is provided by the miRspring software (Humphreys and Suter, 2013). The tool generates a small portable interactive miRNA Sequence Profiling document capable of completely reproducing all the information from a significantly larger mapped sequencing data file in bam format (i.e., from a miRNA-Seq experiment), along with providing miRNA processing statistics. In fact, it is the first software that allows to visualize the processing features, seed distribution and relative expression levels of genomic clustered miRNAs from a whole miRNA data set.
Aside miRNA-specific approaches, other software focuses on small RNAs in general.
The first integrated tool ever developed for the analysis and prediction of several classes of small ncRNAs on RNA-seq data originating from arbitrary sequencing platforms is the web service DARIO (Fasold et al., 2011). The software provides a straighforward interface which allows users to quantify ncRNAs in a completely platform independent way. DARIO annotates reads with information provided by several ncRNA public databases, and excludes mapping loci overlapping with exonic regions, while setting apart those that overlap with introns and intergenic regions for non-annotated ncRNA prediction. An extension of this system to plants has recently been published (Patra et al., 2014).
The web server CPSS (Zhang et al., 2012) takes things a step further. The tool can analyze small RNA deep sequencing data coming from single or two paired samples, with special emphasis on miRNAs. Data are classified into several categories of small ncRNAs according to several referred annotations. Matched mapped reads are then quantified for expression analysis (differential in case of two samples), while unmatched ones are employed to predict novel miRNAs also through miRDeep (Friedländer et al., 2008). CPSS also provides users with the possibility to predict target genes for differentially expressed novel/known miRNAs but, like no precedent approach, it also performs functional enrichment analysis of those targets for further experimental or computational studies.
Differently, ncPRO-seq (Chen et al., 2012), a stand-alone, comprehensive and flexible ncRNA analysis pipeline, systematically investigates all small ncRNA species in a given annotation family in an unbiased way, providing the user with detailed descriptions of read distribution. Furthermore, the tool defines novel small ncRNA families by identifying regions significantly enriched with short reads not classified under any known ncRNA species, allowing the discovery of previously unknown ncRNA- or siRNA-coding regions.
To address the limitations of RNA function prediction methods in classifying ncRNA classes, the machine learning package CoRAL (Leung et al., 2013) classifies RNA transcripts from sRNA-seq data into functional categories by relying on biologically interpretable features more informative than sequence or alignment information, like certain aspects of small RNA biogenesis. Leveraging on the assumption that such biological properties should be consistent within classes of ncRNAs sharing the same molecular function (i.e., across different tissues or organisms), CoRAL was trained in order to identify the most informative features in regard to the molecular mechanisms and metabolic processes of each functional ncRNA class. Based on fragment length, cleavage specificity, and antisense transcription, CoRAL can effectively classify six distinct ncRNA classes among miRNAs and transposon-derived RNAs. Outperforming previous tools such as DARIO and miRDeep2, CoRAL provides the opportunity to annotate ncRNAs in other less well-characterized organisms.
Another tool for ncRNA annotation in NGS data lacking reference genomes is the software RNA-CODE (Yuan and Sun, 2013). As ncRNA homology search takes advantage of both sequence and secondary structure similarity, optimization for NGS data is still widely absent, especially when a reference genome is missing. To compensate for this, RNA-CODE combines secondary structure based homology search with de novo assembly, adjusting the assembly parameters in a family specific fashion. The sofware assumes that true ncRNA reads sequenced from the same gene can be assembled into contigs with significantly high alignment scores against their native families, while reads aligned by chance tend to share poor overlaps and thus are not likely to be assembled. Sensitivity and accuracy of short reads classification is thus greatly improved. Biogenesis-based properties and homology search results are instead employed for ncRNAs, such as miRNAs, which could not as easily be assembled into contigs. The classification results can then be used to quantify the expression levels of different types of ncRNAs, both small and long, in RNA-seq data of non-model organisms.
The works done by the Brown (Salzman et al., 2012) and Sharpless (Jeck et al., 2013) groups are forerunners of a series of algorithmic approaches to effectively identify circRNA, attempting to compensate the non-uniformity of RNA-seq data sets.
Most algorithms have focused on junction read detection whether leveraging on annotated exon boundaries (Salzman et al., 2012), adopting a two-segment alignment for split reads (Memczak et al., 2013) or relying on RNAase-treated sequencing (Jeck et al., 2013). Nevertheless, all these methods are annotation-dependent and unable to detect certain types of circRNAs having complex alignments and/or subject to experimental bias.
A very recent computational tool proven to outperform any precedent approach in the detection of circRNAs from NGS is CIRI (Gao et al., 2015). CIRI is an unbiased, annotation-independent approach employing a de novo algorithm able to accurately detect novel circRNAs based on paired chiastic clipping (PCC) signals combined with a filtering system able to remove false positives. CIRI has been able to specifically identify for the first time the prevalence of intronic/intergenic circRNAs as well as fragments specific to them in the human transcriptome, providing novel targets for further functional studies.
Long ncRNA investigation is a challenging task, as many more NGS reads are required to achieve adequate coverage compared to mRNAs or other types of ncRNAs. Here below, we describe a few recent computational tools which very well represent the general approach employed by several studies so far (Guttman et al., 2010; Cabili et al., 2011; Pauli et al., 2012).
The pipeline employed by Sun et al. (Sun et al., 2012) makes use of a software they have specifically developed to detect novel lncRNA, called lncRScan. The pipeline aims at tackling three of the major technical problems encountered in studying lncRNAs through RNA-seq: eliminating partial transcripts and artifacts in the assembled transcriptome due to RNA-seq-specific issues; identifying lncRNA from the complexity of assemblies; distinguishing lncRNAs from protein-coding mRNAs. After mapping and assembly, the data obtained are compared to a set of combined gene annotations in order to maximize detection and facilitate category labeling of novel transcripts, retaining only multi-exon ones not possessing any annotation for downstream processing. After quality control, the remaining assemblies are given as input to lncRScan for novel lncRNA detection. The tool identifies the candidate lncRNAs through a five-step filtering process: first it organizes input transcripts into five broad categories according to their genomic location in relation to annotated gene transcripts; transcripts longer than 200 nt are selected, filtering out those with open reading frame (ORF) >300 nt; in the last two steps, phylogenetic analysis and potential aminoacidic sequences of the remaining transcripts are performed in order to exclude any protein-coding potential. Performance evaluation of the pipeline has shown its great ability to filter out mRNAs from the candidate set, while revealing a stringent prediction of true lncRNAs from a test set.
iSeeRNA (Sun et al., 2013) is an SVM-based classifier which can accurately and quickly identify lincRNAs from large datasets, employing conservation, ORF- and nucleotide sequences-based features in order to appropriately distinguish lincRNAs from protein-coding transcripts (PCTs). The best classification results on test sets were produced leveraging on 10 features from the three categories mentioned above: sequence conservation score, being lincRNA less conserved than PCTs in general; ORF length and ORF proportion compared to transcript total length; frequencies of seven di- or tri-nucleotide sequences. Homolog search-based features were instead not included due to lack of annotation for novel PCTs which could foster misclassification. Trained in a species-dependent manner, iSeeRNA allows the user, however, to build additional customized SVMs for other species of interest. Supporting file formats widely used by the RNA-Seq assemblers, iSeeRNA can be easily integrated into transcriptome data analysis pipelines.
More recently, Soreq et al. (Soreq et al., 2014) have integrated full profile characterization of lncRNAs into their comprehensive RNA-seq analysis workflow. The pipeline, based on sample specific database construction, is able to analyze count information from RNA-seq data originating from several platforms and mapping analysis methods. After sequence reads have been mapped, their genome coordinates are intersected with those of the largest available database of reconstructed transcript models for lncRNAs, GENECODE (Derrien et al., 2012). Following appropriate filtering, differential expression of the detected lncRNA candidates is performed using the Bioconductor edge-R package (Robinson et al., 2010) which accounts for biological and technical variability as well as moderating the degree of over-dispersion across transcripts, thus improving the reliability of the results.
Musacchia et al. (2015) provide instead a pipeline combining the identification of both coding and long non-coding RNAs in de novo generated transcriptomes, without the support of comparative data. Annocript identifies putative lncRNAs by leveraging on public annotation databases and sequence analysis software to verify lack of protein/domain similarity, lack of long ORFs, and high non-coding potential.
Finally, an innovative approach in the functional annotation of lncRNAs is provided by Jiang et al. (2015). LncRNA2Function provides the first ontology-driven user-friendly web system based on the idea that similar expression patterns across multiple conditions may share similar functions and biological pathways. The tool functionally annotates a single or a set of lncRNAs with the functional terms significantly associated to the set of protein-coding genes significantly co-expressed with the lncRNAs. Standard mapping and assembly are thus followed by the computation of Pearson Correlation Coefficients for all lncRNA–mRNA gene pairs, assigning to each lncRNA a set of significantly co-expressed protein-coding genes which provides the lncRNA with functional and pathway annotations significantly enriched in such set. The tool thus allows to browse the results obtained from an RNA-seq dataset of 19 human normal tissues in order to retrieve the set of lncRNAs associated to a specific functional term, the set of functional terms associated to a lncRNA or assign functional terms to a set of lncRNAs, thus providing a precious resource for lncRNA function investigation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
GN was supported by Italian Foundation for Cancer Research (FIRC – 15046). DV was supported by Italian Foundation for Cancer Research (FIRC – 16572). The authors would like to thank the reviewers for their useful suggestions and Fabio Ferri for his help in editing the manuscript.
An, J., Lai, J., Lehman, M. L., and Nelson, C. C. (2013). miRDeep*: an integrated application tool for miRNA identification from RNA sequencing data. Nucleic Acids Res. 41, 727–737. doi: 10.1093/nar/gks1187
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi:10.1186/gb-2010-11-10-r106
Birney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigo, R., Gingeras, T. R., Margulies, E. H., et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. doi:10.1038/nature05874
Bonnet, E., Wuyts, J., Rouzé, P., and Van de Peer, Y. (2004). Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 20, 2911–2917. doi:10.1093/bioinformatics/bth374
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., et al. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927. doi:10.1101/gad.17446611
Chen, C.-J., Servant, N., Toedling, J., Sarazin, A., Marchais, A., Duvernois-Berthet, E., et al. (2012). ncPRO-seq: a tool for annotation and profiling of ncRNAs in sRNA-seq data. Bioinformatics 28, 3147–3149. doi:10.1093/bioinformatics/bts587
Cordero, F., Beccuti, M., Arigoni, M., Donatelli, S., and Calogero, R. A. (2012). Optimizing a massive parallel sequencing workflow for quantitative miRNA expression analysis. PLoS ONE 7:e31630. doi:10.1371/journal.pone.0031630
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi:10.1101/gr.132159.111
Eddy, S. R. (2001). Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2, 919–929. doi:10.1038/35103511
Elgar, G., and Vavouri, T. (2008). Tuning in to the signals: noncoding sequence conservation in vertebrate genomes. Trends Genet. 24, 344–352. doi:10.1016/j.tig.2008.04.005
Fasold, M., Langenberger, D., Binder, H., Stadler, P. F., and Hoffmann, S. (2011). DARIO: a ncRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res. 39, W112–W117. doi:10.1093/nar/gkr357
Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., and Mello, C. C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. doi:10.1038/35888
Friedländer, M. R., Chen, W., Adamidi, C., Maaskola, J., Einspanier, R., Knespel, S., et al. (2008). Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 26, 407–415. doi:10.1038/nbt1394
Friedländer, M. R., Mackowiak, S. D., Li, N., Chen, W., and Rajewsky, N. (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. doi:10.1093/nar/gkr688
Gao, Y., Wang, J., and Zhao, F. (2015). CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 16, 4. doi:10.1186/s13059-014-0571-3
Guttman, M., Garber, M., Levin, J. Z., Donaghey, J., Robinson, J., Adiconis, X., et al. (2010). Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 28, 503–510. doi:10.1038/nbt.1633
Hardcastle, T. J., and Kelly, K. A. (2010). baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 11:422. doi:10.1186/1471-2105-11-422
Humphreys, D. T., and Suter, C. M. (2013). miRspring: a compact standalone research tool for analyzing miRNA-seq data. Nucleic Acids Res. 41, e147–e147. doi:10.1093/nar/gkt485
Isakov, O., Ronen, R., Kovarsky, J., Gabay, A., Gan, I., Modai, S., et al. (2012). Novel insight into the non-coding repertoire through deep sequencing analysis. Nucleic Acids Res. 40, e86. doi:10.1093/nar/gks228
Jeck, W. R., Sorrentino, J. A., Wang, K., Slevin, M. K., Burd, C. E., Liu, J., et al. (2013). Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19, 141–157. doi:10.1261/rna.035667.112
Jiang, Q., Ma, R., Wang, J., Wu, X., Jin, S., Peng, J., et al. (2015). LncRNA2Function: a comprehensive resource for functional investigation of human lncRNAs based on RNA-seq data. BMC Genomics 16(Suppl. 3):S2. doi:10.1186/1471-2164-16-S3-S2
Jung, C.-H., Hansen, M. A., Makunin, I. V., Korbie, D. J., and Mattick, J. S. (2010). Identification of novel non-coding RNAs using profiles of short sequence reads from next generation sequencing data. BMC Genomics 11:77. doi:10.1186/1471-2164-11-77
Landgraf, P., Rusu, M., Sheridan, R., Sewer, A., Iovino, N., Aravin, A., et al. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414. doi:10.1016/j.cell.2007.04.040
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi:10.1186/gb-2009-10-3-r25
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi:10.1016/0092-8674(93)90529-Y
Leung, Y. Y., Ryvkin, P., Ungar, L. H., Gregory, B. D., and Wang, L.-S. (2013). CoRAL: predicting non-coding RNAs from small RNA-sequencing data. Nucleic Acids Res. 41, e137–e137. doi:10.1093/nar/gkt426
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi:10.1016/j.cell.2004.12.035
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi:10.1186/s13059-014-0550-8
Lu, Z. J., Yip, K. Y., Wang, G., Shou, C., Hillier, L. W., Khurana, E., et al. (2011). Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high-throughput sequencing and array data. Genome Res. 21, 276–285. doi:10.1101/gr.110189.110
Mackowiak, S. D. (2011). Identification of novel and known miRNAs in deep-sequencing data with miRDeep2. Curr. Protoc. Bioinformatics Chapter 12, Unit12.10. doi:10.1002/0471250953.bi1210s36
Malone, J. H., and Oliver, B. (2011). Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 9:34. doi:10.1186/1741-7007-9-34
Mattick, J. S. (2001). Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep. 2, 986–991. doi:10.1093/embo-reports/kve230
Meldrum, C., Doyle, M. A., and Tothill, R. W. (2011). Next-generation sequencing for cancer diagnostics: a practical perspective. Clin. Biochem. Rev. 32, 177–195.
Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. doi:10.1038/nature11928
Musacchia, F., Basu, S., Petrosino, G., Salvemini, M., and Sanges, R. (2015). Annocript: a flexible pipeline for the annotation of transcriptomes able to identify putative long noncoding RNAs. Bioinformatics. doi:10.1093/bioinformatics/btv106
Patra, D., Fasold, M., Langenberger, D., Steger, G., Grosse, I., and Stadler, P. F. (2014). plantDARIO: web based quantitative and qualitative analysis of small RNA-seq data in plants. Front Plant Sci 5:708. doi:10.3389/fpls.2014.00708
Pauli, A., Valen, E., Lin, M. F., Garber, M., Vastenhouw, N. L., Levin, J. Z., et al. (2012). Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 22, 577–591. doi:10.1101/gr.133009.111
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641. doi:10.1016/j.cell.2009.02.006
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi:10.1093/bioinformatics/btp616
Ronen, R., Gan, I., Modai, S., Sukacheov, A., Dror, G., Halperin, E., et al. (2010). miRNAkey: a software for microRNA deep sequencing analysis. Bioinformatics 26, 2615–2616. doi:10.1093/bioinformatics/btq493
Salzman, J., Gawad, C., Wang, P. L., Lacayo, N., and Brown, P. O. (2012). Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 7:e30733. doi:10.1371/journal.pone.0030733
Soreq, L., Guffanti, A., Salomonis, N., Simchovitz, A., Israel, Z., Bergman, H., et al. (2014). Long non-coding RNA and alternative splicing modulations in Parkinson’s leukocytes identified by RNA sequencing. PLoS Comput. Biol. 10:e1003517. doi:10.1371/journal.pcbi.1003517
Sun, K., Chen, X., Jiang, P., Song, X., Wang, H., and Sun, H. (2013). iSeeRNA: identification of long intergenic non-coding RNA transcripts from transcriptome sequencing data. BMC Genomics 14(Suppl. 2):S7. doi:10.1186/1471-2164-14-S2-S7
Sun, L., Zhang, Z., Bailey, T. L., and Perkins, A. C. (2012). Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. Bioinformatics 13, 331. doi:10.1186/1471-2105-13-331
‘t Hoen, P. A. C., Ariyurek, Y., Thygesen, H. H., Vreugdenhil, E., Vossen, R. H. A. M., de Menezes, R. X., et al. (2008). Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res 36, e141.
Todd, G., and Karbstein, K. (2007). RNA takes center stage. Biopolymers 87, 275–278. doi:10.1002/bip.20824
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi:10.1038/nrg2484
Yuan, C., and Sun, Y. (2013). RNA-CODE: a noncoding RNA classification tool for short reads in NGS data lacking reference genomes. PLoS ONE 8:e77596. doi:10.1371/journal.pone.0077596
Zhang, Y., Xu, B., Yang, Y., Ban, R., Zhang, H., Jiang, X., et al. (2012). CPSS: a computational platform for the analysis of small RNA deep sequencing data. Bioinformatics 28, 1925–1927. doi:10.1093/bioinformatics/bts282
Keywords: RNA-seq, miRNA, lncRNA, circRNA, bioinformatics
Citation: Veneziano D, Nigita G and Ferro A (2015) Computational approaches for the analysis of ncRNA through deep sequencing techniques. Front. Bioeng. Biotechnol. 3:77. doi: 10.3389/fbioe.2015.00077
Received: 31 October 2014; Accepted: 14 May 2015;
Published: 03 June 2015
Edited by:
Fengfeng Zhou, Shenzhen Institutes of Advanced Technology, ChinaReviewed by:
Yi Zhao, Chinese Academy of Sciences, ChinaCopyright: © 2015 Veneziano, Nigita and Ferro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dario Veneziano, Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University, 460 W 12th Avenue, Columbus, OH 43210, USA,ZGFyaW8udmVuZXppYW5vQG9zdW1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.