
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioinform. , 20 June 2023
Sec. Protein Bioinformatics
Volume 3 - 2023 | https://doi.org/10.3389/fbinf.2023.1199675
 Nour Nissan1,2
Nour Nissan1,2 Julia Hooker1,2Eric Arezza3Kevin Dick3Ashkan Golshani2
Julia Hooker1,2Eric Arezza3Kevin Dick3Ashkan Golshani2 Benjamin Mimee4
Benjamin Mimee4 Elroy Cober1
Elroy Cober1 James Green3
James Green3 Bahram Samanfar1,2*
Bahram Samanfar1,2*The soybean cyst nematode (SCN) [Heterodera glycines Ichinohe] is a devastating pathogen of soybean [Glycine max (L.) Merr.] that is rapidly becoming a global economic issue. Two loci conferring SCN resistance have been identified in soybean, Rhg1 and Rhg4; however, they offer declining protection. Therefore, it is imperative that we identify additional mechanisms for SCN resistance. In this paper, we develop a bioinformatics pipeline to identify protein–protein interactions related to SCN resistance by data mining massive-scale datasets. The pipeline combines two leading sequence-based protein–protein interaction predictors, the Protein–protein Interaction Prediction Engine (PIPE), PIPE4, and Scoring PRotein INTeractions (SPRINT) to predict high-confidence interactomes. First, we predicted the top soy interacting protein partners of the Rhg1 and Rhg4 proteins. Both PIPE4 and SPRINT overlap in their predictions with 58 soybean interacting partners, 19 of which had GO terms related to defense. Beginning with the top predicted interactors of Rhg1 and Rhg4, we implement a “guilt by association” in silico proteome-wide approach to identify novel soybean genes that may be involved in SCN resistance. This pipeline identified 1,082 candidate genes whose local interactomes overlap significantly with the Rhg1 and Rhg4 interactomes. Using GO enrichment tools, we highlighted many important genes including five genes with GO terms related to response to the nematode (GO:0009624), namely, Glyma.18G029000, Glyma.11G228300, Glyma.08G120500, Glyma.17G152300, and Glyma.08G265700. This study is the first of its kind to predict interacting partners of known resistance proteins Rhg1 and Rhg4, forming an analysis pipeline that enables researchers to focus their search on high-confidence targets to identify novel SCN resistance genes in soybean.
Cultivated soybean (Glycine max (L.) Merr.) is a valuable crop worldwide, regularly used in food, feed, and fuel. Soybean is also an important partner in sustainable agricultural management practices due to its symbiotic relationship with nitrogen-fixing bacteria (Boerema et al., 2016). The reference genome, Williams 82, is ∼1.1 GB with ∼89,500 protein-coding transcripts and 55,589 genes encompassed in 20 chromosomes (Schmutz et al., 2010). The soybean genome is difficult to study due to chromosomal rearrangement, rounds of diploidization, and two major duplication events that occurred 59 and 13 million years ago, making 75% of its genes available in paralogs (Schmutz et al., 2010). Soybean must deal with numerous abiotic stressors, among which are various mineral deficiencies, drought, daylength, and cold weather conditions (Jumrani and Bhatia, 2018). In addition to abiotic stressors which are difficult to control, soybean faces several biotic stressors, including pathogenic stressors such as Fusarium virguliforme (causing sudden death syndrome), Aphis glycines (soybean aphid), Sclerotinia sclerotiorum (causing sclerotinia stem rot), and Heterodera glycines Ichinohe, also known as soybean cyst nematode (SCN) (Bradley et al., 2021).
SCN is one of the most destructive pathogens of soybean, first detected in North America in 1954 in North Carolina (Winstead et al., 1955). SCN attacks soybean roots, thereby creating feeding sites within them called syncytia, and robs nutrients from the plant for its own growth and development (Gheysen and Mitchum, 2011). At the J2 (juvenile) stage, the nematode will live and feed in the syncytia for about 3–4 weeks, until they reach the adult stage. Male adults will leave the roots, while females will continue to feed and grow. At one point, the adult females will push through the roots, releasing pheromones to attract adult males for mating. The still females will then deposit eggs near the root while also keeping some within their body, before hardening into cysts and dying. The cysts, containing viable eggs, are able to remain in the soil for up to 10 years until conditions are favorable for them to emerge and infect more soybean roots (Davis and Tylka, 2000).
At present, there exist two commercially used loci for SCN resistance in soybean, the recessive form of Rhg1 and the dominant form of Rhg4 (Concibido et al., 1997; Glover et al., 2004; Kim et al., 2010; Liu et al., 2012). The Rhg1 locus consists of a 31 kb multi-gene segment coding for an α-SNAP protein (GmSNAP18), a wound-inducible domain protein (WI12; GmWII2), and an amino acid transporter (AAT; GmAAT). All three were shown to be involved in resistance and were mapped to chromosome 18 (Cook et al., 2012; Liu et al., 2017). The Rhg1 locus has two resistance alleles, rhg1-a “Peking-type” and rhg1-b “PI88788-type.” The rhg1-a allele contains a retrotransposon in the GmSNAP18 protein and has a lower copy number for all three proteins (about three or fewer copies), while the rhg1-b allele does not contain a retrotransposon in GmSNAP18 and has a higher copy number for the three proteins (∼4–10 copies). The rhg1-a allele requires Rhg4 for complete resistance, while the rhg1-b allele does not. The Rhg4 gene codes for a cytosolic serine hydroxymethyltransferase (SHMT) protein which confers resistance against SCN (Liu et al., 2012).
Current management strategies against SCN remain challenging as soybean varieties containing resistance alleles at Rhg1 or Rhg4 loci are collapsing as more virulent SCN populations are emerging. Since the human population is expected to reach an all-time high in 2050 and continue growing, the threat that SCN poses to soybean yield is significant, fueling a rise in breeding programs which deal with SCN (Yan and Baidoo, 2018; Shaibu et al., 2020; Nissan et al., 2022). There have been significant advancements in SCN research in the hopes of identifying novel genes involved in resistance, such as fine-mapping studies, methylation, large-scale genomics, transgenics, transcriptomics, and proteomics (Shaibu et al., 2020). There is a lack of knowledge when it comes to Rhg-interacting proteins that trigger the hypersensitive response in soybean, which has been problematic in terms of identifying ways to control SCN.
Protein–protein interactions (PPIs) are critical to cellular functions in living organisms. They participate in many different processes including DNA replication, catalysis of metabolic reactions, DNA transcription, suppression or activation of a protein, and transportation of molecules (Peng et al., 2017). Studying PPIs allows for molecular machinery in cells to be identified (De Las Rivas and Fontanillo, 2012). This is possible because proteins often form complex structures to perform specific functions in an interaction network called the “interactome” instead of functioning as individual units (Cusick et al., 2005). Studying PPI networks has aided in identifying gene function (Zhao et al., 2016; Samanfar et al., 2017; Gligorijević et al., 2018), diseases/allergens (Xu and Li, 2006; Dick et al., 2021a), and pharmaceutical discoveries (Yıldırım et al., 2007; Schoenrock et al., 2015). Primarily small-scale studies have identified PPIs through yeast-two-hybrid (Y2H) experiments, tandem affinity purification and mass spectrometry (TAP-MS), and co-immunoprecipitation (Co-IP) techniques (Bensimon et al., 2012; Stynen et al., 2012). For example, a large-scale comprehensive PPI has been performed for Saccharomyces cerevisiae in a genome consisting of approximately 6,000 genes, using Y2H studies, proteome chips, and a combination of computational and experimental strategies (Uetz et al., 2000; Zhu et al., 2001; Tong et al., 2002). However, limitations begin to arise with the use of wet-laboratory experiments with larger genomes such as soybean, which is composed of 55,589 genes (Torkamaneh et al., 2021). Some of those limitations include labor costs, scale of study, time constraints, and false positive and negative rates (Zhang et al., 2019). Hence, the use of computational predictors of PPIs has become valuable in molecular biology research. These computational approaches supplement and focus the use of wet-laboratory experiments on targeted, high-confidence predictions. In the last decade, there has been an increase in demand for computational tools that can predict a comprehensive interactome, which is the set of all possible pairwise PPIs within or between proteomes. This has become possible due to the emergence of high-performing computer infrastructure and algorithmic optimizations (Dick et al., 2020). The sequence-based PPI prediction methods used in this study exploit information from previously confirmed PPI sets to determine whether two query proteins will physically interact (Li and Ilie, 2017; Dick et al., 2020).
In this study, we use two complementary PPI prediction methods, the latest version of the Protein–protein Interaction Prediction Engine (PIPE), PIPE4, and the Scoring PRotein INTeractions (SPRINT) predictor, to investigate the soybean proteome (Pitre et al., 2006; Li and Ilie, 2017; Dick et al., 2020). These PPI predictors are applied to predict the PPIs of the entire soybean proteome, which enables studying unannotated proteins through a “guilt by association” approach. Such an approach works on the premise that if an unknown protein is found to be interacting with many proteins exhibiting a given function, there is a heightened chance that the unknown protein also shares that function (see Figure 1) (Rao et al., 2014).
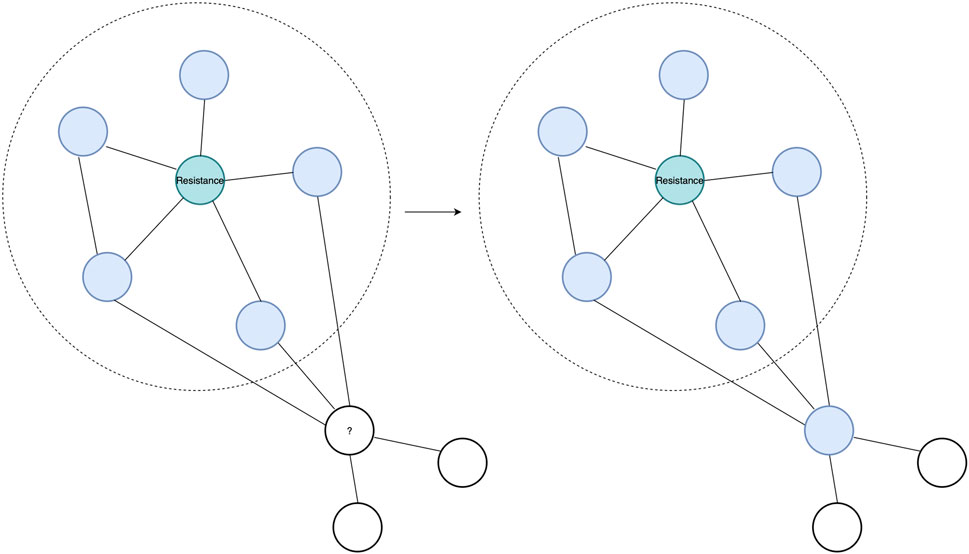
FIGURE 1. Guilt-by-association approach showing a known SCN resistance protein in teal and its top interacting partners highlighted in blue within the dotted circle. An unknown protein, with a question mark, is observed to interact with the same interacting partner as the resistance protein. Hence, through the “guilt by association” approach, the protein with the question mark can be predicted to interact in the resistance protein pathway due to sharing interacting partners with the known resistance protein.
We developed a computational pipeline to identify novel soybean genes possibly involved in the resistance against SCN. Through this analysis, we highlight our most interesting genes by predicting the complete interactome of soybean; we first reveal the direct interactors of Rhg1 and Rhg4 (i.e., the SCN resistance pathway) and, second, discover those unannotated proteins whose interactome overlaps significantly with the pathway.
To identify putative novel genes involved in the resistance pathway to SCN, a data mining pipeline was developed using the longest protein transcript for each of the 55,589 predicted genes. The genes were processed through a sequential cascade of computational analyses. First, PIPE4 and SPRINT were used to predict the entire soybean interactome using the Williams 82 reference genome; decision thresholds are applied to each protein pair within the soybean proteome to predict interactions in the SCN resistance pathway. The resulting candidate list was then refined using GO enrichment of the top candidates using SoyBase’s GO Term Enrichment Tool (https://soybase.org/goslimgraphic_v2/dashboard.php), followed by GO REVIGO (http://revigo.irb.hr) for visualization and further analysis (see Figure 2).
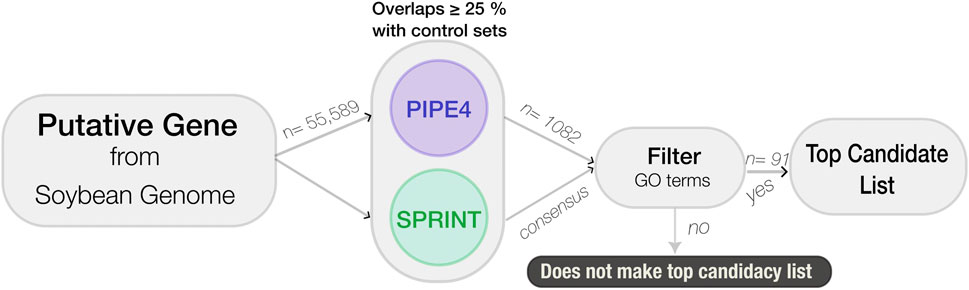
FIGURE 2. Flowchart of the computational large-scale pipeline used to identity novel soybean genes involved in resistance against SCN.
Both PIPE4 and SPRINT were used to predict soybean PPIs for all soybean proteins. Due to the lack of experimental soy–soy PPI data, Arabidopsis thaliana PPI data were used as a cross-species proxy for training PIPE4 and SPRINT predictors (Dick et al., 2020). This training set consisted of 3,027 A. thaliana–A. thaliana confirmed protein interactions between 2,096 proteins. Performing all-to-all soy–soy predictions on 88,647 soy proteins resulted in 3,929,189,628 protein pairs. However, considering only the longest protein sequence isoforms reduces the number of relevant proteins to 56,044 (including Glyma.U proteins), which results in 1,570,492,990 possible interactions. This was performed because both PIPE4 and SPRINT examine the protein sequence, and utilizing the longest sequence will allow the PPI predictors to consider more “windows” for interaction, while removing redundant sequences from the analysis.
Considering the predictions from both PIPE4 and SPRINT, we determined an overlapping set of highly probable candidate PPIs for subsequent analysis. The top 0.07% of soybean-interacting partners (40 genes) were extracted for each of the 55,589 soy proteins (Glyma.U proteins were used to train PPI predictors but were excluded from our search for novel resistant genes). This highly conservative threshold was determined by plotting the rank-order distribution of the top 1,000 predicted interactions and identifying a “knee” in the L-shaped curve where the predicted score plateaus (Figure 3) as motivated by the following work leveraging a similar methodology (Dick and Green, 2018; Dick et al., 2021a). As shown in Figure 3A, there is a notable step-wise decrease in the PIPE4 prediction scores at rank 60, whereas in Figure 3B, there is a notable step-wise decrease in the SPRINT prediction score at rank 40. For consistency within this work, we selected the highly conservative top 40 cutoff values for both methods. Positive control set A (AP) for PIPE4 and (AS) for SPRINT comprise the top 40 ranked interacting partners of Rhg1 (Glyma.18g022400, Glyma.18g022500, and Glyma.18g022600) and Rhg4 (Glyma.08g108900) proteins, resulting in 160 PIPE- and 160 SPRINT-identified soybean proteins overlapping with these positive control sets (Supplementary Table S1 and Figure 4). This work follows the “guilt by association” method and was implemented using Python. Any proteins whose top 40 interactors overlapped by at least 25% with the top 40 interactors of the positive control set AP (and AS when repeated using SPRINT) were kept for further analysis. The top 25% were chosen as both PIPE4 and SPRINT predicted the closest numbers of interacting proteins between both predictors at this percentage threshold (Supplementary Figure S1).
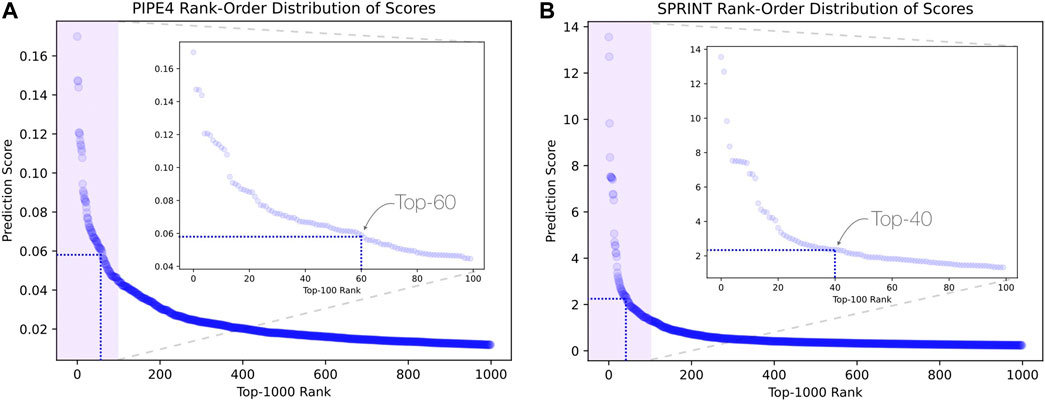
FIGURE 3. L-shaped curves identifying predicted score plateaus using the top 1,000 predicted interactions for (A) PIPE4 and (B) SPRINT.
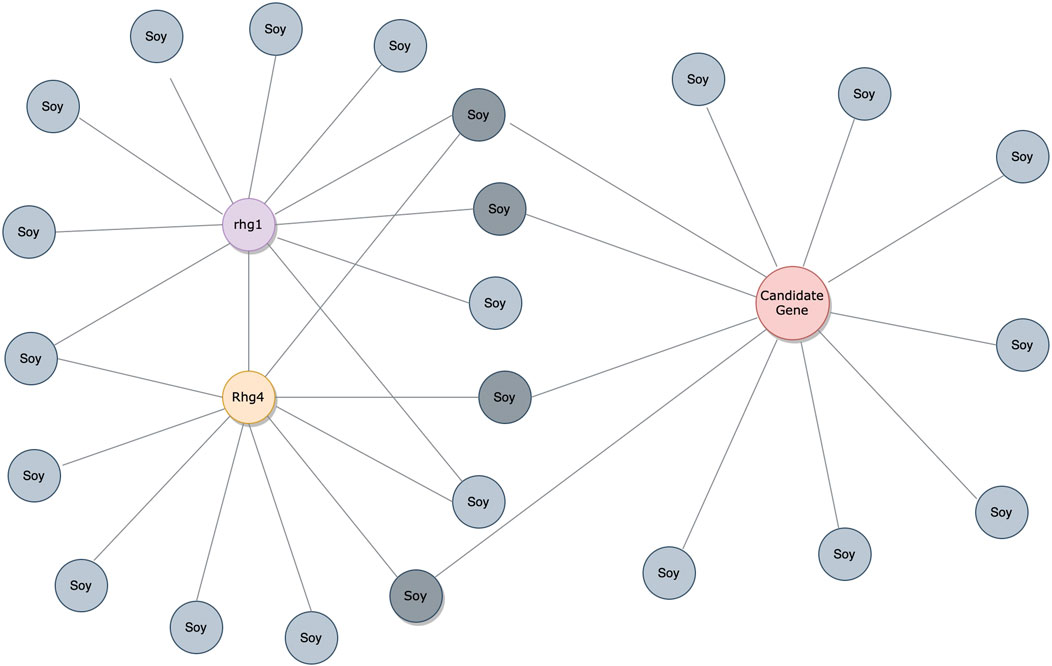
FIGURE 4. PPI analysis through the guilt-by-association approach. The interactions are denoted by gray soy-interacting protein partners. The red protein is a candidate gene with over 25% overlaps in soy-interacting proteins from positive control sets AP/AS.
Finally, to quantify the predictive performance of both PIPE4 and SPRINT within this cross-species prediction schema, we sought to evaluate the performance of the models on unseen experimentally validated PPI pairs. To this end, we extracted all known soy–soy PPI pairs from BioGRID (https://thebiogrid.org, accessed on 18 May 2023) (n = 17) and report the sensitivity of both methods based on the highly conservative top 40 threshold considered in this work (Supplementary Table S2).
Soybean genes in the AP and AS positive control sets, as well as the 1,082 candidate list (Supplementary Table S3), were independently run through the SoyBase GO Term Enrichment Tool (https://www.soybase.org/goslimgraphic_v2/dashboard.php, accessed on 17 February 2023) (Grant et al., 2010) to curate the GO term enrichment of each of the two lists (the 58 overlapping proteins in the AP/AS list and the 1,082 candidate list). The SoyBase GO:BP output was searched for terms related to “defense,” “response,” and “nematode” to encompass terms of interest identified in the QuickGO database (www.ebi.ac.uk/QuickGO/, accessed on 17 February 2023). Terms of interest in the biological process category included but were not limited to terms involved in defense/resistance such as response to nematode (GO:0009624), response to wounding (GO:0009611), defense response (GO:0006952), response to mechanical stimulus (GO:0009612), response to xenobiotic stimulus (GO:0009410), defense response to bacterium (GO:0042742), jasmonic acid and ethylene-dependent systemic resistance (GO:0009861), response to salicylic acid (GO:0009751), response to ethylene (GO:0009723), response to abscisic acid (GO:0009737), and response to jasmonic acid (GO:0009753). Only defense-related annotations remained in the final lists. The enriched GO:BP terms and p-values were then run through REVIGO (http://revigo.irb.hr, accessed on 17 February 2023), a tool used to reduce and visualize large lists of GO terms by scoring mother and daughter ontologies on frequency, relative group size, dispensability, and uniqueness (Supek et al., 2011). REVIGO filtering parameters were set to the medium threshold (0.7), and A. thaliana was used as a reference species. REVIGO scatterplots were created using the log10 size value of the biological process GO terms across all A. thaliana GO terms.
In this study, we employed the AlphaFold2 algorithm to generate precise 3D structural conformations for the proteins of interest, based on their respective amino acid sequences. AlphaFold2, an advanced deep learning model, utilizes a two-step process to predict protein structures (Jumper et al., 2021). First, it employs a neural network trained on a large database of known protein structures to generate protein-specific potentials. These potentials capture the complex relationships between amino acid sequences and their corresponding structures (Jumper et al., 2021). In the second step, the potentials are utilized to optimize the protein structure by minimizing a predefined energy function. The resulting structures are refined iteratively to achieve higher accuracy (Jumper et al., 2021). By leveraging AlphaFold2, we obtained highly accurate 3D structural conformations for the proteins of interest, facilitating a comprehensive understanding of their molecular functions and interactions. We make this predicted structural information available to the broader research community within the GitHub repository associated with this work (https://github.com/earezza/Soybean-Large-Scale-PPI-Analysis).
PIPE4 and SPRINT predicted the top 40 (or top 0.07%) interacting partners of Rhg1 and Rhg4 proteins (Glyma.18g022400, Glyma.18g022500, Glyma.18g022600, and Glyma.08g108900), resulting in the AP and AS positive control sets (Supplementary Table S1) resulting in 58 overlapping proteins.
For the 58 genes found to overlap between both sequence-predictors, we used Gene Ontology (GO) to better understand their roles in relation to SCN resistance or defense response, as well as help clarify the ontologies of the Rhg1 and Rhg4 interactome. SoyBase’s GO term enrichment analysis of the overlapping predicted interacting protein list of genes identified 86 biological process (BP) and molecular function (MF) terms strongly associated with these genes (Supplementary Table S4). The GO analysis was used to search for defense-related annotations. Only those candidate proteins that were strongly associated with defense-related GO terms were retained, resulting in 19 out of the 58 genes. The SoyBase GO terms did not include any hits for “nematode;” however, other defense-related terms were found. Subsequent analysis using REVIGO reduced the GO terms to 68 from 86 (see Figure 5). The terms “defense response” (GO:0006952) and “response to mechanical stimulus” (GO:0009612) were two terms listed with 0.346 and 0.309 dispensability values, respectively (Supplementary Table S5). From the SoyBase GO enrichment data, it was evident that five genes were responsible for both terms (Table 1). In addition to these valuable GO terms, there were other defense-related terms present within the control list including but not limited to “response to xenobiotic stimulus” (GO:0009410) with a dispensability score of 0. Another enriched GO term found in the overlapping proteins in the control list is “defense response to bacterium” (GO:0042742), which contains a dispensability score of 0.488.
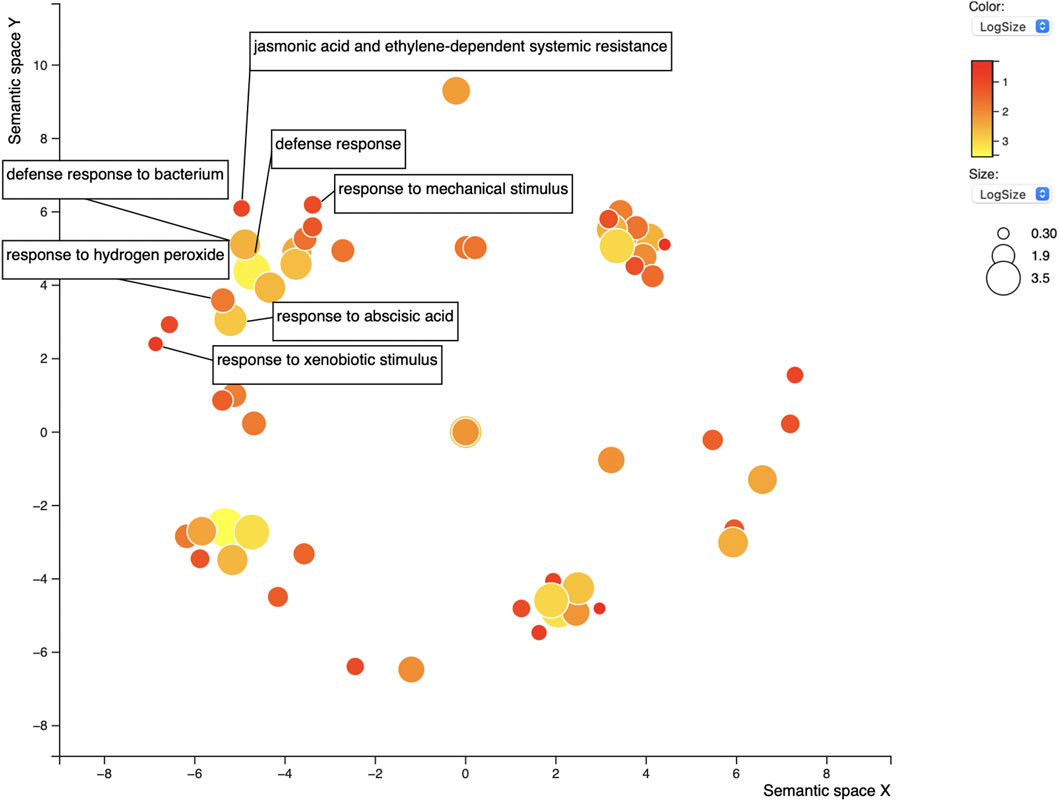
FIGURE 5. REVIGO scatterplot of the 86 GO terms for the 58 overlapping proteins found in positive control lists AP/AS. GO search was performed using medium 0.7 list size and using Arabidopsis thaliana as the species to work with. The size and color of the circles represent logSize value; higher logSize values indicate high numbers of a term and/or its daughter terms within the total database for A. thaliana; terms that are highly represented have larger bubbles.
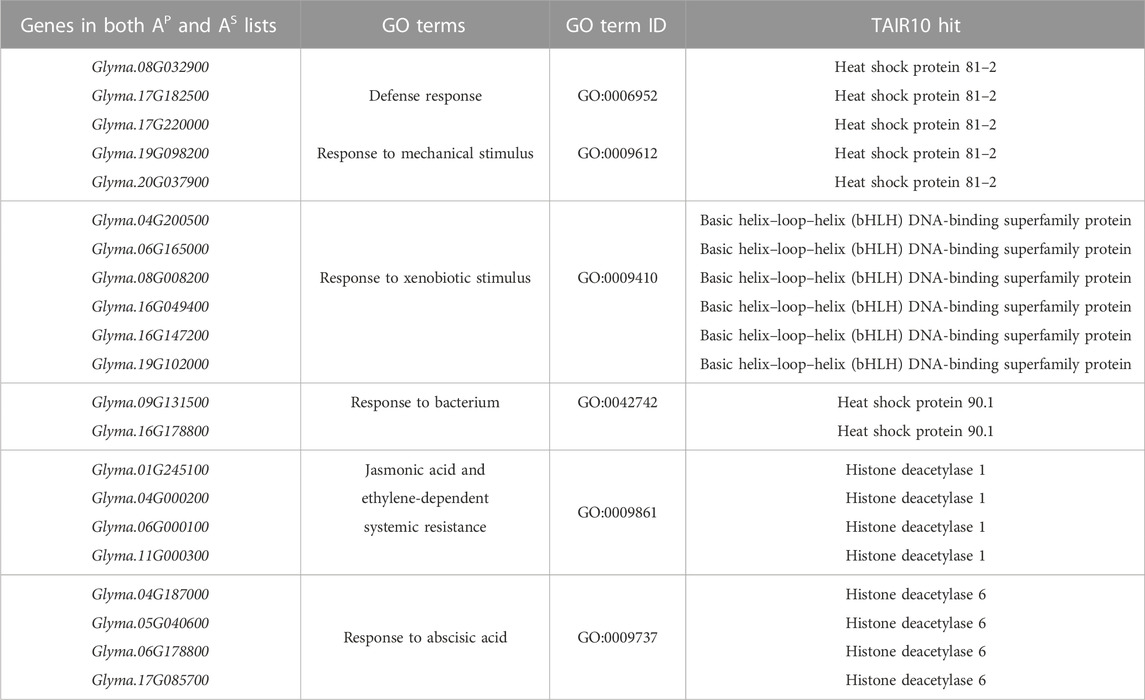
TABLE 1. Top 19 defense-related interacting partners of Rhg1 and Rhg4 proteins predicted by both PIPE4 and SPRINT engines and their corresponding defense-related GO terms.
In addition to the typical defense GO terms, other ontologies related to hormone response that also play a role in plant defense were present, such as “jasmonic acid and ethylene-dependent systemic resistance” (GO:0009861, dispensability: 0.117) and “response to abscisic acid” with four genes (GO:0009737, dispensability: 0.510) (Table 1).
PIPE4 predicted 5,763 genes with 25% or more overlaps in soybean-interacting partners with positive control set AP, while SPRINT predicted 6,153 genes with at least 25% overlaps in soybean-interacting partners with positive control set AS. Comparing PIPE4 and SPRINT results showed that 1,086 genes were common to both with the top four being Rhg1 and Rhg4 genes themselves (Supplementary Table S3).
To visualize and interpret the predicted Rhg1 and Rhg4 partners using PIPE4 and SPRINT, Figure 6 shows a network-based representation that highlights the overlap for both sets: green nodes are the Rhg1 and Rhg4 proteins, yellow nodes are predicted by PIPE4, blue nodes are predicted by SPRINT, and pink nodes represent the overlapping predictions. To better interpret those proteins and their relationships within this network, a fully interactive variant of this network is available at the following link: https://cu-bic.ca/soybean-rgh1-rgh4/.
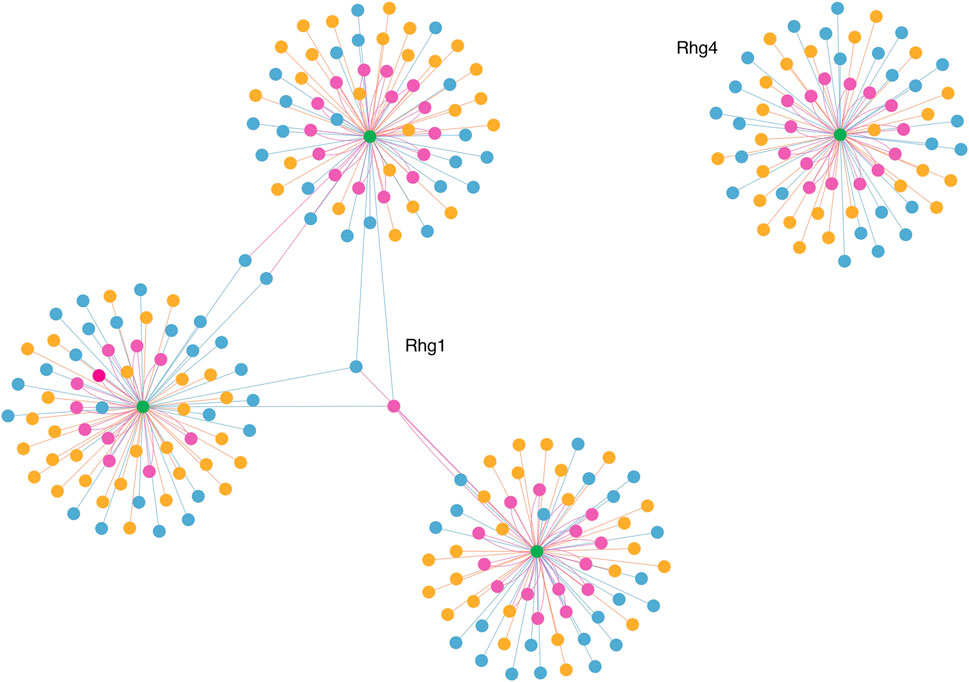
FIGURE 6. Network-based depiction of the Rhg1 and Rhg4 interaction partners by both PIPE4 and SPRINT, as well as their overlapping sets. Green nodes are the Rhg1 and Rhg4 proteins, yellow nodes are predicted by PIPE4, blue nodes are predicted by SPRINT, and pink nodes represent the overlapping predictions. Link for the interactive plot: https://cu-bic.ca/soybean-rgh1-rgh4/.
The set of 1,086 genes (1,082 after excluding the Rhg1 and Rhg4 genes themselves) was assessed for GO term enrichment to refine the search for genes related to SCN infection or defense response. The gene IDs were input into the SoyBase GO Term Enrichment Tool, and the GO enrichment data were extracted. The GO enrichment identified 1,183 unique GO terms from this gene list (Supplementary Table S6). The term “response to nematode” (GO:0009624, dispensability: 0.921) was overrepresented in the GO output; five genes were responsible for this enrichment. Also, among the SoyBase GO term enrichment output, “regulation of nematode larval development” (GO:0061062) was overrepresented among the data and also included one of five genes from the previous GO term. REVIGO filtering reduced the list of GO terms by 30%, resulting in 765 GO terms. The “response to nematode” term was retained after filtering; this is a daughter term of “defense response” (GO:0006952), which was the 15th most frequent GO term (Supplementary Table S7; Figure 7). Of the list of genes, 19 were responsible for the “defense response” term (dispensability: 0). Similarly, “regulation of defense response” (GO:0031347, dispensability: 0.932) was identified and made up a list of six genes, “response to xenobiotic stimulus” (GO:0009410, dispensability: 0.936) was also enriched in six genes, “response to wounding” (GO:0009611, dispensability: 0.922) in 23 genes, “response to mechanical stimulus” (GO:0009612, dispensability: 0.915) three genes, and “innate to immune response” (GO:0045087, dispensability: 0.899) is another important term related to resistance and defense-related genes with three genes being responsible. Finally, “detection of biotic stimulus” (GO:0009595, dispensability: 0.939) is the last GO term of interest overrepresented in this list with eight genes.
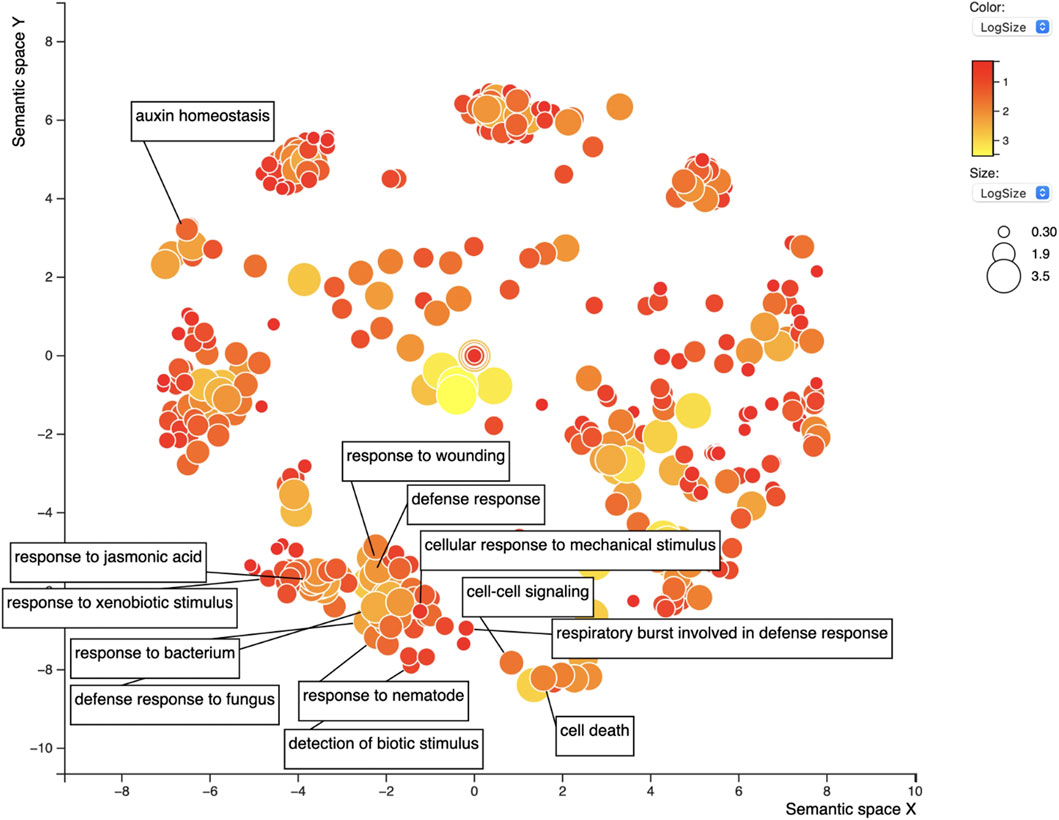
FIGURE 7. REVIGO scatterplot of the 1,183 GO terms for the top 1,082 candidate soybean genes for resistance against SCN. GO search was performed using medium 0.7 list size and using Arabidopsis thaliana as the species to work with. The size and color of the circles represent logSize value; higher logSize values indicate high numbers of a term and/or its daughter terms within the total database for A. thaliana; terms that are highly represented have larger bubbles.
In addition to defense-related GO terms, GO terms related to hormones responsible for defense were also identified within this list including “response to jasmonic acid” (GO:0009753, dispensability: 0.899) enriched in nine genes, “response to ethylene” (GO:0009723, dispensability: 0.904) in 11 genes, “response to salicylic acid” (GO:0009751, dispensability: 0.901) in seven genes, and finally, “response to abscisic acid” (GO:0009737, dispensability: 0.890) enriched in 28 genes. All genes are presented in Table 2.
TABLE 2. Top 91 candidate soybean genes for resistance against SCN identified from the genome-wide computational analysis from the 1,082 gene list and their corresponding defense-related GO terms.
In this research, we used various bioinformatics tools including two machine learning PPI predictors, PIPE4 and SPRINT, to scan the entire soybean genome in search of novel genes involved in resistance against SCN (Li and Ilie, 2017; Dick et al., 2020). We predicted the soybean interactome using Arabidopsis thaliana as a proxy training species, due to the lack of confirmed PPIs in the organism in question. The requirement to use a proxy species will somewhat reduce the accuracy of these methods (Dick et al., 2020). Therefore, we used a highly conservative score threshold, retaining only the top 0.07% of predicted interactions for each protein (i.e., top 40 predictions). Using predictions common to both PIPE4 and SPRINT also provided a conservative approach. The subsequent stages of the analysis pipeline (i.e., threshold filtration and REVIGO filtration) were designed to further filter the list of potential interactors, thereby increasing our confidence in the final list of candidate genes hypothesized to be associated with SCN resistance in soybean.
Early comparisons of the performance of SPRINT to the ancestral PIPE2 algorithm list SPRINT performance as having a sensitivity of 12.92% at a 99.95% specificity compared to PIPE2 having a sensitivity of 6.57% at a 99.95% specificity (Li and Ilie, 2017). In this work, we quantified the performance of the PIPE4 and SPRINT methods using known (experimentally validated physical interactions) G. max-G. max extracted from BioGRID: 17 unseen PPI pairs not used in the training of either method that can be used to quantify the performance of the two methods. From the highly conservative top 40 predictions considered in this work, PIPE4 detected 2/17 pairs, achieving a sensitivity of 11.8%, while SPIRNT detected 4/17 pairs, achieving a sensitivity of 23.5% (Supplementary Table S2). Given the severely limited availability of experimentally validated PPIs (only n = 17 physical interactions), these estimates are extremely conservative and may under-report the performance of both PIPE4 and SPRINT. Numerous recent comparisons of the SPRINT algorithm to the latest PIPE4 algorithm indicate that both methods perform similarly and complement one another in massive-scale interactome studies (Dick et al., 2020; Dick et al., 2021b). Importantly, both methods are tuned to be highly conservative and aim to minimize the false positive rate, making them applicable to interactome-scale screening analysis. At present, PIPE4 and SPRINT predictive performance approaches that of wet-laboratory studies such as tap-tagging and yeast-two-hybrid studies, but on a much faster and larger scale and with lower running costs (Pitre et al., 2006; Li and Ilie, 2017). These two PPI predictors function by using a sequence-based approach utilizing the primary sequence of amino acids and a known dataset of interacting partners. They differ primarily in how short regions of sequence similarity are defined when comparing query proteins to known PPIs. They are both algorithms that automatically learn and extract sequence patterns important to PPIs, learned directly from the training examples of known PPIs (Li and Ilie, 2017; Dick et al., 2020). Since we have confirmed soybean proteins that play a major role in resistance against SCN, Rhg1 and Rhg4, we were able to use PIPE4 and SPRINT to extract the top-ranked predicted interacting soy partners of Rhg1 and Rhg4 to act as positive control groups. By comparing top-ranked interacting partners between these positive control proteins and all other soybean proteins, we can identify candidate soybean proteins that are likely to share resistance-related function with Rhg1 and Rhg4 through the guilt-by-association approach.
To this point in time, researchers have struggled to identify genes involved in the host–pathogen relationship between soybean and SCN as the defense mechanism of soybean against SCN seems to be different from the typical “R gene” type of resistance. This can be seen in the discovery of Rhg1 and Rhg4 genes (Cook et al., 2012; Liu et al., 2012; Liu et al., 2017), as well as the later discovery of a pathogenesis-related protein GmPR08-Bet VI (Glyma.08g230500) as an interacting partner (Lakhssassi et al., 2020). Hence, we posed the question “Can we predict the top interacting partners for Rhg1 and Rhg4 with a high degree of accuracy through a computational large-scale approach, and if so, what kind of genes will we find to be present within that relationship?” To the best of our knowledge, this is the first study to attempt to answer this question on a large scale. By tackling this problem and making our data available for scientists, we can open possibilities for further research on this relationship.
By filtering and visualizing the GO terms of AP/AS positive control lists by first using the SoyBase GO Term Enrichment Tool and then using REVIGO, we identified that our two PPI predictors, PIPE4 and SPRINT, made overlapping predictions of Rhg1 and Rhg4 top interacting proteins with GO terms involved in defense response (GO:0006952) and response to mechanical stimulus (GO:0009612). Seven genes were responsible for these enriched functions (Glyma.17G182500, Glyma.08G032900, Glyma.20G037900, Glyma.17G220000, Glyma.10G098300, Glyma.19G098200, and Glyma.03G114400). As shown in Table 1, our two PPI predictors made common predictions for proteins that interact with Rhg1 and Rhg4 in the broad “defense response” category, also in additional defense-related GO terms including but not limited to response to xenobiotic stimulus (GO:0009410), defense response to bacterium (GO:0042742), and jasmonic acid and ethylene-dependent systemic resistance (GO:0009861) (Table 1). We note here that the genes predicted using both engines have a higher chance of being true predictions and that we may now be one step closer to understanding the soybean–SCN relationship.
The major resistance gene at Rhg1 is an alpha-soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (α-SNAP) that is present in multiple copies in resistant lines (Cook et al., 2012). Normally, this vesicular trafficking chaperone binds SNARE complexes and stimulates their disassembly by activating NSF. However, the resistance allele is defective in interacting with NSF, and the overexpression of α-SNAP in the syncytium leads to the disruption of vesicle trafficking and cytotoxic levels of NSF (Bayless et al., 2016). In our study, 15 proteins were predicted to interact with α-SNAP by both predictors. Many of these were protein kinases. It was shown that mitogen-activated protein kinases were overexpressed in the syncytium, play important signal transduction and membrane trafficking roles, and may be involved in the defense response to nematode infection (McNeece et al., 2019). The second gene at Rhg1, AATRhg1, is a putative amino acid transporter. It was recently shown that AATRhg1 accumulates along the path of nematode invasion and physically interacts with NADPH oxidase (Han et al., 2023). This results in significant reactive oxygen species (ROS) increase in resistant lines. Most of the 11 proteins predicted to interact with AATRhg1, by both predictors in the present work, are heat shock proteins. These proteins are central to the oxidative stress responses and may be partnering with AATRhg1 in SCN resistance. The other main SCN resistance quantitative trait locus (QTL) in soybean, Rhg4, encodes a serine hydroxymethyltransferase (SHMT) (Liu et al., 2012). Our predictions revealed that most of the 15 interactions predicted by both predictors are with proteins involved in the ubiquitin-dependent protein catabolic process. While the mechanism of resistance involving SHMTRhg4 is still not fully understood, the ubiquitin proteasome system is involved in host-defense in many different pathosystems (Kud et al., 2019).
In addition to investigating the positive control sets AP/AS, we were also interested in identifying additional proteins involved in the resistance pathway against SCN. Hence, we posed another question: “If we can predict interacting partners of known resistance genes, can we use those predictions with a guilt-by-association approach to identify novel genes involved in the resistance pathway by scanning the PPI network of the entire soybean proteome?” We wanted to do this on a large scale as current resistant varieties are becoming increasingly susceptible to the pest (Kofsky et al., 2021). We wanted to identify additional genes, through a computational approach, for the possibility of stacking resistance. We identified 1,082 candidates from the entire soybean genome based on the level of overlaps between their interacting partners and the top interacting partners of Rhg1 and Rhg4. Interestingly, by filtering the enriched GO terms, we identified five genes with ontologies related to response to nematode, Glyma.18G029000, Glyma.11G228300, Glyma.08G120500, Glyma.17G152300, and Glyma.08G265700, or regulation to larval development (GO:0061062) (Glyma.08G265700). These offer good targets for future validation studies to characterize their role in resistance against SCN. Among the 1,082 genes, 91 were highlighted (Table 2) based on predicted functions that could be compatible with resistance and will warrant future research, for example, Glyma.19G055000 is a toll-interleukin-1 receptor, nucleotide-binding site, and leucine-rich repeat (TIR-NBS-LRR) disease resistance protein. This class includes many classical plant disease resistance genes. Furthermore, through literature search, it was identified that five out of the top predictions from Tables 1 and 2 had genes present within a ± 50 kb window of recent QTLs and genome-wide association studies, i.e., Glyma.04g007700 (Li et al., 2016), Glyma.06g186200 (Li et al., 2016), Glyma.10g172500 (Tran et al., 2019), Glyma.17g085700 (Li et al., 2016), and Glyma.18g029000 (Chang et al., 2017). Interestingly, two other genes were found within the QTL regions, SCN-2 (Glyma.08g223600) and SCN-3 (Glyma.08g338900) (Swaminathan et al., 2018).
Finally, the predicted structures generated by AlphaFold2 offer significant utility to the broader research community, both in the extension of the research findings herein and more broadly in the realm of host–pathogen biology. These highly accurate 3D structural conformations, available at https://github.com/earezza/Soybean-Large-Scale-PPI-Analysis, serve as valuable resources for scientists investigating the molecular mechanisms underlying plant defense mechanisms. Figure 8 depicts a static view of the folded proteins, and the proteins most relevant within this work and additional structures are given in Supplementary Table S2. By incorporating the predicted structures into their research, scientists can gain insights into putative PPIs, candidate ligand-binding sites, and potential enzymatic activities, facilitating the development of strategies to enhance plant resistance against pathogens. The predicted structures also provide a starting point for experimental studies, allowing for validation and refinement through techniques like X-ray crystallography and cryo-electron microscopy. Overall, the use of AlphaFold2 predictions holds significant promise for advancing our understanding of host–pathogen interactions and contributing to the development of innovative approaches in plant biology. Given the sparsity of known PPIs and/or experimentally determined protein structures within the G. max-G. max proteome, it is our recommendation that subsequent research initiatives leverage these state-of-the-art AI methods to increasingly expand their representation within large-scale consortium databases such as the AlphaFold protein structure database (Varadi et al., 2021).
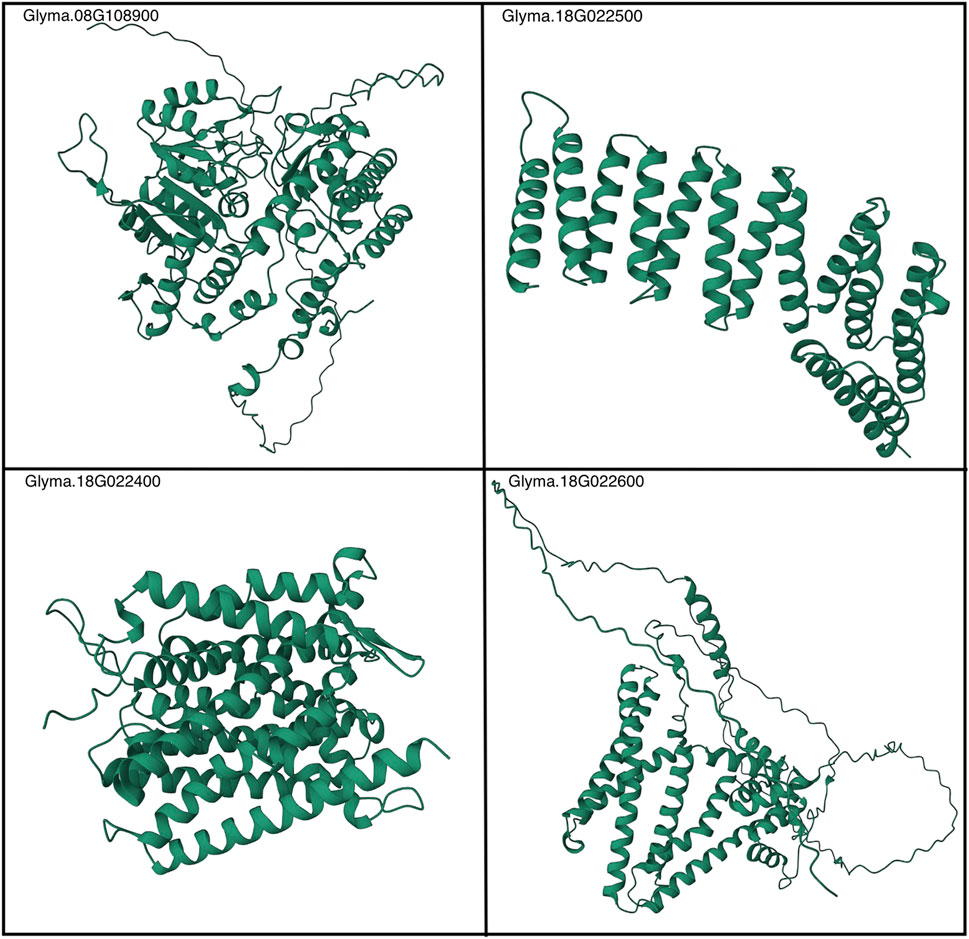
FIGURE 8. AlphaFold2-generated depictions of a static view of Rhg1 and Rhg4 folded proteins. See the following link for more details: https://github.com/earezza/Soybean-Large-Scale-PPI-Analysis.
In this paper, we provide an approach to scan the entire soybean proteome and use a guilt-by-association method, in addition to a multistep workflow, to predict the most likely novel candidates involved in resistance against SCN. This pipeline combined two machine learning tools, PIPE4 and SPRINT, and illustrated the potential of new technological advances to facilitate gene discovery. We believe that these tools can be used to predict other resistance protein-interacting partners and will allow scientists to focus their research in a much more efficient manner to address existing and emergent diseases.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
NN, JH, EA, KD, AG, BM, EC, JG, and BS contributed to the conception and design of the article. NN, BS, and BM interpreted the detailed relevant analysis. JG, EA, and KD modified PIPE4 and SPRINT to work for this analysis. NN, JH, BM, EC, and BS prepared the final draft. All authors contributed to the article and approved the submitted version.
The authors acknowledge Agriculture and Agri-Food Canada and Grain Farmers of Ontario (GFO) for the financial support. NN would like to extend a dedication of this research article to her family and fiancé as they have all encouraged her during the various stages of this research and manuscript editions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2023.1199675/full#supplementary-material
Bayless, A. M., Smith, J. M., Song, J., McMinn, P. H., Teillet, A., August, B. K., et al. (2016). Disease resistance through impairment of α-SNAP–NSF interaction and vesicular trafficking by soybean Rhg1. Proc. Natl. Acad. Sci. [Internet] 113 (47), E7375–E7382. doi:10.1073/pnas.1610150113
Bensimon, A., Heck, A. J. R., and Aebersold, R. (2012). Mass spectrometry-based proteomics and network biology. Annu. Rev. Biochem. 81, 379–405. doi:10.1146/annurev-biochem-072909-100424
Boerema, A., Peeters, A., Swolfs, S., Vandevenne, F., Jacobs, S., Staes, J., et al. (2016). Soybean trade: Balancing environmental and socio-economic impacts of an intercontinental market. PLoS One 11 (5), 01552222–e155313. doi:10.1371/journal.pone.0155222
Bradley, C. A., Allen, T. W., Sisson, A. J., Bergstrom, G. C., Bissonnette, K. M., Bond, J., et al. (2021). Soybean yield loss estimates due to diseases in the United States and Ontario, Canada, from 2015 to 2019. Plant Heal Prog. 22 (4), 483–495. doi:10.1094/php-01-21-0013-rs
Chang, H. X., Hartman, G. L., and Domier, L. L. (2017). Characterization of disease resistance loci in the USDA soybean germplasm collection using genome-wide association studies. Front. Plant Sci. 8 (10), 1139–1151. doi:10.1094/phyto-01-16-0042-fi
Concibido, V., Lange, D., Denny, R., Orf, J., and Young, N. (1997). Genome mapping of soybean cyst nematode resistance genes in ‘Peking’, PI 90763, and PI 88788 using DNA markers. Crop Sci. 37, 258–264. doi:10.2135/cropsci1997.0011183x003700010046x
Cook, D. E., Lee, T. G., Guo, X., Melito, S., Wang, K., Bayless, A. M., et al. (2012). Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338 (6111), 1206–1209. doi:10.1126/science.1228746
Cusick, M. E., Klitgord, N., Vidal, M., and Hill, D. E. (2005). Interactome: Gateway into systems biology. Hum. Mol. Genet. 14, R171–R181. doi:10.1093/hmg/ddi335
Davis, E. L., and Tylka, G. L. (2000). Soybean cyst nematode disease. Plant Heal Instr. doi:10.1094/PHI-I-2000-0725-02
De Las Rivas, J., and Fontanillo, C. (2012). Protein-protein interaction networks: Unraveling the wiring of molecular machines within the cell. Brief. Funct. Genomics 11 (6), 489–496. doi:10.1093/bfgp/els036
Dick, K., and Green, J. R. (2018). Reciprocal perspective for improved protein-protein interaction prediction. Sci. Rep. 8 (1), 11694. doi:10.1038/s41598-018-30044-1
Dick, K., Samanfar, B., Barnes, B., Cober, E. R., Mimee, B., Tan, L. H., et al. (2020). PIPE4: Fast PPI predictor for comprehensive inter- and cross-species interactomes. Sci. Rep. 10 (1), 1–12. doi:10.1038/s41598-019-56895-w
Dick, K., Pattang, A., Hooker, J., Nissan, N., Sadowski, M., Barnes, B., et al. (2021). Human-soybean allergies: Elucidation of the seed proteome and comprehensive protein-protein interaction prediction. J. Proteome Res. 20 (11), 4925–4947. doi:10.1021/acs.jproteome.1c00138
Dick, K., Chopra, A., Biggar, K. K., and Green, J. R. (2021). Multi-schema computational prediction of the comprehensive SARS-CoV-2 vs. human interactome. PeerJ 9, e11117. doi:10.7717/peerj.11117
Gheysen, G., and Mitchum, M. G. (2011). How nematodes manipulate plant development pathways for infection. Curr. Opin. Plant Biol. 14 (4), 415–421. doi:10.1016/j.pbi.2011.03.012
Gligorijević, V., Barot, M., Bonneau, R., and Wren, J. (2018). deepNF: deep network fusion for protein function prediction. Wren J. Editor. Bioinforma. 34, 3873–3881. doi:10.1093/bioinformatics/bty440
Glover, K. D., Wang, D., Arelli, P. R., Carlson, S. R., Cianzio, S. R., and Diers, B. W. (2004). Near isogenic lines confirm a soybean cyst nematode resistance gene from PI 88788 on linkage group J. J. Crop Sci. 44, 936–941. doi:10.2135/cropsci2004.0936
Grant, D., Nelson, R. T., Cannon, S. B., and Shoemaker, R. C. (2010). SoyBase, the USDA-ARS soybean genetics and genomics database. Nucl. Acid. 38 (1), D843–D846. doi:10.1093/nar/gkp798
Han, S., Smith, J. M., Du, Y., and Bent, A. F. (2023). Soybean transporter AATRhg1 abundance increases along the nematode migration path and impacts vesiculation and ROS. Plant Physiol. 192, 133–153. doi:10.1093/plphys/kiad098
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Jumrani, K., and Bhatia, V. S. (2018). Impact of combined stress of high temperature and water deficit on growth and seed yield of soybean. Physiol. Mol. Biol. Plants 24 (1), 37–50. doi:10.1007/s12298-017-0480-5
Kim, M., Hyten, D. L., Bent, A. F., and Diers, B. W. (2010). Fine mapping of the SCN resistance locus rhg1-b from PI 88788. Plant Genome 3 (2). doi:10.3835/plantgenome2010.02.0001
Kofsky, J., Zhang, H., and Song, B-H. (2021). Novel resistance strategies to soybean cyst nematode (SCN) in wild soybean. Sci. Rep. [Internet] 11 (1), 1–13. doi:10.1038/s41598-021-86793-z
Kud, J., Wang, W., Gross, R., Fan, Y., Huang, L., Yuan, Y., et al. (2019). The potato cyst nematode effector RHA1B is a ubiquitin ligase and uses two distinct mechanisms to suppress plant immune signaling. PLOS Pathog. 15 (4), 1–18. doi:10.1371/journal.ppat.1007720
Lakhssassi, N., Piya, S., Bekal, S., Liu, S., Zhou, Z., Bergounioux, C., et al. (2020). A pathogenesis-related protein GmPR08-Bet VI promotes a molecular interaction between the GmSHMT08 and GmSNAP18 in resistance to Heterodera glycines. Plant Biotechnol. J. 18 (8), 1810–1829. doi:10.1111/pbi.13343
Li, Y., and Ilie, L. (2017). Sprint: Ultrafast protein-protein interaction prediction of the entire human interactome. BMC Bioinforma. 18 (1), 485. doi:10.1186/s12859-017-1871-x
Li, Y., Shi, X., Li, H., Reif, J. C., Wang, J., Liu, Z., et al. (2016). Dissecting the genetic basis of resistance to soybean cyst nematode combining linkage and association mapping. Plant Genome 9 (2). doi:10.3835/plantgenome2015.04.0020
Liu, S., Kandoth, P. K., Warren, S. D., Yeckel, G., Heinz, R., Alden, J., et al. (2012). A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens. Nature 492 (7428), 256–260. doi:10.1038/nature11651
Liu, S., Kandoth, P. K., Lakhssassi, N., Kang, J., Colantonio, V., Heinz, R., et al. (2017). The soybean GmSNAP18 gene underlies two types of resistance to soybean cyst nematode. Nat. Commun. 8, 14822. doi:10.1038/ncomms14822
McNeece, B. T., Sharma, K., Lawrence, G. W., Lawrence, K. S., and Klink, V. P. (2019). The mitogen activated protein kinase (MAPK) gene family functions as a cohort during the Glycine max defense response to Heterodera glycines. Plant Physiol. Biochem. 137, 25–41. doi:10.1016/j.plaphy.2019.01.018
Nissan, N., Mimee, B., Cober, E. R., Golshani, A., Smith, M., and Samanfar, B. (2022). A broad review of soybean research on the ongoing race to overcome soybean cyst nematode. Biol. (Basel) 11, 211. doi:10.3390/biology11020211
Peng, X., Wang, J., Peng, W., Wu, F. X., and Pan, Y. (2017). Protein-protein interactions: Detection, reliability assessment and applications. Brief. Bioinform 18 (5), 798–819. doi:10.1093/bib/bbw066
Pitre, S., Dehne, F., Chan, A., Cheetham, J., Duong, A., Emili, A., et al. (2006). Pipe: A protein-protein interaction prediction engine based on the re-occurring short polypeptide sequences between known interacting protein pairs. BMC Bioinforma. 7 (1), 365. doi:10.1186/1471-2105-7-365
Rao, V., Srinivas, K., Sujini, G. N., and Sunand Kumar, G. N. (2014). Protein-protein interaction detection: Methods and analysis. Int. J. Proteomics 2014, 1–12. doi:10.1155/2014/147648
Samanfar, B., Molnar, S. J., Charette, M., Schoenrock, A., Dehne, F., Golshani, A., et al. (2017). Mapping and identification of a potential candidate gene for a novel maturity locus, E10, in soybean. Theor. Appl. Genet. 130 (2), 377–390. doi:10.1007/s00122-016-2819-7
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463 (7278), 178–183. doi:10.1038/nature08670
Schoenrock, A., Burnside, D., Moteshareie, H., Wong, A., Golshani, A., Dehne, F., et al. (2015). “Engineering inhibitory proteins with InSiPS: The in-silico protein synthesizer,” in Proceedings of the international conference for high performance computing, networking, storage and analysis on - SC ’15 (New York, New York, USA: ACM Press), 1–11.
Shaibu, A. S., Li, B., Zhang, S., and Sun, J. (2020). Soybean cyst nematode-resistance: Gene identification and breeding strategies. Crop J. 8 (6), 892–904. doi:10.1016/j.cj.2020.03.001
Stynen, B., Tournu, H., Tavernier, J., and Van Dijck, P. (2012). Diversity in genetic in vivo methods for protein-protein interaction studies: From the yeast two-hybrid system to the mammalian split-luciferase system. Microbiol. Mol. Biol. Rev. 76 (2), 331–382. doi:10.1128/mmbr.05021-11
Supek, F., Bošnjak, M., Škunca, N., and Šmuc, T. (2011). Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 6 (7), e21800. doi:10.1371/journal.pone.0021800
Swaminathan, S., Abeysekara, N. S., Knight, J. M., Liu, M., Dong, J., Hudson, M. E., et al. (2018). Mapping of new quantitative trait loci for sudden death syndrome and soybean cyst nematode resistance in two soybean populations. Theor. Appl. Genet. 131 (5), 1047–1062. doi:10.1007/s00122-018-3057-y
Tong, A. H. Y., Drees, B., Nardelli, G., Bader, G. D., Brannetti, B., Castagnoli, L., et al. (2002). A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295 (5553), 321–324. doi:10.1126/science.1064987
Torkamaneh, D., Laroche, J., Valliyodan, B., O’Donoughue, L., Cober, E., Rajcan, I., et al. (2021). Soybean (Glycine max) haplotype map (GmHapMap): A universal resource for soybean translational and functional genomics. Plant Biotechnol. J. 19 (2), 324–334. doi:10.1111/pbi.13466
Tran, D. T., Steketee, C. J., Boehm, J. D., Noe, J., and Li, Z. (2019). Genome-wide association analysis pinpoints additional major genomic regions conferring resistance to soybean cyst nematode (Heterodera glycines ichinohe). Front. Plant Sci. 10, 401–413. doi:10.3389/fpls.2019.00401
Uetz, P., Glot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., et al. (2000). A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403 (6770), 623–627. doi:10.1038/35001009
Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., et al. (2021). AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50 (D1), D439–D444. doi:10.1093/nar/gkab1061
Winstead, N. N., Skotland, C. B., and Sasser, J. N. (1955). Soybean cyst nematode in North Carolina. Plant Dis. Rep. 39, 9–11.
Xu, J., and Li, Y. (2006). Discovering disease-genes by topological features in human protein–protein interaction network. Bioinformatics 22 (22), 2800–2805. doi:10.1093/bioinformatics/btl467
Yan, G., and Baidoo, R. (2018). Current research status of Heterodera glycines resistance and its implication on soybean breeding. Engineering 4 (4), 534–541. doi:10.1016/j.eng.2018.07.009
Yıldırım, M. A., Goh, K-I., Cusick, M. E., Barabási, A-L., and Vidal, M. (2007). Drug—Target network. Nat. Biotechnol. 25 (10), 1119–1126. doi:10.1038/nbt1338
Zhang, L., Yu, G., Xia, D., and Wang, J. (2019). Protein–protein interactions prediction based on ensemble deep neural networks. Neurocomputing 324, 10–19. doi:10.1016/j.neucom.2018.02.097
Zhao, B., Wang, J., Li, M., Li, X., Li, Y., Wu, F-X., et al. (2016). A new method for predicting protein functions from dynamic weighted interactome networks. IEEE Trans. Nanobioscience 15 (2), 131–139. doi:10.1109/tnb.2016.2536161
Keywords: soybean cyst nematode, computational biology, protein–protein interactions, bioinformatics, SCN resistance
Citation: Nissan N, Hooker J, Arezza E, Dick K, Golshani A, Mimee B, Cober E, Green J and Samanfar B (2023) Large-scale data mining pipeline for identifying novel soybean genes involved in resistance against the soybean cyst nematode. Front. Bioinform. 3:1199675. doi: 10.3389/fbinf.2023.1199675
Received: 03 April 2023; Accepted: 31 May 2023;
Published: 20 June 2023.
Edited by:
Tirso Pons, Spanish National Research Council (CSIC), SpainReviewed by:
Alexander Banguela Castillo, Saarland University, GermanyCopyright © 2023 Nissan, Hooker, Arezza, Dick, Golshani, Mimee, Cober, Green and Samanfar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bahram Samanfar, YmFocmFtLnNhbWFuZmFyQGFnci5nYy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.