 Byron J. Smith
Byron J. Smith Xiangpeng Li
Xiangpeng Li Zhou Jason Shi
Zhou Jason Shi Adam Abate3,4†
Adam Abate3,4† Katherine S. Pollard
Katherine S. Pollard
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioinform., 16 May 2022
Sec. Genomic Analysis
Volume 2 - 2022 | https://doi.org/10.3389/fbinf.2022.867386
This article is part of the Research TopicComputational Methods for Microbiome Analysis, volume 2View all 17 articles
While genome databases are nearing a complete catalog of species commonly inhabiting the human gut, their representation of intraspecific diversity is lacking for all but the most abundant and frequently studied taxa. Statistical deconvolution of allele frequencies from shotgun metagenomic data into strain genotypes and relative abundances is a promising approach, but existing methods are limited by computational scalability. Here we introduce StrainFacts, a method for strain deconvolution that enables inference across tens of thousands of metagenomes. We harness a “fuzzy” genotype approximation that makes the underlying graphical model fully differentiable, unlike existing methods. This allows parameter estimates to be optimized with gradient-based methods, speeding up model fitting by two orders of magnitude. A GPU implementation provides additional scalability. Extensive simulations show that StrainFacts can perform strain inference on thousands of metagenomes and has comparable accuracy to more computationally intensive tools. We further validate our strain inferences using single-cell genomic sequencing from a human stool sample. Applying StrainFacts to a collection of more than 10,000 publicly available human stool metagenomes, we quantify patterns of strain diversity, biogeography, and linkage-disequilibrium that agree with and expand on what is known based on existing reference genomes. StrainFacts paves the way for large-scale biogeography and population genetic studies of microbiomes using metagenomic data.
Intra-specific variation in microbial traits are widespread and are biologically important in human associated microbiomes. Strains of a species may differ in their pathogenicity (Loman et al., 2013), antibiotic resistance (Shoemaker et al., 2001), impacts on drug metabolism (Haiser et al., 2014), and ability to utilize dietary components (Patrick et al., 2010; Ostrowski et al., 2022). Standard methods for analysis of complex microbial communities are limited to coarser taxonomic resolution due to their reliance on slowly evolving marker genes (Case et al., 2007-January) or on genome reference databases lacking diverse strain representation (Nayfach et al., 2020). Approaches that quantify microbiomes at the level of strains may better capture variation in microbial function (Albanese and Donati, 2017), provide insight into ecological and evolutionary processes (Garud and Pollard, 2019), and discover previously unknown microbial etiologies for disease (Yan et al., 2020).
Shotgun metagenomic data can in principle be used to track strains by looking for distinct patterns of alleles observed across single nucleotide polymorphisms (SNPs) within the species. Several tools have recently been developed that count the number of metagenomic reads containing alleles across SNP sites (Nayfach et al., 2016; Costea P. I. et al., 2017; Truong et al., 2017; Beghini et al., 2021; Olm et al., 2021; Shi et al., 2021). Comparisons of the resulting “metagenotypes” across samples has been used to track shared strains (Li et al., 2016; Olm et al., 2021), or to interrogate the biogeography (Costea PI. et al., 2017; Truong et al., 2017) and population genetics of species (Garud et al., 2019). The application of this approach is limited, however, by low sequencing coverage, which results in missing values at some SNP sites, and co-existing mixtures of strains, which introduce ambiguity about the taxonomic source of each metagenomic read.
One promising solution to these challenges is statistical strain deconvolution, which harnesses multiple metagenotypes (e.g., a collection of related samples) to simultaneously estimate the genotypes and relative abundances of strains across samples. Several tools have been developed that take this approach, including Lineage (O’Brien et al., 2014), Strain Finder (Smillie et al., 2018), DESMAN (Quince et al., 2017), and ConStrains (Luo et al., 2015). These methods have been used to track the transmission of inferred strains from donors’ to recipients’ microbiomes after fecal microbiota transplantation (FMT) (Smillie et al., 2018; Chu et al., 2021; Watson et al., 2021; Smith et al., 2022). The application of strain deconvolution has been limited, however, by the computational demands of existing methods, where runtimes scale poorly with increasing numbers of samples, latent strains, and SNPs considered. One reason for this poor scaling is the discreteness of alleles at each SNP, which has led existing methods to use expectation maximization algorithms to optimize model parameters (Smillie et al., 2018), or Markov chain Monte Carlo to sample from a posterior distribution (O’Brien et al., 2014; Luo et al., 2015; Quince et al., 2017).
Here we take a different approach, extending the strain deconvolution framework by relaxing the discreteness constraint and allowing genotypes to vary continuously between alleles. The use of this “fuzzy” genotype approximation makes our underlying model fully differentiable, and allows us to apply modern, gradient-based optimization algorithms to estimate strain genotypes and abundances. Here we show that the resulting tool, StrainFacts, can scale to tens of thousands of samples, hundreds of strains, and thousands of SNPs, opening the door to strain inference in large metagenome collections.
A metagenotype is represented as a count matrix of the number of reads with each allele at a set of SNP sites for a single species in each sample. This can be gathered directly from metagenomic data, for instance by aligning reads to a reference genome and counting the number of reads with each allele at SNP sites. In this study we use GT-Pro (Shi et al., 2021), which instead counts exact k-mers associated with known single nucleotide variants. Although the set of variants at a SNP may include any of the four bases, here we constrain metagenotypes to be biallelic: reference or alternative. For a large majority of SNPs, only two alleles are observed across reference genomes (Shi et al., 2021). Metagenotypes from multiple samples are subsequently combined into a 3-dimensional array.
StrainFacts is based on a generative, graphical model of biallelic metagenotype data (summarized in Supplementary Figure S1) which describes the allele frequencies at each SNP site in each sample (

TABLE 1. Symbols used to describe the StrainFacts model.
The crux of strain deconvolution is taking noisy observations of
StrainFacts does not constrain the elements of
Since true genotypes are in fact discrete, we place a prior on the elements of
For each element of
The underlying allele frequencies
To summarize, our model is as follows (in random variable notation; see Supplementary Figure S1 for a plate diagram):
StrainFacts takes a MAP-based approach to inference on this model, using gradient-based methods to find parameter values that maximize the posterior probability of our model conditioned on the observed counts. We rely heavily on the probabilistic programming framework Pyro (Bingham et al., 2019), which is built on the PyTorch library (Paszke et al., 2019) for numerical methods.
Initial values for
Metagenotype data was simulated in order to enable direct performance benchmarking against ground-truth genotypes and strain compositions. For each independent simulation, discrete genotypes of length
Estimates were evaluated against the simulated ground truth using five different measures of error (see Results).
We applied StrainFacts to data from two previously compiled human microbiome metagenomic datasets: stool samples from a fecal microbiota transplantation (FMT) study described in (Smith et al., 2022, BioProject PRJNA737472) and 20,550 metagenomes from a meta-analysis of publicly available data in (Shi et al., 2021, various accessions). As described in that publication, metagenotypes for gut prokaryotic species were tallied using GT-Pro version 1.0.1 with the default database, which includes up to 1,000 of the highest quality genomes for each species from the Unified Human Gastrointestinal Genome (UHGG) V1.0 (Almeida et al., 2021). This includes both cultured isolates and high-quality metagenomic assemblies. This same database was used as a reference set to which we compared our inferred genotypes. Estimated genomic distances between SNPs were based on the UHGG representative genome.
We describe detailed results for Escherichia coli (id: 102506, MGYG-HGUT-02506), Agathobacter rectalis (id: 102492, MGYG-HGUT-02492), Methanobrevibacter smithii (id: 102163, MGYG-HGUT-02163), and CAG-279 sp1 (id: 102556, MGYG-HGUT-02556). These were selected to demonstrate application of StrainFacts to prevalent gram-positive and gram-negative bacteria in the human gut, the most prevalent archaeon, as well as an unnamed, uncultured, and largely unstudied species. We also describe detailed results for Streptococcus thermophilus (GT-Pro species id: 104345, representative UHGG genome: MGYG-HGUT-04345), selected for its high diversity in one sample of our single-cell sequencing validation.
Of the 159 samples with metagenomes described in the FMT study, we selected two samples for single-cell genomics (which we refer to as the “focal samples”). These samples were obtained from two different study subjects; one is a baseline sample and the other was collected after several weeks of FMT doses as described in (Smith et al., 2022). A full description of the single-cell genomics pipeline is included in the Supplementary Methods, and will be briefly summarized here. For each of the focal samples, microbial cells were isolated from whole feces by homogenization in phosphate buffered saline, 50 μM filter-based removal of large fecal particles, and density gradient separation. After isolating and thoroughly washing the density layer corresponding to the microbiota, this cell suspension was mixed with polyacrylamide precursor solution, and emulsified with a hydrofluoric oil. Aqueous droplets in oil were allowed to gellate before separating the resulting beads from the oil phase and washing. Beads were size selected to between 5 and 25 μM, with the goal of enriching for those encapsulated a single microbial cell. Cell lysis was carried out inside the hydrogel beads by incubating with zymolyase, lysostaphin, mutanolysin, and lysozyme. After lysis, proteins were digested with proteinase K, before thoroughly washing the beads. Tn5 tagmentation and barcode PCR were carried out using the MissionBio Tapestri microfluidics DNA workflow with minor modifications. After amplification, the emulsion was broken and the aqueous phase containing the barcoded amplicons was used for sequencing library preparation with Nextera primers including P5 and P7 sequences followed by Ampure XP bead purification. Libraries were sequenced by Novogene on an Illumina NovaSeq 6000, BioProject PRJNA737472.
Demultiplexed sequence data for each droplet barcode were independently processed with GT-Pro identically to metagenomic sequences. For each barcode, GT-Pro allele counts for a given species were assumed to be representative of a single strain of that species. Horizontal coverage was calculated as the fraction of GT-Pro positions with ≥2 reads, unlike metagenotypes where ≥1 read was used to calculate horizontal coverage. These single-cell genotypes (SCGs) were filtered to those with > 1% horizontal coverage over SNP sites, leaving 87 species with at least one SCG from either of the two focal samples. During analysis, a number of SCGs were found to have nearly identical patterns of horizontal coverage. These may have been formed by merging of droplets during barcoding PCR, which could have resulted in multiple barcodes in the same amplification. To reduce the impact of this artifact, allele counts from multiple SCGs were summed by complete-linkage, agglomerative clustering based on their depth profiles across SNP sites, at a 0.3 cosine dissimilarity threshold.
From GT-Pro metagenotypes, we extracted allele counts for select species and removed SNPs that had < 5% occurance of the minor allele across samples. Species with more than 5,000 SNPs after filtering, were randomly down-sampled without replacement to this number of sites. Samples with less than 5% horizontal coverage were also filtered out.
For all analyses, StrainFacts was run with the following hyperparameters
The number of strains parameterized by our model was chosen as follows. For comparisons to SCGs, the number of strains was set at 30% of the number of samples—e.g. 33 strains were parameterized for S. thermophilus because metagenotypes from 109 samples remained after coverage filtering. For the analysis of thousands of samples described in (Shi et al., 2021), we parameterized our model with 200 strains and increased the numerical precision from 32 to 64 bits. After strain inference using the 5,000 subsampled SNPs, full-length genotypes were estimated post-hoc by conditioning on our estimate of
For computational reproducibility we set fixed seeds for random number generators: 0 for all analyses where we only report one estimate, and 0, 1, 2, 3, and 4 for the five replicate estimates described for simulated datasets. Strain Finder was run with flags --dtol 1 --ntol 2 --max_reps 1. We did not use --exhaustive, Strain Finder’s exhaustive genotype search strategy, as it is much more computationally intensive.
Inferred fuzzy genotypes were discretized to zero or one for downstream analyses. SNP sites without coverage were treated as unobserved. Distances between genotypes were calculated as the masked, normalized Hamming distance, the fraction of alleles that do not agree, ignoring unobserved SNPs. Similarly, SCG genotypes and the metagenotype consensus were discretized to the majority allele. In comparing the distance between SCGs and these inferred genotypes sites missing from either the SCG or the metagenotype were treated as unobserved. Metagenotype entropy, a proxy for strain heterogeneity, was calculated for each sample as the depth weighted mean allele frequency entropy:
where
Where indicated, we dereplicated highly similar strains by applying average-neighbor agglomerative clustering at a 0.05 genotype distance threshold. Groups of these highly similar strains were replaced with a single composite strain with a genotype derived from the majority allele at each SNP site and assigned the sum of strain relative abundances in each sample. Subsequent co-clustering of these dereplicated inferred and reference strains was done in the same way, but at a 0.15 genotype distance threshold. After co-clustering, to test for enrichment of strains in “shared” clusters, we permuted cluster labels and re-tallied the total number of strains found in clusters with both inferred and reference strains. Likewise, to test for enrichment of “inferred-only” clusters we tallied the total number of strains found in clusters without reference strains after this shuffling. By repeating the permutation 9,999 times, we arrived at an empirical null distribution to which we compared our true, observed values to calculate a p-value.
Pairwise linkage disequilibrium (LD) was calculated as the squared Pearson correlation coefficient across genotypes of dereplicated strains. Genome-wide 90th percentile LD, was calculated from a random sample of 20,000 or, if fewer, all available SNP positions. To calculate the 90th percentile LD profile, SNP pairs were binned at either an exact genomic distance or within a window of distances, as indicated. In order to encourage a smooth distance-LD relationship, windows at larger pairwise-distance spanned a larger range. Specifically the ith window covers the span
StrainFacts is implemented in Python3 and is available at https://github.com/bsmith89/StrainFacts and v0.1 was used for all results reported here. Strain Finder was not originally designed to take a random seed argument, necessitating minor modifications to the code. Similarly, we made several modifications to the MixtureS (Li et al., 2021) code allowing us to run it directly on simulated metagenotypes and compare the results to StrainFacts and Strain Finder outputs. Patch files describing each set of changes, as well as all other code and metadata needed to re-run our analyses are available at https://doi.org/10.5281/zenodo.5942586. For reproducibility, analyses were performed using Snakemake (Mölder et al., 2021) and with a Singularity container (Kurtzer et al., 2017) that can be obtained at https://hub.docker.com/repository/docker/bsmith89/compbio.
Runtimes were determined using the Snakemake benchmark: directive, and memory requirements using the GNU time utility, version 1.8 with all benchmarks run on the Wynton compute cluster at the University of California, San Francisco. Across strain numbers and replicates, maximum memory usage for models with 10,000 samples and 1,000 SNPs was, counterintuitively, less than for smaller models, likely because portions of runtime data were “swapped” to disk instead of staying in RAM. We therefore excluded data for these largest models from our statistical analysis of memory requirements.
Inferring the genotypes and relative abundance of strains in large metagenome databases requires a deconvolution tool that can scale to metagenotypes with thousands of SNPs in tens-of-thousands of samples, while simultaneously tracking hundreds of microbial strains. To accomplish this we developed StrainFacts, harnessing fuzzy genotypes to accelerate inference on large datasets. We evaluated the practical scalability of the StrainFacts algorithm by applying it to simulated datasets of increasing size, and comparing its time and memory requirements to Strain Finder, a previously described method for strain inference. While several tools have been developed to perform strain deconvolution (e.g. Lineage O’Brien et al., 2014; and DESMAN Quince et al., 2017), Strain Finder’s model and approach to inference are the most similar to StrainFacts. We therefore selected it for comparison in order to directly assess the value of fuzzy genotypes.
We simulated five replicate metagenotypes for 120 underlying strains in 400 samples, and 250 SNPs, and then applied both StrainFacts and Strain Finder to these data parameterizing them with 120 strains. Both tools use random initializations, which can result in convergence to different optima. We therefore benchmarked runtimes for five independent initializations on each dataset, resulting in 25 total runs for each tool. In this setting, the median runtime for StrainFacts was just 17.2 min, while Strain Finder required a median of 6.4 h. When run on a GPU instead of CPU, StrainFacts was able to fit these data in a median of just 5.1 min.
Since the correct strain number is not known a priori in real-world applications, existing strain inference tools need to be parameterized across a range of plausible strain counts, a step that significantly impacts runtime. To assess performance in this setting, we also fit versions of each model with 50% more strains than the ground-truth, here referred to as the “1.5x parameterization” in contrast to the 1x parameterization already described. In this setting, StrainFacts’ performance advantage was even more pronounced, running in a median of 17.1 min and just 5.3 min on GPU, while Strain Finder required 30.8 h. Given the speed of StrainFacts, we were able to fit an even larger simulation with 2,500 samples and 500 strains. On a GPU, this took a median of 12.6 min with the 1x parameterization and, surprisingly, just 8.9 min with the 1.5x parameterization. We did not attempt to run Strain Finder on this dataset.
We next examined runtime scaling across a range of sample counts between 50 and 2,500. We applied Strain Finder and StrainFacts (both CPU and GPU) to simulated metagenotypes with 250 SNPs, and a fixed 1:5 ratio of strains to samples. Median runtimes for each tool at both the 1x and 1.5x parameterization demonstrate a substantially slower increase for StrainFacts as model size increases (Figure 1A). Strain Finder was faster than StrainFacts on the 1x parameterization of a small simulation with 50 samples and 10 strains: 1.3 min median runtime versus 4 min for StrainFacts on a CPU and 2.8 min on a GPU. However, StrainFacts had faster median runtimes on all other datasets.
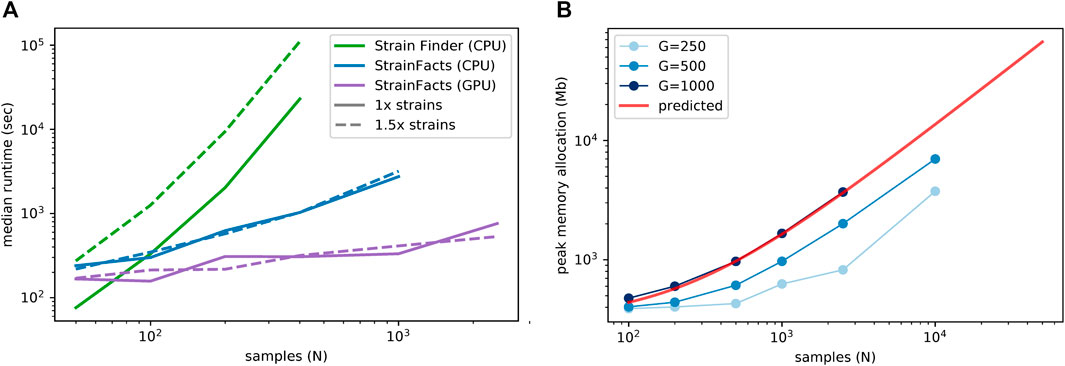
FIGURE 1. Computational scalability of strain inference on simulated data. (A) Runtime (in seconds, log scale) is plotted at a range of sample counts for both Strain Finder and StrainFacts, as well for the latter with GPU acceleration. Throughout, 250 SNPs are considered, and simulated strains are fixed at a 1:5 ratio with samples. Models are specified with this same number of strains (“1x strains”, solid lines) or 50% more (“1.5x strains”, dashed lines). Median of 25 simulation runs is shown. (B) Maximum memory allocation in a model with 100 strains is plotted for StrainFacts models across a range of sample counts (N) and SNP counts (G, line shade). Median of nine replicate runs is shown. Maximum memory requirements are extrapolated to higher numbers of samples for a model with 1,000 SNPs (red line). A version of this panel that includes a range of strain counts is included as Supplementary Figure S2.
Given the good runtime scaling properties of StrainFacts, we next asked if computer memory constraints would limit its applicability to the largest datasets (Figure 1A). A model fitting 10,000 samples, 400 strains, and 500 SNPs had a maximum memory allocation of 7.7 GB, indicating that StrainFacts’ memory requirements are satisfied on most contemporary CPU or GPU hardware and opening the door to even larger models. Using ordinary least squares, we fit the observed memory requirements to the theoretical, asymptomatic expectations,
We next set out to evaluate the accuracy of StrainFacts and to compare it to Strain Finder. We simulated 250 SNPs for 40 strains, generating metagenotypes across 200 samples. For both tools, we specified a model with the true number of strains, fit the model to this data, and compared inferences to the simulated ground-truth. For each of five replicate simulations we performed inference with five independent initializations, thereby gathering 25 inferences for each tool. As in (Smillie et al., 2018), we use the weighted UniFrac distance (Lozupone et al., 2007) as an integrated summary of both genotype and relative abundance error. By this index, StrainFacts and Strain Finder performed similarly well when applied to the simulated data (Figure 2A). We repeated this analysis with the 1.5x parameterization to assess the robustness of inferences to model misspecification, finding that both tools maintained similar performance to the 1x parameterization. By comparison, considering too few strains (the 0.8x parameterization, fitting 32 strains) degraded performance dramatically for both tools, with StrainFacts performing slightly better. Thus, we conclude based on UniFrac distance that StrainFacts is as accurate as Strain Finder and that both models are robust to specifying too many strains.
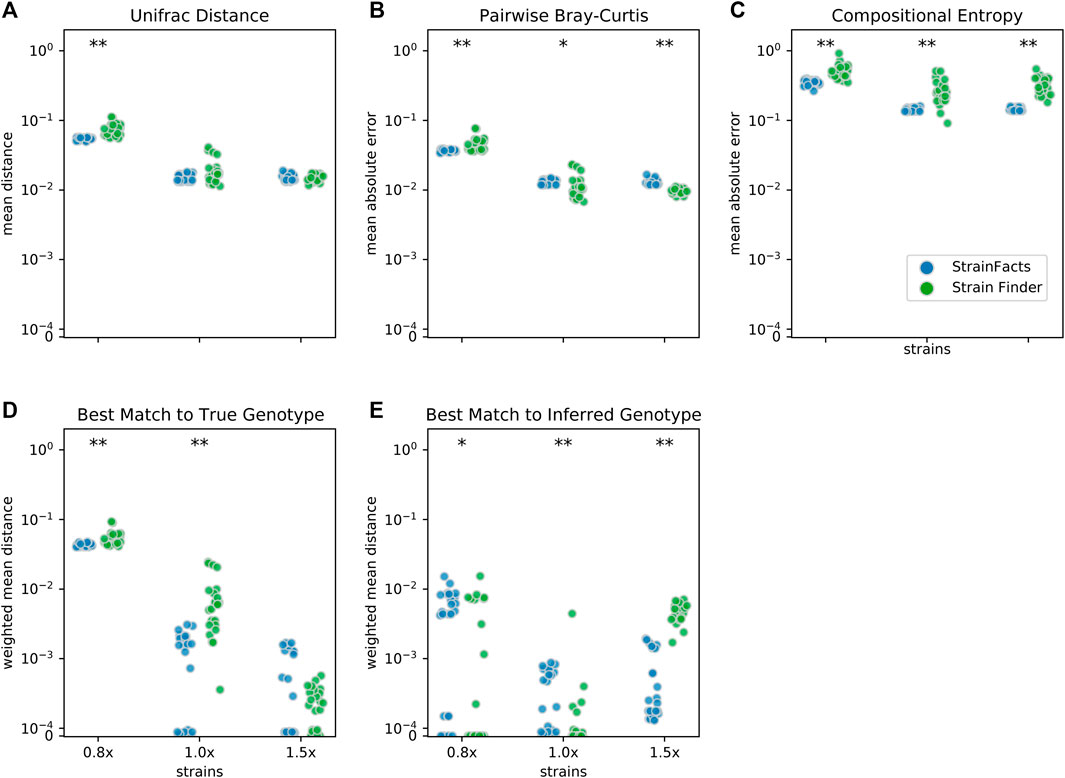
FIGURE 2. Accuracy of strain inference on simulated data. Performance of StrainFacts and Strain Finder are compared across five distinct accuracy indices, with lower scores reflecting better performance on each index. Simulated data had 200 samples, 40 underlying strains, and 250 SNPs. For each tool, 32, 40, and 60 strain models were parameterized (“0.8x”, “1x”, and “1.5x” respectively), and every model was fit with five independent initializations to each simulation. All 25 estimates for each tool-parameterization combination are shown. Scores reflect (A) mean Unifrac distance between simulated and inferred strain compositions, (B) mean absolute difference between all-by-all pairwise Bray-Curtis dissimilarities calculated on simulated versus inferred strain compositions, (C) mean absolute difference in Shannon entropy calculated on simulated versus inferred strain compositions, (D) abundance weighted mean Hamming distance from each ground-truth strain to its best-match inferred genotype, and (E) the reverse: abundance weighted mean Hamming distance from each inferred strain to its best-match true genotype. Markers at the top of each panel indicate a statistical difference between tools at a p < 0.05 (*) or p < 0.001 (**) significance threshold by Wilcoxon signed-rank test. A version of this figure that includes accuracy comparisons to MixtureS is included as Supplementary Figure S3.
To further probe accuracy, we quantified the performance of StrainFacts and Strain Finder with several other measures. First, we evaluated pairwise comparisons of strain composition by calculating the mean absolute error of pairwise Bray-Curtis dissimilarities (Figure 2B). While, with the 1x parameterization, Strain Finder slightly outperformed StrainFacts on this index, the magnitude of the difference was small. This suggests that StrainFacts can be used for applications in microbial ecology that rely on measurements of beta-diversity.
Ideally, inferences should conform to Occam’s razor, estimating “as few strains as possible, but no fewer”. Unfortunately, Bray-Curtis error is not sensitive to the splitting or merging of co-abundant strains and UniFrac error is not sensitive to the splitting or merging of strains with very similar genotypes. To overcome this limitation, we calculated the mean absolute error of the Shannon entropy of the inferred strain composition for each sample (Figure 2C). This score quantifies how accurately inferences reflect within-sample strain heterogeneity. StrainFacts performed substantially better on this score than Strain Finder for all three parameterizations, indicating more accurate estimation of strain heterogeneity.
Finally, we assessed the quality of genotypes reconstructed by StrainFacts compared to Strain Finder using the abundance weighted mean Hamming distance. For each ground-truth genotype, normalized Hamming distance is computed based on the best matching inferred genotype (Figure 2D), then summarized as the mean weighted by the true strain abundance across all samples. We assessed the reverse as well: the abundance weighted mean, best-match Hamming distance for each inferred genotype among the ground-truth genotypes (Figure 2E). These two scores can be interpreted as answers to the distinct questions “how well were the true genotypes recovered?” and “how well do the inferred genotypes reflect the truth?”, respectively. While StrainFacts and Strain Finder performed similarly on these indexes—which tool had higher accuracy varied by score and parameterization—StrainFacts’ accuracy was more stable across the 1x and 1.5x parameterizations. It should be noted that since strain genotypes are only inferred for SNP sites, the genome-wide genotype reconstruction error (which includes invariant sites) will likely be much lower than this Hamming distance. We examine the relationship between genotype distances and average nucleotide identity (ANI) in Supplementary Figure S4 in order to contextualize our simulation results for those more familiar with ANI comparisons.
To expand our performance comparison to a second tool designed for strain inference, we also ran MixtureS on a subset of the simulations. MixtureS estimates strain genotype and relative abundance on each metagenotype individually and therefore does not leverage variation in strain abundance across samples. We found that it performed worse than Strain Finder and Strain Facts on the benchmarks (see Supplementary Figure S3).
Overall, these results suggest that StrainFacts is capable of state-of-the-art performance with respect to several different scientific objectives in a realistic set of simulations. Performance was surprisingly robust to model misspecification with more strains than the simulation. Eliminating the computational demands of a separate model selection step further improves the scaling properties of StrainFacts.
Beyond simulations, we sought to confirm the accuracy of strain inferences in a real biological dataset subject to forms of noise and bias not reflected in the generative model we used for simulations. To accomplish this, we applied a recently developed, single-cell, genomic sequencing workflow to obtain ground-truth, strain genotypes from two fecal samples collected in a previously described, clinical FMT experiment (Smith et al., 2022) from two independent subjects. We ran StrainFacts on metagenotypes derived from these two focal samples as well as the other 157 samples in the same study.
Genotypes that StrainFacts inferred to be present in each of these metagenomes matched the observed SCGs, with a mean, best-match normalized Hamming distance of 0.039. Furthermore, the median distance was just 0.013, reflecting the outsized influence of a small number of SCGs with more extreme deviations. For many species, SCGs also match a consensus genotype—the majority allele at each SNP site in each metagenotype (see Figure 3A). We found a mean distance to the consensus of 0.037 and a median of 0.009. Because this distance excludes sites without observed counts in the metagenotype, we masked these same sites in our inferred genotypes to uniformly contrast the consensus approach to StrainFacts genotypes. Overall, inferred genotypes were similar to the consensus, with a mean, masked distance of 0.031 (median of 0.009). However, the consensus approach fails for species with a mixture of multiple, co-existing strains. When we select only species with a metagenotype entropy of greater than 0.05, an indication of strain heterogeneity, we see that StrainFacts inferences have a distinct advantage, with a mean distance of 0.055 versus 0.069 for the consensus approach (median of 0.018 versus 0.022, p < 0.001). These results validate inferred genotypes in a stool microbiome using single-cell genomics and demonstrate that StrainFacts accounts for strain-mixtures better than consensus genotypes do.
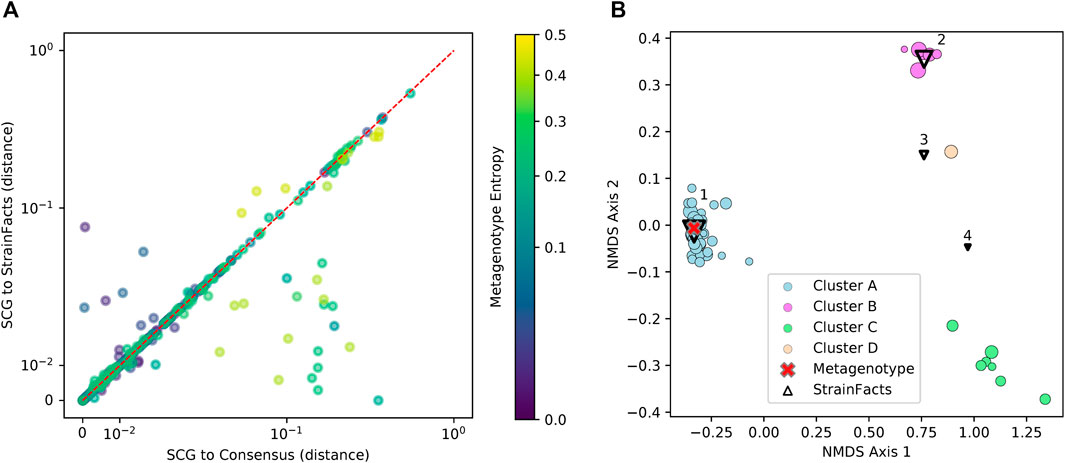
FIGURE 3. Inferred strains reflect genotypes from a single-cell sequencing experiment. (A) Distance between observed SCGs and StrainFacts inferences (X-axis) versus consensus genotypes (Y-axis). Points below and to the right of the red dotted line reflecting an improvement of our method over the consensus, based on the normalized, best-match Hamming distance. Each dot represents an individual SCG reflecting a putative genotype found in the analysed samples. SCGs from all species found in either of the focal samples are represented, and marker colors reflect the metagenotype entropy of that species in the relevant focal sample, a proxy for the potential strain diversity represented. Axes are on a “symmetric” log scale, with linear placement of values below 10–2. (B) A non-metric multidimensional scaling ordination of 68 SCGs and inferred genotypes for one species, S. thermophilus, with notably high strain diversity in one of the two focal samples. Circles represent SCGs, are colored by their assignment to one of four identified clusters, and larger markers indicate greater horizontal coverage. Triangles represent StrainFacts genotypes inferred to be at greater than 1% relative abundance, and larger markers reflect a higher inferred relative abundance. The red cross represents the consensus metagenotype of the focal sample.
Of the 75 species represented in our SCG dataset, one stood out for having numerous SCGs while reflecting a remarkably high degree of strain heterogeneity. Among 68 high-quality SCGs for S. thermophilus, cluster analysis identified four distinct types (here referred to as Clusters A—D), accounting for 48, 7, 6, and one SCGs, respectively (Figure 3B). Independently, StrainFacts inferred four strains in the metagenomic data from the same stool sample, (Strain 1—4) with 57, 32, and 7, and 3% relative abundance, respectively. We explored the concordance between clusters and StrainFacts inferences by assigning a best-match Hamming distance genotype among the inferred strains to each SCG (Table 2). For SCGs in three of the four clusters there was a low median distance to StrainFacts genotypes as well as a perfect 1-to-1 correspondence between strains and clusters. While this genotype concordance was broken for SCGs in cluster B, strain 4 was also inferred to be at the lowest relative abundance, suggesting that there may have been too little information encoded in the metagenotype data to accurately reconstruct that strain’s genotype. While SCG counts and inferred strain fractions do not match perfectly in this sample, this may be due to large differences between SCG and metagenomic sequencing technologies that could result in differentially biased sampling of strains. The SCG cluster with the largest membership was, however, matched with the strain inferred to be at the highest relative abundance. Our findings for S. thermophilus show that StrainFacts’ estimates of genotypes and relative abundances are remarkably accurate for samples with high strain heterogeneity, despite the challenges presented by real biological samples and low abundance strains.
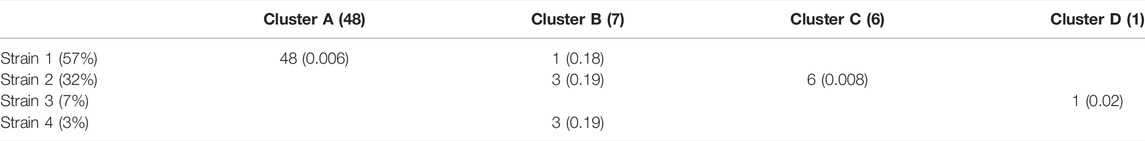
TABLE 2. Concordance among SCGs of cluster assignments and the closest-match StrainFacts inferred genotype, among the four strains inferred to be at greater than 1% relative abundance in the analysed sample. The total number of SCGs in each cluster and the relative abundance of each inferred strain are indicated in parentheses in the column and row labels, respectively. Numbers in each cell indicate the number of SCGs at that intersection and values in parentheses indicate the median normalized Hamming distance of those SCGs to the inferred strain genotype.
Having established the accuracy and scalability of StrainFacts, we applied it to a corpus of metagenotype data derived from 20,550 metagenomes across 44 studies, covering a large fraction of all publicly available human-associated microbial metagenomes (Shi et al., 2021). We performed strain inference on GT-Pro metagenotypes for four species: Escherichia coli, Agathobacter rectalis, Methanobrevibacter smithii, and CAG-279 sp1. E. coli and A. rectalis are two highly prevalent and well studied bacterial inhabitants of the human gut microbiome, and M. smithii is the most prevalent and abundant archaeon detected in the human gut (Scanlan et al., 2008). CAG-279, on the other hand, is an unnamed and little-studied genus and a member of the family Muribaculaceae. This family is common in mice (Lagkouvardos et al., 2019), but to our knowledge does not have representatives cultured from human samples.
For each species, we compared strains inferred by StrainFacts to those represented in the GT-Pro reference database, which is derived from the UHGG (Almeida et al., 2021). In order to standardize comparisons, we dereplicated inferred and reference strains at a 0.05 genotype distance threshold. Interestingly, dereplication had a negligible effect on StrainFacts results, reducing the number of E. coli strains by just 4 (to 119) with no reduction for the three other species. This suggests that the diversity regularization built into the StrainFacts model is sufficient to collapse closely related strains as part of inference.
As GT-Pro only tallies alleles at a fixed subset of SNPs, the relationship between genotype distances and ANI is not fixed. In order to anchor our results to this widely-used measure of genome similarity, we compared the genotype distance to genome-wide ANI for all pairs of genomes in the GT-Pro reference database for all four species. We find that the fraction of positions differing genome wide (calculated as 1—ANI) was nearly proportional to the fraction of genotyped positions differing, but with a different constant of proportionality for each species: E. coli (0.0776, uncentered R2 = 0.994), A. rectalis (0.1069, R2 = 0.990), M. smithii (0.0393, R2 = 0.967), and CAG-279 (0.0595, R2 = 0.991). Additional details of this analysis can be found in Supplementary Figure S4.
E. coli, A. rectalis, and M. smithii all have many genome sequences in GT-Pro reference database, presenting an opportunity to contrast inferred against reference strains. In order to evaluate the concordance between the two (Table 3 and Figure 4), we co-clustered all dereplicated strains (both reference and inferred) at a 0.15 normalized Hamming distance threshold—note, crucially, that this distance reflects a much smaller full-genome dissimilarity, as it is based only on genome positions with polymorphism across metagenomes, ignoring conserved positions.
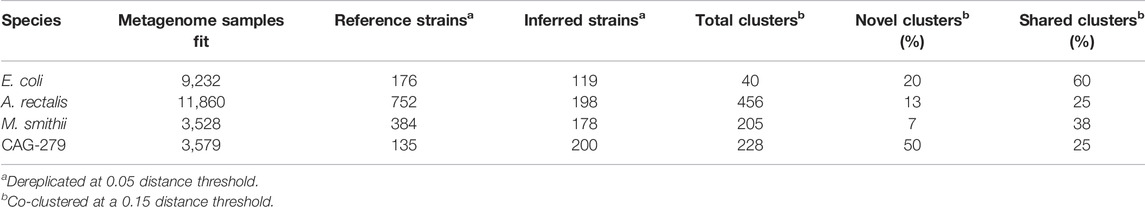
TABLE 3. Dereplication and co-clustering of strains inferred from metagenomes or from a reference database.

FIGURE 4. Concordance between reference and StrainFacts inferred strain genotypes for four prevalent species in the human gut microbiome. Heatmap rows represent consensus genotypes from co-clustering of reference and inferred strains and columns are 3,500 randomly sampled SNP sites (grey: reference and black: alternative allele). Colors to the left of the heatmap indicate clusters with only reference strains (dark purple), only inferred strains (yellow), or both (teal). Rows are ordered by hierarchical clustering built on distances between consensus genotypes and columns are ordered arbitrarily to highlight correlations between SNPs.
For E. coli, we identified 40 strain clusters with 93% of inferred strains and 94% of references falling into clusters containing strains from both sources (“shared” clusters), which is significantly more overlap than expected after random shuffling of cluster labels (p = 0.002 by permutation test). While most metagenome-inferred genotypes are similar to those found in genome reference databases, we observed some clusters composed only of StrainFacts strains, representing novel lineages. However, these strains are no more common than after random permutation (p = 0.81), matching our expectations for this well-studied species.
We next asked if these trends hold for the other species. While A. rectalis had a much greater number of clusters (456), 69% of inferred strains and 45% of reference strains are nonetheless found to be in shared clusters, significantly more than would be expected with random shuffling of cluster labels (p = 0.002 by permutation test). Correspondingly, we do not find evidence for enrichment of inferred strains in novel clusters (p = 0.71). We find similar results for M. smithii and CAG-279—the fraction of strains in shared clusters is significantly greater than after random reassignment (p < 0.001 for both), and there is no evidence for enrichment of inferred strains in novel clusters (p = 1.0 for both). Overall, the concordance between reference and inferred strains supports not only the credibility of StrainFacts’ estimates, but also suggests that our de novo inferences capture a substantial fraction of previously documented strain diversity, even in well studied species.
Going beyond the extensive overlap of strains with reference genomes and StrainFacts inferences, we examined clusters in which references are absent or relatively rare. Visualizing a dendrogram of consensus genotypes from co-clustered strains (Figure 4) we observe some sections of the A. rectalis dendrogram with many novel strains. Similarly, for CAG-279, the sheer number of inferred strains relative to genomes in reference databases means that fully half of all genotype clusters are entirely novel, emphasizing the power of StrainFacts inferences in understudied species. Future work will be needed to determine if these represent new subspecies currently missing from reference databases.
Large metagenomic collections allow us to examine intraspecific microbial diversity at a global scale and among dozens of studies. Towards this end, we identified the most abundant StrainFacts strain of E. coli, A. rectalis, M. smithii, and CAG-279 in stool samples across 34 independent studies. Across all four species, we observe some strains that are distributed globally as well as others that appear specific to one country of origin (Figure 5, Supplementary Figure S5). For example, among the 198 dereplicated, inferred strains of A. rectalis, 75 were found as the dominant strain (i.e. highest relative abundance) in samples collected on three or more continents. While this makes it challenging to consistently discern where a sample was collected based on its dominant strain of a given species, we nonetheless find that studies with samples collected in the United States of America form a distinct cluster, as do those from China, and the two are easily distinguished from one another and from most other studies conducted across Europe and North America (Figure 5). Our observation of a distinct group of A. rectalis strains enriched in samples from China is consistent with previous results (Scholz et al., 2016; Costea PI. et al., 2017; Truong et al., 2017).
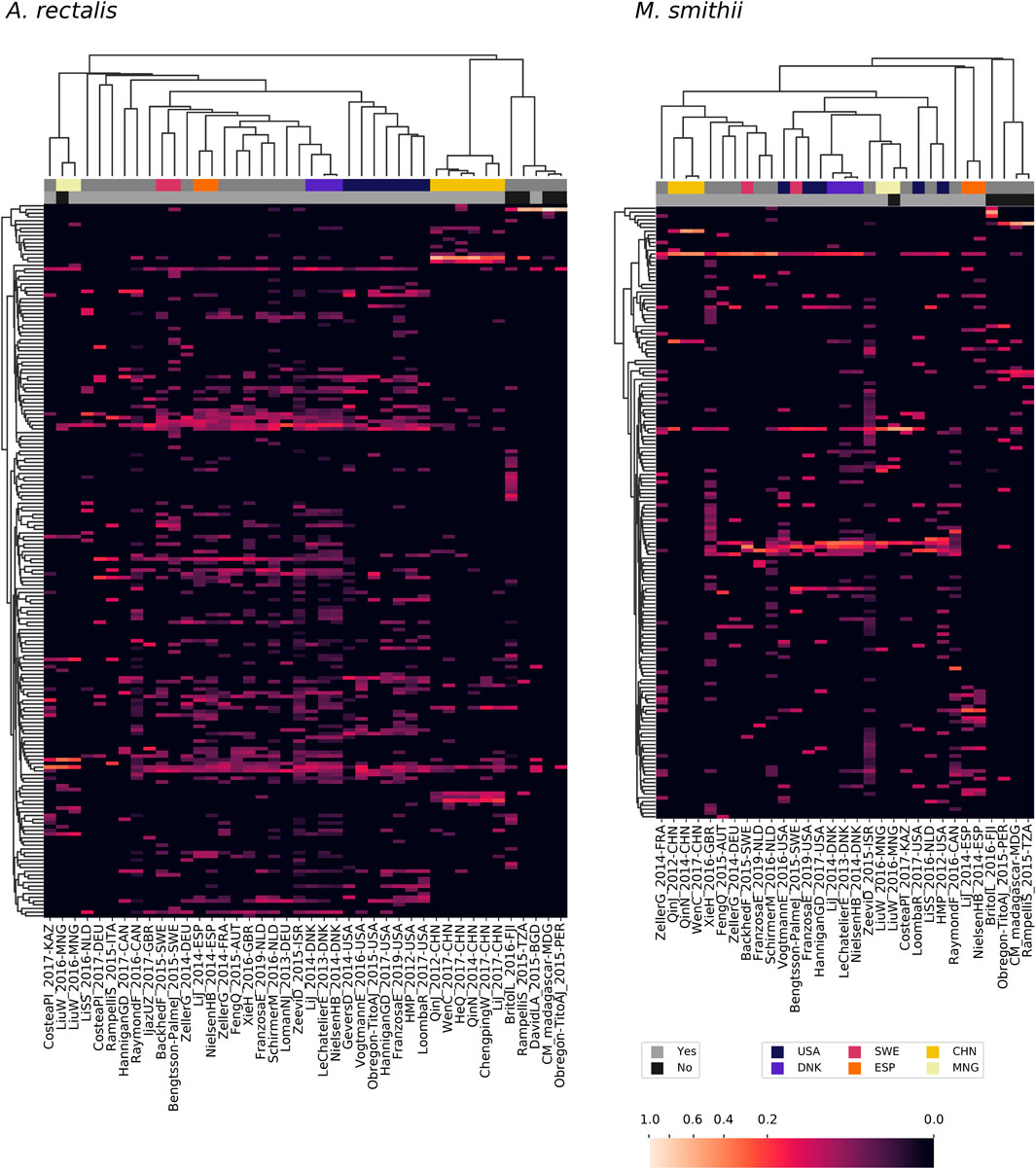
FIGURE 5. Patterns in strain dominance according to geography and lifestyle across thousands of publicly available metagenomes in dozens of independent studies for two common members of the human gut microbiome. Columns represent collections of samples from individual studies and are further segmented by country and lifestyle (westernized or not). Rows represent strains inferred by StrainFacts. Cell colors reflect the fraction of samples in that study segment with that strain as the most abundant member. Study segments are omitted if they include fewer than 10 samples. Row ordering and the associated dendrogram reflect strain genotype distances, while the dendrogram for columns is based on their cosine similarity. Studies with samples collected in several countries with notable clustering for one or more species are highlighted with colors above the heatmap. Additionally, studies from westernized populations are indicated. Both a study identifier and the ISO 3166-ISO country-code are included in the column labels.
These general trends hold across the other three species. In M. smithii, independent studies in the same country often share very similar strain dominance patterns (e.g. see clustering of studies performed in each of China, Mongolia, Denmark, and Spain in Figure 5). In E. coli, while many strains appear to be distributed globally, independent studies from China still cluster together based on patterns in strain dominance (see Supplementary Figure S5). Notably, in CAG-279, studies with individuals in westernized societies do not cluster separately from the five other studies, suggesting that host lifestyle is not highly correlated with specific strains of this species. The variety of dominance patterns across the four species described here suggest that different mechanisms may drive intraspecific biogeography depending on the biology and natural history of the species. As the coverage of diverse microbiomes grows, StrainFacts will enable future studies disentangling the contributions of lifestyle, dispersal limitation and other factors in the global distribution of strains.
Studying the population genetics of host-associated microbes has the potential to elucidate processes of transmission, diversification, and selection with implications for human health and perhaps even our understanding of human origins (Linz et al., 2007; Garud and Pollard, 2019). To demonstrate the application of StrainFacts to the study of microbial evolution, we examined patterns in pairwise LD, here calculated as the squared Pearson correlation coefficient (r2). This statistic can inform understanding of recombination rates in microbial populations (Vos, 2009; Garud et al., 2019). Genome-wide, LD, summarized as the 90th percentile r2 (LD90, Vos et al., 2017), was substantially higher for E. coli (0.24) than A. rectalis (0.04), M. smithii (0.11), or CAG-279 (0.04), perhaps suggesting greater population structure in the species and less panmictic recombination.
We estimated LD distance-decay curves for SNPs, using a single, high-quality reference genome for each species to obtain estimates of pairwise distance between SNP sites. For all four species, adjacent SNPs were much more tightly linked, with an LD90 of > 0.999. LD was still dramatically above background at 50 bases of separation, and fell rapidly with increasing distance (Figure 6). The speed of this decay was different between species, which we characterized with the LD½,90: the distance at which the LD90 was less than 50% of the value for adjacent SNPs (Vos et al., 2017). M. smithii exhibited by far the slowest decay, with a LD½,90 of 520 bases, followed by E. coli at 93 bases, CAG-279 at 66, and A. rectalis at just 28 bases. This variation in decay profiles may reflect major differences in recombination rates across populations.
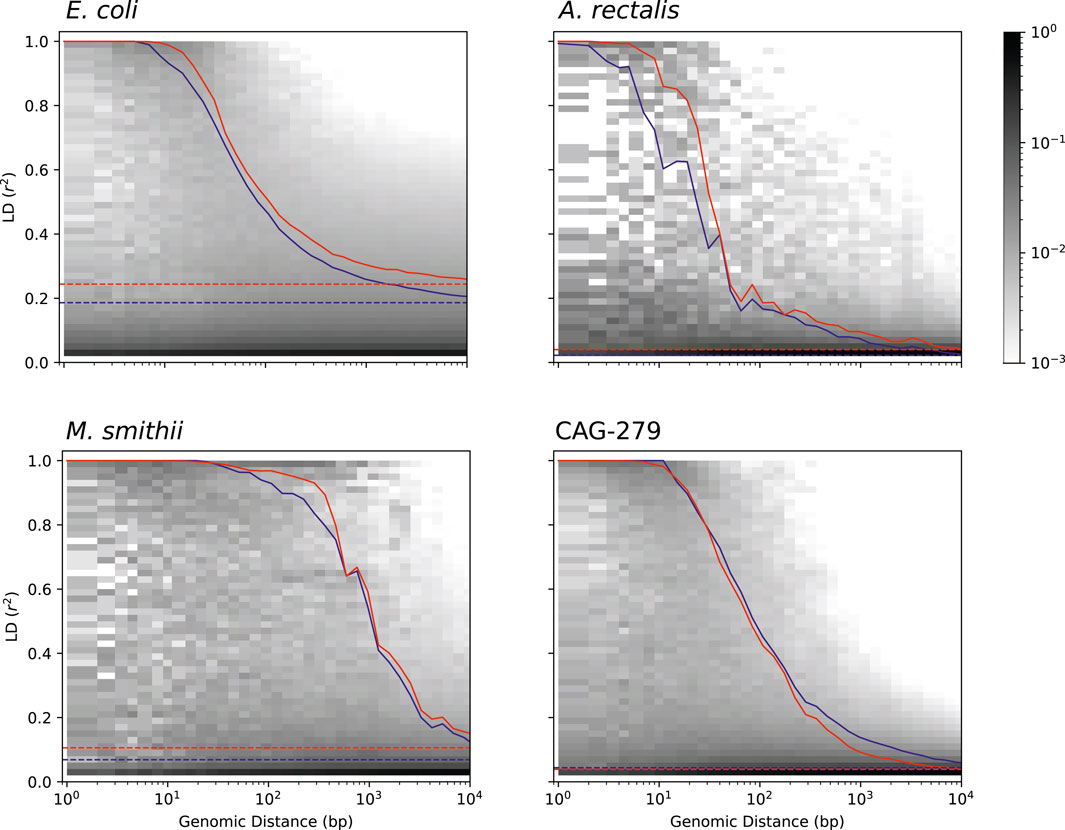
FIGURE 6. Pairwise LD across genomic distance estimated from inferred genotypes for four species. LD was calculated as r2 and genomic distance between polymorphic loci is based on distances in a single, representative genome. The distribution of SNP pairs in each distance window is shown as a histogram with darker colors reflecting a larger fraction of the pairs in that LD bin, and the LD90 for pairs at each distance is shown for inferred strains (red), along with an identical analysis on strains in the reference database (blue). Genome-wide LD90 (dashed lines) is also indicated.
To validate our findings, we ran identical analyses with dereplicated genotypes from genomes in the GT-Pro reference database. Estimates of both LD and the distance-decay relationship are very similar between inferred and reference strains, reinforcing the value of genotypes inferred from metagenomes for microbial population genetics. Interestingly, for three of the four species (E. coli, A. rectalis, and M. smithii), LD estimates from StrainFacts strains were significantly higher than from references (p < 1e-10 for all three by Wilcoxon test), while CAG-279 exhibited a trend towards the reverse (p = 0.85). It is not clear what might cause these quantitative discrepancies, but they could reflect differences in the set of strains in each dataset. Future studies expanding this analysis to additional species will identify patterns in recombination rates across broader microbial diversity.
Here we have described StrainFacts, a novel tool for strain inference in metagenomic data. StrainFacts models metagenotype data using a fuzzy-genotype approximation, allowing us to estimate both the relative abundance of strains across samples as well as their genotypes. To accelerate analysis compared to the current state-of-the-art, we harness the differentiability of our model to apply modern, gradient-based optimization and GPU-parallelization. Consequently, StrainFacts can scale to tens-of-thousands of samples while inferring genotypes for hundreds of strains. On simulated benchmarks, we show that StrainFacts has comparable accuracy to Strain Finder, and we validate strain inferences in vivo against genotypes observed by single-cell genomics. Finally, we apply StrainFacts to a database of tens of thousands of metagenomes from the human microbiome to estimate strains de novo, allowing us to characterize strain diversity, biogeography, and population genetics, without the need for cultured isolates.
Beyond Strain Finder, other alternatives exist for strain inference in metagenomic data. While we do not directly compare to DESMAN, runtimes of several hours have been reported for that tool on substantially smaller simulated datasets (Quince et al., 2017), and hence we believe that StrainFacts is likely the most scalable implementation of the metagenotype deconvolution approach. Still other methods apply regularized regression (e.g. Lasso Albanese and Donati, 2017) to decompose metagenotypes—essentially solving the abundance half of the deconvolution problem but not the genotypes half—or look for previously determined strain signatures (e.g. k-mers Panyukov et al., 2020) based on known strain genotypes. However, both of these require an a priori database of the genotypes that might be present in a sample. An expanding database of strain references can make these approaches increasingly useful, and StrainFacts can help to build this reference.
Several studies avoid deconvolution by directly examining allele frequencies inferred from metagenotypes. While consensus (Truong et al., 2017; Zolfo et al., 2017) or phasing (Garud et al., 2019) approaches can accurately recover genotypes in some cases, their use is limited to low complexity datasets, with sufficient sequencing depth and low strain heterogeneity. Likewise, measuring the dissimilarity of metagenotypes among pairwise samples indicates shared strains (Podlesny and Fricke, 2020), but this approach risks confounding strain mixing with genotype similarity. Finally, assembly (Li et al., 2019) and read-based methods (Cleary et al., 2015) for disentangling strains are most applicable when multiple SNPs can be found in each sequencing read. With ongoing advancements in long-read (Vicedomini et al., 2021) and read-cloud sequencing (Kuleshov et al., 2016; Kang et al., 2018), these approaches will become increasingly feasible. Thus, StrainFacts occupies the same analysis niche as Strain Finder and DESMAN, and it expands upon these reliable approaches by providing a scalable model fitting procedure.
Fuzzy genotypes enable more computationally efficient inference by eliminating the need for discrete optimization. Specifically, we used well-tested and optimized gradient descent algorithms implemented in PyTorch for parameter estimation. In addition, fuzzy genotypes may be more robust to deviations from model assumptions. For instance, an intermediate genotype could be a satisfactory approximation when gene duplications or deletions are present in some strains. While we do not explore the possibility here, fuzzy genotypes may provide a heuristic for capturing uncertainty in strain genotypes. Future work could consider propagating intermediate genotype values instead of discretizing them.
StrainFacts builds on recent advances in metagenotyping, in particular our analyses harnessed GT-Pro (Shi et al., 2021) to greatly accelerate SNP counting in metagenomic reads. While we leave a comparison of StrainFacts performance on the outputs of other metagenotypers to future work, StrainFacts itself is agnostic to the source of input data. It would be straightforward to extend StrainFacts to operate on loci with more than two alleles or to use metagenotypes from a tool other than GT-Pro. It would also be interesting to extend StrainFacts to use SNPs outside the core genome that vary in their presence across strains.
Combined with the explosive growth in publicly available metagenomic data and the development of rapid metagenotyping tools, StrainFacts enables the de novo exploration of intraspecific microbial diversity at a global scale and on well-powered cohorts with thousands of samples. Future applications could examine intraspecific associations with disease, track the history of recombination between microbial subpopulations, and measure the global transmission of host-associated microbes across human populations.
Metagenomic sequencing data from the FMT study are available through the SRA under BioProject PRJNA737472, The two single-cell genomics experiments are also under that project with accessions SRR18748374 and SRR18748375. Publicly available metagenomes are available under various other accessions described in (Shi et al., 2021). Strain genotypes from the GT-Pro reference database are publicly available at https://fileshare.czbiohub.org/s/waXQzQ9PRZPwTdk. All other code and metadata needed to reproduce these results are available at https://doi.org/10.5281/zenodo.5942586.
The studies involving human participants were reviewed and approved by the UCSF Committee on Human Research. The patients/participants provided their written informed consent to participate in this study.
BS: conceptualization, data curation, formal analysis, methodology, software, visualization, writing—original draft, writing—review and editing. XL: investigation, data curation, methodology, writing—original draft, writing—review and editing. ZS: data curation, methodology, writing—review and editing. AA: funding acquisition, supervision. KP: conceptualization, writing—review and editing, funding acquisition, supervision.
BS was supported by the National Institutes of Health grant number 5T32DK007007 and from the UCSF Initiative for Digital Transformation in Computational Biology & Health. BS, ZS, and KP were supported by the National Science Foundation grant number 1563159 and by the Gladstone Institutes. BS, XL, ZS, AA, and KP were supported by the funding from Chan Zuckerberg Biohub.
KP is on the scientific advisory board of Phylagen.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Barbara Engelhardt provided valuable feedback on this project.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2022.867386/full#supplementary-material
Albanese, D., and Donati, C. (2017). Strain Profiling and Epidemiology of Bacterial Species from Metagenomic Sequencing. Nat. Commun. 8, 2260. doi:10.1038/s41467-017-02209-5
Almeida, A., Nayfach, S., Boland, M., Strozzi, F., Beracochea, M., Shi, Z. J., et al. (2021). A Unified Catalog of 204,938 Reference Genomes from the Human Gut Microbiome. Nat. Biotechnol. 39, 105–114. doi:10.1038/s41587-020-0603-3
Beghini, F., McIver, L. J., Blanco-Míguez, A., Dubois, L., Asnicar, F., Maharjan, S., et al. (2021). Integrating Taxonomic, Functional, and Strain-Level Profiling of Diverse Microbial Communities with bioBakery 3. eLife 10, e65088. doi:10.7554/eLife.65088
Bingham, E., Chen, J. P., Jankowiak, M., Obermeyer, F., Pradhan, N., Karaletsos, T., et al. (2019). Pyro: Deep Universal Probabilistic Programming. J. Mach. Learn. Res. 20, 1–6. Available at: http://jmlr.org/papers/v20/18-403.html. (Accessed April 8, 2021). doi:10.48550/arXiv.1810.09538
Case, R. J., Boucher, Y., Dahllöf, I., Holmström, C., Doolittle, W. F., and Kjelleberg, S. (2007). Use of 16S rRNA and rpoB Genes as Molecular Markers for Microbial Ecology Studies. Appl. Environ. Microbiol. 73, 278. doi:10.1128/AEM.01177-06
Chu, N. D., Crothers, J. W., Nguyen, L. T. T., Kearney, S. M., Smith, M. B., Kassam, Z., et al. (2021). Dynamic Colonization of Microbes and Their Functions after Fecal Microbiota Transplantation for Inflammatory Bowel Disease. mBio 12, e0097521. doi:10.1128/mBio.00975-21
Cleary, B., Brito, I. L., Huang, K., Gevers, D., Shea, T., Young, S., et al. (2015). Detection of Low-Abundance Bacterial Strains in Metagenomic Datasets by Eigengenome Partitioning. Nat. Biotechnol. 33, 1053–1060. doi:10.1038/nbt.3329
Costea, P. I., Coelho, L. P., Sunagawa, S., Munch, R., Huerta-Cepas, J., Forslund, K., et al. (2017a). Subspecies in the Global Human Gut Microbiome. Mol. Syst. Biol. 13, 960. doi:10.15252/msb.20177589
Costea, P. I., Hildebrand, F., Arumugam, M., Bäckhed, F., Blaser, M. J., Bushman, F. D., et al. (2017b). Enterotypes in the Landscape of Gut Microbial Community Composition. Nat. Microbiol. 3, 8. doi:10.1038/s41564-017-0072-8
Garud, N. R., Good, B. H., Hallatschek, O., and Pollard, K. S. (2019). Evolutionary Dynamics of Bacteria in the Gut Microbiome within and across Hosts. Plos Biol. 17, e3000102. doi:10.1371/journal.pbio.3000102
Garud, N. R., and Pollard, K. S. (2019). Population Genetics in the Human Microbiome. Trends Genet. 36, 53. doi:10.1016/j.tig.2019.10.010
Haiser, H. J., Seim, K. L., Balskus, E. P., and Turnbaugh, P. J. (2014). Mechanistic Insight into Digoxin Inactivation by Eggerthella Lenta Augments Our Understanding of its Pharmacokinetics. Gut Microbes 5, 233–238. doi:10.4161/gmic.27915
Kang, J. B., Siranosian, B., Moss, E., Andermann, T., and Bhatt, A. (2018). Read Cloud Sequencing Elucidates Microbiome Dynamics in a Hematopoietic Cell Transplant Patient. IEEE Int. Conf. Bioinforma. Biomed. BIBM. 2018, 234. doi:10.1109/bibm.2018.8621297
Kuleshov, V., Snyder, M. P., and Batzoglou, S. (2016). Genome Assembly from Synthetic Long Read Clouds. Bioinformatics 32, i216–i224. doi:10.1093/bioinformatics/btw267
Kurtzer, G. M., Sochat, V., and Bauer, M. W. (2017). Singularity: Scientific Containers for Mobility of Compute. PLOS ONE 12, e0177459. doi:10.1371/journal.pone.0177459
Lagkouvardos, I., Lesker, T. R., Hitch, T. C. A., Gálvez, E. J. C., Smit, N., Neuhaus, K., et al. (2019). Sequence and Cultivation Study of Muribaculaceae Reveals Novel Species, Host Preference, and Functional Potential of This yet Undescribed Family. Microbiome 7, 28. doi:10.1186/s40168-019-0637-2
Li, S. S., Zhu, A., Benes, V., Costea, P. I., Hercog, R., Hildebrand, F., et al. (2016). Durable Coexistence of Donor and Recipient Strains after Fecal Microbiota Transplantation. Science 352, 586–589. doi:10.1126/science.aad8852
Li, X., Saadat, S., Hu, H., and Li, X. (2019). BHap: A Novel Approach for Bacterial Haplotype Reconstruction. Bioinformatics 35, 4624–4631. doi:10.1093/bioinformatics/btz280
Li, X., Hu, H., and Li, X. (2021). MixtureS: A Novel Tool for Bacterial Strain Reconstruction from Reads. Bioinformatics 37, 575. doi:10.1093/bioinformatics/btaa728
Linz, B., Balloux, F., Moodley, Y., Manica, A., Liu, H., Roumagnac, P., et al. (2007). An African Origin for the Intimate Association between Humans and Helicobacter pylori. Nature 445, 915–918. doi:10.1038/nature05562
Loman, N. J., Constantinidou, C., Christner, M., Rohde, H., Chan, J. Z., Quick, J., et al. (2013). A Culture-independent Sequence-Based Metagenomics Approach to the Investigation of an Outbreak of Shiga-Toxigenic Escherichia coli O104:H4. JAMA 309, 1502–1510. doi:10.1001/jama.2013.3231
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and Qualitative β Diversity Measures Lead to Different Insights into Factors that Structure Microbial Communities. Appl. Environ. Microbiol. 73, 1576–1585. doi:10.1128/aem.01996-06
Luo, C., Knight, R., Siljander, H., Knip, M., Xavier, R. J., and Gevers, D. (2015). ConStrains Identifies Microbial Strains in Metagenomic Datasets. Nat. Biotechnol. 33, 1045–1052. doi:10.1038/nbt.3319
Mölder, F., Jablonski, K. P., Letcher, B., Hall, M. B., Tomkins-Tinch, C. H., Sochat, V., et al. (2021). Sustainable Data Analysis with Snakemake. F1000Research 10, 33. doi:10.12688/f1000research.29032.2
Monti, G. S., Mateu-Figueras, G., Pawlowsky-Glahn, V., and Egozcue, J. J. (2011). “The Shifted-Scaled Dirichlet Distribution in the Simplex,” in 4th International Workshop on Compositional Data Analysis, Sant Feliu de Guíxols, Girona Spain.
Nayfach, S., Rodriguez-Mueller, B., Garud, N., and Pollard, K. S. (2016). An Integrated Metagenomics Pipeline for Strain Profiling Reveals Novel Patterns of Bacterial Transmission and Biogeography. Genome Res. 26, 1612–1625. doi:10.1101/gr.201863.115
Nayfach, S., Roux, S., Seshadri, R., Udwary, D., Varghese, N., Schulz, F., et al. (2020). A Genomic Catalog of Earth’s Microbiomes. Nat. Biotechnol. 39, 499. doi:10.1038/s41587-020-0718-6
O'Brien, J. D., Didelot, X., Iqbal, Z., Amenga-Etego, L., Ahiska, B., and Falush, D. (2014). A Bayesian Approach to Inferring the Phylogenetic Structure of Communities from Metagenomic Data. Genetics 197, 925–937. doi:10.1534/genetics.114.161299
Olm, M. R., Crits-Christoph, A., Bouma-Gregson, K., Firek, B. A., Morowitz, M. J., and Banfield, J. F. (2021). inStrain Profiles Population Microdiversity from Metagenomic Data and Sensitively Detects Shared Microbial Strains. Nat. Biotechnol. 39, 727–736. doi:10.1038/s41587-020-00797-0
Ostrowski, M. P., La Rosa, S. L., Kunath, B. J., Robertson, A., Pereira, G., Hagen, L. H., et al. (2022). Mechanistic Insights Into Consumption of the Food Additive Xanthan Gum by the Human Gut Microbiota. Nat. Microbiol. 7 (4), 556–569.
Panyukov, V. V., Kiselev, S. S., and Ozoline, O. N. (2020). Unique K-Mers as Strain-specific Barcodes for Phylogenetic Analysis and Natural Microbiome Profiling. Int. J. Mol. Sci. 21, 944. doi:10.3390/ijms21030944
Paszke, A., Gross, S., Massa, F., Lerer, A., Bradbury, J., Chanan, G., et al. (2019). “PyTorch: An Imperative Style, High-Performance Deep Learning Library,” in Advances In Neural Information Processing Systems(ancouver, Canada: Curran Associates, Inc.). Available at: https://proceedings.neurips.cc/paper/2019/hash/bdbca288fee7f92f2bfa9f7012727740-Abstract.html (Accessed January 30, 2022).
Patrick, S., Blakely, G. W., Houston, S., Moore, J., Abratt, V. R., Bertalan, M., et al. (2010). Twenty-eight Divergent Polysaccharide Loci Specifying within- and Amongst-Strain Capsule Diversity in Three Strains of Bacteroides Fragilis. Microbiology (Reading) 156, 3255–3269. doi:10.1099/mic.0.042978-0
Podlesny, D., and Fricke, W. F. (2020). Microbial Strain Engraftment, Persistence and Replacement after Fecal Microbiota Transplantation. medRxiv 2020, 20203638. doi:10.1101/2020.09.29.20203638
Quince, C., Delmont, T. O., Raguideau, S., Alneberg, J., Darling, A. E., Collins, G., et al. (2017). DESMAN: A New Tool for De Novo Extraction of Strains from Metagenomes. Genome Biol. 18, 181–222. doi:10.1186/s13059-017-1309-9
Scanlan, P. D., Shanahan, F., and Marchesi, J. R. (2008). Human Methanogen Diversity and Incidence in Healthy and Diseased Colonic Groups Using mcrA Gene Analysis. BMC Microbiol. 8, 79. doi:10.1186/1471-2180-8-79
Schmidt, M. N., Winther, O., and Hansen, L. K. (2009). “Bayesian Non-negative Matrix Factorization,” in Independent Component Analysis And Signal Separation Lecture Notes in Computer Science. Editors T. Adali, C. Jutten, J. M. T. Romano, and A. K. Barros (Berlin, Heidelberg: Springer), 540–547. doi:10.1007/978-3-642-00599-2_68
Scholz, M., Ward, D. V., Pasolli, E., Tolio, T., Zolfo, M., Asnicar, F., et al. (2016). Strain-level Microbial Epidemiology and Population Genomics from Shotgun Metagenomics. Nat. Methods 13, 435–438. doi:10.1038/nmeth.3802
Shi, Z. J., Dimitrov, B., Zhao, C., Nayfach, S., and Pollard, K. S. (2021). Fast and Accurate Metagenotyping of the Human Gut Microbiome with GT-Pro. Nat. Biotechnol. 1–10. doi:10.1038/s41587-021-01102-3
Shoemaker, N. B., Vlamakis, H., Hayes, K., and Salyers, A. A. (2001). Evidence for Extensive Resistance Gene Transfer Among Bacteroides Spp. And Among Bacteroides and Other Genera in the Human Colon. Appl. Environ. Microbiol. 67, 561–568. doi:10.1128/AEM.67.2.561-568.2001
Smillie, C. S., Sauk, J., Gevers, D., Friedman, J., Sung, J., Youngster, I., et al. (2018). Strain Tracking Reveals the Determinants of Bacterial Engraftment in the Human Gut Following Fecal Microbiota Transplantation. Cell Host Microbe 23, 229–e5. doi:10.1016/j.chom.2018.01.003
Smith, B. J., Piceno, Y., Zydek, M., Zhang, B., Syriani, L. A., Terdiman, J. P., et al. (2022). Strain-Resolved Analysis in a Randomized Trial of Antibiotic Pretreatment and Maintenance Dose Delivery Mode with Fecal Microbiota Transplant for Ulcerative Colitis. Sci. Rep. 12 (1), 5517. doi:10.1038/s41598-022-09307-5
Truong, D. T., Tett, A., Pasolli, E., Huttenhower, C., and Segata, N. (2017). Microbial Strain-Level Population Structure and Genetic Diversity from Metagenomes. Genome Res. 27, 626–638. doi:10.1101/gr.216242.116
Vicedomini, R., Quince, C., Darling, A. E., and Chikhi, R. (2021). Strainberry: Automated Strain Separation in Low-Complexity Metagenomes Using Long Reads. Nat. Commun. 12, 4485. doi:10.1038/s41467-021-24515-9
Vos, M. (2009). Why Do Bacteria Engage in Homologous Recombination? Trends Microbiol. 17, 226–232. doi:10.1016/j.tim.2009.03.001
Vos, P. G., Paulo, M. J., Voorrips, R. E., Visser, R. G., van Eck, H. J., and van Eeuwijk, F. A. (2017). Evaluation of LD Decay and Various LD-Decay Estimators in Simulated and SNP-Array Data of Tetraploid Potato. Theor. Appl. Genet. 130, 123–135. doi:10.1007/s00122-016-2798-8
Watson, A. R., Füssel, J., Veseli, I., DeLongchamp, J. Z., Silva, M., Trigodet, F., et al. (2021). Adaptive Ecological Processes and Metabolic independence Drive Microbial Colonization and Resilience in the Human Gut. bioRxiv. doi:10.1101/2021.03.02.433653
Yan, Y., Nguyen, L. H., Franzosa, E. A., and Huttenhower, C. (2020). Strain-level Epidemiology of Microbial Communities and the Human Microbiome. Genome Med. 12, 71. doi:10.1186/s13073-020-00765-y
Keywords: metagenomics, strains, microbiome, biogeography, population genetics, model-based inference
Citation: Smith BJ, Li X, Shi ZJ, Abate A and Pollard KS (2022) Scalable Microbial Strain Inference in Metagenomic Data Using StrainFacts. Front. Bioinform. 2:867386. doi: 10.3389/fbinf.2022.867386
Received: 01 February 2022; Accepted: 14 April 2022;
Published: 16 May 2022.
Edited by:
Joao Carlos Setubal, University of São Paulo, BrazilReviewed by:
Guido Zampieri, Università degli Studi di Padova, ItalyCopyright © 2022 Smith, Li, Shi, Abate and Pollard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine S. Pollard, a2F0aGVyaW5lLnBvbGxhcmRAZ2xhZHN0b25lLnVjc2YuZWR1
†ORCID: Byron J. Smith, orcid.org/0000-0002-0182-404X; Adam Abate, orcid.org/0000-0001-9614-4831; Katherine S. Pollard, orcid.org/0000-0002-9870-6196
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.