
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioinform., 13 May 2022
Sec. Genomic Analysis
Volume 2 - 2022 | https://doi.org/10.3389/fbinf.2022.867111
This article is part of the Research TopicComputational Methods for Microbiome Analysis, volume 2View all 17 articles
 Grigorii Sukhorukov1,2*
Grigorii Sukhorukov1,2* Maryam Khalili3Olivier Gascuel4Thierry Candresse3Armelle Marais-Colombel3
Maryam Khalili3Olivier Gascuel4Thierry Candresse3Armelle Marais-Colombel3 Macha Nikolski1,2*
Macha Nikolski1,2*High-throughput sequencing has provided the capacity of broad virus detection for both known and unknown viruses in a variety of hosts and habitats. It has been successfully applied for novel virus discovery in many agricultural crops, leading to the current drive to apply this technology routinely for plant health diagnostics. For this, efficient and precise methods for sequencing-based virus detection and discovery are essential. However, both existing alignment-based methods relying on reference databases and even more recent machine learning approaches are not efficient enough in detecting unknown viruses in RNAseq datasets of plant viromes. We present VirHunter, a deep learning convolutional neural network approach, to detect novel and known viruses in assemblies of sequencing datasets. While our method is generally applicable to a variety of viruses, here, we trained and evaluated it specifically for RNA viruses by reinforcing the coding sequences’ content in the training dataset. Trained on the NCBI plant viruses data for three different host species (peach, grapevine, and sugar beet), VirHunter outperformed the state-of-the-art method, DeepVirFinder, for the detection of novel viruses, both in the synthetic leave-out setting and on the 12 newly acquired RNAseq datasets. Compared with the traditional tBLASTx approach, VirHunter has consistently exhibited better results in the majority of leave-out experiments. In conclusion, we have shown that VirHunter can be used to streamline the analyses of plant HTS-acquired viromes and is particularly well suited for the detection of novel viral contigs, in RNAseq datasets.
Study of viromes at an unprecedented scale has been enabled by the adoption of high-throughput sequencing (HTS) technologies and is now frequently undertaken across an increasing range of host species. In particular, sequencing of plant viromes has become quite common, partly due to its relevance to the agricultural sector. The acquired datasets help to elucidate important questions such as virus spread among host reservoirs and effects of agriculture on the ecosystems and their biodiversity as well as the identification of novel viruses in crops and natural environments (Lefeuvre et al., 2019). These developments are fast advancing our knowledge of viral diversity through the discovery of previously unknown viral species or variants and the identification of new hosts of known viruses (Roossinck et al., 2015; Massart et al., 2017). Following the classification proposed by Stobbe and Roossinck (2014), viruses identified in HTS datasets can be classified into three different groups as follows: 1) viruses that are already known to infect a given host; 2) novel viruses from a known family or known viruses that have not been found previously described to infect a given host; and finally 3) completely novel viruses that share little to no sequence similarity with known viruses already present in the databases.
Using an efficient virus detection method, including for the identification of novel viruses, is essential for efficient disease management. Standard diagnostic tests (ELISA assays and PCR-based assays) depend on specific antibodies or primers and thus require prior knowledge of the virus and of its phylogenetic neighbors. Precise identification of viruses is further complexified by the large diversity encountered in the majority of viral species which is linked to the high mutation rate of these agents. This is particularly true for plant viruses, the majority of which are RNA viruses whose mutation rate is very high (Jenkins et al., 2002). Moreover, the new variants emerging from genomic rearrangements or recombination events can also significantly differ from the parental viruses (Domingo 2010). Also, many of the plant viruses are multihost pathogens, and a single plant can be infected by multiple unrelated viral species (Roossinck, 1997). Such infections by multiple viruses represent an additional challenge for detection since the viral load of different pathogens can be very unequal (Martín and Elena, 2009). Moreover, in most cases, background contamination is currently unavoidable (Kleiner et al., 2015; Maree et al., 2018; Kutnjak et al., 2021). In this context, HTS combined with bioinformatics tools has been shown to be a valuable approach, both for detection of known viruses and for the discovery of novel ones (Maree et al., 2018; Villamor et al., 2019; Mehetre et al., 2021).
Viruses do not have a universal gene marker that could be used for their identification, contrary to the conserved regions of the 16S rRNA and ITS genes, commonly used to classify bacteria and fungi at the genus or species level (Mokili et al., 2012). Moreover, the abundance of viral genomic material in plant sequencing samples can be very low (Massart et al., 2019), due to the dominance of the host material. Hence, specific sample preparation to enrich plant RNA viral-specific sequences is an important step that makes the downstream detection of viruses by bioinformatics methods more reliable. They include approaches providing a high and targeted enrichment of viral sequences, such as the purification of viral double-stranded RNAs (dsRNAs) or that of virion-associated nucleic acids (VANAs) as well as less specific approaches generally affording lower enrichment, such as the sequencing of small interfering RNAs (siRNAs) or inclusion of a ribodepletion step prior to the sequencing of total cellular RNAs (Maree et al., 2018; Kutnjak et al., 2021). As already discussed in a range of reviews, each of these approaches have advantages and weaknesses. In particular, strategies providing high enrichment factors may improve detection sensitivity but often at the cost of introducing biases with the risk of compromising the detection of some particular viruses (Maree et al., 2018; Kutnjak et al., 2021). For example, dsRNA-based approaches are usually poor at detecting DNA viruses, while VANA-based ones may perform poorly for viruses with labile particles.
When interested in known viruses or potentially novel viruses but from a known family, bioinformatics methods that compare the sequenced reads to genomes in public databases are very efficient for virus detection and identification (Stobbe and Roossinck, 2014; Massart et al., 2019). Read-based analysis is thus particularly suited to study viral diversity of sequencing samples in terms of known viral species. Generalistic metagenome analysis tools such as, for example, Kaju (Menzel et al., 2016), Kraken 2 (Wood et al., 2019), and Centrifuge (Kim et al., 2016) show good performance in terms of sensitivity and precision in detection of present known viral species (De Vries et al., 2021).
For the discovery of novel viruses, use of de novo assembly to recover novel viral contigs from sequencing data is an essential step in order to overcome the incompleteness of virus reference databases, annotation errors and, importantly, the limited homology between novel viral sequences and reference genomes (Sutton et al., 2019). The assembly step is a staple of short-read sequencing studies, which are still the vast majority today (Maree et al., 2018; Kutnjak et al., 2021). It represents its own challenges, in particular, for very short reads such as those of siRNAs and for viral populations with multiple and microdiverse variants (Warwick-Dugdale et al., 2019), often leading to microdiversity-associated fragmentation and, sometimes, to chimeras in the resulting contigs (Martinez-Hernandez et al., 2017; Roux et al., 2017), which in turn affects the downstream analysis, including estimation of viral diversity and identification of novel viruses (Nayfach et al., 2021). Popular assembler choices are the generalistic de Bruijn graph assembly metaSPAdes (Nurk et al., 2017) and Trinity, for RNAseq (Grabherr et al., 2011).
Following the recent review (Kutnjak et al., 2021), the methods used to analyze assembled contigs can be grouped into three main categories: 1) alignment and mapping-based methods, 2) protein domain searches, and 3) k-mer-based approaches that can either rely on signatures or leverage machine learning. Among this large plethora of tools, alignment-based methods are widely adopted when working with assembled contigs since they provide a longer sequence for homology search against reference genomes using either BLAST (Altschul et al., 1990) and its derivatives or the amino acid alignment of protein-coding genes predicted from the assembled contigs using DIAMOND (Buchfink et al., 2015). Also, focusing the analysis on coding regions is particularly relevant for RNAseq data since the non-coding sequences of viruses are not highly represented in such samples, even if they can be well conserved in certain viral taxa. However, the main drawback of alignment- or mapping-based approaches lies on the fact that they are both computationally intensive and require expertise for filtering and interpreting the results. As for the generalistic k-mer signature approaches, they remain demanding in terms of memory and are best suited for diversity analysis tasks (Kutnjak et al., 2021).
The emergence of machine learning tools for contig-based analysis of virome sequencing data holds much promise to streamline the discovery of novel viruses in sequencing datasets by both avoiding the time-consuming sequence similarity analyses and modeling even highly divergent sequences. These methods build models based on sequences with known class labels such as “virus” and “host” and learn features that allow them to differentiate between the classes. VirFinder (Ren et al., 2017) and VirSorter2 (Guo et al., 2021) rely on classical machine learning, the former being based on a regularized logistic regression applied to the k-mer frequency matrix extracted from the sequence and the latter on a random forest model built from genomic features. Methods based on deep learning networks have also been proposed for virus detection, such as DeepVirFinder (Ren et al., 2020) and ViraMiner (Tampuu et al., 2019) that both rely on a combination of convolutional neural networks (CNNs) and dense neural networks, and VirNet (Abdelkareem et al., 2018) that relies on a long short-term memory (LSTM) architecture. These three deep learning methods were developed for identification of viral contigs in metagenomic samples and evaluated on bacterial and human metagenomes. However, DeepVirFinder has been recently successfully used in plant-related virome studies (Santos-Medellin et al., 2021).
In this work, we present VirHunter, a deep learning method that uses convolutional neural networks (CNNs), classifies previously assembled contigs to identify potential viral, host, and bacterial (contamination) sequences in RNAseq samples. The hybrid architecture of VirHunter combines a multi-network CNN-based module covering different k-mer sizes with a downstream random forest classifier module. We have trained VirHunter models for three different plant host species: peach, grapevine, and sugar beet. Importantly, we have shown that VirHunter is especially performant for the task of completely novel virus discovery by building 31 leave-out datasets, in which each viral family is excluded from the training dataset, and comparing the results with a standard BLAST-based solution on one side and a state-of-the-art deep learning method, DeepVirFinder, the other side. VirHunter not only systematically outperformed DeepVirFinder in terms of virus detection but also has considerably reduced the False Positive rate. Cross-evaluation has shown that host detection accuracy remained high and decreased slightly when test sequences originated from the plant species were further phylogenetically removed from that used to train the model. We have further evaluated the detection capacity of VirHunter on in silico mutated contigs sampled from the NCBI virus database and have shown that it decreased only slightly with a progressively increased mutation rate (e.g., True Positive rate of 0.898 for 20% mutation rate). Moreover, we generated 12 RNAseq datasets for a range of host species and have shown that VirHunter was not only able to uncover the viruses that were previously identified but also to streamline the analyses by considerably reducing the need for manual curation.
We downloaded all complete and incomplete viral sequences from the NCBI virus database for which the host’s taxonomic id belongs to Viridiplantae on 26/10/2021, which yielded 122,832 sequences. Plant sequences have been downloaded for Prunus persica (peach), Vitis vinifera (grapevine), Beta vulgaris (sugar beet), and Oryza sativa (rice) from the NCBI RefSeq genomes database. On one hand, they consist of the latest available assemblies, GCF_000346465.2, GCF_000003745.3, GCF_000511025.2, and GCF_001433935.1 for peach, grapevine, sugar beet, and rice, respectively, and of the coding region sequences (CDSs), on the other hand. In the absence of the plastid sequence in the reference assembly of the sugar beet, we used the separately available sugar beet plastid sequence (NC_059012.1). All complete representative bacterial genomes have been downloaded from the NCBI RefSeq database on 28/10/2021 using the genome_updater.sh script.
To simulate the discovery of completely unknown viruses that do not have expected similarities with the available data, we constructed virus family leave-out datasets by excluding in turns all the sequences of a given plant viral family from the downloaded virus dataset. The NCBI taxonomy contains 45 viral families. We excluded the Pospiviroidae and the Avsunviroidae families of viroids as they have very small genomes (average length < 1,000). All families with the number of available sequences
Moreover, we generated 12 novel virome-sequencing RNAseq datasets, sampled from peach, grapevine, and sugar beet (see Sample Preparation and Sequencing). Description of these datasets and presence of viruses identified by aligning assembled contigs against the NCBI GenBank database (see Assembly of RNAseq Datasets and Annotation of Viral Contigs) are listed in the Supplementary Table S1.
Total RNAs were extracted from three peach leaf samples, three grapevine phloem scrapping samples, and three sugar beet leaf samples using the CTAB method (Chang et al., 1993), the Spectrum™ Plant Total RNA Kit (Sigma-Aldrich, Saint Quentin-Fallavier, France), and the NucleoSpin RNA plant kit (Macherey-Nagel SAS, Hoerdt, France), respectively. RNAseq libraries were prepared either from total RNAs (peach and grapevine samples), messenger RNAs (grapevine samples), or ribodepleted RNAs (sugar beet samples). High-throughput sequencing was performed on an Illumina platform (Hiseq3000 or NovaSeq600) using a paired-end read length of 2 × 150 bp. Accession numbers for each of the three studies (peach, grapevine, and sugar beet) containing raw FASTQ sequencing files are provided in the Supplementary Table S1.
All of the 12 selected plant virome datasets (see Datasets) were processed with the QIAGEN CLC Genomics Workbench (v.21.0.5). Briefly, reads were first quality-controlled and trimmed using default parameters and then assembled using de novo assembly (word size 50, bubble size 300, and minimum contig length 250). To identify viral contigs present in these datasets, we followed a standard three step BLAST-based approach, see e.g., (Candresse et al., 2018). 1) All contigs were aligned using the CLC built in tBLASTx tool against the NCBI nucleotide non-redundant database limited to taxonomic identifiers of viruses. Contigs having significant hits (e-value below the
Annotation results together with the counts of thus identified viral contigs are listed in the Supplementary Table S1.
To prepare the data for processing by the neural network module, datasets were preprocessed by creating representative one-hot encoded fragments (see Figure 1). Specifically, let us denote the virus dataset by
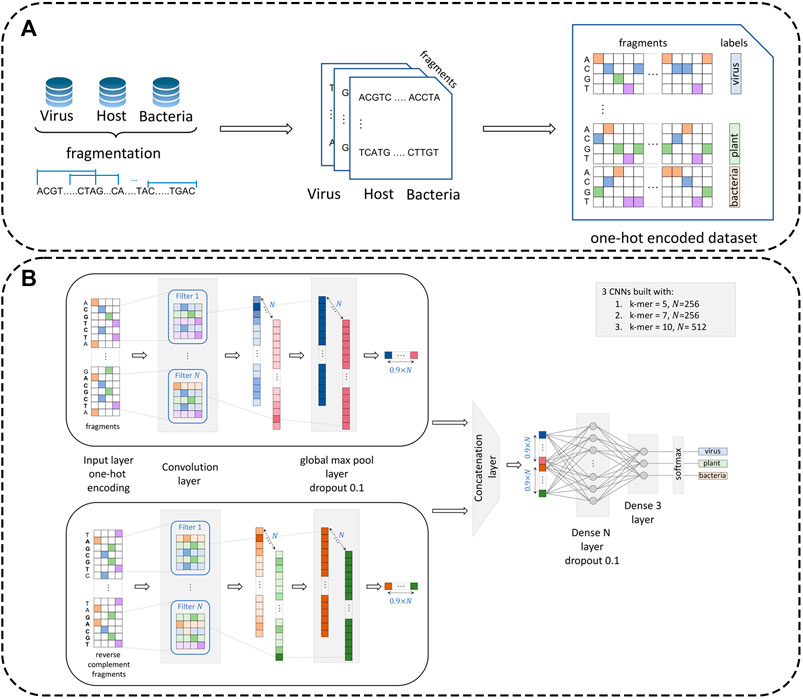
FIGURE 1. Dataset preprocessing procedure and architecture of the multi-CNN module. Panel (A): Reference datasets (virus, plant, and bacteria) are first fragmented with a pre-defined fragment size (
Including plastids in relatively high proportion into the plant dataset
Fragments were further transformed from length
VirHunter architecture was defined with two main components the first component is a multi-path neural network shown in Figure 1, and the second component is a machine learning classification module shown in Figure 2.
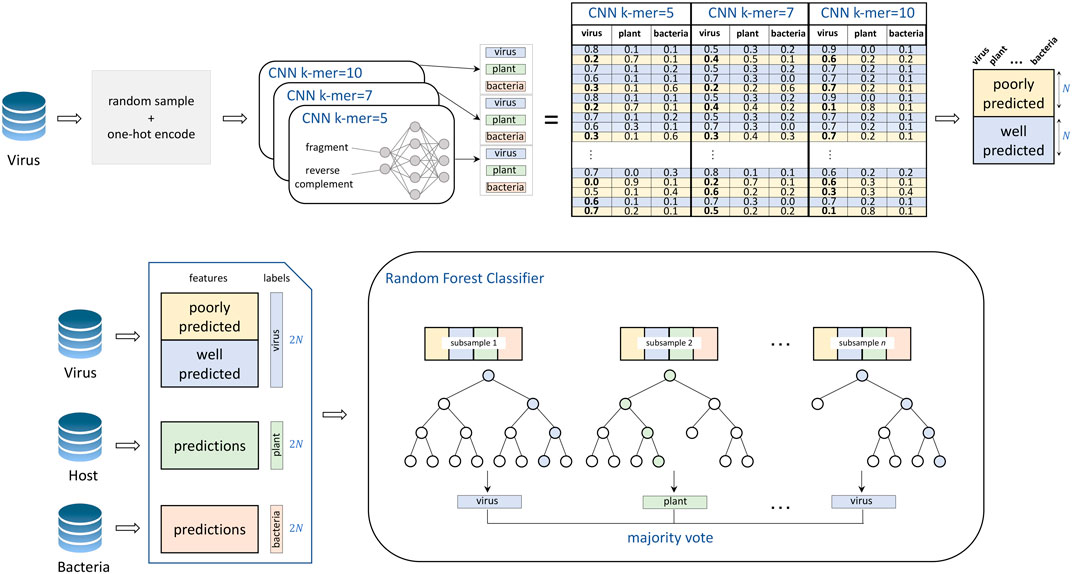
FIGURE 2. Training of the VirHunter’s machine learning module. The individual network predictions are subsetted to contain an equal number of both poorly predicted (prediction value for viral class
1. Neural network component. The neural network module follows a k-mer-based approach. To alleviate a potential difficulty related to the choice of
2. Random Forest component. The second module of the VirHunter implements a random forest classifier (see Figure 2) with the goal to aggregate the predictions from three neural networks. The classifier receives nine real-valued predictions from the multi-network module (three per network) and outputs one of the three classes using the majority vote implementation of random forest. The random forest classifier was chosen over other approaches such as linear regression and simple voting, based on performance (data not shown).
The neural network and machine learning modules were trained separately for each of the three plant host species (peach, grapevine, and sugar beet) and for fragment sizes
The training dataset for the CNN module was built as presented in Data Preprocessing. Training batches with size 512 were prepared in a balanced manner across the three classes (virus, plant, and bacteria) from the training dataset and are split into training and validation with the ratio of 9:1. Each of the three individual networks was trained for
For training and testing the machine learning components, predictions for the three trained networks were obtained on
We verified that overfitting was successfully circumvented by the individual CNN networks that compose the neural network component of our model by comparing the accuracy on validation and test datasets obtained by these individual networks trained on families in the leave-out experiment for peach (see Supplementary Table S9). No significant difference was observed.
VirHunter trained on fragments with
Each fragment of an input contig was preprocessed following the procedure presented in Data Preprocessing. Predictions were produced by the neural network module for each of these one-hot encoded fragments, yielding three probabilities of belonging to a specific class (
VirHunter was trained on GPU (Nvidia Tesla T4) with
Classification results for the viral fragments by VirHunter in this family leave-out experiment are shown in Figure 3 and in Supplementary Tables S2, S3. We compared the capacity of VirHunter to detect novel viruses in the family leave-out setting with the BLAST-based approach on one hand and two state-of-the-art machine learning methods, DeepVirFinder and VirSorter2, on the other hand as also shown in Figure 3. Briefly, each test dataset was aligned using tBLASTx (v2.12.0), preserving one best hit with parameters -max_target_seqs 1 -max_hsps 1, against the respective virus database with the leave-out family removed, and filtered by e-value
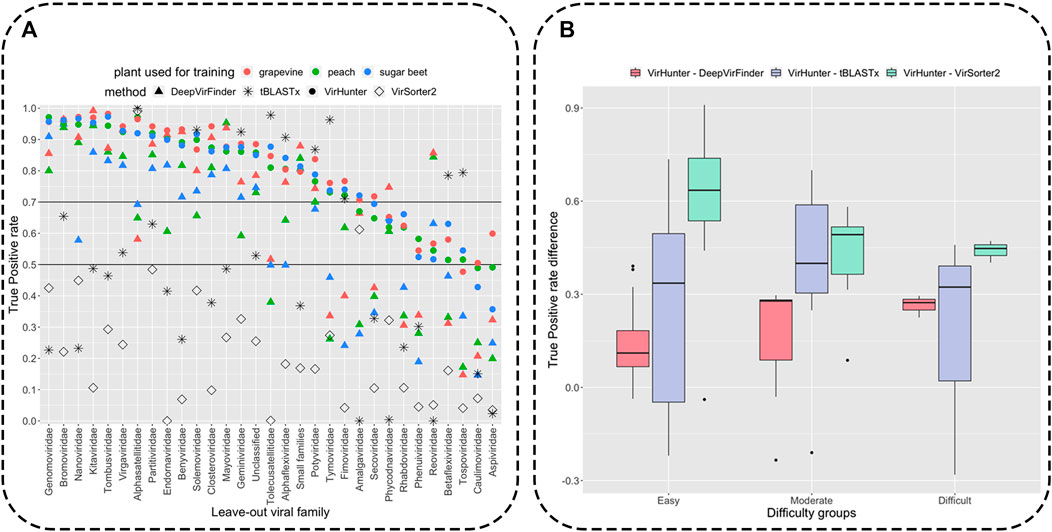
FIGURE 3. Detection of novel viral fragments in the family leave-out setup. Panel (A): Results for the percent of correctly classified fragments (out of
Variability of correct classification was observed for viral fragments of different left-out families for all three methods as shown in Figure 3 (see for detailed results in Supplementary Tables S2–S4). We have split the families into three groups according to the lowest True Positive (TP) rate of VirHunter across the three plant host species: 21 “easy to classify” (TP rate
Of note is also the difference in time requirement for training the VirHunter and DeepVirFinder models. On average, training a full model for one leave-out family for one plant host required 11 h for VirHunter (three CNNs, each for both fragment sizes
Compared to both VirHunter and DeepVirFinder, VirSorter2 has shown poorer performance in the family leave-out setup on all the families except two. Indeed, the TP rate was below
As shown in Figure 3, despite the reasonably permissive filtering criteria, tBLASTx shows best results comparable with VirHunter and for certain families exhibits particularly poor performance relative to the two machine learning methods. For the “easy to classify” families, the difference was mostly in favor of VirHunter, sometimes drastically (see for example, Nanoviridae and Genomoviridae in Panel A and the boxplot in Panel B). In seven cases, tBLASTx outperformed VirHunter, but this difference was mostly marginal (5.8% difference in TP rate on average), the outlier being Tolecusatellitidae and Tymoviridae, where the gain in favor of tBLASTx was the strongest. For the “moderately difficult to classify” families, VirHunter had a higher TP rate than tBLASTx in all cases. For the three “difficult to classify” families, even if VirHunter’s performance was globally low, it still outperformed tBLASTx, with the notable exception of Tospoviridae. Altogether, VirHunter has shown consistently better results than that of tBLASTx, for which the TP rate was frequently below the threshold 0.5 (16 families out of 31).
As for the capacity to correctly classify bacterial fragments, VirHunter has shown a systematically high TP rate, ranging from 0.958 to 0.983, across all the leave-out experiments. As for plant fragments, the TP rate was also satisfactory, sugar beet TP from 0.950 to 0.961, grapevine TP from 0.983 to 0.991, and peach TP from 0.983 to 0.989 (see columns “Bacteria” and “Plant” in Supplementary Table S2).
VirHunter was trained independently with
We cross-evaluated VirHunter’s ability to correctly predict fragments from the plant absent in the training by sampling from the three studied plants, and
As previously described (see VirHunter Outperforms State-of-the art Tools on Family Leave-out Datasets), plant fragments, coming from the same plant that the models were trained on, are consistently well classified for all the three models with the TP rate ranging between 0.95 (“sugar beet” model tested on random fragments from the sugar beet genome) and 0.99 (the “peach” model tested on random fragments from the peach genome) as shown in Table 1. When the plant host species used for training the model is reasonably phylogenetically close to the one of the test datasets, the impact on the TP rate is not very important. For example, the “peach” model tested on random fragments from the grapevine genome still produces the TP rate of 0.9, and the “grapevine” model tested on peach fragments gives the TP rate of 0.836. However, both these models generate a lower TP rate when tested on random fragments from the more phylogenetically distant sugar beet fragments, 0.827 and 0.781, for the “peach” and “grapevine” models, respectively. Similarly, the “sugar beet” model performs less well for both peach and grapevine random fragments, with TP rates of 0.854 and 0.887, respectively.
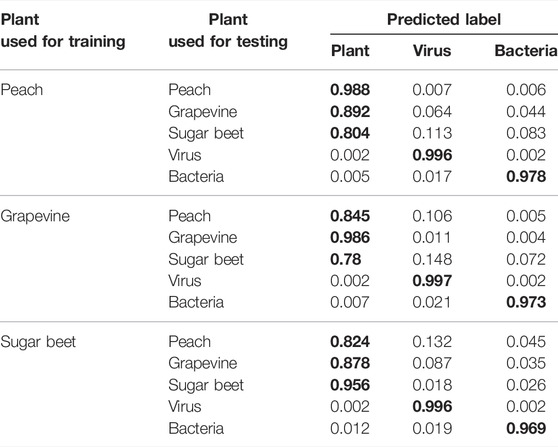
TABLE 1. VirHunter results for prediction of fragments from different plants. Classification results for three plant-specific models of
The three plants used for training models are phylogenetically distant from one another as they belong to different families, sugar beet belongs to the Amaranthaceae family, grapevine belongs to the Vitaceae family, and peach to the Rosaceae family; all the three are eudicots. Out of these three plants, sugar beet is the outlier. Peach and grapevine belong to the Rosids higher clade, while sugar beet belongs to the Caryophyllids higher clade. Given the phylogenetic distance, the lower bound of 0.78 for the true positive rate between these three plants is reasonable.
To evaluate how strongly the performance would be affected if the host of RNAseq dataset was to be from an even further phylogenetically removed plant (belonging to the monocots), we trained a model on the rice (Oryza sativa) dataset that belongs to monocots higher clade. As shown in the Supplementary Table S7, the performance drop was coherent with the increase of the phylogenetic distance (TP rate was 0.766, 0.759, and 0.702 for fragments from peach, grapevine, and sugar beet, respectively); however, the recall remained high for both viral and bacterial fragments. These results highlight that when the host of the RNAseq dataset is phylogenetically highly divergent from any of the plants used to train the available models, a new model for a phylogenetically closer plant has to be trained.
To evaluate the potential quality of VirHunter’s predictions on contigs’ classification, we randomly sampled 10,000 long fragments with
We observed that VirHunter generated highly accurate predictions for long viral fragments with
The capacity of VirHunter to detect novel viral contigs from real RNAseq-sequencing data was evaluated and compared to that of DeepVirFinder and VirSorter2. The 12 virome RNAseq datasets, sampled from peach, grapevine, and sugar beet (see Supplementary Table S1) were assembled as described in Assembly of RNAseq Datasets and Annotation of Viral Contigs. To imitate the novel virus discovery setting, we excluded from the virus dataset those viral species that were annotated as present in the studied plant viromes, and models for each plant species were trained accordingly for VirHunter and DeepVirFinder. For example, to train the “grapevine” model, all viral species present in samples from grapevine (Supplementary Table S1 column “Present viruses”) were deleted from the virus dataset. The same procedure was carried out for training the “peach” and “sugar beet” models. VirSorter2 pretrained models were used following the recommendations in Guo et al. (2021).
The assembled contigs
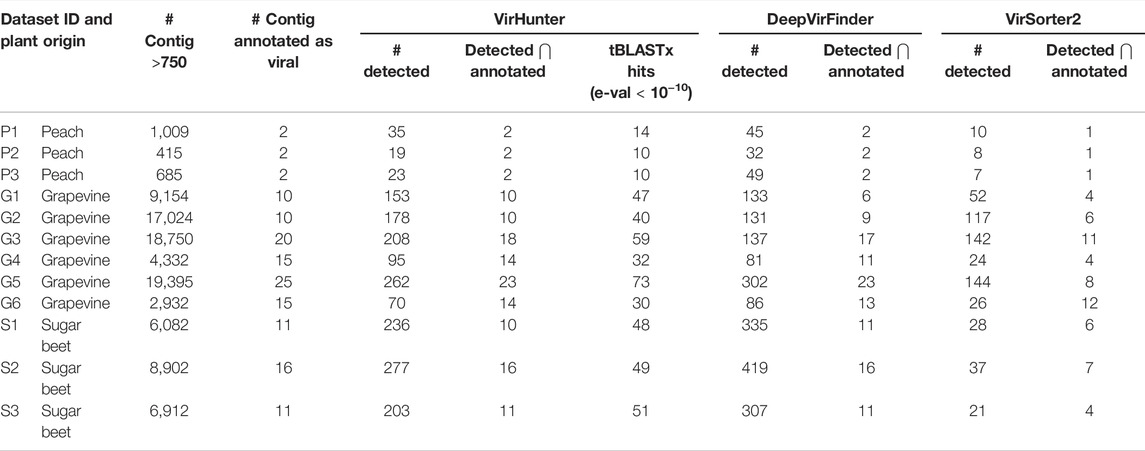
TABLE 2. Performance of VirHunter, DeepVirFinder, and VirSorter2 on 12 RNAseq virome datasets. For each of the 12 datasets shown are the number of contigs that were annotated as being viral by experts and the number of contigs in the initial assembly with length
Moreover, for six datasets (P1, P2, P3, G4, S2, and S3) VirHunter and DeepVirFinder have correctly identified contigs that were previously annotated as viral. For four datasets (G1, G2, G3, and G6), VirHunter was able to discover additional 4, 3, 5, and 1 contigs, respectively. However, for two cases (G5 and S1), DeepVirFinder identified one more annotated contig relative to VirHunter. While VirSorter2 exhibited lower overprediction comparted to VirHunter and DeepVirFinder, its ability to correctly identify viral contigs was low, as it detected at best 60% of the expected viral contigs.
Remember that contigs annotated by experts were all removed from the virus dataset used for the training of VirHunter and DeepVirFinder,
Moreover, it is possible that at least some potentially novel viruses were missed during expert annotation and that the overprediction in columns “# detected” and “tBLASTx hits” (e-val <
High-throughput sequencing (HTS) is capable of broad virus detection for both known and unknown viruses in a variety of hosts and habitats. It has been successfully applied for novel virus discovery in many agricultural crops, leading to the current drive to apply this technology routinely for plant health diagnostics. For this, efficient and precise methods for HTS-based virus detection and discovery are essential.
RNA viruses are the most abundant pathogens infecting plants. However, RNA plant virus detection using HTS presents a number of challenges due to their genetic diversity, lack of conserved regions across viral species, short genome lengths, high mutation rate, and incomplete knowledge present in reference databases. To address this challenge, we developed a novel deep learning method, VirHunter, to detect novel and known plant viruses in RNAseq datasets.
VirHunter is particularly well-suited for the discovery of novel viruses as it was exemplified on 31 synthetic leave-out family datasets, where VirHunter systematically outperformed DeepVirFinder and VirSorter2, reference machine learning tools for virus detection. When compared with the standard tBLASTx approach, we have shown that for most (21 out of 31) leave-out families, VirHunter obtained a higher TP rate. In six cases, tBLASTx was slightly better (5.8% on average). However, there remained four cases where we have seen a much worse performance in VirHunter results. For these specific families, it can be noted that they are particularly well-suited to the alignment-based virus identification, for example, Alphasatellitidae viruses carry high sequence similarity to Geminiviridae (which was confirmed by the majority of tBLASTx hits).
We have shown that the 3-class classification design of VirHunter, accounting for possible bacterial contamination, was justified by evaluating how such contaminating contigs would be classified. Not surprisingly, VirHunter efficiently dealt with bacterial contamination, while DeepVirFinder classified bacteria mostly (65%) as viruses, which should have been “plants” if the goal is to identify viruses. We have also demonstrated that VirHunter is also perfectly suited for the detection of known divergent viruses, by evaluating classification accuracy on contigs with progressively increasing the mutation rate.
Note the fact that VirHunter is designed to be trained separately for a specific plant host species. However, classification of plant contigs still remained reasonable (minimum 0.78 TP rate) when we performed a cross-evaluation by classifying sequences coming from three phylogenetically distant plants (peach, grapevine, and sugar beet) by each of the three models. As expected, VirHunter performed better, when the plants it was trained and tested on were phylogenetically closer: grapevine and peach belong to the same rosids higher clade resulted in better mutual predictions, while sugar beet as an outgroup belonging to the caryophyllids higher clade has shown a relative drop in performance. All these three plants are eudicots (Pin 2012). When the model was trained on an even further phylogenetically distant plant, rice that belongs to monocots and tested on fragments from peach, grapevine, and sugar beet, the classification accuracy of VirHunter was expectedly lower. Together this implies that to classify contigs from an RNAseq experiment, using a pretrained model trained on the exact same plant species as the host of the experimental dataset is not mandatory, but it is preferable to use one trained on a phylogenetically close plant, ideally from the same family and at least belonging to the same eudicots/monocots higher clade. A possible avenue to explore in the future work is the feasibility of transfer learning (Eraslan et al., 2019), to enable fast on-demand retraining for a new plant or building a generalistic plant model.
Finally, we validated VirHunter’s capacity to detect novel viruses on 12 newly acquired RNAseq datasets for peach, grapevine, and sugar beet. In these datasets, VirHunter detected at least 90% (73% for DeepVirFinder and 26% for VirSorter2) of all expert-annotated viral contigs, and in seven datasets it was 100%. Another contribution is the low rate of false positives generated by VirHunter, leaving from 19 to 277 contigs depending on the dataset to be inspected by an expert. These results indicate that VirHunter efficiently reduces the number of contigs requiring manual expert curation.
In conclusion, we have shown that VirHunter can be used to streamline the analyses of plant HTS-acquired viromes and is particularly well suited for the detection of novel viral contigs, in RNAseq datasets.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MN and OG conceptualized the approach. MN and TC designed the study. MN, TC, and AM-C supervised the research. MN and GS contributed to the computational experimental design. GS implemented VirHunter. GS and MK performed genome assembly. TC, AM-C, and MK collected the samples and generated sequencing data. MK and TC performed data annotation. All the authors contributed to writing the manuscript.
This work was supported by the funding from Horizon 2020 Marie Skłodowska-Curie Actions Innovative Training Network (H2020 MSCA-ITN) project “INEXTVIR” (GA 813542).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2022.867111/full#supplementary-material
Abdelkareem, A. O., Khalil, M. I., Elaraby, M., Abbas, H., and Elbehery, A. H. A. (2018). “VirNet: Deep Attention Model for Viral Reads Identification,” in Procs of the 2018 13th Intl Conf. on Computer Engineering and Systems (ICCES), Cairo, Egypt, 18-19 Dec. 2018, 623–626. doi:10.1109/icces.2018.8639400
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215 (3), 403–410. doi:10.1016/S0022-2836(05)80360-2
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 12, 59–60. doi:10.1038/nmeth.3176
Budziszewska, M., and Obrępalska-Stęplowska, A. (2018). The Role of the Chloroplast in the Replication of Positive-Sense Single-Stranded Plant RNA Viruses. Front. Plant Sci. 9, 1776. doi:10.3389/fpls.2018.01776
Candresse, T., Theil, S., Faure, C., and Marais, A. (2018). Determination of the Complete Genomic Sequence of Grapevine Virus H, a Novel Vitivirus Infecting grapevine. Arch. Virol. 163, 277–280. doi:10.1007/s00705-017-3587-7
Chang, S., Puryear, J., and Cairney, J. (1993). A Simple and Efficient Method for Isolating RNA from pine Trees. Plant Mol. Biol. Rep. 11, 113–116. doi:10.1007/BF02670468
de Vries, J. J. C., Brown, J. R., Fischer, N., Sidorov, I. A., Morfopoulou, S., Huang, J., et al. (2021). Benchmark of Thirteen Bioinformatic Pipelines for Metagenomic Virus Diagnostics Using Datasets from Clinical Samples. J. Clin. Virol. 141, 104908. doi:10.1016/j.jcv.2021.104908
Delgado, S., Navarro, B., Serra, P., Gentit, P., Cambra, M. Á., Chiumenti, M., et al. (2019). How Sequence Variants of a Plastid-Replicating Viroid with One Single Nucleotide Change Initiate Disease in its Natural Host. RNA Biol. 16 (7), 906–917. doi:10.1080/15476286.2019.1600396
Edgar, R. C., Taylor, J., Lin, V., Altman, T., Barbera, P., Meleshko, D., et al. (2021). Petabase-scale Sequence Alignment Catalyses Viral Discovery. BioRxiv. 2020-08.
Eraslan, G., Avsec, Ž., Gagneur, J., and Theis, F. J. (2019). Deep Learning: New Computational Modelling Techniques for Genomics. Nat. Rev. Genet. 20, 389–403. doi:10.1038/s41576-019-0122-6
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 29 (7), 644–652. doi:10.1038/nbt.1883
Guglielmi, K. M., Johnson, E. M., Stehle, T., and Dermody, T. S. (2006). Attachment and Cell Entry of Mammalian Orthoreovirus. Curr. Top. Microbiol. Immunol. 309, 1–38. doi:10.1007/3-540-30773-7_1
Guo, J., Bolduc, B., Zayed, A. A., Varsani, A., Dominguez-Huerta, G., and Delmont, T. O. (2021). VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microb. 9 (1), 1–13. doi:10.1186/s40168-020-00990-y
Jenkins, G. M., Rambaut, A., Pybus, O. G., and Holmes, E. C. (2002). Rates of Molecular Evolution in RNA Viruses: A Quantitative Phylogenetic Analysis. J. Mol. Evol. 54, 156–165. doi:10.1007/s00239-001-0064-3
Kim, D., Song, L., Breitwieser, F. P., and Salzberg, S. L. (2016). Centrifuge: Rapid and Sensitive Classification of Metagenomic Sequences. Gen. Res. 26 (12), 1721–1729. doi:10.1101/gr.210641.116
Kleiner, M., Hooper, L. V., and Duerkop, B. A. (2015). Evaluation of Methods to Purify Virus-like Particles for Metagenomic Sequencing of Intestinal Viromes. BMC Genomics 16 (1), 7–15. doi:10.1186/s12864-014-1207-4
Kutnjak, D., Tamisier, L., Adams, I., Boonham, N., Candresse, T., Chiumenti, M., et al. (2021). A Primer on the Analysis of High-Throughput Sequencing Data for Detection of Plant Viruses. Microorganisms 9, 841. doi:10.3390/microorganisms9040841
Lefeuvre, P., Martin, D. P., Elena, S. F., Shepherd, D. N., Roumagnac, P., and Varsani, A. (2019). Evolution and Ecology of Plant Viruses. Nat. Rev. Microbiol. 17, 632–644. doi:10.1038/s41579-019-0232-3
Maree, H. J., Fox, A., Al Rwahnih, M., Boonham, N., and Candresse, T. (2018). Application of HTS for Routine Plant Virus Diagnostics: State of the Art and Challenges. Front. Plant Sci. 9, 1082. doi:10.3389/fpls.2018.01082
Martín, S., and Elena, S. F. (2009). Application of Game Theory to the Interaction between Plant Viruses during Mixed Infections. J. Gen. Virol. 90, 2815–2820. doi:10.1099/vir.0.012351-0
Martinez-Hernandez, F., Fornas, O., Lluesma Gomez, M., Bolduc, B., de la Cruz Peña, M. J., Martínez, J. M., et al. (2017). Single-virus Genomics Reveals Hidden Cosmopolitan and Abundant Viruses. Nat. Commun. 8, 15892. doi:10.1038/ncomms15892
Massart, S., Candresse, T., Gil, J., Lacomme, C., Predajna, L., Ravnikar, M., et al. (2017). A Framework for the Evaluation of Biosecurity, Commercial, Regulatory, and Scientific Impacts of Plant Viruses and Viroids Identified by NGS Technologies. Front. Microbiol. 8, 45. doi:10.3389/fmicb.2017.00045
Massart, S., Chiumenti, M., De Jonghe, K., Glover, R., Haegeman, A., Koloniuk, I., et al. (2019). Virus Detection by High-Throughput Sequencing of Small RNAs: Large-Scale Performance Testing of Sequence Analysis Strategies. Phytopathology 109 (3), 488–497. doi:10.1094/PHYTO-02-18-0067-R
McFadden, G. I. (2001). Primary and Secondary Endosymbiosis and the Origin of Plastids. J. Phycology 37 (6), 951–959. doi:10.1046/j.1529-8817.2001.01126.x
Mehetre, G. T., Leo, V. V., Singh, G., Sorokan, A., Maksimov, I., Yadav, M. K., et al. (2021). Current Developments and Challenges in Plant Viral Diagnostics: A Systematic Review. Viruses 13, 412. doi:10.3390/v13030412
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and Sensitive Taxonomic Classification for Metagenomics With Kaiju. Nature Communications 7 (1), 1–9. doi:10.1038/ncomms11257
Mokili, J. L., Rohwer, F., and Dutilh, B. E. (2012). Metagenomics and Future Perspectives in Virus Discovery. Curr. Opin. Virol. 2, 63–77. doi:10.1016/j.coviro.2011.12.004
Nayfach, S., Camargo, A. P., Schulz, F., Eloe-Fadrosh, E., Roux, S., and Kyrpides, N. C. (2021). CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat. Biotechnol. 39, 578–585. doi:10.1038/s41587-020-00774-7
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a New Versatile Metagenomic Assembler. Genome Res. 27 (5), 824–834. doi:10.1101/gr.213959.116
Pin, P. A. (2012). Life Cycle and Flowering Time Control in Beet. PhD Thesis (Sweden, Umeå: Swedish University of Agricultural Sciences).
Ren, J., Ahlgren, N. A., Lu, Y. Y., Fuhrman, J. A., and Sun, F. (2017). VirFinder: A Novel k-mer Based Tool for Identifying Viral Sequences From Assembled Metagenomic Data. Microb. 5 (1), 1–20. doi:10.1186/s40168-017-0283-5
Ren, J., Song, K., DengAhlgren, C. N. A., Ahlgren, N. A., Fuhrman, J. A., Li, Y., et al. (2020). Identifying Viruses from Metagenomic Data Using Deep Learning. Quant. Biol. 8, 64–77. doi:10.1007/s40484-019-0187-4
Roossinck, M. J., Martin, D. P., and Roumagnac, P. (2015). Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology 105, 716–727. doi:10.1094/PHYTO-12-14-0356-RVW
Roossinck, M. J. (1997). Mechanisms of Plant Virus Evolution. Annu. Rev. Phytopathol 35, 191–209. doi:10.1146/annurev.phyto.35.1.191
Rott, M., Xiang, Y., Boyes, I., Belton, M., Saeed, H., Kesanakurti, P., et al. (2017). Application of Next Generation Sequencing for Diagnostic Testing of Tree Fruit Viruses and Viroids. Plant Dis. 101, 1489–1499. doi:10.1094/PDIS-03-17-0306-RE
Roux, S., Emerson, J. B., Eloe-Fadrosh, E. A., and Sullivan, M. B. (2017). Benchmarking Viromics: an In Silico Evaluation of Metagenome-Enabled Estimates of Viral Community Composition and Diversity. PeerJ 5, e3817. doi:10.7717/peerj.3817
Santos-Medellin, C., Zinke, L. A., Ter Horst, A. M., Gelardi, D. L., Parikh, S. J., and Emerson, J. B. (2021). Viromes Outperform Total Metagenomes in Revealing the Spatiotemporal Patterns of Agricultural Soil Viral Communities. The ISME Journ 15, 1–15. doi:10.1038/s41396-021-00897-y
Shrikumar, A., Greenside, P., and Kundaje, A. (2017). Reverse-complement Parameter Sharing Improves Deep Learning Models for Genomics. bioRxiv. 103663.
Stobbe, A. H., and Roossinck, M. J. (2014). Plant Virus Metagenomics: What We Know and Why We Need to Know More. Front. Plant Sci. 5, 150. doi:10.3389/fpls.2014.00150
Sutton, T. D. S., Clooney, A. G., Ryan, F. J., Ross, R. P., and Hill, C. (2019). Choice of Assembly Software Has a Critical Impact on Virome Characterisation. Microbiome 7 (1), 12–15. doi:10.1186/s40168-019-0626-5
Tampuu, A., Bzhalava, Z., Dillner, J., and Vicente, R. (2019). ViraMiner: Deep Learning on Raw DNA Sequences for Identifying Viral Genomes in Human Samples. PLoS ONE 14, e0222271. doi:10.1371/journal.pone.0222271
Villamor, D. E. V., Ho, T., Al Rwahnih, M., Martin, R. R., and Tzanetakis, I. E. (2019). High Throughput Sequencing for Plant Virus Detection and Discovery. Phytopathology 109 (5), 716–725. doi:10.1094/PHYTO-07-18-0257-RVW
Warwick-Dugdale, J., Solonenko, N., Moore, K., Chittick, L., Gregory, A. C., Allen, M. J., et al. (2019). Long-read Viral Metagenomics Captures Abundant and Microdiverse Viral Populations and Their Niche-Defining Genomic Islands. PeerJ 7, e6800. doi:10.7717/peerj.6800
Keywords: novel virus detection, RNA viruses, plant virome, alignment-free method, deep learning, artificial neural network
Citation: Sukhorukov G, Khalili M, Gascuel O, Candresse T, Marais-Colombel A and Nikolski M (2022) VirHunter: A Deep Learning-Based Method for Detection of Novel RNA Viruses in Plant Sequencing Data. Front. Bioinform. 2:867111. doi: 10.3389/fbinf.2022.867111
Received: 31 January 2022; Accepted: 24 March 2022;
Published: 13 May 2022.
Edited by:
Joao Carlos Setubal, University of São Paulo, BrazilReviewed by:
Deyvid Amgarten, Albert Einstein Israelite Hospital, BrazilCopyright © 2022 Sukhorukov, Khalili, Gascuel, Candresse, Marais-Colombel and Nikolski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grigorii Sukhorukov, Z3JzdWtob3J1a292QGdtYWlsLmNvbQ==; Macha Nikolski, bWFjaGEubmlrb2xza2lAdS1ib3JkZWF1eC5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.