
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Anim. Sci. , 21 March 2023
Sec. Animal Physiology and Management
Volume 4 - 2023 | https://doi.org/10.3389/fanim.2023.1154463
 Macey P. Soltis1
Macey P. Soltis1 Madison T. Henniger1Amanda M. Egert-McLean1
Madison T. Henniger1Amanda M. Egert-McLean1 Brynn H. Voy1Sarah E. Moorey1
Brynn H. Voy1Sarah E. Moorey1 Liesel G. Schnieder1Elizabeth A. Shepherd1
Liesel G. Schnieder1Elizabeth A. Shepherd1 Courtney Christopher2
Courtney Christopher2 Shawn R. Campagna2,3
Shawn R. Campagna2,3 Joe S. Smith4
Joe S. Smith4 Pierre-Yves Mulon4
Pierre-Yves Mulon4 David E. Anderson4
David E. Anderson4 Phillip R. Myer1*
Phillip R. Myer1*The rumen microbiome is a complex microbial network critical to the health and nutrition of its host, due to their inherent ability to convert low-quality feedstuffs into energy. In rumen microbiome studies, samples from the ventral sac are most often collected because of the ease of access and repeatability. However, anatomical musculature demarcates the rumen into five sacs (biogeographical regions), which may support distinct microbial communities. The distinction among the microbes may generate functional variation among the rumen microbiome, thus, specialized tasks within different sacs. The objective of this study was to determine the rumen liquid metabolome and epimural, planktonic, and fiber-adherent bacterial communities among each rumen biogeographical region. It was hypothesized that differences in bacterial species and metabolome would occur due to differing anatomy and physiology associated with the respective regions. To assess this variation, epithelial and content microbial-associated communities were evaluated, as well as the metabolites among various rumen biogeographical regions. A total of 17 cannulated Angus cows were utilized to examine the fiber-adherent (solid fraction), planktonic (liquid fraction), and epimural microbial communities from the cranial, dorsal, caudodorsal blind, caudoventral blind, and ventral sacs. Metagenomic DNA was extracted and sequenced from the hypervariable V4 region of the 16S rRNA gene. Reads were processed using packages ‘phyloseq’ and ‘dada2’ in R. Untargeted metabolomics were conducted on rumen liquid from each sac using UHPLC-HRMS and analyzed in MetaboAnalyst 5.0. An analysis of variance (ANOVA) revealed 13 significant differentially abundant metabolites with pairwise comparisons against the five rumen sacs (P < 0.05). Within the bacterial communities, neither alpha nor beta diversity determined significance against the rumen sacs (P > 0.05), although there was significance against the fraction types (P < 0.05). Utilizing multivariable association analysis with MaAslin2, there were significant differential abundances found in fraction type × location (P < 0.05). Knowledge of similarities among fiber-adherent microbial communities provides evidence that single sac sampling is sufficient for this fraction. However, future projects focusing on either planktonic or epimural fractions may need to consider multiple rumen sac sampling to obtain the most comprehensive analysis of the rumen. Defining these variabilities, especially among the rumen epimural microbiome, are critical to define host-microbiome interactions.
Cattle are foregut fermenters that are essential to the global protein supply due to their unique ability to consume and convert feedstuffs that are not suitable for human consumption into meat and milk (Petri et al., 2020). The conversion to products for human consumption stems from the synergistic relationship between cattle and their resident ruminal microbial communities. This synergistic relationship allows recalcitrant forage particles to be fermented and broken down to produce volatile fatty acids (VFA), providing around 70% of the energy required for cattle (Mizrahi, 2013; Mizrahi et al., 2021; Xue et al., 2022). The extensive, adaptable, and symbiotic rumen microbiome is vital to the ruminal environment and in generating an efficient fermentation vat, breaking down fibrous feedstuffs that cattle would otherwise not be able to utilize (Mann et al., 2018).
The rumen is an intricate organ containing both anatomical complexity and microbial variation. There are a variety of internal and external structures demarcating the rumen into five distinct rumen sacs: the cranial (CS), dorsal (DS), caudodorsal blind (CDBS), caudoventral blind (CVBS), and ventral (VS) sacs, referred to as biogeographical regions. These individual sacs in conjunction with ruminal environmental factors, such as digesta stratification, passage rate, and ruminal contractions, produce different ecological niches within the rumen (Krehbiel, 2014; Weimer, 2015; Schmitz-Esser, 2021). These ecological niches may be grossly observed with more ventral locations comprised of greater liquid digesta components and dorsal locations containing a greater composition of solid feed particles and gaseous space. Due to the variability of rumen content composition, there is evidence as to the differences among specific microbial community structure within the varying fractions of rumen content (Ma et al., 2018; Sbardellati et al., 2020). Specifically, the varying microbial populations are partitioned into three communities: the fiber-adherent, planktonic, and epimural microbial communities (De Mulder et al., 2016; Ji et al., 2017). The interactions and divisions among the fiber-adherent and planktonic communities have been extensively studied (De Mulder et al., 2016), but there has been limited research regarding rumen epimural microbial communities. This may be due to the difficulty of sampling, decreased density, and reduced relative abundance contrasted to the remainder of the rumen microbiome (De Mulder et al., 2016). Even with the multitude of hinderances when investigating the epimural population, this community possesses specialized functions such as scavenging oxygen, recycling host epithelial tissue, and urea transport (Schmitz-Esser, 2021; Na, 2022; Na and Guan, 2022), which are critical in preserving a healthy rumen environment and microbiome. Importantly, the epimural microbes are located along the rumen epithelium, contributing to their ability to influence host gene expression through host-microbiome crosstalk (Petri et al., 2020; Schmitz-Esser, 2021; Na and Guan, 2022). The totality of anatomical, physiological, and microbial variation among the five ruminal locations may highlight the necessity for analysis of the entirety of the rumen to provide a representative and more comprehensive understanding of the rumen microbiome. Further, there may be an indication that sampling from each microbial community further denotes the difference in microbial composition and taxa, but also hints at the interactions allowing the microbiome to continually work synergistically.
The objective of this study was to determine the epimural, planktonic, and fiber-adherent bacterial communities and the rumen liquid metabolome among each of the rumen biogeographical regions. It was hypothesized that differences in the bacterial species and metabolome would occur as a result of the differing anatomy and physiology associated with the respective regions.
This study was conducted at The University of Tennessee following approval of procedures by the campus Institutional Animal Care and Use Committee (2844-0521).
Four-year-old purebred primiparous cannulated Angus cows (n = 17) with average body weight of 714.42 ± 77.23 kg from the University of Tennessee were utilized for sampling. All cows were located at the University of Tennessee East Tennessee Research and Education Center, Blount Unit. Prior to the study, all cows were adapted to the same diet of ad libitum warm and summer season forages (mixed grass with fescue, bermuda and clover) for 9 weeks. This duration was based on previous research demonstrating the rumen microbiome requires 7-10 weeks to stabilize following dietary changes (Clemmons et al., 2019). To determine differences in bacterial communities and the fermentative environment, ruminal solid content, liquid, and epimural samples were taken from each of the five rumen sacs during two sampling days to collect the fiber-adherent, planktonic, and epimural microbial communities, respectively. The sacs sampled included the dorsal sac (DS), cranial sac (CS), caudodorsal blind sac (CDBS), ventral sac (VS), and caudoventral blind sac (CVBS). Rumen liquid content was also retained for metabolomics analyses. Samples were collected and pooled during the two sampling days to reduce individual animal daily variation in the rumen bacterial communities and metabolome. All samples were collected within 10 minutes per animal and within 4 hours after feeding.
For the two sampling days, the hand grab method was used to obtain solid and liquid digesta from the five rumen sacs via the rumen cannula. A handful of rumen content was squeezed through four layers of cheese cloth, where the liquid run-off was collected into a 15 mL conical tube. Approximately 7 mL of rumen liquid was collected on day one to serve as the pool sample, and the remaining approximate 7 mL was collected on the second day of sampling to complete the pool sample. Solid rumen content was collected from the squeezed rumen digesta in the cheesecloth, and a daily sample was collected in a 50 mL conical tube for both sampling days. Samples were pooled to reduce individual animal daily variation. To reduce contamination, samples were first taken from the DS, and the rumen was then partially emptied to gain access to the other sacs. Sampling from the sacs then followed the order of VS, CS, CDBS, CVBS. All samples were flash frozen in liquid nitrogen and stored at -80°C until analysis.
To collect rumen epithelium associated microbes, partial-thickness samples of the rumen epithelium were collected using a single-action Cushing Rongeur (non-serrated jaw, Sontec Instruments Inc, Centennial, CO, USA) and an arm-length plastic sleeve was used to reduce contamination. Rumen epithelium samples from each rumen sac were collected in the following order: CS, DS, VS, CDBS, and CVBS. To collect the sample, rumen contents from the specific sac were removed, and the instrument was advanced until the rumen wall was identified. The jaws of the rongeur were then opened and applied to the epithelial surface, and then closed to collect the sample in a partial thickness bite. The instrument was rinsed with 70% ethanol after each sample was collected and sleeves were changed after each cow. Once the papillae biopsy was extracted, the papillae were taken with sterilized tissue forceps and serially washed four times in 1X PBS (pH 7.4) in a four-quadrant petri dish. After thoroughly washing the papillae, 1 mL of the PBS papillae wash was pipetted into 2 mL cryovials to collect epimural bacteria washed from papillae. Samples were flash frozen in liquid nitrogen and stored at -80°C.
DNA extractions were performed on solid, liquid, and epimural samples. All raw samples were stored at -80°C freezer and were thawed at room temperature prior to extraction.
The DNA extraction protocol was adapted with slight modifications from the Yu and Morrison (Yu and Morrison, 2004) protocol and the Qiagen DNeasy Blood and Tissue kit (QIAGEN, Hilden, Germany). All centrifugation was performed at 16,000 x g and 4°C throughout the protocol. For cell lysis, 0.2 g of sample was added to a 2 mL beaded screw cap tube containing 0.6 mL dry volume mixed with 0.5 mm and 0.1mm ZR BashingBead lysis matrix (Zymo Research, Irvine, CA, USA) and 1 mL of lysis buffer. To obtain 0.2 g of solid sample, forceps were used to obtain 0.1 g of each daily sample to pool. The forceps were sanitized with 70% ethanol between use. Samples were homogenized with the TissueLyser II (QIAGEN, Hilden, Germany) at 21 Hz for 3 min and incubated at 70°C for 15 minutes. Samples were then centrifuged, and the supernatant was transferred to 1.5 mL microcentrifuge tubes. Fresh lysis buffer was added to the bead beating tubes before repeating the previous steps to aspirate the remainder of the supernatant. For nucleic acid precipitation, 260 µL of 10M ammonium acetate was added, followed by 5 minutes on ice and 10 minutes of centrifugation. The supernatant was then equally divided between two tubes and an equal volume of isopropanol was added before incubating on ice for 30 minutes. Samples were centrifuged for 15 minutes to concentrate a nucleic acid pellet. After the supernatant was removed, the nucleic acid pellet was washed with 70% ethanol and allowed to air dry. The time to dry varied based on remaining 70% ethanol observed in the tube after 5 minutes, but samples were allotted no more than 10 minutes to air dry. Following drying, 100 µL of Tris-EDTA (TE) buffer was added to re-constitute the nucleic acid pellet and samples were pooled with their respective sample. The QIAGEN DNeasy Blood and Tissue kit was used (QIAGEN, Hilden, Germany) to complete purification of the nucleic acids. 15 µL of Proteinase K and 200 µL of Buffer AL were added to samples and incubated for 10 minutes. A total of 200 µL of 100% ethanol was added before being transferred to the QIAamp filter column. Samples were centrifuged for one minute and the flow through was discarded. To wash the DNA adhered to the filter, 500 µL of Buffer AW1 was added, incubated at room temperature for 1 min, and centrifuged to remove the flow through. The steps were repeated for Buffer AW2. The column was then centrifuged for 2 minutes to completely dry the column and the flow-through was collected in a 1.5 mL microcentrifuge tube for removal. To elute the DNA, a two-step process was conducted to maximize DNA concentrations. In part one, 70 µL of Buffer AE (QIAGEN, Hilden, Germany) was directly added to the column membrane and incubated at room temperature for 2 minutes. Following incubation, the sample was centrifuged for 1 minute. In part 2, 30 µL of Buffer AE was added to the membrane and incubated and centrifuged like part 1 to elute the DNA. The DNA quality and concentrations were then analyzed on the DeNovix DS-11 Spectrophotometer (DeNovix, Wilmington, DE, USA) before being stored at -20°C until further analysis.
The epimural DNA extraction method was adapted from (Ault-Seay et al., 2022) for bacterial community analysis and Qiagen DNeasy Blood and Tissue kit protocols (QIAGEN, Hilden, Germany). To obtain 1 g of the sample, the samples were thawed and inverted three times before 1 mL was pipetted into 1.5 mL microcentrifuge tubes. Samples were then centrifuged for 10 minutes at 4°C and 4,700 x g where the supernatant was discarded, leaving the pellet in the tube. Pellets were then resuspended in 180 µL of lysis buffer (500mM NaCl, 50 mM Tris-HCl, pH 8.0, 50 mM EDTA, and 4% sodium dodecyl sulfate [SDS]) and vortexed. The homogenized sample was incubated at 37°C for 30 minutes and 25 µL of proteinase K was added to each sample. Following the Qiagen DNeasy Blood and Tissue kit protocol (QIAGEN, Hilden, Germany), samples were incubated at 70°C for 30 minutes and 200 µL of 100% ethanol was added. Samples were transferred to a QIAamp column for DNA purification. After adding Buffers AW1 and AW2 to the column membrane as previously described, samples were centrifuged to dry the column and prevent ethanol contamination. The DNA was eluted as previously described for the solid and liquid samples. The DNA quality and concentrations were then analyzed on the DeNovix DS-11 Spectrophotometer (DeNovix, Wilmington, DE, USA) before being stored at -20°C until PCR amplification and library preparation.
Gel electrophoresis and polymerase chain reaction (PCR) were performed on epimural samples to ensure eluted bacterial DNA was captured versus host DNA, due to lower DNA concentrations among epimural samples. The PCR runs consisted of TE buffer (negative control), a rumen solid sample (positive control), and representative epimural rumen samples. The PCR protocol targeted the 16S V4 hypervariable region, utilizing the forward primer 515F and reverse primer 806R (Apprill et al., 2015) to target the V4 hypervariable region of the gene. The PCR cocktail consisted of PCR Platinum Master mix (12.5 µL per reaction), the forward and reverse primers (0.5 µL of each per reaction), and nuclease free water (8.5 µL per reaction). A total of 22 µL of the PCR cocktail and 3 µL of the template DNA were used for amplification. The thermal cycler (BioRad T100) (Bio-Rad, Berkeley, CA) conditions were set to 94°C for 15 minutes, followed by 35 cycles at 94°C for 30 seconds for denaturation, 55°C for 30 seconds for annealing, 72°C for 1 minute for elongation, with a final extension of 72°C for 5 minutes. Reactions were held at 4°C. A 2% TAE agarose gel was used to confirm bacterial DNA amplicon size and included 5 µL of ethidium bromide. The gel was run at 100 volts (100,000 mAmps) for 45 minutes and observed on the Chemidoc imaging system (Bio-Rad, Berkeley, CA) for visualization.
All library preparation and DNA sequencing was performed at the Genomics Core at The University of Tennessee. Standard operating procedures for the two-step polymerase chain reaction (PCR) library construction was used for these samples. From extracted DNA, the V4 region of the 16S rRNA gene was amplified using primers 515Fb (GTGYCAGCMGCCGCGGTAA) (Parada et al., 2016) and 806Rb (GGACTACNVGGGTWTCTAAT) (Apprill et al., 2015) (Earth Microbiome Project). Primers were slightly modified with the addition of adaptors for Illumina MiSeq sequencing. The initial PCR cocktail included 2 × KAPA HiFi HotStart ReadyMix Taq (Roche, Indianapolis, IN, United States) and 1.5 µM of each primer. Each tube received 2.5 µL of extracted DNA and 22.5 µL of initial PCR cocktail. Thermal cycler conditions consisted of 95°C for 3 minutes, 25 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds, with a final extension of 72°C for 5 minutes. PCR amplification was confirmed on a 2% TAE agarose gel with gel electrophoresis. To purify the PCR product, 20 uL of AMPure XP beads (Agencourt, Beverly, MA, USA) were used while combining with ethanol washes. The final product was eluted in 50 µL of Tris-HCl. To give each sample a unique combination of forward and reverse indexes, Nextera XT indexes (Illumina, Inc., San Diego, CA, United States) were added to each PCR product and run through a reduced-cycle PCR. The reduced-cycle PCR consisted of 95°C for 3 minutes, then 8 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds, and the final extension at 72°C for 5 minutes. For the final PCR purification, 56 µL of AMPure XP beads were used while combining the ethanol washes and the final elution with 25 µL of Tris-HCl. Using the NanoDrop spectrophotometer (Fisher Scientific International, Inc., Hampton, NH, USA), samples were quantified and pooled at approximately equal concentrations. To ensure the product concentrations were approximately equal, they were run on the Agilent Bioanalyzer (Santa Clara, CA, USA) using the standard sensitivity kit. The final library was diluted to 4 pM and pooled with 20% of a 10 pM PhiX library control (Illumina, Inc., San Diego, CA, USA), and run paired-end 250 nucleotides on a v3, 600 cycle flow cell of an Illumina MiSeq sequencer at the University of Tennessee Genomics Core.
Using the pooled liquid digesta, 2 mL were aliquoted into 1.5 mL microcentrifuge tubes. Tubes were centrifuged at 6,000 x g for 15 minutes at 4°C. The supernatants from the two corresponding microcentrifuge tubes were pooled into a 3 mL syringe. The pooled sample was then filtered through a 0.22 µm filter tip (Midsci, St. Louis, MO, USA) into the final microcentrifuge tube. Samples were then stored at -20°C until further analysis.
Metabolomic analysis was performed at the Biological and Small Molecule Mass Spectrometry Core (BSMMSC) at The University of Tennessee, Knoxville, TN (RRID: SCR_021368). Samples were transported and thawed at 4°C for up to an hour before extracting water-soluble metabolites. Polar metabolites were extracted from 100 μL of rumen content using 1.5 mL of metabolomics extraction solvent (4:4:2 HPLC grade acetonitrile/methanol/water with 0.1 M formic acid) (Dridi et al., 2022). The solvent was evaporated under gaseous nitrogen. Once fully dried, metabolites were suspended in 300 µL HPLC grade water. The HPLC grade solvents used in this protocol were all purchased from Fisher Scientific (Hampton, NH, USA). Using ultra high performance liquid chromatography high resolution mass spectrometry (UHPLC-HRMS) (Thermo Scientific, San Jose, CA, USA) metabolites were identified utilizing a previously validated global metabolomics method (Lu et al., 2010). Reverse-phase (RP) ion-pair chromatography was utilized to separate the metabolites based on the polarity of the small molecules. A Synergi Hydro RP column (100 mm x 2.1 mm, 2.6 mm, 100 Å) and an UltiMate 3000 pump (Thermo Fisher) were used for the chromatographic separation. The Exactive™ Plus Orbitrp MS (Thermo Fisher) coupled with the UHPLC system was used for mass analysis. The raw data from Xcalibur MS software (Thermo Electron Corp, Waltham, MA, USA) were converted to mzML format using a tool from MSConverter, ProtroWizaed (Martens et al., 2011; Chambers et al., 2012). Metabolites were identified using MAVEN (Melamud et al., 2010), by comparing the exact mass and retention time (±5ppm) to an in-house library of verified metabolites. Area under the chromatographic curve was integrated for each identified metabolite (Clasquin et al., 2012). The relative metabolite abundances were exported to Excel for statistical analysis.
Each of the rumen liquid samples were thawed at room temperature, vortexed, and the pH probe (Accumet AB15 Basic, Fisher Scientific, Waltham MA) was submerged into the 15 mL tube of rumen liquid from each rumen sac. The pH was then recorded once the pH meter stabilized. Between samples the probe was thoroughly washed with deionized water and the pH was stabilized before the next measurement.
Fastq sequences were evaluated in R Studio (R version 3.6.2). To analyze the quality of the reads, package ‘fastqcr’ v0.1.2 (Kassambara, 2023) was used, which was considered for further parameters downstream. With the filterAndTrim package from ‘phyloseq’ v1.40.0 (McMurdie and Holmes, 2013) sequences were filtered and trimmed with parameters at truncLen of 260, and maximum expected error at 2 for forward reads. Due to poor merging downstream, reverse reads were removed from the data set. After the reads were filtered, the quality was then analyzed for greater than or equal to a quality score of 25 (Q25). If reads were less than Q25, they were removed. To learn error rates, the function learnErrors was utilized from ‘dada2’ package v.1.24.0 (Callahan et al., 2016). Within the dada function, sequences were denoised to produce amplicon sequence variants (ASVs). To remove chimeras from the data set the consensus method from removeBimeraDenovo was used. Taxa were assigned to the SILVA 138.1 using assignTaxonomy with the parameter of bootstrap confidence at a minimum of 80. For downstream analysis, metadata, assigned taxa, and ASV table were merged into the phyloseq object. Utilizing phyloseq, the alpha diversity was measured for observed ASVs, Shannon diversity index, and Chao1 metric. To assess beta diversity the package ‘vegan’ v2.6.2 (Oksanen et al., 2022) was used and the sample counts were ordinated and transformed with the Bray-Curtis Dissimilarity which calculated distances for the principal coordinates analysis (PCoA).
Alpha diversity measurements, observed ASVs, Shannon diversity index, and Chao1, were first visually analyzed for normality and then tested against a Shapiro-Wilks test (W), where normality was determined at W > 0.90 and P > 0.05. The Kruskal-Wallis H test was used to test differences among digesta types and rumen sac. For beta diversity measurements, the sample counts were converted out of total counts and then the distances were calculated by Bray-Curtis Dissimilarity Matrix. Following, a permutational multivariate analysis of variance (PERMANOVA) with 999 permutations using the adonis function from package ‘vegan’ tested for differences between samples (Oksanen et al., 2022). All analyses had significance determined at P < 0.05.
To analyze differential abundances of the bacterial communities among the three rumen digesta types, the five rumen sacs, and digesta type within rumen sac, the MaAsLin2 package v1.10.0 (Mallick et al., 2020) was used. For the analysis, the phyloseq object was rarefied to normalize the data set due to large differences in library size. Following the recommendations from the developers, the data were transformed with the log function and analyzed through a linear model method (LM). This was recommended due to the consistency found across samples (Mallick et al., 2021; Nearing et al., 2022). Taxa present in fewer than 50% of total samples were removed from the model. To test differences in fraction types (planktonic, fiber-adherent, or epimural), individual fraction types were set as the reference level to test all pairwise comparisons. The fixed effects included the fraction type and differences within sac were blocked. To test differential abundances among sacs, fraction types were divided into categories and tested within them. For the individual planktonic, fiber-adherent, and epimural analyses ran, the fixed effects were the rumen sacs. All pairwise comparisons were tested for each sac and digesta type. Multiple testing was addressed using Benjamini-Hochberg (Benjamini and Hochberg, 1995). Significance was set at P > 0.05.
For metabolomics analyses, the data were filtered by interquartile range, log-transformed, and Pareto scaled in MetaboAnalyst prior to analysis (Pang et al., 2021). MetaboAnalyst 5.0 was used to generate partial-least-squares discriminant analysis (PLS-DA) plots and variable importance in projection (VIP) scores. Heatmaps, which displayed log2 fold changes and p-values calculated by a student’s t-test, were generated using R (version 1.0.153).
For the relative pH analyses, a linear model was built in R studio using the ‘lm’ function. The differences in mean pH were tested against the rumen sacs using the ‘Anova’ function from the car package (type II test). If means were significant, then pairwise comparisons were tested using the ‘lht’ function in multiple comparisons using Tukey’s HSD, where alpha was set at 0.05.
Due to low input reads, one sample from the CS in the fiber-adherent microbial community and one sample from the VS in the epimural community were removed. Out of the 253 samples analyzed, 30,176 taxa were identified. The overall output from the Illumina MiSeq averaged 112,045 paired reads with a minimum read count of 55,220 and a maximum read count of 255,381.
To assess alpha diversity, the Kruskal-Wallis H test was used to compare the mean ranks of the rumen fractions and examine the observed ASVs (richness), chao1 (anticipated richness), and Shannon diversity index (richness and evenness). Among fraction types, all measures of richness and evenness differed significantly (P < 0.001) (Figure 1). When analyzing beta diversity, there was a significant difference among fraction types after analysis with a PERMANOVA with 999 permutations (P < 0.05) (Figure 2). After conducting pairwise comparisons of differential abundances of bacterial communities among rumen fraction types collected with MaAsLin2, when epimural was the reference level, there were 469 communities that differed against the planktonic communities, and 307 differences between the communities against the fiber-adherent community (P < 0.05). When the planktonic fraction was used as the reference level, there were 471 differentially abundant bacterial communities against the epimural communities and 494 against the fiber-adherent bacterial communities (P < 0.05). Lastly, with the reference level of fiber-adherent fraction, there were 307 significant differences against the epimural bacterial communities and 490 significant abundance differences against planktonic (P < 0.05) (Table 1).
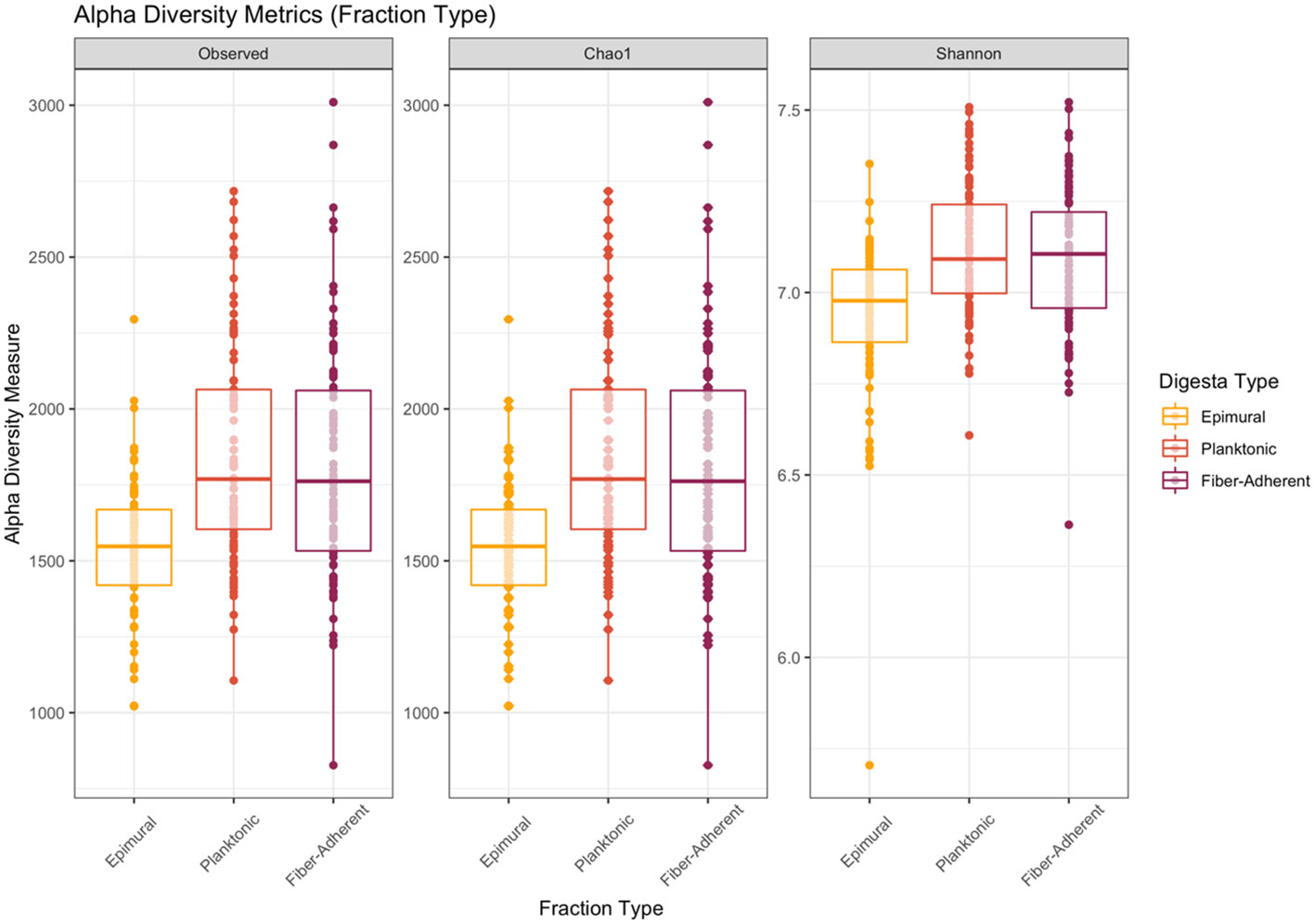
Figure 1 Alpha diversity metrics in the three rumen fractions. Alpha-diversity metrics for bacterial communities. Measures include observed ASVs (richness), Chao1 (expected richness), and Shannon diversity index (richness and evenness) for each digesta fraction. The epimural fraction is represented by orange, the planktonic digesta fraction is represented by red, and the fiber-adherent digesta fraction is represented by purple. With the removal of singletons during data analysis, the Chao1 measurement may appear similar to the observed measurement. Significance was determined at P < 0.001.
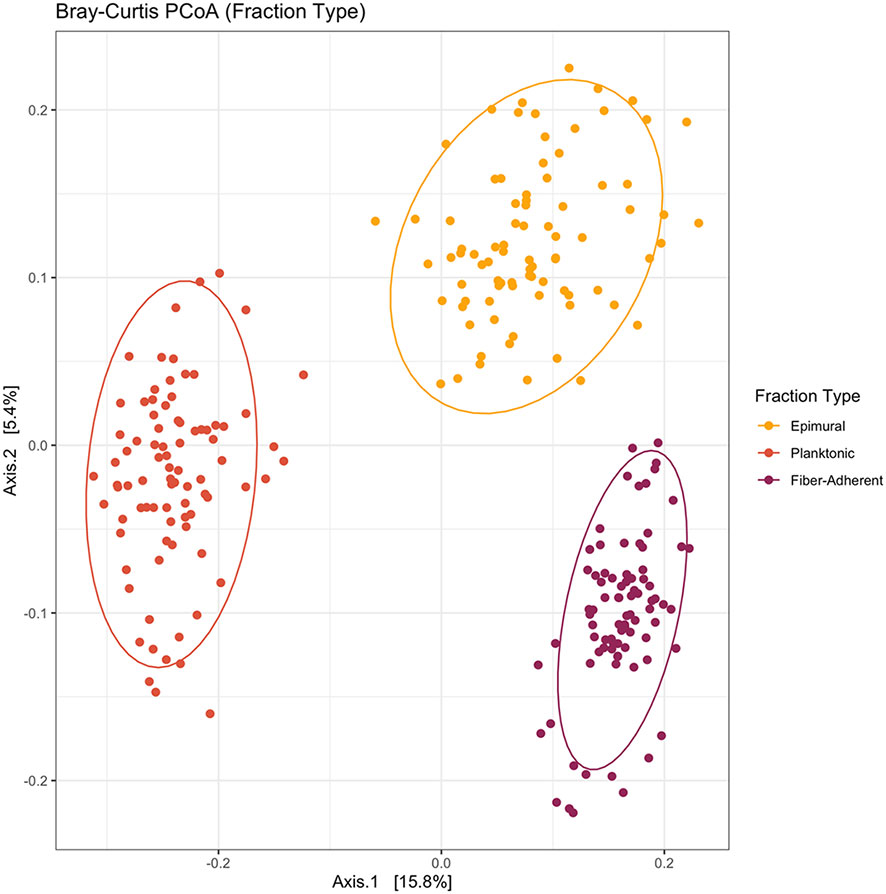
Figure 2 Bray-Curtis principal coordinates analysis among the three fraction types. Beta-diversity was measured using the Bray-Curtis distances and visualized with a principal coordinates analysis (PCoA). The epimural fraction is shown in orange, the planktonic digesta fraction is represented by red, and the fiber-adherent digesta fraction is represented by purple. The circles represent a 95% confidence interval around the means of each fraction type. There was a significant amount of distance between each fraction type (P < 0.05).
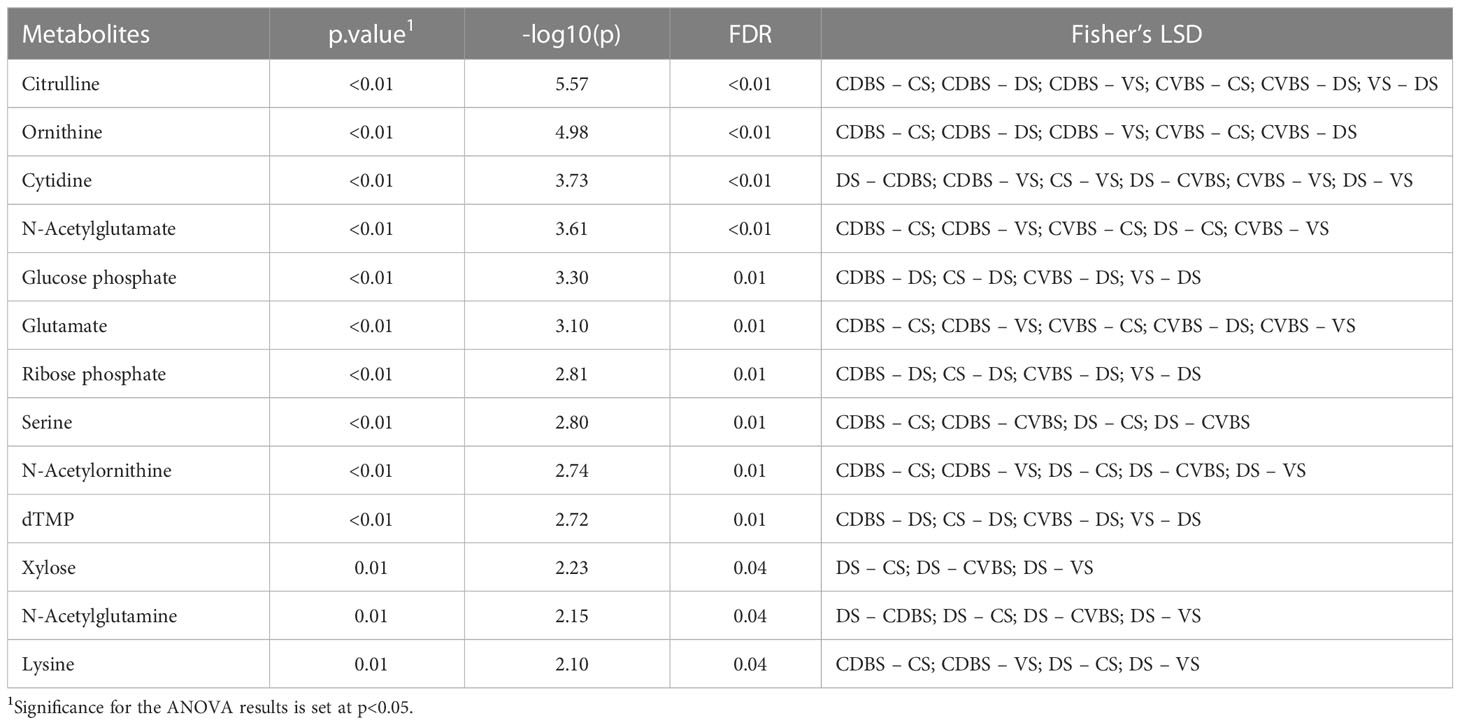
Table 1 Metabolite differences among the five rumen sacs.
Within the 84 fiber-adherent bacterial communities sequenced, 26 phyla and 219 genera were found among the five rumen sacs (Supplementary File 1). The top three phyla having the greatest relative abundance were Firmicutes (47.07%), Bacteroidota (37.50%), and Spirochaetota (3.23%). The remaining top ten phyla making up the greatest relative abundance within the rumen were Fibrobacterota (2.97%), Patescibacteria (2.55%), Verrucomicrobiota (1.93%), Thermoplasmatota (0.84%), Desulfobacterota (0.78%), Euryarchaeota (0.55%), and Proteobacteria (0.53%). The relative abundance of the top ten genera identified in the fiber-adherent community were Prevotella (10.16%), Rikenellaceae RC9 gut group (6.45%), Christensenellaceae R-7 group (5.09%), Succiniclasticum (3.55%), Ruminococcus (3.37%), Fibrobacter (2.97%), Treponema (2.83%), Saccharofermentans (2.57%), Butryvibrio (2.32%), and NK4A214 group (2.24%).
Out of the 85 planktonic samples, 28 phyla and 236 genera were found among the five rumen sacs (Supplementary File 1). The relative abundance of the phyla Bacteroidota (42.54%), Firmicutes (38.20%), and Verrucomicrobiota (7.57%) were the top three greatest identified within the rumen. The remaining top ten phyla were Patescibacteria (3.28%), Fibrobacterota (2.48%), Spirochaetota (1.99%), Proteobacteria (1.30%), Thermoplasmatota (0.69%), Desulfobacterota (0.55%), and Planctomycetota (0.45%). The top three greatest relative abundance of genera identified within the five rumen sacs were Prevotella (14.59%), Rikenellaceae RC9 gut group (4.64%), and Succiniclasticum (2.92%). The remaining seven genera with the greatest relative abundance were Christensenellaceae R-7 group (2.78%), Fibrobacter (2.47%), Prevotellaceae UCG-003 (2.41%), Ruminococcus (1.62%), Candidatus Saccharimonas (1.56%), Prevotellaceae UCG-001 (1.47%), and Treponema (1.40%).
Within the 84 samples collected from the rumen wall, 27 phyla and 297 genera were found among the five rumen sacs (Supplementary File 1). The top ten phyla identified with the greatest relative abundance were Bacteroidota (42.15%), Firmicutes (38.23%), Verrucomicrobiota (4.13%), Patescibacteria (3.02%), Proteobacteria (3.00%), Fibrobacterota (1.89%), Spirochaetota (1.56%), Desulfobacterota (1.25%), Thermoplasmatota (0.99%), and Planctomycetota (0.92%). The greatest relative abundance of genera found were Prevotella (11.17%), Rikenellaceae RC9 gut group (6.26%), and Christensenellaceae R-7 group (4.22%). The remaining genera in the top ten greatest relative abundance were Succiniclasticum (3.66%), Prevotellaceae UCG-001 (2.34%), Ruminococcus (1.95%), Butyvibrio (1.93%), Fibrobacter (1.89%), Prevotellaceae UCG-003 (1.80%), and NK4A214 group (1.77%).
Alpha diversity, beta diversity, and differential abundances among the sacs were initially analyzed by pooling of fiber-adherent, planktonic, and epimural community data, representing the entire bacterial community within the rumen. Richness and evenness did not differ among the five rumen sacs (P > 0.05) (Figure 3) when examining alpha diversity. Further, when analyzing the Bray-Curtis distance matrices, there were no differences among rumen sacs after testing with a PERMANOVA with 999 permutations (P > 0.05) (Figure 4). Measuring differential abundance with MaAsLin2 no significant differences were identified among the rumen sacs (P > 0.05).
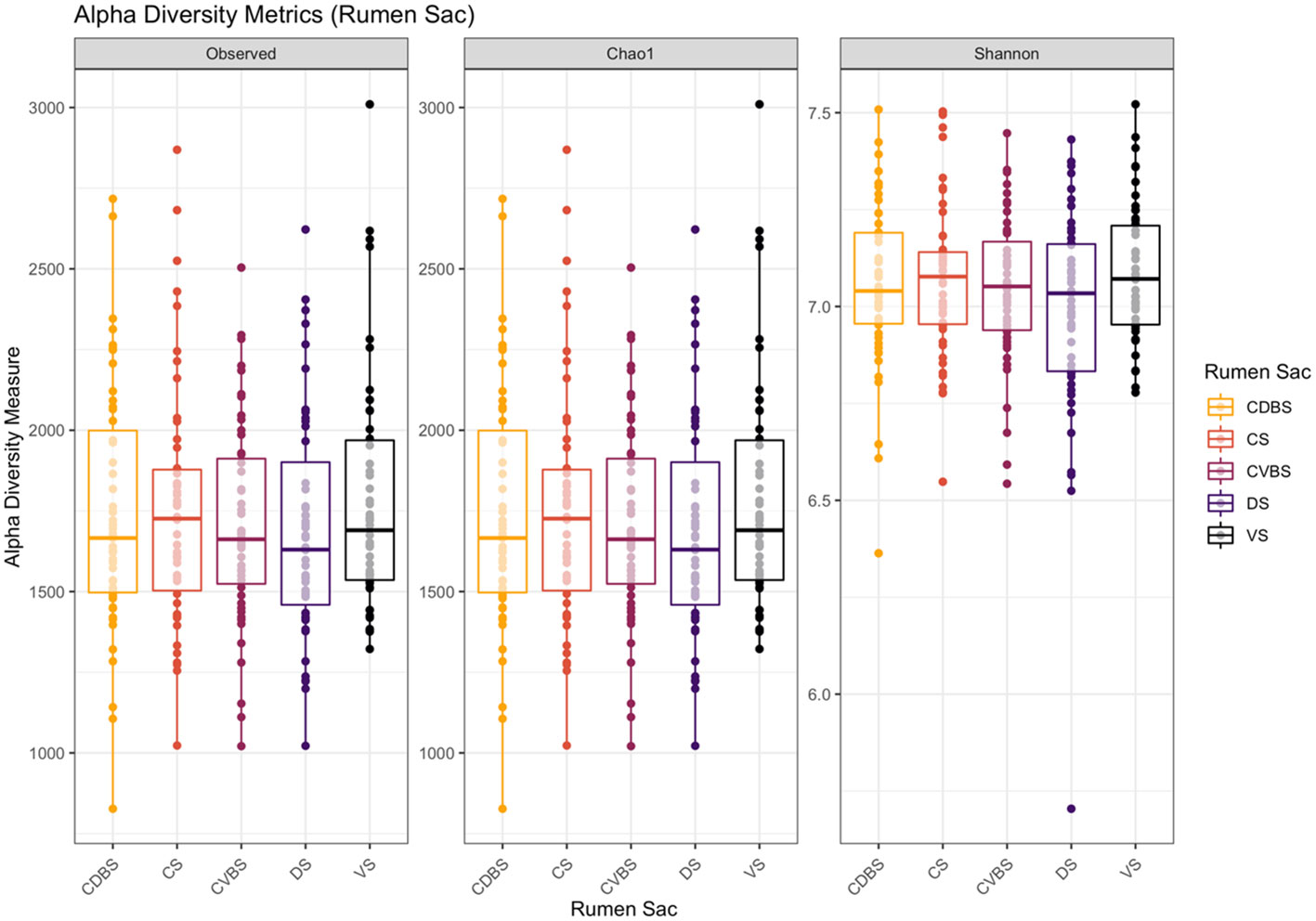
Figure 3 Alpha diversity metrics among the five rumen sacs bacterial communities. Alpha-diversity metrics for bacterial communities within each rumen sac (includes all three fraction types within a rumen sac). Measures include observed ASVs (richness), Chao1 (expected richness), and Shannon diversity index (richness and evenness). The CDBS is denoted by orange, CS is denoted by red, CVBS is denoted by light purple, DS is denoted by dark purple, and VS is denoted by black. With the removal of singletons during data analysis, the Chao1 measurement may appear similar to the observed measurement. No significance was determined among the rumen sacs P > 0.05.
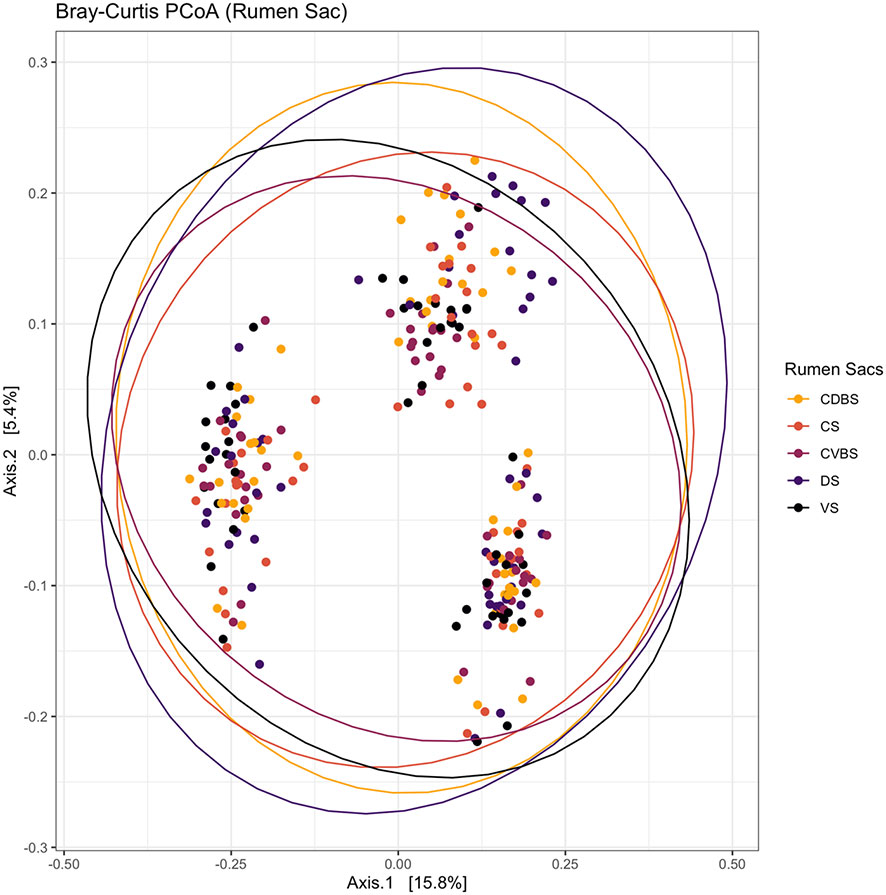
Figure 4 Bray-Curtis principal coordinates analysis among each rumen sac and their bacterial community. Beta-diversity was measured with the Bray-Curtis distances and visualized with a principal coordinates analysis (PCoA). The CDBS is shown in orange, CS is shown in red, CVBS is shown in light purple, DS is shown in dark purple, and VS is shown in black. The circles represent a 95% confidence interval around the means of each rumen sac. No significant distance measurement was determined among the rumen sacs (P > 0.05).
For alpha-diversity metrics, the observed ASVs, chao1, Shannon diversity index were measured for the fiber-adherent community within the rumen sacs to assess for richness and evenness (Figure 5). There were no differences in any alpha diversity measurements when comparing among the five rumen sacs (P > 0.05).

Figure 5 Alpha diversity metrics in the fiber-adherent fraction among the five rumen sacs. Alpha-diversity metrics for fiber-adherent bacterial communities among each of the five rumen sacs. Measures were observed ASVs (richness), Chao1 (expected richness), and Shannon diversity index (richness and evenness). The CDBS is denoted by orange, CS is denoted by red, CVBS is denoted by light purple, DS is denoted by dark purple, and VS is denoted by black. With the removal of singletons during data analysis, the Chao1 measurement may appear similar to the observed measurement. No significance was determined within the fiber-adherent microbial community among the rumen sacs (P > 0.05).
Among the five rumen sacs, no significant differences were observed when testing against the fiber-adherent communities (P > 0.05). The resulting PCoA aids as a visualization of the overlap among the fiber-adherent bacterial communities across all rumen sacs (Figure 6).
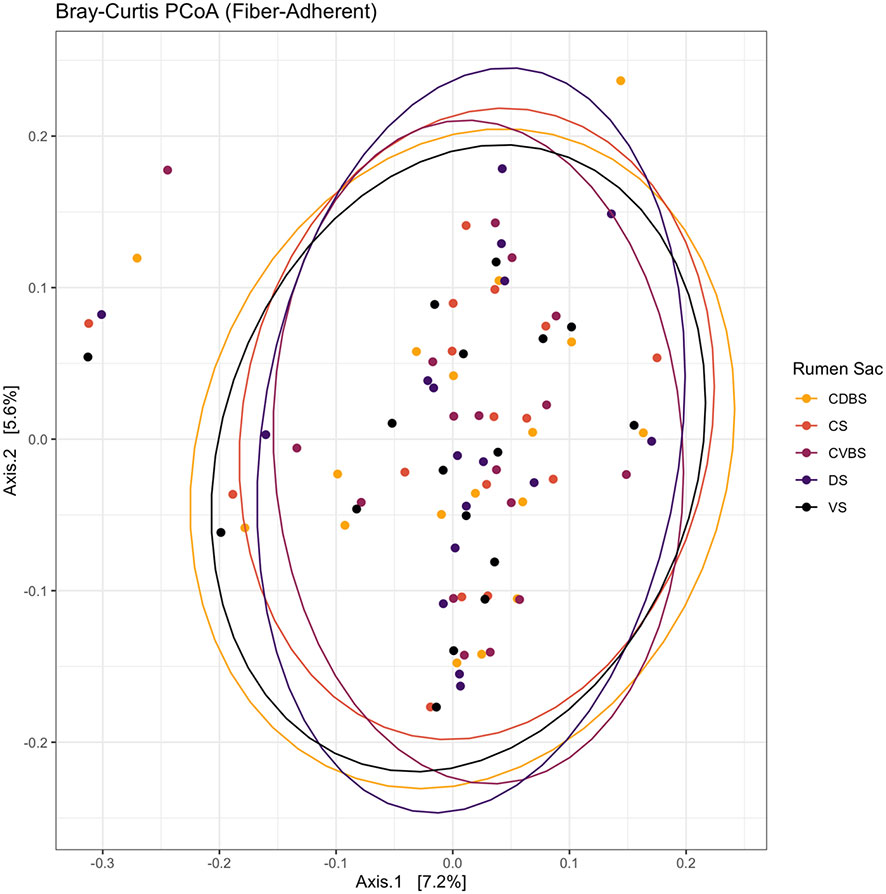
Figure 6 Bray-Curtis principal coordinates analysis of the fiber-adherent fraction among the five rumen sacs. Beta-diversity was measured with the Bray-Curtis distances and visualized with a principal coordinates analysis (PCoA). The CDBS is shown in the orange circles, CS is shown in red circles, CVBS is shown in light purple circles, DS is shown in dark purple circles, and VS is shown in black circles. The circles represent a 95% confidence interval around the means of the fiber-adherent bacterial communities within each rumen sac. No significant amount of distance was determined (P > 0.05).
When using MaAsLin2 to test differential abundances, there were no significant differences among the rumen sacs and the fiber-adherent bacterial communities (P > 0.05; Supplementary File 2).
The alpha-diversity metrics of observed ASVs, chao1, Shannon diversity index were measured and visualized in Figure 7. No significant differences distinguished in richness or evenness among any of the five rumen sacs within the planktonic bacterial communities (P > 0.05).
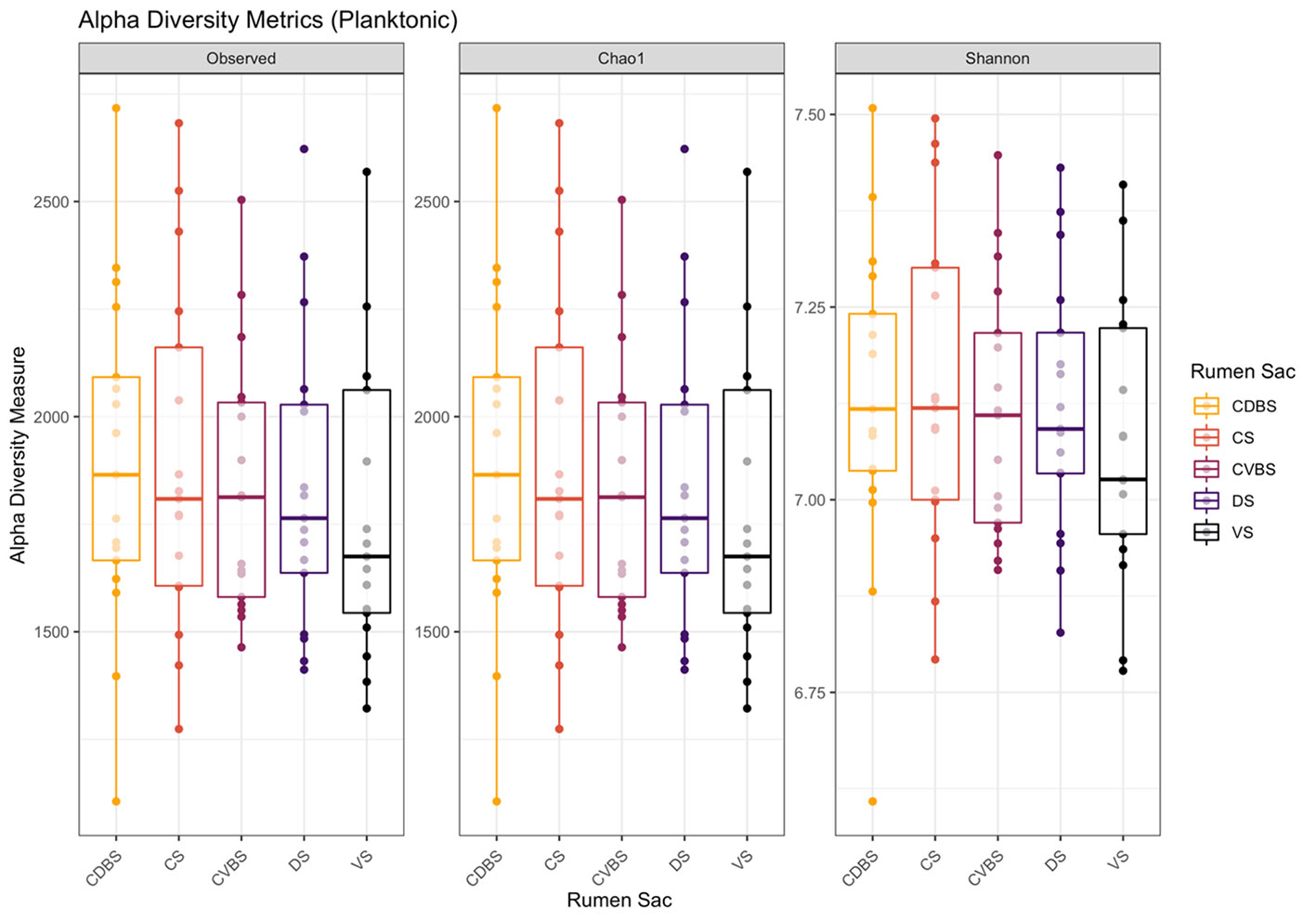
Figure 7 Alpha diversity metrics in the planktonic fraction among the five rumen sacs. Alpha-diversity metrics for the planktonic bacterial communities among each of the five rumen sacs. Measures were observed ASVs (richness), Chao1 (expected richness), and Shannon diversity index (richness and evenness). The CDBS is in orange, CS is in red, CVBS is in light purple, DS in dark purple, and VS is in black. With the removal of singletons during data analysis, the Chao1 measurement may appear similar to the observed measurement. No significance was identified within the planktonic microbial community among the rumen sacs (P > 0.05).
No significant differences were found within the planktonic communities among the five rumen sacs (P > 0.05). This lack of differences is visualized in the PCoA with the overlap among each rumen sac (Figure 8).
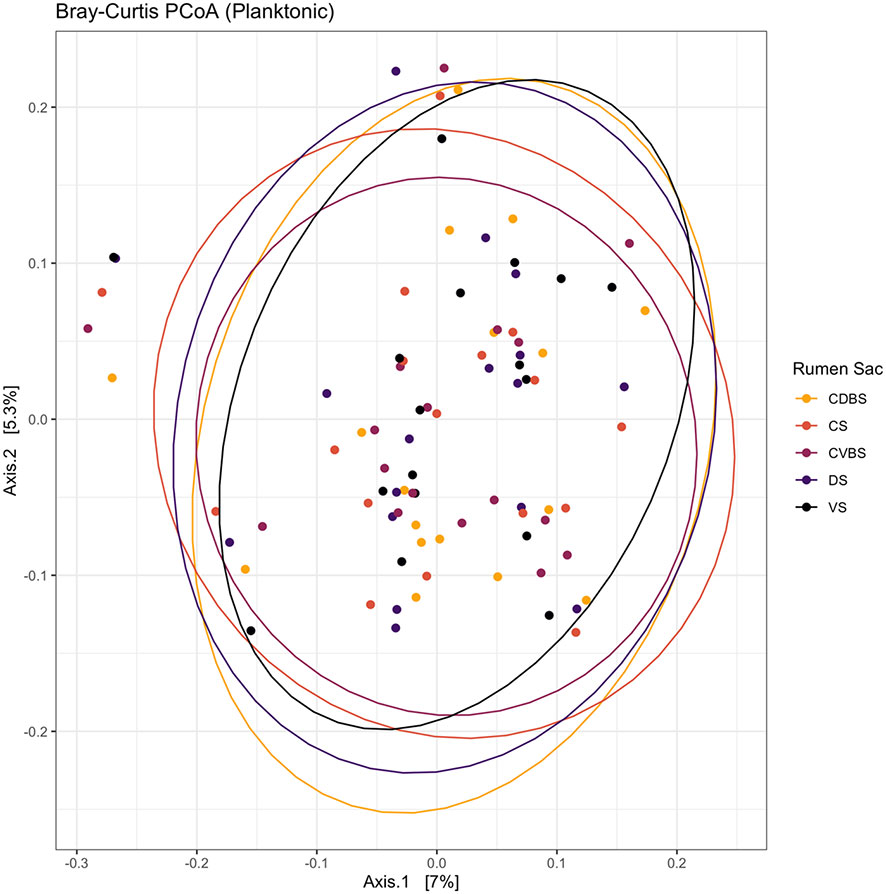
Figure 8 Bray-Curtis principal coordinates analysis of the planktonic fraction among the five rumen sacs. Beta-diversity was measured with the Bray-Curtis distances and visualized through a principal coordinates analysis (PCoA). The CDBS is shown in the orange circles, CS is shown in red circles, CVBS is shown in light purple circles, DS is shown in dark purple circles, and VS is shown in black circles. The circles represent a 95% confidence interval around the means of the planktonic bacterial communities within each rumen sac. No significant distance measurement was found (P > 0.05).
When utilizing pairwise comparisons in MaAslin2 to test differential abundances in bacterial communities among the five rumen sacs within the planktonic samples, 4 ASVs were found to be significantly different when testing against each rumen sac (P < 0.05; FDR < 0.05). The four significant differences among ASVs were only seen when either DS or VS were set as the reference level (i.e., differences only existed between DS and VS). The four ASVs significantly different between the VS and DS were three ASVs belonging to the family Bacteroidales RF16 group and one belonging to genus Christensenellaceae R-7 group (Supplementary File 2).
The epimural alpha-diversity metrics measured were observed ASVs, chao1, Shannon diversity index were measured and visualized in Figure 9. Shannon diversity was significant, measuring richness and evenness among the bacterial communities (P < 0.05). The remaining two measures of observed ASV and Chao1 were not significant (P > 0.05).
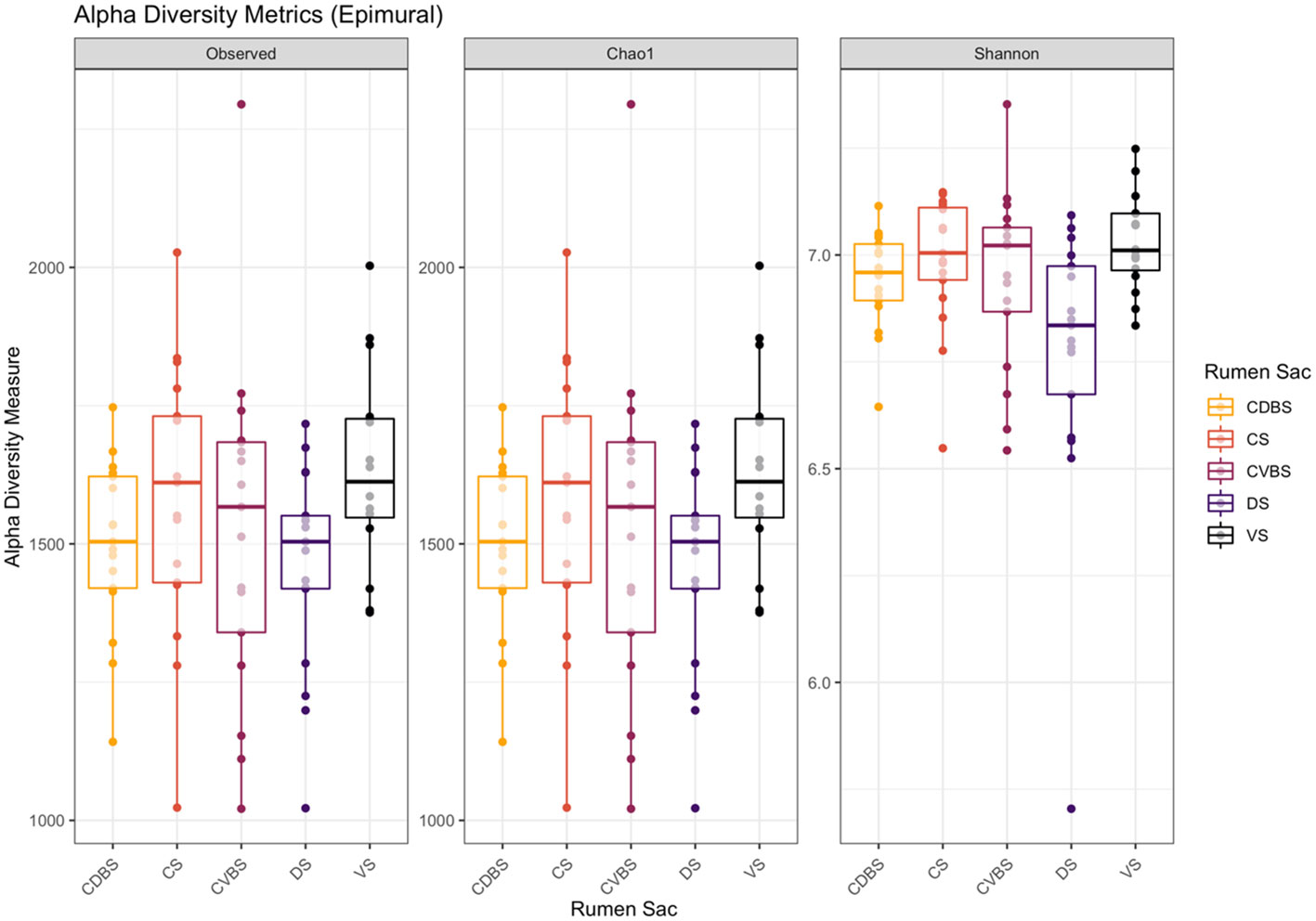
Figure 9 Alpha diversity metrics in the epimural fraction among the five rumen sacs. Alpha-diversity metrics for the epimural bacterial communities among each of the five rumen sacs. Measures include observed ASVs (richness), Chao1 (expected richness), and Shannon diversity index (richness and evenness). The CDBS is in orange, CS is in red, CVBS is in light purple, DS in dark purple, and VS is in black. Significance was determined at P < 0.05 in the measure of Shannon diversity. With the removal of singletons during data analysis, the Chao1 measurement may appear similar to the observed measurement. No significance was determined for the other two measurements (P > 0.05).
When assessing beta diversity among the five rumen sacs, there were significant differences among the epimural communities (P < 0.001). Greater distances among the rumen sacs were observed in the PCoA (Figure 10).
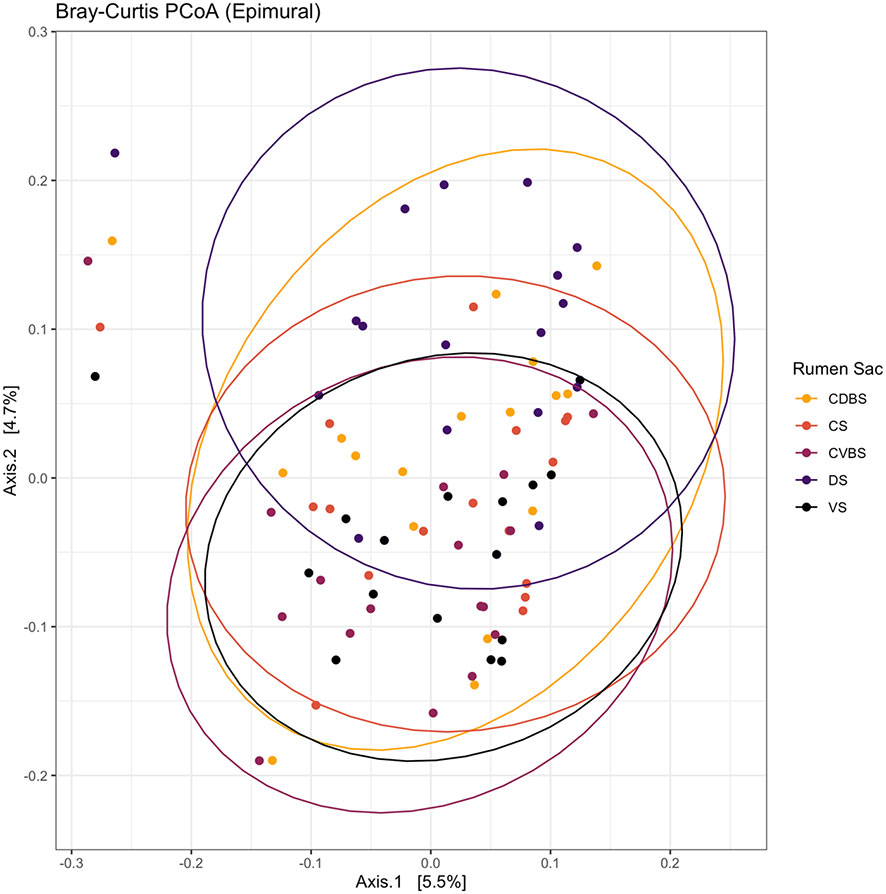
Figure 10 Bray-Curtis principal coordinates analysis of the epimural fraction among the five rumen sacs. Beta-diversity was measured with the Bray-Curtis distances and visualized with a principal coordinates analysis (PCoA). The CDBS is shown in the orange circles, CS is shown in red circles, CVBS is shown in light purple circles, DS is shown in dark purple circles, and VS is shown in black circles. The circles represent a 95% confidence interval around the means of the epimural communities in each rumen sac. Significance was determined at P < 0.05.
Using MaAslin2 to analyze differential abundances among rumen sacs in the epimural bacterial community, a greater number of significant differences were observed among the five rumen sacs contrasted to the other fraction types (Supplementary File 2). Overall, 33 ASVs were found to be significantly differentially abundant among the five rumen sacs and their respective pairwise comparisons (P < 0.05). When the reference level was set at the CDBS, there were five total significant differences where four ASVs were significantly different in the DS, and one ASV was in significantly different in the CVBS. When CS was set as the reference level, there were nine significant ASVs found to be differentially abundant against the DS. There were overall 23 significant differences when the reference level was set at CVBS. With 21 total significant differences against the DS, four ASVs were significantly different against the CDBS, and one ASV in significantly different against the VS. Overall, there were 31 significant differences when the reference level was set at DS, where many ASVs overlapped among the other four rumen sacs. With reference level DS, 24 differences were seen against the CVBS, there were 19 differences when against the VS, there were six differences against the CDBS, there were fourteen significant differences against the CS. There were 14 overall differences seen with the VS set as the reference level. Against the DS there were thirteen ASVs that were differentially abundant and there one ASV that was significantly different against the CVBS.
Global metabolomics analysis was performed on the 85 rumen liquid samples, where 69 metabolites were identified. When visually assessing the metabolites among the five rumen sacs using a partial least squares determinant analysis (PLS-DA), a significant overlap among all five rumen sacs indicated similar metabolic profiles among all five rumen sacs (P < 0.05) (Figure 11). Overall, metabolites detected in the CVBS displayed a greater relative abundance compared to the other four rumen sacs (Figure 12). Among the five rumen sacs, 13 metabolites differed, with the most notable differences between the CDBS and CVBS (Table 1).
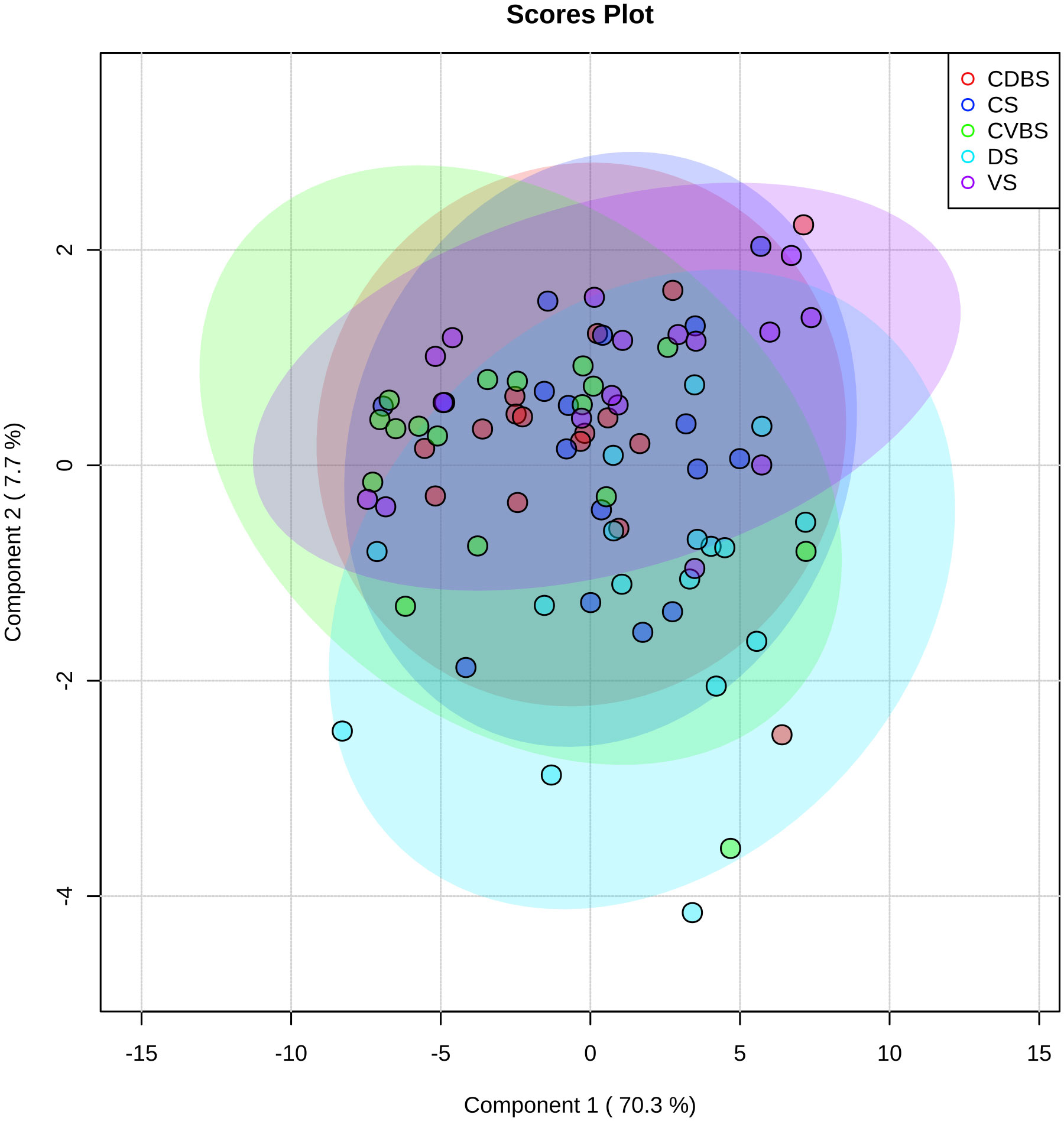
Figure 11 Partial least-square discriminant analysis of metabolites among all five rumen sacs. Partial least-square discriminant analysis of the rumen liquid samples, where each individual cow’s rumen sac is denoted by a dot of a specific color. Purple represents the VS, bright blue represents the DS, green represents the CVBS, dark blue represents CS, and red represents the CDBS. The large transparent circles represent a 95% confidence interval of each rumen sac, where the colors correspond with the dot color. There is significant overlap among each of the rumen sacs, indicating no unique metabolic profile among each rumen sac.
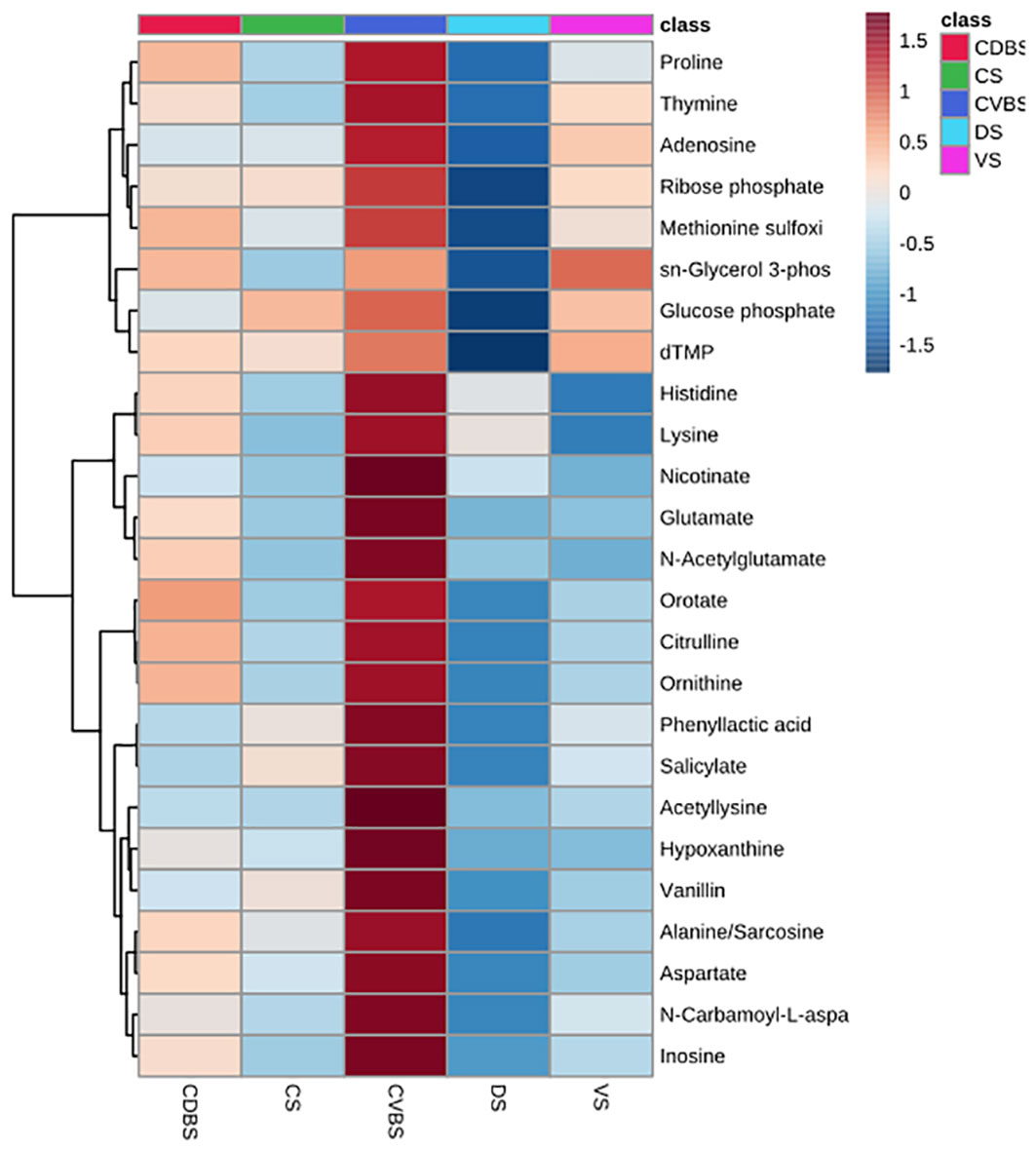
Figure 12 Heat map of top 25 metabolites among the five rumen sacs. Heat map of the top 25 metabolites present among each of the five rumen sacs. The darker the red equates to the greater relative abundance of the metabolite present within the rumen sac and the darker the blue equates to a lesser abundance of the metabolite present within the rumen sac. There was a difference in overall abundance of the metabolites in this heat map in the CVBS among the other four rumen sacs.
Relative pH was measured for the 85 pooled rumen liquid samples. The three significant comparisons were between DS and CDBS, DS and CVBS, and DS and VS (P < 0.05) (Table 2). Relative pH values are listed in Supplementary File 3.
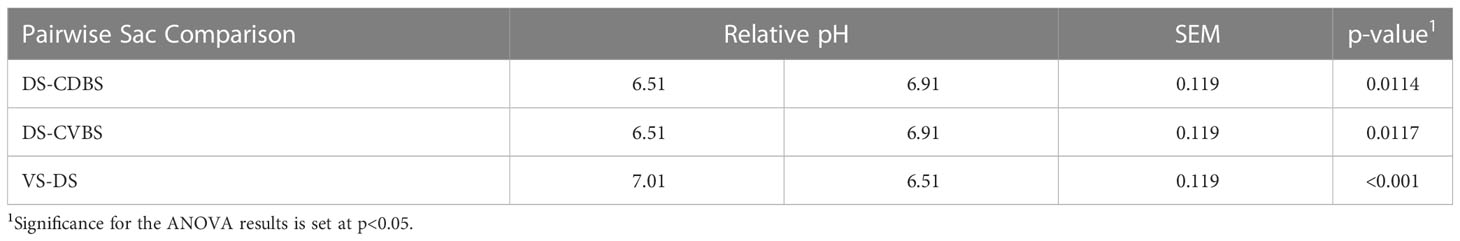
Table 2 Relative pH differences among the five rumen sacs.
To achieve the goal of feeding a growing population, cattle and other ruminants will be relied upon due to their inherent ability to break down human inedible products, utilizing them for maintenance and growth – i.e. meat and milk for human consumption. The importance of the rumen microbiome is well recognized for breaking down plant material into energy for the host (Hobson and Stewart, 1988; Mizrahi et al., 2021), and numerous investigations into the efficiency of cattle have been conducted regarding the rumen microbiome (Li and Guan, 2017; Myer et al., 2017; Elolimy et al., 2018). Many studies have focused solely on the ventral sac (Petri et al., 2013; Neubauer et al., 2019) due to the ease of access and repeatability in sampling this region of the rumen. However, there is evidence that microbial variation exists among the fraction levels within the rumen and among the different rumen sacs (Schären et al., 2017; Sbardellati et al., 2020).
The main objective of this study was to investigate the ruminal biogeographical regions, hypothesizing that differences in bacterial species and metabolome would occur due to differing anatomy and physiology associated with the respective regions. However, neither alpha diversity, beta diversity, nor differential abundance of bacterial communities was significant when analyzing the five rumen sacs, where each rumen sac encompassed the data from each of the three fractions pooled together. When analyzing the three distinct ruminal fractions, significance was measured in alpha diversity, beta diversity, and differential abundance. Historically, the dissimilarity among the ruminal fractions has been observed and, therefore, was an expected outcome (De Mulder et al., 2016; Ji et al., 2017; Skarlupka et al., 2019; Abbas et al., 2020). However, when evaluating alpha diversity, the main differences among the three fractions appeared to be driven by the epimural community.
Fiber-adherent and planktonic communities work closely together during normal ruminal fermentation and digestion due to the contractions and subsequent mixing of the digesta fractions, resulting in a closer spatial relationship between the communities (Schären et al., 2017). The distinct differences noted apart from the rumen content fractions were especially important when evaluating the epimural community. Differences were observed in Shannon diversity, beta diversity, and differential abundances in bacterial species found among the five rumen sacs. Significance in Shannon diversity indicates changes in bacterial species richness and community evenness, whereas no richness metrics were different, which may signify that although richness/abundance may not be influential among the rumen sacs, community diversity and evenness play a larger role in the ruminal environment. This trend has been found in a previous study conducted on cannulated Holstein cows, where epimural samples were taken from four rumen sacs (Sbardellati et al., 2020). When sampled from the CS, CDBS, CVBS and VS, the CDBS had the greatest Shannon diversity (Sbardellati et al., 2020), which was in contrast to our study, where the DS had the lowest Shannon diversity. However, the other four rumen sacs in our study were similar in composition. Had researchers sampled from the DS they may have identified similar results. These data indicate less diversity in the DS, which is likely a result of the lesser extent of feedstuff digestion occurring in this rumen sac. The extent of the dissimilarity of the DS among the other four rumen sacs provides evidence of the DS dominated by distinct species of microbes which may indicate specific duties conducted by the epimural microbial communities in the DS (Sbardellati et al., 2020).
Epimural community dissimilarity among the five rumen sacs is also highlighted by beta diversity distance matrices, indicating different taxonomic composition of bacterial communities within the epimural fraction. Among the sacs, the DS has greater distinction when measured against the other four rumen sacs. Vertical stratification of the rumen contents may drive these changes due to the fiber mat and gaseous phase residing in the DS. The DS is significantly less papillated due to lesser blood flow to this rumen sac (Von Engelhardt and Hales, 1977) and the lesser extent of feedstuffs residing. Therefore, the necessity for papillae to develop for the purpose of microbial by-product absorption is reduced. Also, pillars are not present in the DS to prevent gas from the esophagus during normal feeding/rumination to escape into this sac which further increases gas accumulation (Krehbiel, 2014). With greater accumulation of gas, oxygen scavenging or urea recycling is an important function for DS bacterial species, while fewer microbes support the breakdown of feedstuffs or absorption of nutrients (Ren et al., 2020).
The epimural fraction’s bacterial communities have many other specialized functions including their innate ability to act as an interface between the host and the rumen microbiome, nitrogen cycling, and rumen epithelial tissue recycling (Schären et al., 2017; Ren et al., 2020; Sbardellati et al., 2020). In this study, significant differences were found in bacterial differential abundance determined between the DS and the other four rumen sacs, especially within the genus Campylobacter. Campylobacter is a non-fermentative bacterial species (Hassan et al., 2021) that has been identified in other research as part of the epimural bacterial community and has since been determined as both a nitrate-reducing bacterial species and a responder to oxidative stress (Mann et al., 2018; Ren et al., 2020; Sbardellati et al., 2020; Hassan et al., 2021). In a study investigating the epimural community of three Holstein dairy cows, Campylobacter was a highly abundant genus and had high expression of glutamate dehydrogenase and glutamine synthase, both of which are important in nitrogen metabolism, and thioredoxin reductase, which participates in the oxidative stress response (Mann et al., 2018). It has been observed that increasing nitrate will increase the abundance of Campylobacter. This response to increased nitrate is important for the rumen microbiome so that optimal usage of nitrate is provided and the toxic effects of nitrite accrual in the rumen are avoided (Ao and Emeritus, 2008; Hassan et al., 2021).
The rumen liquid metabolome may further support the distinct rumen biogeographical region of the DS. Thirteen out of 69 total metabolites were determined to be significantly different among the rumen sacs, and although the data were analyzed with pairwise comparisons, it does not imply that a metabolite was different among all rumen sacs. Indeed, four metabolites were significantly different in the DS and one metabolite was significantly different in the VS when compared to the other four rumen sacs. Although only four of the metabolites differed in the DS when compared to the other four rumen sacs, the 13 differentially expressed metabolites consistently had differences in the DS as well. There were numerous significant abundance differences determined among the rumen sacs regarding the epimural bacterial community and metabolome, suggesting that functional variation (Sbardellati et al., 2020) within the rumen sacs may contribute to the dissimilarity in the epimural community. Further, due to distinctions in the regional conditions of each sac, microbe function may vary to maintain ruminal homeostasis. The extent of the environmental distinction in the DS can be further explained by the relative pH measures, where significance was solely determined against the DS indicating microbes in this sac may need to acclimate to this region differently than other regions. This study noted the variability within the epimural community and how biogeographical regions are a major factor when sampling from the rumen. Without examining each rumen sac, valuable data may be overlooked. Research may consider examining these communities due to the importance of the epimural community in maintaining ruminal homeostasis and host-microbe interactions (Zhou et al., 2021).
When examining the entirety of the bacterial communities within the rumen, all three fractions had both Firmicutes and Bacteroidetes as the top two phyla, which have been historically identified in ruminant and mammalian studies (Hart et al., 2018; Sbardellati et al., 2020; Mizrahi et al., 2021). Microbes within these phyla are likely part of the core microbiome of cattle (Wirth et al., 2018). However, in the present study, Spirochaetota was the third most abundant phyla in the fiber-adherent community and Verrucomicrobiota was the third most abundant phyla for both the planktonic and epimural community. A greater abundance of Spirochaetota in the fiber-adherent community is not atypical due to their ability to degrade pectin, hemicellulose, and lignocellulose within the rumen (Gharechahi et al., 2021). The abundance ofthe Spirochaetota phylum in the fiber-adherent bacterial community may be anticipated in this study, as cattle grazed warm season forages. The identification and higher prevalence of Verrucomicrobiota in both the epimural and planktonic microbial communities is infrequent. Verrucomicrobiota have been examined in a lignocellulosic forage studies, where bacteria in this phylum have carbohydrate active enzymes, sulfatases, and peptidases (Gharechahi et al., 2022) which facilitate carbohydrate polysaccharide metabolism and may aid in lignocellulose breakdown within the rumen. In general, having a major phylum in the planktonic community being involved in cellulose degradation is not uncommon (Schären et al., 2017; Tan et al., 2021; Zhou et al., 2021), but it is more unexpected for the epimural community where Proteobacteria are typically detected at greater abundances (De Mulder et al., 2016; Sbardellati et al., 2020; Pacifico et al., 2021; Zhou et al., 2021). Genera within Proteobacteria are comprised of aerobes and facultative anaerobes, and thus are able to participate in oxygen scavenging (De Mulder et al., 2016; Zhou et al., 2021). The ability of Proteobacteria to scavenge oxygen aids in maintaining the anaerobic environment in which oxygen would be toxic to obligate anaerobic microbes (Sadet-Bourgeteau et al., 2010). Proteobacteria was found as the fifth most abundant phyla in the epimural community in this study. Compared to other studies, this may have been due to rumen fill. In the rumen of the cattle used for this study, the DS had a dense lawn of papillae which is far from average (Membrive, 2016). Due to increased fill seen throughout the development of these cattle, the greater papillae may have been higher in prevalence to aid absorption in the DS, causing the shift from the typical microbial phyla profiles.
The absence of differential abundance in bacterial communities within the fiber-adherent fraction among the five rumen sacs may be attributable to the rumen fill. Typically, rumens develop vertical stratification of ruminal contents, where the majority of liquid accumulates in the VS and the fiber mat floats atop liquid due to lower specific gravity, and gas fills the remainder of the dorsal space (Schmitz-Esser, 2021). When the rumen fill is greater than average, there may be greater dispersion of the forage feedstuffs among the rumen sacs which may have forced forages into uncharacteristic rumen sacs. This uncharacteristically high rumen fill may have caused lesser distinction of microbes among sacs that was determined in this project due to the greater homogeneity of forage feedstuffs among the rumen sacs. This rationale is also supported by the limited differences in the planktonic community bacteria, as only four bacteria were different among the sacs. The primary contractions constantly shift and displace rumen liquid (Krehbiel, 2014; Roehe et al., 2016), potentially resulting in a more homogenous rumen liquid among the rumen sacs. Further, the significant differential abundances in the planktonic community were only between the VS and DS; although in the metabolome, stark relative abundance differences were determined between CVBS and VS in the top twenty-five metabolites. Rumen motility and contractions throughout the rumen may influence these metabolite differences. The primary contraction cycle moves digesta in a clockwise fashion around the rumen; thus, higher density feedstuffs settle in more ventral sacs (Krehbiel, 2014; Reece et al., 2015). The feedstuffs settling lower may provide the CVBS with increased time for metabolism and subsequently increasing the abundance of metabolites present in the CVBS (Schmitz-Esser, 2021). It must also be noted the metabolome in the VS is distinct to the CVBS, which is interesting since they are both lower rumen sacs. Research regarding the metabolism and metabolome among the five rumen sacs may facilitate more efficient cattle through better knowledge of how feedstuffs are metabolized throughout the entirety of the rumen.
To date, limited studies have examined the microbial communities and fermentative metabolome among each of the five biogeographical locations and within each of the three fractions within the rumen. There were significant differences found when focusing on the ruminal fraction communities and their differences among the five rumen sacs, further providing evidence that the rumen is not simply a homogeneous fermentation vat. The complex rumen microbiome works with differing functions to break down feedstuffs to synergistically provide energy for the host. Variation within the rumen microbiome based on differing diets exists (Loor et al., 2016; Snelling et al., 2019), but the microbiome is able to adapt to these changes to create an efficient microbiome (Clemmons et al., 2019; Snelling et al., 2019). A majority of the rumen biogeographical differences determined were against the DS and may be due to a multitude of causes, including digesta vertical stratification, papillation, and blood flow. The DS distinction may also further highlight the importance of the epimural community. This study provided further evidence to the necessity for sampling from each of the three ruminal fractions, and the critical need to sample from each of the five rumen sacs, especially when focusing on the epimural fraction.
The rumen is a complex organ, demarcated into five distinct rumen sacs due to distinct anatomical structure, and variable host tissue physiology. Distinctions in anatomical structure are emphasized by microbial and fermentative variability determined through the microbial communities in each rumen sac and fraction type and the rumen metabolome that was investigated in this project. The foremost component differing was driven by the epimural microbial community and particularly when analyzing the DS against the other four rumen fractions. These insights further indicate that the epimural microbes offer the best opportunity of interacting with the host (Schmitz-Esser, 2021; Zhou et al., 2021). Future studies should examine host expression among the rumen biogeographical locations to support the microbial and fermentative differences found in this study, as these data may also help elucidate any impacts on nutrient flow from the rumen, impacting gastrointestinal absorption of nutrients or microbial protein. Despite the differences found, microorganisms around the rumen and within different fractions work together to provide the host with the energy and substrates it needs to create an efficient host. Optimizing the structure of the rumen microbiome may continue to provide data on creating an increasingly efficient host, aiding in feeding the ever-growing global population.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA928747.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee, The University of Tennessee.
MS, AE-M, BV, JS, and P-YM contributed to conception and design of the study. MS organized and conducted the study. MS, SM, LS, CC, SC, and MH performed the statistical analysis. MS wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the USDA-NIFA Hatch/Multistate Project W-3010 - TEN00578: Integrated Approach to Enhance Efficiency of Feed Utilization in Beef Production Systems; Accession Number: 1025826.
The authors would like to thank Brandon Beavers, the University of Tennessee, as well as the East Tennessee AgResearch and Education Center for allowing the use of the facilities and cattle.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fanim.2023.1154463/full#supplementary-material
Supplementary File 1 | Rumen taxa results
Supplementary File 2 | Rumen pairwise comparison results
Supplementary File 3 | pH data
Abbas W., Keel B. N., Kachman S. D., Fernando S. C., Wells J. E., Hales K. E., et al. (2020). Rumen epithelial transcriptome and microbiome profiles of rumen epithelium and contents of beef cattle with and without liver abscesses. J. Anim. Sci. 98 (12), skaa359. doi: 10.1093/jas/skaa359
Ao R. L., Emeritus D. (2008). “The potential of feeding nitrate to reduce enteric methane production in ruminants,” in A report to the department of climate change, Canberra, Australia. (Commonwealth Government of Australia Canberra ACT, Australia).
Apprill A., McNally S., Parsons R., Weber L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microbial. Ecol. 75 (2), 129–137. doi: 10.3354/ame01753
Ault-Seay T. B., Brandt K. J., Henniger M. T., Payton R. R., Mathew D. J., Moorey S. E., et al. (2022). Bacterial communities of the uterus and rumen during heifer development with protein supplementation. Front. Anim. Sci, 3. doi: 10.3389/fanim.2022.903909
Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc: Series B Stat. (Methodol.) 57 (1), 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30 (10), 918–920. doi: 10.1038/nbt.2377
Clasquin M. F., Melamud E., Rabinowitz J. D. (2012). LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr. Protoc. Bioinformatics Chapter 14, Unit14.11. doi: 10.1002/0471250953.bi1411s37
Clemmons B. A., Martino C., Schneider L. G., Lefler J., Embree M. M., Myer P. R. (2019). Temporal stability of the ruminal bacterial communities in beef steers. Sci. Rep. 9 (1), 9522. doi: 10.1038/s41598-019-45995-2
De Mulder T., Goossens K., Peiren N., Vandaele L., Haegeman A., De Tender C., et al. (2016). Exploring the methanogen and bacterial communities of rumen environments: solid adherent, fluid and epimural. FEMS Microbiol. Ecol. 93 (3). doi: 10.1093/femsec/fiw251
Dridi J. S., Greene E. S., Maynard C. W., Brugaletta G., Ramser A., Christopher C. J., et al. (2022). Duodenal metabolic profile changes in heat-stressed broilers. Animals 12 (11), 1337. doi: 10.3390/ani12111337
Elolimy A. A., Abdelmegeid M. K., McCann J. C., Shike D. W., Loor J. J. (2018). Residual feed intake in beef cattle and its association with carcass traits, ruminal solid-fraction bacteria, and epithelium gene expression. J. Anim. Sci. Biotechnol. 9 (1), 67. doi: 10.1186/s40104-018-0283-8
Gharechahi J., Sarikhan S., Han J.-L., Ding X.-Z., Salekdeh G. H. (2022). Functional and phylogenetic analyses of camel rumen microbiota associated with different lignocellulosic substrates. NPJ Biofilms Microbiomes 8 (1), 46. doi: 10.1038/s41522-022-00309-9
Gharechahi J., Vahidi M. F., Bahram M., Han J.-L., Ding X.-Z., Salekdeh G. H. (2021). Metagenomic analysis reveals a dynamic microbiome with diversified adaptive functions to utilize high lignocellulosic forages in the cattle rumen. ISME J. 15 (4), 1108–1120. doi: 10.1038/s41396-020-00837-2
Hart E. H., Creevey C. J., Hitch T., Kingston-Smith A. H. (2018). Meta-proteomics of rumen microbiota indicates niche compartmentalisation and functional dominance in a limited number of metabolic pathways between abundant bacteria. Sci. Rep. 8 (1), 10504. doi: 10.1038/s41598-018-28827-7
Hassan F.-u., Guo Y., Li M., Tang Z., Peng L., X. Liang, et al. (2021). Effect of methionine supplementation on rumen microbiota, fermentation, and amino acid metabolism in In vitro cultures containing nitrate. Microorganisms 9 (8)1717. doi: 10.3390/microorganisms9081717
Hobson P. N., Stewart C. S. (eds). (1988). Rumen microbial ecosystem (Springer Science & Business Media).
Ji S., Zhang H., Yan H., Azarfar A., Shi H., Alugongo G., et al. (2017). Comparison of rumen bacteria distribution in original rumen digesta, rumen liquid and solid fractions in lactating Holstein cows. J. Anim. Sci. Biotechnol. 8 (1), 16. doi: 10.1186/s40104-017-0142-z
Kassambara A.. (2023). fastqcr: Quality Control of Sequencing Data. R package version 0.1.3. Available at: https://CRAN.R-project.org/package=fastqcr.
Krehbiel C. R. (2014). Invited review: Applied nutrition of ruminants: Fermentation and digestive physiology. Prof. Anim. Scientist 30 (2), 129–139. doi: 10.15232/S1080-7446(15)30100-5
Li F., Guan L. L. (2017). Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl. Environ. Microbiol. 83 (9), e00061–e00017. doi: 10.1128/AEM.00061-17
Loor J. J., Elolimy A. A., McCann J. C. (2016). Dietary impacts on rumen microbiota in beef and dairy production. Anim. Front. 6 (3), 22–29. doi: 10.2527/af.2016-0030
Lu W., Clasquin M. F., Melamud E., Amador-Noguez D., Caudy A. A., Rabinowitz J. D. (2010). Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 82 (8), 3212–3221. doi: 10.1021/ac902837x
Ma Z., Wang R., Wang M., Zhang X., Mao H., Tan Z. (2018). Short communication: Variability in fermentation end-products and methanogen communities in different rumen sites of dairy cows. J. Dairy Sci. 101 (6), 5153–5158. doi: 10.3168/jds.2017-14096
Mallick H., Rahnavard A., McIver L. J. (2020). MaAsLin 2: Multivariable Association in Population-scale Meta-omics Studies. R/Bioconductor package. Available at: http://huttenhower.sph.harvard.edu/maaslin2.
Mallick H., Rahnavard A., McIver L. J., Ma S., Zhang Y., Nguyen L. H., et al (2021). Multivariable association discovery in population-scale meta-omics studies. PLOS Computational Biology 17 (11). doi: 10.1371/journal.pcbi.1009442
Mann E., Wetzels S. U., Wagner M., Zebeli Q., Schmitz-Esser S. (2018). Metatranscriptome sequencing reveals insights into the gene expression and functional potential of rumen wall bacteria. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00043
Martens L., Chambers M., Sturm M., Kessner D., Levander F., Shofstahl J., et al. (2011). mzML–a community standard for mass spectrometry data. Mol. Cell. Proteomics MCP 10 (1), R110.000133. doi: 10.1074/mcp.R110.000133
McMurdie P. J., Holmes S. (2013). Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8 (4), e61217. doi: 10.1371/journal.pone.0061217
Melamud E., Vastag L., Rabinowitz J. D. (2010). Metabolomic analysis and visualization engine for LC–MS data. Analytical Chem. 82 (23), 9818–9826. doi: 10.1021/ac1021166
Membrive C. M. B. (2016). Anatomy and physiology of the rumen. In Millen D. D., De Beni Arrigoni M., Lauritano Pacheco R.D. eds. Rumenology. (Cham: Springer International Publishing), 1–38. doi: 10.1007/978-3-319-30533-2_1
Mizrahi I. (2013). “Rumen symbioses,” in The prokaryotes: Prokaryotic biology and symbiotic associations. Eds. Rosenberg E., DeLong E. F., Lory S., Stackebrandt E., Thompson. F. (Berlin, Heidelberg: Springer Berlin Heidelberg), 533–544.
Mizrahi I., Wallace R. J., Morais S. (2021). The rumen microbiome: balancing food security and environmental impacts. Nat. Rev. Microbiol. 19 (9), 553–566. doi: 10.1038/s41579-021-00543-6
Myer P. R., Freetly H. C., Wells J. E., Smith T. P. L., Kuehn L. A. (2017). Analysis of the gut bacterial communities in beef cattle and their association with feed intake, growth, and efficiency1,2,3. J. Anim. Sci. 95 (7), 3215–3224. doi: 10.2527/jas.2016.1059
Na S. W. (2022). Understanding the role of rumen epithelial host-microbial interactions in cattle feed efficiency. Anim. Nutr. 10, 41–53.
Na S. W., Guan L. L. (2022). Understanding the role of rumen epithelial host-microbe interactions in cattle feed efficiency. Anim. Nutr. 10, 41–53. doi: 10.1016/j.aninu.2022.04.002
Nearing J. T., Douglas G. M., Hayes M. G., MacDonald J., Desai D. K., Allward N., et al. (2022). Microbiome differential abundance methods produce different results across 38 datasets. Nat. Comm. 13 (1), 342. doi: 10.1038/s41467-022-28034-z
Neubauer V., Humer E., Mann E., Kröger I., Reisinger N., Wagner M., et al. (2019). Effects of clay mineral supplementation on particle-associated and epimural microbiota, and gene expression in the rumen of cows fed high-concentrate diet. Anaerobe 59, 38–48. doi: 10.1016/j.anaerobe.2019.05.003
Oksanen J., Simpson G., Blanchet F. G., Kindt R., Legendre P., Minchin P., et al. (2022). Vegan: Community ecology package version 2. R package version 2.6-2. Available at: https://CRAN.R-project.org/package=vegan.
Pacifico C., Petri R. M., Ricci S., Mickdam E., Wetzels S. U., Neubauer V., et al. (2021). Unveiling the bovine epimural microbiota composition and putative function. Microorganisms 9 (2), 342. doi: 10.3390/microorganisms9020342
Pang Z., Chong J., Zhou G., de Lima Morais D. A., Chang L., Barrette M., et al. (2021). MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49 (W1), W388–W396. doi: 10.1093/nar/gkab382
Parada A. E., Needham D. M., Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18 (5), 1403–1414. doi: 10.1111/1462-2920.13023
Petri R. M., Neubauer V., Humer E., Kröger I., Reisinger N., Zebeli Q. (2020). Feed additives differentially impact the epimural microbiota and host epithelial gene expression of the bovine rumen fed diets rich in concentrates. Front. Microbiol. 11, 119. doi: 10.3389/fmicb.2020.00119
Petri R. M., Schwaiger T., Penner G. B., Beauchemin K. A., Forster R. J., McKinnon J. J., et al. (2013). Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis. Appl. Environ. Microbiol. 79 (12), 3744–3755. doi: 10.1128/AEM.03983-12
Reece W. O., Erickson H. H., Goff J. P., Uemura E. E. (2015). Dukes' physiology of domestic animals. John Wiley & Sons.
Ren Q., Si H., Yan X., Liu C., Ding L., Long R., et al. (2020). Bacterial communities in the solid, liquid, dorsal, and ventral epithelium fractions of yak (Bos grunniens) rumen. Microbiologyopen 9 (2), e963. doi: 10.1002/mbo3.963
Roehe R., Dewhurst R. J., Duthie C.-A., Rooke J. A., McKain N., Ross D. W., et al. (2016). Bovine host genetic variation influences rumen microbial methane production with best selection criterion for low methane emitting and efficiently feed converting hosts based on metagenomic gene abundance. PloS Genet. 12 (2), e1005846. doi: 10.1371/journal.pgen.1005846
Sadet-Bourgeteau S., Martin C., Morgavi D. P. (2010). Bacterial diversity dynamics in rumen epithelium of wethers fed forage and mixed concentrate forage diets. Veterinary Microbiol. 146 (1), 98–104. doi: 10.1016/j.vetmic.2010.04.029
Sbardellati D. L., Fischer A., Cox M. S., Li W., Kalscheur K. F., Suen G. (2020). The bovine epimural microbiota displays compositional and structural heterogeneity across different ruminal locations. J. Dairy Sci. 103 (4), 3636–3647. doi: 10.3168/jds.2019-17649
Schären M., Kiri K., Riede S., Gardener M., Meyer U., Hummel J., et al. (2017). Alterations in the rumen liquid-, particle- and epithelium-associated microbiota of dairy cows during the transition from a silage- and concentrate-based ration to pasture in spring. Front. Microbiol. 8, 744. doi: 10.3389/fmicb.2017.00744
Schmitz-Esser S. (2021). The rumen epithelial microbiota: Possible gatekeepers of the rumen epithelium and its potential contributions to epithelial barrier function and animal health and performance. Meat Muscle Biol. 4 (2). doi: 10.22175/mmb.11672
Skarlupka J. H., Kamenetsky M. E., Jewell K. A., Suen G. (2019). The ruminal bacterial community in lactating dairy cows has limited variation on a day-to-day basis. J. Anim. Sci. Biotechnol. 10 (1), 1–5. doi: 10.1186/s40104-019-0375-0
Snelling T. J., Auffret M. D., Duthie C.-A., Stewart R. D., Watson M., Dewhurst R. J., et al. (2019). Temporal stability of the rumen microbiota in beef cattle, and response to diet and supplements. Anim. Microbiome 1 (1), 1–14. doi: 10.1186/s42523-019-0018-y
Tan R. S., Zhou M., Li F. (2021). Identifying active rumen epithelial associated bacteria and archaea in beef cattle divergent in feed efficiency using total RNA-seq. Curr. Res. Microbial Sci. 2, 100064. doi: 10.1016/j.crmicr.2021.100064
Von Engelhardt W., Hales J. (1977). Partition of capillary blood flow in rumen, reticulum, and omasum of sheep. Am. J. Physiol-Endocrinol Metab. 232 (1), E53. doi: 10.1152/ajpendo.1977.232.1.E53
Weimer P. J. (2015). Redundancy, resilience, and host specificity of the ruminal microbiota: implications for engineering improved ruminal fermentations. Front. Microbiol. 6, 296. doi: 10.3389/fmicb.2015.00296
Wirth R., Kádár G., Kakuk B., Maróti G., Bagi Z., Szilágyi Á., et al. (2018). The planktonic core microbiome and core functions in the cattle rumen by next generation sequencing. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02285
Xue M.-Y., Wu J.-J., Xie Y.-Y., Zhu S.-L., Zhong Y.-F., Liu J.-X., et al. (2022). Investigation of fiber utilization in the rumen of dairy cows based on metagenome-assembled genomes and single-cell RNA sequencing. Microbiome 10 (1), 11. doi: 10.1186/s40168-021-01211-w
Yu Z., Morrison M. (2004). Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36 (5), 808–812. doi: 10.2144/04365ST04
Keywords: rumen, microbiome, epimural, rumen sacs, ruminal fractions
Citation: Soltis MP, Henniger MT, Egert-McLean AM, Voy BH, Moorey SE, Schnieder LG, Shepherd EA, Christopher C, Campagna SR, Smith JS, Mulon P-Y, Anderson DE and Myer PR (2023) Rumen biogeographical regions and their impact on microbial and metabolome variation. Front. Anim. Sci. 4:1154463. doi: 10.3389/fanim.2023.1154463
Received: 30 January 2023; Accepted: 10 March 2023;
Published: 21 March 2023.
Edited by:
Robin White, Virginia Tech, United StatesCopyright © 2023 Soltis, Henniger, Egert-McLean, Voy, Moorey, Schnieder, Shepherd, Christopher, Campagna, Smith, Mulon, Anderson and Myer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Phillip R. Myer, cG15ZXJAdXRrLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.