 Yue Shang
Yue Shang Sanjay Kumar
Sanjay Kumar Brian Oakley4
Brian Oakley4 Woo Kyun Kim
Woo Kyun Kim- 1St. Boniface Hospital Research Centre, Winnipeg, MB, Canada
- 2Department of Animal Science, University of Manitoba, Winnipeg, MB, Canada
- 3Department of Poultry Science, University of Georgia, Athens, GA, United States
- 4College of Veterinary Medicine, Western University of Health Sciences, Pomona, CA, United States
Sustainable poultry meat and egg production is important to provide safe and quality protein sources in human nutrition worldwide. The gastrointestinal (GI) tract of chickens harbor a diverse and complex microbiota that plays a vital role in digestion and absorption of nutrients, immune system development and pathogen exclusion. However, the integrity, functionality, and health of the chicken gut depends on many factors including the environment, feed, and the GI microbiota. The symbiotic interactions between host and microbe is fundamental to poultry health and production. The diversity of the chicken GI microbiota is largely influenced by the age of the birds, location in the digestive tract and diet. Until recently, research on the poultry GI microbiota relied on conventional microbiological techniques that can only culture a small proportion of the complex community comprising the GI microbiota. 16S rRNA based next generation sequencing is a powerful tool to investigate the biological and ecological roles of the GI microbiota in chicken. Although several challenges remain in understanding the chicken GI microbiome, optimizing the taxonomic composition and biochemical functions of the GI microbiome is an attainable goal in the post-genomic era. This article reviews the current knowledge on the chicken GI function and factors that influence the diversity of gut microbiota. Further, this review compares past and current approaches that are used in chicken GI microbiota research. A better understanding of the chicken gut function and microbiology will provide us new opportunities for the improvement of poultry health and production.
Introduction
The integrity of the gastrointestinal tract (GIT) and the gut microbial community play vital roles in nutrition absorption, development of immunity, and disease resistance. Alterations in the GIT microbial community may have adverse effects on feed efficiency, productivity, and health of chickens (1–3). Understanding the roles of the chicken GI microbiota and understanding the current methods used in microbiome research is essential for improving the poultry GI microbiome. Historically, selective culture-based techniques have been used to identify and characterize the microbial diversity of the avian gut. In the last decade, the use of bacterial 16S ribosomal RNA (rRNA) gene sequencing has dramatically improved our understanding of the composition and diversity of the chicken GI microbiota. Modern high-throughput sequencing approaches are capable of rapidly obtaining a complete census of a bacterial community and are a powerful tool that has led to important new insights into the biological and ecological roles of the GI microbiota. This review aims to summarize avian gut function as well as factors that influence the diversity of the chicken GI microbiota. Furthermore, we have also compared and reviewed past and current approaches used in chicken gut microbiological research.
The Role of Chicken Gastrointestinal Microbiota
The gastrointestinal compartments of chickens are densely populated with complex microbial communities (Bacteria, fungi, Archaea, protozoa, and virus) that are dominated by Bacteria (4). The interactions between the host and the chicken GI bacterial microbiome have been extensively studied and reviewed by many research groups (5–9) and are now considered to play important roles in bird nutrition, physiology and gut development (10, 11).
The gut microbiota can form a protective barrier by attaching to the epithelial walls of the enterocyte and thus reduce the opportunity for the colonization of pathogenic bacteria (12). These bacteria produces vitamins (e.g., vitamin K and vitamin B groups), short chain fatty acids (acetic acid, butyric acid and propionic acid), organic acids (e.g., lactic acid) and antimicrobial compounds (e.g., bacteriocins), lower triglyceride, and induce non-pathogenic immune responses, which provide both nutrition and protection for the animal (2, 12–14). On the other hand, the GI microbiome can also be a source of bacterial pathogens such as Salmonella and Campylobacter which can disseminate to humans or act as a pool for antibiotic resistance and transmission and therefore may pose a serious threat to public health (5, 8, 15).
A normal gut microbial community has benefits and costs to the host (1, 13). The primary benefits that are provided by commensal microbiota are competitive exclusion of pathogens or non-indigenous microbes (13), immune stimulation and programming, and contributions to host nutrition. Earlier reports have established that conventionally raised animals are far less susceptible to pathogens when compared with germ-free animals (16). Furthermore, commensal microbiota can stimulate the development of immune system including the mucus layer, epithelial monolayer, the intestinal immune cells (e.g., cytotoxic and helper T cells, immunoglobulin producing cells and phagocytic cells), and the lamina propria (13, 17, 18). These tissues build barriers between the host and the microbes and combat undesirable gut microorganisms. In the distal gut (i.e., ceca and colon), the microbiota also produces energy and nutrients such as vitamins, amino acids, and short chain fatty acids (SCFA) from the undigested feed, which eventually become available for the host (1, 13). These SCFA have bacteriostatic properties that are capable of eliminating foodborne pathogens, such as Salmonella spp. (19). The SCFA are also a source of energy to the animals and can further stimulate gut epithelial cell proliferation, thus increasing the gastrointestinal absorption surface (13). It has also been established that SCFA production lowers the pH of colon, which inhibits conversion of bile to secondary bile products (20). In addition, gut microbiota also contributes to metabolism of host nitrogenous compounds. For example, cecal bacteria can convert uric acid to ammonia, which is subsequently absorbed by the bird and further used to produce amino-acids such as glutamine (21). Furthermore, some of the nitrogen from the diet gets incorporated into bacterial cellular protein and therefore, bacteria themselves can be a source of proteins/amino-acids (22).
In contrast, commensal microbiota also incurs cost to the host. In the proximal gut (gizzard and small intestine), microbes compete with the host for energy and protein. In both the proximal and distal gut, microbes produce toxic metabolites (e.g., amino acid catabolites) and catabolize bile acids, which may depress growth and decrease fat digestibility of the birds, respectively (1). In the presence of microbiota, the gut mucus layer increases mucin secretion and epithelial cell turnover rate, thereby keeping the GI tract lubricated while preventing microorganisms from invading intestinal epithelial cells of the host. The intestinal immune system is also more developed and secretes IgA, which specifically binds to bacterial epitopes, helps in regulating bacterial composition in the gut (23, 24). While generally beneficial, these processes do increase the demand for energy and protein from the host and therefore have an influence on the growth performance of the birds.
An imbalanced gut microbiota is often referred to as dysbiosis. Dysbiosis can been defined as qualitative and/or quantitative imbalance of normal microbiota in the small intestine, which may lead to a sequential reaction in the GIT, including reduced intestinal barrier function (e.g., thinning of intestinal wall) and poor nutrient digestibility, and therefore, increasing the risk of bacterial translocation and inflammatory responses (25). Both non-infectious and infectious stressors can lead to dysbacteriosis. The non-infectious factors include environmental stressors, nutritional imbalances, dietary changes, mycotoxins, poor management, enzymatic dysfunction, or host genetics (25). Infectious factors include viral or bacterial challenge, coccidiosis, or toxic metabolites produced by harmful microorganisms such as Clostridium perfringens.
The gastrointestinal microbiota can further be classified as the luminal microbiota and the mucosal microbiota (2). The composition of the luminal microbiota is determined by available nutrients, presence of antimicrobial substance and the feed passage rate. The composition of the mucosal-attached microbiota is affected by several host factors, such as expression of specific adhesion sites on the enterocyte membrane, secretion of secretory immunoglobulins, and mucus production rate. The luminal microbiota and the mucosal-associated microbiota of course also influence each other (2) and therefore, it is important to recognize that diet can alter both luminal and mucosal-attached microbiota to influence gut health. To our knowledge, there is no study to date which has compared the taxonomic composition or metabolic functions of these two microbial habitats. However, it would be interesting to study and analyse the variations between the bacterial communities of the mucosa and lumen throughout the different GI sections. Furthermore, studying the mucosal-associated bacterial community will be important to understand the host mucosal responses as any alterations in mucosal immunity may have serious implications on bird's health (26).
The Diversity of Chicken Gut Microbiota
The GI tract of the chicken harbors a diverse bacterial community in which each bacterium is adapted to its own ecological niche and synergistically lives with other bacterial species in the same community. The composition and function of these communities has been shown to vary depending on the age of the birds, location in the GI tract and on the dietary components (6, 18, 27–29).
Bird Age
The age of the birds is one of the most important factors that influences GI bacterial composition, cell density, and metabolic function. Significant changes in the taxonomic composition of gut microbiota have been studied using both DNA finger-printing (30) and high-throughput sequencing approaches (31) and are well-reviewed by many research groups (28, 32–34). Ballou et al. (35) and our recently published data (5) indicates that 1 day post-hatch broiler chicks already have a microbial community in their GIT. There are also successional changes in the composition of the GIT microbiome, due to the replacement and establishment of more stable bacterial taxa, as the bird advances in age (30, 36). Lu et al. (30) discovered that the GIT of chicken at 3 days of age contained L. delbrueckii, C. perfringens and Campylobacter coli, whereas from 7 to 21 days of age, L. acidophilus, Enterococcus, and Streptococcus were more common. At 28 and 49 days of age, the GI tract contains L. crispatus, but the composition is significantly different from other ages (30). In other work, successional changes in the gut microbial community measured with HT-NGS technology has shown that the relative abundance of Clostridium was higher as the bird aged, whereas lactobacilli was low throughout the growth cycle. This variability in results may be due to sample types (feces vs. cecum), and/or conventional microbiological and molecular methods that have limited coverage and accuracy compared to high-throughput NGS platforms which offer higher coverage and depth in determining microbial community. High-throughput sequencing technologies, such as targeted amplicon sequencing and shotgun metagenomic sequencing, have become more common to analyze the gut microbial composition and functions throughout the life span of broilers, but we are still at initial stage of analyses and there is a breach in knowledge regarding host morphological development, and functional properties of the gut microbiome as the bird ages.
Gastrointestinal Tract
The GI tract of the chicken includes the crop, proventriculus, gizzard, duodenum, jejunum, ileum, caeca, large intestine, and cloaca (32). Each GI tract section has different metabolic functions that shape the microbial community (Table 1), and therefore it is important to consider sampling location and study design. The chicken crop harbors 108 to 109 cfu/g bacteria, which is usually dominated by lactobacilli (28, 37). However, large variations in microbial composition among individual broilers fed on the similar diet has been observed by Choi et al. (44) due to difference in time between feeding and sampling. In the gizzard, the concentration of bacteria is similar to the crop, but bacterial fermentation activities are low mainly because of the low pH. The majority of bacteria in the gizzard are lactobacilli, enterococci, lactose-negative enterobacteria, and coliform bacteria (28). Among the small intestinal segments, the bacterial density is the lowest in the duodenum due to short passage time and a dilution of digesta by secreted bile (45). The duodenal bacterial community mainly consists of clostridia, streptococci, enterobacteria, and lactobacilli (46). Ileum microbiota have been studied the most among the small intestine segments. Lu et al. (30) assessed the ileal bacterial community by examining 16S rRNA gene sequences and found Lactobacillus as the major group (70%) followed by members of the family Clostridiaceae (11%), Streptococcus (6.5%) and Enterococcus (6.5%) (30). In corroboration, our recent article also showed lactobacilli as the predominant genus in the ileum (5). Compared to the ileum, the cecum harbors a more diverse, rich and stable microbial community including anaerobes (47, 48). Oakley et al. (18) have documented significant changes in cecal microbial communities from day of hatch to 6 weeks of age in commercial broilers (18, 27) and also significant differences in cecal vs. fecal samples from a single individual (27). Typically, richness and diversity in the cecum increase during these 6 weeks, and the taxonomic composition of the community quickly shifts from Proteobacteria, Bacteroides, and Firmicutes, to almost entirely Firmicutes by 3 weeks of age (18, 27). However, Kumar et al. (5) found that Firmicutes were the most abundant phylum in both ceca and ileum at all the ages (day 0 to day 42) except d 42 in the ceca where Bacteroidetes were abundant. The differences in bacterial composition can be expected due to differences in the nucleic acid extraction protocol, primers, sequencing approach, environmental factors, dietary treatment/ composition, breed, and geographical conditions. In addition to sample types, an adequate sample size is also needed for a proper study design. Higher individual variation in sample types (crop samples) results in higher sample size compared to cecal samples to find the potential differences (49).
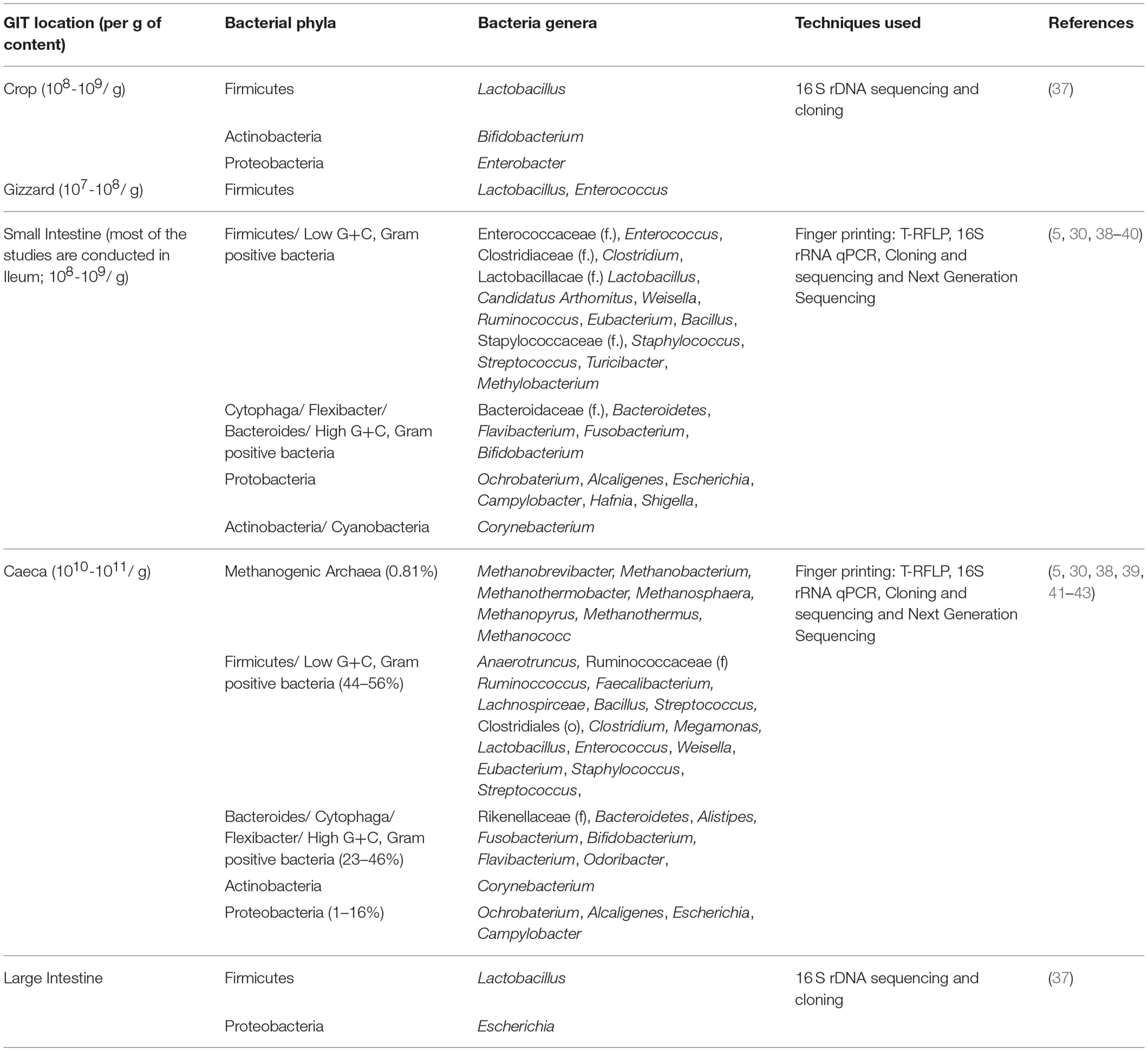
Table 1. Spatial distribution of most common and abundant bacterial taxa (phylum, order (o), family (f), genus) in the gastro-intestinal tract of chickens irrespective of age, diet and technique differences.
Feed processing approaches, feed components and additives are also known to have an effect on the gut microbial community. Knarreborg et al. (50) stated that mash feed lowers the number of Enterococcus spp. and coliforms but increases Lactobacillus spp. and C. perfringens in the broiler ileum, when compared to pellet feed (50). Corn favors low percent G + C clostridia, enterococci and lactobacilli, whereas wheat favors higher percent G + C bifidobacteria (29). Kumar et al. (5) reported low abundance in Firmicutes and high abundance in Bacteroidetes from day 0 to day 42 as birds were shifted from starter diet to finisher diet and argued that members of the phylum Bacteroidetes are vital for fermenting starch to simple sugars. Furthermore, feed supplementation, such as fermentable sugars (prebiotics), can also have an impact on the composition and diversity of chicken gut microbiota.
Prebiotics
The use of prebiotics as dietary modulators has been shown to have positive effects on some bacterial taxa in the colon (51). For example, Fructooligosaccharides (FOS) and Galactooligosaccharides (GOS) increased the population of Bifidobacterium and Lactobacillus (52, 53). In vitro studies have shown that fecal slurries which were incubated with oligofructose and inulin exhibited an increase in bifidobacteria populations in the human large intestine, whereas potential pathogens such as Escherichia coli and Clostridium spp. were maintained at lower levels (54). The majority of bifidobacteria strains (e.g., B. fiagilk, B. thetaiotaomicron, B. vulgatus, B. dktasonk, and B. ovatus) except B. bifidum, can utilize FOS as a growth and fermentation promoter (55). These bacteria secrete ß-fructosidase enzyme that can readily degrade and ferment FOS. However, microorganisms such as E. coli and C. perfringens are not able to exploit FOS as a fermentative carbohydrate source. Rats that were fed dietary FOS have shown a temporary boost in lactic acid-producing bacteria and a long-term elevation in cecal butyric acid (56). Dietary inclusion of FOS reduced C. perfringens and E. coli populations and increased the diversity of Lactobacillus in the broiler GIT (57). Patterson et al. (58) assessed the effects of thermal ketoses oligosaccharides on cecal microbial populations of broiler chickens. The results showed that cecal bifidobacteria and lactobacilli concentrations were increased 24-fold and 7-fold, respectively, in ketoses supplemented diet compared to controls. Another type of prebiotics, mannooligosaccharides (MOS), are proposed to have different mechanisms of action (58). They can (1) bind to potential pathogenic Gram-negative bacteria (e.g., E. coli and Salmonella) which possess type-1 fimbriae (mannose-sensitive lectin), to prevent and dislocate the pathogens from attaching to the gut wall, (2) have immune modulatory effects based on the antigenicity features of mannan and glucan components, (3) modulate intestinal morphology, and (4) enhance the expression of mucin and reduce enterocyte turnover rate (59). The effects of prebiotics on lower GI tract include: (1) serving as food and fermentation sources for cecal and colonic microbiota, (2) production of fermentation end products (e.g., SCFAs), (3) stimulation of saccharolytic fermentation, (4) acidification of the large intestine content, (5) hyperplasia of the cecal and colonic epithelium, (6) stimulation of colonic hormonal peptides secretion, and (7) acceleration of ceco-anal transit (51).
Other than age, GIT location, and prebiotics, breed and sex of the bird can also have a large impact on the intestinal microbiota (34). In addition, it has been well-documented that environmental factors (biosecurity level, housing, litter, feed access, and climate) can also substantially influence the gut bacterial composition. Therefore, data interpretation and outcome of research largely depends on the study design. Best practices for research reporting include providing details regarding host and environmental factors that can enable researchers to do meta-analyses to better understand nutritional, microbiome, and environmental factors that can be modulated to improve bird performance and health.
Discovery of Chicken Gut Microbiota by Molecular Approaches
Classical culture-based methods have historically been widely used to study the chicken gut microbiota. However, these methods are highly selective to cultivable bacteria under specific conditions (60). A majority of bacteria remain uncultured (29). Over 30 years ago, the term “the great plate count anomaly” was coined to reflect laboratory calculations that a very small minority (0.1–1%) of microbial taxa present in a given sample could be cultured (61). Similarly, over 10 years ago, it was observed that of 52 microbial phyla recognized at the time, only half of them had even a single cultivated representative, supporting the description of an “uncultivated majority” (62). Therefore, the richness (number of species) and diversity (number of species weighted by their relative abundance) of intestinal bacteria have been underestimated, and our knowledge of gut microbiota remains incomplete (63).
The development of molecular biotechnology has offered new tools to study the composition, diversity, predicted function and interaction of gut microbiota in different sections of the GI tract. Currently, a variety of molecular techniques are available, each with different strengths and weaknesses. The sample capacity, applications and limitations of some of the most common molecular techniques that can be used to study chicken GI microbial ecology are listed in Table 2. Among these methods, high-throughput sequencing of 16S rRNA gene amplicons has quickly become the method of choice. Although this method had been widely used in other research fields, the first report utilizing high-throughput sequencing of 16S rRNA genes for studying the population of microbial communities and their interactions in the chicken gut was published in 2013 (64).
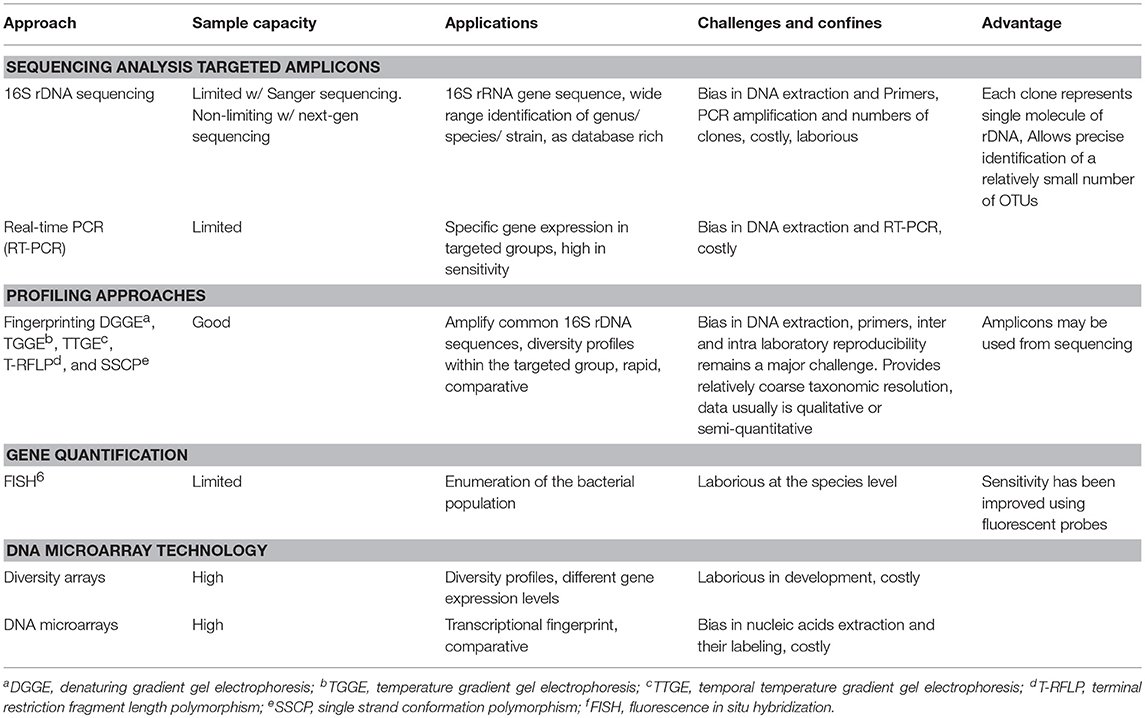
Table 2. 16S rRNA-based molecular approaches for studying microbial ecology in the chicken gut (64–67).
The 16S rRNA molecule is a small subunit of the ribosome that possesses regions of sequence similarity that are highly conserved across all bacteria. To amplify these genes, microbial DNA is extracted from fecal or digesta samples, and broad-range primers, which target conserved regions of the 16S rRNA gene, are used for polymerase chain reaction (PCR) amplification (29). Sequencing of these amplified products (amplicons) can discriminate among bacteria, generally to the genus or species level (68, 65), and the relative abundance of each sequence reflects the relative abundance of that bacterium in the original sample. Thus, sequencing of 16S rRNA genes provides a true census of a bacterial community by defining the types of bacteria present in a sample and their relative abundances. Because of the high richness and diversity of intestinal bacterial communities, it has only been in the last few years that DNA sequencing technology has matured to the point where we can now completely census these complex communities. Beginning in 2008, technical advances in sequencing allowed for several orders of magnitude more sequences to be collected than was previously possible—in a single study the authors deposited as many 16S rRNA sequences in the GenBank database as had been generated historically up to that point (69). With these profound methodological advances and enormous new datasets, it is now possible to easily and accurately take a census of an intestinal sample to determine, for example, how the microbiome responds to different feed additives, husbandry conditions, or disease states (Figure 1).
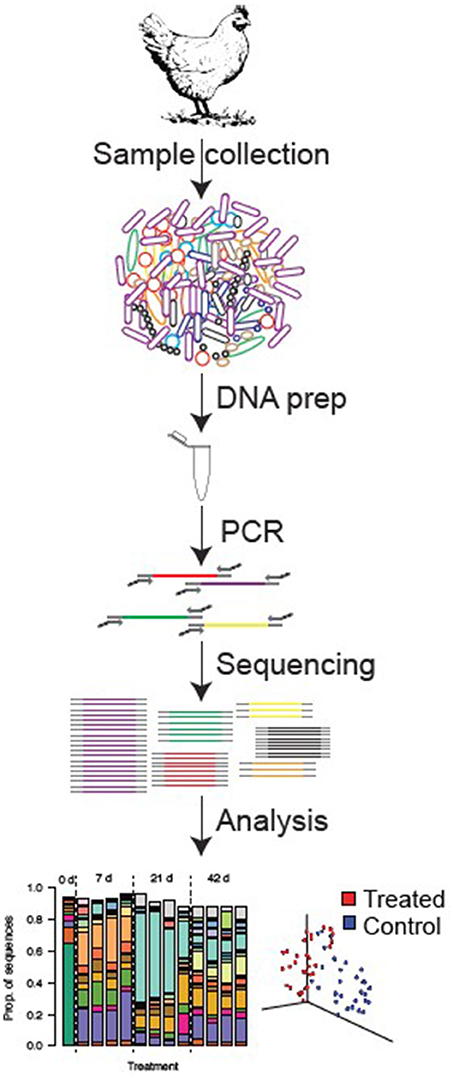
Figure 1. Standard procedure from sample collection to sequencing analysis in poultry gut.
High-throughput or next generation sequencing (NGS), is a powerful tool to investigate the biological and ecological role of gut microbiota (64). NGS has become a convenient, rapid, accurate and inexpensive method for genomic research (70, 66). Current NGS platforms offer high throughput, fast turn-around times, and low costs. Among these platforms the Illumina HiSeq and MiSeq instruments are two of the most frequently used systems in recent chicken gut microbiome and metagenomic research. Despite many advantages, these platforms suffer from limitations including short read assembly and high cost (71). Third-generation sequencing platforms such as single molecule real-time (SMRT) and nanopore sequencing require less time for DNA preparation (no PCR) and are cost effective (71). As these platforms continue to mature, their adoption will surely lead to new understanding of the poultry GI microbiome.
Following sequencing, bioinformatic analyses of sequence data requires open source platforms such as QIIME or mothur which utilize public databases (GreenGenes, Ribosomal Database Project and SILVA 72–76 to perform taxonomic assignment. Predictions of metabolic functions based on taxonomic identities from 16S rRNA gene sequences can be further obtained using algorithms such as PICRUSt and Tax4Fun 77, 78. To catalog the gene functions or analysis of individual genomes, metagenomic or metatranscriptomic approaches (in which genes or transcripts respectively are sequenced directly with no PCR) can be used to provide information on community diversity, structure and metabolic functions, or gene expression (79). Bioinformatic analyses of such datasets are more complex than 16S amplicon data and typically involve a sequence assembler such as Velvet (CLC workbench, Newbler version 3.0, Biospace) or MG-RAST. Bacterial taxa and functional groups can be assigned based on Basic Local Alignment Search Tool (BLAST), and gene functions may be analyzed using either Kyoto Encyclopedia of Genes and Genomes (KEGG) or Cluster of Orthologous genes (COG). In the chicken gut microbiome, metagenomics has been used to study the cecum functions, gut response to pathogen challenge, correlations between microbial response and performance parameters, comparison between fat and lean broiler lines, description on virulome, and antibiotic resistance genes (80). Some of the NGS based studies investigating chicken gut microbial community composition and functions in respect to the dietary responses/ antibiotic treatments are depicted in Table 3. However, it's difficult to compare all these studies because of variation in NGS platforms used, breed, sample type, sampling method etc. Therefore, a standard protocol is needed for studying the chicken gut microbial community, as available for human microbiome, in order to have comparable results. Currently most metagenomic approaches to studying the chicken GIT are still not affordable for most researchers or veterinarians.
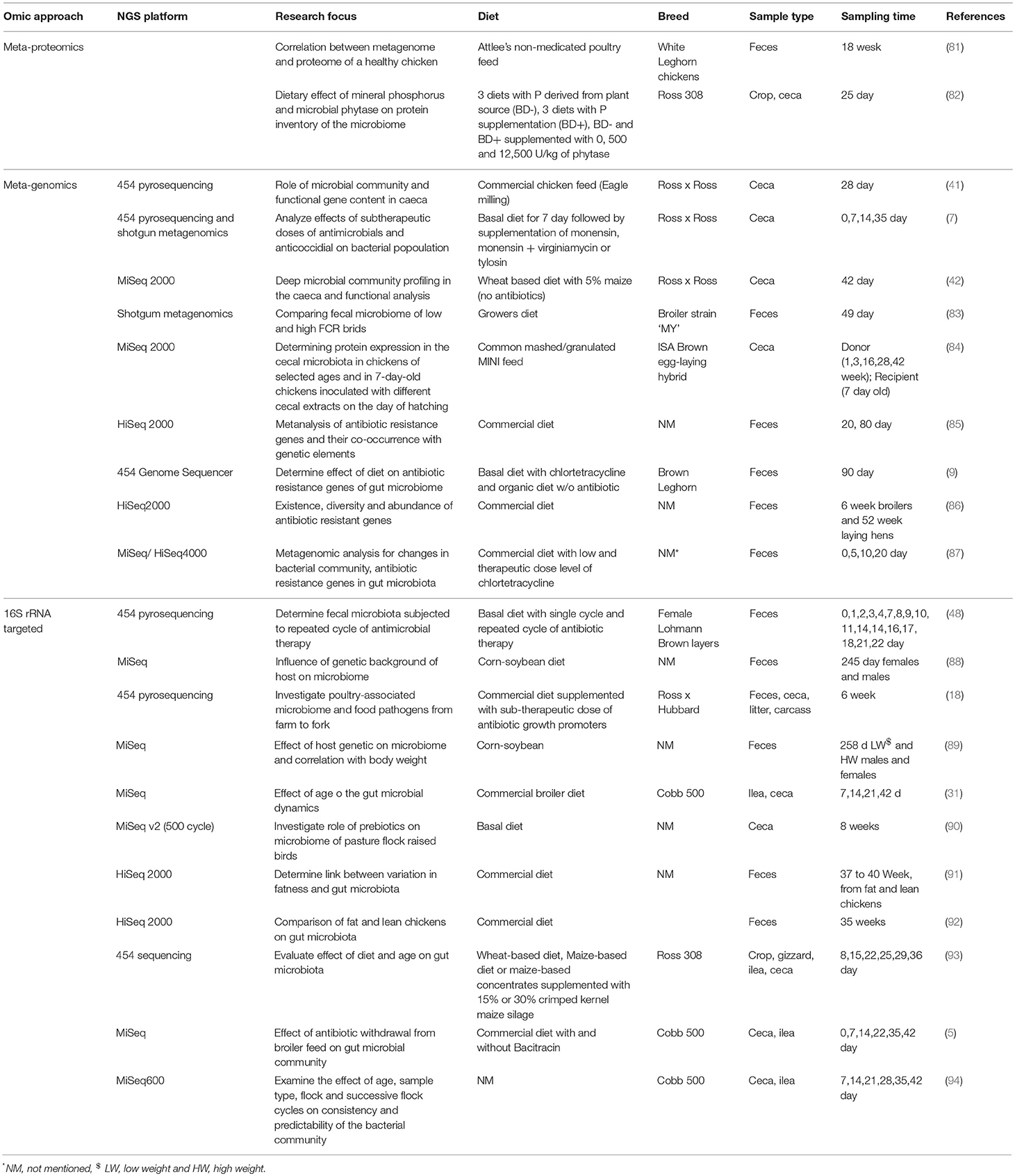
Table 3. : Different omics approaches applied in understanding gut microbial community and functions.
To circumvent some of the confines of sequence-based analysis, proteomic methods have also recently been used to determine the metabolic and functional properties of the microbiome (81, 82). Transcriptomics measures gene transcription in situ, providing an accurate reflection of physiological functions even if utmost care is needed during sampling (71). Since there are limited culture collections for poultry strains, increase in bacterial cultures and proper cataloging of their biochemical and genetic properties will facilitate proteomics and other “omics” approaches.
Conclusion
In recent years, significant progress has been made in understanding the taxonomic composition of the GI microbiome and its contributions to gut health. It is important for future studies to apply multi-omics approaches in order to increase our understanding of the role of the microbiome in nutrition, health, disease, and productivity. Progress in this field will help us to better understand how to manage the gut microbiota based on the environment, diet and physiology changes of the birds, and will further advance our understanding on the modification of microbiota-associated metabolic pathways, thus providing new opportunities for improving overall health of the poultry.
Author Contributions
YS and SK wrote this review manuscript. BO reviewed literature and the manuscript and provided critical suggestion and comments. WK decided a review topic, reviewed literature, and provided critical review and suggestion/comments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gaskins HR, Collier CT, Anderson DB. Antibiotics as growth promotants: Mode of action. Anim Biotechnol. (2002) 13:29–42. doi: 10.1081/ABIO-120005768
2. Jeurissen SH, Lewis F, Van der Klis JD, Mroz Z, Rebel JM, Ter Huurne AA. Parameters and techniques to determine intestinal health of poultry as constituted by immunity, integrity, and functionality. Curr Issues Intest Microbiol. (2002) 3:1–14.
3. Kohl KD. Diversity and function of the avian gut microbiota. J Compar Physiol B Biochem Syst Environ Physiol. (2012) 182:591–602. doi: 10.1007/s00360-012-0645-z
4. Wei S, Morrison M, Yu Z. Bacterial census of poultry intestinal microbiome. Poult Sci Sympos. (2013) 92:671–83. doi: 10.3382/ps.2012-02822
5. Kumar S, Chen C, Indugu N, Werlang GO, Singh M, Kim WK, et al. Effect of antibiotic withdrawal in feed on chicken gut microbial dynamics, immunity, growth performance and prevalence of foodborne pathogens. PloS ONE (2018) 13:e0192450. doi: 10.1371/journal.pone.0192450
6. Pan D, Yu Z. Intestinal microbiome of poultry and its interaction with host and diet. Gut Microbes (2014) 5:108–19. doi: 10.4161/gmic.26945
7. Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ. Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS ONE (2011) 6:e27949. doi: 10.1371/journal.pone.0027949
8. Mancabelli L, Ferrario C, Milani C, Mangifesta M, Turroni F, Duranti S, et al. Insights into the biodiversity of the gut microbiota of broiler chickens. Environ Microbiol. (2016) 18:4727–4738. doi: 10.1111/1462-2920.13363
9. Hegde NV, Kariyawasam S, DebRoy C. Comparison of antimicrobial resistant genes in chicken gut microbiome grown on organic and conventional diet. Veter Anim Sci. (2016) 1:9–16. doi: 10.1016/j.vas.2016.07.001
10. Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature (2011) 474:327–36. doi: 10.1038/nature10213
11. Gerritsen J, Smidt H, Rijkers GT, de Vos WM. Intestinal microbiota in human health and disease: the impact of probiotics. Genes Nutrit. (2011) 6:209–40. doi: 10.1007/s12263-011-0229-7
12. Yegani M, Korver DR. Factors affecting intestinal health in poultry. Poult Sci. (2008) 87:2052–63. doi: 10.3382/ps.2008-00091
13. Dibner JJ, Richards JD. Antibiotic growth promoters in agriculture history and mode of action. Poult Sci. (2005) 84:634–43. doi: 10.1093/ps/84.4.634
14. Apajalahti J. Comparative gut microflora, metabolic challenges, and potential opportunities. J Appl Poult Res. (2005) 14:444–53. doi: 10.1093/japr/14.2.444
15. Zhou W, Wang Y, Lin J. Functional cloning and characterization of antibiotic resistance genes from the chicken gut microbiome. Appl Environ Microbiol. (2012) 78:3028–32. doi: 10.1128/AEM.06920-11
16. Koopman JP, Kennis HM, Mullink JWMA, Prins RA, Stadhouders AM, Boer HD, et al. ‘Normalization’ of germfree mice with anaerobically cultured caecal flora of ‘normal’ mice. Lab Anim (1984) 18:188–94. doi: 10.1258/002367784780891253
17. Shakouri MD, Iji PA, Mikkelsen LL, Cowieson AJ. Intestinal function and gut microflora of broiler chickens as influenced by cereal grains and microbial enzyme supplementation. J Anim Physiol Anim Nutrit. (2009) 93:647–58. doi: 10.1111/j.1439-0396.2008.00852.x
18. Oakley BB, Lillehoj HS, Kogut MH, Kim WK, Maurer JJ, Pedroso A, et al. The chicken gastrointestinal microbiome. FEMS Microbiol Lett. (2014) 360:100–12. doi: 10.1111/1574-6968.12608
19. Ricke SC. Perspectives on the use of organic acids and short chain fatty acids as antimicrobials. Poult Sci. (2003) 82:632–9. doi: 10.1093/ps/82.4.632
20. Christl SU, Bartram HP, Paul A, Kelber E, Scheppach W, Kasper H. Bile acid metabolism by colonic bacteria in continuous culture: effect of starch and pH. Ann Nutrit Metabol. (1997) 41:45–51. doi: 10.1159/000177977
21. Vispo C, Karasov WH. The interaction of avian gut microbes and their host: an elusive symbiosis. In: Gastrointestinal Microbiology Mackie R, White B, editors. New York, NY: Springer. (1997). p. 116–55.
22. Metges CC. Contribution of microbial amino acids to amino acid homeostasis of the host. J Nutrit. (2000) 130:1857S−64S. doi: 10.1093/jn/130.7.1857S
23. Mitchell MA, Moretó M. Absorption function of the small intestine: Adaptions meeting demands. In: Perry GC, editor. Absorption Function of the Small Intestine: Adaptions Meeting Demands. Oxford: CABI Publishing (2006). p. 43–64.
24. Suzuki K, Nakajima A. New aspects of IgA synthesis in the gut. Inter Immunol. (2014) 26:489–94. doi: 10.1093/intimm/dxu059
25. Teirlynck E, Gussem MDE, Dewulf J, Haesebrouck F, Ducatelle R, Van Immerseel F. Morphometric evaluation of “dysbacteriosis” in broilers. Avian pathology (2011) 40:139–44. doi: 10.1080/03079457.2010.543414
26. Borda-Molina D, Vital M, Sommerfeld V, Rodehutscord M, Camarinha-Silva A. Insights into broilers' gut microbiota fed with phosphorus, calcium and phytase supplemented diets. Front Microbiol. (2016) 7:2033. doi: 10.3389/fmicb.2016.02033
27. Kogut MH, Oakley BB. Spatial and temporal changes in the broiler chicken cecal and fecal microbiomes and correlations of bacterial taxa with cytokine gene expression. Front Veter Sci. (2016) 3:11. doi: 10.3389/fvets.2016.00011
28. Rehman HU, Vahjen W, Awad WA, Zentek J. Indigenous bacteria and bacterial metabolic products in the gastrointestinal tract of broiler chickens. Arch Anim Nutrit. (2007) 61:319–35. doi: 10.1080/17450390701556817
29. Apajalahti J, Kettunen A, Graham H. Characteristics of the gastrointestinal microbial communities, with special reference to the chicken. Worlds Poult Sci J. (2004) 60:223–32. doi: 10.1079/WPS20040017
30. Lu J, Idris U, Harmon B, Hofacre C, Maurer JJ, Lee MD. Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Appl Environ Microbiol. (2003) 69:6816–24. doi: 10.1128/AEM.69.11.6816-6824.2003
31. Shaufi MA, Sieo CC, Chong CW, Gan HM, Ho YW. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathol. (2015) 7:4. doi: 10.1186/s13099-015-0051-7
32. Yeoman CJ, Chia N, Jeraldo P, Sipos M, Goldenfeld ND, White BA. The microbiome of the chicken gastrointestinal tract. Anim Health Res Rev. (2012) 13:89–99. doi: 10.1017/S1466252312000138
33. Stanley D, Hughes RJ, Moore RJ. Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Appl Microbiol Biotechnol. (2014) 98:4301–10. doi: 10.1007/s00253-014-5646-2
34. Kers JG, Velkers FC, Fischer EAJ, Hermes GDA, Stegeman JA, Smidt H. Host and Environmental factors affecting the intestinal microbiota in chickens. Front Microbiol. (2018) 9:235 doi: 10.3389/fmicb.2018.00235
35. Ballou AL, Ali RA, Mendoza MA, Ellis JC, Hassan HM, Croom WJ, et al. Development of the chick microbiome: how early exposure influences future microbial diversity. Front Veter Sci. (2016) 3:2. doi: 10.3389/fvets.2016.00002
36. Crhanova M, Hradecka H, Faldynova M, Matulova M, Havlickova H, Sisak F, et al. Immune response of chicken gut to natural colonization by gut microflora and to Salmonella enterica serovar enteritidis infection. Infect Immun. (2011) 79:2755–63. doi: 10.1128/IAI.01375-10
37. Gong J, Si W, Forster RJ, Huang R, Yu H, Yin Y, et al. 16s rRNA gene-based analysis of mucosa-associated bacterial community and phylogeny in the chicken gastrointestinal tracts: From crops to ceca. FEMS Microbiol Ecol. (2007) 59:147–57. doi: 10.1111/j.1574-6941.2006.00193.x
38. Xiao Y, Xiang Y, Zhou W, Chen J, Li K, Yang H. Microbial community mapping in intestinal tract of broiler chicken. Poult Sci. (2017) 96:1387–93. doi: 10.3382/ps/pew372
39. Siegerstetter S-C, Schmitz-Esser S, Magowan E, Wetzels SU, Zebeli Q, Lawlor PG, et al. Intestinal microbiota profiles associated with low and high residual feed intake in chickens across two geographical locations. PLoS ONE (2017) 12:e0187766. doi: 10.1371/journal.pone.0187766
40. Lumpkins BS, Batal AB, Lee MD. Evaluation of the bacterial community and intestinal development of different genetic lines of chickens. Poult Sci. (2010) 89:1614–21. doi: 10.3382/ps.2010-00747
41. Qu A, Brulc JM, Wilson MK, Law BF, Theoret JR, Joens LA, et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ONE (2008) 3:e2945. doi: 10.1371/journal.pone.0002945
42. Sergeant MJ, Constantinidou C, Cogan TA, Bedford MR, Penn CW, Pallen MJ. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS ONE (2014) 9:e91941. doi: 10.1371/journal.pone.0091941
43. Saengkerdsub S, Herrera P, Woodward CL, Anderson RC, Nisbet DJ, Ricke SC. Identification and quantification of methanogenic archaea in adult chicken ceca. Appl Environ Microbiol. (2007) 73:353–6. doi: 10.1128/AEM.01931-06
44. Choi JH, Kim GB, Cha CJ. Spatial heterogeneity and stability of bacterial community in the gastrointestinal tracts of broiler chickens. Poult Sci. (2014) 93:1942–50. doi: 10.3382/ps.2014-03974
45. Shapiro SK, Sarles WB. Microorganisms in the intestinal tract of normal chickens. J Bacteriol. (1949) 58:531–44.
46. Waite DW, Taylor M. Exploring the avian gut microbiota: current trends and future directions. Front Microbiol. (2015) 6:673. doi: 10.3389/fmicb.2015.00673
47. Salanitro JP, Blake IG, Muirhead PA. Studies on the cecal microflora of commercial broiler chickens. Appl Microbiol. (1974) 28:439–47.
48. Videnska P, Sisak F, Havlickova H, Faldynova M, Rychlik I. Influence of Salmonella enterica serovar enteritidis infection on the composition of chicken cecal microbiota. BMC Veter Res. (2013) 9:140. doi: 10.1186/1746-6148-9-140
49. Lagkouvardos I, Overmann J, Clavel T. Cultured microbes represent a substantial fraction of the human and mouse gut microbiota. Gut Microb. (2017) 8:493–503. doi: 10.1080/19490976.2017.1320468
50. Knarreborg A, Simon MA, Engberg RM, Jensen BB, Tannock GW. Effects of dietary fat source and subtherapeutic levels of antibiotic on the bacterial community in the ileum of broiler chickens at various ages. Appl Environ Microbiol. (2002) 68:5918–24. doi: 10.1128/AEM.68.12.5918-5924.2002
51. Gaggia F, Mattarelli P, Biavati B. Probiotics and prebiotics in animal feeding for safe food production. Inter J Food Microbiol. (2010) 141:S15–28. doi: 10.1016/j.ijfoodmicro.2010.02.031
52. Jung SJ, Houde R, Baurhoo B, Zhao X, Lee BH. Effects of galacto-oligosaccharides and a Bifidobacteria lactis-based probiotic strain on the growth performance and fecal microflora of broiler chickens. Poult Sci. (2008) 87:1694–9. doi: 10.3382/ps.2007-00489
53. Xu ZR, Hu CH, Xia MS, Zhan XA, Wang MQ. Effects of dietary fructooligosaccharide on digestive enzyme activities, intestinal microflora and morphology of male broilers. Poult Sci. (2003) 82:1030–6. doi: 10.1093/ps/82.6.1030
54. Nywang X, Gibson GR. Effects of the in vitro fermentation of oligofructose and inulin by bacteria growing in the human large intestine. J Appl Bacteriol. (1993) 75:373–80. doi: 10.1111/j.1365-2672.1993.tb02790.x
55. Hidaka H, Hirayama M. Useful characteristics and commercial applications of fructo-oligosaccharides. Biochem Soc Trans. (1991) 19:561–5. doi: 10.1042/bst0190561
56. Le Blay G, Michel C, Blottière HM, Cherbut C. Prolonged intake of fructo-oligosaccharides induces a short-term elevation of lactic acid-producing bacteria and a persistent increase in cecal butyrate in rats. J Nutrit. (1999) 129:2231–35.
57. Kim HJ, Eom SJ, Park SJ, Cha CJ, Kim GB. Lactobacillus alvi sp. nov., isolated from the intestinal tract of chicken. FEMS Microbiol Lett. (2011) 323:83–7. doi: 10.1111/j.1574-6968.2011.02361.x
58. Patterson JA, Orban JI, Sutton AL, Richards GN. Selective enrichment of bifidobacteria in the intestinal tract of broilers by thermally produced ketoses and effect on broiler performance. Poult Sci. (1997) 76:497–500. doi: 10.1093/ps/76.3.497
59. Yang Y, Iji PA, Choct M. Dietary modulation of gut microflora in broiler chickens: a review of the role of six kinds of alternatives to in-feed antibiotics. World Poult Sci J. (2009) 65:97. doi: 10.1017/S0043933909000087
60. Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. (1998) 180:4765–74.
61. Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Ann Rev Microbiol. (1985) 39:321–46. doi: 10.1146/annurev.mi.39.100185.001541
62. Rappe MS, Giovannoni SJ. The uncultured microbial majority. Ann Rev Microbiol. (2003) 57:369–94. doi: 10.1146/annurev.micro.57.030502.090759
63. Gong J, Forster RJ, Yu H, Chambers JR, Wheatcroft R, Sabour PM, et al. Molecular analysis of bacterial populations in the ileum of broiler chickens and comparison with bacteria in the cecum. FEMS Microbiol Ecol. (2002) 41:171–9. doi: 10.1111/j.1574-6941.2002.tb00978.x
64. Diaz-Sanchez S, Hanning I, Pendleton S, D'souza D. Next generation sequencing: the future of molecular genetics in poultry production and food safety. Poult Sci. (2013) 92:562–72. doi: 10.3382/ps.2012-02741
65. Flint HJ, Leitch ECM, Duncan SH, Walker AW, Patterson AJ, Rincon MT, et al. Molecular approaches to the analysis of gastrointestinal microbial ecosystems. In: Perry GC, editor. Molecular Approaches to the Analysis of Gastrointestinal Microbial Ecosystems. Oxford: CABI Publishing (2006). p. 107–23.
66. Park SH, Hanning I, Perrota A, Bench BJ, Alm E, Ricke SC. Modifying the gastrointestinal ecology in alternatively raised poultry and the potential for molecular and metabolomic assessment. Poult Sci. (2013) 92:546–61. doi: 10.3382/ps.2012-02734
67. Kumar S, Dagar SS, Mohanty AK, Sirohi SK, Puniya M, Kuhad RC, et al. Enumeration of methanogens with a focus on fluorescence in situ hybridization. Naturwissenschaften (2011) 98:457–72. doi: 10.1007/s00114-011-0791-2
68. Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16s ribosomal DNA amplification for phylogenetic study. J Bacteriol. (1991) 173:697–703. doi: 10.1128/jb.173.2.697-703.1991
69. Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods (2008) 5:235–7. doi: 10.1038/nmeth.1184
70. Pettersson E, Lundeberg J, Ahmadian A. Generations of sequencing technologies. Genomics (2009) 93:105–11. doi: 10.1016/j.ygeno.2008.10.003
71. Kumar S, Pitta DW. Revolution in rumen microbiology. In: Puniya AK, Singh R, Kamra DN, ediotrs. Rumen Microbiology: From Evolution to Revolution. New Delhi: Springer (2015). p. 357–79.
72. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods (2010) 7:335–6. doi: 10.1038/nmeth.f.303
73. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. (2009) 75:7537–41. doi: 10.1128/AEM.01541-09
74. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. (2006) 72:5069–72. doi: 10.1128/AEM.03006-05
75. Durso LM, Harhay GP, Smith TP, Bono JL, Desantis TZ, Harhay DM, et al. Animal-to animal variation in fecal microbial diversity among beef cattle. Appl Environ Microbiol. (2010) 76:4858–62. doi: 10.1128/AEM.00207-10
76. Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. (2007) 35:7188–96. doi: 10.1093/nar/gkm864
77. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. (2013) 31:814–21. doi: 10.1038/nbt.2676
78. Abhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics (2015) 31:2882–4. doi: 10.1093/bioinformatics/btv287
79. Zinicola M, Higgins H, Lima S, Machado V, Guard C, Bicalho R Shotgun metagenomic sequencing reveals functional genes and microbiome associated with bovine digital dermatitis. PLoS ONE (2015) 10:e0133674. doi: 10.1371/journal.pone.0133674
80. Borda-Molina D, Seifert J, Camarinha-Silva A. Current perspective of the chicken gastrointestinal tract and its microbiome. Comput Struct Biotechnol J. (2018) 16:131–9. doi: 10.1016/j.csbj.2018.03.002
81. Tang Y, Underwood A, Gielbert A, Woodward MJ, Petrovska L. Metaproteomics analysis reveals the adaptation process for the chicken gut microbiota. Appl Environ Microbiol. (2014) 80:478–85. doi: 10.1128/AEM.02472-13
82. Tilocca B, Witzig M, Rodehutscord M, Seifert J. Variations of phosphorous accessibility causing changes in microbiome functions in the gastrointestinal tract of chickens. PLoS ONE (2016) 11:e0164735. doi: 10.1371/journal.pone.0164735
83. Singh KM, Shah TM, Reddy B, Deshpande S, Rank DN and Joshi CG. Taxonomic and gene-centric metagenomics of the fecal microbiome of low and high feed conversion ratio (FCR) broilers. J Appl Genet. (2014) 55:145–54. doi: 10.1007/s13353-013-0179-4
84. Polansky O, Sekelova Z, Faldynova M, Sebkova A, Sisak F, Rychlik I. Important metabolic pathways and biological processes expressed by chicken cecal microbiota. Appl Environ Microbiol. (2016) 82:1569–76. doi: 10.1128/AEM.03473-15
85. Ma L, Xia Y, Li B, Yang Y, Li LG, Tiedje JM, et al. Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig, chicken, and human feces. Environ Sci Technol. (2016) 50:420–7. doi: 10.1021/acs.est.5b03522
86. Panpan T, Xue J, Lizhi C, Jun L, Lizhi X, Lingwei Z, et al. Metagenome analysis of antibiotic resistance genes in fecal microbiota of chickens. Agri Gene (2017) 5:1–6. doi: 10.1016/j.aggene.2017.06.001
87. Xiong W, Wang Y, Sun Y, Ma L, Zeng Q, Jiang X, et al. Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes. Microbiome (2018) 6:34. doi: 10.1186/s40168-018-0419-2
88. Zhao L, Wang G, Siegel P, He C, Wang H, Zhao W, et al. Quantitative genetic background of the host influences gut microbiomes in chickens. Sci Rep. (2013) 3:1163. doi: 10.1038/srep01163
89. Meng H, Zhang Y, Zhao L, Zhao W, He C, Honaker CF, et al. Body weight selection affects quantitative genetic correlated responses in Gut Microbiota. PLoS ONE (2014) 9:e89862. doi: 10.1371/journal.pone.0089862
90. Park SH, Lee SI, Ricke SC. Microbial populations in naked neck chicken ceca raised on pasture flock fed with commercial yeast cell wall prebiotics via an Illumina MiSeq platform. PLoS ONE (2016) 11:e0151944. doi: 10.1371/journal.pone.0151944
91. Hou Q, Kwok L-Y, Zheng Y, Wang L, Guo Z, Zhang J, Huang W, et al. Differential fecal microbiota are retained in broiler chicken lines divergently selected for fatness traits. Sci Rep. (2016) 6:37376. doi: 10.1038/srep37376
92. Ding J, Zhao L, Wang L, Zhao W, Zhai Z, Leng L, et al. Divergent selection-induced obesity alters the composition and functional pathways of chicken gut microbiota. Genet Select Evolut. (2016) 48:93. doi: 10.1186/s12711-016-0270-5
93. Ranjitkar S, Engberg RM. The influence of feeding crimped kernel maize silage on growth performance and intestinal colonization with Campylobacter jejuni of broilers. Avian Pathol. (2016) 45:253–60. doi: 10.1080/03079457.2016.1146821
Keywords: chicken, gut function, microbiome, prebiotics, DNA sequencing
Citation: Shang Y, Kumar S, Oakley B and Kim WK (2018) Chicken Gut Microbiota: Importance and Detection Technology. Front. Vet. Sci. 5:254. doi: 10.3389/fvets.2018.00254
Received: 23 August 2018; Accepted: 24 September 2018;
Published: 23 October 2018.
Edited by:
Rajesh Jha, University of Hawaii at Manoa, United StatesReviewed by:
Kyung-Woo Lee, Konkuk University, South KoreaSiaka Seriba Diarra, University of the South Pacific, Fiji
Copyright © 2018 Shang, Kumar, Oakley and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Woo Kyun Kim, d2traW1AdWdhLmVkdQ==