 Jinkun Chen1†
Jinkun Chen1† Evann E. Hilt2†
Evann E. Hilt2† Fan Li3†Huan Wu4Zhuojing Jiang1Qinchao Zhang1Jiling Wang1Yifang Wang4Ziqin Li5Jialiang Tang1*
Fan Li3†Huan Wu4Zhuojing Jiang1Qinchao Zhang1Jiling Wang1Yifang Wang4Ziqin Li5Jialiang Tang1* Shangxin Yang2,5*
Shangxin Yang2,5*- 1Shaoxing Center for Disease Control and Prevention, Shaoxing, China
- 2Department of Pathology and Laboratory Medicine, University of California, Los Angeles, Los Angeles, CA, United States
- 3Three Coin Analytics, Inc., Pleasanton, CA, United States
- 4IngeniGen XunMinKang Biotechnology Inc., Shaoxing, China
- 5Zhejiang-California International Nanosystems Institute, Zhejiang University, Hangzhou, China
A novel coronavirus known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the cause of the ongoing Coronavirus Disease 2019 (COVID-19) pandemic. In this study, we performed a comprehensive epidemiological and genomic analysis of SARS-CoV-2 genomes from 10 patients in Shaoxing (Zhejiang Province), a mid-sized city outside of the epicenter Hubei province, China, during the early stage of the outbreak (late January to early February, 2020). We obtained viral genomes with >99% coverage and a mean depth of 296X demonstrating that viral genomic analysis is feasible via metagenomics sequencing directly on nasopharyngeal samples with SARS-CoV-2 Real-time PCR Ct values <28. We found that a cluster of four patients with travel history to Hubei shared the exact same virus with patients from Wuhan, Taiwan, Belgium, and Australia, highlighting how quickly this virus spread to the globe. The virus from another cluster of two family members living together without travel history but with a sick contact of a confirmed case from another city outside of Hubei accumulated significantly more mutations (9 SNPs vs. average 4 SNPs), suggesting a complex and dynamic nature of this outbreak. Our findings add to the growing knowledge of the epidemiological and genomic characteristics of SARS-CoV-2 and offers a glimpse into the early phase of this viral infection outside of Hubei, China.
Introduction
Coronaviruses (CoVs) are a large family of single-stranded RNA viruses that can be isolated from a variety of animals including camels, rats, birds, and bats (1). These coronaviruses can cause a range of disease states in animals including respiratory, enteric, hepatic, and neurological disease (2). Before late 2019, there were six known CoVs capable of infecting humans (Hu-CoVs). The first four Hu-CoVs that cause mild disease are HKU1, NL63, OC43, and 229E and are known to circulate in the human population (3). The other two Hu-CoVs, known as severe acute respiratory syndrome-CoV (SARS-CoV) and middle east respiratory syndrome-CoV (MERS-CoV), caused two previous epidemics in 2003 (4) and 2012 (5), respectively. Both SARS-CoV and MERS-CoV were the results of recent spillover events from animals. These two epidemics highlighted how easy it is for spillover events in CoVs to occur and cause outbreaks in humans.
In December 2019, another spillover event occurred and a seventh Hu-CoV appeared known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), previously named 2019-nCoV (6). SARS-CoV-2 has been spreading rapidly across the world since it was first reported in Wuhan, Hubei province, China (6, 7). The advances and accessibility of sequencing technologies have allowed researchers all over the world to quickly sequence the genome of SARS-CoV-2 (8, 9). Zhou et al. (9) showed that SARS-CoV-2 shared 79.6% sequence identity to SARS-CoV and 96% sequence identity to a bat CoV further supporting the theory of another spillover event.
Genomic analysis of SARS-CoV-2 genomes suggested that there were two major genotypes in the early phase of the outbreak, known as L type and S type, based on almost complete linkage between two SNPs (10). Tang et al. (10) proposed that the S type was more ancient while the L type evolved later and may be more aggressive in replication rates and spreads more quickly. Recent reclassifications of SARS-CoV-2 proposed the use of clade nomenclature and divided all viral genomes into 7 major clades including O, S, L, V, G, GR, and GH, with the S clade corresponding to the S type in the Tang study (11–13). Here we present a comprehensive epidemiological and genomic analysis of SARS-CoV-2 genomes from 10 patients in Shaoxing (Zhejiang Province), a mid-sized city about 500 miles away from Wuhan at the early stages of the outbreak.
Materials and Methods
Study Design and Ethics
Ten remnant nasopharyngeal swab samples collected between 1/27/2020 and 2/7/2020, and tested positive by a SARS-CoV-2 real-time PCR assay with cycle threshold (Ct) values of <28, were included in this study. The samples were de-identified except the associated epidemiological data were retained. Since the patient identification was removed and the samples used in this study were remnant and otherwise would be discarded, the Shaoxing Center for Disease Control and Prevention had determined that the institutional review boards (IRB) approval was waived for this project, and the informed consent form was not required.
SARS-CoV-2 PCR and RNA Sequencing
Total nucleic acid was extracted from the nasopharyngeal swabs using the Total Nucleic Acid Extraction Kit (IngeniGen XMK Biotechnologies, Inc., Zhejiang, China). Real-time PCR was performed by using the IngeniGen XMKbio 2019-nCoV (SARS-CoV-2) RNA Detection kit, which targets the highly specific sequences in the ORF1ab and N genes of the virus, on the ABI 7500 system (ThermoFisher Scientific, Inc., MA, USA). The RNA libraries were constructed using the Ingenigen XMKbio RNA-seq Library Prep Kit (IngeniGen XMK Biotechnologies, Inc., Zhejiang, China). Briefly, DNase was used to remove residual human DNA and the RNA was fragmented, followed by double-strand cDNA synthesis, end-repair, dA-tailing, and adapter ligation. Sequencing was performed by using the 2 × 75 bp protocol on the Nextseq 550 system (Illumina, Inc., CA, USA). Sequencing data have been deposited to NCBI SRA under BioProject PRJNA638211, and to GISAID with accessions EPI_ISL_463889 and EPI_ISL_463894 to 463902.
Data Analysis
Quality control and trimming of paired-end reads was performed using custom Python scripts as follows: (1) trim 3′ adapters; (2) trim reads at ambiguous bases; (3) filter reads shorter than 40 bp; (4) filter reads with average quality score <20. Host-derived reads were removed by alignment against the GRCh38.p13 genome reference using bowtie2 (v2.3.4.3) (14) with default parameters. The retained reads were then mapped to 163 published SARS-CoV-2 reference genomes obtained from GISAID (https://www.gisaid.org/CoV2020/, accessed March 2, 2020) by bowtie2 (v2.3.4.3) with default parameters. Snippy (v4.5.0) was used for variant and indel calling, and core SNP alignment against the Wuhan-Hu-1 (NC_045512.2) reference (6), FastTree (v2.1.3) which infers approximately-maximum-likelihood phylogenetic trees (15) was used for tree construction using default parameters, and Figtree (v1.4.4) (16) was used to visualize the resulting phylogenetic tree. HISAT2 (v2.2.0) and StringTie (v2.1.3) were used for RNA-Seq alignment and transcript assembly to identify novel isoforms (17, 18). Additional statistical analyses and visualizations were performed using the “ggplot2” package in the R statistical environment (v3.6). The clade and lineage nomenclature was determined by using pipeline pangolin (https://github.com/hCoV-2019/pangolin) as described previously (13).
Results
Epidemiology of Shaoxing Patients
All 10 patients presented with symptoms (fever and cough) consistent with COVID-19 in late January and early February of 2020. The patients can be categorized into two epidemiologic groups with either a travel history to the Hubei province or sick contact with a confirmed case (Table 1). There was one case where we were unable to obtain a travel or exposure history (Shaoxing-8).
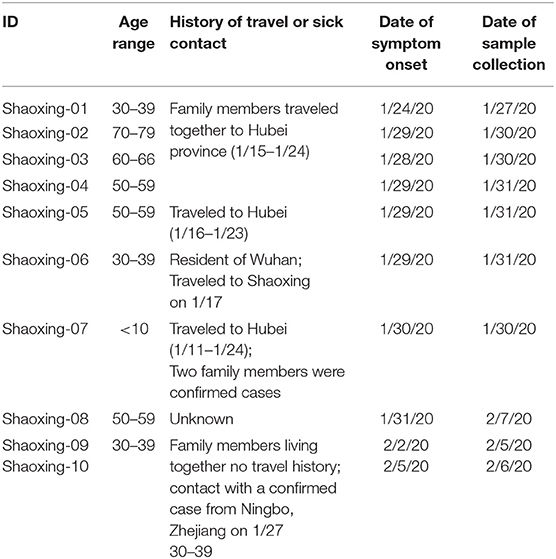
Table 1. Epidemiological history of the 10 Shaoxing patients.
There are two apparent clusters in these 10 patients. The first cluster involves four patients who are relatives and traveled together to Hubei province for a wedding in late January. The first patient in this cluster had symptom onset on their last day in Hubei province while the other three patients had symptom onset 4–5 days after coming back to Shaoxing (Table 1). The second cluster involves two patients who are family members that live together and did not travel to Hubei province. One of the family members (Shaoxing-09) had a sick contact with a confirmed case who visited her but lived in Ningbo, a more populated city in Zhejiang province (Table 1).
Metagenomic Sequencing
The patients were confirmed to have SARS-CoV-2 infection by a commercial Real-time PCR assay. The average Ct values for the 10 patient samples were 23.17 for ORF1ab and 24.54 for N (Table 2). Metagenomic sequencing was performed to recover the full viral genome. The total number of sequence reads per samples ranged from 10.4 to 27.5 million with an average of 17.1 million. A small percentage of these reads mapped to SARS-CoV-2 RNA genome and we did not identify any novel transcripts or fusion events, nor did we detect any insertion or deletion (Table 2). The range of sequence reads that mapped to SARS-CoV-2 RNA was 2,413–163,158 with an average of 49,066. We observed a clear negative correlation between the Ct values of each gene (ORF1ab and N) and the log value of SARS-CoV-2 RNA reads (Figure 1). However, the linearity is not significant (R2 = 0.6628, 0.5595 for ORF1ab and N, respectively), indicating that the number of RNA reads measured by metagenomics sequencing are only semi-quantitative and cannot be interpreted directly as viral loads.
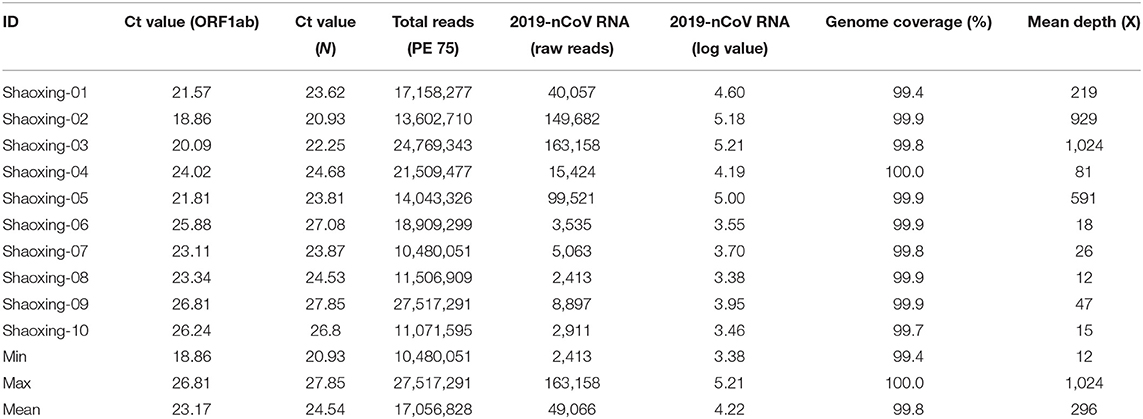
Table 2. Summary of sequencing results of 10 Shaoxing patient samples.
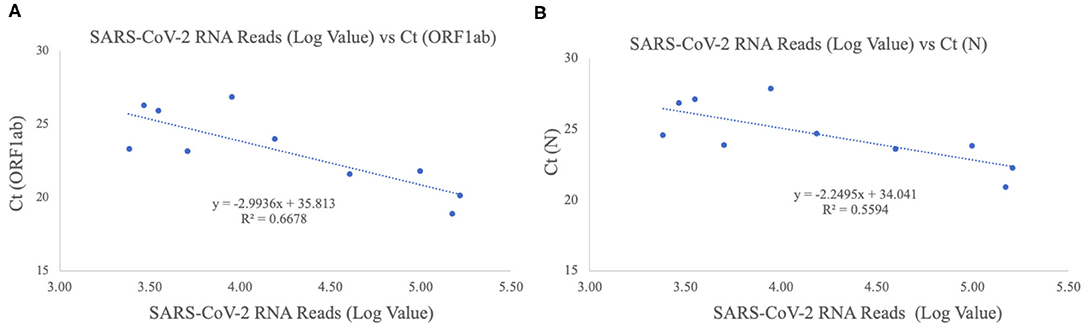
Figure 1. Correlation of Ct values and SARS-CoV-2 RNA reads. (A) ORF1ab gene target. The X-axis plots the log value of the SARS-CoV-2 RNA reads while the Y-axis plots the Ct values for the ORF1ab gene for the 10 Shaoxing patients. (B) N gene target. The X-axis plots the log value of the SARS-CoV-2 RNA reads while the Y-axis plots the Ct values for the N gene for the 10 Shaoxing patients.
With a large variation in the SARS-CoV-2 RNA mapped reads, we were still able to obtain excellent coverage and depth when each genome was mapped to the first SARS-CoV-2 genome, Wuhan-Hu-1 [(6); Figure 2A]. The coverage for all genomes was above 99% and the mean depth for the genomes ranged from 12X to 1024X (Table 2, Figure 2B). Genomes sequenced to a relatively low mean depth (12X to 47X) were still able to be genotyped successfully but our results suggest that SARS-CoV-2 read counts of at least 15,000 yield sufficiently high depth to characterize even low prevalence or rare mutations.
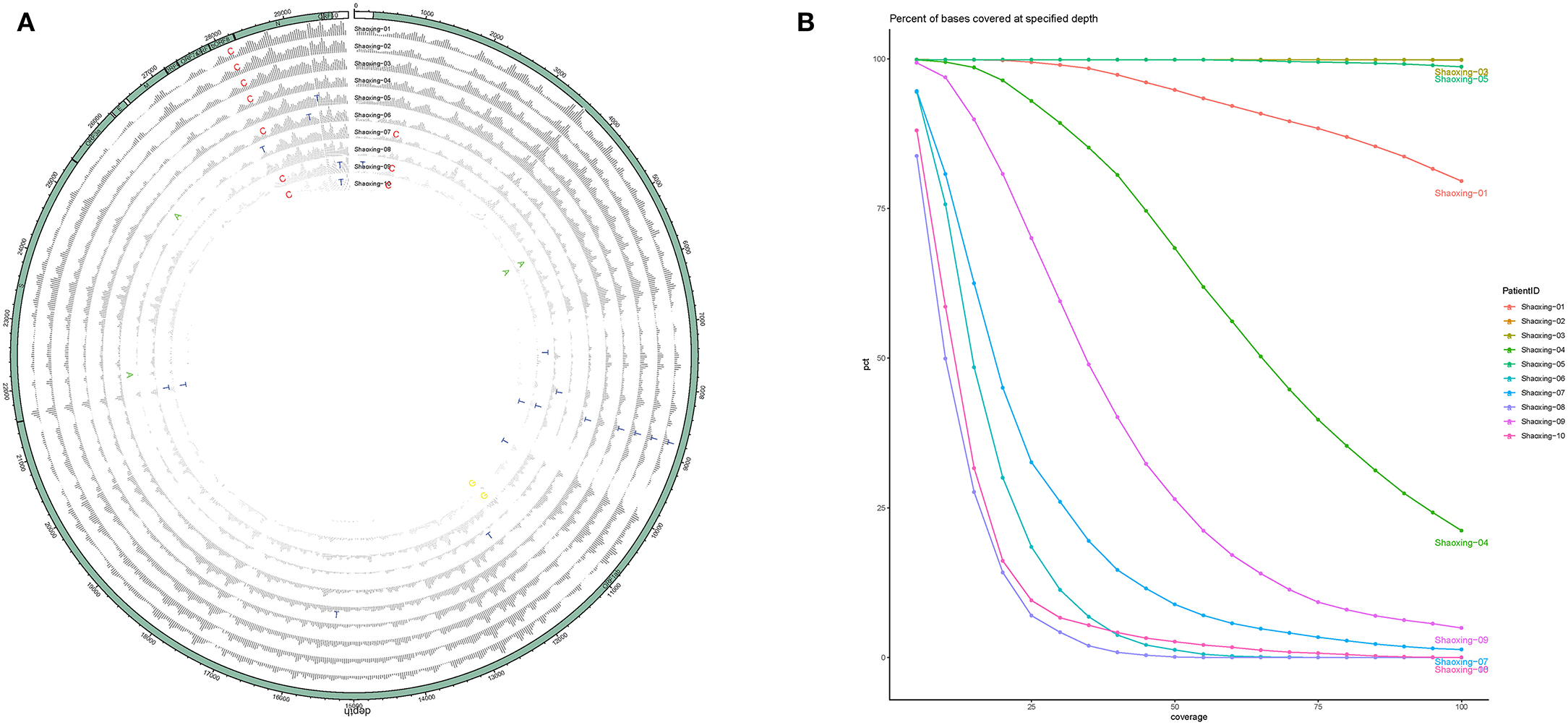
Figure 2. Coverage and depth. (A) Coverage and depth map. Normalized depth at each position along the genome are shown for each sample, with SNPs marked by the alternate allele. (B) Depth ratio. The X-axis plots the log value of the depth for each genome while the Y-axis plots the cumulative percentage of bases covered to the specified depth.
Estimation of Mutation Rate
To determine the single nucleotide polymorphisms (SNPs) of SARS-CoV-2 in these 10 patients, we mapped each genome to the original Wuhan-Hu-1 reference which was collected on December 31, 2019 (6). The genomes contained a fairly moderate number of SNPs (mean of 4 SNPs, range 1-9) (Table 3), consistent with previous reports of relatively low mutation rates (19). The genomes with the largest number of SNPs came from individuals who had contact with a confirmed case from Ningbo, Zhejiang and no travel history to the Hubei province (Table 3, Shaoxing-9 and 10).

Table 3. Summary of genomic descriptions for the Shaoxing SARS-CoV-2 genomes.
Using the SNP analysis, we calculated the various mutation rates using the number of days between the date that the sample was collected and the date the Wuhan-Hu-1 sample was collected. The mutation rate (SNP per day) ranged from 0.03 to 0.25 (Table 3). We used this mutation rate to calculate the nucleotide substitution per site per day and the nucleotide substitution per site per year. We saw an average mutation rate of 3.74 × 10−6 nucleotide substitution per site per day and an average mutation rate of 1.37 × 10−3 nucleotide substitution per site per year (Table 3).
We investigated each SNP to determine if there were any non-synonymous mutations in genes important to the virus lifecycle. Several non-synonymous mutations were found in the following genes: orf1ab, orf3, N, orf8, and orf10 (Table 4A). These mutations were identified with sufficient confidence as at least 5X depth was achieved in all the SNPs (Table 4B, highlighted in orange), and at least 4X depth was achieved at the positions of these SNPs in all samples (Table 4B, unhighlighted). In 7 samples, we identified C8782T and T28144C mutations which are the landmark events of the S clade (Table 4A). We did not identify other GISAID clade defining mutations outlined by Mercatelli and Giorgi (12). No non-synonymous mutations were found in the S gene, which encodes the spike protein that's critical for viral binding to human receptor ACE2 (9). Notably in the cluster of the two family members (Shaoxing-9 and−10), the two viruses are closely related but not identical. Shaoxing-9 was infected first and then transmitted to Shaoxing-10, whose virus gained a non-synonymous mutation C9962T in the ORF1ab gene (Table 4). This could be explained by the sequential transmission, however, we could not rule out a possibility of intra-host viral heterogeneity in the two patients.
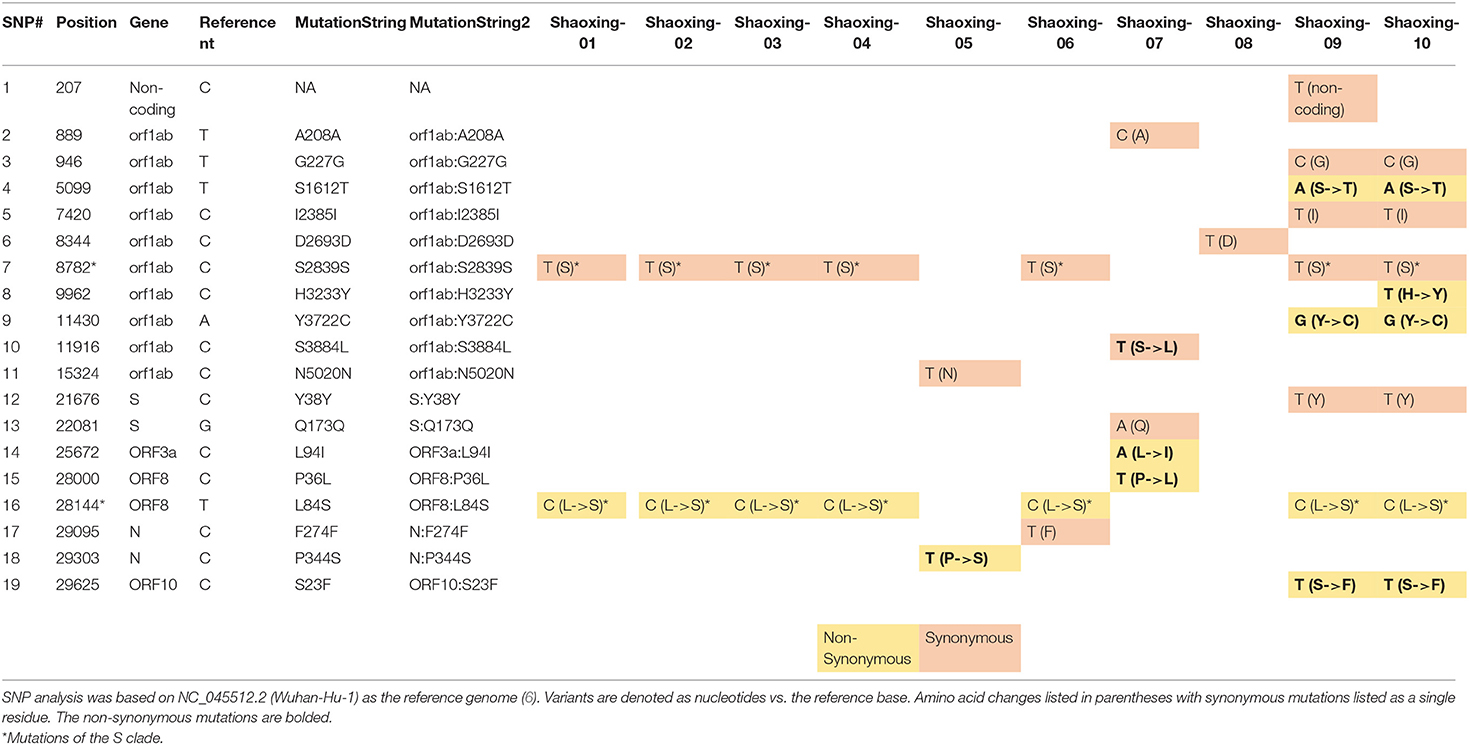
Table 4A. Summary of SNPs in the 10 SARS-CoV-2 genomes.
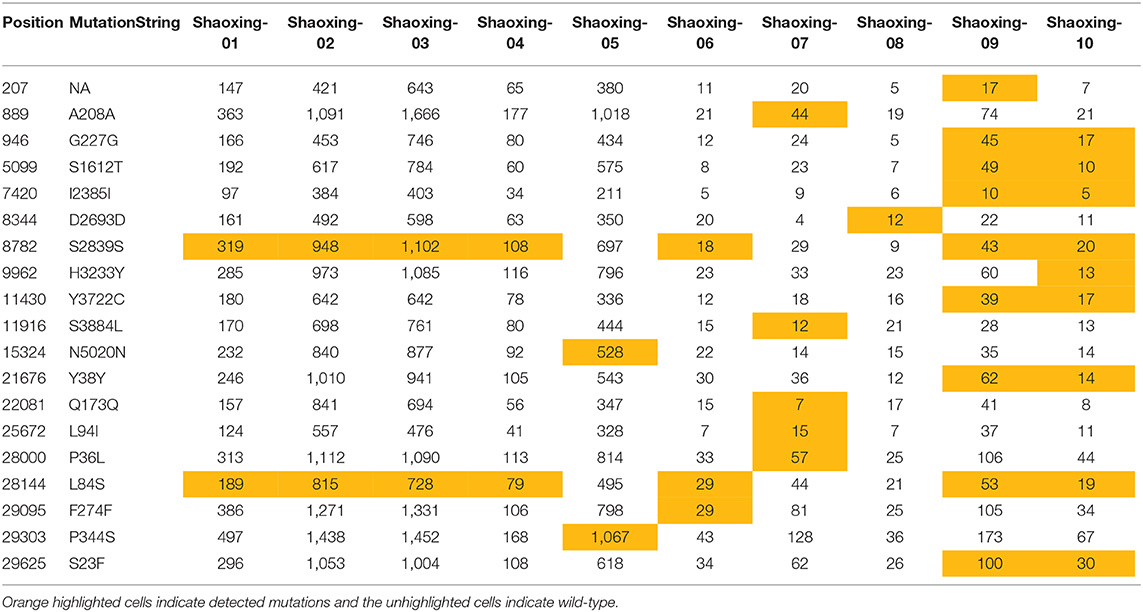
Table 4B. Depth of SNP in the 10 SARS-CoV-2 genomes.
SARS-CoV-2 Genotype and Phylogenetic Characteristics
Previous reports demonstrate that SARS-CoV-2 has two genotypes known as L type and S type in the early phase of the outbreak (10); however, recent classification has divided the SARS-CoV-2 genomes into 7 different clades (O, S, L, V, G, GR, GH) (12). We decided to compare our 10 SARS-CoV-2 genomes to 163 other SARS-CoV-2 genomes obtained from GISAID (8) published by mid-March. Although the majority of the SARS-CoV-2 genomes obtained from GISAID in the early phase of the pandemic belonged to the L clade (Figure 3, green), the 10 Shaoxing SARS-CoV-2 genomes (Figure 3, black boxes) were distributed throughout these genomes with more of them classified in the S clade (Figure 3, orange). The more detailed phylogenetic tree with bootstrap values is shown in Supplementary Figure 1.

Figure 3. Phylogenetic comparison of SARS-CoV-2 genomes. Phylogenetic comparison of 163 published genomes from GISAID (8) and the 10 Shaoxing genomes (boxed out). Genomes are color-coded based on their clade.
Interestingly, four of the Shaoxing SARS-CoV-2 genomes (Shaoxing−1 to−4) were identical to six other GISAID SARS-CoV-2 genomes (Figure 3, Cluster 1). These six other genomes were isolated from patients all over the world: two from Wuhan, two from Taiwan, one from Belgium, and one from Australia (Figure 3, Cluster 1). Shaoxing-6 is identical to five other genomes isolated in Shenzhen, Guangdong Province in Southern China (Figure 3, Cluster 2). Notably, in all 10 Shaoxing patients, we found no virus with D614G Spike gene mutation, which was shown to start spreading in Europe in early February, and rapidly become the dominant form in the rest of the world out of China (20).
Discussion
In this study, we sequenced the SARS-CoV-2 genome from 10 patient samples from Shaoxing, Zhejiang, China. Using metagenomic sequencing, we were able to obtain above 99% coverage and an average depth of 296X for all 10 SARS-CoV-2 genomes. Although not statistically significant, there does appear to be a clear negative correlation between the Ct values of both gene targets and the log count of SARS-CoV-2 RNA sequence reads acquired by metagenomics sequencing. This suggests that the log value of RNA sequence reads by metagenomics sequencing may be used as a semi-quantitative measurement for SARS-CoV-2 viral loads.
The rapid spread of this virus is highlighted by the fact that four SARS-CoV-2 genomes from Shaoxing individuals were identical to six other SARS-CoV-2 genomes from patients all over the world. Our data support recent publications that the virus had spread rapidly around the world especially in Europe before the United States (21–23).
Overall, we did not see a large number of SNPs in these SARS-CoV-2 genomes. The greatest number of SNPs seen was 9 and these two SARS-CoV-2 genomes were from individuals with no travel history to Hubei province (Table 3, Shaoxing-9 and 10). Instead, Shaoxing-9 and 10 had contact with a confirmed case from Ningbo, another city outside of Hubei. We can use these data to infer that the virus accumulated more mutations when it was spread to another city outside of Hubei first before coming to Shaoxing, compared to the virus from people traveled to Shaoxing directly from Hubei.
We combined epidemiologic data with the SNP analysis to estimate the mutation rate of the SARS-CoV-2 from these 10 patients. We saw an average mutation rate of 1.37 × 10−3 nucleotide substitution per site per year for SARS-CoV-2, which is consistent with other reports on the mutation rate of SARS-CoV-2(19, 24) and SARS-CoV-1 with a reported mutation rate of 0.80–2.38 × 10−3 nucleotide substitution per site per year (25). These data demonstrate that SARS-CoV-2 is similar in the mutation rate as other coronaviruses.
The major limitation of this study is that we only had 10 samples analyzed due to the requirement of sufficient SARS-CoV-2 RNA from a metagenomic sample. However, with the development of SARS-CoV-2 probe enrichment or multiplex PCR protocols, this type of viral sequencing analysis may be applied to samples with lower viral loads, thereby enabling more complete molecular epidemiological surveillance. In addition, the Ct value cut-off of 28 established in this study may not be directly applicable to other real-time PCR assays due to the technical differences. Inevitably, exclusion of samples with lower viral load could introduce bias in the genomic surveillance of SARS-CoV-2 and potentially lead to missed identification of important genotypes. Last, we did not analyze or predict the potential biological changes that may be caused by the identified mutations, such as alterations in RNA secondary structure, protein stability, interaction with host proteins, and codon usage, etc.
In summary, we demonstrated that a full viral genomic analysis is feasible via metagenomics sequencing directly on nasopharyngeal samples, which allows retrospective molecular surveillance on SARS-CoV-2 to understand the dynamics of the outbreak in the early phase. The identical virus found in patients in Shaoxing, a mid-sized city outside of Hubei, China, and patients in Europe and Australia was striking. Our analysis added to the growing body of evidence that SARS-CoV-2 spread extremely quickly around the globe as early as January. Although only 10 patients were included in this study, we found numerous mutations (both synonymous and non-synonymous) across the entire viral genome. Our study contributed to the understanding of the SARS-CoV-2 evolution in the early phase of the COVID-19 pandemic.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The raw FASTQ files have been deposited to NCBI SRA under BioProject PRJNA638211, and the snippy-based consensus genome sequences have been deposited to GISAID. They can be found with accessions EPI_ISL_463889 and EPI_ISL_463894 to EPI_ISL_463902.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author Contributions
JC, EH, FL, ZL, JT, and SY conceived and planned the experiments. JC, ZJ, QZ, JW, and YW carried out the experiments. EH, HW, and FL performed the data analysis. JC and EH took the lead in writing the manuscript. ZL, JT, and SY supervised the research project. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Funding
This study was funded by Shaoxing IngeniGen XMK Biotechnologies, Inc.
Conflict of Interest
HW and YW were employed by the company Shaoxing IngeniGen XMK Biotechnologies. FL was the Chief Executive Officer of the company Three Coin Analytics. The authors declare that this study received funding from Shaoxing IngeniGen XMK Biotechnologies. The funder had the following involvement with this study: providing sequencing data and preliminary bioinformatics analysis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Yong-Zhen Zhang (Fudan University) and Eddie Holmes (University of Sydney) for sharing the sequence of the first SARS-CoV-2 isolate in a very timely manner. We would also like to thank Fanchao Meng, Bin Hu, Haihao Shou, and Yuanyuan Cai from Shaoxing IngeniGen XMK Biotechnologies, Inc. for their technical assistance. This manuscript has been released as a pre-print at MedRxiv: Chen et al. (26).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2020.567621/full#supplementary-material
Supplementary Figure 1. Phylogenetic tree of SARS-CoV-2 genomes with bootstrap values.
References
1. Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R. Features, evaluation and treatment coronavirus (COVID-19). In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC.
2. Weiss SR, Leibowitz JL. Coronavirus pathogenesis. Adv Virus Res. (2011) 81:85–164. doi: 10.1016/B978-0-12-385885-6.00009-2
3. Corman VM, Muth D, Niemeyer D, Drosten C. Hosts and sources of endemic human coronaviruses. Adv Virus Res. (2018) 100:163–88. doi: 10.1016/bs.aivir.2018.01.001
4. Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. (2003) 300:1394–9. doi: 10.1126/science.1085952
5. Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. (2012) 367:1814–20. doi: 10.1056/NEJMoa1211721
6. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
7. Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. (2020) 395:470–3. doi: 10.1016/S0140-6736(20)30185-9
8. Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill. (2017) 22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494
9. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
10. Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. (2020) 7:1012–23. doi: 10.1093/nsr/nwaa036
11. Han AX, Parker E, Scholer F, Maurer-Stroh S, Russell CA. Phylogenetic clustering by linear integer programming (PhyCLIP). Mol Biol Evol. (2019) 36:1580–95. doi: 10.1093/molbev/msz053
12. Mercatelli D, Giorgi F. Geographic and genomic distribution of SARS-CoV-2 mutations. Preprints. (2020). doi: 10.20944/preprints202004.0529.v1
13. Rambaut A, Holmes EC, Hill V, O'Toole Á, Mccrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv [preprint]. (2020). doi: 10.1038/s41564-020-0770-5
14. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. (2012) 9:357–9. doi: 10.1038/nmeth.1923
15. Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
16. Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. (2017) 8:28–36. doi: 10.1111/2041-210X.12628
17. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols. (2016) 11:1650–67. doi: 10.1038/nprot.2016.095
18. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. (2019) 37:907–15. doi: 10.1038/s41587-019-0201-4
19. Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, et al. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. (2020) 92: 667–4. doi: 10.1002/jmv.25762
20. Korber B, Fischer W, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv [preprint]. (2020). doi: 10.1101/2020.04.29.069054
21. Deng X, Gu W, Federman S, Du Plessis L, Pybus O, Faria N, et al. A genomic survey of SARS-CoV-2 reveals multiple introductions into northern California without a predominant lineage. medRxiv [preprint]. (2020). doi: 10.1101/2020.03.27.20044925
22. Gonzalez-Reiche AS, Hernandez MM, Sullivan M, Ciferri B, Alshammary H, Obla A, et al. Introductions and early spread of SARS-CoV-2 in the New York City area. medRxiv [preprint]. (2020). doi: 10.1101/2020.04.08.20056929
23. Schuchat A. Public health response to the initiation adn spread of the pandemic COVID-19 in the United States. MMWR Morbid Mortail Weekly Rep. (2020) 551–6. doi: 10.15585/mmwr.mm6918e2
24. Li X, Wang W, Zhao X, Zai J, Zhao Q, Li Y, et al. Transmission dynamics and evolutionary history of 2019-nCoV. J Med Virol. (2020) 92:501–11. doi: 10.1002/jmv.25701
25. Zhao Z, Li H, Wu X, Zhong Y, Zhang K, Zhang YP, et al. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol. (2004) 4:21. doi: 10.1186/1471-2148-4-21
Keywords: 2019-nCoV, COVID-19, genotype, metagenomic sequencing, mutation rate, transmission, genomic epidemiology, SARS-CoV-2
Citation: Chen J, Hilt EE, Li F, Wu H, Jiang Z, Zhang Q, Wang J, Wang Y, Li Z, Tang J and Yang S (2020) Epidemiological and Genomic Analysis of SARS-CoV-2 in 10 Patients From a Mid-Sized City Outside of Hubei, China in the Early Phase of the COVID-19 Outbreak. Front. Public Health 8:567621. doi: 10.3389/fpubh.2020.567621
Received: 30 May 2020; Accepted: 14 August 2020;
Published: 18 September 2020.
Edited by:
Roger Hewson, Public Health England, United KingdomReviewed by:
Federico Manuel Giorgi, University of Bologna, ItalyJosé Eduardo Levi, University of São Paulo, Brazil
Copyright © 2020 Chen, Hilt, Li, Wu, Jiang, Zhang, Wang, Wang, Li, Tang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jialiang Tang, OTkyNDg4OTA0QHFxLmNvbQ==; Shangxin Yang, c2hhbmd4aW55YW5nQG1lZG5ldC51Y2xhLmVkdQ==
†These authors have contributed equally to this work