 Dongguang Wang
Dongguang Wang Yao Ma2†
Yao Ma2† Yonggang Zhang
Yonggang Zhang Hong Fan
Hong Fan- 1Department of Respiratory and Critical Care Medicine, West China Hospital, West China School of Medicine, Sichuan University, Chengdu, China
- 2The Center of Gerontology and Geriatrics, West China Hospital, West China School of Medicine, Sichuan University, Chengdu, China
- 3Department of Periodical Press, West China Hospital, Sichuan University, Chengdu, China
Diabetes mellitus is a systematic metabolic disease characterized by persistent hyperglycemia, which complications often involve multiple organs and systems including vessels, kidneys, retinas, and nervous system. Idiopathic pulmonary fibrosis is a chronic, progressive, fibrotic disease with usual interstitial pneumonia patterns. With in-depth research, diabetic related lung injury has been confirmed, and the lung is also considered as one of the targeted organs of diabetes, which mainly manifests as the pulmonary fibrosis. Based on that, this review discusses the association between diabetes mellitus and idiopathic pulmonary fibrosis from clinical findings to possible mechanisms.
Introduction
Idiopathic pulmonary fibrosis (IPF) is defined as a chronic, progressive, fibrosing interstitial lung disease with the histologic appearance of usual interstitial pneumonia (UIP) (1). The conservative estimate of incidence on IPF is 3~9 cases per 100,000 people per year for Europe and North America (2). The prognosis for patients with IPF is quite poor, with a median survival of 3~5 years from the initial diagnosis if not treated (1). However, the etiology remains little understood.
Diabetes mellitus (DM) is a systemic, metabolic disorder characterized by insulin deficiency or resistance, chronic hyperglycemia, and micro- and macro-vascular bed damage. In the long run, diabetes deeply affects multiple organs throughout the body, especially kidney, retina, heart, and brain, making loss of function of these crucially important organs and bringing about death (3–6). The lung consists of abundant alveolar-capillary network and connective tissue, suggesting that it may be targeted by diabetic micro-vascular damage. Unfortunately, the correlation has often been disregarded and there is lack of research and evidence considering lung as a target for the diabetic impairment. In recent years, several studies have revealed that hyperglycemia could lead to interstitial fibrotic changes and alveolar microangiopathy (7–10). Here, we review the present clinical and experimental evidence, aiming to explore the role of diabetes on idiopathic pulmonary fibrosis (Table 1).
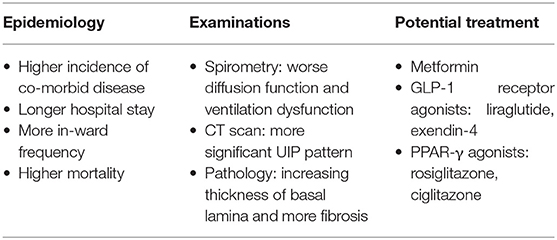
Table 1. Characteristics in diabetic pulmonary fibrosis patients.
Epidemiology
Since Schuyler et al. first demonstrated loss of pulmonary elastic recoil in the diabetics and proposed the abnormalities might be manifestations of extensive abnormal collagen and elastin depositions in the 1970's (11), more and more research has investigated the link between diabetes and idiopathic pulmonary fibrosis. Recent epidemiological research in populations with different genetic background are more prone to the opinion that diabetes is an independent risk factor for idiopathic pulmonary fibrosis, with the prevalence of IPF accompanied by DM estimated to be 10~42%, and the reported result is still consistent even if excluding the interference of patients treated with glucocorticoids (12–18). Following the poor prognosis in IPF patients, diabetes were reported to be a risk factor with an obviously higher mortality in this population (HR 2.5, 95%CI 1.04~5.9) (19). In addition, other studies showed a higher incidence of co-morbid diseases, longer hospital length of stay, and more frequent hospital admission and readmission in IPF patients with diabetes, which also indicated a worse prognosis (12, 15). However, the results above came from studies conducted in several countries with small populations, and amounts of cases included were restricted, more large-sample studies are needed for the overall prognosis assessment of diabetic pulmonary fibrosis in the near future.
Clinical Characteristics
Spirometry
Spirometry is regarded one of the most common measuring tools used to evaluate the severity of respiratory disease. Pulmonary function test on IPF patients mainly shows a ventilatory dysfunction and decreased diffusion capacity. Several longitudinal clinical studies have explored changes of pulmonary function in diabetic individuals. A prospective observational study from Europe found a visible decline of lung volumes and airflow limitation (decreased FVC, FEV1, VC, and PEF) in type 2 diabetes patients (20). Likewise, a 5-years prospective study in Japan showed that diabetes was associated with the restrictive pulmonary function impairment (multivariable adjusted HR 1.57, 95%CI 1.04~2.36) (21). What's more, the recent results from meta-analyses supported the epidemiological evidence as well, with one showing that diabetes free from overt pulmonary disease was associated with impaired pulmonary function in a restrictive pattern and the other finding a lower FVC than FEV1 and DLCO in diabetic individuals (22, 23). While the reduction of spirometric parameters in diabetes seems to be consistent with that caused by IPF, inferences regarding casual effects cannot be made due to lack of evidence of pulmonary fibrosis from these diabetic populations. For this purpose, more longitudinal diabetic research is needed to evaluate the dynamic changes of pulmonary fibrosis indexes in the future.
Imagings
Apart from spirometry, computed tomography (CT) examination is also essential for differential diagnosis of pulmonary lesions. Imaging in patients with IPF usually manifests as UIP pattern including reticular and honeycomb patterns. Kim et al. revealed that IPF patients with diabetes were more likely found the UIP pattern on high resolution computed tomography (HRCT) than those without diabetes (15). Similar results was also demonstrated from a Chinese study, in which more significant presence of funicular and reticular changes were found in diabetic participates compared to normal controls without type 1 diabetes (8). Unfortunately, there is still a lack of enough prospective data to confirm the imaging changes in diabetic patients.
Pathological Changes
Histologically, the basal lamina (BL) is an extracellular scaffold located between parenchymal cells and connective tissue, by its presence compensating new cells normally apoptotic or damaged during the injury (24). If the integrity of basal lamina is destroyed, the repair in most organs results in pathologic fibrosis and loss of organ function. Over the past decades, several studies focused on diabetes had demonstrated a wide alterations of BL in different organs including lungs. Vracko et al. found a thickness of basal lamina both in epithelial and capillary of alveoli from diabetic individuals, and similar morphological changes were verified by Matsubara and other researchers in the 1990's (7, 25–27). Recently, experiments performed in induced diabetic murine models have also confirmed the clinical findings. Diabetic mice and rats were both found a significant thickening of alveolar septa and more fibrosis than non-diabetic controls (8, 28, 29). Since the collagen is the main component of basal lamina, increasing thickness of basal lamina may be the microscopic appearance of idiopathic pulmonary fibrosis (30), and as a result, the pathological changes mentioned above further strengthen the link between diabetes and idiopathic pulmonary fibrosis.
Possible Pathogenesis
The exact pathogenesis of IPF has not been fully elucidated. Traditionally, the evolution of IPF could be divided into three stages: (1) initiation stage, oxidative stress damage from all causes is the initiation factor of IPF; (2) progression stage, inflammation of alveoli, activation of immune cells, and secretion of various pro-inflammatory factors, causing injuries of epithelial, endothelial, and interstitial cells, collagenous tissues, and basement membranes; (3) outcome stage, the formation of pulmonary fibrosis, characterized by proliferation of fibroblasts and myofibroblasts, deposition of extracellular matrix (ECM) and destruction of structure in the lung tissues, eventually leading to chronic respiratory failure. Epithelial-mesenchymal transition (EMT), with characteristics of the increase of fibrotic markers such as α-smooth muscle actin (α-SMA) and vimentin, and the reduction of epithelial markers such as E-cadherin, has been affirmed the crucial tache in pathological pulmonary fibrosis (31). Hyperglycemia promotes to the development of pulmonary fibrosis by participating in all these processes in diabetic animals.
Advanced Glycation End-Products (AGEs) and the Receptor for Advanced Glycation End-Products (RAGEs)
The distribution of AGEs in wide range of organs is limited at a low basal level under normal conditions, while diabetes induces AGEs to accumulate in the lungs (32, 33). Using the streptozotocin (STZ) induced diabetic mice, researchers detected an extensive accumulation of AGEs and pathological hyperplasia of extracellular matrix and interstitial connective tissue in the lung (34, 35). RAGEs, a cell surface receptor for AGEs, have been detected as an antagonist for the clearance of AGE proteins. Cumulative evidence has confirmed a spontaneously pulmonary fibrosis-like alteration in RAGE (-/-) mice (36, 37). RAGEs blocked the activation of Smad2, ERK, and JNK signals, playing a negative role on transforming growth factor beta (TGF-β) induced EMT in A549 cells (38, 39). However, in contradiction to this conclusion, He et al. found a decreased expression of RAGEs and fibroblast lesions in RAGE (-/-)-mice (40), and overexpression of RAGEs increased the oxidative stress and mRNA expression of elastin both in fibroblasts and co-cultured lung epithelial cells (41). Thus, the need of further studies on the function of RAGEs in pulmonary fibrosis is clear now.
Sirtuins (Sirt)
Sirt are class of NAD+ dependent proteins, mediating the process of secretion of insulin, cell cycle, and cell apoptosis. Shaikh reviewed the role of sirtuin family (Sirt1-7), out of which Sirt1, Sirt3, Sirt6, and Sirt7 exerted a positive effect on IPF (42). Overexpressions of Sirt inhibit the oxidative stress, pro-inflammatory cytokines IL-1β and p21 expressions, TGF-β1/Smad3 signaling pathway and mitochondrial DNA damage, elucidating a potential therapeutic approach to IPF. Oppositely, Talakatta observed an EMT and up-regulation of N-cadherin, Sirt3, and Sirt7 levels in diabetic cells exposed to high concentration of glucose (43), indicating that more studies should be carried out to demonstrate the exact role of Sirt on diabetic pulmonary fibrosis.
Pro-Inflammatory and Pro-Fibrotic Factors
Connective tissue growth factor (CTGF) is a downstream mediator of the profibrotic properties of TGF-β, playing an important role in tissue remodeling and fibrosis. Up-regulated expression of CTGF is observed in several fibrotic organs such as kidney, heart, liver, skin, and lung (44). It was reported an increased expression level of CTGF in the lung tissues by the STZ induced type 1-like diabetic rat models, thereby indicating the adverse influence of diabetes on pulmonary fibrosis (8, 45). Besides, fibronectin, angiotensin II (Ang II), plasminogen activator inhibitor-1 (PAI-1) and other pro-inflammatory or pro-fibrotic factors were also found increased in diabetic mouse lungs, and they widely participated in the process of diabetic lung fibrosis by a variety of mechanisms (8, 46).
Endoplasmic Reticulum Stress (ERS)
The endoplasmic reticulum (ER) is a kind of multi-functional organelle which is essential for the cellular homeostasis. Excessive demand on the ER or conditions could destruct the function of ER may cause the accumulation of unfolded or misfolded proteins, triggering ERS and activation of the unfolded protein response (UPR) (47). It has been demonstrated that ERS related markers in the lung were activated by bleomycin both in vitro and in vivo and treatment with ERS inhibitors resulted in the reduction of fibroblast proliferation and improvement of lung functions, suggesting a potential role of ERS for the pathogenesis of IPF (48, 49). Also, ERS is found associated with the β cell failure in type 1 and type 2 diabetes (47, 50). However, the role of ERS in diabetic pulmonary fibrosis is still unclear. Whether hyperglycemia could act as a stimulus inducing ERS in the process of diabetic pulmonary fibrosis or not remains to be further studied.
To conclude, activation of multiple pathways in the high-glucose environment results in abnormal intracellular stress and cytokine expressions, consequently causing loss of basal laminae integrity in alveolar-capillary barrier, failure of reepithelialization and reendothelialization, and epithelial-mesenchymal transition, and eventually, leading to destroyed lung architecture and pathological pulmonary fibrosis.
Potential Application of Anti-Diabetic Drugs in IPF
Metformin
At present, the therapeutic strategies and effects of IPF remain limited, although several tyrosine kinase inhibitors such as pirfenidone and nintedanib have come into our attention in recent years. Metformin is a classic oral anti-diabetic agent, which has been demonstrated anti-inflammatory, anti-angiogenetic and anti-fibrotic properties in animal models as well. In experimental murine models, studies support the role of metformin to reverse the established pulmonary fibrosis by facilitating deactivation and apoptosis of myofibroblasts via activating AMPK and ameliorating TGF-β signaling pathways (51–53). However, the pharmacokinetic data of oral metformin suggest that much of the concentration is in the gut, even after absorption. Besides, there is evidence suggesting that high dose of intravenous metformin has disconcerting effects on rats and inhibiting the viability of blood-brain barrier cell lines (54, 55). Thus, a pinpoint delivery of metformin to the lung such as inhalational drug delivery seems to be an attractive idea in future experimental studies.
Glucagon Like Peptide-1 (GLP-1) Receptor Agonists
GLP-1 is an essential hormone for the regulation of insulin secretion, carbohydrate metabolism and appetite. GLP-1 receptor, a G protein coupled receptor, predominantly localized in β-cells of pancreas and smooth muscle cells of arteries and arterioles in kidney and lung, is mainly used as the target for anti-diabetic drugs (56). In recent years, several studies also found the anti-pulmonary fibrotic effect of GLP-1 receptor agonist in animals. Gou et al. demonstrated that liraglutide treatment significantly alleviated bleomycin (BLM)-induced lung damage and fibrosis in mice through inactivation of nuclear factor kappa-B (NF-κB) (57). Similarly, Oztay et al. found exendin-4 ameliorating hyperglycemia-mediated lung injury by reducing oxidative stress and stimulating cell proliferation (58).
Peroxisome Proliferator-Activated Receptor-Gamma (PPAR-γ) Agonists
PPAR-γ belongs to nuclear hormone receptor superfamily, with action in many activities including alterations of metabolic and inflammatory responses. Recent studies also found an efficacy of PPAR-γ agonists in BLM-induced pulmonary fibrosis (59, 60). Rosiglitazone and ciglitazone were found inhibiting the fibrotic changes in TGF-β1-mediated EMT of alveolar epithelial cells, differentiation of myofbroblasts, and production of collagen in murine models, suggesting a therapeutic role of PPAR-γ ligands for fibrotic pulmonary disease (61, 62).
Summary
Currently, incidences of diabetes and idiopathic pulmonary fibrosis are rising year by year, causing a heavy burden on patients. Objective evidence has found a link between the two diseases. Relying on limited research, we speculate that the sustained hyperglycemia in the body promotes the development of pulmonary fibrosis, through either directly damaging the alveolar epithelial cells (AECs) or participating in the generation of other pro-inflammatory and pro-fibrotic factors. Although the lung is not the main organ of diabetic complications, patients suffering from both diseases are reported a worsen prognosis. Increasing number of studies have found an anti-fibrotic effect of some anti-diabetic agents, and more attention and deeper studies on mechanism and intervention are needed for diabetic pulmonary fibrosis as well as an integrated follow-up system.
Author Contributions
DW, YM, XT, YZ, and HF collaboratively conceptualized this manuscript. DW wrote the first draft of this manuscript. YM and XT wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
This study was supported by National Key R&D Program of China (2017YFC1309703) and 1.3.5 project for disciplines of excellence-Clinical Research Incubation Project, West China Hospital, Sichuan University (2019HXFH008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
IPF, idiopathic pulmonary fibrosis; UIP, usual interstitial pneumonia; DM, diabetes mellitus; FVC, forced vital capacity; FEV1, forced expiratory volume in 1 s; VC, vital capacity; PEF, peak expiratory flow; DLCO, diffusing capacity of the lung for carbon monoxide; HR, hazard ratio; CI, confidence interval; CT, computed tomography; HRCT, high resolution computed tomography; BL, basal lamina; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; α-SMA, α-smooth muscle actin; AGEs, advanced glycation end-products; RAGEs, receptor for advanced glycation end-products; STZ, streptozotocin; ERK, extracellular regulated protein kinase; JNK, c-Jun N-terminal kinase; TGF-β, transforming growth factor beta; mRNA, messenger RNA; Sirt, sirtuins; NAD, nicotinamide adenine dinucleotide; CTGF, connective tissue growth factor; Ang-II, angiotensin II; PAI-1, plasminogen activator inhibitor-1; ERS, endoplasmic reticulum stress; ER, endoplasmic reticulum; UPR, unfolded protein response; AMPK, adenosine 5‘-monophosphate (AMP)-activated protein kinase; GLP-1, glucagon like peptide-1; BLM, bleomycin; NF-κB, nuclear factor kappa-B; PPAR-γ, peroxisome proliferator-activated receptor-gamma; AECs, alveolar epithelial cell.
References
1. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. American thoracic society and Latin American thoracic diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2018) 198:e44–68. doi: 10.1164/rccm.201807-1255ST
2. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. (2015) 46:795–806. doi: 10.1183/09031936.00185114
3. Powell DW, Kenagy DN, Zheng S, Coventry SC, Xu J, Cai L, et al. Associations between structural and functional changes to the kidney in diabetic humans and mice. Life Sci. (2013) 93:257–64. doi: 10.1016/j.lfs.2013.06.016
4. Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med. (2012) 366:1227–39. doi: 10.1056/NEJMra1005073
5. Szuszkiewicz-Garcia MM, Davidson JA. Cardiovascular disease in diabetes mellitus: risk factors and medical therapy. Endocrinol Metab Clin North Am. (2014) 43:25–40. doi: 10.1016/j.ecl.2013.09.001
6. Toth C. Diabetes and neurodegeneration in the brain. Handb Clin Neurol. (2014) 126:489–511. doi: 10.1016/B978-0-444-53480-4.00035-7
7. Weynand B, Jonckheere A, Frans A, Rahier J. Diabetes mellitus induces a thickening of the pulmonary basal lamina. Respiration. (1999) 66:14–9. doi: 10.1159/000029331
8. Hu Y, Ma Z, Guo Z, Zhao F, Wang Y, Cai L, et al. Type 1 diabetes mellitus is an independent risk factor for pulmonary fibrosis. Cell Biochem Biophys. (2014) 70:1385–91. doi: 10.1007/s12013-014-0068-4
9. Ali MO. Pulmonary complications in diabetes mellitus. Mymensingh Med J. (2014) 23:603–5. doi: 10.1016/s0025-6196(12)61752-2
10. Klein OL, Kalhan R, Williams MV, Tipping M, Lee J, Peng J, et al. Lung spirometry parameters and diffusion capacity are decreased in patients with Type 2 diabetes. Diabet Med. (2012) 29:212–9. doi: 10.1111/j.1464-5491.2011.03394.x
11. Schuyler MR, Niewoehner DE, Inkley SR, Kohn R. Abnormal lung elasticity in juvenile diabetes mellitus. Am Rev Respir Dis. (1976) 113:37–41.
12. Sherbini N, Feteih MN, Wali SO, Alamoudi OS, Al-Faifi SM, Khalid I. Idiopathic pulmonary fibrosis in Saudi Arabia: demographic, clinical and survival data from two tertiary care hospitals. Ann Thorac Med. (2014) 9:168–72. doi: 10.4103/1817-1737.134073
13. Enomoto T, Usuki J, Azuma A, Nakagawa T, Kudoh S. Diabetes mellitus may increase risk for idiopathic pulmonary fibrosis. Chest. (2003) 123:2007–11. doi: 10.1378/chest.123.6.2007
14. Pedraza-Serrano F, Jimenez-Garcia R, Lopez-de-Andres A, Hernandez-Barrera V, Sanchez-Munoz G, Puente-Maestu L, et al. Characteristics and outcomes of patients hospitalized with interstitial lung diseases in Spain, 2014 to 2015. Medicine. (2019) 98:e15779. doi: 10.1097/MD.0000000000015779
15. Kim YJ, Park JW, Kyung SY, Lee SP, Chung MP, Kim YH, et al. Clinical characteristics of idiopathic pulmonary fibrosis patients with diabetes mellitus: the national survey in Korea from 2003 to 2007. J Korean Med Sci. (2012) 27:756–60. doi: 10.3346/jkms.2012.27.7.756
16. Garcia-Sancho Figueroa MC, Carrillo G, Perez-Padilla R, Fernandez-Plata MR, Buendia-Roldan I, Vargas MH, et al. Risk factors for idiopathic pulmonary fibrosis in a Mexican population. A case-control study. Respir Med. (2010) 104:305–9. doi: 10.1016/j.rmed.2009.08.013
17. Gribbin J, Hubbard R, Smith C. Role of diabetes mellitus and gastro-oesophageal reflux in the aetiology of idiopathic pulmonary fibrosis. Respir Med. (2009) 103:927–31. doi: 10.1016/j.rmed.2008.11.001
18. Ehrlich SF, Quesenberry CP Jr, Van Den Eeden SK, Shan J, Ferrara A. Patients diagnosed with diabetes are at increased risk for asthma. chronic obstructive pulmonary disease. Pulmonary fibrosis and pneumonia but not lung cancer. Diabetes Care. (2010) 33:55–60. doi: 10.2337/dc09-0880
19. Hyldgaard C, Hilberg O, Bendstrup E. How does comorbidity influence survival in idiopathic pulmonary fibrosis? Respir Med. (2014) 108:647–53. doi: 10.1016/j.rmed.2014.01.008
20. Davis WA, Knuiman M, Kendall P, Grange V, Davis TM, Fremantle Diabetes S. Glycemic exposure is associated with reduced pulmonary function in type 2 diabetes: the fremantle diabetes study. Diabetes Care. (2004) 27:752–7. doi: 10.2337/diacare.27.3.752
21. Sonoda N, Morimoto A, Tatsumi Y, Asayama K, Ohkubo T, Izawa S, et al. A prospective study of the impact of diabetes mellitus on restrictive and obstructive lung function impairment: The Saku study. Metabolism. (2018) 82:58–64. doi: 10.1016/j.metabol.2017.12.006
22. van den Borst B, Gosker HR, Zeegers MP, Schols AM. Pulmonary function in diabetes: a metaanalysis. Chest. (2010) 138:393–406. doi: 10.1378/chest.09-2622
23. Klein OL, Krishnan JA, Glick S, Smith LJ. Systematic review of the association between lung function and type 2 diabetes mellitus. Diabet Med. (2010) 27:977–87. doi: 10.1111/j.1464-5491.2010.03073.x
24. Vracko R. Basal lamina scaffold-anatomy and significance for maintenance of orderly tissue structure. Am J Pathol. (1974) 77:314–46.
25. Vracko R, Thorning D, Huang TW. Basal lamina of alveolar epithelium and capillaries: quantitative changes with aging and in diabetes mellitus. Am Rev Respir Dis. (1979) 120:973–83.
26. Matsubara T, Hara F. The pulmonary function and histopathological studies of the lung in diabetes mellitus. Nihon Ika Daigaku Zasshi. (1991) 58:528–36.
27. Watanabe K, Senju S, Toyoshima H, Yoshida M. Thickness of the basement membrane of bronchial epithelial cells in lung diseases as determined by transbronchial biopsy. Respir Med. (1997) 91:406–10. doi: 10.1016/S0954-6111(97)90254-7
28. Popov D, Simionescu M. Structural and transport property alterations of the lung capillary endothelium in diabetes. Ital J Anat Embryol. (2001) 106:405–12. doi: 10.1183/09031936.97.10081850
29. Carlson EC, Audette JL, Veitenheimer NJ, Risan JA, Laturnus DI, Epstein PN. Ultrastructural morphometry of capillary basement membrane thickness in normal and transgenic diabetic mice. Anat Rec A Discov Mol Cell Evol Biol. (2003) 271:332–41. doi: 10.1002/ar.a.10038
30. Todd NW, Atamas SP, Luzina IG, Galvin JR. Permanent alveolar collapse is the predominant mechanism in idiopathic pulmonary fibrosis. Expert Rev Respir Med. (2015) 9:411–8. doi: 10.1586/17476348.2015.1067609
31. Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L525–34. doi: 10.1152/ajplung.00163.2007
32. Yang J, Xue Q, Miao L, Cai L. Pulmonary fibrosis: a possible diabetic complication. Diabetes Metab Res Rev. (2011) 27:311–7. doi: 10.1002/dmrr.1175
33. Sanchez E, Lecube A, Betriu A, Hernandez C, Lopez-Cano C, Gutierrez-Carrasquilla L, et al. Subcutaneous advanced glycation end-products and lung function according to glucose abnormalities: the ILERVAS Project. Diabetes Metab. (2018) 45:595–8. doi: 10.1016/j.diabet.2018.04.002
34. Popov D, Hasu M, Costache G, Stern D, Simionescu M. Capillary and aortic endothelia interact in situ with nonenzymatically glycated albumin and develop specific alterations in early experimental diabetes. Acta Diabetol. (1997) 34:285–93. doi: 10.1007/s005920050090
35. Usuki J, Enomoto T, Azuma A, Matsuda K, Aoyama A, Kudoh S, et al. Influence of hyperglycemia to the severity of pulmonary fibrosis. Chest. (2001) 120:71S. doi: 10.1378/chest.120.1_suppl.S71
36. Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, Fattman CL, et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol. (2008) 172:583–91. doi: 10.2353/ajpath.2008.070569
37. Kumar V, Fleming T, Terjung S, Gorzelanny C, Gebhardt C, Agrawal R, et al. Homeostatic nuclear RAGE-ATM interaction is essential for efficient DNA repair. Nucleic Acids Res. (2017) 45:10595–613. doi: 10.1093/nar/gkx705
38. Ding H, Ji X, Chen R, Ma T, Tang Z, Fen Y, et al. Antifibrotic properties of receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. (2015) 35:34–41. doi: 10.1016/j.pupt.2015.10.010
39. Song JS, Kang CM, Park CK, Yoon HK, Lee SY, Ahn JH, et al. Inhibitory effect of receptor for advanced glycation end products (RAGE) on the TGF-beta-induced alveolar epithelial to mesenchymal transition. Exp Mol Med. (2011) 43:517–24. doi: 10.3858/emm.2011.43.9.059
40. He M, Kubo H, Ishizawa K, Hegab AE, Yamamoto Y, Yamamoto H, et al. The role of the receptor for advanced glycation end-products in lung fibrosis. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L1427–36. doi: 10.1152/ajplung.00075.2007
41. Al-Robaiy S, Weber B, Simm A, Diez C, Rolewska P, Silber RE, et al. The receptor for advanced glycation end-products supports lung tissue biomechanics. Am J Physiol Lung Cell Mol Physiol. (2013) 305:L491–500. doi: 10.1152/ajplung.00090.2013
42. Shaikh SB, Prabhu A, Bhandary YP. Targeting anti-aging protein sirtuin (Sirt) in the diagnosis of idiopathic pulmonary fibrosis. J Cell Biochem. (2018) 120:6878–85. doi: 10.1002/jcb.28033
43. Talakatta G, Sarikhani M, Muhamed J, Dhanya K, Somashekar BS, Mahesh PA, et al. Diabetes induces fibrotic changes in the lung through the activation of TGF-beta signaling pathways. Sci Rep. (2018) 8:11920. doi: 10.1038/s41598-018-30449-y
44. Gressner OA, Gressner AM. Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. (2008) 28:1065–79. doi: 10.1111/j.1478-3231.2008.01826.x
45. Wang CM, Hsu CT, Niu HS, Chang CH, Cheng JT, Shieh JM. Lung damage induced by hyperglycemia in diabetic rats: the role of signal transducer and activator of transcription 3 (STAT3). J Diabetes Complications. (2016) 30:1426–33. doi: 10.1016/j.jdiacomp.2016.07.005
46. Yang J, Tan Y, Zhao F, Ma Z, Wang Y, Zheng S, et al. Angiotensin II plays a critical role in diabetic pulmonary fibrosis most likely via activation of NADPH oxidase-mediated nitrosative damage. Am J Physiol Endocrinol Metab. (2011) 301:E132–44. doi: 10.1152/ajpendo.00629.2010
47. Engin F. ER stress and development of type 1 diabetes. J Investig Med. (2016) 64:2–6. doi: 10.1097/JIM.0000000000000229
48. Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su K, et al. Involvement of ER stress, PI3K/AKT activation. and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci Rep. (2017) 7:14272. doi: 10.1038/s41598-017-14612-5
49. Delbrel E, Soumare A, Naguez A, Label R, Bernard O, Bruhat A, et al. HIF-1alpha triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells. a key event in pulmonary fibrosis. Sci Rep. (2018) 8:17939. doi: 10.1038/s41598-018-36063-2
50. Papa FR. Endoplasmic reticulum stress pancreatic beta-cell degeneration and diabetes. Cold Spring Harb Perspect Med. (2012) 2:a007666. doi: 10.1101/cshperspect.a007666
51. Gamad N, Malik S, Suchal K, Vasisht S, Tomar A, Arava S, et al. Metformin alleviates bleomycin-induced pulmonary fibrosis in rats: pharmacological effects and molecular mechanisms. Biomed Pharmacother. (2018) 97:1544–53. doi: 10.1016/j.biopha.2017.11.101
52. Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med. (2018) 24:1121–7. doi: 10.1038/s41591-018-0087-6
53. Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J, et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res. (2016) 17:107. doi: 10.1186/s12931-016-0420-x
54. Peuler JD. Opposing adrenergic actions of intravenous metformin on arterial pressure in female spontaneously hypertensive rats. Cardiovasc Res. (1999) 43:237–47. doi: 10.1016/S0008-6363(99)00051-6
55. Prasad S, Sajja RK, Kaisar MA, Park JH, Villalba H, Liles T, et al. Role of Nrf2 and protective effects of metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol. (2017) 12:58–69. doi: 10.1016/j.redox.2017.02.007
56. Pyke C, Heller RS, Kirk RK, Orskov C, Reedtz-Runge S, Kaastrup P, et al. GLP-1 receptor localization in monkey and human tissue: novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. (2014) 155:1280–90. doi: 10.1210/en.2013-1934
57. Gou S, Zhu T, Wang W, Xiao M, Wang XC, Chen ZH. Glucagon like peptide-1 attenuates bleomycin-induced pulmonary fibrosis. Involving the inactivation of NF-kappaB in mice. Int Immunopharmacol. (2014) 22:498–504. doi: 10.1016/j.intimp.2014.07.010
58. Oztay F, Sancar-Bas S, Gezginci-Oktayoglu S, Ercin M, Bolkent S. Exendin-4 partly ameliorates - hyperglycemia-mediated tissue damage in lungs of streptozotocin-induced diabetic mice. Peptides. (2018) 99:99–107. doi: 10.1016/j.peptides.2017.12.007
59. Jin GY, Bok SM, Han YM, Chung MJ, Yoon KH, Kim SR, et al. Effectiveness of rosiglitazone on bleomycin-induced lung fibrosis: Assessed by micro-computed tomography and pathologic scores. Eur J Radiol. (2012) 81:1901–6. doi: 10.1016/j.ejrad.2010.12.061
60. Yu W, Mi L, Long T. Efficacies of rosiglitazone and retinoin on bleomycin-induced pulmonary fibrosis in rats. Exp Ther Med. (2017) 14:609–15. doi: 10.3892/etm.2017.4555
61. Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L891–901. doi: 10.1152/ajplung.00333.2007
Keywords: diabetic pulmonary fibrosis, diabetic complications, diabetes mellitus, idiopathic pulmonary fibrosis, risk factors, potential mechanisms
Citation: Wang D, Ma Y, Tong X, Zhang Y and Fan H (2020) Diabetes Mellitus Contributes to Idiopathic Pulmonary Fibrosis: A Review From Clinical Appearance to Possible Pathogenesis. Front. Public Health 8:196. doi: 10.3389/fpubh.2020.00196
Received: 02 September 2019; Accepted: 29 April 2020;
Published: 03 June 2020.
Edited by:
Liang-Jun Yan, University of North Texas Health Science Center, United StatesCopyright © 2020 Wang, Ma, Tong, Zhang and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yonggang Zhang, jebm_zhang@yahoo.com; Hong Fan, fanhongfan@qq.com
†These authors have contributed equally to this work