 Rainald Schmidt-Kastner
Rainald Schmidt-Kastner Sinan Guloksuz
Sinan Guloksuz Thomas Kietzmann4
Thomas Kietzmann4 Jim van Os
Jim van Os Bart P. F. Rutten
Bart P. F. Rutten- 1Integrated Medical Science Department, C.E. Schmidt College of Medicine, Florida Atlantic University (FAU), Boca Raton, FL, United States
- 2Department of Psychiatry and Neuropsychology, School for Mental Health and Neuroscience, Maastricht University Medical Centre, Maastricht, Netherlands
- 3Department of Psychiatry, Yale School of Medicine, New Haven, CT, United States
- 4Faculty of Biochemistry and Molecular Medicine and Biocenter Oulu, University of Oulu, Oulu, Finland
- 5Department of Psychiatry, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht University, Utrecht, Netherlands
- 6Department of Psychosis Studies, Institute of Psychiatry, King’s College London, King’s Health Partners, London, United Kingdom
Obstetric complications (OCs) can induce major adverse conditions for early brain development and predispose to mental disorders, including schizophrenia (SCZ). We previously hypothesized that SCZ candidate genes respond to ischemia-hypoxia as part of OCs which impacts neurodevelopment. We here tested for an overlap between SCZ genes from genome-wide association study (GWAS) (n=458 genes from 145 loci of the most recent GWAS dataset in SCZ) and gene sets for ischemia-hypoxia response. Subsets of SCZ genes were related to (a) mutation-intolerant genes (LoF database), (b) role in monogenic disorders of the nervous system (OMIM, manual annotations), and (c) synaptic function (SynGO). Ischemia-hypoxia response genes of the brain (IHR genes, n=1,629), a gene set from RNAseq in focal brain ischemia (BH, n=2,449) and genes from HypoxiaDB (HDB, n=2,289) were overlapped with the subset of SCZ genes and tested for enrichment with Chi-square tests (p < 0.017). The SCZ GWAS dataset was enriched for LoF (n=112; p=0.0001), and the LoF subset was enriched for IHR genes (n=25; p=0.0002), BH genes (n=35; p=0.0001), and HDB genes (n=23; p=0.0005). N=96 genes of the SCZ GWAS dataset (21%) could be linked to a monogenic disorder of the nervous system whereby IHR genes (n=19, p=0.008) and BH genes (n=23; p=0.002) were found enriched. N=46 synaptic genes were found in the SCZ GWAS gene set (p=0.0095) whereby enrichments for IHR genes (n=20; p=0.0001) and BH genes (n=13; p=0.0064) were found. In parallel, detailed annotations of SCZ genes for a role of the hypoxia-inducible factors (HIFs) identified n=33 genes of high interest. Genes from SCZ GWAS were enriched for mutation-intolerant genes which in turn were strongly enriched for three sets of genes for the ischemia-hypoxia response that may be invoked by OCs. A subset of one fifth of SCZ genes has established roles in monogenic disorders of the nervous system which was enriched for two gene sets related to ischemia-hypoxia. SCZ genes related to synaptic functions were also related to ischemia-hypoxia. Variants of SCZ genes interacting with ischemia-hypoxia provide a specific starting point for functional and genomic studies related to OCs.
Introduction
A core concept of contemporary psychiatric research is that multiple genes interact with environmental factors to increase the risk of psychosis spectrum disorder, including schizophrenia (1–4). Obstetric complications (OCs) are established risk factors for schizophrenia (5–7) while it is generally thought that perturbations of oxygen and substrate delivery (i.e., ischemia-hypoxia) during OCs affect the developing brain and impair neuronal development during critical phases (8, 9). Immune mechanisms and neuroinflammation are also considered as major prenatal risk factors for schizophrenia (10), and inflammation typically invokes hypoxic challenges (11). Furthermore, evidence for links between vascular factors, including angiogenesis, and schizophrenia has been reported (12–14).
We previously hypothesized that candidate genes for schizophrenia interact with ischemia-hypoxia during OCs (9). To explore this hypothesis, we used our “ischemia-hypoxia response” (IHR) database that combined information for changes at the mRNA level after brain ischemia-hypoxia from multiple experimental studies (15, 16). Candidate genes for schizophrenia were then annotated using the IHR gene database (9, 17). Thereby, the assumptions were made that those genes observed experimentally in the adult brain are likely expressed in the developing brain, and that genes transcribed in mice or rats are also expressed in the human brain. The concept was extended to the expression or function in vascular cells, because ischemia-hypoxia will immediately invoke vascular responses and genetic mechanisms affecting vessels may cause ischemia-hypoxia. The hypothesis was that genetic variants of schizophrenia genes annotated as IHR genes and/or vascular genes may alter responsiveness to ischemia-hypoxia during OCs. In fact, our hypothesis has received support from studies where the interaction between SNPs for selected candidate genes were linked to IHR genes and OCs in relation to schizophrenia risk (18–20).
The initial analysis (9) was based on a selection from SzGene which collected more than 700 candidate genes for schizophrenia (21). A bias within SzGene toward genes of major interest for brain damage and neurodegeneration would have inflated the importance of IHR genes. Genome-wide studies such as GWAS are now considered as standard for the hypothesis-free analysis of polygenic disorders such as schizophrenia. Therefore, it was timely to examine the genes in loci defined by GWAS in schizophrenia (22, 23) in relation to ischemia-hypoxia. To do this, we linked the schizophrenia GWAS dataset to ischemia-hypoxia using the following approaches to form relevant subsets. 1) A large set of mutation-intolerant genes (LoF) was recently described (24), and it was reported that common schizophrenia alleles in GWAS are enriched for mutation-intolerant genes (23). It was hypothesized that IHR genes should be also enriched in datasets of LoF if they are essential for the response to ischemic-hypoxic challenges. Then, tests were performed for an overlap between schizophrenia genes from GWAS, mutation-intolerant schizophrenia genes, and IHR genes. 2) Schizophrenia genes from GWAS were annotated for known monogenic disorders of the nervous system that affect development and function, and then tested for overlap with IHR genes. 3) Since schizophrenia genes emerging from genomic studies are closely related to synaptic functions (25, 26), a novel database of synaptic proteins [SynGO; (27)] was used to define IHR genes related to synaptic functions and tested for the overlap with schizophrenia genes. 4) Studies were supplemented by analyzing a recently proposed set of 104 schizophrenia candidate genes from the original 108-loci study using a complex combination of parameters (28). 5) To test for a more general involvement of neurodevelopmental disease mechanisms, a gene set related to developmental delay and autism spectrum disorder (29) was tested for enrichment as well. To further explore a to link to ischemia-hypoxia, a gene set from a recent RNAseq study in focal brain ischemia (30) and the HDB database (31) were employed.
Materials and Methods
An overview of the analysis is provided in Figure 1.

Figure 1 Graphic presentation of the annotation process. GWAS data for schizophrenia (SCZ GWAS) were taken from Pardinas et al. [ref. (23)] and subsets of genes were formed for Loss of Function [LoF; ref. (24)], for Monogenic Disorders (Monogenic Dis.; described under Materials and Methods), for synaptic genes collected in SynGO [ref. (27)] and for a subset described by Wang et al., 2019 [ref.(28)]. Gene sets related to Ischemia-Hypoxia were taken from our database named “ischemia-hypoxia response genes” [IHR Genes; refs. (16, 17)], from the RNAseq study of Bhattarai et al., 2019 [BH Genes; ref. (30)] and from the public database named HypoxiaDB [HDB Genes; ref. (31)]. Overlapping genes were determined, and enrichment tested using Chi-square tests.
Databases for IHR
The database of “ischemia-hypoxia response” (IHR) genes contains manually curated genes (n=1,629) from n=24 microarray studies in experimental brain ischemia-hypoxia. This cumulative database was first described in a study of retinal genes (15) and an update has been reported (16). Most of the genes are upregulated (higher mRNA levels) in experimental studies. The definition of “response” relates to regulation of gene expression but also includes other mechanisms, i.e. mRNA splicing, stability, and degradation. The curation included alignment of mouse or rat gene symbols with human gene symbols; this was initially achieved by fusing the IHR gene list with a human gene list (32) and then by resolving conflicts by detailed searches in Entrez Gene and HomoloGene. BLAST searches for the microarray probe were used in some cases. We estimate that the combined IHR genes (n=1,629) represent 9% of genes potentially expressed during brain development; this was based on a conservative estimate of n=20k for the total number of protein-coding genes in the genome, the report for >90% of genes being expressed in the developing human brain (33), and on the assumption of genome-wide coverage of the microarrays. Then enrichment in a specific schizophrenia gene set was estimated by comparing the observed overlap with the randomly expected overlap set at 9%.
A recent study of focal brain ischemia in mice has used RNAseq analysis at three different time points (30); these data were combined as “BH-genes,” n=2,449 (14%). Since this dataset uses the sensitive RNAseq method and a single experimental model, it was considered as a replication test for the microarray-based, cumulative dataset of IHR genes.
HDB is a database related to proteomic findings for hypoxia in general with n=2,289 entries [12%; ref. (31)]. This database is composed of findings made at the protein level across multiple organs (31) and therefore is independent of expression in the brain. For convenience, this dataset was named “HDB genes” in this analysis.
Selection of Subsets of GWAS-Based Schizophrenia Genes
N=458 genes from 145 loci of the recent GWAS dataset in schizophrenia were retrieved (23). Subsets of genes were then selected using specific annotations.
The database of loss of function (LoF) (24) was used to retrieve mutation-intolerant genes (n=3,230).
A manual annotation of GWAS-based schizophrenia genes (n=458 genes from 145 loci) was carried out for monogenic disorders that are known to affect development and function of the nervous system. Gene symbols were searched in OMIM™ (https://www.omim.org), the DDD-database (34), or DisGeNET (35); this was supplemented by abstract searches in PubMed. Genes were included when a single gene mutation has been described to cause a neurodevelopmental disorder, intellectual disability, neurological disease, or diseases of the retina and hearing nerve.
SynGO (27) was used to define synaptic proteins (n=1,112) among GWAS-based schizophrenia genes.
A subset of schizophrenia genes from GWAS analysis was defined in a recent genomic study that used a Bayesian framework to integrate multiomics data and gene networks [n=104 genes; ref. (28)].
Neurodevelopmental Disorders
To test whether the overlap with ischemia-hypoxia can be extended to neurodevelopmental disorders, a collection of genes defined by de novo mutations in developmental delay and autism spectrum disorder (n=253) was adopted, named DD/ASD (29).
Exploration of Hypoxia-Inducible Factors
Hypoxia-inducible factors (HIFs) are major transcriptional regulators of the hypoxia response (36, 37), including the brain (38). For annotation of schizophrenia genes, information for HIF-targets and HIF-regulators was compiled from multiple databases and literature.
Analyses
Subsets of schizophrenia genes (as defined above) and DD/ASD were overlapped with the three gene sets for response to ischemia-hypoxia. Testing for enrichment was performed by comparing the observed number of shared genes to the randomly expected number of genes using Chi-square tests (p < 0.017 was considered as statistically significant, given the three comparisons). As additional test for a link to synaptic genes, the IHR gene dataset was subjected to annotation by DAVID Bioinformatics (https://david.ncifcrf.gov).
Results
The overall goal of this study was to uncover links between experimentally defined genes of the IHR (as collected in three databases, i.e., IHR, BH, and HDB) and genes from schizophrenia GWAS. Following the analysis of the total gene set, four subsets of genes from schizophrenia GWAS were formed and tested for enrichment. For comparison, a gene set for neurodevelopmental disorders was subjected to the same analysis.
Total Gene Set From Schizophrenia GWAS
Some genes well-known from experimental studies of brain ischemia-hypoxia stood out following the annotation of the total gene set from schizophrenia GWAS for IHR genes (Table 1). N=51 IHR genes, n= 59 BH genes, and n=48 HDB genes were overlapping with schizophrenia genes; however, none of these individual datasets showed enrichment in the total schizophrenia GWAS dataset. Therefore, subsets of the GWAS datasets were formed using criteria specified above and tested for enrichment with gene sets related to ischemia-hypoxia.
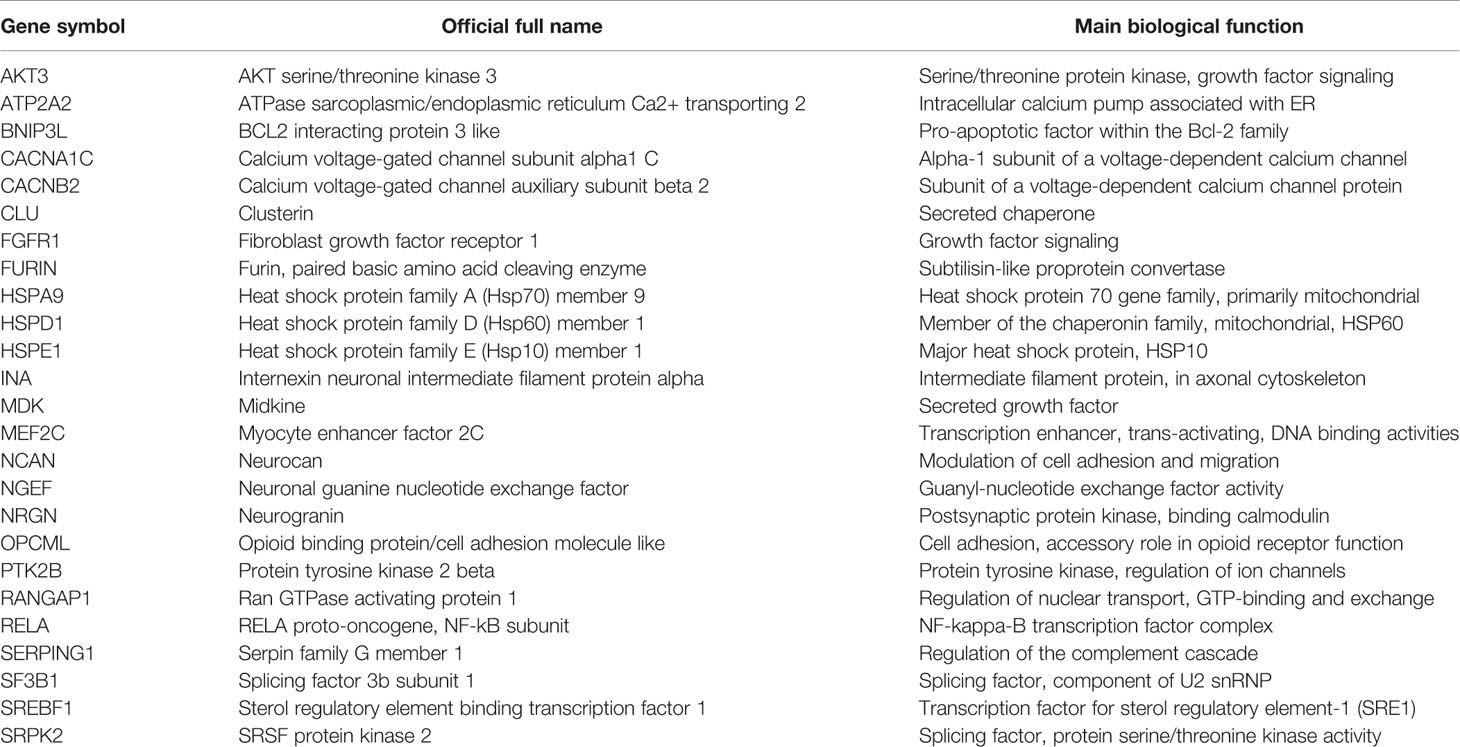
Table 1 Selected schizophrenia genes derived from genome-wide association study (GWAS) matching well-recognized ischemia-hypoxia response (IHR) genes of the brain.
Subset for LoF
LoF genes were analyzed with the rationale that these are high-risk genes for schizophrenia (23). When the overall LoF database was compared with the IHR gene database, enrichment was found (n=452 or 28%; Chi-square p=0.0001), indicating that a large portion of IHR genes are potentially mutation-intolerant. The schizophrenia GWAS dataset as such was enriched with N=112 LoF genes (p=0.0001), as to be expected (23); thereby, n=25 IHR genes were among the LoF genes for schizophrenia, indicating enrichment (p=0.0002). When BH genes (30) were tested, n=35 genes were matched to LoF genes in schizophrenia (p=0.0001). Further, n=23 HDB genes were overlapping LoF genes in schizophrenia (p=0.0005). Taken together, a subset of schizophrenia genes can be defined by the combination of LoF and potential for change in expression levels during ischemic-hypoxic challenges.
Subset for Monogenic Disorders
In a separate approach, GWAS-derived schizophrenia genes were selected if a mutation was known to affect development or function of the nervous system. This annotation lead to n=96 genes (21%) of the schizophrenia GWAS dataset linked to a monogenic disorder of the nervous system. Therein, genes from the IHR dataset (n=19; p=0.008) and BH dataset (n=23; p=0.002) were enriched in the subset of schizophrenia genes matching a monogenic disorder, whereas the HDB dataset showed only a trend (n=16; p=0.05).
Subset for Synaptic Proteins
In view of multiple findings related to neurotransmission and synaptic functions in the literature for genomics of schizophrenia (25), synaptic genes within the schizophrenia GWAS dataset were analyzed in the next step. To do this, a gene/protein set curated for synaptic functions (n=1,112) was downloaded from SynGO (27). First, we tested whether synaptic genes are enriched among the IHR genes and found n=270 (24%; p=0.0001) shared genes which indicates that synaptic genes are strongly involved in the response to ischemia-hypoxia as such. For confirmation, a DAVID Bioinformatics analysis of the complete IHR gene set was run; the Gene Ontology (GO) term “synapse” (GO:0045202) was found highly enriched (p=2.2E-25, Bonferroni corrected p=1.22E-22). Next, we tested whether synaptic genes are enriched in the schizophrenia GWAS gene set as such and found n=46 shared genes (p=0.0095). Finally, when overlapping these n=46 schizophrenia genes listed in SynGO with IHR genes, n=20 were matched (p=0.0001); n=13 for BH genes (p=0.0064); and n=6 for HDB genes (p=0.6).
Subset of Multiomics Data
Subsequently, additional gene sets derived from schizophrenia GWAS data were searched for confirmation, and the dataset of n=104 genes related to schizophrenia by multiomics studies (28) was selected. Enrichment was found for IHR genes (n=31; p=0.0001), but not for BH genes (n=27; p=0.03) or HDB genes (n=22; p=0.095).
Neurodevelopmental Disorders
Furthermore, it was explored whether the role of IHR genes can be extended more broadly to neurodevelopment disorders, and a gene set for DD/ASD was analyzed (n=253) (29). Thereby, only 14/235 of the DD/ASD genes were overlapping with the schizophrenia GWAS dataset, indicating an independent dataset. Strong overlap was observed for the DD/ASD gene set with IHR genes (n=47; p=0.002), the BH dataset (n=72; p=0.0001), and HDB genes (n=59; p=0.0007).
Exploration of HIFs
HIFs are major regulators of gene expression in response to hypoxia (36, 37). Therefore, an initial survey of GWAS-based schizophrenia genes for links to HIF regulation was performed using expert curation (38). Specific annotations for HIF-related genes in the schizophrenia GWAS dataset yielded n=33 genes (Table 2).

Table 2 Annotation of schizophrenia genes derived from genome-wide association study (GWAS) for a role of hypoxia-inducible factors (HIFs).
Discussion
Gene-environment interactions (G × E) play a key role in the neurodevelopmental model of schizophrenia. OCs are established environmental factors linked to the risk of schizophrenia which may involve ischemic-hypoxic events in the developing brain (5–7). The goal of this study was to apply datasets of experimentally defined genes responding to ischemia-hypoxia (IHR genes) to a large set of GWAS-defined schizophrenia genes. Thereby, the annotation for IHR genes opens the possibility that a matched schizophrenia gene is subject to regulation by ischemic-hypoxic periods occuring during OCs (i.e. during neurodevelopment). Genetic variation captured by GWAS may modify the ability of such genes to respond when challenged by ischemia-hypoxia; an oligogenic or polygenic abnormal response could negatively affect the developing brain. A graphic presentation of the G × E concept related to ischemia-hypoxia including the present analyses is provided in Figure 2.
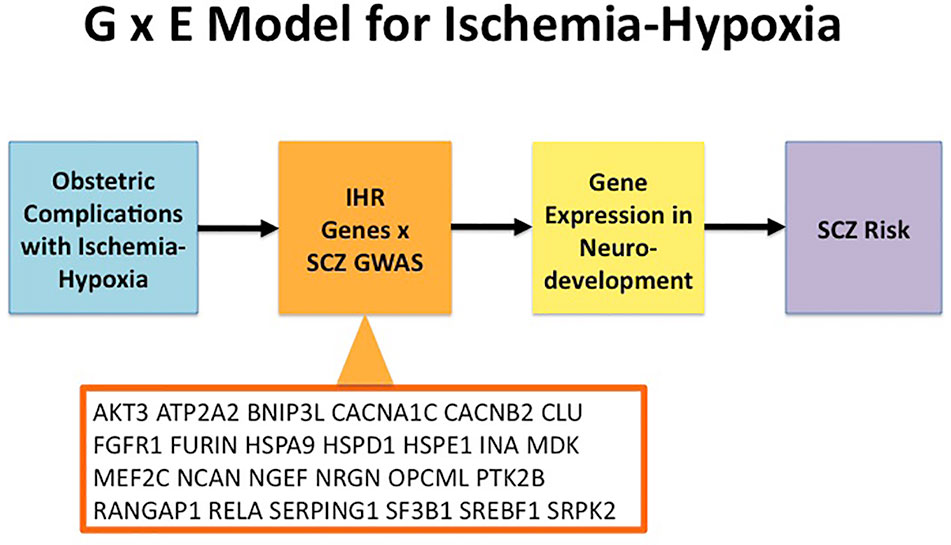
Figure 2 Model for Gene × Environment (G × E) interactions in relation to ischemia-hypoxia and Schizophrenia. Obstetric complications can cause ischemia-hypoxia that activates ischemia-hypoxia response (IHR) genes. A subset of genes from GWAS in schizophrenia (SCZ) overlap with IHR genes as shown here for the genes listed in Table 1. Subsequently, ischemia-hypoxia induces changes in gene expression for the overlapping SCZ genes during neurodevelopment. In turn, disturbances of neurodevelopment increase the risk of SCZ. GWAS, genome-wide association study; IHR, ischemia-hypoxia response; SCZ, schizophrenia.
While the overall gene set from the most recent GWAS (23) was not enriched for IHR genes, subsets of schizophrenia genes closely related to disease mechanisms were enriched based on a neurodevelopmental model. Genes from schizophrenia GWAS were enriched for mutation-intolerant genes (23), which in turn were strongly enriched for IHR genes. The enrichment of hypoxia-responsive genes among mutation-intolerant schizophrenia genes was replicated with two additional datasets (BH genes, HDB genes). A separate analysis showed that one fifth of genes from schizophrenia GWAS can be linked to monogenic disorders of the nervous system, which were also enriched for IHR genes and BH genes. We propose that the interaction between genetic variants for schizophrenia and effects of ischemia-hypoxia may affect the same cellular functions as the LoF or known mutations, although to a lesser degree. Thus, schizophrenia risk genes with a link to mutations affecting brain development and function provide a specific starting point for functional and experimental studies. In support, it was shown that a set of DD/ASD genes selected based on exome sequencing (29) was also enriched for IHR, BH and HBD genes.
Multiple pathway studies on genomic data in schizophrenia have converged on synaptic mechanisms (25, 26). Using a gene set for synaptic genes [SynGO; ref. (27)], enrichment was confirmed here for the genes in the GWAS loci for schizophrenia. In addition, it was shown with two independent methods that IHR genes are enriched in synaptic genes, indicating the involvement of the synaptic machinery in the response to brain ischemia-hypoxia. Subsequently, an enrichment for IHR and BH genes among the schizophrenia genes related to synaptic functions was detected. This subset of genes is proposed as suitable for functional in vitro studies of gene variants under hypoxic conditions.
A limitation of the present analysis is that IHR genes are a compilation from several experimental microarray studies of ischemia and hypoxia in animal models. Enrichments for three subsets of schizophrenia genes could be replicated with a large transcriptomics dataset derived by RNAseq in a focal brain ischemia model [“BH genes”; ref. (30)]. The analyses of enrichment for transcriptomics data was based on the assumptions that 18k genes are expressed in the developing brain and that the microarrays and RNAseq provided genome-wide coverage. Responses to ischemia and hypoxia mostly involve changes in transcription (“gene expression”) that have been described in multiple, independent experiments. Alterations of pre-mRNA splicing during ischemia-hypoxia also need to be considered (30). Since mRNA stability is also influenced by hypoxia (39), mRNA degradation may variably occur during early stages of ischemic neuronal damage. Genetic variants of schizophrenia risk genes may then influence gene expression, pre-mRNA splicing and/or mRNA stability after ischemia-hypoxia. Abnormal increases and decreases of the expressed proteins can alter neurodevelopmental trajectories, and impairment of protective responses can develop. A caveat is that a set of brain-expressed genes was tested against genomic data for a brain disorder; indeed, recent studies with gene sets obtained for different neuronal populations under physiological conditions showed enrichment with genomic data from schizophrenia (40). The dataset HDB was compiled across several organs on the basis of protein expression (31), and only the link for the subset of LoF genes was significant, but not for the subset for monogenic disorders or synaptic proteins. At this point, it remains unclear whether this finding was related to the use of protein studies which may be less sensitive than mRNA studies.
It remains possible that IHR genes are markers for highly responsive genes in the brain, because ischemia-hypoxia is a very strong stimulus for gene regulation; weaker stimuli such as abnormal neuronal activity could evoke regulation of the same genes that could be relevant for the pathophysiology of schizophrenia.
To address the issue of specificity, HIFs as major regulators of gene expression in response to hypoxia were analyzed (36, 37). The importance of low oxygen levels (relative hypoxia) and HIF regulation in neurodevelopment has been documented in great detail in experimental studies (38). Therefore, we here performed a survey of GWAS-based genes for links to HIF regulation and found several relevant genes, e.g. EP300 (11). Interestingly, a recent combined analysis of gene expression and GWAS data in schizophrenia showed a strong signal for HIF1A in the dorso-lateral prefrontal cortex (41). In the future, it will be of interest to see whether SNPs identified by GWAS in schizophrenia can be aligned with regulatory regions responding to HIFs.
A point of consideration is that our analysis has been focused on neurodevelopment under the assumption that gene x hypoxia interactions occur in the brain (9). A recent report has shifted the emphasis to gene expression in the placenta in relation to risk of schizophrenia (42). Evidently, the placenta is the central organ for oxygen supply to the embryo and fetus, and ischemic-hypoxic events of the brain during OCs often originate in placental dysfunction. The same gene variants related to ischemia-hypoxia will be expressed in the developing brain and placenta; therefore complex interactions can be envisioned that remain to be addressed. The novel observation that the schizophrenia GWAS dataset contains genes for monogenic disorders of neurodevelopment which overlap with IHR genes supports a role for brain mechanisms. Furthermore, overlap could be also shown for a set of DD/ASD genes which are specifically involved during neurodevelopment.
Vascular factors have been implicated in the emergence of schizophrenia during adolescence and early adulthood (12–14). Abnormalities in lactate levels and brain pH have been measured in manifest schizophrenia (43) which points to abnormalities in glycolytic pathways. Thus, a complementary view could be that the interaction between schizophrenia genes and ischemia-hypoxia (or closely related metabolic perturbations) occurs before or at the onset of psychosis during adolescence. However, direct evidence for vascular dysfunction or overt pathology of cerebral vessels in schizophrenia is presently not available. On the contrary, detailed stereological studies of the capillary system in chronic schizophrenia have not provided evidence for loss of microvessels (44), although larger vessels remain to be studied. It is also conceivable that episodic vascular events become important only on the background of a genetic susceptibility to impaired energy metabolism. In this respect, mitochondrial function has to be integrated into a late-onset model (45, 46).
Finally, the importance of gene variants may only become manifest under conditions of cellular stress (47). Thus, G × E designs using hypoxia are needed to put the hypothesis to the test for schizophrenia-related genes based on GWAS data. Studies capturing multiple medical conditions across the life-span, including detailed pre- and perinatal observations, are needed (48). Even then, the occurrence of ischemia-hypoxia in the fetal brain during OCs has to be assumed, based on experimental studies of maternal-fetal pathophysiology. Expression studies in animal models or cell culture of human neurons may provide new insight. In vivo neonatal models of ischemia-hypoxia have demonstrated regulation of some schizophrenia-related genes (49), but this approach does not capture variation associated with risk of schizophrenia. Studies on developing neurons derived from human iPSCs and exposed to variations in oxygen conditions may shed further light on functional effects of SNPs identified in GWAS for schizophrenia. Interestingly, an initial study using human iPSCs and CRISPR editing has explored a putative causal SNP in FURIN (50) which is an IHR gene and was captured by the present analysis of HIF targets.
A recent study looked for phenotype-specific enrichment of Mendelian disorder genes near GWAS loci across multiple complex traits (51). This provided the rationale for annotating genes in GWAS loci for monogenic (Mendelian) disorders of the nervous system that are known to affect development and function. A broad net was cast by searching for neurodevelopmental and neurological disorders, including disorders of the retina and hearing nerve. Many of these disorders may be traced back to effects of the mutations on early neural development. In the future, it can be tested whether SNPs found in GWAS studies of schizophrenia impact the same developmental process or neuronal function as a known mutation of the same gene.
In conclusion, the analysis of subsets of GWAS-defined schizophrenia genes related to putative disease mechanisms showed significant overlap with gene sets related ischemia-hypoxia, thereby providing cumulative evidence for a role of the hypoxia response in the etiopathogenesis of psychotic disorders. Further studies are warranted to define the genetic risk associated with ischemic-hypoxic events during OCs. Thereby, improved detection of abnormal blood flow and decreased oxygenation in the fetal brain is needed that could lead to novel treatment options.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
Ethics approval and written informed consent was not required as per local legislation and national guidelines.
Author Contributions
RS-K wrote the first draft and all authors provided feedback. RS-K generated databases for ischemia-hypoxia response and carried out the enrichment analyses. TK provided expert annotations of HIF genes.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
RS-K was supported by IMSD, COM. BR was funded by a VIDI award (no. 91718336) from the Netherlands Scientific Organization. BR, JO, and SG were funded by the grant agreement HEALTH‐F2‐2010‐241909 from the European Community’s Seventh Framework Programme (project EU-GEI). TK was supported by grants from the Finnish Center of International Mobility, Finnish Academy of Sciences (SA296027), Jane and AatosErkko Foundation, Finnish Cancer Foundation, Sigrid Juselius Foundation, Biocenter Oulu, and University of Oulu.
References
1. van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature (2010) 468:203–12. doi: 10.1038/nature09563
2. van Os J, Pries KL, Delespaul P, Kenis G, Luykx JJ, Lin BD, et al. Replicated evidence that endophenotypic expression of schizophrenia polygenic risk is greater in healthy siblings of patients compared to controls, suggesting gene-environment interaction. The EUGEI study. Psychol Med (2019). doi: 10.1017/S003329171900196X
3. Weinberger DR. Thinking about schizophrenia in an era of genomic medicine. Am J Psychiatry (2019) 176:12–20. doi: 10.1176/appi.ajp.2018.18111275
4. Guloksuz S, Pries LK, Delespaul P, Kenis G, Luykx JJ, Lin BD, et al. Examining the independent and joint effects of molecular genetic liability and environmental exposures in schizophrenia: results from the EUGEI study. World Psychiatry (2019) 18:173–82. doi: 10.1002/wps.20629
5. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry (2002) 159:1080–92. doi: 10.1176/appi.ajp.159.7.1080
6. Mittal VA, Ellman LM, Cannon TD. Gene-environment interaction and covariation in schizophrenia: the role of obstetric complications. Schizophr Bull (2008) 34:1083–94. doi: 10.1093/schbul/sbn080
7. Fatemi SH, Folsom TD. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull (2009) 35:528–48. doi: 10.1093/schbul/sbn187
8. Van Erp TG, Saleh PA, Rosso IM, Huttunen M, Lönnqvist J, Pirkola T, et al. Contributions of genetic risk and fetal hypoxia to hippocampal volume in patients with schizophrenia or schizoaffective disorder, their unaffected siblings, and healthy unrelated volunteers. Am J Psychiatry (2002) 159:1514–20. doi: 10.1176/appi.ajp.159.9.1514
9. Schmidt-Kastner R, van Os J, Esquivel G, Steinbusch HW, Rutten BP. An environmental analysis of genes associated with schizophrenia: hypoxia and vascular factors as interacting elements in the neurodevelopmental model. Mol Psychiatry (2012) 17:1194–205. doi: 10.1038/mp.2011.183
10. Allswede DM, Cannon TD. Prenatal inflammation and risk for schizophrenia: A role for immune proteins in neurodevelopment. Dev Psychopathol (2018) 30:1157–78. doi: 10.1017/S0954579418000317
11. Mendonça DB, Mendonça G, Aragão FJ, Cooper LF. NF-κB suppresses HIF-1α response by competing for P300 binding. Biochem Biophys Res Commun (2011) 404:997–1003. doi: 10.1016/j.bbrc.2010.12.098
12. Hanson DR, Gottesman II. Theories of schizophrenia: a genetic-inflammatory-vascular synthesis. BMC Med Genet (2005) 6:7. doi: 10.1186/1471-2350-6-7
13. Moises HW, Wollschläger D, Binder H. Functional genomics indicate that schizophrenia may be an adult vascular-ischemic disorder. Transl Psychiatry (2015) 5:e616. doi: 10.1038/tp.2015.103
14. Katsel P, Roussos P, Pletnikov M, Haroutunian V. Microvascular anomaly conditions in psychiatric disease. Schizophrenia - angiogenesis connection. Neurosci Biobehav Rev (2017) 77:327–39. doi: 10.1016/j.neubiorev.2017.04.003
15. Schmidt-Kastner R, Yamamoto H, Hamasaki D, Parel JM, Schmitz C, Dorey CK, et al. Hypoxia-regulated components of the U4/U6.U5 tri-small nuclear riboprotein complex: possible role in autosomal dominant retinitis pigmentosa. Mol Vis (2008) 14:125–35.
16. Schmidt-Kastner R. Genomic approach to selective vulnerability of the hippocampus in brain ischemia-hypoxia. Neuroscience (2015) 309:259–79. doi: 10.1016/j.neuroscience.2015.08.034
17. Schmidt-Kastner R, van Os J WM, Steinbusch H, Schmitz C. Gene regulation by hypoxia and the neurodevelopmental origin of schizophrenia. Schizophr Res (2006) 84:253–71. doi: 10.1016/j.schres.2006.02.022
18. Nicodemus KK, Marenco S, Batten AJ, Vakkalanka R, Egan MF, Straub RE, et al. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol Psychiatry (2008) 13:873–7. doi: 10.1038/sj.mp.4002153
19. Joo EJ, Lee KY, Jeong SH, Roh MS, Kim SH, Ahn YM, et al. AKT1 Gene polymorphisms and obstetric complications in the patients with schizophrenia. Psychiatry Invest (2009) 6:102–7. doi: 10.4306/pi.2009.6.2.102
20. Ursini G, Cavalleri T, Fazio L, Angrisano L, Iacovelli L, Porecelli A, et al. BDNF rs6265 methylation and genotype interact on risk for schizophrenia. Epigenetics (2016) 11:11–23. doi: 10.1080/15592294.2015.1117736
21. Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet (2008) 40:827–34. doi: 10.1038/ng.171
22. Ripke S, Neale BM, Corvin A, Walters JT, Farh KH, Holsmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511:421–7. doi: 10.1038/nature13595
23. Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet (2018) 50:381–9. doi: 10.1038/s41588-018-0059-2
24. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature (2016) 536:285–91. doi: 10.1038/nature19057
25. Pocklington AJ, O’Donovan M, Owen MJ. The synapse in schizophrenia. Eur J Neurosci (2014) 39:1059–67. doi: 10.1111/ejn.12489
26. Hall J, Trent S, Thomas KL, O’Donovan MC, Owen MJ. Genetic risk for schizophrenia: convergence on synaptic pathways involved in plasticity. Biol Psychiatry (2015) 77:52–8. doi: 10.1016/j.biopsych.2014.07.011
27. Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, et al. SynGO: An evidence-based, expert-curated knowledge base for the synapse. Neuron (2019) 103:217–234.e214. doi: 10.1016/j.neuron.2019.05.002
28. Wang Q, Chen R, Cheng F, Wei Q, Ji Y, Yang H, et al. A Bayesian framework that integrates multi-omics data and gene networks predicts risk genes from schizophrenia GWAS data. Nat Neurosci (2019) 22:691–9. doi: 10.1038/s41593-019-0382-7
29. Coe BP, Stessman HAF, Sulovari A, Geisheker MR, Bakken TE, Lake A, et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet (2019) 51:106–16. doi: 10.1038/s41588-018-0288-4
30. Bhattarai S, Aly A, Garcia K, Ruiz D, Pontarelli F, Dharap A. Deep sequencing reveals uncharted isoform heterogeneity of the protein-coding transcriptome in cerebral ischemia. Mol Neurobiol (2019) 56:1035–43. doi: 10.1007/s12035-018-1147-0
31. Khurana P, Sugadev R, Jain J, Singh SB. HypoxiaDB: a database of hypoxia-regulated proteins. Database (Oxford) (2013), bat074. doi: 10.1093/database/bat074
32. Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet (2011) 43:838–46. doi: 10.1038/ng.909
33. Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature (2014) 508:199–206.
34. Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature (2015) 519:223–8. doi: 10.1038/nature14135
35. Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, et al. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res (2017) 45:D833–9. doi: 10.1093/nar/gkw943
36. Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE (2005) 2005(306):re12. doi: 10.1126/stke.3062005re12
37. Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE (2007) 2007:cm8. doi: 10.1126/stke.4072007cm8
38. Kietzmann T, Knabe W, Schmidt-Kastner R. Hypoxia and hypoxia-inducible factor modulated gene expression in brain: involvement in neuroprotection and cell death. Eur Arch Psychiatry Clin Neurosci (2001) 251:170–8. doi: 10.1007/s004060170037
39. Fortenbery GW, Sarathy B, Carraway KR, Mansfield KD. Hypoxic stabilization of mRNA is HIF-independent but requires mtROS. Cell Mol Biol Lett (2018) 23:48. doi: 10.1186/s11658-018-0112-2
40. Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. Genetic identification of brain cell types underlying schizophrenia. Nat Genet (2018) 50:825–33. doi: 10.1038/s41588-018-0129-5
41. Huckins LM, Dobbyn A, Ruderfer DM, Hoffman G, Wang W, Pardiñas AF, et al. Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat Genet (2019) 51:659–74. doi: 10.1038/s41588-019-0364-4
42. Ursini G, Punzi G, Chen Q, Marenco S, Robinson JF, Porcelli A, et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat Med (2018) 24:792–801. doi: 10.1038/s41591-018-0021-y
43. Dogan AE, Yuksel C, Du F, Chouinard VA, Öngür D. Brain lactate and pH in schizophrenia and bipolar disorder: a systematic review of findings from magnetic resonance studies. Neuropsychopharmacology (2018) 43:1681–90. doi: 10.1038/s41386-018-0041-9
44. Kreczmanski P, Heinsen H, Mantua V, Woltersdorf F, Masson T, Ulfig N, et al. Microvessel length density, total length, and length per neuron in five subcortical regions in schizophrenia. Acta Neuropathol (2009) 117:409–21. doi: 10.1007/s00401-009-0482-7
45. Ben-Shachar D. Mitochondrial multifaceted dysfunction in schizophrenia; complex I as a possible pathological target. Schizophr Res (2017) 187:3–10. doi: 10.1016/j.schres.2016.10.022
46. Sullivan CR, O’Donovan SM, McCullumsmith RE, Ramsey A. Defects in bioenergetic coupling in schizophrenia. Biol Psychiatry (2018) 83:739–50. doi: 10.1016/j.biopsych.2017.10.014
47. Singleton A, Hardy J. The Evolution of Genetics: Alzheimer’s and Parkinson’s Diseases. Neuron (2016) 90:1154–63. doi: 10.1016/j.neuron.2016.05.040
48. Lipner E, Murphy SK, Ellman LM. Prenatal maternal stress and the cascade of risk to schizophrenia spectrum disorders in offspring. Curr Psychiatry Rep (2019) 21:99. doi: 10.1007/s11920-019-1085-1
49. Paparelli A, Iwata K, Wakuda T, Iyegbe C, Murray RM, Takei N. Perinatal asphyxia in rat alters expression of novel schizophrenia risk genes. Front Mol Neurosci (2017) 10:341. doi: 10.3389/fnmol.2017.00341
50. Schrode N, Ho SM, Yamamuro K, Dobbyn A, Huckins L, Matos MR, et al. Synergistic effects of common schizophrenia risk variants. Nat Genet (2019) 51:1475–85. doi: 10.1038/s41588-019-0497-5
Keywords: schizophrenia, gene-environment (G-E) interaction, obstetric complications, ischemia, hypoxia, HIF, gene expression, synapse
Citation: Schmidt-Kastner R, Guloksuz S, Kietzmann T, van Os J and Rutten BPF (2020) Analysis of GWAS-Derived Schizophrenia Genes for Links to Ischemia-Hypoxia Response of the Brain. Front. Psychiatry 11:393. doi: 10.3389/fpsyt.2020.00393
Received: 15 October 2019; Accepted: 17 April 2020;
Published: 12 May 2020.
Edited by:
Andreas Menke, University Hospital Wuerzburg, GermanyReviewed by:
Stefania Schiavone, University of Foggia, ItalyMario Herrera-Marschitz, University of Chile, Chile
Copyright © 2020 Schmidt-Kastner, Guloksuz, Kietzmann, van Os and Rutten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rainald Schmidt-Kastner, rainald@ix.netcom.com