 Livia De Picker
Livia De Picker Manuel Morrens
Manuel Morrens
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Psychiatry , 01 May 2020
Sec. Schizophrenia
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.00362
This article is part of the Research Topic The Role of Inflammation in the Etiology and Treatment of Schizophrenia View all 12 articles
Positron emission tomography using ligands targeting translocator protein 18 kDa (TSPO PET) is an innovative method to visualize and quantify glial inflammatory responses in the central nervous system in vivo. Compared to some other neuropsychiatric disorders, findings of TSPO PET in schizophrenia and related psychotic disorders have been considerably more heterogeneous. Two conflicting meta-analyses have been published on the topic within the last year: one asserting evidence for decreased TSPO uptake, while the other observed increased TSPO uptake in a selection of studies. In this paper, we review and discuss five hypotheses which may explain the observed variability of TSPO PET findings in psychotic illness, namely that (1) an inflammatory phenotype is only present in a subgroup of psychosis patients; (2) heterogeneity is caused by interference of antipsychotic medication; (3) interference of other clinical confounders in the study populations (such as age, sex, BMI, smoking, and substance use); or (4) methodological variability between studies (such as choice of tracer and kinetic model, genotyping, study power, and diurnal effects); and (5) the glial responses underlying changes in TSPO expression are themselves heterogeneous and dynamic. Finally, we propose four key recommendations for future research proposals to mitigate these different causes of heterogeneity.
Heterogeneity seems to come with the territory of psychiatry, and the study of positron emission tomography (PET) imaging of immune alterations in the central nervous system using nuclear ligands targeting translocator protein 18 kDA (TSPO) in psychotic disorders is no different. Ten years after the first studies were published, mixed results have remained an obstinate problem. In 2017, we published the first systematic review on TSPO PET imaging in psychotic illness (1). In that year, a series of negative studies had just emerged (2–4), bluntly sobering the initial enthusiasm that had accompanied early positive results (5, 6). Today, confusion and disappointment are tangibly present as even meta-analyses are openly contradicting each other: While Marques et al. found a significant increase in TSPO binding, based mainly on studies using first-generation TSPO tracer [11C]PK11195 (cfr. Table 1), Plaven-Sigray et al. found very strong evidence of decreased levels of TSPO in their meta-analysis using single-participant data of second-generation tracer studies (18, 19). To date, 14 studies have measured TSPO tracer binding in schizophrenia-spectrum disorders, of which seven were conducted in patients within the first 5 years of diagnosis (cfr. Table 1). Three studies included at ultra-high risk for psychosis (2, 4, 10). We have previously highlighted variability in terms of study population, tracer, kinetic modeling, and outcome measures (1). In this paper, we will discuss five potential sources of heterogeneity in TSPO PET imaging in psychotic illness.
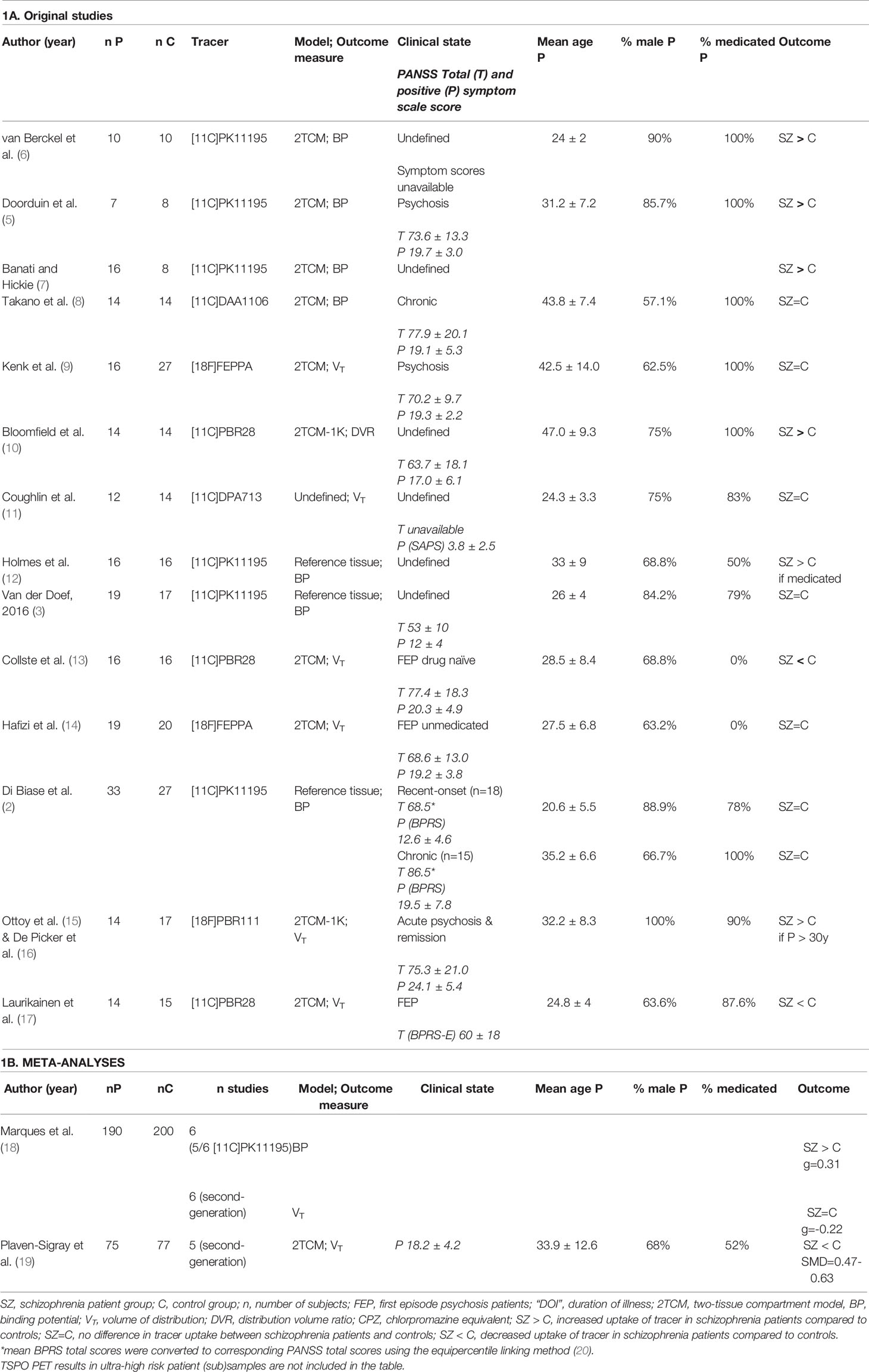
Table 1 PET studies with TSPO tracer in patients with psychotic illness versus controls.
It has been proposed that one or more different “immunophenotypes” (i.e., immune-inflammatory biotypes) may exist within the psychotic population (21). While this hypothesis has not been studied within the central nervous system, a recently published meta-analysis of 35 studies of peripheral immune markers among first-episode psychosis did not point towards the existence of subgroups (22). However, pro-inflammatory cytokines IL-6 and IFN-γ are elevated in first-episode patients who do not respond to antipsychotic treatment relative to those who do respond, raising the possibility that this treatment-resistant subgroup, could involve specific CNS immune changes (23). This hypothesis has yet to be tested with TSPO PET imaging.
For obvious ethical and practical reasons, most patients suffering from psychotic illness will be or have previously been exposed to antipsychotic medication. Antipsychotics are known to differentially affect microglial activation (increased with haloperidol and olanzapine, but reduced with risperidone) (24, 25) and TSPO binding (increased with clozapine, no significant changes with sulpiride) in rats (26). The interpretation of TSPO PET results in schizophrenia is therefore confounded by a medication effect of unknown size. Prior to the use of TSPO PET, two [3H]PK11195 autoradiography studies have found the number of TSPO binding sites in peripheral platelet cells to demonstrate a 30% decrease in patients who had been (chronically) medicated with antipsychotics relative to unmedicated patients and age-matched controls (27–29). This contradicts findings of two [11C]PK11195 PET studies who found that unmedicated patients had relatively lower TSPO levels compared with antipsychotic-treated patients (2, 12). However, as the TSPO imaging in these unmedicated psychotic patients took place very early in the illness course, during the prodromal stage or early in the first psychotic symptoms while the medicated patients were older and had a longer duration of illness, they cannot differentiate between medication and dynamic effects of illness state and progression (16). Plaven-Sigray et al. did not find a difference in TSPO levels between drug-free and medicated patients in their meta-analysis (19).
A recent multicentric study confirmed significant positive correlations between age and TSPO binding in the frontal and temporal cortex of 140 healthy volunteers, and a significant positive correlation with age in all brain regions in male subjects (30). It is therefore very likely that age may also have an impact on TSPO binding in disease states. In our own work, a significant interaction with age was found to influence the TSPO binding of patients over the longitudinal course of a psychotic episode (cfr. Figure 1) (15, 16). While such a dynamic interaction with age offers a compelling explanation for some of the discrepancies between different study cohorts of psychosis patients, this finding deserves independent replication before drawing firm conclusions.
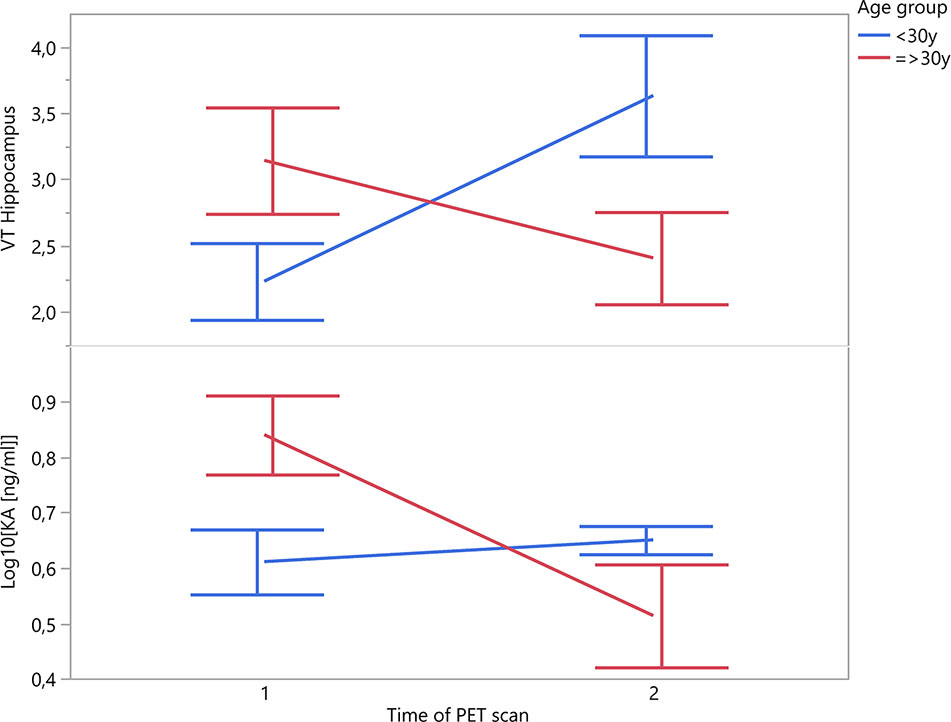
Figure 1 Patients’ change in TSPO binding in hippocampal region and plasma KA over the time course of a psychotic episode (n = 10; time interval 12 ± 4 weeks).
Not unimportantly in the predominantly male schizophrenia patient group, the same multicentric study has indicated that females show significantly higher TSPO binding in all regions compared to males. TSPO mediates the rate-limiting step of steroidogenesis. An alteration in TSPO levels could thus affect the production of neurosteroids in the brain, among which female sex hormones, independently of peripheral endocrine sources (31). Finally, many patients—especially those with longer duration of illness and exposure to atypical antipsychotics—also tend towards higher BMIs, which correlate inversely with TSPO uptake (30).
In a cohort of healthy controls, smokers had a 15.5–17.0% decreased TSPO uptake, both in the satiated state and after a night of abstention, compared to nonsmokers (32, 33). If chronic cigarette smoking leads to a global reduction in TSPO binding, this could markedly influence the results of cohorts of psychotic patients, known to be active smokers at a much higher prevalence than the general population (32, 33). A similar effect is true for cannabis use, which is also highly prevalent among schizophrenia patients. Most studies of TSPO PET in schizophrenia will include patients that have a history of cannabis use if they remain abstinent during their study participation and/or are inpatients with restricted access to illicit substances. A cohort of long-term cannabis users (using cannabis at least 4 times per week for 12 months and/or meeting criteria for cannabis use disorder) had a 23.3% increased TSPO binding compared to non-users (34). Given the long biological half-life of cannabinoids, these effects may very well persist into abstinence, although this has not yet been studied. It is unclear what the effect would be of concomitant use of nicotine and cannabis products, and if vaping of nicotine- and/or THC-containing liquids exerts the same effect. In our own work, smoking status and cannabis use were not identified as significant confounders; however, the unequal distribution of (nicotine and/or cannabis) smokers between cohorts could mask such an effect.
Two studies of TSPO PET in alcohol use disorder patients have demonstrated 10–20% decreased TSPO binding in recently detoxified patients (35, 36). Intriguingly, increased binding was shown with acute alcohol exposure in a non-human primate “binge drinking” model—persisting for several months after abstinence (37). Abuse of methamphetamine (but not cocaine) has also been demonstrated to significantly affect TSPO binding (38, 39). Ideally, future studies should aim to match cohorts for smoking status, as well as current and prior substance use.
A major limitation of TSPO PET imaging studies is that the quantification of data is a complex task. Methodological issues related to this process have been extensively discussed by other authors (19, 40–44). In summary, because microglia are distributed ubiquitously throughout the entire brain, a traditional reference tissue approach is problematic as no brain region of interest can be relied upon to be devoid of specific signal (45). Advanced methods using cluster analysis have been devised to determine a suitable reference region (43). Until these methods have been thoroughly validated for multiple ligands, an arterial input function remains the gold standard to reliably quantify the TSPO radioligand signal. A concern is that (radial) arterial sampling is invasive and arduous to patients, leading to higher inclusion bias and drop-out rates. A total of six different TSPO tracers have been applied in the clinical study of schizophrenia/psychotic illness. The advantages and disadvantages as well as the specific sensitivity of each tracer to TSPO protein have been reviewed elsewhere (46, 47). First-generation TSPO radiotracers, such as [11C]PK11195, exhibit poor extraction and lower signal-to-noise than second-generation tracers (e.g., [11C]PBR28, [11C]DPA-713, [18F]PBR111, or [18F]FEPPA) and tend to use outcome measure non-displaceable binding (BP: tracer binding in region of interest relative to other “reference” brain regions) instead of “gold standard” outcome measure volume of distribution (VT: total amount of tracer in region of interest relative to blood). Marques et al. found a significant increased TSPO level with studies using BP as outcome measure (with five out of six studies using first-generation tracer [11C]PK11195) (18), but the methodological validity of these findings are controversial (44)—recent findings suggest that at least half of the variability in [11C]PK11195 studies is due to measurement error (44). Other sources of methodological variation and potential bias are the correction for plasma free fraction and ligand binding to plasma proteins such as α1-acid glycoprotein, variability in outcome measurements, the inclusion of an additional endothelial compartment to the traditional two-compartment model (13, 27, 42).
In 2010, Owen et al. warned that second-generation ligand [11C]PBR28 does not produce a specific binding signal in approximately 14% of healthy volunteers (48). One year later, they demonstrated that all second-generation TSPO ligands in clinical use recognize high affinity (HABs, 66% of the Caucasian population), mixed affinity (MABs, 29%), and low affinity binders (LABs, 5%) in brain tissue in vitro (49). A single nucleotide polymorphism (rs6971) in exon 4 of the TSPO gene causes an alanine-to-threonine substitution affecting the ligand-binding affinity of TSPO. Prior genotyping of subjects for this polymorphism (with exclusion of LAB and stratification between MAB and HAB) is therefore required to reliably quantify TSPO binding with second-generation radiotracers. One Japanese study by Takano et al. (8) was published before this knowledge became available and therefore failed to correct for this important confounder yet the prevalence of the low-binding allele is estimated at only 4% in this demographic (8, 50). Some TSPO studies in psychotic illness have reported significant findings in one genotype group, but not in the other, thereby confusing overall interpretation. For instance, Marques et al. found no significant difference in HAB, but a significant decrease of TSPO binding in MAB subjects with second-generation tracers (18).
The relatively small patient sample sizes in TSPO PET studies of schizophrenia patients have often been criticized (44). Yet the mean sample size in TSPO PET studies across 41 neuropsychiatric diagnoses is not significantly different from studies in schizophrenia/psychotic illness (17.4 ± 10.9 all diagnoses versus 19.9 ± 9.0 in psychotic illness; De Picker et al., in prep). However, as mentioned above, in second-generation TSPO tracers’ stratification of study groups by genotype is required. To compensate for this loss of study power, the sample size in studies of second-generation TSPO tracers has been on average 46% larger than with [11C]PK11195 (12.7 ± 1.9 vs. 19.0 ± 1.4) across diagnostic categories, but only 10% larger in studies of psychotic illness (18.4 ± 3.4 vs. 20.3 ± 2.7; De Picker et al., in prep). As most of the studies using second-generation tracers were published in the last 5 years, at an average study completion time of 4–5 years, power calculations have probably been based on the effect size estimates of the earlier [11C]PK11195 studies (published in 2008–2009), which in retrospect may have reported inflated effect sizes (44). We therefore cannot exclude the possibility that some of the later second-generation ligand studies have been underpowered.
Specific immune cells and cytokines show a 24-hour circadian variation in plasma and CSF—similar diurnal changes may also exist in TSPO binding (51). A 18.5 ± 23.9% higher VT was observed in grey matter of healthy subjects in the afternoon compared to the morning of the same day (52).
TSPO is expressed at low levels at the outer mitochondrial membrane of various cell types, including microglia, astrocytes, and vascular endothelial cells throughout the brain and increases sharply in response to neuronal injury and inflammation. TSPO is often considered a biomarker of “neuroinflammation” or “microglial activation”, yet novel findings have indicated this notion is erroneous and it is more appropriate to equate TSPO binding to glial responses in general. Firstly, “neuroinflammation” is essentially a spectrum of still ill-defined physiological functions and dynamic response patterns which varies with the type and course of a pathological condition. Contingent upon the integrity of the blood-brain barrier (BBB) and a condition’s regional focus, distinct patterns of TSPO upregulation can ensue in different brain pathologies. Secondly, our knowledge on the cellular mechanisms of neuroinflammation is suboptimal (1). Studies in animal models have compellingly demonstrated the increased TSPO signal in brain pathology is derived from both microglial cells and astrocytes, in a dynamic temporal interplay (summarized by Guilarte, 2019) (27). Following exposure to a neurotoxic substance, an early microglial response at 2 weeks is followed by a later astrocytic activation and further increase in TSPO levels at 3–4 weeks. Upon removal of the toxic compound, the TSPO signal gradually decreases (50% decrease after 6 weeks), with the astrocytic signal enduring after the microglial response has already subsided (27). It is also largely unknown how central and peripheral inflammatory responses cross-talk with each other. TSPO levels have been demonstrated to increase 30% within 1 hour and 60% after 4 hours following a classical immune challenge, correlating with an increase in blood levels of inflammatory cytokines as well as sickness symptoms (53, 54). Yet in some auto-immune conditions, increased peripheral cytokines were found to be inversely correlated with TSPO binding. Likewise, reduced prefrontal TSPO levels were found in an infection-mediated neurodevelopmental mouse model, accompanied with increases in inflammatory cytokines and schizophrenia-relevant behavioral abnormalities (55).
Given the considerable intra- and inter-individual variability in symptomatology, treatment response and illness course among patients with psychotic disorders, cross-sectional studies clearly do not provide an accurate representation of the dynamic nature of glial responses. TSPO levels in psychotic illness could be differentially altered in specific symptomatic states (i.e., acute psychotic syndrome, negative symptoms) or stages throughout the illness course (i.e., prodromal, relapsing-remitting, chronic, and treatment-resistant), depending on the differential recruitment from different cellular sources. Both microglia and astrocytes have been implicated in post-mortem research of schizophrenia patients. Kynurenic acid (KA), an astrocyte-derived neuroinhibitory tryptophan neurometabolite, is at the core of the hypothesis linking neuroinflammation to psychotic illness. KA reduces striatal extracellular dopamine through antagonism of α7nAch- and NMDA receptors (56). Increased central and decreased peripheral levels of KA have been found in schizophrenia (57). In our own work, the evolution of the TSPO expression over the course of a psychotic episode differed in subjects under the age of 30 compared to those who were older (mean scan interval 12.3 ± 4.6 weeks) (16). Interestingly, in these same subjects, plasma KA levels mimicked this effect (cfr Figure 1; unpublished data). Even if these findings only concern a relatively small sample size of n = 10 patients, they corroborate the interesting question whether dynamic TSPO changes could be related to a differential recruitment of microglial and astrocytic populations in an age-dependent or illness-specific pattern.
Finally, regardless of the cellular source, TSPO is not functionally involved in neuroimmune signaling and therefore also does not reliably identify pro- versus anti-inflammatory processes. A consistent downregulation of TSPO emerged in macrophages activated to a pro-inflammatory, or “M1” phenotype. Conversely, stimulation of macrophages to an “M2” phenotype with IL-4, dexamethasone or TGF-β1 did not alter TSPO expression (58). However, findings derived from the study of macrophages or rodent microglia cannot be reliably extrapolated to the behavior of human microglial cells in vivo (59, 60). Even in those neuroinflammatory conditions accompanied by clear and unequivocal TSPO upregulation, better understanding of the functional meaning of these glial responses is crucial before we can jump to therapeutic avenues. It has been proposed that because neuroinflammation plays a central role in the progression of neurodegenerative diseases, drugs such as minocycline and cyclooxygenase (COX) inhibitors could be beneficial for their in vivo anti-inflammatory—hence neuroprotective—properties. The underlying assumption that increased glial responses are pathological and detrimental to the brain is however simply not true. This has been painfully demonstrated in a clinical trial of 15 patients who had suffered moderate-to-severe traumatic brain injury, randomized to receive either minocycline 200 mg per day or no drug for 12 weeks. While minocycline effectively reduced the glial activity on TSPO PET, it also increased markers of neurodegeneration (61). It appears in the specific case of traumatic brain injury the observed glial responses are of a reparative rather than a pathological nature. On the other hand, both minocycline and COX-2 inhibitor celecoxib have been shown to be clinically beneficial when used as add-on treatment to antipsychotics in psychotic patients, even though the TSPO PET results in psychotic patients have been ambivalent (1).
Even at the level of simple nosology, clinicians and researchers in psychotic disorders understand that diagnostic categories do not represent valid underlying constructs and are more likely to encompass a wider range of disease entities or subgroups, emerging as a similar clinical syndrome. It therefore does not come as a surprise that heterogeneity also springs up in the research on its underlying neurobiology. In fact, as has recently been demonstrated by Brugger et al. in their meta-analyses of intra-individual variance of regional brain structure (62) and PET imaging of striatal dopaminergic transmission (63), this heterogeneity does not have to be an insurmountable challenge, and can instead be turned into an interesting research question in and of itself. Importantly, Plaven-Sigray et al. have noted that, when all clinical and technical confounders were accounted for, the heterogeneity between studies in their meta-analysis was actually quite low (19). This highlights the importance of using a suitable study design which serves to minimize the variability caused by methodological problems. Still, the emergence of heterogeneous results in the study of TSPO in psychotic illness over the last 5 years has paradoxically raised new questions about the nature and the dynamics of the underlying glial responses and how they relate to TSPO binding in schizophrenia and other illnesses.
Compared to other neuroinflammatory conditions, psychotic illnesses come with some built-in disadvantages. They cannot rely as much on animal or other preclinical models to translationally evaluate the role of neuroinflammatory and glial mechanisms. There is no consistent illness-free region which could serve as reference in imaging studies, and confounding effects of medication and substance use are difficult to eradicate. Furthermore, because the underlying pathophysiology of schizophrenia—specifically, the sequence of causal events in the development of the disorder—is largely unknown, immune and glial mechanisms could be subject to disease-specific, state-specific, and age-specific alterations. Given the considerable cost and effort involved in executing these studies, often taking 4 to 5 years to complete, research groups as well as funding agencies may be reluctant to invest in the field of TSPO PET imaging in psychotic illness any further until we find a way to obtain more consistent results. It is very likely that the true cause of the observed heterogeneity is multifactorial in nature, with several or all of the abovementioned hypotheses contributing to some extent. We therefore advocate future research proposals to take into consideration the following recommendations to mitigate the different reasons for heterogeneity:
1. A consensus on the optimal methodology for TSPO PET imaging in psychotic disorders, which can be applied and replicated in further studies, needs to be reached. Because of the low reliability and sensitivity of (R)-[11C]PK11195 outcomes, preference should be given to second-generation tracers with arterial input function kinetic modeling. In terms of reporting, we advocate for adherence to the guidelines on the content and format of PET brain data publications which were recently provided in a consensus paper (64). Specifically for TSPO PET, both 2TCM and 2TCM-1K VT outcome measures should be reported. Cohorts should be genotyped for rs6971 and power calculations should establish adequate sample sizes for each genotype subgroup;
2. Control cohorts should be matched for age, sex, BMI, smoking status, and prior substance use (up to 3 months before PET scan) as well as genotype and time of scan. If matching for substance use is not possible, a careful history of any substance use in the last 3 months prior to the scan needs to be reported;
3. Longitudinal studies are needed to track the evolution of TSPO expression changes through different phases of the illness, as well as before and after the initiation of antipsychotic medication;
4. Studies should stratify patients according to relevant subgroups such as immunophenotypes, treatment-resistant or ultra-high-risk individuals, and different age groups. Findings should be corrected for (lifetime) cumulative medication exposure.
The datasets generated for this study are available on request to the corresponding author.
LP devised the main conceptual ideas and proof outline. MM aided in interpreting the results and deciding on the final scope of the manuscript. LP wrote the manuscript in consultation with MM.
The authors have received funding from University Hospital Duffel/University of Antwerp to cover open access publication fees.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. De Picker LJ, Morrens M, Chance SA, Boche D. Microglia and Brain Plasticity in Acute Psychosis and Schizophrenia Illness Course: A Meta-Review. Front Psychiatry (2017) 8:238. doi: 10.3389/fpsyt.2017.00238
2. Di Biase MA, Zalesky A, O’Keefe G, Laskaris L, Baune BT, Weickert CS, et al. PET imaging of putative microglial activation in individuals at ultra-high risk for psychosis, recently diagnosed and chronically ill with schizophrenia. Trans Psychiatry (2017) 7(8):e1225. doi: 10.1038/tp.2017.193
3. van der Doef TF, de Witte LD, Sutterland AL, Jobse E, Yaqub M, Boellaard R, et al. In vivo (R)-[(11)C]PK11195 PET imaging of 18kDa translocator protein in recent onset psychosis. NPJ Schizophr (2016) 2:16031. doi: 10.1038/npjschz.2016.31
4. Hafizi S, Da Silva T, Gerritsen C, Kiang M, Bagby RM, Prce I, et al. Imaging Microglial Activation in Individuals at Clinical High Risk for Psychosis: an In Vivo PET Study with [(18)F]FEPPA. Neuropsychopharmacol : Off Publ Am Coll Neuropsychopharmacol (2017) 42(13):2474–81. doi: 10.1038/npp.2017.111
5. Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nuclear Med : Off Publication Soc Nuclear Med (2009) 50(11):1801–7. doi: 10.2967/jnumed.109.066647
6. van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry (2008) 64(9):820–2. doi: 10.1016/j.biopsych.2008.04.025
7. Banati R, Hickie IB. Therapeutic signposts: using biomarkers to guide better treatment of schizophrenia and other psychotic disorders. Med J Australia (2009) 190(4 Suppl):S26–32.
8. Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, et al. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int J Neuropsychopharmacol (2010) 13(7):943–50. doi: 10.1017/s1461145710000313
9. Kenk M, Selvanathan T, Rao N, Suridjan I, Rusjan P, Remington G, et al. Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr Bull (2015) 41(1):85–93. doi: 10.1093/schbul/sbu157
10. Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, et al. Microglial Activity in People at Ultra High Risk of Psychosis and in Schizophrenia: An [(11)C]PBR28 PET Brain Imaging Study. Am J Psychiatry (2016) 173(1):44–52. doi: 10.1176/appi.ajp.2015.14101358
11. Coughlin JM, Wang Y, Ambinder EB, Ward RE, Minn I, Vranesic M, et al. In vivo markers of inflammatory response in recent-onset schizophrenia: a combined study using [(11)C]DPA-713 PET and analysis of CSF and plasma. Trans Psychiatry (2016) 6:e777. doi: 10.1038/tp.2016.40
12. Holmes SE, Hinz R, Drake RJ, Gregory CJ, Conen S, Matthews JC, et al. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: a [(11)C](R)-PK11195 positron emission tomography study. Mol Psychiatry (2016) 21(12):1672–9. doi: 10.1038/mp.2016.180
13. Collste K, Plaven-Sigray P, Fatouros-Bergman H, Victorsson P, Schain M, Forsberg A, et al. Lower levels of the glial cell marker TSPO in drug-naive first-episode psychosis patients as measured using PET and [(11)C]PBR28. Mol Psychiatry (2017) 22(6):850–6. doi: 10.1038/mp.2016.247
14. Hafizi S, Tseng HH, Rao N, Selvanathan T, Kenk M, Bazinet RP, et al. Imaging Microglial Activation in Untreated First-Episode Psychosis: A PET Study With [(18)F]FEPPA. Am J Psychiatry (2017) 174(2):118–24. doi: 10.1176/appi.ajp.2016.16020171
15. Ottoy J, De Picker L, Verhaeghe J, Deleye S, Wyffels L, Kosten L, et al. (18)F-PBR111 PET Imaging in Healthy Controls and Schizophrenia: Test-Retest Reproducibility and Quantification of Neuroinflammation. J Nuclear Med : Off Publication Soc Nuclear Med (2018) 59(8):1267–74. doi: 10.2967/jnumed.117.203315
16. De Picker L, Ottoy J, Verhaeghe J, Deleye S, Wyffels L, Fransen E, et al. State-associated changes in longitudinal [(18)F]-PBR111 TSPO PET imaging of psychosis patients: Evidence for the accelerated ageing hypothesis? Brain Behav Immun (2019) 77:46–54. doi: 10.1016/j.bbi.2018.11.318
17. Laurikainen H, Vuorela A, Toivonen A, Reinert-Hartwall L, Trontti K, Lindgren M, et al. Elevated serum chemokine CCL22 levels in first-episode psychosis: associations with symptoms, peripheral immune state and in vivo brain glial cell function. Trans Psychiatry (2020) 10(1):94. doi: 10.1038/s41398-020-0776-z
18. Marques TR, Ashok AH, Pillinger T, Veronese M, Turkheimer FE, Dazzan P, et al. Neuroinflammation in schizophrenia: meta-analysis of in vivo microglial imaging studies. psychol Med (2019) 49(13):2186–96. doi: 10.1017/S0033291718003057
19. Plaven-Sigray P, Matheson GJ, Collste K, Ashok AH, Coughlin JM, Howes OD, et al. Positron Emission Tomography Studies of the Glial Cell Marker Translocator Protein in Patients With Psychosis: A Meta-analysis Using Individual Participant Data. Biol Psychiatry (2018) 84(6):433–42. doi: 10.1016/j.biopsych.2018.02.1171
20. Leucht S, Rothe P, Davis JM, Engel RR. Equipercentile linking of the BPRS and the PANSS. Eur Neuropsychopharmacol : J Eur Coll Neuropsychopharmacol (2013) 23(8):956–9. doi: 10.1016/j.euroneuro.2012.11.004
21. Leboyer M, Berk M, Yolken RH, Tamouza R, Kupfer D, Groc L. Immuno-psychiatry: an agenda for clinical practice and innovative research. BMC Med (2016) 14(1):173. doi: 10.1186/s12916-016-0712-5
22. Pillinger T, Osimo EF, Brugger S, Mondelli V, McCutcheon RA, Howes OD. A Meta-analysis of Immune Parameters, Variability, and Assessment of Modal Distribution in Psychosis and Test of the Immune Subgroup Hypothesis. Schizophr Bull (2019) 45(5):1120–33. doi: 10.1093/schbul/sby160
23. Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry (2014) 75(5):e11–3. doi: 10.1016/j.biopsych.2013.06.011
24. Zhu F, Zheng Y, Ding YQ, Liu Y, Zhang X, Wu R, et al. Minocycline and risperidone prevent microglia activation and rescue behavioral deficits induced by neonatal intrahippocampal injection of lipopolysaccharide in rats. PloS One (2014) 9(4):e93966. doi: 10.1371/journal.pone.0093966
25. Cotel MC, Lenartowicz EM, Natesan S, Modo MM, Cooper JD, Williams SC, et al. Microglial activation in the rat brain following chronic antipsychotic treatment at clinically relevant doses. Eur Neuropsychopharmacol : J Eur Coll Neuropsychopharmacol (2015) 25(11):2098–107. doi: 10.1016/j.euroneuro.2015.08.004
26. Danovich L, Veenman L, Leschiner S, Lahav M, Shuster V, Weizman A, et al. The influence of clozapine treatment and other antipsychotics on the 18 kDa translocator protein, formerly named the peripheral-type benzodiazepine receptor, and steroid production. Eur Neuropsychopharmacol : J Eur Coll Neuropsychopharmacol (2008) 18(1):24–33. doi: 10.1016/j.euroneuro.2007.04.005
27. Guilarte TR. TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharmacol Ther (2019) 194:44–58. doi: 10.1016/j.pharmthera.2018.09.003
28. Weizman R, Tanne Z, Karp L, Tyano S, Gavish M. Peripheral-type benzodiazepine-binding sites in platelets of schizophrenics with and without tardive dyskinesia. Life Sci (1986) 39(6):549–55. doi: 10.1016/0024-3205(86)90512-6
29. Gavish M, Weizman A, Karp L, Tyano S, Tanne Z. Decreased peripheral benzodiazepine binding sites in platelets of neuroleptic-treated schizophrenics. Eur J Pharmacol (1986) 121(2):275–9. doi: 10.1016/0014-2999(86)90500-5
30. Tuisku J, Plaven-Sigray P, Gaiser EC, Airas L, Al-Abdulrasul H, Bruck A, et al. Effects of age, BMI and sex on the glial cell marker TSPO - a multicentre [(11)C]PBR28 HRRT PET study. Eur J Nuclear Med Mol Imaging (2019) 46(11):2329–38. doi: 10.1007/s00259-019-04403-7
31. Da Pozzo E, Costa B, Martini C. Translocator protein (TSPO) and neurosteroids: implications in psychiatric disorders. Curr Mol Med (2012) 12(4):426–42. doi: 10.2174/156652412800163451
32. Brody AL, Hubert R, Enoki R, Garcia LY, Mamoun MS, Okita K, et al. Effect of Cigarette Smoking on a Marker for Neuroinflammation: A [(11)C]DAA1106 Positron Emission Tomography Study. Neuropsychopharmacol : Off Publ Am Coll Neuropsychopharmacol (2017) 42(8):1630–9. doi: 10.1007/s11910-017-0733-2
33. Brody AL, Gehlbach D, Garcia LY, Enoki R, Hoh C, Vera D, et al. Effect of overnight smoking abstinence on a marker for microglial activation: a [(11)C]DAA1106 positron emission tomography study. Psychopharmacology (2018) 235(12):3525–34. doi: 10.1007/s00213-018-5077-3
34. Da Silva T, Hafizi S, Watts JJ, Weickert CS, Meyer JH, Houle S, et al. In Vivo Imaging of Translocator Protein in Long-term Cannabis Users. JAMA Psychiatry (2019). doi: 10.1001/jamapsychiatry.2019.2516
35. Hillmer AT, Sandiego CM, Hannestad J, Angarita GA, Kumar A, McGovern EM, et al. In vivo imaging of translocator protein, a marker of activated microglia, in alcohol dependence. Mol Psychiatry (2017) 22(12):1759–66. doi: 10.1038/mp.2017.10
36. Kalk NJ, Guo Q, Owen D, Cherian R, Erritzoe D, Gilmour A, et al. Decreased hippocampal translocator protein (18 kDa) expression in alcohol dependence: a [(11)C]PBR28 PET study. Trans Psychiatry (2017) 7(1):e996. doi: 10.1038/tp.2016.264
37. Saba W, Goutal S, Auvity S, Kuhnast B, Coulon C, Kouyoumdjian V, et al. Imaging the neuroimmune response to alcohol exposure in adolescent baboons: a TSPO PET study using (18) F-DPA-714. Addict Biol (2018) 23(5):1000–9. doi: 10.1111/adb.12548
38. Narendran R, Lopresti BJ, Mason NS, Deuitch L, Paris J, Himes ML, et al. Cocaine abuse in humans is not associated with increased microglial activation: an 18-kDa translocator protein positron emission tomography imaging study with [11C]PBR28. J Neurosci : Off J Soc Neurosci (2014) 34(30):9945–50. doi: 10.1523/jneurosci.0928-14.2014
39. Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, et al. Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci : Off J Soc Neurosci (2008) 28(22):5756–61. doi: 10.1523/jneurosci.1179-08.2008
40. Turkheimer FE, Rizzo G, Bloomfield PS, Howes O, Zanotti-Fregonara P, Bertoldo A, et al. The methodology of TSPO imaging with positron emission tomography. Biochem Soc Trans (2015) 43(4):586–92. doi: 10.1042/bst20150058
41. Schain M, Zanderigo F, Ogden RT, Kreisl WC. Non-invasive estimation of [(11)C]PBR28 binding potential. NeuroImage (2018) 169:278–85. doi: 10.1016/j.neuroimage.2017.12.002
42. Veronese M, Reis Marques T, Bloomfield PS, Rizzo G, Singh N, Jones D, et al. Kinetic modelling of [(11)C]PBR28 for 18 kDa translocator protein PET data: A validation study of vascular modelling in the brain using XBD173 and tissue analysis. J Cereb Blood Flow Metab : Off J Int Soc Cereb Blood Flow Metab (2018) 38(7):1227–42. doi: 10.1177/0271678x17712388
43. Garcia-Lorenzo D, Lavisse S, Leroy C, Wimberley C, Bodini B, Remy P, et al. Validation of an automatic reference region extraction for the quantification of [(18)F]DPA-714 in dynamic brain PET studies. J Cereb Blood Flow Metab : Off J Int Soc Cereb Blood Flow Metab (2018) 38(2):333–46. doi: 10.1177/0271678x17692599
44. Plaven-Sigray P, Cervenka S. Meta-analytic studies of the glial cell marker TSPO in psychosis - a question of apples and pears? Psychol Med (2019) 49(10):1624–8. doi: 10.1017/S003329171800421X
45. Sridharan S, Raffel J, Nandoskar A, Record C, Brooks DJ, Owen D, et al. Confirmation of Specific Binding of the 18-kDa Translocator Protein (TSPO) Radioligand [(18)F]GE-180: a Blocking Study Using XBD173 in Multiple Sclerosis Normal Appearing White and Grey Matter. Mol Imaging Biol (2019) 21(5):935–44. doi: 10.1007/s11307-019-01323-8
46. Best L, Ghadery C, Pavese N, Tai YF, Strafella AP. New and Old TSPO PET Radioligands for Imaging Brain Microglial Activation in Neurodegenerative Disease. Curr Neurol Neurosci Rep (2019) 19(5):24. doi: 10.1007/s11910-019-0934-y
47. Cumming P, Burgher B, Patkar O, Breakspear M, Vasdev N, Thomas P, et al. Sifting through the surfeit of neuroinflammation tracers. J Cereb Blood Flow Metab : Off J Int Soc Cereb Blood Flow Metab (2018) 38(2):204–24. doi: 10.1177/0271678x17748786
48. Owen DR, Howell OW, Tang SP, Wells LA, Bennacef I, Bergstrom M, et al. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metab : Off J Int Soc Cereb Blood Flow Metab (2010) 30(9):1608–18. doi: 10.1038/jcbfm.2010.63
49. Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nuclear Med : Off Publication Soc Nuclear Med (2011) 52(1):24–32. doi: 10.2967/jnumed.110.079459
50. Mizrahi R, Rusjan PM, Kennedy J, Pollock B, Mulsant B, Suridjan I, et al. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [(18)F]-FEPPA. J Cereb Blood Flow Metab : Off J Int Soc Cereb Blood Flow Metab (2012) 32(6):968–72. doi: 10.1038/jcbfm.2012.46
51. Agorastos A, Hauger RL, Barkauskas DA, Moeller-Bertram T, Clopton PL, Haji U, et al. Circadian rhythmicity, variability and correlation of interleukin-6 levels in plasma and cerebrospinal fluid of healthy men. Psychoneuroendocrinology (2014) 44:71–82. doi: 10.1016/j.psyneuen.2014.02.020
52. Collste K, Forsberg A, Varrone A, Amini N, Aeinehband S, Yakushev I, et al. Test-retest reproducibility of [(11)C]PBR28 binding to TSPO in healthy control subjects. Eur J Nuclear Med Mol Imaging (2016) 43(1):173–83. doi: 10.1007/s00259-015-3149-8
53. Sandiego CM, Gallezot JD, Pittman B, Nabulsi N, Lim K, Lin SF, et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci U S A (2015) 112(40):12468–73. doi: 10.1073/pnas.1511003112
54. Hannestad J, Gallezot JD, Schafbauer T, Lim K, Kloczynski T, Morris ED, et al. Endotoxin-induced systemic inflammation activates microglia: [(1)(1)C]PBR28 positron emission tomography in nonhuman primates. NeuroImage (2012) 63(1):232–9. doi: 10.1016/j.neuroimage.2012.06.055
55. Notter T, Coughlin JM, Sawa A, Meyer U. Reconceptualization of translocator protein as a biomarker of neuroinflammation in psychiatry. Mol Psychiatry (2018) 23(1):36–47. doi: 10.1038/mp.2017.232
56. Wu HQ, Rassoulpour A, Schwarcz R. Kynurenic acid leads, dopamine follows: a new case of volume transmission in the brain? J Neural Transmission (2007) 114(1):33–41. doi: 10.1007/s00702-006-0562-y
57. Plitman E, Iwata Y, Caravaggio F, Nakajima S, Chung JK, Gerretsen P, et al. Kynurenic Acid in Schizophrenia: A Systematic Review and Meta-analysis. Schizophr Bull (2017) 43(4):764–77. doi: 10.1093/schbul/sbw221
58. Narayan N, Mandhair H, Smyth E, Dakin SG, Kiriakidis S, Wells L, et al. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ‘M1’ human macrophages. PloS One (2017) 12(10):e0185767. doi: 10.2174/1871527316666171004125107
59. Owen DR, Narayan N, Wells L, Healy L, Smyth E, Rabiner EA, et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J Cereb Blood Flow Metab : Off J Int Soc Cereb Blood Flow Metab (2017) 37(8):2679–90. doi: 10.1016/j.pnpbp.2017.05.007
60. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol (2013) 39(1):3–18. doi: 10.1111/nan.12011
61. Scott G, Zetterberg H, Jolly A, Cole JH, De Simoni S, Jenkins PO, et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain : A J Neurol (2018) 141(2):459–71. doi: 10.1093/brain/awx339
62. Brugger SP, Howes OD. Heterogeneity and Homogeneity of Regional Brain Structure in Schizophrenia: A Meta-analysis. JAMA Psychiatry (2017) 74(11):1104–11. doi: 10.1001/jamapsychiatry.2017.2663
63. Brugger SP, Angelescu I, Abi-Dargham A, Mizrahi R, Shahrezaei V, Howes OD. Heterogeneity of Striatal Dopamine Function in Schizophrenia: Meta-analysis of Variance. Biol Psychiatry (2019). doi: 10.1016/j.biopsych.2019.07.008
64. Knudsen GM, Ganz M, Appelhoff S, Boellaard R, Bormans G, Carson RE, et al. Guidelines for the content and format of PET brain data in publications and archives: A consensus paper. J Cereb Blood Flow Metab : Off J Int Soc Cereb Blood Flow Metab (2020) 271678X20905433. doi: 10.1177/0271678X20905433
Keywords: translocator protein, schizophrenia, psychosis, microglia, positron emission tomography, neuroinflammation, astrocytes
Citation: De Picker L and Morrens M (2020) Perspective: Solving the Heterogeneity Conundrum of TSPO PET Imaging in Psychosis. Front. Psychiatry 11:362. doi: 10.3389/fpsyt.2020.00362
Received: 08 November 2019; Accepted: 09 April 2020;
Published: 01 May 2020.
Edited by:
Ole Köhler-Forsberg, Aarhus University, DenmarkReviewed by:
Sina Hafizi, University of Manitoba, CanadaCopyright © 2020 De Picker and Morrens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Livia De Picker, bGl2aWEuZGVwaWNrZXJAdWFudHdlcnAuYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.