 Alexei M. Bygrave
Alexei M. Bygrave Kasyoka Kilonzo
Kasyoka Kilonzo Dimitri M. Kullmann
Dimitri M. Kullmann David M. Bannerman
David M. Bannerman Dennis Kätzel
Dennis Kätzel- 1Department of Neuroscience, Johns Hopkins University, Baltimore, MD, United States
- 2Institute of Applied Physiology, Ulm University, Ulm, Germany
- 3UCL Queen Square Institute of Neurology, University College London, London, United Kingdom
- 4Department of Experimental Psychology, University of Oxford, Oxford, United Kingdom
Hypofunction of N-methyl-D-aspartate glutamate receptors (NMDARs), whether caused by endogenous factors like auto-antibodies or mutations, or by pharmacological or genetic manipulations, produces a wide variety of deficits which overlap with—but do not precisely match—the symptom spectrum of schizophrenia. In order to understand how NMDAR hypofunction leads to different components of the syndrome, it is necessary to take into account which neuronal subtypes are particularly affected by it in terms of detrimental functional alterations. We provide a comprehensive overview detailing findings in rodent models with cell type–specific knockout of NMDARs. Regarding inhibitory cortical cells, an emerging model suggests that NMDAR hypofunction in parvalbumin (PV) positive interneurons is a potential risk factor for this disease. PV interneurons display a selective vulnerability resulting from a combination of genetic, cellular, and environmental factors that produce pathological multi-level positive feedback loops. Central to this are two antioxidant mechanisms—NMDAR activity and perineuronal nets—which are themselves impaired by oxidative stress, amplifying disinhibition. However, NMDAR hypofunction in excitatory pyramidal cells also produces a range of schizophrenia-related deficits, in particular maladaptive learning and memory recall. Furthermore, NMDAR blockade in the thalamus disturbs thalamocortical communication, and NMDAR ablation in dopaminergic neurons may provoke over-generalization in associative learning, which could relate to the positive symptom domain. Therefore, NMDAR hypofunction can produce schizophrenia-related effects through an action on various different circuits and cell types.
Introduction: The Glutamate Hypothesis of Schizophrenia
Schizophrenia is characterized by a wide range of symptoms, classically grouped into positive (hallucinations, delusions, disordered thought), negative (social withdrawal, anhedonia, apathy), and cognitive (deficits of attention, working memory, cognitive flexibility) domains (1, 2). Despite decades of research, the circuit basis of schizophrenia remains elusive, hampering progress in diagnosis and treatment.
No major breakthrough has been made in drug discovery for schizophrenia treatments since the introduction of the most effective atypical antipsychotic clozapine in 1971 (3, 4). All antipsychotic drugs currently approved for schizophrenia therapy have in common that they decrease signaling through dopamine D2 receptors (D2Rs), and are most effective on positive symptoms. However, negative and cognitive symptoms are largely resistant to currently available medication, and even positive symptoms respond only partially to the usual antipsychotic drugs, which in turn have significant side effects (1). Therefore, different treatment targets—not relying on dopamine receptor antagonism—are urgently required.
In that context, the so-called “glutamate hypothesis”—the assumption that aberrant glutamatergic signaling is at the core of schizophrenia pathology—has sparked the most hope. The only two new specific targets tested in phase III clinical trials in the last two decades in schizophrenia patients—the metabotropic glutamate receptor type 2/3 (mGluR2/3) (5–8) and the glycine transporter 1 (GlyT1) (9), which affects levels of the N-methyl-D-aspartate (NMDA) receptor (NMDAR) co-agonist glycine—result from work pointing to abnormal glutamatergic transmission in schizophrenia. A third target currently under investigation, D-amino acid oxidase (DAAO) (10), which also indirectly affects occupancy of the NMDAR co-agonist site, is also based on the “glutamate hypothesis.” A central pillar of this hypothesis is that hypofunction of NMDARs causes some or all of the symptoms of schizophrenia (11–13). NMDAR hypofunction may be a precursor to deficits in synaptic plasticity which have also been strongly linked to the disorder (14). The core question for preclinical research in this context is whether the NMDAR itself, or molecules that affect its activity, are appropriate drug targets in established schizophrenia. In turn, this can only be evaluated when the molecular phenomenon of NMDAR hypofunction is spelled out mechanistically in the context of neural circuits—i.e. when it is clarified which cell types in different brain structures are affected by NMDAR hypofunction and how such altered signaling could cause different aspects of the complex symptoms characterizing the disease.
Linking NMDAR Hypofunction to Schizophrenia: Genetic Evidence
The glutamatergic synapse, and in particular the NMDAR, is one of the most prominent points of convergence of genetic risk factors for schizophrenia (15–17). Reduced expression of the obligatory NMDAR subunit GluN1 (also known as NR1; encoded by GRIN1) and also of GluN2C (NR2C, encoded by GRIN2C) has been reported in post-mortem prefrontal cortex tissue of schizophrenia patients (18, 19). In addition, the GRIN2A and GRIN2B genes, encoding the GluN2A (NR2A) and GluN2B (NR2B) subunit of the NMDAR respectively, are identified schizophrenia risk genes (Figure 1) (16, 17).
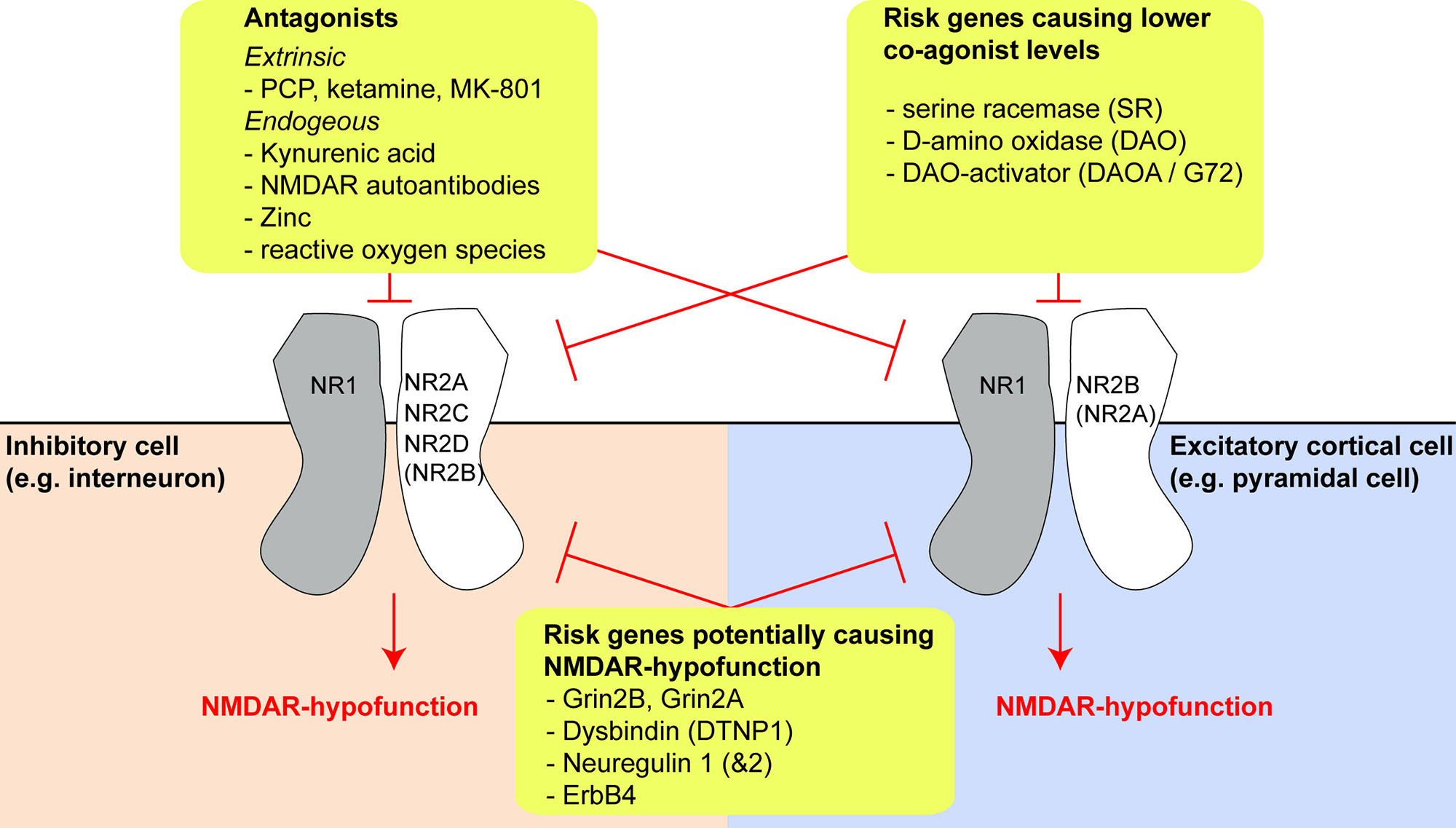
Figure 1 Risk genes and molecules producing N-methyl-D-aspartate receptor (NMDAR) hypofunction and their effects on different cell types. Overview of three different categories of risk genes or NMDAR-antagonistic molecules which may produce NMDAR hypofunction (yellow boxes). The expression of different GluN2-subunits by different cell types may be a key driver of differential vulnerability of specific types of neurons in schizophrenia. The extent of GluN2B expression in interneurons and GluN2A expression in pyramidal cells is uncertain, as conflicting results exist in the literature and public databases, but they are likely to be small, at least in prefrontal cortex (see main text).
At least one GRIN2B risk allele (rs1805502) is associated with decreased expression of GluN1 in patients (18). Moreover, exome-sequencing studies revealed that rare damaging mutations in GRIN1, GRIN2A, and GRIN2B, which are expected to cause NMDAR hypofunction directly, are found in samples from schizophrenia patients, but not controls (20, 21).
Experiments in mice provide further support for a link between schizophrenia and hypofunction of GluN2A and GluN2B: Global ablation of Grin2A causes spatial working/short-term memory deficits (without impairing basic spatial processing) (22). Further, pharmacological blockade of GluN2B alone is sufficient to cause behavioral abnormalities in rodents, in particular increased locomotor activity, which has been likened to psychosis (23, 24), as well as reduced cognitive flexibility (25), but does not recapitulate the entire profile of schizophrenia, at least as far as the disease can be modeled in rodents. For instance, pharmacological GluN2B blockade does not affect pre-pulse inhibition and may increase motivation, working memory, processing speed, sustained attention, and also motor impulsivity (23, 24, 26–29)—note, however, that these pharmacological results can contrast strongly with the schizophrenia-related deficits seen with cell type–specific Grin2B-ablation in mice (see below).
Risk genes other than those encoding NMDAR subunits may affect the production or degradation of D-amino acids that act as obligatory co-agonists at the NMDAR, especially of D-serine (Figure 1). This includes the genes encoding the D-serine producing enzyme serine racemase (SR), the D-serine degrading enzyme D-amino acid oxidase (DAAO, encoded by DAO), and its activator DAOA, also termed G72 (30). Four other risk genes—dysbindin (DTNP1), neuregulin 1 (NRG1), neuregulin 2 (NRG2), and the NRG1/2-receptor ErbB4 (ERBB4)—are implicated in regulating NMDAR function and synaptic plasticity, and their expression is altered in post-mortem tissue in schizophrenia: downregulation of DTNP1 in hippocampal and cortical tissue has been reported in schizophrenia patients, and Dtnp1 knockout in mice leads to reduced expression of NMDARs and correlates with working memory impairments (31, 32). Increased neuregulin 1–ErbB4 signaling has been reported in the frontal cortex in patients and suppresses NMDAR activation (33, 34), modulating GluN2B subunits in particular (35). Two studies also found increased neuregulin 1 expression in post-mortem tissue from prefrontal cortex (36) and hippocampus (37) of schizophrenia patients, although a third did not detect this pattern (35).
Similarly, neuregulin 2 expression leads to internalization of GluN2B-containing NMDARs in cortical interneurons, but is also itself decreased by NMDAR activation (38). Neuregulin 2 expression is, however, reduced in schizophrenia patients (36), and Nrg2 knockout in mice leads to a wide range of schizophrenia-related and partly clozapine-responsive deficits (reduced prefrontal dopamine, novelty- and amphetamine-induced hyperlocomotion, and impairments of T-maze working memory, sociability, and pre-pulse inhibition) (39). Finally, exome sequencing has revealed rare schizophrenia-related damaging mutations in the gene encoding the tyrosine-kinase FYN, which regulates NMDAR-trafficking (20, 21).
Linking NMDAR Hypofunction to Schizophrenia: Pharmacological Evidence
Support for the NMDAR hypofunction hypothesis of schizophrenia also comes from the observation that ketamine and phencyclidine (PCP), which are use-dependent non-competitive blockers of the NMDAR-channel, induce cognitive, negative, and positive symptoms of the disease (and increased dopamine signaling) in humans and rodents (13). The finding that dopaminergic agonists, in contrast, may only reproduce positive symptoms is central to the proposal that NMDAR hypofunction is causally upstream of dopaminergic aberrations in schizophrenia (11, 40, 41). Importantly though, both PCP and ketamine have also been reported to act as agonists on D2Rs as well, they display a similar affinity for D2Rs as for NMDARs and a considerably higher affinity for the high dopamine-affinity functional state D2RHigh (42–45). Such D2R-agonism could contribute synergistically to the psychotomimetic effects of PCP and ketamine. Also, low-affinity binding of both drugs to 5-HT2 receptors has been suggested (42) but has not been confirmed by others (46). Low-affinity binding of PCP to the serotonin transporter (SERT) has also been found (47, 48), but the contribution to its psychotomimetic effects are unclear. Finally, ketamine—through its main metabolite hydroxynorketamine—also increases expression of prefrontal and hippocampal AMPA glutamate receptor subunits, alters striatal BDNF signaling, and—just like ketamine itself—increases Gs-protein mediated signaling, including cAMP response element binding (CREB) protein levels, independently from NMDARs (49, 50).
The similarity between the psychological effects of NMDAR blockers in humans and symptoms of schizophrenia was described in a large review of over two dozen studies, which report the effects of PCP when used as an anesthetic or recreationally, or when administered experimentally in schizophrenia patients (51). This evidence was complemented with several subsequent controlled studies using ketamine (11, 52–54). Some studies showed that ketamine (55) or PCP (51, 56–58) may re-institute psychotic symptoms in therapeutically stabilized patients with schizophrenia. Notably, in healthy humans, ketamine induces a resting state connectivity pattern (especially involving the prefrontal cortex) that resembles the brain state of the prodrome and early-stage schizophrenia, rather than the chronic brain state in schizophrenia (59).
Furthermore, there is evidence that maternal PCP abuse during pregnancy (putatively causing NMDAR hypofunction in the developing brain) increases the risk for developing schizophrenia later in life (60). Complementary rodent studies show that NMDARs have a crucial role in virtually all processes of cellular neuronal development (61, 62), leading to alterations of neuronal circuits (63) and behavior later in life (64).
Schizophrenia Does Not Equal NMDAR Blockade: a Need to Refine the Framework
While the evidence summarized above supports an association between NMDAR hypofunction and schizophrenia, several recent studies call for qualification of some earlier conclusions.
Firstly, the specific deficits seen with PCP and ketamine do not precisely recapitulate the specific symptoms seen in patients with schizophrenia. The apparent discrepancy might, at least in part, result from over-generalization during symptom classification, for instance when referring too airily to “symptom domains.” One recent study evaluated the experiences reliably induced by ketamine—as reported by recreational drug users; the experience items of the questionnaire used in this study had previously been matched to distinct psychiatric disorders by professional psychiatrists. Notably, ketamine was not reported to reliably induce experiences that cluster specifically within any of the cognitive, positive, or negative domains of schizophrenia as opposed to other psychiatric disorders (e.g. depression) (65). In fact, non-glutamatergic drugs such as psilocybin and amphetamine provided a better match (65).
Another study demonstrated that the psychotic experiences induced by ketamine, especially body image disturbances and “out-of-body” experiences—also described in previous PCP-studies (51, 66)—do not correspond to psychotic symptoms seen in schizophrenia (66). The effects seen after higher doses of PCP (e.g. 10 mg PCP i.v.) may be better categorized as “delirium” as defined by DSM-V (66, 67). There is also evidence that the use of ketamine as an anaesthetic seems to be tolerated by schizophrenic patients at least as well as, if not better than, depressed subjects (68). The specificity of symptom-matching required to evaluate the suitability of this pharmacological model also raises a note of caution for preclinical research, which does not even come close to the level of precision in measuring psychological deficits achieved in humans.
Secondly, there are questions surrounding data from mouse models designed to exhibit reduced NMDAR function. So-called NMDAR hypomorph mice which have a 90–95% reduction of NMDAR levels exhibit a very wide range of behavioral and physiological abnormalities not restricted to those related to schizophrenia (69–74). Indeed, there were no behavioral assays on which these mice performed normally. Thus, these mice exhibit a global behavioral impairment, and it is impossible to determine what psychological processes are disrupted in these mice. For instance, they also show altered ultrasonic vocalizations and increased repetitive behavior which are more suggestive of autism (75). Likewise, electrophysiological biomarkers of both schizophrenia and autism are found (69, 75). In essence, these mice may not show a specific schizophrenia-related endophenotypic profile.
Thirdly, although the identification of anti-NMDAR encephalitis as an important neurological disease with marked psychotic features (76, 77) at first sight provides a direct link to schizophrenia (78), the syndromes exhibit important differences. These patients develop an autoimmune disorder associated with antibodies targeting the extracellular domain of the obligatory NR1 (GluN1) subunit present in all NMDARs, leading to receptor clustering and internalization (79). In primary neuronal cultures, patient antibodies can reduce the surface expression of NR1 in both excitatory and inhibitory neurons (80). Some patients experience symptom patterns that are remarkably similar to schizophrenia—including positive symptoms such as hallucinations, delusions, and agitation; deficits in attention, executive functioning, working, and short-term memory present in around 50% of patients (77, 81–83). A further common endophenotype of both diseases is increased delta oscillations (84, 85). However, these schizophrenia-like symptoms do not occur in all patients with anti-NMDAR encephalitis, and negative symptoms are rather rare (86). Furthermore, deficits unrelated to schizophrenia such as hyper-religiosity, strong speech reduction and disintegration, high anxiety and impulsivity, epileptic seizures, breathing problems, and autonomic instability are common. Given the frequent occurrence of rigidity, mutism, or dystonia (83), there might be parallels to catatonic schizophrenia (a sub-category, which has, however, been separated from schizophrenia in DSM-V), but, overall, NMDAR encephalitis appears to extend beyond and is not a perfect match for schizophrenia (86). This conclusion is supported by the very different temporal trajectories of pathologies in the two disorders with a rapid onset of symptoms in NMDAR encephalitis (86) but a more prolonged (likely neurodevelopmental) disease course over multiple years in schizophrenia (87).
A final note of caution derives from the limited clinical success of pharmacological enhancement of NMDAR function. Several phase III clinical trials evaluating the GlyT1 inhibitor bitopertin, which elevates extrasynaptic levels of the NMDAR co-agonist glycine, have failed to show significant symptom improvements (9, 88). Moreover, a meta-analysis of the therapeutic efficacy of glycine, D-serine, and other NMDAR co-agonists or positive modulators revealed that—while these drugs had small-to-medium significant beneficial effects when given to non-clozapine-treated schizophrenia patients (with glycine having qualitatively a larger effect-size than D-serine)—none of the approaches was superior to therapy with clozapine (89); for a review, see (90). In line with these results, knockout mice lacking serine racemase failed to show key deficits expected for a mouse model of schizophrenia, including no impairment of social interaction or various forms of short-term memory (91). Furthermore, the evidence that the glycine-binding site can be activated by these drugs is actually quite weak. Indeed, the lack of strong effects might be due to the fact that this binding site is already largely saturated (92, 93). On the other hand, the DAAO inhibitor sodium benzoate, which blocks degradation of D-serine, has shown promising effects on positive and negative symptoms (10). Several clinical trials with DAAO inhibitors are underway. Providing a potential proof of concept for DAAO inhibitors, genetic ablation of DAAO in mice results in enhancement of multiple forms of short-term memory—but not long-term memory (94, 95).
In summary, numerous studies link NMDAR hypofunction to schizophrenia-related deficits in humans and animal models, but the actual nature and extent of its contribution to the causation of human schizophrenia remains uncertain. The effects of global NMDAR antagonism exceed the symptom spectrum of schizophrenia. This discrepancy might be explained if NMDAR hypofunction in human schizophrenia is only partial (i.e. weaker than, for example, in anti-NMDAR encephalitis), and potentially causes aberrations across many years of learning and brain development, reflective of the neurodevelopmental nature of schizophrenia (87). Another possibility explored in the remainder of this review is that schizophrenia might be associated with NMDAR hypofunction in only some cell types of the brain, but not others. These alternative explanations are, of course, not mutually exclusive.
Does NMDAR Blockade Exert Its Effects Primarily Through Interneurons?
Pharmacological blockade of NMDARs by ketamine or PCP leads to an increase in extracellular glutamate levels (96). Elevated glutamate in the hippocampus and prefrontal cortex has also been suggested to occur in patients with schizophrenia, measured using magnetic resonance spectroscopy (MRS) (97), although the relationship between glutamate detected using MRS and extracellular (synaptic or extrasynaptic) levels of glutamate remains uncertain. In rodents, NMDAR blockade has been reported to lead to increased activity of excitatory neurons and release of glutamate (98–101). This counterintuitive finding, that the blockade of a major excitatory ionotropic receptor leads to an elevated activity of cortical excitatory cells releasing glutamate, has been explained by a preferential decrease of NMDAR-mediated excitatory drive on to inhibitory cells within the circuit, leading to a net disinhibition of excitatory cells (78, 99) (Figure 2).
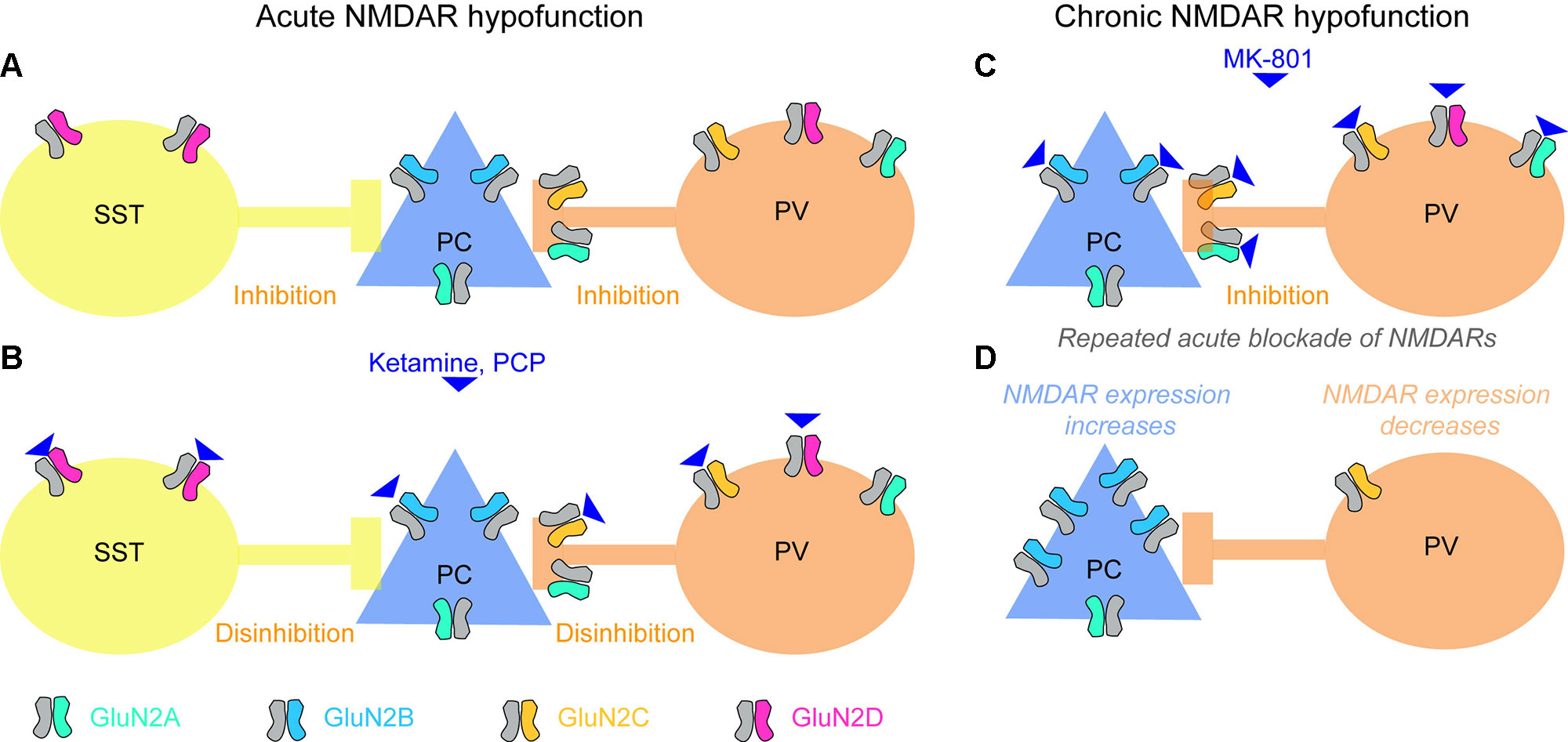
Figure 2 Effects of acute and chronic blockade of NMDARs. (A) Baseline state with GluN2 NMDA receptor subunit types most prevalent in each of the three cortical neuron classes (somatostatin-positive interneuron, SST; glutamatergic pyramidal cells, PCs; parvalbumin-positive interneuron, PV) color-coded. SST and PV interneurons provide inhibition to pyramidal cells. (B) PCs express mainly GluN2B-containing NMDARs that are less sensitive to ketamine and strongly blocked by magnesium ions at normal (hyperpolarized) resting membrane potential; in some areas they might also express GluN2A subunits, which are similar in terms of sensitivity to magnesium and ketamine. NMDARs containing GluN2C (on PV cells) and GluN2D (on PV and SST cells) (78, 102–106), in contrast, may be more sensitive to ketamine (107), and are hardly blocked by magnesium (107–109), therefore contributing to the glutamate-mediated excitation of these cells even at non-depolarized resting potential. [Although note that a recent RNAseq study questions the notion that GluN2C/D are strongly expressed in neocortical PV cells (110, 111)]. Consequently, when ketamine is applied, glutamatergic excitation of SST and PV cells could be strongly reduced, while excitation of PCs is not altered much, and the activity in the circuit increases due to disinhibition. Note, that—as an important qualification of this model—NMDARs are only weakly expressed by the somata of PV cells, but they occur in their presynaptic terminals, and their blockade reduces release of their inhibitory neurotransmitter GABA (see main text). Also, this model is complicated by the fact that the affinities of different GluN2-subunits differ for the two NMDAR blockers frequently used to model schizophrenia, ketamine and MK-801, and, furthermore, that the relevant literature is inconsistent. While one study reported nearly equal efficacies of MK-801 for all GluN2-subunits (112), another study reported nearly equal efficacies for ketamine but a 10-fold higher efficacy of MK-801 on GluN2A/B relative to GluN2C/D (108, 113). A more recent study demonstrated a high selectivity of ketamine for GluN2C/D over GluN2A/B, and additionally reported that the discrepancy with the earlier study might have been due to lack of Mg2+ (114, 107). Also note that a further blocker applied frequently in both humans and animals, phencyclidine (PCP), seems to cause schizophrenia-related abnormalities preferentially by blockade of GluN2D-NMDARs (115), similarly to ketamine (116). (C, D) Repeated blockade of NMDARs on pyramidal cells and PV interneurons (C) results in an adaptive increase of NMDAR expression in PCs, but a reduction of NMDAR expression in PV cells (D) (117). This means that the contribution that NMDARs make to the excitation of these two neuron classes shifts, which could be responsible for the observed overactivity of the hippocampus produced by chronic ketamine, and could make the circuit prone to different responses (e.g. leading to catatonic states of schizophrenia) when global blockade of NMDARs occurs again (see main text). Note, however, that the adaptation shown in (C, D) has been shown in prefrontal cortex using MK-801, while the mechanisms in acute blockade (A, B) largely reflect data collected in the hippocampus and usage of ketamine or PCP.
So far, however, this local disinhibitory mechanism has only been demonstrated in the CA1-network of the hippocampus (101). The observed increase of excitatory activity in prefrontal cortex (mPFC) (99) appears to be a consequence of NMDAR blockade in the hippocampus or/and thalamus, rather than local disinhibition in the mPFC (101, 118–121).
Both acute and chronic application of ketamine in mice increases glutamate release in the ventral hippocampus, corresponding to hyperactivity of the anterior hippocampus in patients (98). However, while acute blockade of NMDARs leads to elevated dopamine release in the prefrontal cortex (122, 123), repeated treatment blunts prefrontal dopamine release and—in contrast to acute application—also decreases mismatch-negativity and both the power and coherence of gamma oscillations (124). Given that schizophrenia patients also show reduced prefrontal dopamine release (125), the adaptation to NMDAR hypofunction, rather than—or in addition to—NMDAR hypofunction itself, could be important to the pathophysiology of the disease.
Localizing Schizophrenia-Related NMDAR Hypofunction to Parvalbumin-Positive Interneurons
To date the most influential version of the NMDAR hypofunction hypothesis of schizophrenia localizes this defect to interneurons that express the calcium-buffer protein parvalbumin (PV, encoded by PVALB), which are typically fast-spiking and implicated in gamma oscillations (41). PV interneurons appear to react uniquely when NMDAR hypofunction is induced globally: For example, in rodents PV expression is reduced following MK-801 (126–128), PCP (129), or ketamine treatment (130, 131) [although see (132)], potentially due to hypermethylation of the Pvalb promoter (133). This recapitulates the reduced expression of PV found post-mortem in cortical tissue from schizophrenia patients (134, 135), which is expected to alter GABAergic output from PV interneurons given that PV buffers calcium during the high-frequency firing of these interneurons (41, 136, 137). Also, acute NMDAR antagonism by MK-801 leads to a chronic reduction of NMDAR expression in PV interneurons but not in the vast majority of other neurons (117). Furthermore, increased expression of NRG1 in transgenic mice, reproducing an endophenotype in patients (36, 37), leads to reduced NMDAR expression in PV-positive basket cells of the hippocampus, although CCK-positive cells also show this pattern (138). Interestingly, the NRG1-receptor ErbB4, encoded by a schizophrenia risk gene (16), is preferentially expressed in PV and other hippocampal interneurons, but not pyramidal cells (139), and may regulate NMDAR expression also in response to the NRG1-homolog NRG2, which is expressed by those interneurons themselves (38).
From NMDAR Hypofunction to PV Interneuron Hypofunction
The PV cell population itself includes several subtypes, and there is continuing uncertainty regarding which population and which brain region (the hippocampus, the prefrontal cortex, the neocortex as a whole, or other brain areas such as the thalamus) is the most relevant. The most prominent account (41), which has become known as the “Lisman/Grace model,” assumes that NMDAR hypofunction in PV interneurons of the CA1-subfield of the human anterior hippocampus (corresponding to the rodent ventral hippocampus) is the key starting point of schizophrenia pathology (Figure 3).
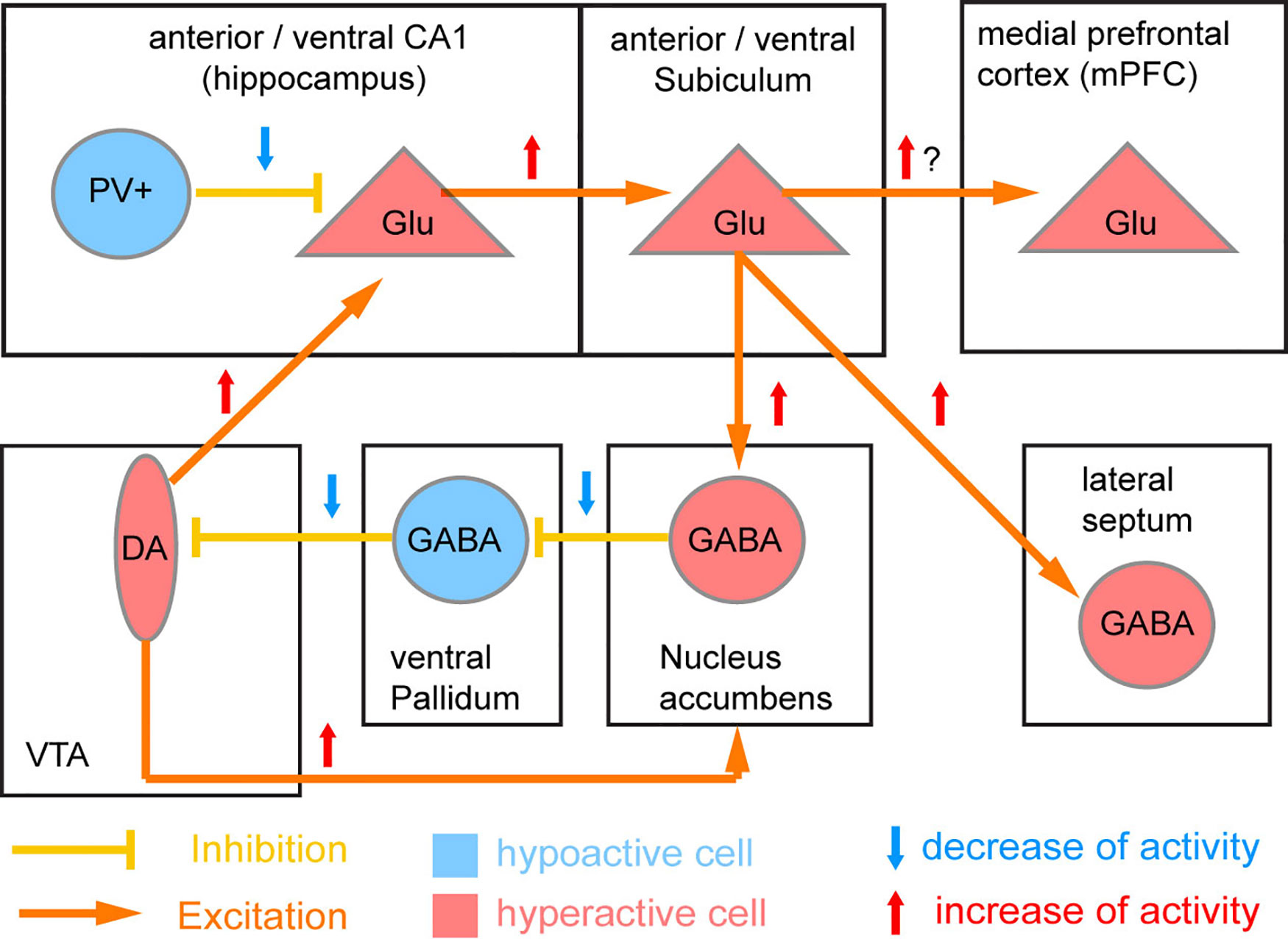
Figure 3 Circuit model of NMDAR hypofunction induced deficit of PV neurons. The original model by Lisman et al. (41) proposed that parvalbumin-positive (PV) interneurons of the CA1 region of the human anterior (rodent ventral) hippocampus is the primary location of NMDAR hypofunction in schizophrenia. Reduced NMDAR activity in these PV cells is supposed to lead to a downregulation of PV and Gad67 expression and thereby of the GABAergic output of PV cells and resulting disinhibition of surrounding excitatory glutamatergic (Glu) cells. The resulting disinhibition of CA1 excitatory glutamatergic projection neurons would lead to overactivation of the output stage of the hippocampus (subiculum) as seen in patients (98), and through a disynaptic loop through the basal ganglia cause a hyperactivity of ventral tegmental area (VTA) dopamine (DA) neurons innervating the nucleus accumbens (potentially causing psychosis) and also the hippocampus (positive feedback loop).
From the outset, this seems to be an unlikely scenario, since PV cells have very low expression of NMDA receptors compared to other neuron types in this region (140). In fact, PV interneurons across the brain (in common with other medial ganglionic eminence–derived interneurons) have relatively small NMDAR-mediated currents and, conversely, a high synaptic calcium current through inward-rectifying calcium-permeable AMPA-receptors (141). This implies that a reduction of NMDAR expression might not be expected to reduce the excitation or calcium influx in PV cells appreciably. However, it is important to note that there are input-specific differences in the magnitude of NMDAR currents recorded in CA1 PV interneurons (142). More specifically, Schaffer collateral feed-forward inputs onto PV-basket cells—as recorded in (141)—have vastly lower NMDAR currents compared to feedback inputs from within CA1 (142, 143). This likely has consequences for dendritic computations in PV interneurons, as has been shown with NMDAR-dependent supra-linear summation of local CA1 inputs (143).
However, a key twist in this model (which posits a deficit of NMDAR signaling in PV cells as a core pathological mechanism, Figure 3) is the assumption that NMDARs in such PV interneurons serve primarily as sensors of the overall glutamatergic activity present in a local circuit—thereby obviating the need for a receptor to be present in large quantities in order to have strong effects (41). Lower activation of NMDARs and a resulting decrease in calcium entry could signal reduced overall excitatory network activity and lead to a homeostatic downregulation of inhibitory output from PV cells in order to maintain the excitation/inhibition balance. Reduced expression of the primary GABA-producing enzyme GAD67 (encoded by GAD1) and PV could then reflect adaptive reductions of inhibitory output, and are indeed seen in post-mortem cortical tissue from schizophrenia patients (134, 144, 145). While the number of neocortical PV interneurons does not seem to decrease in schizophrenia, about half of them lack GAD67 (at least in prefrontal and anterior cingulate cortex) (135, 146, 147), and may thereby lose their ability to exert an inhibitory influence on the network (148, 149). Notably, the expression of GAD67 and PV seems to be constantly changing even under physiological conditions, depending on sensory experience and memory formation (150). This implies that a pathological situation that decouples NMDAR activity in PV cells from actual glutamate release in the circuit could lead to a maladaptive reduction of GABA release from PV interneurons (41).
The nature of this decoupling is unclear. Post-mortem findings in schizophrenia have reported reduced NMDAR expression in multiple cell types, including interneurons, but notably PV cells were not disproportionately affected (146, 151, 152). However, the GluN2C subunit, which is much more prominently expressed in cortical PV interneurons than pyramidal cells according to some studies (102, 103, 117)—but not others (110, 111)—is indeed downregulated in the prefrontal cortex (18). Alternatively, a decrease in levels of NMDAR co-agonists (D-serine, glycine; for example due to genetic changes in the schizophrenia risk genes SR, DAO, or DAOA/G72) or increased levels of endogenous NMDAR blockers such as zinc or kynurenic acid, could play a role (Figure 1). The tryptophan-degradation metabolite kynurenic acid, which is also an endogenous non-specific antagonist of the NMDAR glycine site, exhibits elevated levels in the cortex (153) and cerebrospinal fluid (154) in patients with schizophrenia. Application of kynurenic acid induces spatial working memory deficits in rodents (155), while blockade of its production enhances cognition (156) and reduces firing activity of ventral tegmental area (VTA) dopamine neurons, as would be predicted from a specific augmenting action on inhibitory neurons of the ventral hippocampus (41, 157). However, it is unclear why such global NMDAR hypofunction should affect PV cells with relative specificity.
In the Lisman/Grace model (Figure 3), at the circuit level the proposed key downstream consequences of the resulting disinhibition of CA1 pyramidal cells are hyperactivity of the anterior hippocampal output stage, the subiculum, and excess activation of dopamine neurons in the VTA resulting from a disinhibitory loop through the nucleus accumbens and the ventral pallidum (41). Virtually all of those physiological aberrations have been documented in patients with schizophrenia, and the control of VTA dopamine neurons by the subiculum has been established in rodents (158, 159). However, it is as yet unclear if such a hyperactivity of the hippocampal output actually results from a decrease of inhibitory output from CA1 PV interneurons. While direct chemogenetic silencing of hippocampal PV interneurons failed to evoke behavioral correlates of elevated VTA-dopamine activity—elevated locomotion and amplification of amphetamine-induced locomotion—inhibition of other hippocampal interneurons (specified by GAD2-expression) was able to do so (160). Also, putative blockade of GluN2A-containing NMDARs, which are particularly prominent in PV interneurons (117, 161), does not appear to cause hyperlocomotion at all (23). On the other hand, mouse models in which hippocampal PV interneurons are permanently reduced in numbers did recapitulate core deficits of schizophrenia, including hippocampal hyperactivity and increased novelty-induced hyperlocomotion which was partially resistant to anti-dopaminergic treatment (162, 163). It should be noted that although the characteristic increase of locomotion (a rodent correlate of positive symptoms) seen with stimulation of the ventral subiculum is usually attributed to increased VTA dopaminergic activity (41, 159), the locomotion-inducing effect of NMDAR blockers may not entirely require intact dopamine release given that it is also apparent in dopamine-depleted rodents (164–167).
Other versions of this hypothesis propose that NMDAR hypofunction is instead localized to prefrontal PV interneurons (134, 145, 168–170). Decreased PV expression has so far been documented in neocortical tissue from schizophrenia patients rather than in the hippocampus (146–148). It has been suggested that weakening of the output of prefrontal PV interneurons would lead to less synchronized output from prefrontal projection neurons targeting a specific subset of dopamine and GABAergic neurons in the VTA (168). Given their known target specificity, this would be expected to result in reduced activation of prefrontal-projecting (mesocortical) dopaminergic neurons and concomitant disinhibitory activation of (mesolimbic) dopamine cells projecting to the nucleus accumbens, thereby giving rise to the well-documented co-existence of hypofrontality and striatal hyperdopaminergia in schizophrenia (168). However, a direct demonstration of this mechanism is so far lacking. Related accounts focused rather on the link between NMDARs in PV interneurons and gamma oscillations, which are relevant to various cognitive functions and are disrupted in schizophrenia (145, 169–171).
Finally, a recent study showed that NMDARs are expressed presynaptically by prefrontal PV interneurons innervating pyramidal cells, and that their activation can enhance evoked GABA release (i.e. PV cell–mediated inhibition), and their blockade by MK-801 reduced inhibitory currents in pyramidal cells, if (and only if) glutamate was released by surrounding neurons (172). This finding could resolve the long-standing puzzle as to how acute application of NMDAR antagonists could lead to an effective PV disinhibition of pyramidal cells, despite the fact that NMDARs might not contribute significantly to the somatodendritic excitation of PV interneurons.
Genetic Modeling of NMDAR Hypofunction in Cortical Interneurons
In order to assess whether NMDAR hypofunction in PV-positive interneurons—during both postnatal development and adulthood—can indeed cause core aberrations of schizophrenia, several laboratories have now deleted the obligatory NMDAR subunit GluN1 (and thereby all NMDARs) selectively from PV-positive interneurons in mice. The principal method to achieve this is to cross a mouse line in which an exon of the GluN1-encoding gene Grin1 is floxed, to a mouse line that expresses Cre-recombinase selectively in PV-positive interneurons. In the resulting double-transgenic mice, NMDARs are ablated only in neurons where Cre is expressed. The time at which this occurs depends on when the promoter that drives Cre is activated and on the location of the lox-sites within the Grin1 gene because recombination is a stochastic event whose probability increases the closer the lox-sites are to each other (78, 173). Because the average lifetime of an individual NMDAR-molecule in the post-synaptic membrane is not known, it is difficult to estimate precisely the actual time course of the removal of those NMDARs that had been produced before the gene disruption. In practice, strategies to delete NMDARs from PV interneurons have placed Cre under the control of the promoter of either the Ppp1r2 gene, with expression onset at postnatal day (P) 7, or the PV gene (Pvalb) with an estimated onset between P10 and P14 in neocortex (174), generating Grin1ΔPpp1r2 or Grin1ΔPV mice, respectively. In our experiments using the Pvalb promoter, a significantly reduced NMDAR-mediated current in PV cells could be measured at 2 months of age, although some PV interneurons did still show NMDAR-mediated currents, despite the comprehensive coverage of the PV cell population by the genetic driver line that was used (175).
PV-Specific NMDAR Ablation Alone Does Not Reliably Induce a Schizophrenia-Related Spectrum of Deficits
Surprisingly, selective genetic deletion of NMDARs in PV interneurons in mice has, by and large, not supported the model that NMDAR hypofunction in these cells is sufficient to mimic schizophrenia. Table 1 summarizes the results of the behavioral assessment of these mouse lines published to date. In our hands, using either the Ppp1r2-Cre (176) or the more commonly used Pvalb-Cre (175) line to drive GluN1 ablation, there were hardly any behavioral deficits across a wide spectrum of rodent correlates of positive, negative, and cognitive symptoms when the mice were raised in enriched environments (that is, involving a shelter, such as a house and/or tube, and nesting material). This is largely supported by the results from three other labs that have used the same Pvalb-Cre mouse line, albeit in combination with a floxed-GluN1 line that featured a larger distance between lox-sites (171, 177–179); see Table 1. A possible exception is a deficit in alternation-based spatial working memory assays, which has been seen in either strong (180) or very subtle (171, 175, 176) forms by some labs but not by others (177, 178).
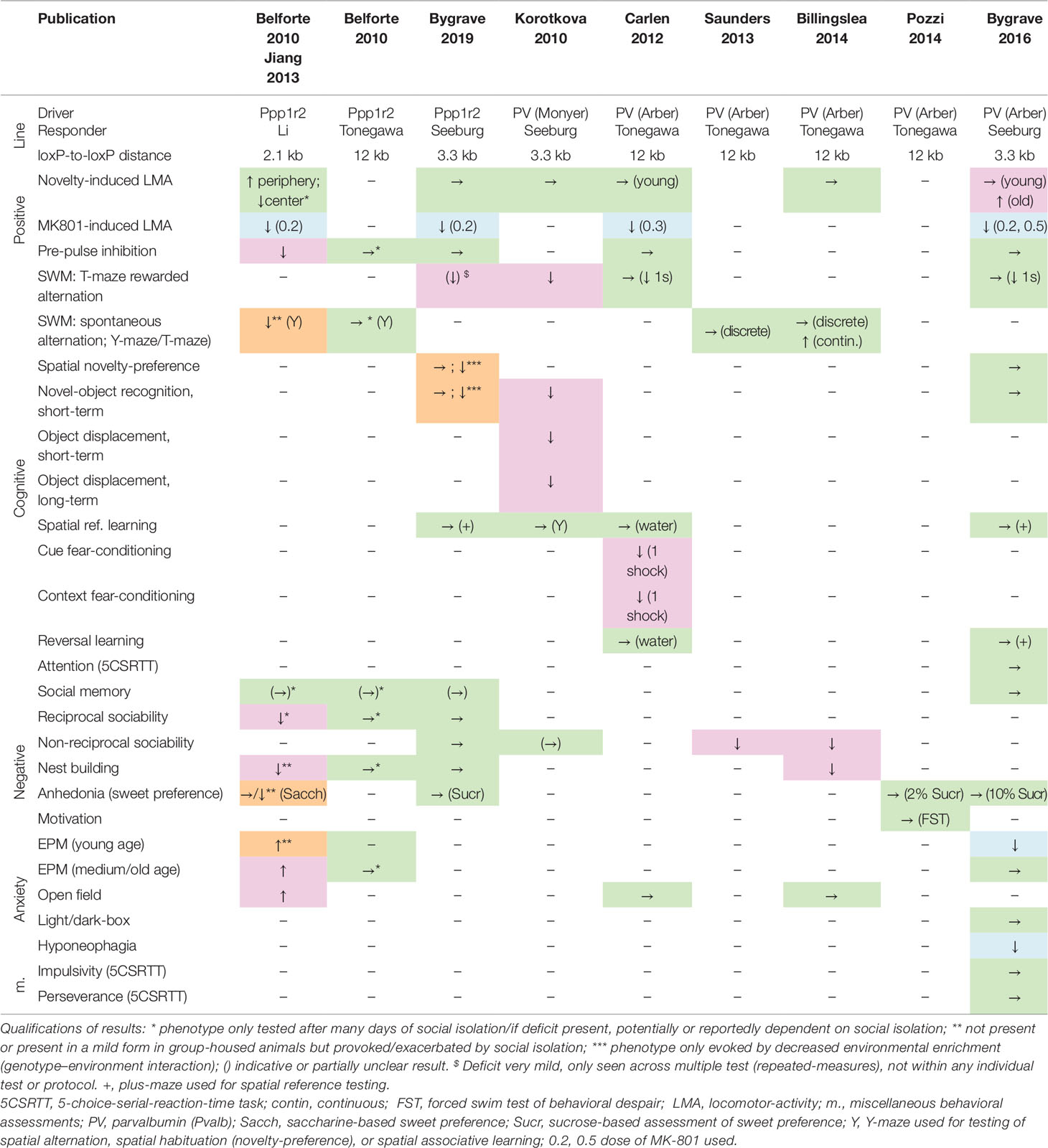
Table 1 Deficits induced by putative genetic N-methyl-D-aspartate receptor (NMDAR) ablation in parvalbumin interneurons. Results of behavioral tests conducted in the indicated studies (top row) measuring rodent correlates of schizophrenia in the positive, cognitive, and negative domain as well as anxiety (see left two columns) in distinct double-transgenic conditional knockout lines (stated in rows 2–4). Green →, no change; magenta, schizophrenia-related deficit; orange, deficit provoked or exacerbated by environmental stress; blue, apparent improvement of function or opposite of the expected. ↑, increase of behavioral measure; ↓, decrease of behavioral measure. Studies: Belforte 2010 (173), Billingslea 2014 (178), Bygrave 2016 (175), Bygrave 2019 (176), Carlen 2012 (171), Jiang 2013 (181), Korotkova 2010 (180), Saunders 2013 (177), Pozzi 2014 (179). Cre-Driver lines: Ppp1r2 (Jax# 012686) (173), PV (Monyer) (182, 183), PV (Arber, Jax# 008069) (175, 184, 185). Floxed-Grin1 responder lines: Seeburg (186), Li (187), Tonegawa (Jax# 005246) (188).
However, there are some technical differences among the published studies using mouse models with NMDAR knockout in PV interneurons (Table 1). Firstly, they differ partly with respect to the deployed Cre-driver lines. While most recent studies (171, 175, 177–179) used a widely adopted targeted PV-Cre 3’UTR knock-in line (184) that covers the vast majority of PV neurons in neocortex and hippocampus with high specificity (175, 185), the Ppp1r2-Cre line (173) as well as the PV-Cre line (180, 182, 183) used in earlier studies are BAC-transgenic lines that did not target the PV cell population comprehensively. The Ppp1r2-Cre line had originally been reported to express in roughly 70% of the cortical PV cell population, while—conversely—around 75% of the targeted cells were PV-positive (173). A recent re-evaluation in neocortex, however, reported that fewer than 40% of neurons targeted by Ppp1r2-Cre are PV-positive, 10–16% are reelin-positive and 8–12% are somatostatin (SST)-positive (40). In addition, a small fraction of neurons in the hippocampus were also Gad67-negative, and expression in such putative pyramidal cells was reported to expand strongly, especially in CA1, by the ∼20th week of age (173). In general, therefore, the Ppp1r2-Cre line cannot be considered a strictly PV-specific driver.
NMDAR Hypofunction on PV Interneurons and Environmental Stressors Interact to Produce Behavioral Deficits
Another important reason for the potential differences between previous results in different labs, even with the same line, is that NMDAR hypofunction in PV interneurons may interact with environmental risk factors in order to be pathophysiologically harmful. Environmental stressors such as drug abuse, social stress, or even urbanicity can influence both symptoms and treatment success in patients with schizophrenia (189–194). There are now two studies showing that both post-weaning long-term social isolation (181) and a long-term reduction of environmental enrichment starting in early adulthood (176) may provoke or exacerbate various schizophrenia-related deficits in Grin1ΔPpp1r2 mice. A further study demonstrated elevated novelty-induced locomotion in Grin1ΔPV mice with increasing age (175). Jiang et al. demonstrated that genetic deletion of NMDARs renders PV interneurons more susceptible to oxidative stress which may, in turn, be evoked by environmental stressors such as social isolation (181). It is therefore possible that the susceptibility to stress that NMDAR deletion confers to PV interneurons interacts with differences in housing conditions or other stress-inducing manipulations and thereby causes the differences observed at the behavioral level across studies. Notably, as marked in Table 1, the vast majority of testing in the first study in Grin1ΔPpp1r2 mice was conducted in mice that had been socially isolated for at least a week, and sometimes for many weeks, before testing (173). Indeed, for two of these deficits—anhedonia and decreased alternation-based working memory—their dependence on social isolation was subsequently demonstrated explicitly (181, 195). Bygrave et al. demonstrated that even seemingly minor changes in animal housing conditions—as likely exist between different laboratories (reflecting e.g. different regulatory conditions in different parts of the world)—are sufficient to provoke deficits in Grin1ΔPpp1r2 mice, which are otherwise absent in highly enriched conditions (176).
Oxidative Stress and Zinc Release Constitute Positive Feedback Loops Amplifying NMDAR Hypofunction and Disinhibition
There is evidence that increased oxidative stress in the brain is involved in the pathophysiology of schizophrenia (196, 197) and, due to their high metabolic demands, fast-spiking PV interneurons are thought to be particularly sensitive to oxidative stress (198) (Figure 4). In patients with schizophrenia, levels of the antioxidant glutathione (GSH) have been shown to be reduced (199), whereby GSH levels correlated with the severity of negative symptoms of the disease (200). In the rodent neonatal ventral hippocampus lesion model of schizophrenia, early administration of N-acetylcysteine (NAC), a precursor to GSH, is able to prevent oxidative stress and rescue associated electrophysiological and behavioral abnormalities (201).
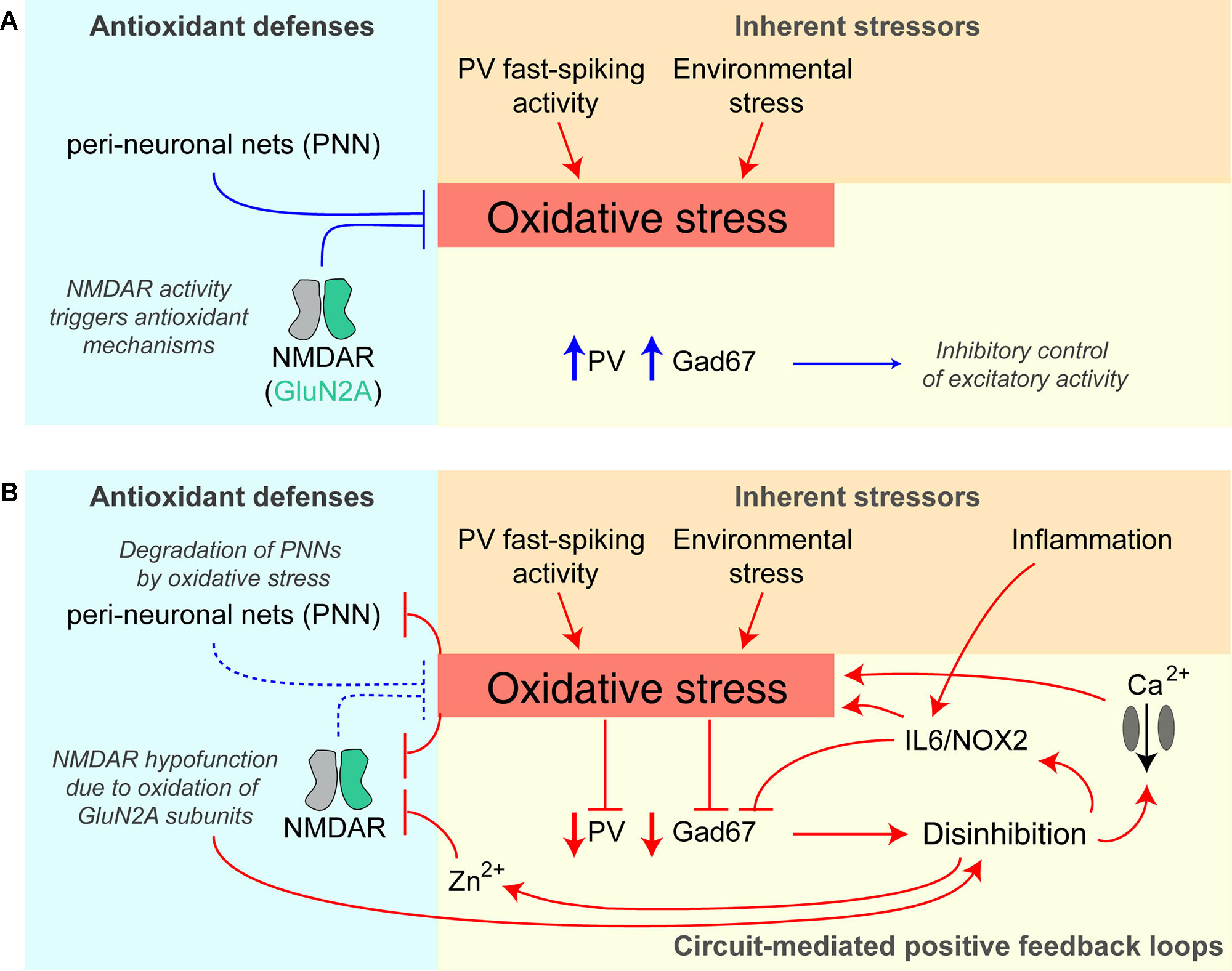
Figure 4 Potential positive feedback loops provoking PV interneuron dysfunction and oxidative stress. (A) Healthy baseline state; parvalbumin-positive (PV) interneurons are prone to higher oxidative stress due to their fast-spiking activity and resulting higher metabolism, but perineuronal nets (PNNs) around them and signaling triggered by NMDARs provide antioxidant defenses. They express high levels of PV and Gad67 (producing the inhibitory neurotransmitter GABA) and therefore provide intact inhibition to the neurons in the surrounding circuit. (B) Once oxidative stress prevails, e.g. evoked by additional environmental stressors or hypofunction (downregulation/decreased activation) of PV-NMDARs, multiple positive feedback loops are triggered that further amplify oxidative stress and decrease PV interneuron function: the prior protection mechanisms are themselves reduced by oxidative stress—perineuronal nets are degraded by oxidative stress, and specifically, GluN2A-containing NMDARs (that are prominent in PV cells) become oxidized and thereby hypofunctional. Also, the resulting reduction of Gad67 and GABAergic inhibitory output leads to disinhibition of excitatory neurons of the circuit, and via IL6/NOX2 and calcium influx through voltage-gated calcium channels increases oxidative stress further. Disinhibition also provokes more glutamate release entailing co-release of zinc ions (Zn2+), which block preferentially GluN2A-NMDARs. See (195, 198, 202) for details. Lines with arrows indicate enhancement; lines with vertical line-endings indicate suppression or degradation.
Intriguingly, there appear to be reciprocal interactions between NMDAR hypofunction and oxidative stress (198) (Figure 4). NMDARs are themselves sensitive to oxidative stress because GluN1 and GluN2A-NMDAR subunits contain redox-sensitive cysteine residues that can form disulfide bonds resulting in reduced receptor currents (203). Conversely, the activation of NMDARs can promote the transcription of antioxidant genes (204, 205). This coupling of NMDAR activation and regulation of the antioxidant system is thought to equip metabolically active neurons with sufficient antioxidant defenses (198). Therefore, NMDAR hypofunction could reduce such antioxidant mechanisms, and the ensuing oxidative stress then further reduces the conductance of GluN1/GluN2A-containing NMDARs, effectively creating a positive feedback loop. PV interneurons would be particularly vulnerable to this vicious cycle because of their relatively high content of GluN2A-containing NMDARs (117) and their high metabolic activity (206).
A second pathological positive feedback loop involves perineuronal nets (PNNs), which provide an antioxidant defense system, particularly around PV interneurons, but are themselves damaged by oxidative stress. PNNs are reduced in post-mortem tissue from schizophrenia patients, as well as in a variety of related rodent models (206–208).
Beyond cellular mechanisms, NMDAR hypofunction may also induce oxidative stress through circuit-wide effects. Through initiating disinhibition of cortical circuits, NMDAR antagonists can boost levels of the pro-inflammatory cytokine IL-6, with subsequent activation of NADPH oxidase (Nox2) and the production of H2O2 [reviewed in (209, 210)]. Therefore, repeated or chronic global NMDAR hypofunction—as induced by multiple applications of ketamine, but not by a single injection—leads to an IL-6/Nox2–mediated reduction of GABAergic inhibition from PV interneurons specifically (202, 211). Complementing this finding, interneuron-specific deletion of NMDARs increases reactive oxygen species (ROS), albeit in both inhibitory and excitatory cortical neurons (181). Notably, IL-6 is also increased by immune challenges (212) which in this way—and particularly during development—may contribute to disinhibition independently from (but synergistically with) an NMDAR-dependent mechanism (Figure 4).
Environmental stressors, in turn, may lead to oxidative stress if NMDARs on PV interneurons are hypofunctional, thereby provoking damage to cortical circuits that lead to schizophrenia-related deficits. Interestingly, chronic treatment with the antioxidant apocynin was reported to improve many social isolation–induced behavioral deficits in Grin1ΔPpp1r2 mice, including nest building, spontaneous alternation, and pre-pulse inhibition, while also normalizing the production of ROS (181).
A similar circuit-level pathological positive feedback loop may revolve around zinc: zinc is co-released with glutamate by glutamatergic synapses and reduces the open probability (at low concentrations) and blocks (at high concentrations) preferentially GluN2A-containing NMDARs (213–215). In this way, disinhibition in the circuit leads to more zinc release, causing GluN2A-NMDAR hypofunction in PV interneurons, and thereby more disinhibition (Figure 4).
NMDAR Hypofunction in PV Cells as a Risk Factor for Schizophrenia
A model that emerges from this work is that NMDAR hypofunction in PV interneurons is likely to be a risk factor for developing schizophrenia, but may not be sufficient on its own. Other (risk) factors, such as environmental stress (potentially causing oxidative stress at the cellular level), or acutely occurring NMDAR hypofunction in other cell types of the circuit [see below and (175)] need to be present and to interact with the cellular consequences of reduced NMDAR expression in PV interneurons to provoke symptoms of schizophrenia (Figure 4). Moreover, it is interesting to note that NMDAR knockout in PV interneurons provokes molecular and electrophysiological endophenotypes of schizophrenia potentially predisposing the circuit to future insults. For example, Grin1ΔPpp1r2 mice show reduced expression of GAD67 (as also seen in patients with schizophrenia (173) and excess amphetamine-induced accumbal dopamine release aside blunted prefrontal dopamine release (40). The same dopaminergic phenotype was seen in Grin1ΔPV mice (40).
Does NMDAR Hypofunction in PV Interneurons Induce Schizophrenia Only When Occurring During Development?
Against this backdrop of discrepancies between studies (Table 1), it has also been proposed that NMDAR hypofunction in PV cells may cause schizophrenia-related deficits only if it occurs at a young age but not if it begins after adolescence (173). For example, NMDAR blockade by ketamine leads to reduced PV expression (as also seen in neocortical and hippocampal post-mortem tissue from schizophrenia patients) if the drug is administered during adolescence but not in adulthood (126). It has also been reported that juvenile NMDAR ablation in PV interneurons in mice leads to schizophrenia-related deficits, while ablation later in life does not (78, 216). According to this view, the P7-onset of the Ppp1r2-promoter activation, potentially in combination with the 2.1-kB-short distance between lox-P sites used in the Belforte et al. study (173), which reported a wide range of deficits, might result in NMDAR hypofunction in a critical juvenile developmental window, while the ∼P14-onset of the PV-Cre promoter (174), potentially in combination with a slightly (3.3 kB) (175) or even a much larger distance between loxP sites (12 kB) (177, 178) achieves significant NMDAR hypofunction only in adulthood and is therefore less effective in causing deficits (Table 1).
Indeed, some evidence aligns directly with this view. Most importantly, two studies from the Nakazawa lab have shown directly that NMDAR ablation which is stochastically delayed by using the 12-kB-long inter-loxP distance responder line does not lead to a schizophrenia-related phenotype even when combined with long-term social isolation (see Table 1), while ablation of GluN1 at an earlier time point using the 2.1 kB inter-loxP distance responder line does (40, 173).
However, when looking across the large body of studies as a whole (Table 1), the argument is not always empirically supported. For example, when using the same early-onset Ppp1r2-Cre driver line and only a slightly larger distance between loxP sites, we (176) obtained a rather different phenotype from the originally published one (173).
Acute Global NMDAR Hypofunction in Mice With Chronic PV-Specific NMDAR Hypofunction as a Model of Catatonic Schizophrenia
A single exposure to the NMDAR blocker MK-801 reduces NMDAR expression in PV interneurons but increases expression of NMDARs in all neocortical neurons combined (i.e. presumably in excitatory cells, Figure 2) (117). This means that a brain that has previously been exposed to an NMDAR blocker resembles a mouse line with NMDAR hypofunction in PV cells. In such a brain, the balance of NMDAR-mediated contribution to excitation is shifted between the excitatory and PV inhibitory populations of neurons, and when a NMDAR blocker is applied to such a brain—i.e. when global NMDAR hypofunction occurs—the consequences are expected to be different. In line with these considerations, we found that MK-801 may induce cognitive and negative deficits of schizophrenia in Grin1ΔPV mice at concentrations lower than those required in wild type animals to produce similar effects (175). Again, this argues that NMDAR hypofunction in PV interneurons is a risk factor—rather than a direct cause—of schizophrenia.
However, the most pronounced phenotype observed with MK-801 application at moderate doses in Grin1ΔPV mice was the induction of catalepsy, with mice often remaining in the same posture for several minutes. Such cataleptic episodes alternated abruptly with phases of increased locomotion and various forms of stereotypic movement, including circling and repetitive head-shaking (175). Those symptoms are most akin to catatonia—a condition that can occur in, but is not specific for, schizophrenia. Catatonic episodes can last for days or even weeks and can be resistant to antipsychotic drugs (217). We also observed pronounced prefrontal delta-frequency (4 Hz) oscillations induced by MK-801 application in these mice (175). An abnormal enhancement of delta oscillations in the awake state has also been described in patients with schizophrenia (85), and it would be intriguing to investigate their presence in patients with catatonic schizophrenia, specifically. These considerations suggest that PV-specific NMDAR hypofunction may be a necessary pre-condition particularly for catatonic schizophrenia patients. This predicts that repeated episodes of pronounced NMDAR antagonism may produce first the pre-condition (downregulation of NMDARs specifically in PV cells) and subsequently the trigger (global NMDAR hypofunction) of catatonic episodes.
The Role of NMDAR Hypofunction in Cortical Excitatory Cells
Most studies relevant to the potential consequences of NMDAR hypofunction in excitatory cortical cells have not been designed to study the relevance to schizophrenia, but rather to investigate the role of NMDAR-dependent synaptic plasticity in various forms of learning and memory. Despite early demonstrations that associative spatial learning in the Morris water maze (MWM)—but not non-spatial, visual discrimination learning—was impaired by pharmacological blockade of NMDARs (218), it was subsequently shown that rats treated with NMDAR antagonists were in fact perfectly capable of acquiring the MWM task if they were given drug-free pre-training prior to subsequent spatial testing (219–221).
Likewise, the evidence from genetically modified mouse studies that NMDARs on hippocampal principal cells are required for associative memory formation is equivocal at best. Initially, genetically targeted NMDAR ablation (Table 2) focused on subsets of cell types such as the principal cells of CA1 (188) and CA3 (222, 223). It was reported that ablation of NMDARs specifically from the CA1 hippocampal subfield impaired associative spatial memory in the water maze (188). However, the first Cre-Driver line (T29-1, CamKIIα-Cre) used in this study to target CA1 excitatory cells was later found to express across the vast majority of excitatory cells of the hippocampus, neocortex, and peri- and enthorhinal cortex (224, 225). It was subsequently demonstrated that NMDARs in CA1 and/or dentate gyrus are necessary for making correct choices related to ambiguous cues associated with overlapping or competing memories, rather than mediating associative spatial memory formation or recall as such (226). NMDARs in CA3 are likewise not essential for associative spatial memory formation, but have been claimed to support memory recall with incomplete cues as well as performance on a delayed match-to-place (serial reversal) memory task (222, 223).
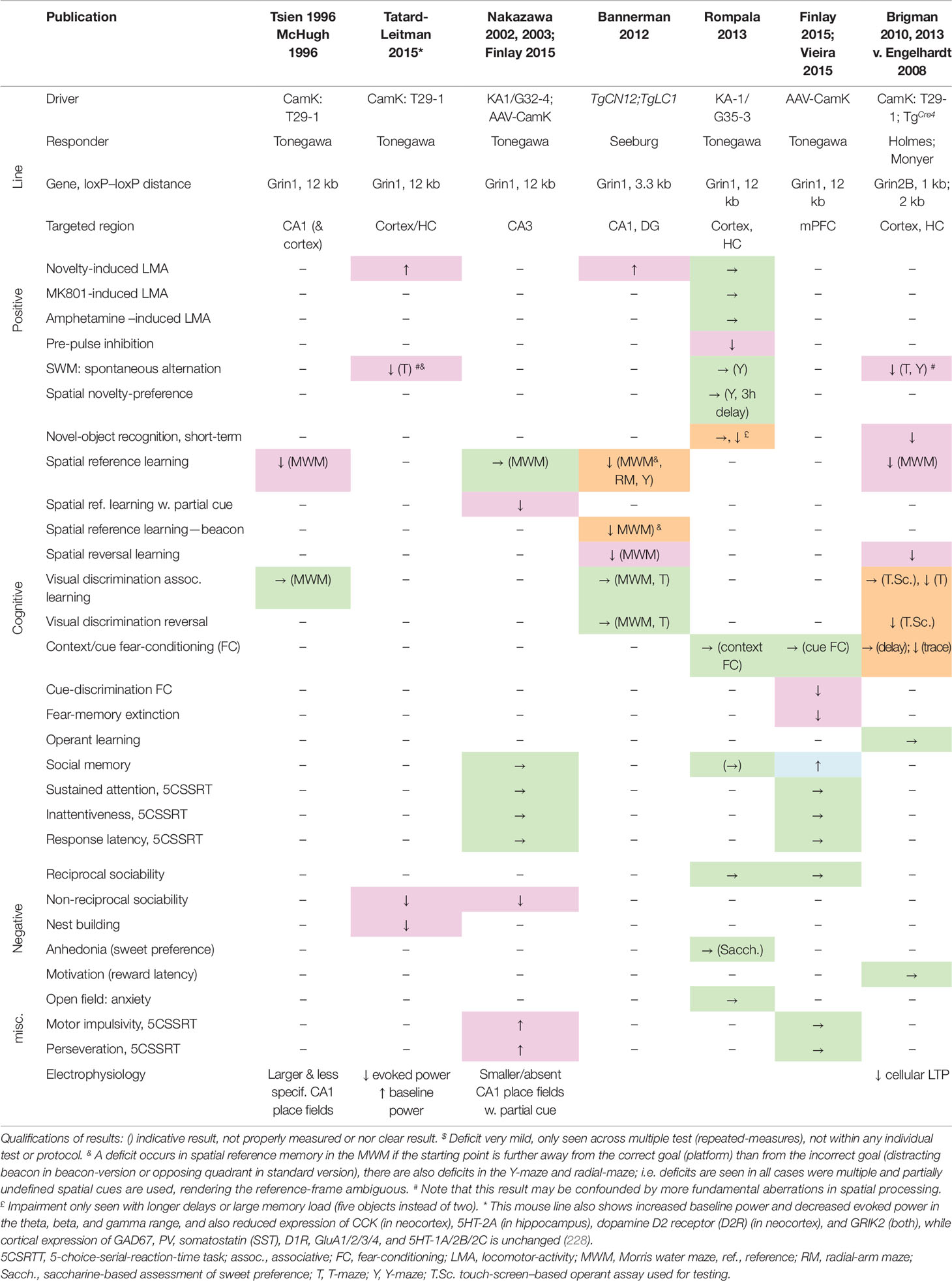
Table 2 Deficits induced by putative genetic NMDAR ablation in excitatory cells of the cortex, including hippocampus. Results of behavioral tests conducted in the indicated studies (top row) measuring rodent correlates of schizophrenia in the positive, cognitive, and negative domain as well as anxiety (see left two columns) in distinct double-transgenic conditional knockout lines (stated in rows 2–4). Green →, no change; magenta, schizophrenia-related deficit; orange, deficit seen in specific test phases or with specific test conditions but not others; blue, apparent improvement of function or opposite of the expected. ↑, increase of behavioral measure; ↓, decrease of behavioral measure. Studies: Tsien 1996 (188), McHugh 1996 (227), Tatard-Leitman 2015 (228), Nakazawa 2002, 2003 (222, 223), Finlay 2015 (229), Bannerman 2012 (226), Rompala 2013 (230), Vieira 2015 (231), Brigman 2010, 2013 (224, 232). Cre-Driver lines: T29-1/CamK-Cre (225) [originally assumed to target CA1 pyramidal cells, but later shown to target excitatory cells across neocortex and hippocampus, e.g. (224); TgCre4/CamK-Cre (233); KA1/G32-4-Cre expresses in CA3 pyramidal cells (222); TgCN12;TgLC1 expresses in excitatory cells of CA1 and dentate gyrus (226); G35-3-Cre expresses in excitatory neurons of the hippocampus and the frontal, parahippocampal, and sensory cortex, esp. in layers 2/3 (230). AAV-CamK, Cre is expressed from a locally injected AAV-vector and driven by the CamKIIα-promoter (229). Floxed-Grin1 and -Grin2B responder lines: Tonegawa (Jax# 005246) (188), Seeburg (186), Holmes (224), Monyer (234).
A later re-assessment of a mouse line with NMDAR ablation in cortical excitatory cells (Grin1ΔCamKIIα mice) across a wide range of schizophrenia-related behaviors and endophenotypes demonstrated that this line shows impairments across the major symptom domains related to schizophrenia (see Table 2), including robust deficits in alternation-based spatial working memory, nest building, social interaction, and novelty-induced hyperlocomotion (228). Grin1ΔCamKIIα mice also displayed electrophysiological endophenotypes relevant to schizophrenia (235–237), including an elevated power of baseline gamma, theta, and beta oscillations and decrease of stimulus-evoked oscillations in these frequency bands (228). Interestingly, pyramidal cells were also more excitable in response to current injections. In contrast to the phenotype induced by pharmacological or PV-specific NMDAR hypofunction, in mice with NMDAR hypofunction in excitatory cells the expression of PV and Gad67 was not altered in the neocortex or hippocampus (228). This demonstrates that several behavioral and electrophysiological endophenotypes seen in schizophrenia could be caused by NMDAR hypofunction in excitatory pyramidal cells alone and do not require reduced Gad67 expression in PV cells. Notably, the level of cortical Gad67 expression also varies strongly among patients with schizophrenia (148), suggesting that its reduction might not be a necessary element of disease development.
In a similar approach, following ablation of GluN2B from excitatory cells across the forebrain (Grin2BΔCamKIIα mice), a broad range of profound memory deficits were found, including chance-level performance in novel-object recognition and alternation-based spatial working memory, but also in spatial long-term memory tasks, and in visual discrimination learning (234). In contrast, when GluN2B-ablation was restricted to CA1 and dentate gyrus excitatory cells, basic spatial associative learning was left intact, although deficits in spatial reversal learning and (to a lesser extent) alternation-based working memory remained (234).
However, there are two caveats to these observations: firstly, on the one hand, some deficits seen after ablation of Grin1 or Grin2B in excitatory cells (Table 2) may be difficult to reconcile with the symptom-profile of schizophrenia, including a wide range of deficits in basic long-term associative spatial memory tasks, fear extinction and cue-discrimination in fear-conditioning, as well as enhanced social memory (188, 226, 229, 231). Note, however, that these learning abnormalities could reflect abnormal credit assignment with inappropriate associations between cues resulting from aberrant assignment of salience, which is suspected to be a core deficit in schizophrenia (238–240).
Secondly, a study using a different Cre line that expresses in a wide range of cortical excitatory cells (although lacking expression in deeper layers in some areas of neocortex and in all layers of some parts of neocortex, e.g. retrosplenial cortex, and in a considerable proportion of hippocampal pyramidal cells), found almost no schizophrenia-related deficits (230). Therefore, it is probably a rather specific subset of excitatory cells, which likely include—but are not limited to—hippocampal pyramidal neurons, in which NMDAR hypofunction may lead to schizophrenia-related deficits.
The Role Of NMDAR Hypofunction In Thalamic Neurons And The Importance Of GluN2C
A third potential location of NMDAR hypofunction promoting schizophrenia pathogenesis is the thalamus—especially its reticular nucleus (RTN), whose neurons are GABAergic, PV-positive, and rely largely on GluN2C-containing NMDARs for their excitatory drive (78, 114). GluN2C-NMDARs are also expressed in the glutamatergic cells of the relay and unspecific nuclei of the thalamus (241).
Work by John Lisman’s group supports a model in which blockade of GluN2C-containing NMDARs in both the excitatory relay cells and the inhibitory PV-positive RTN cells of the thalamus might be the key mechanism by which NMDAR antagonists like ketamine produce increased delta oscillations. Ketamine—at a psychotomimetic dose in humans—strongly blocks GluN2C-containing receptors, but affects GluN2A/2B-containing NMDARs to a lesser degree, since the former are about three times more sensitive to this drug compared to the latter (114). Additionally, the channel is relatively insensitive to Mg2+ ions, allowing it to provide a tonic excitatory drive at the resting potential of thalamic neurons (78, 114, 107). GluN2C-NMDAR blockade by ketamine would be expected to hyperpolarize both cell types, thereby activating T-type calcium channels Cav3.3 (slow) in PV-positive RTN cells and Cav3.1 channels (fast) in glutamatergic relay cells (242, 243), inducing endogenous depolarization-cycles that translate into thalamocortical delta oscillations. This hypothesis is supported by the finding that prefrontal delta oscillations are induced by ketamine and are (abnormally) present in the wake state of patients with schizophrenia (85, 114).
We also observed strong prefrontal delta oscillations after application of MK-801 in Grin1ΔPV mice (175). A combination of reduced activation of inhibitory, PV-positive RTN cells due to NMDAR ablation in Grin1ΔPV mice on one hand, and an additional blockade of remaining NMDARs in thalamic relay cells by MK-801 on the other, could lead to hyperpolarization and activation of Cav3.1 channels in relay cells, and hence the induction of delta oscillations [as observed in (175)]. Also note that Cav3.3 is a schizophrenia risk gene, and the risk-associated mutation renders this channel hypofunctional (244), thereby potentially producing weaker activation of inhibitory RTN cells. Notably, local infusion of MK-801 specifically into the mediodorsal nucleus of the thalamus (at high doses) and into the RTN (already at low doses) causes increased glutamate release in prefrontal cortex in rodents (118, 119, 121). It should also be noted that specific nuclei in the human (but not the rodent) thalamus also contain PV-positive interneurons and projection neurons, and a degeneration of the latter has been described in post-mortem tissue from schizophrenia patients (245, 246).
NMDAR Hypofunction in Dopamine Cells, Striatal Neurons, and SST Interneurons
Other candidate cell types which have been implicated in schizophrenia and suggested as a locus of NMDAR hypofunction are SST-positive interneurons (247) and VTA dopamine neurons (78). In contrast to neocortical PV interneurons and pyramidal cells (117), both of these cell types contain GluN2D-NMDARs (78) which—like GluN2C-containing NMDARs—have a higher affinity for ketamine and are largely insensitive to the Mg2+-block at resting membrane potential (78); these properties imply that NMDAR antagonists would preferentially reduce excitatory drive in those cells, as opposed to neurons that contain mainly GluN2A/B-NMDARs. Grin1ΔSST mice still need to be evaluated comprehensively regarding schizophrenia-related deficits, but a first analysis showed that they do not demonstrate the elevated accumbal and blunted prefrontal dopamine release after amphetamine application and elevated amphetamine-induced locomotion seen in Grin1ΔPpp1r2 and Grin1ΔPV mice (40).
A further locus of NMDAR hypofunction might be VTA dopamine neurons. These cells are a putative hub of schizophrenia pathology and feature a surprising decrease in expression of NMDARs in a prominent schizophrenia mouse model with striatal D2R overexpression (248). Selective ablation of NMDARs from dopamine neurons does not induce core endophenotypes of schizophrenia such as deficits of working memory, short-term object memory, sociability, or pre-pulse inhibition, nor elevated novelty- or amphetamine-induced locomotor activity (249, 250). However, NMDAR knockout from dopamine neurons causes inappropriate generalization in fear-learning across cues as well as impaired visual discrimination and spatial reference learning (250, 251).
Finally, NMDAR ablation has been experimentally confined to medium spiny neurons (MSNs) of the basal ganglia using the RGS9-Cre driver line. Mice lacking all NMDARs (i.e. Grin1) in MSNs display deficits in motor learning [on the rotarod (187)], while mice lacking GluN2B-containing NMDARs in MSNs have difficulties with visual discrimination learning (i.e. long-term associative memory) and its reversal, while simple operant learning is intact (232). It is unclear to what extent these deficits are relevant to schizophrenia, and the overall behavioral profile suggests that striatal MSNs are not a major site of NMDAR hypofunction in the pathogenesis of this disease.
Deficits of accurate discrimination between sensory stimuli and resulting over-generalization during learning—seen with NMDAR ablation in the VTA and the MSNs—might, however, point to aberrations of learning processes that could contribute to aberrant salience (239) and the formation of inappropriate associations and delusions in humans (252).
Conclusions
In summary, NMDAR hypofunction in multiple specific neuron types may contribute to the causation of symptoms in schizophrenia. Indeed, given that genetically driven schizophrenia will potentially produce NMDAR hypofunction across all cells, it may be unrealistic to think that deficits in any one cell type may explain the disorder. It is possible that acute blockade of NMDARs by NMDAR antagonists such as ketamine causes schizophrenia-related symptoms by preferentially reducing the activity of neurons that express NMDARs containing GluN2D (SST interneurons, dopamine neurons) and GluN2C (thalamic relay cells, PV-positive cells of the thalamic RTN and cortex, and presynaptic terminals of PV interneurons). NMDAR hypofunction in the chronic disease state, in contrast, is probably localized somewhat differently, and GluN2A/B-containing NMDARs likely play a role as well. While there is little compelling evidence that NMDAR hypofunction in PV-positive interneurons causes schizophrenia directly, it is likely a key risk factor for this disease. It renders PV interneurons more prone to oxidative stress which may be exacerbated by inflammation, environmental stress, or interactions with other risk genes.
The consequences of NMDAR hypofunction in other interneurons, such as SST and CCK cells still remain to be comprehensively assessed. NMDAR ablation in dopamine or striatal MSNs, in contrast, does not seem to induce a schizophrenia-related spectrum of deficits, although the observed specific aberrations in valuation and over-generalization during learning might contribute to positive symptoms. Nevertheless, some of the symptoms of schizophrenia—especially in the cognitive domain—may be caused by NMDAR hypofunction in the thalamus and in specific subsets of cortical pyramidal cells. We speculate that treatments that increase NMDAR function, reduce oxidative stress, or directly boost the function of PV and other interneurons, could conceivably be beneficial in these cases, especially if applied in the prodrome or early in schizophrenia (253). Given the limited success of previous approaches to enhance NMDAR-function rather directly, it is likely that any future success will depend on a better understanding of the interacting role(s) of NMDARs in different cell types within multiple different neural circuits.
Author Contributions
AB, KK, and DK conducted literature research and prepared the manuscript, which was revised by all authors.
Funding
The research that was conducted by the authors and is discussed in this review was funded by a Sir Henry Wellcome Postdoctoral Fellowship (DK, DMK; grant number 098896), a predoctoral OXION fellowship from the Wellcome Trust (AMB), a predoctoral LGFG fellowship from the state of Baden-Württemberg (KK), the John Fell Fund of the Oxford University Press (DK, DMB), the DFG (DK), grants from the Medical Research Council (DMK, DMB), and an Investigator Award from the Wellcome Trust (DMK). The publication of this article is funded by a grant from the Deutsche Forschungsgemeinschaft (DFG, KA/4594/2-1) and Ulm University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Drs. Simon Butt (Oxford), Norbert Weiss (Prague), Johannes Hell (Davis), John Lisman (deceased), and Alexander Sartorius (Mannheim) for helpful advice, and Simonas Masiulis (Oxford) and Elizabeth Nicholson (London) for contributing essential data sets to our previous publications in this field.
References
1. Mueser KT, McGurk SR. Schizophrenia. Lancet (2004) 363:2063–72. doi: 10.1016/S0140-6736(04)16458-1
2. Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet (2016) 388:86–97. doi: 10.1016/S0140-6736(15)01121-6
3. Hyman SE. Revolution Stalled. Sci Transl Med (2012) 4:155cm11–155cm11. doi: 10.1126/scitranslmed.3003142
4. Leucht S, Cipriani A, Spineli L, Mavridis D, Örey D, Richter F, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet (2013) 382:951–62. doi: 10.1016/S0140-6736(13)60733-3
5. Downing AM, Kinon BJ, Millen BA, Zhang L, Liu L, Morozova MA, et al. A double-blind, placebo-controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC Psychiatry (2014) 14:351. doi: 10.1186/s12888-014-0351-3
6. Adams DH, Zhang L, Millen BA, Kinon BJ, Gomez J-C. Pomaglumetad methionil (LY2140023 Monohydrate) and Aripiprazole in patients with Schizophrenia: a Phase 3, Multicenter, double-blind comparison. Schizophr Res Treat (2014) 758212. doi: 10.1155/2014/758212
7. Stauffer VL, Millen BA, Andersen S, Kinon BJ, LaGrandeur L, Lindenmayer JP, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res (2013) 150:434–41. doi: 10.1016/j.schres.2013.08.020
8. Adams DH, Kinon BJ, Baygani S, Millen BA, Velona I, Kollack-Walker S, et al. phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC Psychiatry (2013) 13:143. doi: 10.1186/1471-244X-13-143
9. Bugarski-Kirola D, Iwata N, Sameljak S, Reid C, Blaettler T, Millar L, et al. Efficacy and safety of adjunctive bitopertin versus placebo in patients with suboptimally controlled symptoms of schizophrenia treated with antipsychotics: results from three phase 3, randomised, double-blind, parallel-group, placebo-controlled, multicentre studies in the SearchLyte clinical trial programme. Lancet Psychiatry (2016) 3:1115–28. doi: 10.1016/S2215-0366(16)30344-3
10. Lane H-Y, Lin C-H, Green MF, Hellemann G, Huang C-C, Chen P-W, et al. Add-on Treatment of Benzoate for Schizophrenia: a randomized, double-blind, placebo-controlled trial of d-amino acid oxidase inhibitor. JAMA Psychiatry (2013) 70:1267–75. doi: 10.1001/jamapsychiatry.2013.2159
11. Coyle JT. NMDA Receptor and Schizophrenia: a brief history. Schizophr Bull (2012) 38:920–6. doi: 10.1093/schbul/sbs076
12. Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D. Has an Angel Shown the Way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull (2012) 38:958–66. doi: 10.1093/schbul/sbs069
13. Javitt DC. Twenty-five Years of Glutamate in Schizophrenia: Are We There Yet? Schizophr Bull (2012) 38:911–3. doi: 10.1093/schbul/sbs100
14. Forsyth JK, Lewis DA. Mapping the consequences of impaired synaptic plasticity in schizophrenia through development: an integrative model for diverse clinical features. Trends Cognit Sci (2017) 21:760–78. doi: 10.1016/j.tics.2017.06.006
15. Ruderfer DM, Charney AW, Readhead B, Kidd BA, Kähler AK, Kenny PJ, et al. Polygenic overlap between schizophrenia risk and antipsychotic response: a genomic medicine approach. Lancet Psychiatry (2016) 3:350–7. doi: 10.1016/S2215-0366(15)00553-2
16. Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511:421–7. doi: 10.1038/nature13595
17. Allen NC, Bagade S, McQueen MB, Ioannidis JPA, Kavvoura FK, Khoury MJ, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet (2008) 40:827–34. doi: 10.1038/ng.171
18. Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry (2013) 18:1185–92. doi: 10.1038/mp.2012.137
19. Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-d-aspartate receptor expression levels in schizophrenia: how can we link molecular abnormalities to mismatch negativity deficits?. Biol Psychol (2016) 116:57–67. doi: 10.1016/j.biopsycho.2015.10.013
20. Tsavou A, Curtis D. In-silico investigation of coding variants potentially affecting the functioning of the glutamatergic N-methyl-D-aspartate receptor in schizophrenia. Psychiatr Genet (2019) 29:44–50. doi: 10.1097/YPG.0000000000000216
21. Curtis D, Coelewij L, Liu S-H, Humphrey J, Mott R. Weighted burden analysis of exome-sequenced case-control sample implicates synaptic genes in Schizophrenia aetiology. Behav Genet (2018) 48:198–208. doi: 10.1007/s10519-018-9893-3
22. Bannerman DM, Niewoehner B, Lyon L, Romberg C, Schmitt WB, Taylor A, et al. NMDA Receptor subunit NR2A is required for rapidly acquired spatial working memory but not incremental spatial reference memory. J Neurosci (2008) 28:3623–30. doi: 10.1523/JNEUROSCI.3639-07.2008
23. Gilmour G, Pioli EY, Dix SL, Smith JW, Conway MW, Jones WT, et al. Diverse and often opposite behavioural effects of NMDA receptor antagonists in rats: implications for “NMDA antagonist modelling” of schizophrenia. Psychopharmacol (Berl) (2009) 205:203–16. doi: 10.1007/s00213-009-1530-7
24. Higgins GA, Ballard TM, Huwyler J, Kemp JA, Gill R. Evaluation of the NR2B-selective NMDA receptor antagonist Ro 63-1908 on rodent behaviour: evidence for an involvement of NR2B NMDA receptors in response inhibition. Neuropharmacology (2003) 44:324–41. doi: 10.1016/s0028-3908(02)00402-1
25. Dalton GL, Ma LM, Phillips AG, Floresco SB. Blockade of NMDA GluN2B receptors selectively impairs behavioral flexibility but not initial discrimination learning. Psychopharmacol (Berl) (2011) 216:525–35. doi: 10.1007/s00213-011-2246-z
26. Smith JW, Gastambide F, Gilmour G, Dix S, Foss J, Lloyd K, et al. A comparison of the effects of ketamine and phencyclidine with other antagonists of the NMDA receptor in rodent assays of attention and working memory. Psychopharmacol (Berl) (2011) 217:255–69. doi: 10.1007/s00213-011-2277-5
27. Jiménez-Sánchez L, Campa L, Auberson YP, Adell A. The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modeling Schizophrenia and treating refractory depression. Neuropsychopharmacology (2014) 39:2673–80. doi: 10.1038/npp.2014.123
28. Higgins GA, Silenieks LB, MacMillan C, Sevo J, Zeeb FD, Thevarkunnel S. Enhanced attention and impulsive action following NMDA receptor GluN2B-selective antagonist pretreatment. Behav Brain Res (2016) 311:1–14. doi: 10.1016/j.bbr.2016.05.025
29. Higgins GA, Ballard TM, Enderlin M, Haman M, Kemp JA. Evidence for improved performance in cognitive tasks following selective NR2B NMDA receptor antagonist pre-treatment in the rat. Psychopharmacol (Berl) (2005) 179:85–98. doi: 10.1007/s00213-005-2203-9
30. Lin E, Lin C-H, Lai Y-L, Huang C-H, Huang Y-J, Lane H-Y. Combination of G72 genetic variation and g72 protein level to detect Schizophrenia: machine learning approaches. Front Psychiatry (2018) 9:566. doi: 10.3389/fpsyt.2018.00566
31. Karlsgodt KH, Robleto K, Trantham-Davidson H, Jairl C, Cannon TD, Lavin A, et al. Reduced dysbindin expression mediates N-Methyl-D-Aspartate receptor hypofunction and impaired working memory performance. Biol Psychiatry (2011) 69:28–34. doi: 10.1016/j.biopsych.2010.09.012
32. Weickert CS, Rothmond DA, Hyde TM, Kleinman JE, Straub RE. Reduced DTNBP1 (dysbindin-1) mRNA in the hippocampal formation of Schizophrenia patients. Schizophr Res (2008) 98:105–10. doi: 10.1016/j.schres.2007.05.041
33. Hahn C-G, Wang H-Y, Cho D-S, Talbot K, Gur RE, Berrettini WH, et al. Altered neuregulin 1–erbB4 signaling contributes to NMDA< receptor hypofunction in schizophrenia. Nat Med (2006) 12:824–8. doi: 10.1038/nm1418
34. Li B, Woo R-S, Mei L, Malinow R. The neuregulin-1 receptor ErbB4 controls glutamatergic synapse maturation and plasticity. Neuron (2007) 54:583–97. doi: 10.1016/j.neuron.2007.03.028
35. Bjarnadottir M, Misner DL, Haverfield-Gross S, Bruun S, Helgason VG, Stefansson H, et al. Neuregulin1 (NRG1) signaling through fyn modulates NMDA receptor phosphorylation: differential synaptic function in NRG1+/– Knock-Outs compared with wild-type mice. J Neurosci (2007) 27:4519–29. doi: 10.1523/JNEUROSCI.4314-06.2007
36. Hashimoto R, Straub RE, Weickert CS, Hyde TM, Kleinman JE, Weinberger DR. Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Mol Psychiatry (2004) 9:299–307. doi: 10.1038/sj.mp.4001434
37. Law AJ, Lipska BK, Weickert CS, Hyde TM, Straub RE, Hashimoto R, et al. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc Natl Acad Sci (2006) 103:6747–52. doi: 10.1073/pnas.0602002103
38. Vullhorst D, Mitchell RM, Keating C, Roychowdhury S, Karavanova I, Tao-Cheng J-H, et al. A negative feedback loop controls NMDA receptor function in cortical interneurons via neuregulin 2/ErbB4 signalling. Nat Commun (2015) 6:7222. doi: 10.1038/ncomms8222
39. Yan L, Shamir A, Skirzewski M, Leiva-Salcedo E, Kwon OB, Karavanova I, et al. Neuregulin-2 ablation results in dopamine dysregulation and severe behavioral phenotypes relevant to psychiatric disorders. Mol Psychiatry (2018) 23:1233–43. doi: 10.1038/mp.2017.22
40. Nakao K, Jeevakumar V, Jiang SZ, Fujita Y, Diaz NB, Pretell Annan CA, et al. Schizophrenia-like dopamine release abnormalities in a mouse model of NMDA receptor hypofunction. Schizophr Bull (2019) 45:138–47. doi: 10.1093/schbul/sby003
41. Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci (2008) 31:234–42. doi: 10.1016/j.tins.2008.02.005
42. Kapur S, Seeman P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D 2 and serotonin 5-HT 2 receptors—implications for models of schizophrenia. Mol Psychiatry (2002) 7:837–44. doi: 10.1038/sj.mp.4001093
43. Seeman P, Ko F, Tallerico T. Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol Psychiatry (2005) 10:877–83. doi: 10.1038/sj.mp.4001682
44. Seeman P, Guan H-C. Phencyclidine and glutamate agonist LY379268 stimulate dopamine D2High receptors: D2 basis for schizophrenia. Synap N Y N (2008) 62:819–28. doi: 10.1002/syn.20561
45. Seeman P, Guan H-C, Hirbec H. Dopamine D2High receptors stimulated by phencyclidines, lysergic acid diethylamide, salvinorin A, and modafinil. Synap N Y N (2009) 63:698–704. doi: 10.1002/syn.20647
46. Rabin RA, Doat M, Winter JC. Role of serotonergic 5-HT2A receptors in the psychotomimetic actions of phencyclidine. Int J Neuropsychopharmacol (2000) 3:333–8. doi: 10.1017/S1461145700002091
47. Goodman CB, Thomas DN, Pert A, Emilien B, Cadet JL, Carroll FI, et al. RTI-4793-14, a new ligand with high affinity and selectivity for the (+)-MK801-insensitive [3H]1-]1-(2-thienyl)cyclohexyl]piperidine binding site (PCP site 2) of guinea pig brain. Synap N Y N (1994) 16:59–65. doi: 10.1002/syn.890160107
48. Roth BL, Gibbons S, Arunotayanun W, Huang X-P, Setola V, Treble R, et al. The ketamine analogue methoxetamine and 3- and 4-Methoxy analogues of phencyclidine are high affinity and selective ligands for the glutamate NMDA receptor. PloS One (2013) 8:e59334. doi: 10.1371/journal.pone.0059334
49. Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature (2016) 533:481–6. doi: 10.1038/nature17998
50. Wray NH, Schappi JM, Singh H, Senese NB, Rasenick MM. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol Psychiatry (2018). doi: 10.1038/s41380-018-0083-8
51. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry (1991) 148:1301–8. doi: 10.1176/ajp.148.10.1301
52. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry (1994) 51:199–214. doi: 10.1001/archpsyc.1994.03950030035004
53. Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, et al. Ketamine-Induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology (1999) 20:106–18. doi: 10.1016/S0893-133X(98)00067-0
54. Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, et al. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry (2000) 48:627–40. doi: 10.1016/S0006-3223(00)00976-8
55. Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in Schizophrenia. Neuropsychopharmacology (1995) 13:9–19. doi: 10.1016/0893-133X(94)00131-I
56. Ban TA, Lohrenz JJ, Lehmann HE. Observations on the action of Sernyl-a new psychotropic drug. Can Psychiatr Assoc J (1961) 6:150–7. doi: 10.1177/070674376100600307
57. Domino EF, Luby ED. Phencyclidine/Schizophrenia: one view toward the past, the other to the future. Schizophr Bull (2012) 38:914–9. doi: 10.1093/schbul/sbs011
58. Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenomimetic drug; sernyl. AMA Arch Neurol Psychiatry (1959) 81:363–9. doi: 10.1001/archneurpsyc.1959.02340150095011
59. Anticevic A, Corlett PR, Cole MW, Savic A, Gancsos M, Tang Y, et al. N-Methyl-D-Aspartate Receptor antagonist effects on prefrontal cortical connectivity better model early than chronic Schizophrenia. Biol Psychiatry (2015) 77:569–80. doi: 10.1016/j.biopsych.2014.07.022
60. Deutsch SI, Mastropaolo J, Rosse RB. Neurodevelopmental consequences of early exposure to phencyclidine and related drugs. Clin Neuropharmacol (1998) 21:320–32.
61. du Bois TM, Huang X-F. Early brain development disruption from NMDA receptor hypofunction: relevance to schizophrenia. Brain Res Rev (2007) 53:260–70. doi: 10.1016/j.brainresrev.2006.09.001
62. Contestabile A. Roles of NMDA receptor activity and nitric oxide production in brain development. Brain Res Rev (2000) 32:476–509. doi: 10.1016/S0165-0173(00)00018-7
63. Kjaerby C, Hovelsø N, Dalby NO, Sotty F. Phencyclidine administration during neurodevelopment alters network activity in prefrontal cortex and hippocampus in adult rats. J Neurophysiol (2017) 118:1002–11. doi: 10.1152/jn.00081.2017
64. Lim AL, Taylor DA, Malone DT. Consequences of early life MK-801 administration: long-term behavioural effects and relevance to schizophrenia research. Behav Brain Res (2012) 227:276–86. doi: 10.1016/j.bbr.2011.10.052
65. Carhart-Harris RL, Brugger S, Nutt DJ, Stone JM. Psychiatry’s next top model: cause for a re-think on drug models of psychosis and other psychiatric disorders. J Psychopharmacol (Oxf) (2013) 27:771–8. doi: 10.1177/0269881113494107
66. Greifenstein F, DeVAULT M, Yoshitake J, Gajewski J. A study of a 1-aryl cyclo hexyl amine for anesthesia*. Anesth Analg (1958) 37:283–94.
67. Vollenweider FX, Kometer M. The neurobiology of psychedelic drugs: implications for the treatment of mood disorders. Nat Rev Neurosci (2010) 11:642–51. doi: 10.1038/nrn2884
68. Janke C, Bumb JM, Aksay SS, Thiel M, Kranaster L, Sartorius A. Ketamin als Anästhetikum bei der Elektrokrampftherapie. Anaesthesist (2015) 64:357–64. doi: 10.1007/s00101-015-0027-5
69. Gandal MJ, Sisti J, Klook K, Ortinski PI, Leitman V, Liang Y, et al. GABAB-mediated rescue of altered excitatory-inhibitory balance, gamma synchrony and behavioral deficits following constitutive NMDAR-hypofunction. Transl Psychiatry (2012) 2:e142. doi: 10.1038/tp.2012.69
70. Barkus C, Dawson LA, Sharp T, Bannerman DM. GluN1 hypomorph mice exhibit wide-ranging behavioral alterations. Genes Brain Behav (2012) 11:342–51. doi: 10.1111/j.1601-183X.2012.00767.x
71. Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to Schizophrenia. Cell (1999) 98:427–36. doi: 10.1016/s0092-8674(00)81972-8
72. Moy SS, Nikolova VD, Riddick NV, Baker LK, Koller BH. Preweaning sensorimotor deficits and adolescent hypersociability in grin1 knockdown mice. Dev Neurosci (2012) 34:159–73. doi: 10.1159/000337984
73. Bodarky CL, Halene TB, Ehrlichman RS, Banerjee A, Ray R, Hahn C-G, et al. Novel environment and GABA agonists alter event-related potentials in N-Methyl-d-aspartate NR1 hypomorphic and wild-type mice. J Pharmacol Exp Ther (2009) 331:308–18. doi: 10.1124/jpet.109.150938
74. Halene TB, Ehrlichman RS, Liang Y, Christian EP, Jonak GJ, Gur TL, et al. Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for Schizophrenia. Genes Brain Behav (2009) 8:661–75. doi: 10.1111/j.1601-183X.2009.00504.x
75. Gandal MJ, Anderson RL, Billingslea EN, Carlson GC, Roberts TPL, Siegel SJ. Mice with reduced NMDA receptor expression: more consistent with autism than schizophrenia?. Genes Brain Behav (2012) 11:740–50. doi: 10.1111/j.1601-183X.2012.00816.x
76. Dalmau J, Tüzün E, Wu H, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic Anti–N-methyl-D-aspartate receptor encephalitis associated with Ovarian teratoma. Ann Neurol (2007) 61:25–36. doi: 10.1002/ana.21050
77. Kayser MS, Dalmau J. Anti-NMDA Receptor encephalitis in psychiatry. Curr Psychiatry Rev (2011) 7:189–93. doi: 10.2174/157340011797183184
78. Nakazawa K, Jeevakumar V, Nakao K. Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr (2017) 3:7. doi: 10.1038/s41537-016-0003-3
79. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of Anti-NMDA receptor encephalitis. J Neurosci (2010) 30:5866–75. doi: 10.1523/JNEUROSCI.0167-10.2010
80. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti–N-Methyl-D-Aspartate Receptor encephalitis. Ann Neurol (2014) 76:108–19. doi: 10.1002/ana.24195
81. Finke C, Kopp UA, Prüss H, Dalmau J, Wandinger K-P, Ploner CJ. Cognitive deficits following anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry (2012) 83:195–8. doi: 10.1136/jnnp-2011-300411
82. Wandinger K-P, Saschenbrecker S, Stoecker W, Dalmau J. Anti-NMDA-receptor encephalitis: a severe, multistage, treatable disorder presenting with psychosis. J Neuroimmunol (2011) 231:86–91. doi: 10.1016/j.jneuroim.2010.09.012
83. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol (2011) 10:63–74. doi: 10.1016/S1474-4422(10)70253-2
84. Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush. Neurology (2012) 79:1094–100. doi: 10.1212/WNL.0b013e3182698cd8
85. Schulman JJ, Cancro R, Lowe SI, Lu F, Walton KD, Llinás RR. Imaging of thalamocortical dysrhythmia in neuropsychiatry. Front Hum Neurosci (2011) 5:69. doi: 10.3389/fnhum.2011.00069
86. Al-Diwani A, Handel A, Townsend L, Pollak T, Leite MI, Harrison PJ, et al. The psychopathology of NMDAR-antibody encephalitis in adults: a systematic review and phenotypic analysis of individual patient data. Lancet Psychiatry (2019) 6:235–46. doi: 10.1016/S2215-0366(19)30001-X
88. Bugarski-Kirola D, Blaettler T, Arango C, Fleischhacker WW, Garibaldi G, Wang A, et al. Bitopertin in negative symptoms of Schizophrenia—Results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry (2017) 82:8–16. doi: 10.1016/j.biopsych.2016.11.014
89. Singh SP, Singh V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs (2011) 25:859–85. doi: 10.2165/11586650-000000000-00000
90. MacKay M-AB, Paylor JW, Wong JTF, Winship IR, Baker GB, Dursun SM. Multidimensional connectomics and treatment-resistant Schizophrenia: linking phenotypic circuits to targeted therapeutics. Front Psychiatry (2018) 9:537. doi: 10.3389/fpsyt.2018.00537
91. DeVito LM, Balu DT, Kanter BR, Lykken C, Basu AC, Coyle JT, et al. Serine racemase deletion disrupts memory for order and alters cortical dendritic morphology. Genes Brain Behav (2011) 10:210–22. doi: 10.1111/j.1601-183X.2010.00656.x
92. Mothet J-P, Parent AT, Wolosker H, Brady RO, Linden DJ, Ferris CD, et al. d-Serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc Natl Acad Sci (2000) 97:4926–31. doi: 10.1073/pnas.97.9.4926
93. Czepita D, Daw NW, Reid SN. Glycine at the NMDA receptor in cat visual cortex: saturation and changes with age. J Neurophysiol (1996) 75:311–7. doi: 10.1152/jn.1996.75.1.311
94. Pritchett D, Hasan S, Tam SKE, Engle SJ, Brandon NJ, Sharp T, et al. d-amino acid oxidase knockout (Dao –/–) mice show enhanced short-term memory performance and heightened anxiety, but no sleep or circadian rhythm disruption. Eur J Neurosci (2015) 41:1167–79. doi: 10.1111/ejn.12880
95. Pritchett D, Taylor AM, Barkus C, Engle SJ, Brandon NJ, Sharp T, et al. Searching for cognitive enhancement in the Morris water maze: better and worse performance in D-amino acid oxidase knockout (Dao –/–) mice. Eur J Neurosci (2016) 43:979–89. doi: 10.1111/ejn.13192
96. Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry (2012) 17:664–5. doi: 10.1038/mp.2011.171
97. Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK. Nature of glutamate alterations in Schizophrenia: a meta-analysis of proton magnetic resonance spectroscopy studies. JAMA Psychiatry (2016) 73:665–74. doi: 10.1001/jamapsychiatry.2016.0442
98. Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron (2013) 78:81–93. doi: 10.1016/j.neuron.2013.02.011
99. Homayoun H, Moghaddam B. NMDA Receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci (2007) 27:11496–500. doi: 10.1523/JNEUROSCI.2213-07.2007
100. Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U.S.A. (2004) 101:8467–72. doi: 10.1073/pnas.0308455101
101. Jodo E. The role of the hippocampo-prefrontal cortex system in phencyclidine-induced psychosis: a model for schizophrenia. J Physiol-Paris (2013) 107:434–40. doi: 10.1016/j.jphysparis.2013.06.002
102. Furlanis E, Traunmüller L, Fucile G, Scheiffele P. Landscape of ribosome-engaged transcript isoforms reveals extensive neuronal-cell-class-specific alternative splicing programs. Nat Neurosci (2019) 22:1709–17. doi: 10.1038/s41593-019-0465-5
104. Perszyk RE, DiRaddo JO, Strong KL, Low C-M, Ogden KK, Khatri A, et al. GluN2D-Containing N-methyl-d-Aspartate Receptors mediate synaptic transmission in hippocampal interneurons and regulate interneuron activity. Mol Pharmacol (2016) 90:689–702. doi: 10.1124/mol.116.105130
105. Swanger SA, Vance KM, Acker TM, Zimmerman SS, DiRaddo JO, Myers SJ, et al. A novel negative allosteric modulator selective for GluN2C/2D-containing NMDA receptors inhibits synaptic transmission in hippocampal interneurons. ACS Chem Neurosci (2018) 9:306–19. doi: 10.1021/acschemneuro.7b00329
106. von Engelhardt J, Bocklisch C, Tönges L, Herb A, Mishina M, Monyer H. GluN2D-containing NMDA receptors-mediate synaptic currents in hippocampal interneurons and pyramidal cells in juvenile mice. Front Cell Neurosci (2015) 9. doi: 10.3389/fncel.2015.00095
107. Kotermanski SE, Johnson JW. Mg2+ imparts NMDA receptor subtype selectivity to the alzheimer’s drug memantine. J Neurosci (2009) 29:2774–9. doi: 10.1523/JNEUROSCI.3703-08.2009
108. Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol (2007) 7:39–47. doi: 10.1016/j.coph.2006.08.011
109. Kuner T, Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ Block in NMDA Receptor channels. J Neurosci (1996) 16:3549–58. doi: 10.1523/JNEUROSCI.16-11-03549.1996
110. Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature (2018) 563:72–8. doi: 10.1038/s41586-018-0654-5
111. Allen Institute. RNA-Seq Mouse Data Navigator:: Allen Brain Atlas: Cell Types. (2018). http://celltypes.brain-map.org/rnaseq/mouse/v1-alm.
112. Monaghan DT, Larsen H. NR1 and NR2 Subunit Contributions to N-Methyl-d-aspartate Receptor channel blocker pharmacology. J Pharmacol Exp Ther (1997) 280:614–20.
113. Yamakura T, Mori H, Masaki H, Shimoji K, Mishina M. Different sensitivities of NMDA receptor channel subtypes to non-competitive antagonists. Neuroreport (1993) 4:687–90. doi: 10.1097/00001756-199306000-00021
114. Khlestova E, Johnson JW, Krystal JH, Lisman J. The role of GluN2C-containing NMDA receptors in ketamine’s psychotogenic action and in Schizophrenia models. J Neurosci (2016) 36:11151–7. doi: 10.1523/JNEUROSCI.1203-16.2016
115. Hagino Y, Kasai S, Han W, Yamamoto H, Nabeshima T, Mishina M, et al. Essential role of NMDA receptor channel ε4 subunit (GluN2D) in the effects of phencyclidine, but not methamphetamine. PloS One (2010) 5:e13722. doi: 10.1371/journal.pone.0013722
116. Sapkota K, Mao Z, Synowicki P, Lieber D, Liu M, Ikezu T, et al. GluN2D N-Methyl-d-Aspartate Receptor subunit contribution to the stimulation of brain activity and gamma oscillations by Ketamine: implications for Schizophrenia. J Pharmacol Exp Ther (2016) 356:702–11. doi: 10.1124/jpet.115.230391
117. Xi D, Zhang W, Wang HX, Stradtman GG, Gao WJ. Dizocilpine (MK-801) induces distinct changes of N-methyl-D-aspartic acid receptor subunits in parvalbumin-containing interneurons in young adult rat prefrontal cortex. Int J Neuropsychopharmacol (2009) 12:1395–408. doi: 10.1017/S146114570900042X
118. Fukuyama K, Hasegawa T, Okada M. Cystine/Glutamate antiporter and aripiprazole compensate NMDA antagonist-induced dysfunction of thalamocortical L-Glutamatergic transmission. Int J Mol Sci (2018) 19:3645. doi: 10.3390/ijms19113645
119. Fukuyama K, Kato R, Murata M, Shiroyama T, Okada M. Clozapine Normalizes a Glutamatergic transmission abnormality induced by an impaired NMDA Receptor in the thalamocortical pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules (2019) 9:234. doi: 10.3390/biom9060234
120. Jodo E, Suzuki Y, Katayama T, Hoshino K-Y, Takeuchi S, Niwa S-I, et al. Activation of medial prefrontal cortex by phencyclidine is mediated via a Hippocampo-prefrontal pathway. Cereb Cortex (2005) 15:663–9. doi: 10.1093/cercor/bhh168
121. Okada M, Fukuyama K, Kawano Y, Shiroyama T, Ueda Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc–. Pharmacol Res Perspect (2019) 7:e00457. doi: 10.1002/prp2.457
122. Jentsch JD, Redmond DE, Elsworth JD, Taylor JR, Youngren KD, Roth RH. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of Phencyclidine. Science (1997) 277:953–5. doi: 10.1126/science.277.5328.953
123. Jentsch JD, Tran A, Le D, Youngren KD, Roth RH. Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology (1997) 17:92–9. doi: 10.1016/S0893-133X(97)00034-1
124. Ahnaou A, Huysmans H, Biermans R, Manyakov NV, Drinkenburg WHIM. Ketamine: differential neurophysiological dynamics in functional networks in the rat brain. Transl Psychiatry (2017) 7:e1237. doi: 10.1038/tp.2017.198
125. Slifstein M, de Giessen E, Snellenberg JV, Thompson JL, Narendran R, Gil R, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in Schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry (2015) 72:316–24. doi: 10.1001/jamapsychiatry.2014.2414
126. Honeycutt JA, Chrobak JJ. Parvalbumin loss following chronic sub-anesthetic NMDA antagonist treatment is age-dependent in the hippocampus: Implications for modeling NMDA hypofunction. bioRxiv (2018). doi: 10.1101/399188.399188.
127. Rujescu D, Bender A, Keck M, Hartmann AM, Ohl F, Raeder H, et al. A pharmacological model for psychosis based on N-methyl-D-aspartate Receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry (2006) 59:721–9. doi: 10.1016/j.biopsych.2005.08.029
128. Braun I, Genius J, Grunze H, Bender A, Möller H-J, Rujescu D. Alterations of hippocampal and prefrontal GABAergic interneurons in an animal model of psychosis induced by NMDA receptor antagonism. Schizophr Res (2007) 97:254–63. doi: 10.1016/j.schres.2007.05.005
129. Abdul-Monim Z, Neill JC, Reynolds GP. Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J Psychopharmacol (Oxf) (2007) 21:198–205. doi: 10.1177/0269881107067097
130. Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, et al. Ketamine-induced loss of phenotype of fast-spiking interneurons Is mediated by NADPH-Oxidase. Science (2007) 318:1645–7. doi: 10.1126/science.1148045
131. Keilhoff G, Becker A, Grecksch G, Wolf G, Bernstein HG. Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience (2004) 126:591–8. doi: 10.1016/j.neuroscience.2004.03.039
132. Benneyworth MA, Roseman AS, Basu AC, Coyle JT. Failure of NMDA receptor hypofunction to induce a pathological reduction in PV-positive GABAergic cell markers. Neurosci Lett (2011) 488:267–71. doi: 10.1016/j.neulet.2010.11.043
133. Fachim HA, Srisawat U, Dalton CF, Harte MK, Marsh S, Neill JC, et al. Subchronic administration of phencyclidine produces hypermethylation in the parvalbumin gene promoter in rat brain. Epigenomics (2016) 8:1179–83. doi: 10.2217/epi-2016-0050
134. Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci (2005) 6:312–24. doi: 10.1038/nrn1648
135. Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, et al. gene expression deficits in a subclass of gaba neurons in the prefrontal cortex of subjects with Schizophrenia. J Neurosci (2003) 23:6315–26. doi: 10.1523/JNEUROSCI.23-15-06315.2003
136. Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B, et al. Developmental changes in parvalbumin regulate presynaptic Ca2+ Signaling. J Neurosci (2005) 25:96–107. doi: 10.1523/JNEUROSCI.3748-04.2005
137. Vreugdenhil M, Jefferys JGR, Celio MR, Schwaller B. Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol (2003) 89:1414–22. doi: 10.1152/jn.00576.2002
138. Kotzadimitriou D, Nissen W, Paizs M, Newton K, Harrison PJ, Paulsen O, et al. Neuregulin 1 Type I overexpression is associated with reduced NMDA Receptor–Mediated synaptic signaling in hippocampal interneurons expressing PV or CCK. eNeuro (2018) 5. doi: 10.1523/ENEURO.0418-17.2018
139. Fazzari P, Paternain AV, Valiente M, Pla R, Luján R, Lloyd K, et al. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature (2010) 464:1376–80. doi: 10.1038/nature08928
140. NyÍri G, Stephenson FA, Freund TF, Somogyi P. Large variability in synaptic n-methyl-d-aspartate receptor density on interneurons and a comparison with pyramidal-cell spines in the rat hippocampus. Neuroscience (2003) 119:347–63. doi: 10.1016/S0306-4522(03)00157-X
141. Matta JA, Pelkey KA, Craig MT, Chittajallu R, Jeffries BW, McBain CJ. Developmental origin dictates interneuron AMPA and NMDA receptor subunit composition and plasticity. Nat Neurosci (2013) 16:1032–41. doi: 10.1038/nn.3459
142. Roux NL, Cabezas C, Böhm UL, Poncer JC. Input-specific learning rules at excitatory synapses onto hippocampal parvalbumin-expressing interneurons. J Physiol (2013) 591:1809–22. doi: 10.1113/jphysiol.2012.245852
143. Cornford JH, Mercier MS, Leite M, Magloire V, Häusser M, Kullmann DM. Dendritic NMDA receptors in parvalbumin neurons enable strong and stable neuronal assemblies. Elife (2019) 8: e49872. doi: 10.7554/eLife.49872
144. Lewis DA, Hashimoto T, Morris HM. Cell and receptor type-specific alterations in markers of GABA neurotransmission in the prefrontal cortex of subjects with schizophrenia. Neurotox Res (2008) 14:237–48. doi: 10.1007/BF03033813
145. Dienel SJ, Lewis DA. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol Dis (2018). doi: 10.1016/j.nbd.2018.06.020
146. Woo T, Walsh J, Benes F. Density of glutamic acid decarboxylase 67 messenger rna–containing neurons that express the n-methyl-d-aspartate receptor subunit nr2a in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry (2004) 61:649–57. doi: 10.1001/archpsyc.61.7.649
147. Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, et al. Cortical deficits of glutamic acid decarboxylase 67 expression in Schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry (2011) 168:921–9. doi: 10.1176/appi.ajp.2011.11010052
148. Lewis DA, Curley AA, Glausier J, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in Schizophrenia. Trends Neurosci (2012) 35:57–67. doi: 10.1016/j.tins.2011.10.004
149. Frankle WG, Cho RY, Prasad KM, Mason NS, Paris J, Himes ML, et al. In vivo measurement of GABA transmission in healthy subjects and Schizophrenia patients. Am J Psychiatry (2015) 172:1148–59. doi: 10.1176/appi.ajp.2015.14081031
150. Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature (2013) 504:272–6. doi: 10.1038/nature12866
151. Bitanihirwe BKY, Lim MP, Kelley JF, Kaneko T, Woo TUW. Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry (2009) 9:71. doi: 10.1186/1471-244X-9-71
152. Woo T-UW, Shrestha K, Lamb D, Minns MM, Benes FM. N-Methyl-D-Aspartate Receptor and calbindin-containing neurons in the anterior cingulate cortex in Schizophrenia and bipolar disorder. Biol Psychiatry (2008) 64:803–9. doi: 10.1016/j.biopsych.2008.04.034
153. Schwarcz R, Rassoulpour A, Wu H-Q, Medoff D, Tamminga CA, Roberts RC. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry (2001) 50:521–30. doi: 10.1016/S0006-3223(01)01078-2
154. Erhardt S, Blennow K, Nordin C, Skogh E, Lindström LH, Engberg G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett (2001) 313:96–8. doi: 10.1016/S0304-3940(01)02242-X
155. Chess AC, Simoni MK, Alling TE, Bucci DJ. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophr Bull (2007) 33:797–804. doi: 10.1093/schbul/sbl033
156. Potter MC, Elmer GI, Bergeron R, Albuquerque EX, Guidetti P, Wu H-Q, et al. Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology (2010) 35:1734–42. doi: 10.1038/npp.2010.39
157. Kegel ME, Johansson V, Wetterberg L, Bhat M, Schwieler L, Cannon TD, et al. Kynurenic acid and psychotic symptoms and personality traits in twins with psychiatric morbidity. Psychiatry Res (2017) 247:105–12. doi: 10.1016/j.psychres.2016.11.017
158. Grace AA. Dopamine system dysregulation by the hippocampus: implications for the pathophysiology and treatment of Schizophrenia. Neuropharmacology (2012) 62:1342–8. doi: 10.1016/j.neuropharm.2011.05.011
159. Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci (2007) 27:11424–30. doi: 10.1523/jneurosci.2847-07.2007
160. Nguyen R, Morrissey MD, Mahadevan V, Cajanding JD, Woodin MA, Yeomans JS, et al. Parvalbumin and GAD65 interneuron inhibition in the ventral hippocampus induces distinct behavioral deficits relevant to schizophrenia. J Neurosci (2014) 34:14948–60. doi: 10.1523/JNEUROSCI.2204-14.2014
161. Picard N, Takesian AE, Fagiolini M, Hensch TK. NMDA 2A receptors in parvalbumin cells mediate sex-specific rapid ketamine response on cortical activity. Mol Psychiatry (2019). doi: 10.1038/s41380-018-0341-9
162. Gilani AI, Chohan MO, Inan M, Schobel SA, Chaudhury NH, Paskewitz S, et al. Interneuron precursor transplants in adult hippocampus reverse psychosis-relevant features in a mouse model of hippocampal disinhibition. Proc Natl Acad Sci (2014) 111:7450–5. doi: 10.1073/pnas.1316488111
163. Grimm CM, Aksamaz S, Schulz S, Teutsch J, Sicinski P, Liss B, et al. Schizophrenia-related cognitive dysfunction in the Cyclin-D2 knockout mouse model of ventral hippocampal hyperactivity. Transl Psychiatry (2018) 8:212. doi: 10.1038/s41398-018-0268-6
164. Chartoff EH, Heusner CL, Palmiter RD. Dopamine is not required for the hyperlocomotor response to NMDA receptor antagonists. Neuropsychopharmacology (2005) 30:1324–33. doi: 10.1038/sj.npp.1300678
165. Carlsson M, Carlsson A. The NMDA antagonist MK-801 causes marked locomotor stimulation in monoamine-depleted mice. J Neural Transm (1989) 75:221–6. doi: 10.1007/bf01258633
166. Swanson CJ, Schoepp DD. The Group II metabotropic glutamate receptor agonist (–)-2-Oxa-4-aminobicyclo[3.1.0.]hexane-4,6-dicarboxylate (LY379268) and clozapine reverse phencyclidine-induced behaviors in monoamine-depleted rats. J Pharmacol Exp Ther (2002) 303:919–27. doi: 10.1124/jpet.102.038422
167. Steinpreis RE, Salamone JD. The role of nucleus accumbens dopamine in the neurochemical and behavioral effects of phencyclidine: a microdialysis and behavioral study. Brain Res (1993) 612:263–70. doi: 10.1016/0006-8993(93)91671-E
168. Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology (2012) 62:1574–83.
169. Jadi MP, Behrens MM, Sejnowski TJ. Abnormal gamma oscillations in N-Methyl-D-Aspartate receptor hypofunction models of Schizophrenia. Biol Psychiatry (2016) 79:716–26. doi: 10.1016/j.biopsych.2015.07.005
170. Cohen SM, Tsien RW, Goff DC, Halassa MM. The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr Res (2015) 167:98–107. doi: 10.1016/j.schres.2014.12.026
171. Carlén M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry (2012) 17:537–48. doi: 10.1038/mp.2011.31
172. Pafundo DE, Miyamae T, Lewis DA, Gonzalez-Burgos G. Presynaptic Effects of N-Methyl-D-Aspartate Receptors enhance parvalbumin cell–mediated inhibition of pyramidal cells in mouse prefrontal cortex. Biol Psychiatry (2018) 84:460–70. doi: 10.1016/j.biopsych.2018.01.018
173. Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci (2010) 13:76–83. doi: 10.1038/nn.2447
174. Butt SJB, Sousa VH, Fuccillo MV, Hjerling-Leffler J, Miyoshi G, Kimura S, et al. The requirement of Nkx2-1 in the temporal specification of cortical interneuron subtypes. Neuron (2008) 59:722–32. doi: 10.1016/j.neuron.2008.07.031
175. Bygrave AM, Masiulis S, Nicholson E, Berkemann M, Sprengel R, Harrison P, et al. Knockout of NMDA-receptors from parvalbumin interneurons sensitizes to schizophrenia-related deficits induced by MK-801. Transl Psychiatry (2016) 6:e778. doi: 10.1038/tp.2016.44
176. Bygrave AM, Masiulis S, Kullmann DM, Bannerman DM, Kätzel D. Gene-environment interaction in a conditional NMDAR-Knockout model of Schizophrenia. Front Behav Neurosci (2019) 12. doi: 10.3389/fnbeh.2018.00332
177. Saunders JA, Tatard-Leitman VM, Suh J, Billingslea EN, Roberts TP, Siegel SJ. Knockout of NMDA receptors in parvalbumin interneurons recreates autism-like phenotypes. Autism Res Off J Int Soc Autism Res (2013) 6:69–77. doi: 10.1002/aur.1264
178. Billingslea EN, Tatard-Leitman VM, Anguiano J, Jutzeler CR, Suh J, Saunders JA, et al. Parvalbumin cell ablation of NMDA-R1 causes increased resting network excitability with associated social and self-care deficits. Neuropsychopharmacology (2014) 39:1603–13. doi: 10.1038/npp.2014.7
179. Pozzi L, Dorocic IP, Wang X, Carlén M, Meletis K. Mice Lacking NMDA Receptors in parvalbumin neurons display normal depression-related behavior and response to antidepressant action of NMDAR antagonists. PloS One (2014) 9:e83879. doi: 10.1371/journal.pone.0083879
180. Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA Receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron (2010) 68:557–69. doi: 10.1016/j.neuron.2010.09.017
181. Jiang Z, Rompala GR, Zhang S, Cowell RM, Nakazawa K. Social isolation exacerbates Schizophrenia-like phenotypes via Oxidative Stress in Cortical Interneurons. Biol Psychiatry (2013) 73:1024–34. doi: 10.1016/j.biopsych.2012.12.004
182. Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron (2007) 53:591–604. doi: 10.1016/j.neuron.2007.01.031
183. Murray AJ, Sauer J-F, Riedel G, McClure C, Ansel L, Cheyne L, et al. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci (2011) 14:297–9. doi: 10.1038/nn.2751
184. Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A Developmental switch in the response of DRG neurons to ETS transcription factor signaling. PloS Biol (2005) 3:e159. doi: 10.1371/journal.pbio.0030159
185. Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature (2009) 459:698–702. doi: 10.1038/nature07991
186. Niewoehner B, Single FN, Hvalby, Jensen V, Meyer zum Alten Borgloh S, Seeburg PH, et al. Impaired spatial working memory but spared spatial reference memory following functional loss of NMDA receptors in the dentate gyrus. Eur J Neurosci (2007) 25:837–46. doi: 10.1111/j.1460-9568.2007.05312.x
187. Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci (2006) 103:15254–9. doi: 10.1073/pnas.0601758103
188. Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell (1996) 87:1327–38. doi: 10.1016/s0092-8674(00)81827-9
189. Corcoran C, Mujica-Parodi L, Yale S, Leitman D, Malaspina D. Could stress cause psychosis in individuals vulnerable to Schizophrenia?. CNS Spectr (2002) 7:33–42. doi: 10.1017/s1092852900022240
190. van Os J, Kenis G, Rutten BPF. The environment and schizophrenia. Nature (2010) 468:203–12. doi: 10.1038/nature09563
191. Réthelyi JM, Benkovits J, Bitter I. Genes and environments in schizophrenia: the different pieces of a manifold puzzle. Neurosci Biobehav Rev (2013) 37:2424–37. doi: 10.1016/j.neubiorev.2013.04.010
192. Linszen DH, Dingemans PM, Nugter MA, van der Does AJW, Scholte WF, Lenior MA. Patient attributes and expressed emotion as risk factors for psychotic relapse. Schizophr Bull (1997) 23:119–30. doi: 10.1093/schbul/23.1.119
193. Parker G, Hadzi-Pavlovic D. Expressed emotion as a predictor of schizophrenic relapse: an analysis of aggregated data. Psychol Med (1990) 20:961–5. doi: 10.1017/s0033291700036655
194. Peterson EC, Docherty NM. Expressed emotion, attribution, and control in parents of Schizophrenic patients. Psychiatry Interpers Biol Process (2004) 67:197–207. doi: 10.1521/psyc.67.2.197.35959
195. Jiang Z, Cowell RM, Nakazawa K. Convergence of genetic and environmental factors on parvalbumin-positive interneurons in schizophrenia. Front Behav Neurosci (2013) 7:116. doi: 10.3389/fnbeh.2013.00116
196. Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol (2009) 19:220–30. doi: 10.1016/j.conb.2009.05.001
197. Herken H, Uz E, Özyurt H, Söğüt S, Virit O, Akyol Ö. Evidence that the activities of erythrocyte free radical scavenging enzymes and the products of lipid peroxidation are increased in different forms of schizophrenia. Mol Psychiatry (2001) 6:66–73. doi: 10.1038/sj.mp.4000789
198. Hardingham GE, Do KQ. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat Rev Neurosci (2016) 17:125–34. doi: 10.1038/nrn.2015.19
199. Do KQ, Trabesinger AH, Kirsten-Krüger M, Lauer CJ, Dydak U, Hell D, et al. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci (2000) 12:3721–8. doi: 10.1046/j.1460-9568.2000.00229.x
200. Matsuzawa D, Hashimoto K. Magnetic resonance spectroscopy study of the antioxidant defense system in Schizophrenia. Antioxid Redox Signal (2010) 15:2057–65. doi: 10.1089/ars.2010.3453
201. Cabungcal JH, Counotte DS, Lewis E, Tejeda HA, Piantadosi P, Pollock C, et al. Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron (2014) 83:1073–84. doi: 10.1016/j.neuron.2014.07.028
202. Behrens MM, Sejnowski TJ. Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex?. Neuropharmacology (2009) 57:193–200. doi: 10.1016/j.neuropharm.2009.06.002
203. Lipton SA, Choi Y-B, Takahashi H, Zhang D, Li W, Godzik A, et al. Cysteine regulation of protein function – as exemplified by NMDA-receptor modulation. Trends Neurosci (2002) 25:474–80. doi: 10.1016/S0166-2236(02)02245-2
204. Papadia S, Soriano FX, Léveillé F, Martel M-A, Dakin KA, Hansen HH, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci (2008) 11:476–87. doi: 10.1038/nn2071
205. Baxter PS, Bell KFS, Hasel P, Kaindl AM, Fricker M, Thomson D, et al. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat Commun (2015) 6:6761. doi: 10.1038/ncomms7761
206. Cabungcal J-H, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, et al. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci (2013) 110:9130–5. doi: 10.1073/pnas.1300454110
207. Steullet P, Cabungcal J-H, Coyle J, Didriksen M, Gill K, Grace AA, et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol Psychiatry (2017) 22:936–43. doi: 10.1038/mp.2017.47
208. Paylor JW, Lins BR, Greba Q, Moen N, de Moraes RS, Howland JG, et al. Developmental disruption of perineuronal nets in the medial prefrontal cortex after maternal immune activation. Sci Rep (2016) 6:37580. doi: 10.1038/srep37580
209. Wang X, Pinto-Duarte A, Sejnowski TJ, Behrens MM. How Nox2-Containing NADPH oxidase affects cortical Circuits in the NMDA receptor antagonist model of Schizophrenia. Antioxid Redox Signal (2013) 18:1444–62. doi: 10.1089/ars.2012.4907
210. Steullet P, Cabungcal JH, Monin A, Dwir D, O’Donnell P, Cuenod M, et al. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: a “central hub” in schizophrenia pathophysiology?. Schizophr Res (2014). doi: 10.1016/j.schres.2014.06.021
211. Behrens MM, Ali SS, Dugan LL. Interleukin-6 mediates the increase in NADPH-Oxidase in the ketamine model of Schizophrenia. J Neurosci (2008) 28:13957–66. doi: 10.1523/JNEUROSCI.4457-08.2008
212. Meyer U. Developmental neuroinflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2013) 42:20–34. doi: 10.1016/j.pnpbp.2011.11.003
213. Monaghan DT, Jane DE. Pharmacology of NMDA Receptors. In: Van Dongen AM. (Editor). Biology of the NMDA Receptor. Front in Neurosci. (Boca Raton (FL): CRC Press/Taylor & Francis).
214. Chen N, Moshaver A, Raymond LA. Differential sensitivity of RecombinantN-Methyl-d-Aspartate Receptor subtypes to zinc inhibition. Mol Pharmacol (1997) 51:1015–23. doi: 10.1124/mol.51.6.1015
215. Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1–NR2A Receptors. J Neurosci (1997) 17:5711–25. doi: 10.1523/JNEUROSCI.17-15-05711.1997
216. Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ. Hippocampal GABAergic inhibitory interneurons. Physiol Rev (2017) 97:1619–747. doi: 10.1152/physrev.00007.2017
217. Rasmussen SA, Mazurek MF, Rosebush PI. Catatonia: our current understanding of its diagnosis, treatment and pathophysiology. World J Psychiatry (2016) 6:391–8. doi: 10.5498/wjp.v6.i4.391
218. Morris RGM, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature (1986) 319:774–6. doi: 10.1038/319774a0
219. Bannerman DM, Good MA, Butcher SP, Ramsay M, Morris RGM. Distinct components of spatial learning revealed by prior training and NMDA receptor blockade. Nature (1995) 378:182–6. doi: 10.1038/378182a0
220. Bannerman DM, Sprengel R, Sanderson DJ, McHugh SB, Rawlins JNP, Monyer H, et al. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat Rev Neurosci (2014) 15:181–92. doi: 10.1038/nrn3677
221. Saucier D, Cain DP. Spatial learning without NMDA receptor-dependent long-term potentiation. Nature (1995) 378:186–9. doi: 10.1038/378186a0
222. Nakazawa K, Quirk MC, Chitwood RA, Watanabe M, Yeckel MF, Sun LD, et al. Requirement for Hippocampal CA3 NMDA Receptors in associative memory recall. Science (2002) 297:211–8. doi: 10.1126/science.1071795
223. Nakazawa K, Sun LD, Quirk MC, Rondi-Reig L, Wilson MA, Tonegawa S. Hippocampal CA3 NMDA Receptors are crucial for memory acquisition of one-time experience. Neuron (2003) 38:305–15. doi: 10.1016/S0896-6273(03)00165-X
224. Brigman JL, Wright T, Talani G, Prasad-Mulcare S, Jinde S, Seabold GK, et al. Loss of GluN2B-Containing NMDA Receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci (2010) 30:4590–600. doi: 10.1523/JNEUROSCI.0640-10.2010
225. Rondi-Reig L, Petit GH, Tobin C, Tonegawa S, Mariani J, Berthoz A. Impaired sequential egocentric and allocentric memories in forebrain-Specific–NMDA receptor knock-out mice during a new task dissociating strategies of navigation. J Neurosci (2006) 26:4071–81. doi: 10.1523/JNEUROSCI.3408-05.2006
226. Bannerman DM, Bus T, Taylor A, Sanderson DJ, Schwarz I, Jensen V, et al. Dissecting spatial knowledge from spatial choice by hippocampal NMDA receptor deletion. Nat Neurosci (2012) 15:1153–9. doi: 10.1038/nn.3166
227. McHugh TJ, Blum KI, Tsien JZ, Tonegawa S, Wilson MA. Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice. Cell (1996) 87:1339–49. doi: 10.1016/s0092-8674(00)81828-0
228. Tatard-Leitman VM, Jutzeler CR, Suh J, Saunders JA, Billingslea EN, Morita S, et al. Pyramidal cell selective ablation of N-Methyl-D-Aspartate Receptor 1 causes increase in cellular and network excitability. Biol Psychiatry (2015) 77:556–68. doi: 10.1016/j.biopsych.2014.06.026
229. Finlay JM, Dunham GA, Isherwood AM, Newton CJ, Nguyen TV, Reppar PC, et al. Effects of prefrontal cortex and hippocampal NMDA NR1-subunit deletion on complex cognitive and social behaviors. Brain Res (2015) 1600:70–83. doi: 10.1016/j.brainres.2014.10.037
230. Rompala GR, Zsiros V, Zhang S, Kolata SM, Nakazawa K. Contribution of NMDA Receptor hypofunction in prefrontal and cortical excitatory neurons to Schizophrenia-like phenotypes. PloS One (2013) 8:e61278. doi: 10.1371/journal.pone.0061278
231. Vieira PA, Corches A, Lovelace JW, Westbrook KB, Mendoza M, Korzus E. Prefrontal NMDA receptors expressed in excitatory neurons control fear discrimination and fear extinction. Neurobiol Learn Mem (2015) 119:52–62. doi: 10.1016/j.nlm.2014.12.012
232. Brigman JL, Daut RA, Wright T, Gunduz-Cinar O, Graybeal C, Davis MI, et al. GluN2B in corticostriatal circuits governs choice learning and choice shifting. Nat Neurosci (2013) 16:1101–10. doi: 10.1038/nn.3457
233. Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Villalba AM, et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet (2002) 31:47–54. doi: 10.1038/ng882
234. von Engelhardt J, Doganci B, Jensen V, Hvalby Ø, Göngrich C, Taylor A, et al. Contribution of hippocampal and extra-hippocampal NR2B-Containing NMDA Receptors to performance on spatial learning tasks. Neuron (2008) 60:846–60. doi: 10.1016/j.neuron.2008.09.039
235. Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci (2010) 11:100–13. doi: 10.1038/nrn2774
236. Bachiller A, Poza J, Gómez C, Molina V, Suazo V, Hornero R. A comparative study of event-related coupling patterns during an auditory oddball task in schizophrenia. J Neural Eng (2015) 12:016007. doi: 10.1088/1741-2560/12/1/016007
237. Andreou C, Leicht G, Nolte G, Polomac N, Moritz S, Karow A, et al. Resting-state theta-band connectivity and verbal memory in schizophrenia and in the high-risk state. Schizophr Res (2015) 161:299–307. doi: 10.1016/j.schres.2014.12.018
238. Spitzer M. A neurocomputational approach to delusions. Compr Psychiatry (1995) 36:83–105. doi: 10.1016/S0010-440X(95)90103-5
239. Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry (2003) 160:13–23. doi: 10.1176/appi.ajp.160.1.13
240. Barkus C, Sanderson DJ, Rawlins JNP, Walton ME, Harrison PJ, Bannerman DM. What causes aberrant salience in schizophrenia? A role for impaired short-term habituation and the GRIA1 (GluA1) AMPA receptor subunit. Mol Psychiatry (2014) 19:1060–70. doi: 10.1038/mp.2014.91
241. Vukadinovic Z. NMDA receptor hypofunction and the thalamus in schizophrenia. Physiol Behav (2014) 131:156–9. doi: 10.1016/j.physbeh.2014.04.038
242. Weiss N, Zamponi GW. T-type calcium channels: From molecule to therapeutic opportunities. Int J Biochem Cell Biol (2019) 108:34–9. doi: 10.1016/j.biocel.2019.01.008
243. Liu X-B, Murray KD, Jones EG. Low-threshold calcium channel subunit Ca(v) 3.3 is specifically localized in GABAergic neurons of rodent thalamus and cerebral cortex. J Comp Neurol (2011) 519:1181–95. doi: 10.1002/cne.22567
244. Andrade A, Hope J, Allen A, Yorgan V, Lipscombe D, Pan JQ. A rare schizophrenia risk variant of CACNA1I disrupts Cav3.3 channel activity. Sci Rep (2016) 6:34233. doi: 10.1038/srep34233
245. Danos P, Baumann B, Bernstein HG, Franz M, Stauch R, Northoff G, et al. Schizophrenia and anteroventral thalamic nucleus: selective decrease of parvalbumin-immunoreactive thalamocortical projection neurons. Psychiatry Res (1998) 82:1–10. doi: 10.1016/s0925-4927(97)00071-1
246. Dixon G, Dissanaike S, Harper CG. Parvalbumin-immunoreactive neurons in the human anteroventral thalamic nucleus. Neuroreport (2000) 11:97–101. doi: 10.1097/00001756-200001170-00020
247. Alherz F, Alherz M, Almusawi H. NMDAR hypofunction and somatostatin-expressing GABAergic interneurons and receptors: a newly identified correlation and its effects in schizophrenia. Schizophr Res Cognit (2017) 8:1–6. doi: 10.1016/j.scog.2017.02.001
248. Krabbe S, Duda J, Schiemann J, Poetschke C, Schneider G, Kandel ER, et al. Increased dopamine D2 receptor activity in the striatum alters the firing pattern of dopamine neurons in the ventral tegmental area. Proc Natl Acad Sci (2015) 112:E1498–506. doi: 10.1073/pnas.1500450112
249. Zweifel LS, Argilli E, Bonci A, Palmiter RD. Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron (2008) 59:486–96. doi: 10.1016/j.neuron.2008.05.028
250. Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, et al. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci (2009) 106:7281–8. doi: 10.1073/pnas.0813415106
251. Zweifel LS, Fadok JP, Argilli E, Garelick MG, Jones GL, Dickerson TMK, et al. Activation of dopamine neurons is critical for aversive conditioning and prevention of generalized anxiety. Nat Neurosci (2011) 14:620–6. doi: 10.1038/nn.2808
252. Fletcher PC, Frith CD. Perceiving is believing: a Bayesian approach to explaining the positive symptoms of schizophrenia. Nat Rev Neurosci (2009) 10:48–58. doi: 10.1038/nrn2536
Keywords: schizophrenia, psychosis, N-methyl-D-aspartate receptor, N-methyl-D-aspartate receptor hypofunction, parvalbumin, ketamine, MK-801, catatonic schizophrenia
Citation: Bygrave AM, Kilonzo K, Kullmann DM, Bannerman DM and Kätzel D (2019) Can N-Methyl-D-Aspartate Receptor Hypofunction in Schizophrenia Be Localized to an Individual Cell Type? Front. Psychiatry 10:835. doi: 10.3389/fpsyt.2019.00835
Received: 29 July 2019; Accepted: 21 October 2019;
Published: 21 November 2019.
Edited by:
Bernat Kocsis, Harvard Medical School, United StatesReviewed by:
Margarita Behrens, Salk Institute for Biological Studies, United StatesVasileios Kafetzopoulos, Harvard Medical School, United States
Copyright © 2019 Bygrave, Kilonzo, Kullmann, Bannerman and Kätzel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dennis Kätzel, ZGVubmlzLmthZXR6ZWxAdW5pLXVsbS5kZQ==