
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Psychiatry , 08 October 2019
Sec. Molecular Psychiatry
Volume 10 - 2019 | https://doi.org/10.3389/fpsyt.2019.00706
 Xiaodan Li1,2†Yuncong Zhang3†Luxi Wang1,2Yunqing Lin3Zhaomin Gao1,2Xiaolei Zhan1,2Yan Huang1,2Caihong Sun1,2
Xiaodan Li1,2†Yuncong Zhang3†Luxi Wang1,2Yunqing Lin3Zhaomin Gao1,2Xiaolei Zhan1,2Yan Huang1,2Caihong Sun1,2 Dong Wang3,4
Dong Wang3,4 Shuang Liang1,2*
Shuang Liang1,2* Lijie Wu1,2*
Lijie Wu1,2*Autism spectrum disorder (ASD) is a set of complex neurodevelopmental disorders with etiology that remains elusive. Although there is a mounting body of investigation in different brain regions related to ASD, our knowledge about the common and distinct perturb condition between them is at the threshold of accumulation. In this study, based on protein–protein interactions, post-mortem transcriptome analysis was performed with corpus callosum (CC) and prefrontal cortex (PFC) samples from ASD individuals and controls. Co-expression network analysis revealed that a total of seven (four for CC set, three for PFC set) core dysfunctional modules strongly enriched for known ASD-risk genes. Three quarters of them in CC set (M4, M6, M29) significantly enriched for genes annotated by genetically associated variants in our previous whole genome sequencing data. We further determined transcriptional and post-transcriptional regulation subnetwork for each ASD-correlated module, including 47 pivot transcription factors, 130 pivot miRNAs, and 7 pivot lncRNAs. Moreover, there were significantly more interactions between CC-M4, -M6, and PFC-M2, mainly involved in synaptic functions and neuronal development. Our integrated multifactor analysis of ASD brain transcriptome profile illustrated underlying common and distinct molecular mechanisms and the module crosstalk between CC and PFC, helping to shed light on the molecular neuropathological underlying ASD.
Autism spectrum disorder (ASD) is a group of neurodevelopmental disorders characterized by high degree of clinical and genetic heterogeneity. The Diagnostic and Statistical Manual of Mental Disorders-Fifth Edition (DSM-5) defines ASD by deficits in social communication and interactions, as well as by repetitive behaviors and restrictive interests, with onset in early development. Currently, 1 of every 59 children in the United States is diagnosed with ASD (1), whereas the prevalence of ASD in China is 8.3 per 10,000, which is likely underestimated due to the strict diagnosis criteria (2). Consistent with the primary features of ASD, many studies demonstrated the association between brain abnormalities and the disease, yet no clear and common molecular or pathology mechanisms have proved to be responsible for this disease so far. Animal experiments showed that abnormal brain regions were mainly involved in the temporal lobe, cerebellar cortex, frontal lobe, hypothalamus, and the striatum by 26 different ASD mouse models (3). Postmortem and structural magnetic resonance imaging studies have highlighted the frontal lobes, amygdala and cerebellum as pathological area in ASD (4). Although several genetic and environmental risk factors have been identified, we do not have a firm understanding of the pathophysiological basis of this complex disease by now.
Among the human brain regions implicated in the pathophysiology of ASD, the prefrontal cortex (PFC), as the center of the highest-order cognitive functions, has always been focused on due to its role in the cognitive, decision-making and social behavior (5). Previous studies have confirmed that some microglial activation in ASD was associated with a neuron-specific reaction in the dorsolateral prefrontal cortex (6). The corpus callosum (CC), predominantly populated by oligodendrocyte cells, has been the largest white matter tract in the human brain, interconnecting homologous association areas of both hemispheres (7). Dysplasia of the CC often involves in social impairments similar to those seen in high-function people with ASD, encompassing diminished social self-awareness, difficulty in imaging the social perspective of others, poor conversation skills and restricted verbal expression of emotional experience (8). Though its contribution to cognition and behavior remains unclear, the indirect relationship between the volume of CC in anatomy and ASD severity suggested its susceptibility of etiology (9).
Furthermore, brain function is governed by precise regulation of gene expression across its anatomically distinct structures. However, the regulation programs of gene expression in human are controlled by thousands of transcription factors (TFs), cofactors and chromatin regulators, involving transcriptional and post-transcriptional levels. Dysregulation of these programs may cause a broad range of diseases (2, 10). Transcription Factor 4 (TCF4) binding sites were found in a large number of neuronal genes that were implicated as genetic risk factors for common neurodevelopmental disorders (11). Previous studies found miR-146a up-regulation was the most common miRNA dysregulation event in neurodevelopmental disorders such as ASD, epilepsy, and intellectual disability (12). Long non-coding RNA (lncRNA) Shank2-AS was abnormally expressed in patients with ASD and might affect the structure and growth of neurons by regulating its sense strand gene Shank2 expression (13).
We reasoned that distinct pathogenic mechanisms in CC and PFC area in ASD might converge on common pathways that are not yet well understood. Since systems biology made it possible to study larger and more intricate systems than before, gene set and network-based analysis became powerful tools for evaluating putative genetic risk factors and dysfunctional modules for diseases. RNA-sequencing data of CC (GSE62098) (14) have provided DEGs between ASD children and typically developing controls and was used to confirm the involvement of a protein interaction module (with GO enrichment for synaptic transmission) in ASD. Li et al. (14) examined the expression specificity of the module in the CC with immunochemical method and showed that the human CC was predominantly populated by oligodendrocyte cells. Then multiple genomic data further revealed a significant involvement of this module in the development of oligodendrocyte cells in mouse brain. For PFC data (GSE102741), Wright et al. (15) revealed that seven histamine genes (SNORA74A, SNORA53, SNORD17, SNORA54, SNORA74B, SNORD114-23, and RP6-206I17.3) showed altered expressions in ASD children, suggesting that the histaminergic system might also be altered in ASD. However, few efforts have been made to conduct a comprehensive approach to decipher tissue specific pathogenic mechanism of ASD.
To this end, weighted gene co-expression network analysis (WGCNA) was performed in this paper based on integrated RNA-seq data, protein–protein interactions (PPIs) and TF-, ncRNA-target interactions. In our previous study, we have conducted a comprehensive scan of genomic variance differences among three pairs of monozygotic twins with whole genome sequencing (WGS) (16). Here we extend our genomic analysis to the transcriptome profile of ASD-affected individuals aimed at identifying common and distinct transcriptional alterations in dysfunctional modules of CC and PFC, which may prove to be a crucial step to better understand ASD.
Gene expression profile data of 24 corpus callosum samples [GSE62098 (14), 12 ASDs, 12 controls] and 52 prefrontal cortex samples [GSE102741 (15), 13 ASDs, 39 controls] were downloaded from the NCBI Gene Expression Omnibus (GEO) (17) database. For GSE62098, original samples were requested from Autism Speak’s Autism Tissue Program (www.atpportal.org) and NICHD Brain and Tissue Bank (http://medschool.umaryland.edu/btbank/). For GSE102741, samples were collected at National Institute of Mental Health brain collection (18), the University of Maryland Brain and Tissue Bank (www.medschool.umaryland.edu/btbank/) and the Stanley Medical Research Institute sample characterization (www.stanleyresearch.org). Subjects with evidence of drug use, alcohol abuse or psychiatric illness were excluded from the control cohort. Details of clinical characterization, neuropathological examinations and toxicological analyses are available on their respective websites.
Two sets of data were processed separately. The quality control was performed by Fast-QC (version 0.11.5). Then, filtered reads were used to map to the hg38 genome reference genome (GRCH38) using HISAT2 (version 2.1.0) (19) with default parameters. Fragments Per Kilobase transcriptome per Million (FPKM) reads values and raw counts of gene expression were calculated within StringTie (20) (version 1.3.3). Based on raw counts tables, differentially expressed genes (DEGs) were ranked and filtered by DESeq2 (R package) (21) for further analysis (|Fold Change| > 1.2, p-value < 0.05).
WGCNA was applied to derive gene networks based on all pair-wise gene expression correlations between genes (22). A human protein–protein physical interaction (PPI) subnetwork (combined score ≥ 900) was firstly extracted from STRING database (v10.5) (23), consisting of 9,635 proteins and 170,876 interactions. Then, DEGs and their interactors in the PPI network were identified to construct weighted gene correlation network. Finally, the expression profile of genes in the weighted gene correlation network was input to WGCNA for co-expression modules. For GSE62098 (CC set), a power of 12 was chosen in this study and the parameters minModuleSize = 20, minimum height = 0.15 were set to cut tree. For GSE102741 (PFC set), power = 8, minModuleSize = 20, minimum height = 0.15 were set to cut tree. For each co-expression module, nodes with KME value > 0.8 were considered as hub genes.
TF-target interactions (785 TFs, 2,489 target genes) were recruited from AnimalTFDB v3 (24) and TRRUST v2 (25). For post-transcriptional regulation, miRNA- (1179 miRNAs, 6,677 target genes) and lncRNA-target interactions (93 lncRNAs, 7,744 target genes) were recruited from RAID v2.0 (26). If a factor (i) has >1 interactions and (ii) the number of its target nodes significantly enriched for the module (hypergeometric test, p < 0.05) (27), the factor was identified as a pivot regulator.
First, we compiled a list of 3,352 protein coding genes differently expressed between CC (white matter) and PFC (frontal lobe) using the Allen Human Brain Atlas (28) (|Fold Change| ≥4, p-value < 0.05), 814 were on STRING PPI network. Tissue-specific modules were defined as modules of which specific expression parts containing DEGs. We counted the number of interactions between each tissue-specific module. Then by keeping the number of nodes unchanged compared to the corresponding tissue-specific module genes on PPI network, paired gene sets were randomly sampled for 100,000 times and also counted the number of interactions of each pair, which allowed us to assign the p value. Significant crosstalk module pair was defined as a pair of gene sets from CC and PFC set separately which of significantly more interactions between each other than random pairs (permutation test, p < 0.05).
Gene ontology biological processes (GO-BP), Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were carried out with R package clusterProfiler (29). For pivot miRNAs, functional annotations were performed on DIANA-miRPath v3 (30).
We performed a differential expression (DE) analysis to determine the gene significantly expressed in ASD compared to controls, and a total of 653 and 720 DEGs were identified for further analysis, in CC and PFC data respectively (|Fold change| > 1.2 and p-value < 0.05, Supplementary Table S1). Since interacted genes imply co-expression (31), human protein–protein interaction (PPI) subnetwork based on DEGs containing 3,492 interactors for CC set and 3,243 for PFC set were constructed for additional analysis (see Materials and Methods section). Then based on WGCNA, 30 (CC set) and 28 (PFC set) dysfunctional modules were initially determined (Figure 1). Taking the effectiveness of the modules into account, modules containing DEGs and size less than 500 genes were kept for further analysis (Supplementary Table S2).
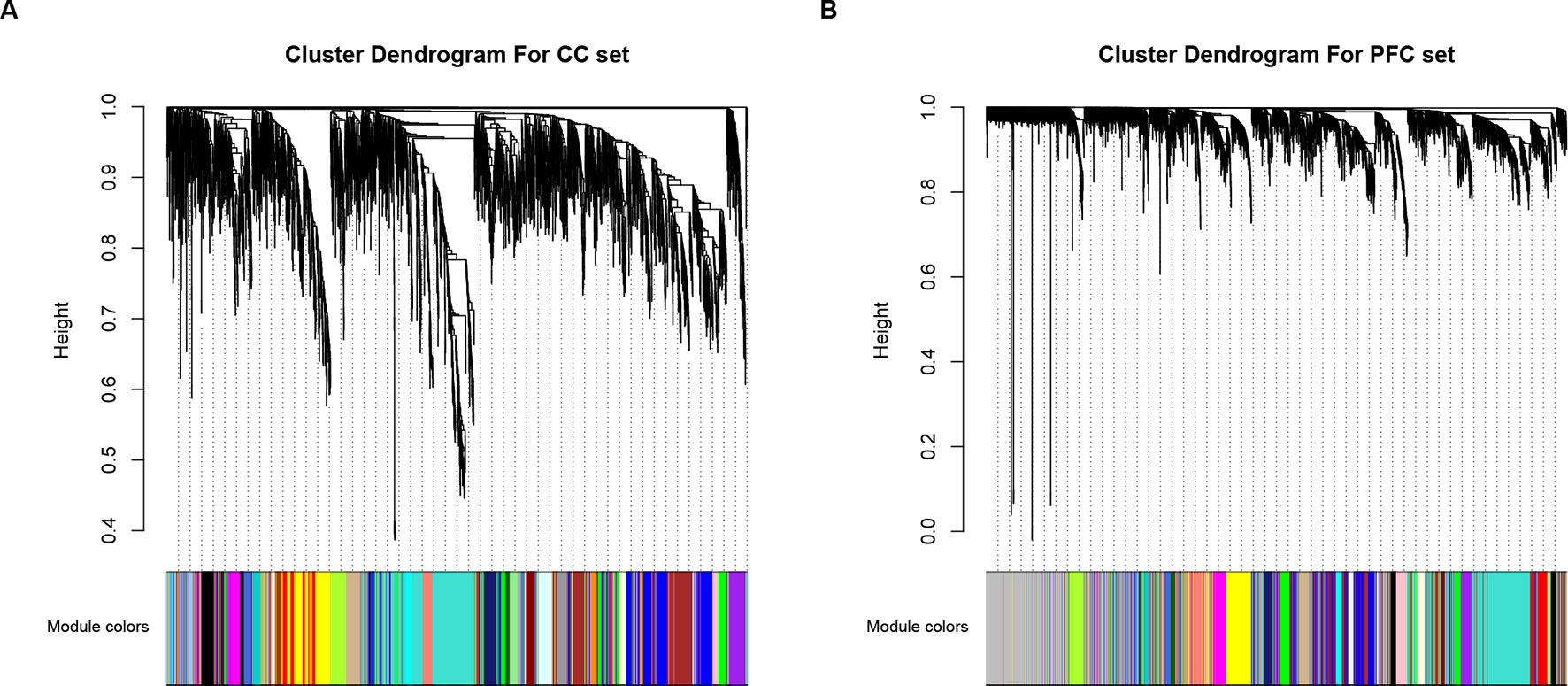
Figure 1 Visualization of WGCNA results. Clustering dendrograms of genes within (A) CC and (B) PFC subset, X-axis represents genes and Y-axis represents height of the gene tree. Total of 30 (CC set) and 28 (PFC set) co-expression modules corresponding to different color bars while grey bars represent genes not included in any co-expression module.
In order to detect whether there were common or distinct contributors perturbed in the functional modules, remained modules were further filtered by genomic variation burden and correlation with ASD. Based on our previous study (16), discordant variations in monozygotic twin (DVMT) including single nucleotide variants (SNVs), small insertions and deletions (InDels), and copy number variations (CNVs) presented in at least two twin pairs were filtered as putative ASD risk sites. A subset of 1,714 protein coding genes annotated by three types of DVMT were used to determine the genomic variance burden in the present study and made further analysis. First, we calculated the number of genes involved in ASD susceptibility to determine whether any of co-expression modules were associated with ASD, using SFARI Gene list (https://gene.sfari.org/autdb/). M4, M6, M17, and M29 for CC set and M2, M6, and M8 for PFC set showed significant enrichment, which were referred to be dysfunctional modules (p < 0.05). Next, we confirmed the results above by calculating overlaps between module nodes and DVMT genes using our previous WGS data, which were significantly converged into M4, M6, M7, M9, M29 (CC set), and M5, M11, M24 (PFC set) (p < 0.05). Notably, most of ASD-associated modules (M4, M6, and M29) of CC set showed DVMT significance, while none of that was observed with PFC set (Table 1, Supplementary Table S3).
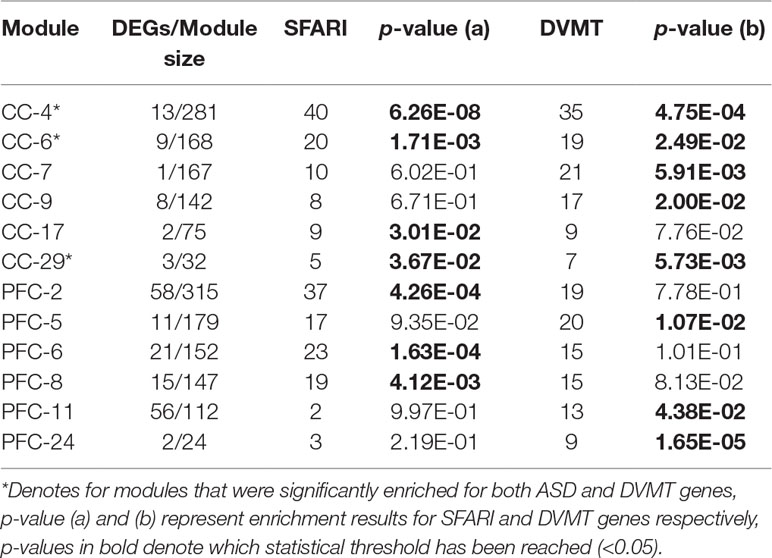
Table 1 Co-expression modules significantly enriched for SFARI and/or DVMT genes.
Functional enrichment analysis was performed for each dysfunctional module and revealed multiple biological processes of gene ontology (GO-BP) critical to major functions for both sets (Figure 2, Supplementary Table S4), including cognition, learning or memory, long term depression, nervous system development, and synaptic function. KEGG pathway analysis showed that common pathways like neuroactive ligand–receptor interaction, calcium signaling, MAPK and PI3K-Akt signaling were observed. We found that 28% (20/72) and 29% (9/31) of genes involved in neuroactive ligand–receptor interaction, and in CC and PFC set respectively, have been reported to be related to ASD. There were also module-specific terms like RNA transport and localization involved in M17 of CC set, protein ubiquitination and regulation of cell-substrate adhesion in M6 and M8 of PFC set, respectively. The ubiquitin–proteasome system has been considered to be a major non-lysosomal proteolytic process that regulates the levels of cellular proteins related to synaptic plasticity and long-term memory, and to ASD (32, 33).
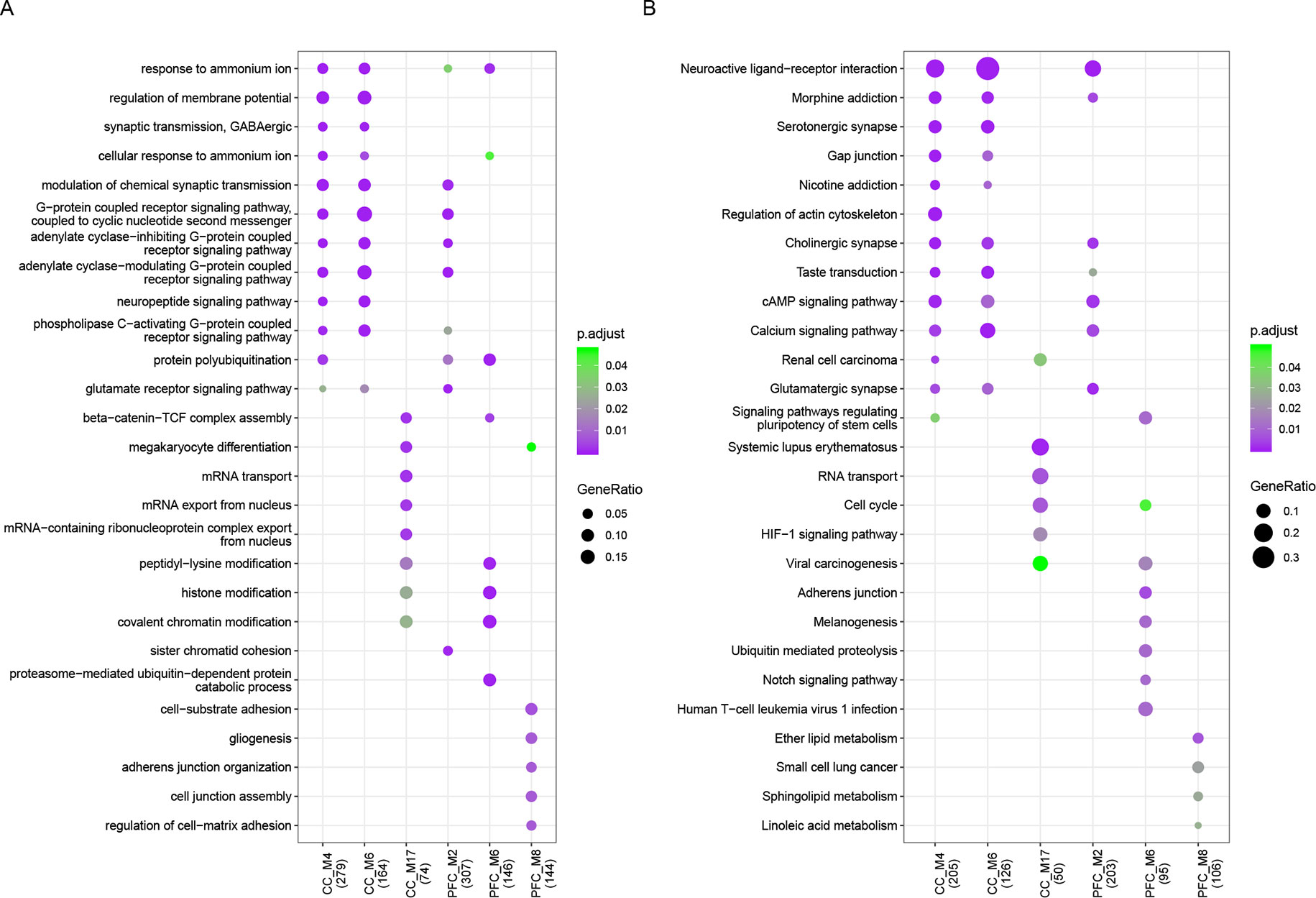
Figure 2 Functional enrichment results. (A) GO biological process and (B) KEGG pathway enrichment results of ASD-related dysfunctional modules which enriched for at least one GO term or pathway with p.adjust < 0.05. The node size represents gene ratio and color represents p.adjust value.
Hub genes are those that show most connections in the network and of great use in identifying genes with significant biologically meaning. In line with functional enrichment results, CACNA1C, one of the hubs of CC M6 presented in both SFARI and WGS set, in which genetic variation have been associated with ASD, Major Depressive Disorder, Schizophrenia as well as some undiagnosable psychiatric illness (34). Other hub genes (GABBR2, GRM7, MEF2C, SCN2A, KMT2C, and DAGLA) dysfunction have been reported to contribute to ASD and other psychiatric disorders such as Attention-Deficit Hyperactivity Disorder (ADHD) (35), Huntington’s disease (36), and Rett-syndrome (37). These observations supported the existence of shared dysfunction convergence but might distinct mechanism under CC and PFC in ASD.
We determined pivotal regulators by transcriptional and post-transcriptional level regulations among modules significantly enriched for genomic variants and/or known ASD-risk genes (see Materials and Methods section, Supplementary Table S5). Then, regulator-DEG and DEG-interactor subnetworks were constructed (Figure 3).
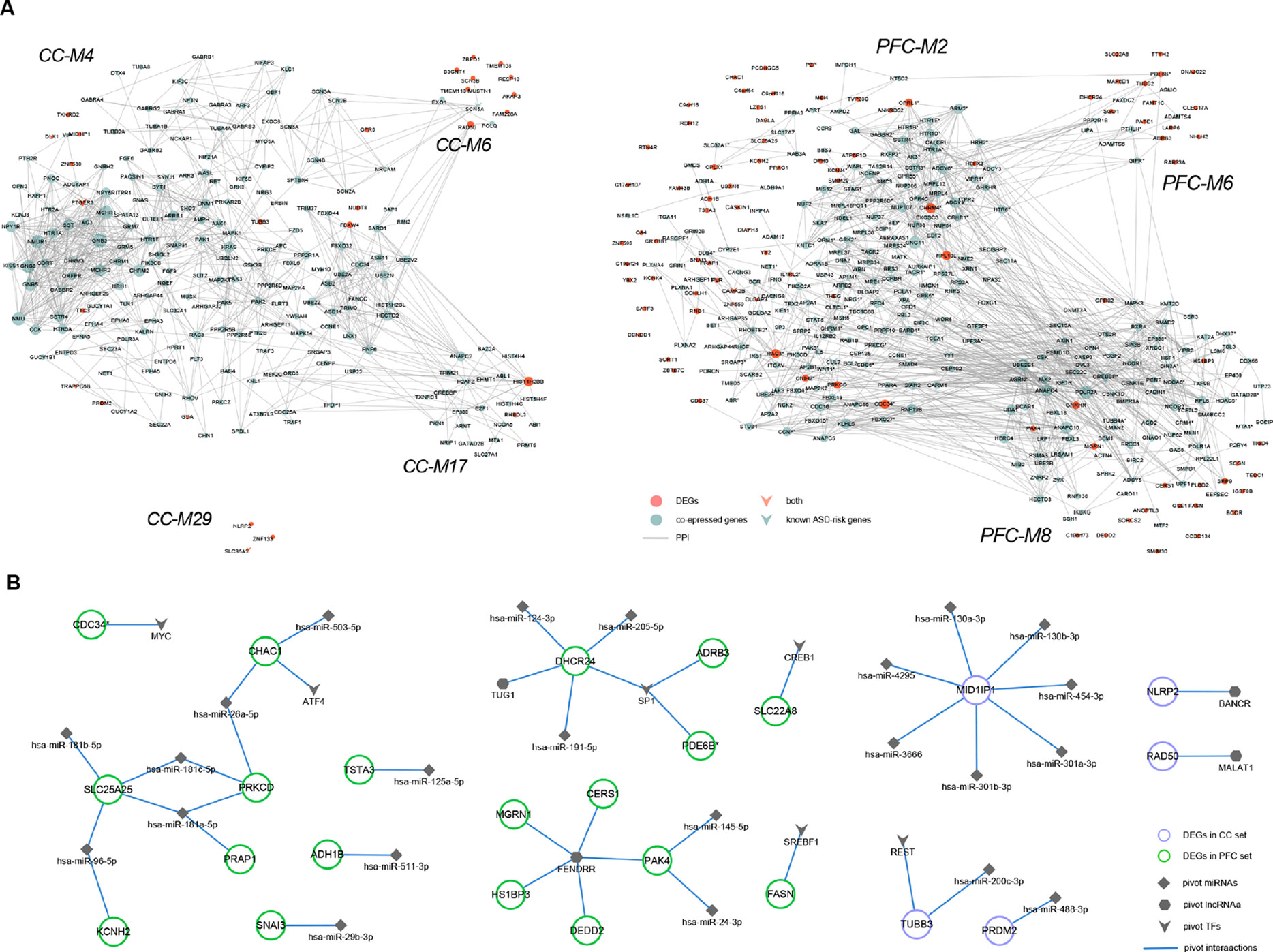
Figure 3 Regulatory subnetworks. (A) DEGs and their direct or indirect interactors within ASD-related modules (CC-M4, -M6, -M17, M29, and PFC-M2, -M6, -M8). The node size is proportional to degree. (B) The transcriptional and post-transcriptional pivot regulator-DEG subnetworks of ASD-related modules, containing 6 pivot TFs, 4 pivot lncRNAs, and 23 pivot miRNAs.
Recent whole exome sequencing (WES) has revealed substantial overlap in ASD-risk genes and cancer (38). The transcription factor MYC, regulating both CC-M4 and PFC-M2, is a cell growth regulator which strongly oncogenic, and estimated to contribute to most cancers (39). Transcription factor SP1, regulating CC-M6, -M7, and PFC-M5, -M8, has been reported dysfunctional in the anterior cingulate gyrus (ACG) of ASD brain and the potential ASD candidate gene PTEN (in PFC-M5) regulated by SP1 was shown to be over-expressed (40, 41). CREB1, down-regulated at PFC, can regulate CC-M4, -M9, and PFC-M8, assumed to be linked to cognition and behavior related to butyrate dysregulation in ASD (42). Moreover, de novo deletion within CREB1 was observed in a girl with developmental delay, autistic traits and Rett-like features (43). Pivot TFs E2F1, regulating CC-M17 while down-regulated at PFC, was involved in cell cycle regulation and apoptosis as well as functionally related to obesity which was consistent with the function enrichment results of M17 (44).
LncRNAs are more abundant in the human brain and are involved in neurodevelopment and neurodevelopmental disorders, including ASD (45). It was observed that most pivotal lncRNAs examined in this study have been reported related to neurodegenerative disease and cancers. TUG1, regulating M7 of CC set and M2, M5, and M8 of PFC set, was reported to be a tumor suppressor in breast cancer and high correlations were found between ASD prevalence and the incidence of in situ breast cancer (46, 47). FENDRR, regulating CC M4, M7, and PFC M6, is a kind of endothelial gene critical for vascular development that could inhibit breast cancer cell proliferation (48). Moreover, UCA1, regulating CC M6, M17, and PFC M6, has been identified as a pivotal regulator in the tumorigenesis of glioma which represent the most common solid tumor of childhood (49, 50).
MicroRNAs are post-transcriptional regulators that play key roles in brain development, synapse formation and fine-tuning of genes underlying synaptic plasticity and memory formation (51). We perform GO enrichment analysis and found that pivot miRNAs regulated DEGs were significantly enriched for GO terms like cellular protein modification process, neurotrophin TRK receptor signaling pathway, nervous system development, and axon guidance. Furthermore, KEGG pathway analysis showed that both sets were convergent into pathways like ErbB signaling, long-term depression, Estrogen signaling pathway and so on, which were closely related to ASD (52, 53). Taken all together, these results indicated that pivot regulators might play an important role in the disease process directly or indirectly.
Modules do not act alone. We further analyzed the module interactions between CC and PFC set (Figure S1). To this end, CC-M4, -M6, and PFC-M2 were determined as tissue-specific modules using the Allen Human Brain Atlas and module pairs PFC M2-CC M4, and PFC M2-CC M6 were found to be connected with each other significantly with more interactions than random gene set pairs (p = 5.00E–05 and 2.50E–04, respectively) (see Materials and Methods section). All of these three modules were most significantly enriched for “neuroactive ligand–receptor interaction” pathway and participated in neuro system development, indicating the association of ASD pathogenic mechanism between CC and PFC.
Although the symptoms of ASD are the most striking among the behavioral and functional manifestations of affected individuals, however, findings are profoundly heterogeneous and the etiology of ASD is still unclear. The corpus callosum plays a central role in mediating signal communication between the brain hemispheres through the axons extending from different cortical layers (54), whereas the prefrontal cortex plays an essential role in the organization and control of goal-directed thought and behavior (55). Recent advances have reported ASD clinical heterogeneous based on typical brain functional regions (56, 57). Most genetic abnormalities are difficult to verify at the level of variation, however significance is repeatedly observed at the gene and pathway levels. (58).
In this study, we compared the transcriptome signatures of CC and PFC across ASD-affected individuals versus healthy controls. WGCNA was performed to identify co-expression patterns of DEGs and their protein–protein interactors. Then, based on DVMT and SFARI genes, 12 of 49 (6/24 for CC set, 6/25 for PFC set) dysfunctional modules significantly enriched for genomic variants and/or known ASD susceptibility genes were identified as ASD-correlated modules and kept for additional analysis. GO-BP and KEGG pathway enrichment analysis showed that pathophysiological process of both CC and PFC in ASD seem to converge on specific molecular dysregulation, mainly for synaptic and neuronal signaling pathways, which in line with earlier studies for de novo mutations associated with ASD (59–61). In addition, PFC M5, which showed significantly enriched for DVMT but not SFARI genes, shared most ASD-related GO terms and pathways with CC M4 and M6, supporting its potential role in the disorder. Moreover, we identified pivot regulators that might perturb gene regulation or affect gene function by constructing multi factor-mediated regulation subnetworks.
Cross-talk between dysfunctional modules revealed that modules can not only affect each other through PPIs but be perturbed directly or indirectly by multi-regulators. Remarkably, we found that there were significantly more interactions between CC-M4, -M6 and PFC-M2. These three modules mainly converged on synaptic and neuronal developmental functions indicated the close correlation between different brain area, which also highlighted the importance of integrative strategy to ASD. Collectively, our system-level analysis of the ASD brain transcriptome demonstrated the existence of common and distinct molecular abnormalities in CC and PFC for the first time, and implicated different distribution of genomic variants burden as underlying mechanisms of neuronal dysfunction for the disorder.
There are also limitations to this study. First, our analysis is biased by the available data. Gene transcriptional profiling in ASD is mainly performed on post-mortem brain samples, but the availability of human ASD brain tissues often represents a significant challenge. Given this fact, we can only use tissues from different individuals as sample sources, which may reduce the comparability to some extent. Second, the SFARI and DVMT gene list, although comprehensive, is likely to have potential curation bias, further confirmations are needed. Third, gene expression studies highlight just one aspect of gene regulation. We have considered some additional regulatory levels (TFs, miRNAs, and lncRNAs), nevertheless, post-translational regulation mechanisms that affect the development of the disease are still under investigation.
Gene expression profile GSE62098 and GSE102741 were downloaded from the NCBI Gene Expression Omnibus (GEO).
XL and YZ contributed to data analysis and wrote the manuscript. LWa, YL, and YH contributed to plot pictures and tables. ZG, CS and XZ contributed to data collection. DW and SL were responsible for the study design and SL revised the manuscript. LWu supervised the whole study.
This study was funded by grants from the National Natural Science Foundation of China (81973068), the China Postdoctoral Science Foundation (2014M561375), and the Scientific Research Fund of Heilongjiang Postdoctoral Program (LBH-Z14153).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by Province Key Laboratory of Children development and genetic research in Harbin Medical University, Heilongjiang, China.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2019.00706/full#supplementary-material
1. Eshraghi RS, Deth RC, Mittal R, Aranke M, Kay SS, Moshiree B, et al. Early disruption of the microbiome leading to decreased antioxidant capacity and epigenetic changes: Implications for the rise in autism. Front Cell Neurosci (2018) 12:256. doi: 10.3389/fncel.2018.00256
2. Jin Z, Yang Y, Liu S, Huang H, Jin X. Prevalence of DSM-5 autism spectrum disorder among school-based children aged 3–12 years in Shanghai, China. J Autism Dev Disord (2018) 48(7):2434–43. doi: 10.1007/s10803-018-3507-z
3. Ellegood J, Anagnostou E, Babineau BA, Crawley JN, Lin L, Genestine M, et al. Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol Psychiatry (2015) 20(1):118–25. doi: 10.1038/mp.2014.98
4. Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci (2008) 31(3):137–45. doi: 10.1016/j.tins.2007.12.005
5. Teffer K, Semendeferi K. Human prefrontal cortex: evolution, development, and pathology. Prog Brain Res (2012) 195:191–218. doi: 10.1016/B978-0-444-53860-4.00009-X
6. Morgan JT, Chana G, Abramson I, Semendeferi K, Courchesne E, Everall IP. Abnormal microglial–neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Res (2012) 1456:72–81. doi: 10.1016/j.brainres.2012.03.036
7. van der Knaap LJ, van der Ham IJ. How does the corpus callosum mediate interhemispheric transfer? A review. Behav Brain Res (2011) 223(1):211–21. doi: 10.1016/j.bbr.2011.04.018
8. Paul LK, Corsello C, Kennedy DP, Adolphs R. Agenesis of the corpus callosum and autism: a comprehensive comparison. Brain (2014) 137(Pt 6):1813–29. doi: 10.1093/brain/awu070
9. Giuliano A, Saviozzi I, Brambilla P, Muratori F, Retico A, Calderoni S. The effect of age, sex and clinical features on the volume of Corpus Callosum in pre-schoolers with Autism Spectrum Disorder: a case-control study. Eur J Neurosci (2018) 47(6):568–78. doi: 10.1111/ejn.13527
10. Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell (2013) 152(6):1237–51. doi: 10.1016/j.cell.2013.02.014
11. Forrest MP, Hill MJ, Kavanagh DH, Tansey KE, Waite AJ, Blake DJ. The Psychiatric Risk Gene Transcription Factor 4 (TCF4) regulates neurodevelopmental pathways associated with schizophrenia, autism, and intellectual disability. Schizophr Bull (2018) 44(5):1100–10. doi: 10.1093/schbul/sbx164
12. Nguyen LS, Fregeac J, Bole-Feysot C, Cagnard N, Iyer A, Anink J, et al. Role of miR-146a in neural stem cell differentiation and neural lineage determination: relevance for neurodevelopmental disorders. Mol Autism (2018) 9:38. doi: 10.1186/s13229-018-0219-3
13. Luo T, Liu P, Wang XY, Li LZ, Zhao LP, Huang J, et al. (2018). Effect of the autism-associated lncRNA Shank2-AS on architecture and growth of neurons. J Cell Biochem 120(2):1754–62. doi: 10.1002/jcb.27471
14. Li J, Shi M, Ma Z, Zhao S, Euskirchen G, Ziskin J, et al. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol Syst Biol (2014) 10:774. doi: 10.15252/msb.20145487
15. Wright C, Shin JH, Rajpurohit A, Deep-Soboslay A, Collado-Torres L, Brandon NJ, et al. Altered expression of histamine signaling genes in autism spectrum disorder. Transl Psychiatry (2017) 7(5):e1126. doi: 10.1038/tp.2017.87
16. Huang Y, Zhao Y, Ren Y, Yi Y, Li X, Gao Z, et al. Identifying genomic variations in monozygotic twins discordant for autism spectrum disorder using whole-genome sequencing. Mol Ther Nucleic Acids (2019) 14:204–11. doi: 10.1016/j.omtn.2018.11.015
17. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res (2013) 41(Database issue):D991–995. doi: 10.1093/nar/gks1193
18. Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, et al. Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol Psychiatry (2006) 60(6):650–8. doi: 10.1016/j.biopsych.2006.06.019
19. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods (2015) 12(4):357–60. doi: 10.1038/nmeth.3317
20. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol (2015) 33(3):290–5. doi: 10.1038/nbt.3122
21. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
22. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics (2008) 9:559. doi: 10.1186/1471-2105-9-559
23. Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res (2017) 45(D1):D362–d368. doi: 10.1093/nar/gkw937
24. Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, Guo AY. (2019). AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res 47(D1):D33–8. doi: 10.1093/nar/gky822
25. Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res (2018) 46(D1):D380–d386. doi: 10.1093/nar/gkx1013
26. Yi Y, Zhao Y, Li C, Zhang L, Huang H, Li Y, et al. RAID v2.0: an updated resource of RNA-associated interactions across organisms. Nucleic Acids Res (2017) 45(D1):D115–d118. doi: 10.1093/nar/gkw1052
27. Ulitsky I, Shamir R. Pathway redundancy and protein essentiality revealed in the Saccharomyces cerevisiae interaction networks. Mol Syst Biol (2007) 3:104. doi: 10.1038/msb4100144
28. Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, et al. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res (2013) 41(Database issue):D996–d1008. doi: 10.1093/nar/gks1042
29. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics (2012) 16(5):284–7. doi: 10.1089/omi.2011.0118
30. Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, et al. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res (2015) 43(W1):W460–466. doi: 10.1093/nar/gkv403
31. Fraser HB, Hirsh AE, Wall DP, Eisen MB. Coevolution of gene expression among interacting proteins. Proc Natl Acad Sci U S A (2004) 101(24):9033–8. doi: 10.1073/pnas.0402591101
32. Jarome TJ, Helmstetter FJ. The ubiquitin–proteasome system as a critical regulator of synaptic plasticity and long-term memory formation. Neurobiol Learn Mem (2013) 105:107–16. doi: 10.1016/j.nlm.2013.03.009
33. Crider A, Pandya CD, Peter D, Ahmed AO, Pillai A. Ubiquitin–proteasome dependent degradation of GABAAalpha1 in autism spectrum disorder. Mol Autism (2014) 5:45. doi: 10.1186/2040-2392-5-45
34. Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM, et al. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol (2012) 99(1):1–14. doi: 10.1016/j.pneurobio.2012.06.001
35. Kim DS, Burt AA, Ranchalis JE, Wilmot B, Smith JD, Patterson KE, et al. Sequencing of sporadic Attention-Deficit Hyperactivity Disorder (ADHD) identifies novel and potentially pathogenic de novo variants and excludes overlap with genes associated with autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet (2017) 174(4):381–9. doi: 10.1002/ajmg.b.32527
36. Philpott AL, Fitzgerald PB, Bailey NW, Churchyard A, Georgiou-Karistianis N, Cummins TD. A GABBR2 gene variant modifies pathophysiology in Huntington’s disease. Neurosci Lett (2016) 620:8–13. doi: 10.1016/j.neulet.2016.03.038
37. Yoo Y, Jung J, Lee YN, Lee Y, Cho H, Na E, et al. GABBR2 mutations determine phenotype in rett syndrome and epileptic encephalopathy. Ann Neurol (2017) 82(3):466–78. doi: 10.1002/ana.25032
38. Crawley JN, Heyer WD, LaSalle JM. Autism and cancer share risk genes, pathways, and drug targets. Trends Genet (2016) 32(3):139–46. doi: 10.1016/j.tig.2016.01.001
39. Zaytseva O, Quinn LM. (2017). Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling. Genes (Basel) 8(4):118. doi: 10.3390/genes8040118
40. Thanseem I, Anitha A, Nakamura K, Suda S, Iwata K, Matsuzaki H, et al. Elevated transcription factor specificity protein 1 in autistic brains alters the expression of autism candidate genes. Biol Psychiatry (2012) 71(5):410–8. doi: 10.1016/j.biopsych.2011.09.020
41. Spinelli L, Black FM, Berg JN, Eickholt BJ, Leslie NR. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet (2015) 52(2):128–34. doi: 10.1136/jmedgenet-2014-102803
42. Rose S, Bennuri SC, Davis JE, Wynne R, Slattery JC, Tippett M, et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl Psychiatry (2018) 8(1):42. doi: 10.1038/s41398-017-0089-z
43. Jang DH, Chae H, Kim M. Autistic and Rett-like features associated with 2q33.3-q34 interstitial deletion. Am J Med Genet A (2015) 167a(9):2213–8. doi: 10.1002/ajmg.a.37119
44. Denechaud PD, Fajas L, Giralt A. E2F1, a novel regulator of metabolism. Front Endocrinol (Lausanne) (2017) 8:311. doi: 10.3389/fendo.2017.00311
45. Tang J, Yu Y, Yang W. Long noncoding RNA and its contribution to autism spectrum disorders. CNS Neurosci Ther (2017) 23(8):645–56. doi: 10.1111/cns.12710
46. Kao HT, Buka SL, Kelsey KT, Gruber DF, Porton B. The correlation between rates of cancer and autism: an exploratory ecological investigation. PLoS One (2010) 5(2):e9372. doi: 10.1371/journal.pone.0009372
47. Fan S, Yang Z, Ke Z, Huang K, Liu N, Fang X, et al. Downregulation of the long non-coding RNA TUG1 is associated with cell proliferation, migration, and invasion in breast cancer. Biomed Pharmacother (2017) 95:1636–43. doi: 10.1016/j.biopha.2017.09.076
48. Li Y, Zhang W, Liu P, Xu Y, Tang L, Chen W, et al. Long non-coding RNA FENDRR inhibits cell proliferation and is associated with good prognosis in breast cancer. Onco Targets Ther (2018) 11:1403–12. doi: 10.2147/OTT.S149511
49. Zhao W, Sun C, Cui Z. A long noncoding RNA UCA1 promotes proliferation and predicts poor prognosis in glioma. Clin Transl Oncol (2017) 19(6):735–41. doi: 10.1007/s12094-016-1597-7
50. Sun Y, Jin JG, Mi WY, Zhang SR, Meng Q, Zhang ST. Long noncoding RNA UCA1 targets miR-122 to promote proliferation, migration, and invasion of glioma Cells. Oncol Res (2018) 26(1):103–10. doi: 10.3727/096504017X14934860122864
51. Fregeac J, Colleaux L, Nguyen LS. The emerging roles of MicroRNAs in autism spectrum disorders. Neurosci Biobehav Rev (2016) 71:729–38. doi: 10.1016/j.neubiorev.2016.10.018
52. Kaphzan H, Hernandez P, Jung JI, Cowansage KK, Deinhardt K, Chao MV, et al. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol Psychiatry (2012) 72(3):182–90. doi: 10.1016/j.biopsych.2012.01.021
53. Kwan V, Unda BK, Singh KK. Wnt signaling networks in autism spectrum disorder and intellectual disability. J Neurodev Disord (2016) 8:45. doi: 10.1186/s11689-016-9176-3
54. Ribolsi M, Daskalakis ZJ, Siracusano A, Koch G. Abnormal asymmetry of brain connectivity in schizophrenia. Front Hum Neurosci (2014) 8:1010. doi: 10.3389/fnhum.2014.01010
55. Zhou Y, Fan L, Qiu C, Jiang T. Prefrontal cortex and the dysconnectivity hypothesis of schizophrenia. Neurosci Bull (2015) 31(2):207–19. doi: 10.1007/s12264-014-1502-8
56. Chaudhry A, Chung BH, Stavropoulos DJ, Araya MP, Ali A, Heon E, et al. Agenesis of the corpus callosum, developmental delay, autism spectrum disorder, facial dysmorphism, and posterior polymorphous corneal dystrophy associated with ZEB1 gene deletion. Am J Med Genet A (2017) 173(9):2467–71. doi: 10.1002/ajmg.a.38321
57. Koehler L, Fournel A, Albertowski K, Roessner V, Gerber J, Hummel C, et al. Impaired odor perception in autism spectrum disorder is associated with decreased activity in olfactory cortex. Chem Senses (2018) 43(8):627–34. doi: 10.1093/chemse/bjy051
58. Luo W, Zhang C, Jiang YH, Brouwer CR. Systematic reconstruction of autism biology from massive genetic mutation profiles. Sci Adv (2018b) 4(4):e1701799. doi: 10.1126/sciadv.1701799
59. O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature (2012) 485(7397):246–50. doi: 10.1038/nature10989
60. Ben-David E, Shifman S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry (2013) 18(10):1054–6. doi: 10.1038/mp.2012.148
Keywords: autism spectrum disorder, corpus callosum, prefrontal cortex, protein interaction network, WGCNA
Citation: Li X, Zhang Y, Wang L, Lin Y, Gao Z, Zhan X, Huang Y, Sun C, Wang D, Liang S and Wu L (2019) Integrated Analysis of Brain Transcriptome Reveals Convergent Molecular Pathways in Autism Spectrum Disorder. Front. Psychiatry 10:706. doi: 10.3389/fpsyt.2019.00706
Received: 23 April 2019; Accepted: 02 September 2019;
Published: 08 October 2019.
Edited by:
Hao Lin, University of Electronic Science and Technology of China, ChinaReviewed by:
Ju Wang, Tianjin Medical University, ChinaCopyright © 2019 Li, Zhang, Wang, Lin, Gao, Zhan, Huang, Sun, Wang, Liang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuang Liang, bGlhbmdzaHVhbmdAZW1zLmhyYm11LmVkdS5jbg==; Lijie Wu, d3VsaWppZWh5ZEAxMjYuY29t
†These two authors contributed equally to the paper
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.