 Audrey Thurm
Audrey Thurm Cristan Farmer
Cristan Farmer Emma Salzman
Emma Salzman Catherine Lord3
Catherine Lord3 Somer Bishop
Somer Bishop- 1Neurodevelopmental and Behavioral Phenotyping Service, Office of the Clinical Director, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, United States
- 2UCSF Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, CA, United States
- 3Semel Institute of Neuroscience and Human Behavior, David Geffen School of Medicine at UCLA, Los Angeles, CA, United States
The topic of this special issue on secondary versus idiopathic autism allows for discussion of how different groups may come to manifest autism spectrum disorder (ASD) or ASD-like symptoms despite important etiological differences. A related issue is that, because many of the social communication deficits that define ASD represent a failure to acquire developmentally expected skills, these same deficits would be expected to occur to some extent in all individuals with intellectual disability (ID). Thus, regardless of etiology, ASD symptoms may appear across groups of individuals with vastly different profiles of underlying deficits and strengths. In this focused review, we consider the impact of ID on the diagnosis of ASD. We discuss behavioral distinctions between ID and ASD, in light of the diagnostic criterion mandating that ASD should not be diagnosed if symptoms are accounted for by ID or general developmental delay. We review the evolution of the autism diagnosis and ASD diagnostic tools to understand how this distinction has been conceptualized previously. We then consider ways that operationalized criteria may be beneficial for making the clinical distinction between ID with and without ASD. Finally, we consider the impact of the blurred diagnostic boundaries between ID and ASD on the study of secondary versus idiopathic ASD. Especially pertinent to this discussion are findings that a diagnosis of ID in the context of an ASD diagnosis may be one of the strongest indicators that an associated condition or specific etiological factor is present (i.e., secondary autism).
Introduction
Whereas the rest of this special issue is devoted to specific factors that relate to secondary versus idiopathic autism spectrum disorder (ASD), in this focused review, we discuss a diagnostic concern that cuts across this discussion: When and how should ASD be diagnosed in the presence of intellectual disability (ID) of varying degrees? This discussion is relevant because of the high rate of ID in what is being termed “secondary” or non-idiopathic ASD, similar to what was previously described as “complex” ASD; see Miles et al. (1), where specific genetic etiologies are identified as contributing to the manifestation of ASD. In fact, genes associated with ASD are often the same genes that are associated with ID (2, 3), validating both the phenotypic and genotypic overlap between these conditions. In addition, the rate of genetic abnormality associated with ASD is significantly higher in the presence of comorbid ID (4, 5). Thus, when it comes to discussions of primary/idiopathic versus secondary autism, considering the role of ID in diagnosis and phenotyping is critical.
In this review, we focus on the impact of ID on the diagnosis of ASD. A few important trends underscore the importance of reviewing the clinical distinction between ID and ASD. One of these trends is the very large increase in ASD prevalence, accompanied by a strikingly similar decline in ID (6–8) (see Figure 1). The other related trend is the increase in research on genetic conditions, many of them previously considered to be disorders associated with ID (e.g., Fragile X Syndrome and Williams Syndrome), where increasingly high rates of ASD features and/or diagnoses are reported (6, 9). These trends lead to questions about how context and measurement may influence changes in diagnostic practice and also suggest inconsistencies in how individual clinicians and researchers distinguish ID from ASD.
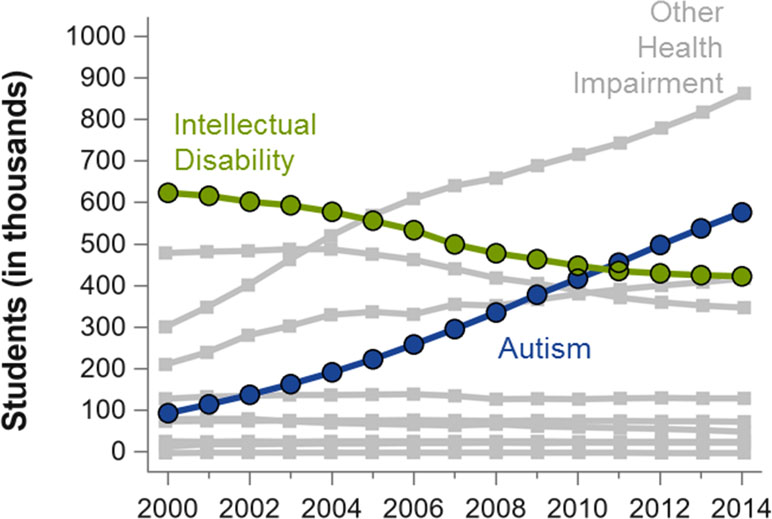
Figure 1 Number of students (in thousands) in the US who receive special education services pursuant to the Individuals with Disabilities Education Act, adapted from a previous publication (6). Numbers are plotted by the beginning of academic year (X-axis) and by diagnostic group, which are mutually exclusive. Other diagnoses not explicitly labeled include deaf-blindness, developmental delay, emotional disturbance, hearing impairment, multiple disabilities, and orthopedic impairment. The most common diagnoses, specific learning disabilities (in 2014, n = 2,278), and speech or language impairment (in 2014, n = 1,332) are not shown. Figure produced from data obtained from the U.S. Department of Education (7).
Before proceeding, it is important to underscore the reasons for understanding how and when a distinction is made between ASD and ID, and when the two conditions are diagnosed simultaneously. As we approach the realization of treatments for the root cause of specific neurodevelopmental problems, such as preventative gene therapy (10), we must identify specific endpoints that will be used to evaluate their efficacy. The considerable etiologic and phenotypic overlap between ASD and ID makes the identification of relevant endpoints that are likely to be changed as a result of treatment (such as social communication abilities or IQ) difficult. More proximal to the everyday lives of individuals with these conditions, similar issues must be considered with respect to behavioral therapy goals and even educational categorization for classroom placements. Finally, from a theoretical perspective, exploration of the differences between diagnoses of ID and ASD begets questions about how to operationalize criteria for the ASD diagnosis itself. That is, the concept of deficits in social communication implies that these are unexpected or significantly more impairing than delays observed in the context of the individual’s other functional abilities. Whereas ID is associated with general deficits across developmental domains, ASD is in fact defined by the observation that social communication deficits are particularly impairing.
Historical Context
Kanner’s first description of children with autistic disorder included delay in intellectual development as an associated feature in several of the cases described (11). Moving forward to formal recognition of autism as a disorder in the Diagnostic and Statistical Manual of Mental Disorders, Third Edition (DSM-III), criterion C specified gross deficits in language, and criterion D stipulated peculiar patterns of speech, if present (12). Thus, early descriptions of ASD assumed that a significant proportion of children would be minimally verbal, which overlaps considerably with ID (13). In fact, earlier epidemiological reports indicated that as many as 70% of individuals with ASD had co-occurring ID (14, 15). It was only recently that epidemiological reports reversed this trend, with some suggesting that ID was present in as few as 30% of children with ASD (6, 16). However, methdology may artificially deflate these rates (17). Recent studies have found that both minimally verbal and ID subsets of the ASD population are disporpotionately and increasingly underincluded in ASD treatment studies (18), neuroimaging studies (19), and ASD research in general (20).
The systematic exclusion of individuals with ID in ASD-focused research may affect the validity of current estimates for rates of ID in ASD (20). Further, it raises concerns about how clinicians are trained to diagnose ASD in the context of ID. Studies up to the early 2000s often focused on how to differentiate ASD from ID (without ASD), as this was a predominant referral question (21–24). However, recent literature on ASD screening and diagnosis has shifted to reflect the changing referral trends, focusing more on differential diagnosis among children with average IQ (e.g., differentiating between ASD and ADHD or ASD and language disorder) (25, 26). Studies that have continued to explore differentiation of ASD and ID are primarily focused on genetic syndromes (27–29), since ASD geneticists are looking to specific genetic conditions with increased rates of ASD for clues about how to derive treatments for ASD symptoms (30). In addition, ASD-focused research within rare genetic syndromes has been fueled to some extent by specialized funding resources targeting ASD.
Diagnostic and Statistical Manual of Mental Disorders (DSM)
While diagnoses akin to ID were in earlier versions, beginning with DSM-III (12), the American Psychiatric Association introduced a multiaxial conceptualization. This system was maintained in DSM-IV, which placed mental retardation (the term previously used for ID) on Axis II. ID therefore served as a sort of “add on” that could be assigned in the presence of many other Axis I diagnoses, including ASD. This had an advantage of encouraging clinicians to consider each axis, and provided a clear structure for reporting different, though potentially associated, difficulties. However, this structure effectively invalidated a conceptualization of ID as a primary or autonomous mental disorder. While other organizations already had nosology for ID as a distinct condition (31, 32), the DSM-5 rid the multiaxial system so ID could be recognized as a primary diagnosis (33).
ID is now included as a freestanding diagnosis within the Neurodevelopmental Disorders section of the DSM-5. It may therefore be diagnosed alongside any other neurodevelopmental disorder (although cautions are given relating to diagnostic distinctions for several conditions, including ASD). The DSM-5 defines ID based on deficits in intellectual functioning and deficits in adaptive functioning. The option to assign ID as its own diagnosis, not in the presence of ASD or any other neurodevelopmental disorder, requires that clinicians have sufficient understanding of the clinical manifestation of ID. Given the shift in composition of ASD referrals to include a significant proportion of individuals with borderline range, average or above-average IQ, clinicians working with the “contemporary” ASD population may be far less experienced with ID (especially in the severe to profound range) than previous generations. It is worth noting that “With or Without Intellectual Impairment” is included as a specifier to be considered when assigning an ASD diagnosis under DSM-5; however, while this specifier can be made independently of an ID diagnosis, this specifier is redundant when an independent diagnosis of ID is made.
Specifiers for ID severity differentiate individuals based on ability to function independently across conceptual, social, and practical domains. The conceptualization of intellectual functioning as a generally normal distribution suggests that the majority of people with ID should be in the mild range, a supposition borne out by the data (34). Individuals with mild ID typically achieve verbal fluency and are often able to function with independence in at least some domains, whereas those with profound ID (representing a very small percentage of those diagnosed with ID) require 24-h care for all activities of daily living (35). Of particular relevance to the diagnosis of ASD in ID is the fact that social function is included in the conceptualization of ID severity, as it is expected that degrees of ID will be associated with increasing immaturity in social interactions. As described in the DSM-5, verbal or nonverbal modes of social communication may be observed, but the content will be limited. Expectations for social abilities decrease as severity increases; in or beyond the moderate range of ID, “individuals may not perceive or interpret social cues accurately” (p. 35) (33). Thus, as described below, clinicians are presented with the difficult task of determining when observed social deficits are attributable to an individual’s ID, and when an additional diagnosis of ASD is warranted.
At all levels of ID, the social communication deficits and interfering repetitive behaviors/restricted interests that confer a comorbid diagnosis of ASD result in even greater reductions in functional independence than if the individual only had a diagnosis of ID. This is reflected in studies showing that adaptive behavior skills are lower than expected based on IQ for individuals with ASD (36–39). Furthermore, data from clinical populations suggest that the distribution of ID severity among people with ASD is skewed downward, such that the rate of severe-to-profound ID is higher among those with ASD and ID than in those with ID alone (40). This is consistent with the detrimental effects of ASD on both attainment and measurement of cognitive and adaptive abilities (41, 42). With respect to the severity of ID, the fact that individuals with both ASD and ID are particularly likely to have a specific genetic etiology, and that severe to profound ID is more common in such cases of rare genetic syndromes than in the general ID population (43), results in a theoretical and non-hereditary “bump” in the normal distribution at the very low end of IQ (44).
Diagnosing ASD in the Context of ID
Diagnosing mental conditions in individuals with ID presents challenges in general. For this reason, a separate diagnostic manual has been created to provide guidance about when DSM-5 criteria should be modified for the ID population (45). The Diagnostic Manual-Intellectual Disability-2 highlights complexities in distinguishing ASD in individuals with ID, but does not indicate any adaptations for the ASD diagnostic criteria beyond utilizing the DSM-5 requirement that deficits “exceed impairment consistent with the level of intellectual disability” (p. 131) (45). Similarly, Criterion E of the DSM-5 ASD criteria requires that “disturbances are not better explained by intellectual disability or global developmental delay” (p. 51) (33), though it does not specify how this should be determined. Thus, neither manual gives instructions about how or when ID may or may not “explain” symptoms of ASD. In addition, as discussed later, the ability to make such determinations may depend on the severity of ID and the age of the individual, as certain distinctions may be increasingly challenging at more severe levels of ID and/or at certain ages/developmental periods.
Research on specific genetic conditions associated with ASD has highlighted some of these diagnostic challenges, because ID is much more common when a genetic etiology is identified than in the idiopathic ASD population (46). There are examples from several genetic syndromes, including Fragile X Syndrome (47, 48), Dup15q Syndrome; (49), and Smith–Lemli–Opitz Syndrome (50), wherein lower IQ is associated with increased rates of ASD diagnosis. An exception may be when the ID is so severe that clinicians apparently find it impossible to identify relative deficits in social communication and play, given the extremely low mental age (51).
Variability in rates of ASD diagnosis within and across genetic syndromes may also depend on the clinician’s degree of reliance on standardized instruments versus clinical judgment (9). Moss and Howlin (28) provide a comprehensive review of the literature with respect to diagnosing ASD in individuals with genetic syndromes, urging even more caution when attempting to differentiate clinically relevant symptoms of ASD from ID. Results of studies that rely exclusively on scores or cutoffs from standardized measures without following manual-based instructions to carefully consider expert clinical judgment are likely to be especially misleading (52, 53). It is essential that clinicians use all available information about the individual to decide whether social communication skills that are below chronological age expectations are also lower than mental age/developmental expectations. This is challenging, especially when there are discrepancies in various aspects of mental age (e.g., verbal, nonverbal), when the individual’s mental age is lower than the minimum mental age assumed for the measure itself (27, 54), or when the clinical picture is complicated by comorbid sensory or other medical concerns. Therefore, depending on the study methodology and the clinical training and experience of the diagnosticians, rates of ASD diagnosis can vary tremendously between samples of individuals with the same syndrome.
ASD Diagnostic Assessment of Individuals With ID
When considering if and how to make a diagnosis of ASD in an individual with ID, it is imperative that severity of ID be considered carefully. The mental age associated with ID in the severe to profound range of ID may not exceed 18 months, an age before which it may not be possible to assess certain abilities (e.g., spoken language, which is not expected to develop until 12 to 18 months of age). Thus, while there is no agreed-upon mental age that automatically triggers the exception described in the DSM-5 criterion E that “disturbances are not better explained by intellectual disability or global developmental delay” (33), diagnosticians need to keep in mind the possibility that severe ID might prevent valid assessment of certain ASD symptoms.
One convincing reason for not advising a minimum mental age requirement for the diagnosis of ASD is the need to diagnose children as young as possible. ASD is diagnosed as early as the second year of life (55), so requiring that toddlers meet a mental age threshold of 18 or 24 months (for example) could preclude an ASD diagnosis in very young toddlers and/or those with even slight cognitive delays (56). In addition, the symptoms of ASD may themselves deflate cognitive scores, creating circularity where young children with ASD cannot be diagnosed with ASD because their ASD has caused their test scores to be artificially low. The limited social responsiveness, interfering repetitive behaviors, and other behavior problems (e.g., hyperactivity, aggression, and inattention) common in children with ASD complicate cognitive testing in this group (57), potentially leading to lower scores (58). On the other hand, given longitudinal data showing that early (i.e., at or before age 3 years) nonverbal IQ scores below 70 usually persist into adulthood (41), clinicians need to be careful not to dismiss the prognostic significance of low IQ scores in young children with ASD.
Although diagnostic criteria do not explicitly state a developmental level under which a diagnosis of ASD is not advisable, ASD-focused research studies have begun to introduce specific thresholds for inclusion. The Simons Simplex Collection (SSC) required a mental age of 18 months for probands with ASD (59), and a large case–control cohort study of young children excluded children with mental ages under 24 months from analyses of the data (60). These decisions were based on research findings that widely used diagnostic instruments for ASD (i.e., the Autism Diagnostic Interview-Revised and the Autism Diagnostic Observation Schedule) are far less specific when used for individuals with nonverbal mental ages below 15 months (21), as well as recommendations from the test developers that the instruments be used cautiously (or not at all) with children with very low mental ages (61, 62). Such limitations in age and developmental level extend to other commonly used measures of ASD symptoms. The manual for the Social Responsiveness Scale states that all validation data were obtained from individuals with IQs above 70; therefore, “additional clinical judgment” is needed to interpret scores for individuals with ID (63). Both a thorough review of autism screening and diagnostic instruments (64) and a recent review of diagnostic instruments for preschoolers (65) suggested caution regarding how IQ can affect performance of these measures in general, with several examples of lower specificity in ID groups. For example, although limited sensitivity and specificity data for diagnostic measures such as the Diagnostic Interview for Social and Communication Disorders (DISCO) (66) and the Developmental, Dimensional and Diagnostic Interview (67) exist, two studies of the DISCO showed over-classification of individuals with low IQ (66, 68).
A common approach in research has been to proceed with an abundance of caution by restricting the participation of individuals with ID, although this reduces the generalizability of results. However, research on genetic syndromes cannot realistically employ mental age cutoffs for inclusion. They can employ minimum mental age cutoffs for individuals they deem capable of completing ASD diagnostic instruments, and apply even more stringent thresholds regarding whether scores from diagnostic instruments should be considered valid (28), but these practices have not been consistently adopted up to this point.
In addition to the complicating factor of cognitive impairments, individuals with ID associated with genetic conditions are likely to have physical disabilities. Sensory impairments affecting vision and hearing are among the most frequently identified, along with other neurologic conditions that include ataxia and epilepsy (69). The implications of this are twofold. First, these comorbidities confer further limitations to the standardization (and therefore validity) of ASD diagnostic instruments and are themselves often difficult to differentiate from symptoms of ASD. Second, individuals with such sensory impairments are at increased risk of ASD (70, 71). Interestingly, the DSM-5 states that it may be difficult to specify the severity of ID in individuals with sensory impairments due to limitations in administering and interpreting standardized measures in this population (33), but there is no consideration of sensory impairments in the differential diagnosis section pertaining to ASD criteria. As a result, there have been recent efforts to develop specific autism diagnostic measures that can be validated in samples of individuals with ID and specified sensory impairments (72).
Assessment of Individuals With Mild to Moderate ID
Focusing on those individuals with ID who are in the mental age range appropriate for standardized ASD diagnostic instruments, one way of conceptualizing the diagnostic features of ID + ASD (versus ID alone) would be to separate basic social communication skills, which are relatively ASD-specific, from the more advanced social communication skills that may be difficult for individuals with a range of neurodevelopmental disorders, including ID (73). Analyses of instruments like the ADI-R and ADOS indicate that nonverbal communication behaviors that typically emerge in early development, such as eye contact, facial expressions, gestures, as well as the capacity to share enjoyment and participate in simple, back and forth games (e.g., peek-a-boo), are most specific to ASD (74, 75). This is consistent with older literature showing that, compared to children with ASD, children with ID show greater capacity for joint attention behaviors, showing and directing attention, range of directed affect, as well as use of socially appropriate eye gaze to social communication purposes (76–78).
Although these deficits in basic social communication behaviors tend to be relatively more specific to ASD, their diagnostic utility may decrease over time, as individuals with and without ASD acquire more skills. Consequently, as individuals progress in age and developmental level, and achieve higher levels of language, ASD instruments will necessarily focus more on higher-level social communication skills (e.g., conversation, ability to modify behavior to match social expectations). This complicates assessment of social communication in individuals with ID with fluent language abilities; for many individuals with mild to moderate ID who do not have ASD, their language skills are at least commensurate with nonverbal ability, and it would not be uncommon for them to be capable of completing Module 3 or 4 of the ADOS, for example (79). However, ASD-focused assessment tools for individuals with fluent language were developed primarily with non-ID populations (61, 62, 80, 81), reflecting the fact that most individuals with ASD who are capable of producing complex sentences also have average or above average nonverbal IQ (82). This also highlights the problem of identifying appropriate comparison groups for ASD research, because the degree of language deficit (or nonverbal-verbal IQ discrepancy) that occurs in some individuals with ASD does not occur as commonly outside of ASD (83). Therefore, in addition to expanding standardization of tools such as the ADOS to capture individuals at older ages who are not able to verbally respond to modules traditionally designed for an older population (84), it will also be important to further develop our understanding of individuals with ID without ASD. Specifically, assessing large groups of individuals with ID without ASD will be critical to develop tools that can detect subtle differences in higher-level social communication skills even if the groups differ significantly in language and/or nonverbal cognitive skills.
Severe to Profound ID
The data required to inform the differentiation of ASD from ID, especially in the severe to profound range, are limited. For individuals with severe or profound ID, the term profound intellectual and multiple disabilities (PIMD) is often used, to reflect the accompanying physical disabilities and/or sensory deficits (e.g., vision and/or hearing impairments) (85). Individuals with PIMD are the least common generally, but this group is overrepresented in many genetic conditions implicated in the etiology of ASD (46). Despite the interest in measuring ASD symptoms in this group, the fact remains that standardized measures of ASD symptoms are generally not appropriate for this purpose (21, 61, 62). ASD diagnostic measures were designed to identify individuals with ASD primarily, and therefore they assume profiles of ability that are more traditionally characteristic of ASD. For example, motor skills tend to be relatively preserved in ASD even among those with very low IQ and language abilities (86, 87), as are other basic sensory abilities such as vision and hearing. As a result, standardized diagnostic instruments like the ADI-R and ADOS were not validated for individuals with interfering vision, hearing, or mobility limitations or other significant motor impairments (e.g., ataxia, dystonia), which are common co-occurring problems for many individuals with PIMD, especially those with genetic syndromes and/or neurological conditions (e.g., epilepsy).
Restrictions placed on use of standardized diagnostic tools have had downstream effects on our ability to conceptualize ASD in the context of ID. As the use of these tools in research on ASD and neurodevelopmental disorders becomes more widespread, results are fed back to inform clinical and policy decisions, shaping diagnostic systems like DSM-5. Thus, an unintended consequence of the fact that these tools were not designed or validated for individuals with PIMD is that people with PIMD may not be included in ASD research (other than studies that focus on specific genetic conditions), and empirical data about which behaviors best differentiate ASD in the context of severe to profound ID are not available. The need to operationalize specific differences between ID with and without ASD is clearly apparent, but the data to do this are limited in part by who was able to undergo valid standardized assessment of social communication and repetitive behaviors using currently available tools. Fortunately, there have been recent attempts to actually create diagnostic tools that are specifically tailored and standardized in samples of individuals with ID that include severe-to-profound ID (88), as well as modifications of the ADOS that are more appropriate for minimally verbal and older individuals with a nonverbal mental age of at least 18 months (84).
Application of Criterion E: Clinical Guidelines
Best practice clearly encourages clinical judgment over prescriptive algorithms for the differentiation of ASD and ID at various chronological and mental age levels. Nevertheless, the following guidelines may be helpful to consider:
a) When evaluating an individual with ID for a potential diagnosis of ASD, it is necessary to be aware of the child’s cognitive ability (based on IQ or developmental test scores) and to understand any sources or clinical manifestations other than cognitive ability that may have influenced those scores. Thus, the diagnosing clinician should either directly administer the measure of cognitive functioning or obtain a detailed account of the behaviors that contributed to the IQ scores. Along with measurements of adaptive behavior, this information will provide context not only for the individual’s developmental level, but also for the behavioral, motor, and/or sensory impairments that may be contributing to the overall presentation.
b) An assessment of potential motor and sensory impairments (e.g., vision, hearing) should be incorporated into the differential diagnosis, especially given the increased prevalence of these impairments in individuals with a neurogenetic condition and/or severe to profound ID. If sensory or motor impairments are present, it is important to consider tools that are validated in these particular populations, and also to consider using a team approach to diagnosis that includes experts in the relevant sensory and/or motor deficits.
c) Given that Criterion E states “to make comorbid diagnoses of ASD and ID, social communication should be below that expected for general developmental level,” the diagnostician must evaluate his/her ability to determine whether observed deficits are consistent with expectations for the individual’s developmental level.
i. The clinician must consider the behaviors expected at a given developmental level. However, the chronological age of the child is also relevant, as the effect of life experiences on behaviors cannot be ignored. For example, expectations for an 18-month mental age differ in the context of a 4-year-old versus a teenager. As a result of more life experiences, the teenager may be able to sit for longer periods or time; have more expertise with certain devices, objects, or toys; and be more compliant with routines and simple daily living tasks.
ii. In the case of young children, the DSM-5 warns that distinguishing ID from ASD may be particularly difficult. At very young ages, it may be difficult, or even impossible, to determine social communication “delay” from “deviance,” since uneven developmental profiles are more subtle when children are too young to have developed many skills.
iii. Related to the above, it is important to distinguish between when a symptom is present, but criterion E is met (the disturbance is accounted for by global developmental delay or ID so it should be considered “not applicable”), including when it is not possible to determine whether a symptom is present. In other words, each symptom should be considered “present,” “absent,” or “not applicable.” For instance, if motor impairments such as ataxia or motor apraxia interfere with a child’s ability to make purposeful movements such as pointing or other gestures, a rating of “not applicable” may accurately capture the criterion of deficits in specific nonverbal communication behaviors. Similarly, as discussed previously with respect to minimum mental age requirements, it may not be possible to judge presence/absence of a symptom if an individual has not achieved certain developmental skills. In such cases, a child may be observed to “grow into” his/her ASD symptoms in later childhood, once he/she acquires the cognitive, language, and/or motor skills required to assess social communication and play. A simple example of this would include a child who lacks necessary motor or cognitive skills to play with toys in any capacity, but then begins exhibiting highly repetitive play once these skills develop.
d) In the case of older individuals, the developmental trajectory may be particularly useful. Specifically, the clinician should consider whether social communication delays have always been approximately commensurate with other domains of development, or whether social communication delays have at times been an isolated or more significant impairment. It is also important for clinicians to consider that ASD diagnostic status may change. For example, as noted above, it may not be possible to judge the presence of ASD in a child with very significant ID until he/she attains a certain mental age equivalent. On the other hand, in cases of mild to moderate ID, clinicians must be careful when assigning ASD diagnoses in later childhood, adolescence, or adulthood. It is unlikely for any individual (with or without ID) to suddenly manifest social communication deficits and repetitive behaviors beyond the early childhood period that warrant an ASD diagnosis. In cases when ASD is suddenly being considered in an older individual with ID, the clinician must carefully consider whether observed social communication difficulties (e.g., difficulty related to same-aged peers, minor conversational difficulties, mild social disinhibition) are simply a reflection of social immaturity attributable to their ID or potentially changes in mental state in reaction to life transitions (e.g., more limited social exposure post high school).
e) While the administration of autism screening and diagnostic tools may be helpful in a variety of situations, clinicians must first consider the consequences of scoring and interpreting the results. For both research and clinical purposes, interpretations of the data may be significantly limited by characteristics commonly observed in individuals with ID, which may affect scoring on autism symptom tools. Examples are ataxia, recent onset of independent walking, significant dysarthria or motor apraxia, and visual or hearing impairments. Thus, even if the clinician intends to rely upon qualitative behavioral observations, the use of a given tool in any capacity may still be problematic if the tool is not appropriate for an individual.
f) When applying clinical judgment, biases and other motivations must be considered. The use of a multidisciplinary team may be most helpful to allow for multiple observers and perspectives. For clinicians whose primary training is in ASD, it may be necessary to seek substantial consultation when an individual presents with certain medical problems, neurological issues, motor impairments, etc. that are less common in individuals with ASD without ID. Clinicians must recognize that standardized tools perform differently in different populations and that their own biases and/or idiosyncrasies in how they normally “weight” certain scores or observations will not apply in the same way to all clinical groups. Further, clinicians must have sufficient understanding of the individual’s social history, including factors relating to family, socio-cultural, and service-related needs that may be important in teasing out caregiver’s responses to questions about the individual’s ability to respond to different social situations.
Research Implications
There are important research implications for the preceding clinical discussion. The differentiation between ID and ASD is perhaps especially important in the research setting, where studies of genetic conditions associated with ID and ASD are proliferating (89). In order to decide between ASD features versus ID as a primary outcome of a trial, for example, the research team must be able to adequately assess both and understand which is more prominent. Further, this discussion highlights the importance of systematic inclusion of individuals with all levels of intellectual ability in the development and validation of instruments, diagnostic or otherwise, for measuring ASD symptoms. As mentioned earlier, some studies may choose to use measures even if they are not validated for ID, but for research purposes only. Even if this tiered approach is employed, some members of multidisciplinary teams may not be privy to the important issues that they must consider when interpreting scores from instruments not validated in individuals with ID. Thus, it is essential that a knowledgeable member of the study team is involved in any analysis or report that includes such phenotypic findings. Finally, the difficulty of differentiating ASD and ID highlights the need for further research on the operationalization of DSM-5 ASD criteria (and other classification systems) in the context of moderate-to-profound ID.
Conclusion
As diagnostic tools for ASD are distributed and used more widely, and as awareness (and prevalence) of ASD continues to rise, researchers and clinicians are increasingly faced with extremely complex referrals for ASD diagnostic assessment. Indeed, the complicating presence of ID has been recognized since diagnosis of autism was first proposed. Further, ID is as heterogeneous as ASD, so strategies that may be helpful in distinguishing ASD from ID in individuals of a certain age and/or level of severity may not be applicable to others. Nevertheless, the specific considerations outlined above may be helpful to enhance both research and practice when evaluating and treating children with neurodevelopmental disorders.
Although the focus of this work was to help clarify the distinction between ID and ID + ASD, we must also emphasize the commonalities of the two conditions, which indicate that many interventions may be beneficial regardless of which neurodevelopmental disorder is diagnosed. Early intervention services (90) and services focused on improving communication (91) are indicated for children with any significant developmental delay and should be initiated at the time this concern is identified, rather than waiting for a diagnosis of ASD or ID to be made (92). Although, depending on locality, service provisions may differ considerably between individuals diagnosed with ASD versus ID, the perception is that individuals with ASD are generally afforded more comprehensive services (93, 94). This has motivated many parents to seek out an additional ASD diagnosis in order to secure additional services and has further muddled the question of when and for whom a comorbid ASD diagnosis is appropriate. To advance our understanding of the diagnostic distinctions between ID with and without ASD, there is an urgent need for changes in service provision that emphasize individual needs over diagnostic classification.
Data Availability
The datasets for this manuscript are not publicly available because consents allow for limited data sharing. Requests to access the datasets should be directed to Somer Bishop,
Author Contributions
AT and SB initially drafted this manuscript, and CL, CF, and ES provided extensive editing and specific contributions.
Funding
This work was partially supported by the Intramural Research Program of the NIMH (1ZICMH002961) to AT and CF, as well as R01HD093012 to SB.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Statement
CL and SB receive royalties from Western Psychological Services for sales of ASD diagnostic instruments [CL for the Autism Diagnostic Interview-Revised, the Autism Diagnostic Observation Schedule (ADOS), and the ADOS-2; SB for the ADOS-2].
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank all of the families who have participated in research that contributed to this work.
References
1. Miles J, Takahashi T, Bagby S, Sahota P, Vaslow D, Wang C, et al. Essential versus complex autism: definition of fundamental prognostic subtypes. Am J Med Genet A (2005) 135(2):171–80. doi: 10.1002/ajmg.a.30590
2. Casanova EL, Sharp JL, Chakraborty H, Sumi NS, Casanova MF. Genes with high penetrance for syndromic and non-syndromic autism typically function within the nucleus and regulate gene expression. Mol Autism (2016) 7(1):18. doi: 10.1186/s13229-016-0082-z
3. Zhu X, Need AC, Petrovski S, Goldstein DB. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci (2014) 17(6):773–81. doi: 10.1038/nn.3713
4. Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 Risk Loci. Neuron (2015) 87(6):1215–33. doi: 10.1016/j.neuron.2015.09.016
5. Robinson EB, Samocha KE, Kosmicki JA, McGrath L, Neale BM, Perlis RH, et al. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc Nat Acad Sci U S A (2014) 111(42):15161–5. doi: 10.1073/pnas.1409204111
6. Polyak A, Kubina RM, Girirajan S. Comorbidity of intellectual disability confounds ascertainment of autism: implications for genetic diagnosis. Am J Med Genet B Neuropsychiatr Genet (2015) 168(7):600–8. doi: 10.1002/ajmg.b.32338
7. U.S. Department of Education. National Center for Education Statistics. Digest of Education Statistics, 2016 (NCES 2017-094). Chapter 2 2018; Retrieved 25 March 2019 from https://nces.ed.gov/fastfacts/display.asp?id=64.
8. King M, Bearman P. Diagnostic change and the increased prevalence of autism. Int J Epidemiol (2009) 38(5):1224–34. doi: 10.1093/ije/dyp261
9. Richards C, Jones C, Groves L, Moss J, Oliver C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry (2015) 2(10):909–16. doi: 10.1016/S2215-0366(15)00376-4
10. Benger M, Kinali M, Mazarakis ND. Autism spectrum disorder: prospects for treatment using gene therapy. Mol Autism (2018) 9:39. doi: 10.1186/s13229-018-0222-8
12. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 3rd ed. Washington, DC: American Psychiatric Publishing (1980).
13. Bal VH, Katz T, Bishop SL, Krasileva K. Understanding definitions of minimally verbal across instruments: evidence for subgroups within minimally verbal children and adolescents with autism spectrum disorder. J Child Psychol Psychiatry (2016) 57(12):1424–33. doi: 10.1111/jcpp.12609
14. Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry (2005) 66 Suppl 10:3–8.
15. Ritvo ER, Freeman BJ, Pingree C, Mason-Brothers A, Jorde L, Jenson WR, et al. The UCLA-University of Utah epidemiologic survey of autism: prevalence. Am J Psychiatry (1989) 146(2):194–9. doi: 10.1176/ajp.146.2.194
16. Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 Sites, United States, 2014. Morbidity and mortality weekly report Surveillance summaries (Washington, DC: 2002). 2018;67(6):1–23. doi: 10.15585/mmwr.ss6706a1
17. Dykens E, Lense M. Intellectual disabilities and autism spectrum disorder: A cautionary note. In: Amaral GDDHG DG, editor. Autism spectrum disorders. Oxford University Press (2011). p. 261–9. doi: 10.1093/med/9780195371826.003.0018
18. Stedman A, Taylor B, Erard M, Peura C, Siegel M. Are children severely affected by autism spectrum disorder underrepresented in treatment studies? An analysis of the literature. J Autism Dev Disord (2019) 49(4):1378–90. doi: 10.1007/s10803-018-3844-y
19. Jack A, Pelphrey K. Annual Research Review: understudied populations within the autism spectrum—current trends and future directions in neuroimaging research. J Child Psychol Psychiatry (2017) 58(4):411–35.
20. Russell G, Mandy W, Elliott D, White R, Pittwood T, Ford T. Selection bias on intellectual ability in autism research: a cross-sectional review and meta-analysis. Mol Autism (2019) 10:9. doi: 10.1186/s13229-019-0260-x
21. Risi S, Lord C, Gotham K, Corsello C, Chrysler C, Szatmari P, et al. Combining information from multiple sources in the diagnosis of autism spectrum disorders. J Am Acad Child Adolesc Psychiatry (2006) 45(9):1094–103. doi: 10.1097/01.chi.0000227880.42780.0e
22. Steinhausen HC, Metzke CW. Differentiating the behavioural profile in autism and mental retardation and testing of a screener. Eur Child Adolesc Psychiatry (2004) 13(4):214–20. doi: 10.1007/s00787-004-0400-4
23. Osterling JA, Dawson G, Munson JA. Early recognition of 1-year-old infants with autism spectrum disorder versus mental retardation. Dev Psychopathol (2002) 14(2):239–51. doi: 10.1017/S0954579402002031
24. Ventola P, Kleinman J, Pandey J, Wilson L, Esser E, Boorstein H, et al. Differentiating between autism spectrum disorders and other developmental disabilities in children who failed a screening instrument for ASD. J Autism Dev Disord (2007) 37(3):425–36. doi: 10.1007/s10803-006-0177-z
25. Craig F, Lamanna AL, Margari F, Matera E, Simone M, Margari L. Overlap between autism spectrum disorders and attention deficit hyperactivity disorder: searching for distinctive/common clinical features. Autism Res (2015) 8(3):328–37. doi: 10.1002/aur.1449
26. Reisinger LM, Cornish KM, Fombonne E. Diagnostic differentiation of autism spectrum disorders and pragmatic language impairment. J Autism Dev Disord (2011) 41(12):1694–704. doi: 10.1007/s10803-011-1196-y
27. Hepburn SL, Moody EJ. Diagnosing autism in individuals with known genetic syndromes: clinical considerations and implications for intervention. Int Rev Res Dev Dis (2011) 40:229–59. doi: 10.1016/B978-0-12-374478-4.00009-5
28. Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res (2009) 53(10):852–73. doi: 10.1111/j.1365-2788.2009.01197.x
29. Soorya L, Leon J, Trelles MP, Thurm A. Framework for assessing individuals with rare genetic disorders associated with profound intellectual and multiple disabilities (PIMD): the example of Phelan McDermid syndrome. Clin Neuropsychol (2018) 32(7):1226–55. doi: 10.1080/13854046.2017.1413211
30. Simons VIP Consortium. Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron (2012) 73(6):1063–7. doi: 10.1016/j.neuron.2012.02.014
31. MacMillan DL, Gresham FM, Siperstein GN. Conceptual and psychometric concerns about the 1992 AAMR definition of mental retardation. Am J Ment Retard (1993) 98(3):325–35.
32. Werry JS. ICD 9 & DSM III classification for the clinician. J Child Psychol Psychiatry (1985) 26(1):1–6. doi: 10.1111/j.1469-7610.1985.tb01624.x
33. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington, VA: American Psychiatric Publishing (2013). doi: 10.1176/appi.books.9780890425596
34. Bourke J, de Klerk N, Smith T, Leonard H. Population-based prevalence of intellectual disability and autism spectrum disorders in Western Australia: a comparison with previous estimates. Medicine (2016) 95(21):e3737. doi: 10.1097/MD.0000000000003737
35. Tasse MJ, Balboni G, Navas P, Luckasson R, Nygren MA, Belacchi C, et al. Developing behavioural indicators for intellectual functioning and adaptive behaviour for ICD-11 disorders of intellectual development. J Intellect Disabil Res (2019) 63(5):386–407. doi: 10.1111/jir.12582
36. Ashwood KL, Tye C, Azadi B, Cartwright S, Asherson P, Bolton P. Brief report: adaptive functioning in children with ASD, ADHD and ASD + ADHD. J Autism Dev Disord (2015) 45(7):2235–42. doi: 10.1007/s10803-014-2352-y
37. Pugliese CE, Anthony L, Strang JF, Dudley K, Wallace GL, Kenworthy L. Increasing adaptive behavior skill deficits from childhood to adolescence in autism spectrum disorder: role of executive function. J Autism Dev Disord (2015) 45(6):1579–87. doi: 10.1007/s10803-014-2309-1
38. Kanne SM, Gerber AJ, Quirmbach LM, Sparrow SS, Cicchetti DV, Saulnier CA. The role of adaptive behavior in autism spectrum disorders: implications for functional outcome. J Autism Dev Disord (2011) 41(8):1007–18. doi: 10.1007/s10803-010-1126-4
39. Duncan AW, Bishop SL. Understanding the gap between cognitive abilities and daily living skills in adolescents with autism spectrum disorders with average intelligence. Autism (2015) 19(1):64–72. doi: 10.1177/1362361313510068
40. Fombonne E. Epidemiological trends in rates of autism. Mol Psychiatry (2002) 7 Suppl 2:S4–6. doi: 10.1038/sj.mp.4001162
41. Bishop SL, Farmer C, Thurm A. Measurement of nonverbal IQ in autism spectrum disorder: scores in young adulthood compared to early childhood. J Autism Dev Disord (2015) 45(4):966–74. doi: 10.1007/s10803-014-2250-3
42. Bal VH, Kim SH, Cheong D, Lord C. Daily living skills in individuals with autism spectrum disorder from 2 to 21 years of age. Autism (2015) 19(7):774–84. doi: 10.1177/1362361315575840
43. Kurki MI, Saarentaus E, Pietilainen O, Gormley P, Lal D, Kerminen S, et al. Contribution of rare and common variants to intellectual disability in a sub-isolate of Northern Finland. Nat Commun (2019) 10(1):410. doi: 10.1101/332023
44. Reichenberg A, Cederlöf M, McMillan A, Trzaskowski M, Kapra O, Fruchter E, et al. Discontinuity in the genetic and environmental causes of the intellectual disability spectrum. Proc Natl Acad Sci U S A (2016) 113(4):1098–103. doi: 10.1073/pnas.1508093112
45. Fletcher R, Barnhill J, Cooper A. DM-ID-2: diagnostic manual, intellectual disability: a textbook of diagnosis of mental disorders in persons with intellectual disability. Kingston, NY: NADD (2017).
46. Rosti RO, Sadek AA, Vaux KK, Gleeson JG. The genetic landscape of autism spectrum disorders. Dev Med Child Neurol (2014) 56(1):12–8. doi: 10.1111/dmcn.12278
47. McDuffie A, Thurman A, Hagerman R, Abbeduto L. Symptoms of autism in males with fragile X syndrome: a comparison to nonsyndromic ASD using current ADI-R scores. J Autism Dev Disord (2015) 45(7):1925–37. doi: 10.1007/s10803-013-2013-6
48. McDuffie A, Abbeduto L, Lewis P, Kover S, Kim J, Weber A, et al. Autism spectrum disorder in children and adolescents with fragile X syndrome: within-syndrome differences and age-related changes. Am J Intellect Dev Disabil (2010) 115(4):307–26. doi: 10.1352/1944-7558-115.4.307
49. DiStefano C, Gulsrud A, Huberty S, Kasari C, Cook E, Reiter LT, et al. Identification of a distinct developmental and behavioral profile in children with Dup15q syndrome. J Neurodev Disord (2016) 8(1):19. doi: 10.1186/s11689-016-9152-y
50. Thurm A, Tierney E, Farmer C, Albert P, Joseph L, Swedo S, et al. Development, behavior, and biomarker characterization of Smith-Lemli-Opitz syndrome: an update. J Neurodev Disord (2016) 8:12. doi: 10.1186/s11689-016-9145-x
51. Oberman LM, Boccuto L, Cascio L, Sarasua S, Kaufmann WE. Autism spectrum disorder in Phelan–McDermid syndrome: initial characterization and genotype–phenotype correlations. Orphanet J Rare Dis (2015) 10:105. doi: 10.1186/s13023-015-0323-9
52. Klusek J, Martin GE, Losh M. Consistency between research and clinical diagnoses of autism among boys and girls with fragile X syndrome. J Intellect Disabil Res (2014) 58(10):940–52. doi: 10.1111/jir.12121
53. Wenger TL, Miller JS, DePolo LM, de Marchena AB, Clements CC, Emanuel BS, et al. 22q11.2 duplication syndrome: elevated rate of autism spectrum disorder and need for medical screening. Mol Autism (2016) 7:27. doi: 10.1186/s13229-016-0097-5
54. Harris JC. The origin and natural history of autism spectrum disorders. Nat Neurosci (2016) 19(11):1390–1. doi: 10.1038/nn.4427
55. Zwaigenbaum L, Bryson S, Lord C, Rogers S, Carter A, Carver L, et al. Clinical assessment and management of toddlers with suspected autism spectrum disorder: insights from studies of high-risk infants. Pediatrics (2009) 123(5):1383–91. doi: 10.1542/peds.2008-1606
56. Landa R, Garrett-Mayer E. Development in infants with autism spectrum disorders: a prospective study. J Child Psychol Psychiatry (2006) 47(6):629–38. doi: 10.1111/j.1469-7610.2006.01531.x
57. Akshoomoff N. Use of the mullen scales of early learning for the assessment of young children with autism spectrum disorders. Child Neuropsychol (2006) 12(4-5):269–77. doi: 10.1080/09297040500473714
58. Nader A-M, Courchesne V, Dawson M, Soulières I. Does WISC-IV underestimate the intelligence of autistic children? J Autism Dev Disord (2016) 46(5):1582–9. doi: 10.1007/s10803-014-2270-z
59. Lord C, Petkova E, Hus VB, Gan W, Lu F, Martin DM, et al. A multisite study of the clinical diagnosis of different autism spectrum disorders. Arch Gen Psychiatry (2012) 69(3):306–13. doi: 10.1001/archgenpsychiatry.2011.148
60. Wiggins LD, Reynolds A, Rice CE, Moody EJ, Bernal P, Blaskey L, et al. Using standardized diagnostic instruments to classify children with autism in the study to explore early development. J Autism Dev Disord (2015) 45(5):1271–80. doi: 10.1007/s10803-014-2287-3
61. Lord C, Rutter M, DiLavore PC, Risi S, Gotham K, Bishop SL. Autism Diagnostic Observation Schedule, Second Edition (ADOS-2) Modules 1-4. Los Angeles, California: Western Psychological Services (2012).
62. Rutter M, LeCouteur A, Lord C. Autism Diagnostic Interview-Revised (ADI-R). Los Angeles, CA: Western Psychological Services (2003).
63. Constantino JN. Social Responsiveness Scale, 2nd edition manual. Los Angeles, CA: Western Psychological Services (2012).
64. Charman T, Gotham K. Measurement issues: screening and diagnostic instruments for autism spectrum disorders—Lessons from research and practice. Child Adolesc Ment Health (2013) 18(1):52–63. doi: 10.1111/j.1475-3588.2012.00664.x
65. Randall M, Egberts KJ, Samtani A, Scholten R, Hooft L, Livingstone N, et al. Diagnostic tests for autism spectrum disorder (ASD) in preschool children. Cochrane Database Syst Rev (2018) 7. doi: 10.1002/14651858.CD009044.pub2
66. Leekam SR, Libby SJ, Wing L, Gould J, Taylor C. The diagnostic interview for social and communication disorders: algorithms for ICD-10 childhood autism and Wing and Gould autistic spectrum disorder. J Child Psychol Psychiatry (2002) 43(3):327–42. doi: 10.1111/1469-7610.00024
67. Skuse D, Warrington R, Bishop D, Chowdhury U, Lau J, Mandy W, et al. The developmental, dimensional and diagnostic interview (3di): a novel computerized assessment for autism spectrum disorders. J Am Acad Child Adolesc Psychiatry (2004) 43(5):548–58. doi: 10.1097/00004583-200405000-00008
68. Maljaars J, Noens I, Scholte E, van Berckelaer-Onnes I. Evaluation of the criterion and convergent validity of the Diagnostic Interview for Social and Communication Disorders in young and low-functioning children. Autism (2012) 16(5):487–97. doi: 10.1177/1362361311402857
69. Kinnear D, Morrison J, Allan L, Henderson A, Smiley E, Cooper S-A. Prevalence of physical conditions and multimorbidity in a cohort of adults with intellectual disabilities with and without Down syndrome: cross-sectional study. BMJ Open (2018) 8(2):e018292. doi: 10.1136/bmjopen-2017-018292
70. Rydzewska E, Hughes-McCormack LA, Gillberg C, Henderson A, MacIntyre C, Rintoul J, et al. Prevalence of sensory impairments, physical and intellectual disabilities, and mental health in children and young people with self/proxy-reported autism: observational study of a whole country population. Autism (2019) 23(5):1201–9. doi: 10.1177/1362361318791279
71. Hoevenaars-van den Boom MA, Antonissen AC, Knoors H, Vervloed MP. Differentiating characteristics of deafblindness and autism in people with congenital deafblindness and profound intellectual disability. J Intellect Disabil Res (2009) 53(6):548–58. doi: 10.1111/j.1365-2788.2009.01175.x
72. de Vaan G, Vervloed MPJ, Peters-Scheffer NC, van Gent T, Knoors H, Verhoeven L. Assessing autism spectrum disorder in people with sensory impairments combined with intellectual disabilities. J Dev Phys Disabil (2018) 30(4):471–87. doi: 10.1007/s10882-018-9597-x
73. Bishop SL, Havdahl KA, Huerta M, Lord C. Subdimensions of social-communication impairment in autism spectrum disorder. J Child Psychol Psychiatry (2016) 57(8):909–16. doi: 10.1111/jcpp.12510
74. Bishop S, Gahagan S, Lord C. Re-examining the core features of autism: a comparison of autism spectrum disorder and fetal alcohol spectrum disorder. J Child Psychol Psychiatry (2007) 48(11):1111–21. doi: 10.1111/j.1469-7610.2007.01782.x
75. Grzadzinski R, Dick C, Lord C, Bishop S. Parent-reported and clinician-observed autism spectrum disorder (ASD) symptoms in children with attention deficit/hyperactivity disorder (ADHD): implications for practice under DSM-5. Mol Autism (2016) 7:7. doi: 10.1186/s13229-016-0072-1
76. Lord C, Pickles A. Language level and nonverbal social-communicative behaviors in autistic and language-delayed children. J Am Acad Child Adolesc Psychiatry (1996) 35(11):1542–50. doi: 10.1097/00004583-199611000-00024
77. Mundy P, Sigman M, Kasari C. Joint attention, developmental level, and symptom presentation in autism. Dev Psychopathol (1994) 6(3):389–401. doi: 10.1017/S0954579400006003
78. Wing L, Gould J. Severe impairments of social interaction and associated abnormalities in children: epidemiology and classification. J Autism Dev Disord (1979) 9(1):11–29. doi: 10.1007/BF01531288
79. Klein-Tasman BP, van der Fluit F, Mervis CB. Autism spectrum symptomatology in children with Williams syndrome who have phrase speech or fluent language. J Autism Dev Disord (2018) 48(9):3037–50. doi: 10.1007/s10803-018-3555-4
80. Hus V, Lord C. The Autism Diagnostic Observation Schedule, Module 4: revised algorithm and standardized severity scores. J Autism Dev Disord (2014) 44(8):1996–2012. doi: 10.1007/s10803-014-2080-3
81. Gotham K, Risi S, Pickles A, Lord C. The Autism Diagnostic Observation Schedule: revised algorithms for improved diagnostic validity. J Autism Dev Disord (2007) 37(4):613–27. doi: 10.1007/s10803-006-0280-1
82. Anderson DK, Liang JW, Lord C. Predicting young adult outcome among more and less cognitively able individuals with autism spectrum disorders. J Child Psychol Psychiatry (2014) 55(5):485–94. doi: 10.1111/jcpp.12178
83. Charman T, Pickles A, Simonoff E, Chandler S, Loucas T, Baird G. IQ in children with autism spectrum disorders: data from the Special Needs and Autism Project (SNAP). Psychol Med (2011) 41(3):619–27. doi: 10.1017/S0033291710000991
84. Tager-Flusberg H, Plesa Skwerer D, Joseph RM, Brukilacchio B, Decker J, Eggleston B, et al. Conducting research with minimally verbal participants with autism spectrum disorder. Autism (2017) 21(7):852–61. doi: 10.1177/1362361316654605
85. Soorya L, Leon J, Trelles MP, Thurm A. Framework for assessing individuals with rare genetic disorders associated with profound intellectual and multiple disabilities (PIMD): the example of Phelan McDermid Syndrome. Clin Neuropsychol (2018), 32(7):1226–55. doi: 10.1080/13854046.2017.1413211
86. Perry A, Flanagan HE, Dunn Geier J, Freeman NL. Brief Report: the Vineland adaptive behavior ccales in young children with autism spectrum disorders at different cognitive levels. J Autism Dev Disord (2009) 39(7):1066–78. doi: 10.1007/s10803-009-0704-9
87. Bishop SL, Thurm A, Farmer C, Lord C. Autism spectrum disorder, intellectual disability, and delayed walking. Pediatrics (2016) 137(3):1–8. doi: 10.1542/peds.2015-2959
88. Mutsaerts CG, Heinrich M, Sterkenburg PS, Sappok T. Screening for ASD in adults with ID—moving toward a standard using the DiBAS-R and the ACL. J Intellect Disabil Res (2016) 60(5):512–22. doi: 10.1111/jir.12290
89. Baker E, Jeste SS. Diagnosis and management of autism spectrum disorder in the era of genomics: rare disorders can pave the way for targeted treatments. Pediatr Clin North Am (2015) 62(3):607–18. doi: 10.1016/j.pcl.2015.03.003
90. Reddy A, Graves C, Augustyn M. Parents seek early intervention services for a two-year-old without autism. J Dev Behav Pediatr (2011) 32(8):616–8. doi: 10.1097/DBP.0b013e318231066f
91. Simacek J, Dimian AF, McComas JJ. Communication intervention for young children with severe neurodevelopmental disabilities via telehealth. J Autism Dev Disord (2017) 47(3):744–67. doi: 10.1007/s10803-016-3006-z
92. National Research C. Educating children with autism. Washington DC: National Academies Press (2001).
93. Lord C, Bishop SL. Autism spectrum disorders: diagnosis, prevalence, and services for children and families. Social Policy Report. Soc Res Child Dev (2010) 24(2). doi: 10.1002/j.2379-3988.2010.tb00063.x
Keywords: differential diagnosis, developmental delay, intellectual disability, autism spectrum disorder, DSM-5
Citation: Thurm A, Farmer C, Salzman E, Lord C and Bishop S (2019) State of the Field: Differentiating Intellectual Disability From Autism Spectrum Disorder. Front. Psychiatry 10:526. doi: 10.3389/fpsyt.2019.00526
Received: 31 March 2019; Accepted: 03 July 2019;
Published: 30 July 2019.
Edited by:
Manuel Fernando Casanova, University of South Carolina, United StatesReviewed by:
Mathijs Vervloed, Radboud University Nijmegen, NetherlandsJane McCarthy, The University of Auckland, New Zealand
Copyright © 2019 Thurm, Farmer, Salzman, Lord and Bishop. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Audrey Thurm, YXRodXJtQG1haWwubmloLmdvdg==