 Antonio Rampino1,2Aleksandra Marakhovskaia3Tiago Soares-Silva3Silvia Torretta1Federica Veneziani1
Antonio Rampino1,2Aleksandra Marakhovskaia3Tiago Soares-Silva3Silvia Torretta1Federica Veneziani1 Jean Martin Beaulieu3*
Jean Martin Beaulieu3*- 1Department of Basic Medical Science, Neuroscience and Sense Organs, University of Bari “Aldo Moro”, Bari, Italy
- 2Azienda Ospedaliero-Universitaria Consorziale Policlinico di Bari, Bari, Italy
- 3Department of Pharmacology and Toxicology, University of Toronto, Toronto, ON, Canada
Antipsychotic drugs targeting dopamine neurotransmission are still the principal mean of therapeutic intervention for schizophrenia. However, about one third of people do not respond to dopaminergic antipsychotics. Genome wide association studies (GWAS), have shown that multiple genetic factors play a role in schizophrenia pathophysiology. Most of these schizophrenia risk variants are not related to dopamine or antipsychotic drugs mechanism of action. Genetic factors have also been implicated in defining response to antipsychotic medication. In contrast to disease risk, variation of genes coding for molecular targets of antipsychotics have been associated with treatment response. Among genes implicated, those involved in dopamine signaling mediated by D2-class dopamine receptor, including DRD2 itself and its molecular effectors, have been implicated as key genetic predictors of response to treatments. Studies have also reported that genetic variation in genes coding for proteins that cross-talk with DRD2 at the molecular level, such as AKT1, GSK3B, Beta-catenin, and PPP2R2B are associated with response to antipsychotics. In this review we discuss the relative contribution to antipsychotic drug responsiveness of candidate genes and GWAS identified genes encoding proteins involved in dopamine responses. We also suggest that in addition of these older players, a deeper investigation of new GWAS identified schizophrenia risk genes such as FXR1 can provide new prospects that are not clearly engaged in dopamine function while being targeted by dopamine-associated signaling molecules. Overall, further examination of genes proximally or distally related to signaling mechanisms engaged by medications and associated with disease risk and/or treatment responsiveness may uncover an interface between genes involved in disease causation with those affecting disease remediation. Such a nexus would provide realistic targets for therapy and further the development of genetically personalized approaches for schizophrenia.
Introduction
Whole genome association studies (GWAS) have identified several single nucleotide polymorphisms (SNPs) associated with enhanced risk for schizophrenia (1). These studies underscore the extraordinary polygenicity of schizophrenia and raises the hope that understanding the developmental causes of the disease may lead to new therapies (2, 3). Yet, most of these SNPs were not found in genes traditionally associated to the mechanism of action of existing antipsychotic drugs and it remains unclear whether it will be practically possible to intervene on pathways involved in disease causation.
Alternatively, several genetic factors have been shown to modulate the severity of schizophrenia symptoms and treatment responsiveness. Twins studies have demonstrated that, like pathophysiology and risk for schizophrenia, response to antipsychotic medication is a heritable trait (4, 5). Furthermore, reports have highlighted that schizophrenia risk, pathophysiology and response to treatments likely share a common genetic background (6). Based on such evidence, GWAS are currently considered a powerful tool to identify new molecular targets for antipsychotic treatments, while confirming the involvement of the already established ones. Genes and related proteins belonging to dopaminergic signaling have consistently been indicated as key elements for antipsychotics treatment responsiveness by both hypothesis- and data-driven pharmacogenetic studies.
The neurotransmitter dopamine is involved in the regulation of several cerebral functions including, reward, mood, sensory motor gating, affect and locomotor functions, learning and motivation (7, 8). Importantly, dopamine receptors, especially the D2 dopamine receptor (DRD2), are major pharmacological targets of all existing antipsychotic drugs (9–11) (Table 1). Several dopamine producing neuron populations exist in the adult brain. Cells from the nigrostriatal pathway project from the substantia nigra pars compacta to the caudate nucleus and the putamen in the striatum (12, 13). Dopamine neurons from the mesolimbic pathway (14) project from the ventral tegmental area to the ventral striatum, amygdala, and several cortical areas (e.g., pre-frontal cortex) expressing dopamine receptors (15). Dopamine neurons from the infundibular nucleus of the hypothalamus are also involved in the dopamine-mediated regulation of the pituitary gland (16). Among these neuronal networks, the mesolimbic pathway has received the most attention in the context of schizophrenia.
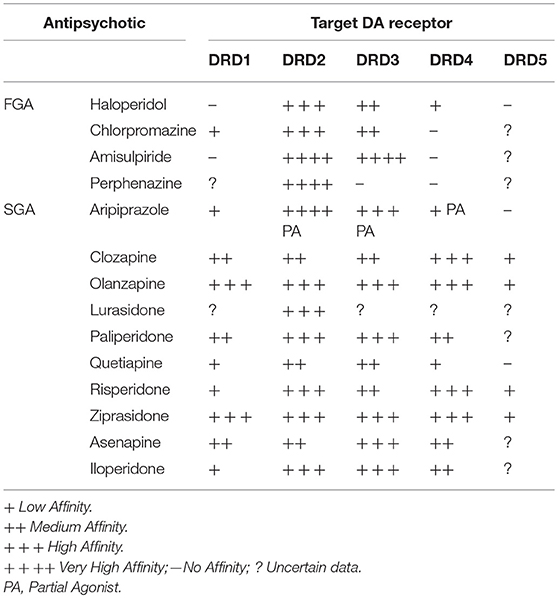
Table 1. List of main First-Generation (FGA) and Second-Generation (SGA) Antipsychotics with respective target Dopamine (DA) Receptors (D1–D5).
Here, we will provide a brief overview of the biological regulation of dopamine and of the cellular mechanisms engaged by its receptors; for readers that would want a more complete description of the biology of dopamine receptor we would suggest more exhaustive reviews (7, 17, 18). We will then examine how molecular pathways involved in dopamine function intersect with genetic factors for antipsychotic drug responsiveness and, if applicable, disease causation. To evaluate this, two investigators independently conducted a systematic PubMed search (June 2018) for studies of antipsychotic medication responsiveness involving dopamine signaling cascade and related genetics. Combinations of the following keywords were used: Antipsychotics (APs), first-generation APs, second-generation APs, AP response, AP pharmaco-genetics, AP pharmaco-genomics, schizophrenia risk, schizophrenia genetics, schizophrenia genomics, schizophrenia polygenic risk score, dopamine, dopamine receptors, dopamine signaling. Finally, we will underscore how a newly discovered pathway may provide new avenues to investigate the genetic of antipsychotic drug responsiveness.
Dopamine Homeostasis
During dopamine synthesis, the amino acid tyrosine undergoes hydroxylation to levodopa that is catalyzed by the enzyme tyrosine hydroxylase (TH) (Figure 1). This is followed by a decarboxylation of levodopa to dopamine by the enzyme dopa-decarboxylase (DDC) (19). In adult neurons, dopamine is loaded into synaptic vesicles by the vesicular monoamine transporter 2/solute carrier family 18 member A2 (VMAT2/SCL18A2) (20). Following synaptic release, dopamine can either be preferentially up-taken or degraded, depending upon neuronal circuit. The major transporter in charge of dopamine reuptake is the dopamine transporter/solute carrier family 6 member 3 (DAT1/SLC6A3) (21). This transporter is the key element in the mechanism removing extracellular dopamine in striatal structures. However, dopamine has also been shown to be reuptaken by other transporters such as the norepinephrine transporter/solute carrier family 6 member 2 (DAT/SLC6A2) in brain regions expressing low levels of the SLC6A3 gene product (22, 23). The major enzyme in charge of dopamine degradation is the Catechol-O-methyltransferase (COMT), which methylates dopamine into 3-metoxytyramine (3-MT). Degradation by COMT is the principal mechanism of dopamine clearance in the pre-frontal cortex. Dopamine can also be oxidized into 3,4-Dihydroxyphenyl-acetic acid (DOPAC) by the monoamine oxidases (MAOA and MAOB). The final step of dopamine metabolism involves the production of homovanillic acid (HVA) from 3-MT by monoamine oxidases or from DOPAC by COMT (24).
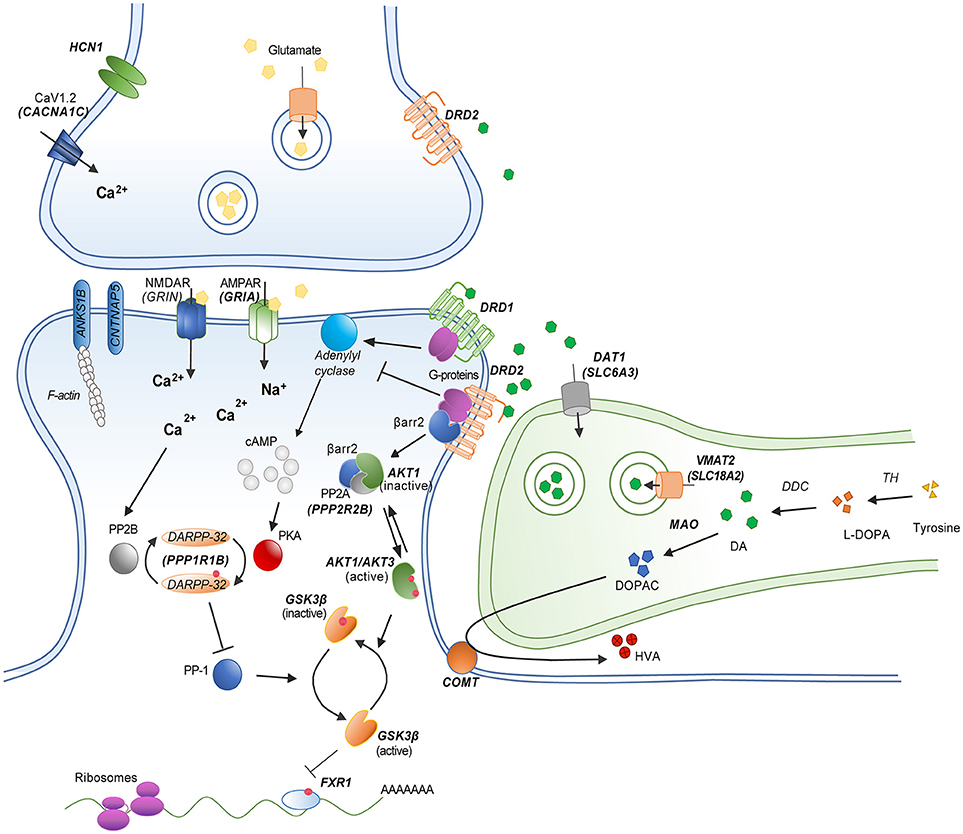
Figure 1. Schematic model of a striatal dopamine/glutamate synapse showing the functional relationship between different dopamine related gene products. Genes implicated in candidate gene studies and/or GWAS are indicated in bold. Red dots represent phosphorylation events.
Dopamine Receptors Signaling Mechanisms
Dopamine exerts its effects via the stimulation of five different G-protein coupled dopamine receptors (Figure 1). These receptors are divided in two groups based on their coupling and gene structures (17). D1-class dopamine receptors DRD1 and DRD5 are mostly coupled to Gαs G-proteins and encoded by genes that are devoid of introns. D2-class dopamine receptors DRD2, DRD3, and DRD4, are encoded by genes that comprise introns and are generally coupled to Gαi/o G-proteins. D1-class receptors mediate post-synaptic responses to dopamine. In contrast D2-class receptors can both mediate post-synaptic responses and act as presynaptic auto-receptors to limit dopamine synthesis and release (19). Of note, the DRD2 gene encodes two splice variants of the receptor. The long isoform (D2L) is mostly expressed on post-synaptic neurons while the short isoform (D2S) is preferentially expressed by pre-synaptic dopamine neurons (25).
Activation of D1-class receptors results in an increased production of the second messenger cyclic Adenosine Monophosphate (cAMP) by class 3 adenylyl cyclases (ADCY) (26). Activation of D2-class receptor results in a reduction of cAMP by inhibiting this same mechanism (27). Major downstream effectors of dopamine receptors are cAMP-dependent protein kinases A (PKA) (7). PKA are holoenzymes comprised of a catalytic subunit and different regulatory subunits. Catalytic subunits are encoded by the genes PRKACA, PRKACB, and PRKACG. Regulatory subunits are encoded by the PRKAR1A, PRKAR1B, PRKAR2A, and PRKAR2B genes in humans. Regulation of PKA activity by dopamine receptors is involved in several cellular processes including, among others, the regulation of gene expression by transcription factors and the regulation of ionotropic receptors for various neurotransmitters. Among several targets of interest, the activity of cAMP response elements binding proteins family of leucine zipper transcription factors (i.e., CREB1) can be modulated by dopamine. Subunits of AMPA and NMDA ionotropic glutamate receptors (i.e., GRIN1, GRIA1, GRIA4) are also regulated by PKA downstream of dopamine receptors (7, 18). Finally, the protein phosphatase 1 regulatory subunit 1B (PPP1R1B/DARPP-32) has been shown to be regulated by dopamine and cAMP and to play a role in the balance of phosphorylation/dephosphorylation of several PKA substrates involved in dopamine receptor signaling and the integration of metabotropic (slow) and ionotropic (fast) neurotransmission (28, 29).
The signaling of dopamine receptors is not restricted to the regulation of cAMP production. Some receptors have been reported to have the possibility to couple to GαQ G-proteins to regulate intracellular inositol and calcium signaling (30, 31). Furthermore, activation of Gβγ G-protein subunits by DRD2 results in neuronal hyperpolarization by regulating the activity of L and N-Type calcium channels (LTCC and NTCC) and G-protein gated inwardly rectifying potassium channels (e.g., GIRK2/KCNJ6) (7). Furthermore, DRD2 modulates neuronal function by acting on G-protein independent mechanisms. Following their activation, dopamine receptors are phosphorylated by G-protein receptor kinases (e.g., GRK2, GRK6) (32). This leads to the recruitment of beta-arrestins (ARBB1 and ARBB2), which inactivate G-protein coupling, stimulate receptor internalization and mediate additional cell signaling functions (33, 34). In the case DRD2, the recruitment of ARBB2 results in the formation of a protein complex that favors the inactivation of protein kinases from the Akt family (AKT1, AKT2, AKT3) by protein phosphatase 2 holoenzymes (i.e., PPP2R2B, PPP2CA, PPP2CB). The inactivation of AKT kinases downstream of DRD2 releases the inhibition of glycogen kinase 3 family proteins (GSK3A, GSK3B) thus increasing their activity (35, 36). The nature of the GSK3 substrates involved in DRD2 signaling is still unclear. One frequently investigated potential target is the canonical wingless (WNT) signaling transcription factor beta-catenin (CTNNB1) (37). However, beta-catenin does not appear to be a major determinant of antipsychotic drug action downstream of DRD2 in mice (38). The RNA binding protein fragile-X-mental-retardation-autosomal-homolog-1 (FXR1) is a recently identified GSK3B substrate (39) that has been shown to modulate ionotropic glutamate receptor functions in vivo (40, 41). It is thus possible that FXR1 may contribute to the regulation of neuronal functions by GSK3-mediated DRD2 signaling (42).
Candidate Gene Studies
Candidate gene studies have often been focused on the role of dopamine receptors and related genes (namely DRD1, DRD2, DRD3, DRD4, and DRD5) as modulators of antipsychotic drug responsiveness and schizophrenia symptoms. In particular, genetics studies on schizophrenia endo-phenotypes, i.e., behavioral and/or neuroimaging phenotypes associated with the disorder, have suggested that variations in genes coding for this cluster of receptors, especially those coding for DRD2, DRD3, and DRD4, are associated with cognitive deficits and related brain pre-frontal cortex malfunction, which are typical phenotypes of schizophrenia. Candidate gene investigations have identified allelic variation in SNPs of functional relevance to D2, D3, and D4 receptors to be associated with response to antipsychotic medication (Table 2).

Table 2. Genes and corresponding mutation implicated in antipsychotic response/resistance by Candidate gene studies or GWAS.
Genetic studies on the role of DRD2 genetic variation in the pathophysiology and risk for schizophrenia and response to antipsychotics have been motivated by the evidence that DRD2 antagonism is a key mechanism of action of antipsychotic agents to the extent that occupancy of this receptor subtype is correlated with antipsychotic potency (Table 1). The DRD2 locus is the only dopamine receptor gene for which common variants have been associated to schizophrenia risk by GWAS (1). Candidate gene studies on functional polymorphisms of DRD2 gene, involving SNPs that affect DRD2 gene expression and/or DRD2 Long/Short transcription isoform ratio, along with DRD2 membrane density, have also pointed to a role of such genetic variation in schizophrenia. For example, rs1800497 (also known as Taq1A) is a missense variant determining a Glu-to-Lys substitution at position 713 of DRD2 gene and associated with the gene mRNA stability and expression (68), striatal dopamine receptor density (69), and modification in DRD2 binding in human striatum (70). Interestingly, rs1800497 has been associated with performance in Working Memory, a prototypical endophenotypes of schizophrenia, mainly subtended by pre-frontal cortex activity (71). Similarly, rs1801028, a SNP occurring in DRD2 gene and determining a Ser-to-Cys substitution at position 311 of DRD2 gene (Ser311Cys) has been shown to alter the physiology and function of DRD2 receptor (72) and has consistently been associated with risk for schizophrenia in a number of studies since 1994 (73)—for a meta-analysis see: (74). Another example has been provided by rs1076560, a SNP located in intron 6 of the DRD2 gene, whose T allele has been reported to shift the splicing of DRD2 transcript from D2S to D2L isoforms (75). Rs1076560 has been associated with risk for schizophrenia (76), behavior and brain activity during cognitive and emotion processing in healthy controls and patients with schizophrenia (77), with efficiency of pre-fronto-striatal activity during Working Memory (78) and levels of striatal dopamine (79).
Since DRD2 neuronal signaling is mediated by a number of partner molecular elements downstream of the receptor, including proteins belonging to cAMP-dependent and cAMP-independent pathways, such as AKT1, GSK3B, PPP2R2B, and CTNNB1, not surprisingly also genes coding for such proteins have been associated with phenotypes that are relevant to schizophrenia pathology. For example, rs1130233, a cis-eQTL of AKT1 gene that interacts with rs1076560 in affecting pre-frontal AKT1 mRNA expression levels along with level of phosphorylation of the AKT1 kinase target GSK3beta, interacts with rs1076560 also on cingulate cortex activity during attentional control, another cognitive function typically impaired by schizophrenia (67). Similarly, rs12630592 (80) and rs609412 (81), two eQTLs of GSK3beta and PPP2R2B gene, respectively, have been associated with cognitive performance and related brain activity critically implicated in the disorder.
Functional variation of DRD2 gene has also been studied as predictor of antipsychotic response, (for a review see: (82). In general, dopamine receptors variants associated with reduced expression of the receptor protein or altered functioning were also associated with poorer response to antipsychotic drugs, confirming that these receptors mediate antipsychotic activity. Rs1800497 has been associated with response to nemonapride (43), haloperidol (44), risperidone (45), and clozapine (48). Similarly, rs1801028 has been associated with response to risperidone (47), while interaction between rs1076560 and rs1130233 has been implicated in response to olanzapine as measured by variation in Positive and Negative Symptoms Scale total scores after 56 days of stable dosage treatment (67). Other SNPs in DRD2 have been associated with antipsychotics response, including rs1079597, also known as Taq1B, whose B1 allele has been associated with reduced DRD2 protein density in the striatum, and B2 allele has been implicated in response to clozapine (48). Further mutations have been associated with clinical response to chlorpromazine (49) and aripiprazole (46).
DRD3 has systematically been studied by pharmacogenetics since many antipsychotic drugs show high affinity for the D3 dopamine receptor (83). A missense polymorphism in exon 1 of DRD3 leading to a serine to glycine substitution at amino acid position 9 (rs6280) has been associated with altered DRD3 dopamine affinity and density in some brain areas (84) along with response to first-generation antipsychotics and lack of response to clozapine (51). Some studies (52) found rs6280 to be associated with greater acute positive symptom remission after olanzapine treatment. The same effects were observed for different SNPs in linkage disequilibrium with rs6280. Further studies have also implicated rs6280 in response to the second-generation antipsychotic aripiprazole (53).
Evidence has also established a correlation between response to antipsychotics and functional genetic variation of the DRD4 gene. In particular, since DRD4 is targeted by clozapine, the most effective antipsychotic drug currently available (85), studies have particularly focused on the effect of genetic variability of corresponding gene on response to such a drug. One of the numerous polymorphisms occurring in DRD4 gene (namely, rs4646984), is a 48 bp Variable Number Tandem Repeat (48.bp VNTR) falling within the third exon of the gene and resulting in a different length of the third cytoplasmatic loop of DRD4 protein. Importantly, the longer repeat version of the protein has been associated with reduced clozapine binding, thus suggesting the 48 bp VNTR to be an interesting candidate polymorphism to study as predictor of clinical response to this antipsychotic. However, while some studies have reported an effect of rs4646984 on responsiveness to various antipsychotics including clozapine (54–57) several other studies also reported no associations, as reviewed in Zhang and Malhotra (83).
Along with genes coding for dopamine receptors, other genes belonging to the dopaminergic system have also been implicated in pathophysiology and risk for schizophrenia, hence becoming candidate genes for as regulators of response to antipsychotics (Table 2). The COMT and SLC6A3 genes, have been particularly relevant within this framework.
Studies have consistently demonstrated that a common polymorphism in COMT, rs4680 or Val108Met, is responsible for a functional variation in enzymatic activity with the Met/Met condition characterized by lower activity (hence, reduced pre-frontal dopamine clearance) as compared to the Val/Val condition. A large number of studies have investigated the genetic association between rs4680 and the diagnosis of schizophrenia with mixed results—for a meta-analysis see: (86). In line with the role of COMT in dopamine turnover, pharmacogenetic investigations have reported an association between rs4680 allelic variations and the clinical response to antipsychotics acting on dopamine receptors. An early study found that individuals with schizophrenia carrying a met/met genotype were less responsive to first-generation antipsychotics (58). The val/val genotype has also been associated with reduced response of negative symptoms to olanzapine (59) while the met/met genotype has been associated with increased response of cognitive symptoms to clozapine (60). A number of additional studies, then, confirmed the impact of the COMT Val158Met polymorphism on the efficacy of first-generation (61, 62) and second-generation antipsychotics (63, 64, 87).
Studies have also implicated SLC6A3, coding for the dopamine transporter, in schizophrenia phenotypes and response to medication. A functional Variable Number of Repeats polymorphism at 3' end of this gene (3'-VNTR) has been described (88, 89). Alleles of this polymorphism range from 3 to 11 repeats, with the 9- and 10-repeat alleles by far the most common (88, 89). As compared with the 9-repeat allele, the 10-repeat allele has been associated with increased SLC6A3 gene expression both in vitro (90, 91) and in human striatum (92). Furthermore, studies have reported that the 10-repeat allele is associated with more focused cortical activity during memory and attention, two critical endophenotypes of schizophrenia (93–98). Evidence has also documented an interaction between DRD2 rs1076560 and SLC6A3 3'-VNTR on further imaging phenotypes associated with the disorder, such as pre-fronto-striatal activity and volume in humans (77).
Although not directly targeted by antipsychotic medications, the dopamine transporter, may influence dopamine-signaling intensity by virtue of its function of dopamine clearance at the synaptic level and contribute to treatment outcome. Consistently, a number of studies have reported association between polymorphisms in the gene coding for this transporter, including 3'-VNTR and level of response to first and second-generation antipsychotics (65, 99).
Genome Wide Association Studies
GWAS have allowed for detection of genetic variants associated with a specific disease without any a priori hypothesis on its pathophysiology (hypothesis-free or data-driven studies). Such an approach has confirmed the polygenic nature of risk for schizophrenia since it has identified more than one hundred genetic variants associated with the disorder at genome wide statistical level of significance (i.e., p < 10−8) (1). Furthermore, studies attempting at collapsing GWAS multigenic risk in so called Polygenic Risk Scores (PRSs) have probed these scores to predict behavioral and neuroimaging phenotypes of schizophrenia (81). Nevertheless, since GWAS and PRSs do not provide any specific insights into the biological role of genetic variation associated with a disease, different approaches have been developed in order to dissect risk into biologically meaningful pathways and probe the impact of pathway-specific risk variants and PRSs onto pathophysiology of schizophrenia and related phenotypes (81).
Several genes, including DRD2, have been associated with schizophrenia by GWAS, even though, most findings have implicated different variants from those detected by candidate gene approaches (1). Nonetheless, as for candidate genes investigation, risk variants identified by GWAS have been implicated in response to antipsychotic medication, confirming the hypothesis that schizophrenia pathophysiology and response to treatment share genetic bases that are partially involving dopamine signaling. A prototypical example is provided by rs2514218, a SNP in DRD2 gene that was associated with schizophrenia in a large GWAS published by the Psychiatric Genomics Consortium (1). Zhang and collaborators (50) examined whether genotype at this SNP could predict response to 12 weeks of risperidone or aripiprazole treatment in a cohort of patients with first episode of psychosis and found that homozygotes for the risk (C) allele at this SNP had significantly greater reduction in positive symptoms after treatment with either risperidone or aripiprazole as compared to the T allele carriers.
From a pharmacogenomics perspective, a pivotal role in the study of antipsychotics response has been played by the Clinical Antipsychotics Trials of Intervention Effectiveness (CATIE) (100), a multicenter research project promoted by the USA National Institute of Health and investigating the effectiveness of first and second-generation antipsychotics. Within the CATIE, a number of GWAS have been conducted on both treatment response and adverse reaction (Table 2).
The first GWAS (66) found a SNP (rs17390445) located on Chromosome 4 in an intergenic position, to be associated with response of schizophrenia positive symptoms to the second-generation antipsychotic ziprasidone, while another SNP in the same intergenic region approached, but did not reach, genome wide significance. Both rs17390445 and this second SNP functions remain unknown, but involvement in dopamine signaling cannot be excluded. Interestingly, in a different study, the same group looked at neurocognition as an outcome of antipsychotics response (101) and found a weak association with SNPs in DRD2 gene. This same study identified SNPs associated with olanzapine and risperidone response in Ankyrin Repeat and Sterile Alpha Motif Domain-Containing Protein 1B (ANKS1B) and in the Contactin-Associated protein-Like 5 genes (CNTNAP5) which, are not directly connected with dopamine signaling, but are involved in interneuron communication.
GWAS were also used to explore the clinical response to new antipsychotics partially exerting their pharmacological action by blocking dopamine transmission. For example, two of these studies (102, 103) investigated genome wide association with response to Iloperidone, an antagonist of D2 and D3 dopamine receptors also blocking noradrenergic and serotonergic neuronal signaling. None of the SNPs reaching genome wide significance were directly involved in dopaminergic system, even though one of the genes associated, GRIA4, codes for AMPA 4 glutamate receptor, that may impact on dopamine neurotransmission by mediation of glutamate signaling. In fact, evidence suggests that aberrant glutamatergic function may alter dopamine system function in psychotic disorders (104, 105) and dopamine exert several of its biological actions by modulating ionotropic AMPA and NMDA glutamate receptor functions (7, 29). Furthermore, dopamine D2/D3 receptor availability is linked to the severity of psychotic symptoms induced by glutamatergic antagonism (106) suggesting that factors influencing glutamate signaling may contribute to dopamine dysregulation and symptoms or response to dopamine-mediated treatments (107). Interestingly, a study (108) looking at drugs targeting proteins encoded by genes GWAS associated with schizophrenia, found that antipsychotics are the only medication surviving enrichment procedures and that, two of the four genes associated with antipsychotic response were either directly implicated in glutamatergic signaling (namely, GRIN2A) or indirectly related to dopaminergic system (i.e., AKT3). The remaining two genes, CACNA1C and HCN1, respectively coding for LTCC Cav 1.2 and Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, have also been implicated in dopamine signaling modulation (109, 110).
GWAS-based pharmacogenomics studies on antipsychotic response have also used PRS strategies in order to explore the cumulative role of genetic risk on pharmacological effects. For example, one study (111) showed a PRS based on schizophrenia-associated SNPs reported by the Psychiatric Genomics Consortium (1) to predict clozapine response, in that clozapine responders had higher PRS as compared to non-responders. Furthermore, a study using a multistage GWAS-PRS-Pathway Analysis approach (6) to detect genetic variation associated with response to the second-generation antipsychotic lurasidone, suggested associated variants to belong to functional categories of relevance to neuronal transmission and being, at least partially, involved in dopamine signaling by impacting on glutamate an serotonin systems modulation.
An Interesting New Candidate Gene, the Fragile X Autosomal HOMOLOG 1
At first glance, most schizophrenia risk SNPs identified by GWAS are not associated to genes encoding products generally related to dopamine transmission or cellular responses to antipsychotic drugs (1). However, some of these loci may still encode proteins regulated by therapeutic agents. Investigating such genes in the context of known mechanisms of antipsychotic actions may thus be a fruitful avenue for future studies.
For example, the RNA binding protein FXR1 encode on chromosome 3q26.33 represents an interesting new candidate among the genetic risks factors for schizophrenia identified by GWAS (1). FXR1 belongs to the fragile X proteins family, which comprises three RNA-binding proteins members—Fragile X mental retardation protein (FMR1/FMRP), and two Fragile X related proteins—FXR1 and FXR2 (112, 113). All three proteins play a role in regulating the development of several tissues including the brain. Furthermore, these proteins are also implicated in the regulation of neuronal functions (114–116).
The Fragile X mental retardation syndrome is a primary monogenic cause of autistic spectrum disorders (ASDs) and is caused by mutations in FMR1 gene, which is highly expressed in neurons and plays a crucial role in regulating synaptic plasticity (117–119). Both FXR1 and FXR2 have also been recently shown to participate in synaptic plasticity (40, 41, 120). Proteins from this family comprise homologous amino-terminal regions containing a tandem Tudor domain which can mediate binding to methylated lysine on histones (121). The amino-terminal region of these proteins also includes a nuclear localization and export signals (NLS and NES) that mediate shuttling between nucleus and cytoplasm (122). The medial regions of FMR1, FXR1, and FXR2 comprise RNA-binding KH domain and RGG box that allow RNA-binding (123). Finally, the three proteins sequences are most divergent at the carboxyl-terminal domain, which seem to functionally distinguish members of the family (124). All three fragile X family proteins participate to the regulation of mRNA translation, degradation, and are associated with ribosomes (116, 123, 125).
FMR1 is by far the most studied of these three proteins. Interestingly, an exome study of rare genetic variants in schizophrenia identified an enrichment for gene encoding mRNA that are associated to FMR1 (126). Furthermore, a postmortem study has shown altered protein levels of FMR1 targets in the frontal cortex of subjects with schizophrenia or bipolar disorder (127). However, SNPs in FMR1 are not associated to schizophrenia as defined by a GWAS studies of common genetic risk variants (1). In contrast to FMR1, FXR1 came out of the shadow after its identification as one of the top 30 potential genetic risk factor for schizophrenia (1). For a long time FXR1 was considered either a protein functionally redundant to FMR1 (125) or as its muscle specific homolog (115). Both FXR1 and FMR1 have been shown to interact together but can also have mutually exclusive functions and cellular localization (128). It would thus be premature to conclude that changes in FMR1 targets identified in schizophrenia results from altered FXR1 functions since these two proteins are not biologically equivalent.
The FXR1 gene encodes seven major alternatively spliced mRNA variants (Figure 2, mouse gene used as an example), three of which are expressed specifically in muscles and testis (115, 129). Characterization of the schizophrenia risk allele in the FXR1 locus (rs34796896) has shown it to be in linkage disequilibrium with splicing quantitative trait loci (sQTL) SNP (rs1805564) identified in postmortem human dorsolateral pre-frontal cortex (DLPFC). Altered splicing in the locus of exon 15 is associated and functionally changed in presence of the rs34796896 schizophrenia risk allele (130). The expression of exon 15 containing isoforms of FXR1 has only been demonstrated in muscle and testicular tissues by means of western blot. However, the presence of exon 15 RNAs has been detected by sequencing in other tissues, thus suggesting that minor amounts of the protein carrying this exon could be expressed outside of the muscle and testis (130). Similar non-canonical expression of intronic sequence possessing novel isoform of FXR1 in CD4+ T cells was also shown by means of mass spectrometry as a confirmation of RNA-sequencing data (131).
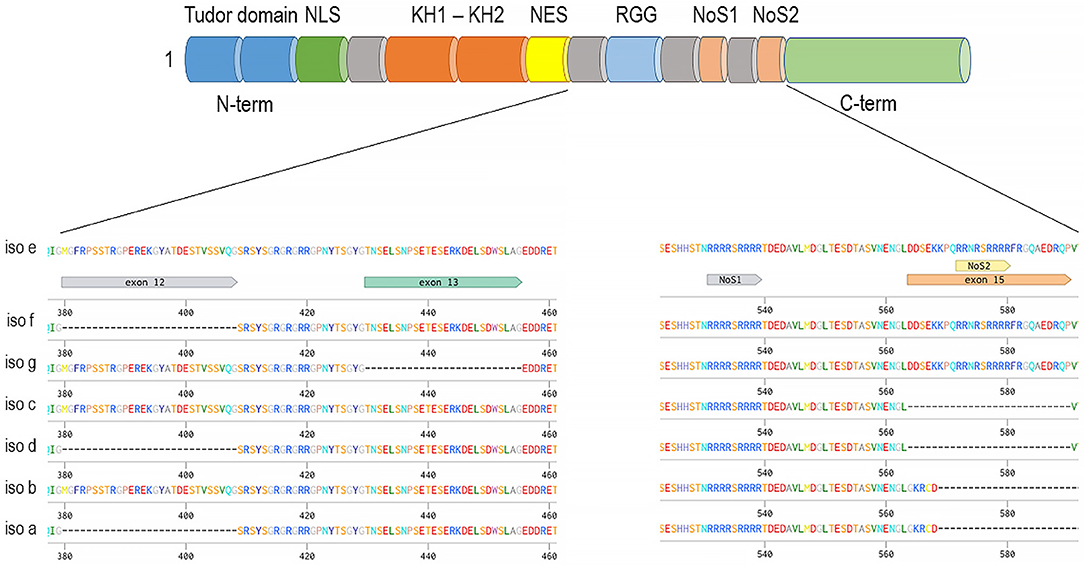
Figure 2. Fxr1 gene (mouse gene depicted as an example) alternatively spliced mRNAs and common domain structure on example of the longest isoform—e. The presence of exon 15 provides an additional nucleolar targeting signal.
Exon 15 provides an additional nucleolar-targeting signal (NoS2), which is necessary for efficient shuttling between cytoplasm and nucleoli. This shuttling was demonstrated in Cos-7 cell line for the isoform e of FXR1 as well as for FXR2 but not for FXR1 isoform d or FMR1 (132). Lately, nucleolar localization was also demonstrated for FMRP in Hela cells where FMRP expression is high (133). Interestingly, unlike other family members that exhibit a similar cytoplasmic localization between fetal and adult tissues, FXR1 has been reported to have nuclear localization only during brain and muscle development (114, 115). Thus, it is possible that alteration in the nuclear or nucleolar localization of FXR1, regulated by the NoS2 of exon 15, may contribute to increase the risk to develop schizophrenia.
FXR1 may also contribute to DRD2 and GSK3β mediated signaling (42). Indeed, GSK3β, which is activated following DRD2 stimulation and inactivated by several antipsychotics (134), is able to directly phosphorylate Fxr1 and regulate its expression in a negative manner (39). Furthermore, chronic treatment with lithium and valproate resulting in an inhibition of GSK3β activity also increased Fxr1 expression in the mouse striatum and pre-frontal cortex (39). Interestingly, GSK3β and FXR1 are not affected by chronic lithium or valproate treatment in mice lacking ARBB2 (39). Furthermore, lithium also fails to engage AKT-GSK3 mediated signaling in mice lacking either ARBB2 of DRD2 (135, 136), which suggests an engagement of the DRD2-ARBB2-AKT-GSK3 pathway in the regulation of Fxr1 levels.
A functional interaction has been reported between SNPs affecting the relative expression of the FXR1 (rs496250) and GSK3B (rs12630592) genes (39). In healthy humans, this interaction has been shown to affect amygdala response to emotional faces, as measured by functional Magnetic Resonance Imaging (fMRI). The same interaction was observed on measures of trait Emotional Stability as conceived within the Big Five Model of Personality (39). Finally, interaction between these functional SNPs has been replicated and showed to affect symptoms severity and, putatively, treatment responsiveness in bipolar patients (137). Evidence for a contribution of FXR1 to dopamine signaling and responsiveness to psychiatric disorders remains preliminary. However, the existing observations would support that FXR1 is one example of a gene for which further investigation of contribution to schizophrenia, DRD2 signaling, and antipsychotics drug responsiveness is warranted. Further examination of other GWAS identified loci may allow finding additional candidate genes of interest with more distal relations to dopamine neurotransmission.
Conclusion
Genetic investigations of risk factors for schizophrenia and determinants of drug responsiveness revealed a very multi-genic landscape for both indications. This suggests that schizophrenia arises from a combination of multiple genetic and socio-environmental hits occurring during development. This is also in line with the variable profile of drug responsiveness observed at the clinical level. This picture can be discouraging as several risk factors may only participate to disease at a pre-onset stage or, contribute to ubiquitous functions across different organs, thus preventing their practical or ethical use as therapeutic targets. Nonetheless, the overlap of schizophrenia risk and genes affecting responsiveness to existing drugs also points toward a nexus of biological mechanisms engaged by medication and those contributing to disease etiology. This supports the idea that disease remediation can intersect with disease causation and help compensate for developmental insults, even during adulthood. The prevalence of genes involved in dopamine transmission among those associated with treatment responsiveness is flagrant. However, dopamine is an “old player” in schizophrenia therapy and the prevalence of dopamine-associated genes probably results from a bias induced by the mechanism of action of therapeutic agents. The further examination of genes that are more distal to dopamine signaling and are nonetheless associated with schizophrenia risk and drug responsiveness may provide a new roster of “prospects” that can be realistically targeted for therapies whether alone, or in conjunction with older dopamine-targeting therapeutics. Furthermore, the integrated investigation of new and old variants associated both to genetic risk and treatment responsiveness can provides the bases for the development of personalized treatment protocols for schizophrenia.
Author Contributions
All authors wrote the manuscript. JB also provided funds for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from Canada Institutes of Health Research (CIHR, MOP-136916, PJT-148568) to JB. JB is Canada Research Chair in Molecular Psychiatry (Tier1), NARSAD independent investigator and a One-Mind Rising Star awardee. AR has been supported by Fondazione con il Sud, grant Sviluppo del Capitale Umano ad Alta Qualificazione.
References
1. Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511:421–7. doi: 10.1038/nature13595
2. Mccarroll SA, Feng G, Hyman SE. Genome-scale neurogenetics: methodology and meaning. Nat Neurosci. (2014) 17:756–63. doi: 10.1038/nn.3716
3. Hyman SE. The daunting polygenicity of mental illness: making a new map. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20170031. doi: 10.1098/rstb.2017.0031
4. Vojvoda D, Grimmell K, Sernyak M, Mazure CM. Monozygotic twins concordant for response to clozapine. Lancet (1996) 347:61. doi: 10.1016/S0140-6736(96)91594-9
5. Mata I, Madoz V, Arranz MJ, Sham P, Murray RM. Olanzapine: concordant response in monozygotic twins with schizophrenia. Br J Psychiatry (2001) 178:86. doi: 10.1192/bjp.178.1.86
6. Li J, Yoshikawa A, Brennan MD, Ramsey TL, Meltzer HY. Genetic predictors of antipsychotic response to lurasidone identified in a genome wide association study and by schizophrenia risk genes. Schizophr Res. (2018) 192:194–204. doi: 10.1016/j.schres.2017.04.009
7. Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. (2011) 63:182–217. doi: 10.1124/pr.110.002642
8. Berke JD. What does dopamine mean? Nat Neurosci. (2018) 21:787–93. doi: 10.1038/s41593-018-0152-y
9. Snyder SH, Taylor KM, Coyle JT, Meyerhoff JL. The role of brain dopamine in behavioral regulation and the actions of psychotropic drugs. Am J Psychiatry (1970) 127:199–207. doi: 10.1176/ajp.127.2.199
10. Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. (2004) 3:353–9. doi: 10.1038/nrd1346
11. Comai S, Tau M, Pavlovic Z, Gobbi G. The psychopharmacology of aggressive behavior: a translational approach: part 2: clinical studies using atypical antipsychotics, anticonvulsants, and lithium. J Clin Psychopharmacol. (2012) 32:237–60. doi: 10.1097/JCP.0b013e31824929d6
12. Anden NE, Carlsson A, Dahlstroem A, Fuxe K, Hillarp NA, Larsson K. Demonstration and mapping out of nigro-neostriatal dopamine neurons. Life Sci. (1964) 3:523–30. doi: 10.1016/0024-3205(64)90161-4
13. Lavoie B, Smith Y, Parent A. Dopaminergic innervation of the basal ganglia in the squirrel monkey as revealed by tyrosine hydroxylase immunohistochemistry. J Comp Neurol. (1989) 289:36–52. doi: 10.1002/cne.902890104
14. Phillipson OT. Afferent projections to the ventral tegmental area of Tsai and interfascicular nucleus: a horseradish peroxidase study in the rat. J Comp Neurol. (1979) 187:117–43. doi: 10.1002/cne.901870108
15. Khlghatyan J, Quintana C, Parent M, Beaulieu JM. High sensitivity mapping of cortical dopamine D2 receptor expressing neurons (2018b). Cereb Cortex. doi: 10.1093/cercor/bhy261. [Epub ahead of print].
16. Bosse R, Fumagalli F, Jaber M, Giros B, Gainetdinov RR, Wetsel WC, et al. Anterior pituitary hypoplasia and dwarfism in mice lacking the dopamine transporter. Neuron (1997) 19:127–38. doi: 10.1016/S0896-6273(00)80353-0
17. Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. (1998) 78:189–225. doi: 10.1152/physrev.1998.78.1.189
18. Beaulieu JM, Espinoza S, Gainetdinov RR. Dopamine receptors - IUPHAR Review 13. Br J Pharmacol. (2015) 172:1–23. doi: 10.1111/bph.12906
19. Carlsson A. Perspectives on the discovery of central monoaminergic neurotransmission. Annu Rev Neurosci. (1987) 10:19–40. doi: 10.1146/annurev.ne.10.030187.000315
20. Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, et al. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron (1997) 19:1285–96. doi: 10.1016/S0896-6273(00)80419-5
21. Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. (1993) 14:43–9. doi: 10.1016/0165-6147(93)90029-J
22. Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci. (2002) 22:389–95. doi: 10.1523/JNEUROSCI.22-02-00389.2002
23. Smith CC, Greene RW. CNS dopamine transmission mediated by noradrenergic innervation. J Neurosci. (2012) 32:6072–80. doi: 10.1523/JNEUROSCI.6486-11.2012
24. Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. (2004) 56:331–49. doi: 10.1124/pr.56.3.1
25. De Mei C, Ramos M, Iitaka C, Borrelli E. Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr Opin Pharmacol. (2009) 9:53–8. doi: 10.1016/j.coph.2008.12.002
26. Kebabian JW, Greengard P. Dopamine-sensitive adenyl cyclase: possible role in synaptic transmission. Science (1971) 174:1346–9. doi: 10.1126/science.174.4016.1346
27. Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature (1979) 277:93–6. doi: 10.1038/277093a0
28. Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. (2004) 44:269–96. doi: 10.1146/annurev.pharmtox.44.101802.121415
29. Castello J, Lefrancois B, Flajolet M, Greengard P, Friedman E, Rebholz H. CK2 regulates 5-HT4 receptor signaling and modulates depressive-like behavior. Mol Psychiatry (2018) 23:872–82. doi: 10.1038/mp.2017.240
30. Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, et al. Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. (2004) 279:35671–8. doi: 10.1074/jbc.M401923200
31. Medvedev IO, Ramsey AJ, Masoud ST, Bermejo MK, Urs N, Sotnikova TD, et al. D1 dopamine receptor coupling to PLCbeta regulates forward locomotion in mice. J Neurosci. (2013) 33:18125–33. doi: 10.1523/JNEUROSCI.2382-13.2013
32. Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD, et al. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron (2003) 38:291–303. doi: 10.1016/S0896-6273(03)00192-2
33. Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. (2004) 27:107–44. doi: 10.1146/annurev.neuro.27.070203.144206
34. Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev. (2010) 62:305–30. doi: 10.1124/pr.109.002436
35. Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. (2004) 101:5099–104. doi: 10.1073/pnas.0307921101
36. Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell (2005) 122:261–73. doi: 10.1016/j.cell.2005.05.012
37. Ochs SM, Dorostkar MM, Aramuni G, Schon C, Filser S, Poschl J, et al. Loss of neuronal GSK3beta reduces dendritic spine stability and attenuates excitatory synaptic transmission via beta-catenin. Mol Psychiatry (2015) 20:482–9. doi: 10.1038/mp.2014.55
38. Urs NM, Snyder JC, Jacobsen JP, Peterson SM, Caron MG. Deletion of GSK3beta in D2R-expressing neurons reveals distinct roles for beta-arrestin signaling in antipsychotic and lithium action. Proc Natl Acad Sci USA. (2012) 109:20732–7. doi: 10.1073/pnas.1215489109
39. Del'Guidice T, Latapy C, Rampino A, Khlghatyan J, Lemasson M, Gelao B, et al. FXR1P is a GSK3beta substrate regulating mood and emotion processing. Proc Natl Acad Sci USA. (2015) 112:E4610–9. doi: 10.1073/pnas.1506491112
40. Cook D, Nuro E, Jones EV, Altimimi HF, Farmer WT, Gandin V, et al. FXR1P limits long-term memory, long-lasting synaptic potentiation, and de novo GluA2 translation. Cell Rep. (2014) 9:1402–16. doi: 10.1016/j.celrep.2014.10.028
41. Khlghatyan J, Evstratova A, Chamberland S, Marakhovskaia A, Bahremand A, Toth K, et al. Mental illnesses-associated Fxr1 and its negative regulator Gsk3beta are modulators of anxiety and glutamatergic neurotransmission. Front Mol Neurosci. (2018a) 11:119. doi: 10.3389/fnmol.2018.00119
42. Khlghatyan J, Beaulieu JM. Are FXR family proteins integrators of dopamine signaling and glutamatergic neurotransmission in mental illnesses? Front Synaptic Neurosci. (2018) 10:22. doi: 10.3389/fnsyn.2018.00022
43. Suzuki A, Mihara K, Kondo T, Tanaka O, Nagashima U, Otani K, et al. The relationship between dopamine D2 receptor polymorphism at the Taq1 A locus and therapeutic response to nemonapride, a selective dopamine antagonist, in schizophrenic patients. Pharmacogenetics (2000) 10:335–41. doi: 10.1097/00008571-200006000-00007
44. Schafer M, Rujescu D, Giegling I, Guntermann A, Erfurth A, Bondy B, et al. Association of short-term response to haloperidol treatment with a polymorphism in the dopamine D(2) receptor gene. Am J Psychiatry (2001) 158:802–4. doi: 10.1176/appi.ajp.158.5.802
45. Ikeda M, Yamanouchi Y, Kinoshita Y, Kitajima T, Yoshimura R, Hashimoto S, et al. Variants of dopamine and serotonin candidate genes as predictors of response to risperidone treatment in first-episode schizophrenia. Pharmacogenomics (2008) 9:1437–43. doi: 10.2217/14622416.9.10.1437
46. Shen YC, Chen SF, Chen CH, Lin CC, Chen SJ, Chen YJ, et al. Effects of DRD2/ANKK1 gene variations and clinical factors on aripiprazole efficacy in schizophrenic patients. J Psychiatr Res. (2009) 43:600–6. doi: 10.1016/j.jpsychires.2008.09.005
47. Lane HY, Lee CC, Chang YC, Lu CT, Huang CH, Chang WH. Effects of dopamine D2 receptor Ser311Cys polymorphism and clinical factors on risperidone efficacy for positive and negative symptoms and social function. Int J Neuropsychopharmacol. (2004) 7:461–70. doi: 10.1017/S1461145704004389
48. Hwang R, Shinkai T, De Luca V, Muller DJ, Ni X, Macciardi F, et al. Association study of 12 polymorphisms spanning the dopamine D(2) receptor gene and clozapine treatment response in two treatment refractory/intolerant populations. Psychopharmacology (2005) 181:179–87. doi: 10.1007/s00213-005-2223-5
49. Wu S, Xing Q, Gao R, Li X, Gu N, Feng G, et al. Response to chlorpromazine treatment may be associated with polymorphisms of the DRD2 gene in Chinese schizophrenic patients. Neurosci Lett. (2005) 376:1–4. doi: 10.1016/j.neulet.2004.11.014
50. Zhang JP, Robinson DG, Gallego JA, John M, Yu J, Addington J, et al. Association of a schizophrenia risk variant at the DRD2 locus with antipsychotic treatment response in first-episode psychosis. Schizophr Bull. (2015) 41:1248–55. doi: 10.1093/schbul/sbv116
51. Jonsson EG, Burgert E, Crocq MA, Gustavsson JP, Forslund K, Mattila-Evenden M, et al. Association study between dopamine D3 receptor gene variant and personality traits. Am J Med Genet B Neuropsychiatr Genet. (2003) 117B:61–5. doi: 10.1002/ajmg.b.10009
52. Adams DH, Close S, Farmen M, Downing AM, Breier A, Houston JP. Dopamine receptor D3 genotype association with greater acute positive symptom remission with olanzapine therapy in predominately caucasian patients with chronic schizophrenia or schizoaffective disorder. Hum Psychopharmacol. (2008) 23:267–74. doi: 10.1002/hup.930
53. Chen SF, Shen YC, Chen CH. Effects of the DRD3 Ser9Gly polymorphism on aripiprazole efficacy in schizophrenic patients as modified by clinical factors. Prog Neuropsychopharmacol Biol Psychiatry (2009) 33:470–4. doi: 10.1016/j.pnpbp.2009.01.007
54. Hwu HG, Hong CJ, Lee YL, Lee PC, Lee SF. Dopamine D4 receptor gene polymorphisms and neuroleptic response in schizophrenia. Biol Psychiatry (1998) 44:483–7. doi: 10.1016/S0006-3223(98)00134-6
55. Cohen BM, Ennulat DJ, Centorrino F, Matthysse S, Konieczna H, Chu HM, et al. Polymorphisms of the dopamine D4 receptor and response to antipsychotic drugs. Psychopharmacology (1999) 141:6–10. doi: 10.1007/s002130050799
56. Zhao AL, Zhao JP, Zhang YH, Xue ZM, Chen JD, Chen XG. Dopamine D4 receptor gene exon III polymorphism and interindividual variation in response to clozapine. Int J Neurosci. (2005) 115:1539–47. doi: 10.1080/00207450590957863
57. Pai P, Arathil P, Kotambail A, Nair R, Gupta M, Moily NS, et al. Association of GRIN1, ABCB1, and DRD4 genes and response to antipsychotic drug treatment in schizophrenia. Psychiatr Genet. (2015) 25:135–6. doi: 10.1097/YPG.0000000000000079
58. Illi A, Mattila KM, Kampman O, Anttila S, Roivas M, Lehtimaki T, et al. Catechol-O-methyltransferase and monoamine oxidase A genotypes and drug response to conventional neuroleptics in schizophrenia. J Clin Psychopharmacol. (2003) 23:429–34. doi: 10.1097/01.jcp.0000088916.02635.33
59. Bertolino A, Caforio G, Blasi G, Rampino A, Nardini M, Weinberger DR, et al. COMT Val158Met polymorphism predicts negative symptoms response to treatment with olanzapine in schizophrenia. Schizophr Res. (2007) 95:253–5. doi: 10.1016/j.schres.2007.06.014
60. Woodward ND, Jayathilake K, Meltzer HY. COMT val108/158met genotype, cognitive function, and cognitive improvement with clozapine in schizophrenia. Schizophr Res. (2007) 90:86–96. doi: 10.1016/j.schres.2006.10.002
61. Anttila S, Illi A, Kampman O, Mattila KM, Lehtimaki T, Leinonen E. Interaction between NOTCH4 and catechol-O-methyltransferase genotypes in schizophrenia patients with poor response to typical neuroleptics. Pharmacogenetics (2004) 14:303–7. doi: 10.1097/00008571-200405000-00005
62. Molero P, Ortuno F, Zalacain M, Patino-Garcia A. Clinical involvement of catechol-O-methyltransferase polymorphisms in schizophrenia spectrum disorders: influence on the severity of psychotic symptoms and on the response to neuroleptic treatment. Pharmacogenomics J. (2007) 7:418–26. doi: 10.1038/sj.tpj.6500441
63. Fijal BA, Kinon BJ, Kapur S, Stauffer VL, Conley RR, Jamal HH, et al. Candidate-gene association analysis of response to risperidone in African-American and white patients with schizophrenia. Pharmacogenomics J. (2009) 9:311–8. doi: 10.1038/tpj.2009.24
64. Gupta M, Bhatnagar P, Grover S, Kaur H, Baghel R, Bhasin Y, et al. Association studies of catechol-O-methyltransferase (COMT) gene with schizophrenia and response to antipsychotic treatment. Pharmacogenomics (2009a) 10:385–97. doi: 10.2217/14622416.10.3.385
65. Bilic P, Jukic V, Vilibic M, Savic A, Bozina N. Treatment-resistant schizophrenia and DAT and SERT polymorphisms. Gene (2014) 543:125–32. doi: 10.1016/j.gene.2014.03.050
66. Mcclay JL, Adkins DE, Aberg K, Stroup S, Perkins DO, Vladimirov VI, et al. Genome-wide pharmacogenomic analysis of response to treatment with antipsychotics. Mol Psychiatry (2011b) 16:76–85. doi: 10.1038/mp.2009.89
67. Blasi G, Napolitano F, Ursini G, Taurisano P, Romano R, Caforio G, et al. DRD2/AKT1 interaction on D2 c-AMP independent signaling, attentional processing, and response to olanzapine treatment in schizophrenia. Proc Natl Acad Sci USA. (2011) 108:1158–63. doi: 10.1073/pnas.1013535108
68. Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, et al. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. (2003) 12:205–16. doi: 10.1093/hmg/ddg055
69. Jonsson EG, Nothen MM, Grunhage F, Farde L, Nakashima Y, Propping P, et al. Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers. Mol Psychiatry (1999) 4:290–6. doi: 10.1038/sj.mp.4000532
70. Thompson J, Thomas N, Singleton A, Piggott M, Lloyd S, Perry EK, et al. D2 dopamine receptor gene (DRD2) Taq1 A polymorphism: reduced dopamine D2 receptor binding in the human striatum associated with the A1 allele. Pharmacogenetics (1997) 7:479–84. doi: 10.1097/00008571-199712000-00006
71. Soderqvist S, Matsson H, Peyrard-Janvid M, Kere J, Klingberg T. Polymorphisms in the dopamine receptor 2 gene region influence improvements during working memory training in children and adolescents. J Cogn Neurosci. (2014) 26:54–62. doi: 10.1162/jocn_a_00478
72. Itokawa M, Arinami T, Futamura N, Hamaguchi H, Toru M. A structural polymorphism of human dopamine D2 receptor, D2(Ser311–>Cys). Biochem Biophys Res Commun. (1993) 196:1369–75.
73. Arinami T, Itokawa M, Enguchi H, Tagaya H, Yano S, Shimizu H, et al. Association of dopamine D2 receptor molecular variant with schizophrenia. Lancet (1994) 343:703–4. doi: 10.1016/S0140-6736(94)91581-4
74. Glatt SJ, Jonsson EG. The Cys allele of the DRD2 Ser311Cys polymorphism has a dominant effect on risk for schizophrenia: evidence from fixed- and random-effects meta-analyses. Am J Med Genet B Neuropsychiatr Genet. (2006) 141B, 149–54. doi: 10.1002/ajmg.b.30273
75. Zhang Y, Bertolino A, Fazio L, Blasi G, Rampino A, Romano R, et al. Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc Natl Acad Sci USA. (2007) 104:20552–7. doi: 10.1073/pnas.0707106104
76. Allen NC, Bagade S, Mcqueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. (2008) 40:827–34. doi: 10.1038/ng.171
77. Blasi G, Lo Bianco L, Taurisano P, Gelao B, Romano R, Fazio L, et al. Functional variation of the dopamine D2 receptor gene is associated with emotional control as well as brain activity and connectivity during emotion processing in humans. J Neurosci. (2009) 29:14812–9. doi: 10.1523/JNEUROSCI.3609-09.2009
78. Bertolino A, Fazio L, Caforio G, Blasi G, Rampino A, Romano R, et al. Functional variants of the dopamine receptor D2 gene modulate prefronto-striatal phenotypes in schizophrenia. Brain (2009) 132:417–25. doi: 10.1093/brain/awn248
79. Bertolino A, Taurisano P, Pisciotta NM, Blasi G, Fazio L, Romano R, et al. Genetically determined measures of striatal D2 signaling predict prefrontal activity during working memory performance. PLoS ONE (2010) 5:e9348. doi: 10.1371/journal.pone.0009348
80. Blasi G, Napolitano F, Ursini G, Di Giorgio A, Caforio G, Taurisano P, et al. Association of GSK-3beta genetic variation with GSK-3beta expression, prefrontal cortical thickness, prefrontal physiology, and schizophrenia. Am J Psychiatry (2013) 170:868–76. doi: 10.1176/appi.ajp.2012.12070908
81. Rampino A, Di Carlo P, Fazio L, Ursini G, Pergola G, De Virgilio C, et al. Association of functional genetic variation in PP2A with prefrontal working memory processing. Behav Brain Res. (2017) 316:125–30. doi: 10.1016/j.bbr.2016.08.054
82. Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. Am J Psychiatry (2010) 167:763–72. doi: 10.1176/appi.ajp.2009.09040598
83. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. (2011) 7:9–37. doi: 10.1517/17425255.2011.532787
84. Jeanneteau F, Funalot B, Jankovic J, Deng H, Lagarde JP, Lucotte G, et al. A functional variant of the dopamine D3 receptor is associated with risk and age-at-onset of essential tremor. Proc Natl Acad Sci USA. (2006) 103:10753–8. doi: 10.1073/pnas.0508189103
85. Asenjo Lobos C, Komossa K, Rummel-Kluge C, Hunger H, Schmid F, Schwarz S, et al. Clozapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. (2010) 10:CD006633. doi: 10.1002/14651858.CD006633
86. Gonzalez-Castro TB, Hernandez-Diaz Y, Juarez-Rojop IE, Lopez-Narvaez ML, Tovilla-Zarate CA, Fresan A. The role of a Catechol-O-Methyltransferase (COMT) Val158Met genetic polymorphism in schizophrenia: a systematic review and updated meta-analysis on 32,816 subjects. Neuromol Med. (2016) 18:216–31. doi: 10.1007/s12017-016-8392-z
87. Gupta M, Chauhan C, Bhatnagar P, Gupta S, Grover S, Singh PK, et al. Genetic susceptibility to schizophrenia: role of dopaminergic pathway gene polymorphisms. Pharmacogenomics (2009b) 10:277–91. doi: 10.2217/14622416.10.2.277
88. Vandenbergh DJ, Persico AM, Hawkins AL, Griffin CA, Li X, Jabs EW, et al. Human dopamine transporter gene (DAT1) maps to chromosome 5p15.3 and displays a VNTR. Genomics (1992a) 14:1104–6. doi: 10.1016/S0888-7543(05)80138-7
89. Vandenbergh DJ, Persico AM, Uhl GR. A human dopamine transporter cDNA predicts reduced glycosylation, displays a novel repetitive element and provides racially-dimorphic TaqI RFLPs. Brain Res Mol Brain Res. (1992b) 15:161–6. doi: 10.1016/0169-328X(92)90165-8
90. Mill J, Asherson P, Browes C, D'souza U, Craig I. Expression of the dopamine transporter gene is regulated by the 3' UTR VNTR: evidence from brain and lymphocytes using quantitative RT-PCR. Am J Med Genet. (2002) 114:975–9. doi: 10.1002/ajmg.b.10948
91. Vanness SH, Owens MJ, Kilts CD. The variable number of tandem repeats element in DAT1 regulates in vitro dopamine transporter density. BMC Genet. (2005) 6:55. doi: 10.1186/1471-2156-6-55
92. Heinz A, Goldman D, Jones DW, Palmour R, Hommer D, Gorey JG, et al. Genotype influences in vivo dopamine transporter availability in human striatum. Neuropsychopharmacology (2000) 22:133–9. doi: 10.1016/S0893-133X(99)00099-8
93. Fossella J, Sommer T, Fan J, Wu Y, Swanson JM, Pfaff DW, et al. Assessing the molecular genetics of attention networks. BMC Neurosci. (2002) 3:14. doi: 10.1186/1471-2202-3-14
94. Cornish KM, Manly T, Savage R, Swanson J, Morisano D, Butler N, et al. Association of the dopamine transporter (DAT1) 10/10-repeat genotype with ADHD symptoms and response inhibition in a general population sample. Mol Psychiatry (2005) 10:686–98. doi: 10.1038/sj.mp.4001641
95. Bertolino A, Blasi G, Latorre V, Rubino V, Rampino A, Sinibaldi L, et al. Additive effects of genetic variation in dopamine regulating genes on working memory cortical activity in human brain. J Neurosci. (2006) 26:3918–22. doi: 10.1523/JNEUROSCI.4975-05.2006
96. Johanson CE, Frey KA, Lundahl LH, Keenan P, Lockhart N, Roll J, et al. Cognitive function and nigrostriatal markers in abstinent methamphetamine abusers. Psychopharmacology (2006) 185:327–38. doi: 10.1007/s00213-006-0330-6
97. Schott BH, Seidenbecher CI, Fenker DB, Lauer CJ, Bunzeck N, Bernstein HG, et al. The dopaminergic midbrain participates in human episodic memory formation: evidence from genetic imaging. J Neurosci. (2006) 26:1407–17. doi: 10.1523/JNEUROSCI.3463-05.2006
98. Caldu X, Vendrell P, Bartres-Faz D, Clemente I, Bargallo N, Jurado MA, et al. Impact of the COMT Val108/158 Met and DAT genotypes on prefrontal function in healthy subjects. Neuroimage (2007) 37:1437–44. doi: 10.1016/j.neuroimage.2007.06.021
99. Xu M, Xing Q, Li S, Zheng Y, Wu S, Gao R, et al. Pharacogenetic effects of dopamine transporter gene polymorphisms on response to chlorpromazine and clozapine and on extrapyramidal syndrome in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2010) 34:1026–32. doi: 10.1016/j.pnpbp.2010.05.017
100. Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. (2005) 353:1209–23. doi: 10.1056/NEJMoa051688
101. Mcclay JL, Adkins DE, Aberg K, Bukszar J, Khachane AN, Keefe RS, et al. Genome-wide pharmacogenomic study of neurocognition as an indicator of antipsychotic treatment response in schizophrenia. Neuropsychopharmacology (2011a) 36:616–26. doi: 10.1038/npp.2010.193
102. Lavedan C, Licamele L, Volpi S, Hamilton J, Heaton C, Mack K, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry (2009) 14:804–19. doi: 10.1038/mp.2008.56
103. Volpi S, Potkin SG, Malhotra AK, Licamele L, Lavedan C. Applicability of a genetic signature for enhanced iloperidone efficacy in the treatment of schizophrenia. J Clin Psychiatry (2009) 70:801–9. doi: 10.4088/JCP.08m04391
104. Mcguire P, Howes OD, Stone J, Fusar-Poli P. Functional neuroimaging in schizophrenia: diagnosis and drug discovery. Trends Pharmacol Sci. (2008) 29:91–8. doi: 10.1016/j.tips.2007.11.005
105. Howes O, Mccutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol. (2015) 29:97–115. doi: 10.1177/0269881114563634
106. Vernaleken I, Klomp M, Moeller O, Raptis M, Nagels A, Rosch F, et al. Vulnerability to psychotogenic effects of ketamine is associated with elevated D2/3-receptor availability. Int J Neuropsychopharmacol. (2013) 16:745–54. doi: 10.1017/S1461145712000764
107. Stevenson JM, Reilly JL, Harris MS, Patel SR, Weiden PJ, Prasad KM, et al. Antipsychotic pharmacogenomics in first episode psychosis: a role for glutamate genes. Transl Psychiatry (2016) 6:e739. doi: 10.1038/tp.2016.10
108. Ruderfer DM, Charney AW, Readhead B, Kidd BA, Kahler AK, Kenny PJ, et al. Polygenic overlap between schizophrenia risk and antipsychotic response: a genomic medicine approach. Lancet Psychiatry (2016) 3:350–7. doi: 10.1016/S2215-0366(15)00553-2
109. Difrancesco JC, Difrancesco D. Dysfunctional HCN ion channels in neurological diseases. Front Cell Neurosci. (2015) 6:174. doi: 10.3389/fncel.2015.00071
110. Terrillion CE, Dao DT, Cachope R, Lobo MK, Puche AC, Cheer JF, et al. Reduced levels of Cacna1c attenuate mesolimbic dopamine system function. Genes Brain Behav. (2017) 16:495–505. doi: 10.1111/gbb.12371
111. Frank J, Lang M, Witt SH, Strohmaier J, Rujescu D, Cichon S, et al. Identification of increased genetic risk scores for schizophrenia in treatment-resistant patients. Mol Psychiatry (2015) 20:913. doi: 10.1038/mp.2015.52
112. Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell (1993) 74:291–8. doi: 10.1016/0092-8674(93)90420-U
113. Siomi MC, Siomi H, Sauer WH, Srinivasan S, Nussbaum RL, Dreyfuss G. FXR1, an autosomal homolog of the fragile X mental retardation gene. EMBO J. (1995) 14:2401–8. doi: 10.1002/j.1460-2075.1995.tb07237.x
114. Tamanini F, Willemsen R, Van Unen L, Bontekoe C, Galjaard H, Oostra BA, et al. Differential expression of FMR1, FXR1 and FXR2 proteins in human brain and testis. Hum Mol Genet. (1997) 6:1315–22. doi: 10.1093/hmg/6.8.1315
115. Dube M, Huot ME, Khandjian EW. Muscle specific fragile X related protein 1 isoforms are sequestered in the nucleus of undifferentiated myoblast. BMC Genet. (2000) 1:4. doi: 10.1186/1471-2156-1-4
116. Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell (2001) 107:489–99. doi: 10.1016/S0092-8674(01)00566-9
117. Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell (1991) 65:905–14. doi: 10.1016/0092-8674(91)90397-H
118. Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell (2011) 146:247–61. doi: 10.1016/j.cell.2011.06.013
119. Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. (2012) 7:219–45. doi: 10.1146/annurev-pathol-011811-132457
120. Zhang J, Hou L, Klann E, Nelson DL. Altered hippocampal synaptic plasticity in the FMR1 gene family knockout mouse models. J Neurophysiol. (2009) 101:2572–80. doi: 10.1152/jn.90558.2008
121. Adams-Cioaba MA, Guo Y, Bian C, Amaya MF, Lam R, Wasney GA, et al. Structural studies of the tandem Tudor domains of fragile X mental retardation related proteins FXR1 and FXR2. PLoS ONE (2010) 5:e13559. doi: 10.1371/journal.pone.0013559
122. Eberhart DE, Malter HE, Feng Y, Warren ST. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet. (1996) 5:1083–91. doi: 10.1093/hmg/5.8.1083
123. Siomi MC, Zhang Y, Siomi H, Dreyfuss G. Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol Cell Biol. (1996) 16:3825–32. doi: 10.1128/MCB.16.7.3825
124. Kirkpatrick LL, Mcilwain KA, Nelson DL. Comparative genomic sequence analysis of the FXR gene family: FMR1, FXR1, and FXR2. Genomics (2001) 78:169–77. doi: 10.1006/geno.2001.6667
125. Ascano MJr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature (2012) 492:382–6. doi: 10.1038/nature11737
126. Leonenko G, Richards AL, Walters JT, Pocklington A, Chambert K, Al Eissa MM, et al. Mutation intolerant genes and targets of FMRP are enriched for nonsynonymous alleles in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. (2017) 174:724–31. doi: 10.1002/ajmg.b.32560
127. Folsom TD, Thuras PD, Fatemi SH. Protein expression of targets of the FMRP regulon is altered in brains of subjects with schizophrenia and mood disorders. Schizophr Res. (2015) 165:201–11. doi: 10.1016/j.schres.2015.04.012
128. El Fatimy R, Davidovic L, Tremblay S, Jaglin X, Dury A, Robert C, et al. Tracking the fragile X mental retardation protein in a highly ordered neuronal ribonucleoparticles population: a link between stalled polyribosomes and RNA granules. PLoS Genet. (2016) 12:e1006192. doi: 10.1371/journal.pgen.1006192
129. Huot ME, Mazroui R, Leclerc P, Khandjian EW. Developmental expression of the fragile X-related 1 proteins in mouse testis: association with microtubule elements. Hum Mol Genet. (2001) 10:2803–11. doi: 10.1093/hmg/10.24.2803
130. Takata A, Matsumoto N, Kato T. Genome-wide identification of splicing QTLs in the human brain and their enrichment among schizophrenia-associated loci. Nat Commun. (2017) 8:14519. doi: 10.1038/ncomms14519
131. Mitchell CJ, Getnet D, Kim MS, Manda SS, Kumar P, Huang TC, et al. A multi-omic analysis of human naive CD4+ T cells. BMC Syst Biol. (2015) 9:75. doi: 10.1186/s12918-015-0225-4
132. Tamanini F, Kirkpatrick LL, Schonkeren J, Van Unen L, Bontekoe C, Bakker C, et al. The fragile X-related proteins FXR1P and FXR2P contain a functional nucleolar-targeting signal equivalent to the HIV-1 regulatory proteins. Hum Mol Genet. (2000) 9:1487–93. doi: 10.1093/hmg/9.10.1487
133. Taha MS, Nouri K, Milroy LG, Moll JM, Herrmann C, Brunsveld L, et al. Subcellular fractionation and localization studies reveal a direct interaction of the fragile X mental retardation protein (FMRP) with nucleolin. PLoS ONE (2014) 9:e91465. doi: 10.1371/journal.pone.0091465
134. Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. (2009) 49:327–47. doi: 10.1146/annurev.pharmtox.011008.145634
135. Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, et al. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell (2008) 132:125–36. doi: 10.1016/j.cell.2007.11.041
136. Del'Guidice T, Beaulieu JM. Selective disruption of dopamine D2-receptors/beta-arrestin2 signaling by mood stabilizers. J Recept Signal Transduct Res. (2015) 35:224–32. doi: 10.3109/10799893.2015.1072976
Keywords: dopamine, risk factors, antipsychotic agents, genetic variants, schizophrenia therapy
Citation: Rampino A, Marakhovskaia A, Soares-Silva T, Torretta S, Veneziani F and Beaulieu JM (2019) Antipsychotic Drug Responsiveness and Dopamine Receptor Signaling; Old Players and New Prospects. Front. Psychiatry 9:702. doi: 10.3389/fpsyt.2018.00702
Received: 27 June 2018; Accepted: 03 December 2018;
Published: 09 January 2019.
Edited by:
Mirko Manchia, Università degli Studi di Cagliari, ItalyReviewed by:
Kimberly Frances Raab-Graham, Wake Forest School of Medicine, United StatesStefano Comai, Vita-Salute San Raffaele University, Italy
Copyright © 2019 Rampino, Marakhovskaia, Soares-Silva, Torretta, Veneziani and Beaulieu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean Martin Beaulieu, bWFydGluLmJlYXVsaWV1QHV0b3JvbnRvLmNh