 Leszek Rudzki
Leszek Rudzki Agata Szulc
Agata Szulc
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 29 May 2018
Sec. Psychopharmacology
Volume 9 - 2018 | https://doi.org/10.3389/fpsyt.2018.00205
This article is part of the Research TopicPeripheral Markers of Immune Response in Major Psychiatric Disorders: Where Are We Now and Where Do We Want to Be?View all 12 articles
Interaction between the gastrointestinal tract (GI) and brain functions has recently become a topic of growing interest in psychiatric research. These multidirectional interactions take place in the so-called gut-brain axis or more precisely, the microbiota-gut-brain axis. The GI tract is the largest immune organ in the human body and is also the largest surface of contact with the external environment. Its functions and permeability are highly influenced by psychological stress, which are often a precipitating factor in the first episode, reoccurrence and/or deterioration of symptoms of psychiatric disorders. In recent literature there is growing evidence that increased intestinal permeability with subsequent immune activation has a major role in the pathophysiology of various psychiatric disorders. Numerous parameters measured in this context seem to be aftermaths of those mechanisms, yet at the same time they may be contributing factors for immune mediated psychopathology. For example, immune activation related to gut-derived bacterial lipopolysaccharides (LPS) or various food antigens and exorphins were reported in major depression, schizophrenia, bipolar disorder, alcoholism and autism. In this review the authors will summarize the evidence and roles of such parameters and their assessment in major psychiatric disorders.
In the last two decades, significant progress has been made in our understanding of the role of the immune system and inflammatory processes in the pathogenesis of psychiatric disorders. A recent discovery, published in NATURE (1), that the central nervous system (CNS) has its own lymphatic system is a spectacular yet thought-provoking realization; that in the vast oceans of exponentially growing amounts of scientific data, there are still major “unknowns,” which could redefine “the bigger picture.” Thanks to the synthesis of philosophy that “you cannot see the forest while looking at the leaf” along with recent fascinating discoveries of microbiotic and psychoneuroimmune complexities of the microbiota-gut-brain axis, we are now able to take a few steps back to have another, broader look at the role of the GI tract in various inflammatory, autoimmune and numerous psychiatric disorders.
The role of the GI tract in the pathogenesis of psychiatric disorders came into the scientific debate at the beginning of twentieth century (2). Buscaino reported various inflammatory changes in the GI tract in the post mortem examination of 82 patients suffering from schizophrenia. Fifty percent of those patients had manifestations of gastritis, 88% enteritis and 92% colitis (2, 3). Asperger also noted connections between celiac disease and psychotic disorders in his work (4). Baruk in his extensive work on schizophrenia pointed out the significant role of the GI tract, intestinal toxins and infection in the context of schizophrenia and catatonia (5–8). In 1979 Dohan suggested a fascinating hypothesis that “Basic biological defect in schizophrenia is genetic impairment (e.g., via defective enzymes or, receptors) of the gut and other barrier systems which eases the passage of food-derived neuroactive polypeptides from gut lumen to brain cells” (9). In this hypothesis he suggested that impairment of both intestinal and blood-brain-barrier (BBB) could contribute to the pathogenesis of schizophrenia.
Nowadays extensive data has revealed the indisputable role of immunity and inflammation in psychiatric disorders (10–22). The GI tract with its gut-associated lymphoid tissue (GALT) is the largest immune organ of the human organism and it produces 70–80% of immune cells. Consequently, its role in psychopathology is no longer controversial and it is drawing a lot of attention in neuroscience.
Connotation of the word Stress usually relates to its psychological perspective. It is mostly perceived as the feeling of fear, threat, anger, frustration, hatred, insecurity, abandonment, and unpredictability. Stress reaction may also take the form of the fight-flight-freeze response. However, stress is non-specific and for the human organism it has a much broader meaning. Inflammation, viral, bacterial or parasitic infections, injury, exposure to various toxins, radiation, oxidative and nitrosative stress, and excessive physical training are also recognized as stress by the human organism. The body's reaction to various stressors is relatively uniform, whether it is facing psychological or physical stressors. On one hand, stress may activate the immune system and inflammatory response, e.g., via an elevated level of pro-inflammatory cytokines, and the trafficking of immune cells between blood and tissues. This activation is preparing the organism to “face and fight” potential threats. On the other hand, stress response leads to the activation of the hypothalamic–pituitary–adrenal axis (HPA) and to the increased secretion of anti-inflammatory adrenal hormone, cortisol. This “safety switch” is supposed to prevent an excessive activation of potentially destructive inflammatory response (23–25). Interestingly, all of the stressors mentioned above can directly or indirectly lead to increased intestinal permeability and its various immune and psychopathological consequences. The GI tract forms the largest surface, about 300 m2, of interaction between the internal and external environment of the human body (26). The intestinal barrier constitutes of one layer epithelium composed of enterocytes interconnected by protein junctional complexes—tight junctions (zonulae occludentes). Permeation of molecules from the intestinal lumen is both transcellular and paracellular, and the opening of tight junctions regulate the latter (27). Moreover, the mucosal layer and intestinal microbiota are also crucial elements of this barrier and they are determining its permeability (28, 29). Psychological stress mediated by corticoliberin (CRH) (26, 30–33), proinflammatory cytokines e.g., IL-1β (34), TNF-α (35, 36), INF-γ (37), dysbiosis (38, 39), small intestine bacterial overgrowth (SIBO) (40), bacterial, parasitic or fungal infections (41, 42), oxidative and nitrosative stress (32, 43), the nuclear factor NF-κB (44), prolonged strenuous exercise (45, 46), heat stress (47), alcohol (38, 48–50), food additives (51), certain drugs e.g., non-steroidal anti-inflammatory drugs (NSAIDs) (52, 53) or antibiotics (54–57) can cause the loss of intestinal barrier integrity leading to its excessive permeability (Figure 1). This may lead to subsequent immune activation with further consequences in the CNS (39). It is worth mentioning that the structure and mechanics of the BBB are in many ways similar to the GI barrier (58), and BBB permeability may also be compromised by analogous factors to those compromising the GI barrier e.g., psychological stress (59), pro-inflammatory cytokines (60–66), oxidative and nitrosative stress (63, 67, 68) (Figure 1).
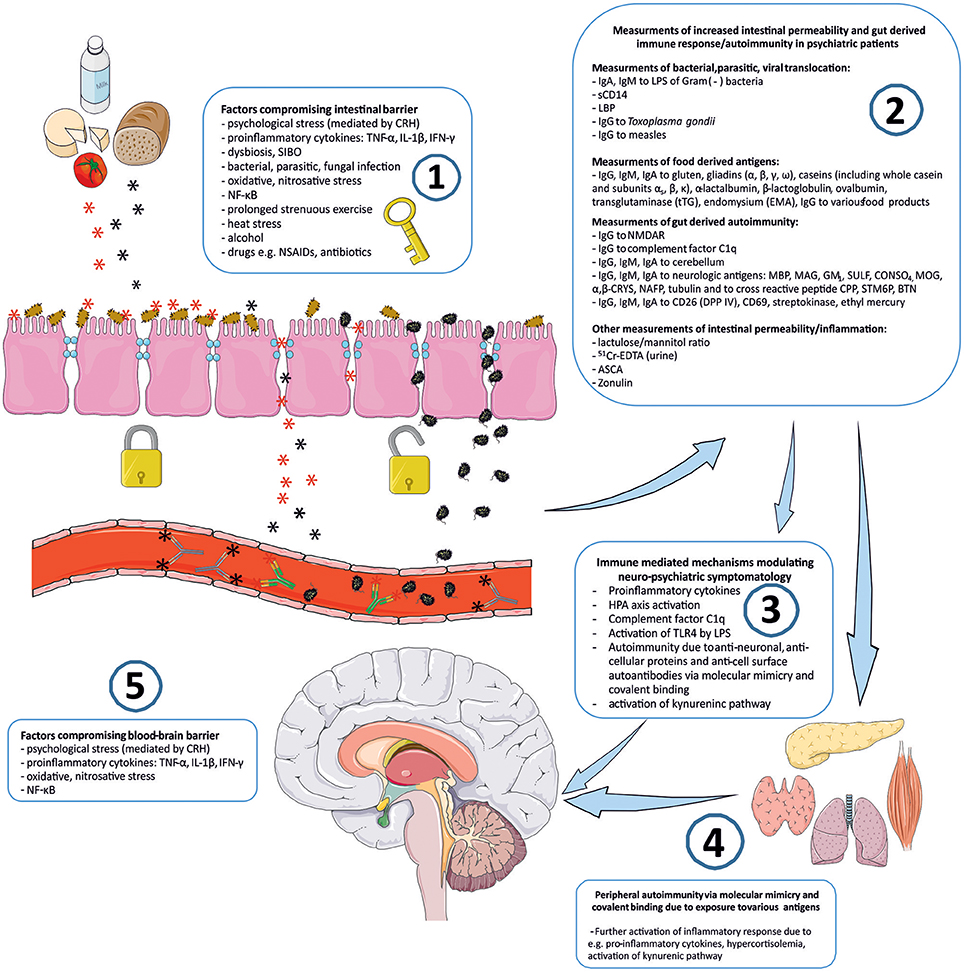
Figure 1. “Immune Gate” of psychopathology – mechanisms of gut derived immune activation leading to psychiatric manifestations. Image generated using Servier Medical Art. 1: Various detrimental factors compromising intestinal barrier lead to increased intestinal permeability. 2: Increased intestinal permeability is as a source of food derived and microbial, bacterial, parasitic antigens, and subsequent activation of inflammatory response with production of immunoglobulins against those antigens. Other markers of increased intestinal permeability. 3: Detrimental for the brain immune consequences of gut derived antigens modulate neuro-psychiatric symptomatology. 4: Peripheral autoimmunity (via molecular mimicry, covalent binding) due to various gut derived antigens and further activation of inflammatory response detrimental to CNS via e.g., pro-inflammatory cytokines, activation, and changes in kynurenic pathway metabolism. 5: Factors compromising blood-brain-barrier contribute to the periphery-derived activation of inflammatory response and its psychiatric and neurological manifestations. CRH, corticoliberin; SIBO, small intestine bacterial overgrowth; NF-κB, nuclear factor kappa B; NSAIDs, non-steroidal anti-inflammatory drugs; IgA, IgE, IgG, IgM, immunoglobulin A, E, G, M; LPS, bacterial lipopolysaccharide; LBP, lipopolysaccharide binding protein; sCD14, soluble CD14; NMDAR, N-methyl-D-aspartate glutamate receptor; MBP, myelin basic protein; MAG, myelin-associated glycoprotein; GM1, ganglioside; SULF, sufatide; CONSO4, chondroitin sulfate; MOG, myelin oligodendrocyte glycoprotein; α,β-CRYS, α,β-crystallin; NAFP, neurofilament proteins; CPP, Chlamydia pneumoniae; STM6P, streptococcal M protein; BTN, milk butyrophilin; DPP IV, dipeptidyl peptidase IV (synonym of CD26); 51Cr-EDTA, chromium ethylenediaminetetraacetic acid; ASCA, IgG to Saccharomyces cerevisiae; TLR4, Toll-like receptor 4.
There are various mechanisms for how increased intestinal permeability and gut-derived antigens can have immune-mediated consequences for the brain and behavior. Measurements of these mechanisms could serve as potential biomarkers for the involvement of the gut-brain axis in the psychopathology of patients and could also have significant value in broader therapeutic approach.
Bacterial lipopolysaccharides—LPS (a glycolipid complexes found in the outer membrane of Gram negative bacteria) have a profound influence on immunity and brain function, and upstream activation of Toll-like receptor 4 (TLR4) by LPS has a crucial role in these interactions (69, 70). Therefore, LPS challenge represents a laboratory model of inducing transient, low-grade inflammation with subsequent behavioral changes in animals and sickness behaviors in human subjects (71). Usually this challenge is performed through intravenous or intramuscular injection of LPS. However, some of the major consequences of the various stressors discussed above, including psychological stress, are dysfunction of the gut barrier, increased permeability, translocation of enteric bacteria from the intestinal lumen, and subsequent activation of the inflammatory response by LPS (29) (Figure 1). It was demonstrated that chronic psychological stress increased intestinal permeability of E. coli via follicle associated epithelium, by more than 30-fold and permeability to the antigenic protein horseradish peroxide (HRP), almost 4-fold. Moreover, serum corticosterone was significantly increased after 10 days of chronic stress and this was accompanied by a 3-fold increase in the number of mucosal mast cells. In this study acute stress also increased permeability to HRP (72). Bacterial LPS are able to induce enzymes of the kynurenic pathway, e.g., indoleamine 2,3 dioxygenase (IDO), the first step enzyme converting tryptophan toward the kynurenine pathway. It was demonstrated in animal studies that peripheral LPS challenge resulted in increased expression of brain pro-inflammatory cytokines and neuroinflammatory glial cellular markers. This was also accompanied with increased activity of brain kynurenic pathway enzymes with subsequent activation of neurotoxic branch of this pathway toward synthesis of detrimental kynurenines (73). In healthy humans administration of LPS resulted in increased body temperature, malaise, increased levels of cortisol and pro-inflammatory cytokines e.g., TNF-α, soluble TNF receptors, IL-6, IL-1, and IL-1 receptor antagonist. Furthermore, this low-grade immune response was accompanied by increased anxiety, depressed mood and a decrease in verbal and non-verbal memory functions (74, 75). Also, administering bacterial LPS had a dose dependent negative impact on cognitive functions (76). Moreover, bacterial translocation and LPS are known to induce monocyte activation and their trafficking into the CNS. This is considered to be a crucial mechanism of action in the pathogenesis of human immunodeficiency virus (HIV)-associated dementia (77).
Measurements of antibodies against LPS of Gram negative enterobacteria can be used as surrogate marker for the assessment of intestinal permeability. Due to increased intestinal permeability we can expect increased bacterial translocation and increased blood concertation of immunoglobulins against various enterobacteria. This approach was initially used by Maes et al. in assessing patients with chronic fatigue syndrome (CFS) (78) (Table 1). Patients suffering from this disorder had increased prevalence and median values of serum IgA against LPS of enterobacteria compared to those of healthy controls and patients with partial CFS. Additionally, IgA levels were significantly correlated to the severity of CFS, such as irritable bowel, muscular tension, fatigue, the inability to concentrate and failing memory. These results suggest that increased intestinal permeability to enterobacteria are involved in the etiology of CFS. In another study patients with CFS had increased levels of LPS, along with elevated levels of surrogate markers of bacterial translocation namely, soluble CD14 (sCD14) and lipopolysaccharide binding protein (LBP) (79). In the same study, profiling of gut microbial diversity by sequencing 16s ribosomal ribonucleic acid (rRNA) genes from stool, revealed reduction in diversity and abundance of bacteria belonging to Firmicutes phylum, and reduction of anti-inflammatory with the concurrent increase of pro-inflammatory bacterial species in CFS patients. Results of this study indicate that besides increased intestinal microbial translocation, dysbiosis of gut microflora may also play a role in inflammatory symptoms of CFS.
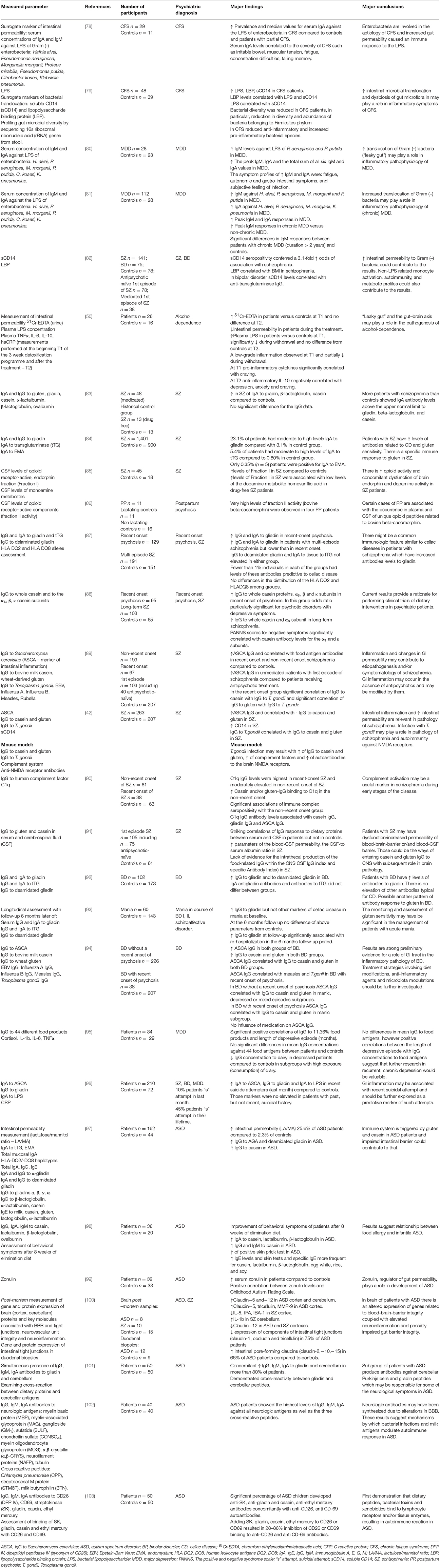
Table 1. Major research on the gut derived immunity in psychiatric disorders.
Furthermore, the role of increased intestinal permeability to enteric bacteria, also known as “leaky gut,” was demonstrated in two consecutive studies and it was suggested to be a potential pathophysiological mechanism of major depression (80, 81). In these studies (Table 1) serum concentrations of IgM and IgA were measured against LPS of six different enterobacteria: Hafnia alvei, Pseudomonas aeruginosa, Morganella morganii, Pseudomonas putida, Citrobacter koseri, and Klebsiella pneumonia. Levels of immunoglobulins were significantly higher in depressed patients compared to the control group. Interestingly there were also significantly higher IgM responses in patients suffering from chronic depression (duration > 2 years) when compared to patients with non-chronic depression and controls. This suggests that patients with chronic depression may also have increased intestinal permeability to a larger extent than non-chronic patients. This conclusion seems to be particularly valuable in further understanding of the mechanisms of treatment-resistant and chronic depression.
Excess alcohol consumption is a known factor for compromising the gut barrier, leading to increased intestinal permeability to macromolecules and bacterial endotoxins (38, 48–50). Furthermore, translocation of gut-derived bacterial toxins and related inflammatory response are believed to play a significant role in the development of progressive alcoholic liver injury (104–106). In alcohol-dependent patients, a 3 week alcohol detoxification programme resulted in significant improvement in intestinal barrier function, assessed using urine 51Cr-EDTA and plasma LPS levels (50). Additionally, parameters of systemic inflammations (TNF-α, IL-6, IL-10, hsCRP) partially decreased during withdrawal and inflammatory parameters correlated with depressive symptoms and alcohol-craving (Table 1). These results suggest that the gut-brain axis could also have significance in the pathogenesis of alcohol dependence and in affective symptomatology observed in this disorder.
Another group of parameters believed to be contributing factors in psychopathology are related to food derived antigens and exorphins (Figure 1). The current literature covering this topic is mostly focused on the involvement of glutens, caseins, and exorphins in disorders such as psychoses, schizophrenia, bipolar disorder, and autistic spectrum disorders (ASD) (Table 1). Glutens are storage proteins of various grass-related grains. They consist of numerous protein components and can be divided into two main fractions: the soluble in aqueous alcohols gliadins, and insoluble glutenins (107). Bovine milk is another source of food derived antigens and it consists of two major group of proteins: caseins and whey proteins. Caseins account for 80% of bovine milk proteins and they are divided into a further four major groups: αs1, αs2, β, and κ caseins (108). Some milk and gluten proteins can be a source of exorphins, which are peptides with morphine-like activity due to their ability to bind to opioid μ-receptors e.g., in the CNS or gastrointestinal tract (109). Moreover, exorphins can stimulate T-cells and induce peptide-specific T cell responses that may result in further activation of inflammation, including elevated concentrations of pro-inflammatory cytokines and autoimmunity (103). Furthermore, in animal study, consumption of β-casein derived peptides, β-casomorphins, resulted with inflammatory immune response in gut (110). Thus, food-derived compounds may influence immunity and brain function due to their antigenic, pro-inflammatory qualities and/or their abilities to behave as ligands of various opioid receptors. Previously high levels of β-casomorphin-like opioid peptides were observed in CSF and serum of patients with postpartum psychosis (86). Also, patients with schizophrenia had increased opioid activity in CSF (85) and exorphins were found in the urine of untreated patients with schizoaffective disorder (111).
Numerous studies report that patients experiencing psychiatric and neurologic symptoms have abnormal reactions to food-derived antigens. For instance, celiac disease (CD)—an autoimmune disorder and widely recognized manifestation of gluten sensitivity, has various psychiatric, and neurologic manifestations and is considered to be a gut-brain axis “flagship” condition (112, 113). Various epidemiological studies have shown substantial association of schizophrenia with CD (114–116). However, there are also various examples of abnormal responses to food antigens which go beyond the presence of antibodies to deamidated epitopes of gliadin and tissue transglutaminase (tTG) which are characteristic immune responses observed in CD (117). It was previously demonstrated that patients with schizophrenia had increased IgA to gliadin, β-lactoglobulin and casein (83). A large study of 1401 schizophrenia patients from the CATIE study (clinical Antipsychotic Trials of Intervention Effectiveness), and 900 controls, revealed that 23.1% of patients had moderate-to-high levels of IgA to gliadin (IgA-AGA) compared with 3.1% in the control group (84). Moderate-to-high levels of antibodies to tTG were also observed in 5.4% of patients with schizophrenia compared with the 0.80% in the control group. Only 0.35% (n = 5) patients were positive for IgA to endomysium (EMA). Results of this study revealed that patients with schizophrenia have higher levels of antibodies related to CD and gluten sensitivity and that there is also a specific immune response to gluten in this population. In another study, patients with the recent-onset of psychosis and patients with multi-episode schizophrenia had increased levels of IgG and IgA antibodies to gliadin compared with controls (87). However, these patients did not have increased IgG to deamidated gliadin or IgA antibodies to tTG, and < 1% of patients had levels of antibodies symptomatic for CD. These results point to the existence of different immune mechanisms in schizophrenia compared to those observed in CD. Moreover, increased IgA antibody levels to gliadin, β-lactoglobulin, and casein were observed in schizophrenia (83). Elevated IgG to whole casein and αs, β, κ casein subunits was demonstrated in patients with recent-onset of psychosis. In contrast, in the group suffering from long-term schizophrenia there was an increase in IgG to whole casein and αs subunit (88). Interestingly, in this study Positive and Negative Syndrome Scale (PANNS) scores for negative symptoms significantly correlated with casein antibody concentration to subunits α and β. In recent years, a novel syndrome of gluten intolerance, known as non-celiac gluten sensitivity (NCGS) or gluten sensitivity (GS), has gained recognition (118). Patients with NCGS do not develop typical antibodies of CD, however they experience various physical and behavioral symptoms after gluten consumption. The most common symptoms are IBS-like symptoms, chronic fatigue, headache, bone and joint pain, numbness of hand and feet, erythema, muscle contractions, and depression. Patients may also experience hyperactivity, disturbed attention and it is likely that NCGS may contribute to symptoms of other psychiatric disorders (119).
Anti-Saccharomyces cervisiae IgG antibodies (ASCA), typically increased in Crohn's disease or ulcerative colitis, is a marker of GI inflammation (94). It was demonstrated that levels of ASCA IgG were significantly elevated in patients with schizophrenia compared to the control group, and ASCA significantly correlated with antibody levels to gluten and casein in the same patients. Interestingly, in this study authors revealed significant correlations between IgG to Toxoplasma gondii and IgG to food antigens in recent-onset schizophrenia. They suggested that infection with this parasite could result in increased permeability of the intestinal barrier with subsequent increased absorption of food antigens (89). Infection with T. gondii is also a known risk factor for the development of schizophrenia. Severance et al. demonstrated a fascinating association between T. gondii infection with gut-derived inflammation, increased intestinal permeability, allergy to food antigens and development of anti-NMDA receptor autoantibodies (42). In this study, patients with schizophrenia had increased levels of ASCA, which correlated with antibody levels to gluten and casein. Moreover, these patients had increased levels of soluble CD14—a marker of intestinal microbial translocation previously mentioned above. Infection with T. gondii also correlated with antibodies to food antigens. In further investigation of these clinical observations, using a mouse model, the same authors demonstrated that T. gondii infection may result in the elevation of IgG to casein and gluten, activation of complement system and increased levels of autoantibodies to the brain NMDA receptors.
Another group of molecules receiving a lot of attention in psychiatry research and neuroscience recently is the complement system of the immune system. The complement system is a protein complex involved in the recognition, opsonisation and lysis of various antigens. These proteins are also involved in synapse development, neuronal pruning and neurodegeneration, and are present in the human CNS, where they are mostly produced by activated microglia (120–122). Involvement of the complement system was demonstrated in various neurodegenerative disorders such as Alzheimer's disease (123, 124), Huntington's disease (125), Parkinson's disease (126), Pick's disease (127) and amyotrophic lateral sclerosis (ALS) (128). Moreover, activation of the complement system has been demonstrated in schizophrenia (129–133) and autism (134–136), and it is believed that the complement system could contribute to symptomatology of those disorders. For instance, association of C1qB gene polymorphism with schizophrenia was demonstrated in an Armenian population and it was suggested that the“C1qB gene may be considered as a relevant candidate for susceptibility to schizophrenia.” Interestingly, Severance et al. suggested the hypothesis that food antigens could be the source of activation of the complement system, and that these antigens could bind and activate the C1q, the first component in classical activation of the complement system. These authors demonstrated increased binding of casein and/or gluten IgG to C1q in patients with non-recent onset schizophrenia compared to controls and that levels of C1q-casein/gluten-related immune complexes and C1q correlated with ASCA. The authors of this study suggested “complement activation may be a useful biomarker to diagnose schizophrenia early during the course of the disease” (90). Furthermore, an upregulation of cerebral C1q was demonstrated in response to latent T. gondii infection and it was hypothesized that “complement activity may aid in the clearance of this parasite from the CNS and in so doing, have consequences for the connectivity of neighboring cells and synapses” (137).
As suggested by Dohan in 1979, besides increased permeability of intestinal barrier, dysfunction of barrier systems within the CNS could be another contributing factor for heightened transit of food-derived antigens and neuroactive polypeptides from the intestinal lumen to the CNS (9, 138). In line with this hypothesis, striking correlations between serum and cerebrospinal fluid (CSF) IgG to wheat gluten and bovine milk casein were demonstrated in antipsychotic-naïve schizophrenia patients compared to healthy controls (91). In the same study, there was a lack of intrathecal, local CNS production of IgG to food antigens, which supported the hypothesis that these antigens were derived from the periphery and were required to cross to the CNS via defective BBB (91). Previously the dysfunction of BBB was also reported in psychotic and affective disorders, and autism (100, 139–141).
Bipolar affective disorder is another severe psychiatric disorder in which compromised gut barrier has been demonstrated (Table 1) (92–94, 96). Patients with this disorder had elevated serum concentrations of IgG to gliadin and deamidated gliadin in comparison to controls. There was no difference in IgA to gliadin and to tTG between patients and control group (92). In a follow-up study, patients with manic symptoms had increased baseline IgG to gliadin, which normalized after 6 months of treatment (93). In the same study, re-hospitalized patients during a 6-month follow-up period were more likely to have increased IgG to gliadin at the follow-up. Analogically to schizophrenia there is also evidence for increased GI inflammatory parameters in patients with bipolar disorder. Patients with this disorder were demonstrated to have increased levels of ASCA along with IgG to casein and gluten, and ASCA correlated with IgG to these food antigens compared to controls (94). ASCA were also correlated with IgG to T. gondii and measles in patients who experienced recent-onset of psychosis in the course of bipolar disorder.
In a study performed by our group, we measured IgG against 44 different food products in patients with Major Depressive Disorder (MDD). We found significant positive correlations of IgG to 11.36% of food products with the length of depressive episode (months). We did not observe significant differences in mean IgG concentrations against 44 food antigens between patients and the control group, however most of our patients experienced the first episode of MDD, which could have significantly influenced our results. The conclusion of the study was that “it could be valuable to further explore a potential role for increased intestinal permeability to food antigens with subsequent IgG responses in patients with chronic, recurrent depression, and in patients with gastrointestinal, and extra-intestinal autoimmune diseases with co-morbid depression” (118).
GI inflammation and increased intestinal permeability may play also a significant role in suicidal symptomatology. In a recent pilot study it was demonstrated that recent suicidal attempters (within the last month) in the course of major depression, bipolar disorder and schizophrenia had increased IgA to ASCA, IgG to gliadin and increased IgA to LPS compared to a healthy control group (96). Moreover, association between the number of suicide attempts and the levels of IgM antibodies to T. gondii and cytomegalovirus (CMV) was demonstrated in individuals with serious mental illness previously (142).
Increased levels of antibodies against food antigens has also been demonstrated in ASD (Table 1). In general, this disorder is characterized by high comorbidity of various gastrointestinal abnormalities e.g., constipation, diarrhea, reflux, esophagitis, gastritis, duodenitis, enterocolitis, lymphoid nodular hyperplasia, increased intestinal permeability, impaired detoxification (for example, defective sulfation of phenolic amines), SIBO, dysbiosis with bacterial overgrowth and yeast overgrowth (143–145). For instance, patients with ASD had increased parameters of intestinal permeability measured with lactulose/mannitol ratio (LA/MA) and they had elevated levels IgG to AGA, deamidated gliadin and IgG to casein (97). Moreover, increased concentrations of IgA antibodies to casein, lactalbumin and β-lactoglobulin, and IgG and IgM to casein were demonstrated in infantile autism and 8 weeks of an elimination diet identified by a positive skin test, resulted in marked improvement in behavioral symptoms (98). Furthermore, ASD patients had increased concentrations of zonulin, a physiological regulator of gut epithelium permeability via modulation of tight junctions opening between enterocytes (99). Also, Fiorentino et al. demonstrated in a post mortem study that 75% of ASD patients had reduced expression of components of intestinal tight junctions (claudin-1, occluding, and tricellulin), and 66% of patients had elevated expression of pore-forming claudins (claudin−2,−10,−15) compared to the control group (100). Moreover, in the same study, brain samples from patients with ASD revealed alterations in genes expression related to BBB stability, coupled with elevated neuroinflammation. However, patients with ASD may exhibit additional dysfunction of GI tract, which adds to the complexity of gut-brain-axis involvement in this disorder. For instance, decreased activity of carbohydrate digestive enzymes (disaccharidases or glucoamylase) was found in 58.3% of children with ASD (146) and multiple studies demonstrated association between disaccharidases deficiencies and intestinal inflammatory changes (147). The most frequent finding was a low lactase level. This enzyme has a role in the hydrolysis of lactose to glucose and galactose, and the latter is essential for the synthesis of brain galactolipids. Consequently, malabsorption of disaccharides is believed to play a role in the behavioral problems observed in non-verbal ASD patients. Also, decreased activity of GI enzymes e.g., dipeptidyl peptidase IV (DPPIV) has been suggested to be the cause of inadequate digestion of caseins including casomorphins and glutens including gliadomorphins in ASD (103, 148, 149) and those exorphins were demonstrated to exhibit pro-inflammatory properties (103, 150). “Leakiness” of both the intestinal and blood-brain barriers, observed in autistic patients, could result in easier access of neuroactive peptides and food derived antigens to the CNS which could have pro-inflammatory and neurobehavioral consequences. Also, decreased activity of DPP IV was demonstrated in depressed patients and DPP IV activity correlated with immune-inflammatory markers such as, number of CD4+T cells and CD4+/CD8+ T cell ratio (151, 152). Interestingly, therapeutic effects of an enzyme-based therapy for autism have also been reported and are believed to be due to the improvement of digestion of caseins, glutens and exorphins (153, 154).
A role of increased gut barrier permeability in the pathogenesis of autoimmune disorders has previously been described by Fasano et al. (155–157). Consequently, increased intestinal permeability was demonstrated in various autoimmune disorders e.g., celiac diseases, type 1 diabetes, asthma, multiple sclerosis, inflammatory bowel diseases, ankyloses spondylitis, and it is believed that gut-derived molecular mimicry could be a pathogenic factor of autoimmunity observed in those conditions (Figure 1). Various autoimmune disorders are known risk factors of major depression, schizophrenia, and psychotic disorders (158, 159) and comorbidity of autoimmune diseases is associated with a 45% increased risk of schizophrenia (160). Moreover, it is believed that autoantibodies could play a significant role in the pathogenesis of depression and that autoimmune and depressive disorders may share common pathogenic factors (161–166). Interestingly, intracerebroventricular injection of human anti-ribosomal P antibodies induced depressive behavior in mice (167). Also, in major depression and schizophrenia, increased concentrations of various autoantibodies to cellular proteins e.g., α7 nicotinic and dopamine receptors, cardiolipin, parietal cells (PCA), smooth muscle actin, antinuclear (ANA) and anti-thyroid gland (TGA) was demonstrated (168–170). Presence of serotonin autoantibodies was also revealed in patients with schizoaffective psychoses, chronic alcoholism and rheumatoid arthritis (171). In both schizophrenia and mood disorders, increased levels of autoantibodies to hypothalamus, hippocampus and cerebellum, and anti-nuclear antibodies was demonstrated (172). Also the presence of various other autoantibodies in schizophrenia has been previously reviewed (173). Recently, there has been a lot of scientific attention focused on neurologic and psychiatric manifestations related to various cell surface autoantibodies such as antibodies to N-methyl-D-aspartate glutamate receptor (NMDAR), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), voltage-gated potassium channel (VGKC), γ-aminobutyric acid-B receptor (GABABR), the glycine receptor (GlyR) and metabotropic glutamate receptor 5 (mGluR5) (174). These antibodies could be associated with cancer, however more commonly they are non-paraneoplastic with the source of autoimmunity remaining unknown (175, 176). Co-occurrence of these autoantibodies was shown to be associated with various psychiatric symptoms e.g., psychosis, mania, agitation, emotional lability, anxiety, aggression, compulsive behavior, personality change, confusion, memory impairment, and amnesia (174). Antibodies to the NMDA receptor and to the VGKC were described in patients with schizophrenia (177–179). Respectively, Lennox et al. suggested that a sub-group of patients with a diagnosis of schizophrenia may actually suffer from undiagnosed NMDAR encephalitis (180). Interestingly, it was demonstrated that GI inflammation and increased intestinal permeability caused by T. gondii infection, a known risk factor of schizophrenia, resulted in the development of anti-NMDA receptor antibodies in laboratory animals. This infection also resulted in increased levels of anti-gluten and anti-casein IgG antibodies along with increased concentrations of complement factors which play a crucial role in neurodevelopment and neuronal pruning (42). In a recent breakthrough research, which clearly links gut-derived antigens with neuronal autoimmunity, Lambert and Vojdani demonstrated that patients with antibody reactivity to specific food proteins had higher co-occurrence of various tissue antibodies compared to controls without such food reactivities (181). More precisely, 35% of the control group (negative for IgG against glutens) and 64% of patients (positive for IgG against gluten) were reactive against tissues. Thirty percent of the control group (negative for dairy proteins antibodies) and 73% of patients (positive for dairy antibodies) were reactive against tissues. Twenty two percent of the control group (negative for IgG against wheat germ agglutinin—WGA) and 76% of patients (positive for IgG against WGA) were reactive against tissues. Furthermore, authors demonstrated that of all three groups of food antigens assessed (gluten, dairy, and lectin/agglutinin family proteins), autoimmune reactivity to neurological tissues was the highest in the patient group. It should be noted that this study did not provide specific information on patients' inclusion criteria and their diagnoses, therefore similar research in psychiatric patients is required.
Analogous immune mechanisms were also described in ASD where concomitant increase of IgG, IgM, IgA to gliadin, and cerebellum was demonstrated in more than 80% of autistic patients (101) (Table 1). Furthermore, the same study revealed a cross-reactivity between gliadin and cerebellar peptides. Moreover, it was suggested that autoimmunity due to bacterial infections and exposure to milk antigens may be a pathogenic factor in autism.
Voidani et al. also demonstrated that in individuals with predisposing HLA molecules, infectious agents including superantigens [for e.g., bacterial streptokinase (SK)], heat shock protein (HSP-60), dietary proteins (for e.g., gliadin and casein), and xenobiotics [for e.g., ethyl mercury (thimerosal derivate)] bind to different enzymes or cell surface receptors e.g., CD26 (DPP-IV) and CD69, and induce autoantibodies against HLA peptides (103, 148) (Table 1). In this study a significant percentage of ASD children developed anti-SK, anti-gliadin and casein, anti-ethyl mercury antibodies concomitantly with anti-CD26, anti-CD69 autoantibodies. Furthermore, adding SK, gliadin, casein, ethyl mercury to CD26 or CD69 resulted in 28–86% inhibition of CD26 or CD69 binding to anti-CD26 and anti-CD69 antibodies.
So far, two main mechanisms have been identified for food protein-induced autoimmunity in various tissues; firstly molecular mimicry, also known as cross-reactivity and secondly, covalent binding of food derived lectins and agglutinins to human tissues (181) (Figure 1). In molecular mimicry, there is case of “mistaken identity” between specific food antigens and human tissue due to high molecular homology e.g., amino acid homology of gliadin or dairy proteins with human tissue. In some scenarios (e.g., increased intestinal permeability) (Figure 1), antibodies against these food antigens are produced and the immune system can “mistake” these mimicking antigens for host tissue and react against it. For example, such reactions between gliadin and milk proteins with cerebellar tissue, and myelin have been reported previously (101, 182–184). In case of the second mechanism mentioned above, numerous plant lectins and agglutinins can covalently bind to various tissues and in response, the immune system may react against the new structure, as well as the surrounding tissue (181).
Another component of the microbiota-gut brain axis with a crucial role determining intestinal permeability and immunity of both the GI tract and the CNS is intestinal microbiota. In the last few years, there has been significant progress in our understanding of the role of bacteria in brain function and behavior and these mechanisms have been reviewed extensively elsewhere (185–189). Microbiota play a significant role in maintaining the psycho-neuro-immunological balance by various mode-of-actions, such as the modulation of the immune and neuroendocrine systems, e.g., hypothalamic-pituitary adrenal axis (HPA), changes of the tryptophan (TRP) metabolism in the serotonin and kynurenic axes, production and metabolism of multiple neuroactive compounds e.g., short-chain fatty acids (SCFAs) and neurotransmitters. Beneficial bacteria also influence neurogenesis and the expression of neurotransmitters' receptors in the CNS (186, 187). Microbiota are also believed to be key regulators of neuroinflammation and to modulate mucosal innate and adaptive immune responses during infection, inflammation and autoimmunity (188). For instance, it was demonstrated that gastrointestinal microbiota have a significant function in the maturation and immune function of microglia (190). These bacteria also influence blood-brain barrier permeability (191). Furthermore, as natural guardians of the gut epithelium, intestinal microbiota, have a crucial role in the maintenance and modulation of gut epithelium barrier and in the regulation of various gut-associated lymphoid tissue (GALT) functions (29, 192). Key downstream effects of these beneficial microbes include their ability to decrease concentration of pro-inflammatory cytokines and the nuclear factor, NF-κB, increase concentrations of anti-inflammatory cytokines, and changes in tryptophan, and kynurenines levels (192–196). Since pro-inflammatory cytokines, NF-κB, and zonulin have a crucial role in increase of intestinal permeability, various microbiota, due to ability to modulate those parameters, have a protective effects on intestinal barrier (192–194, 197). Those bacteria have a beneficial influence on the composition of intestinal tight junctions proteins, inhibit adherence of pathogens to intestinal barrier, increase mucin production by epithelial goblet cells, increase secretory IgA (sIgA) and antimicrobial β-defensin secretion into the luminal mucous, what enhances intestinal barrier (198).
There is growing evidence to support the therapeutic effects of microbiota and probiotics on the symptoms of anxiety, low mood and depression, CFS, and cognitive functions (186, 199–215) and beneficial bacteria have recently earned a general name of psychobiotics (216).
Altered composition of gut microbes was demonstrated in various psychiatric disorders including CFS (79), MDD (217–220), ASD (221–225), schizophrenia and bipolar disorder (226), and alcoholism (38). In MDD altered proportions of Prevotella and Klebsiella bacterial genus were consistent with the Hamilton depression rating scale (220). Additionally, fecal microbiota transplant from patients with major depression to germ free (GF) mice resulted in depression-like behavior in recipient mice (227). Small intestine bacterial overgrowth (SIBO) is another abnormality of intestinal flora observed in ASD and alcoholism (145, 228). It was hypothesized that these alterations could play a significant role in psychopathology and assessment of flora composition could become a clinical marker in psychiatry. Interestingly, psychiatric pharmacotherapy was shown to influence composition of intestinal microbiota. For example, antipsychotic medication such as olanzapine and risperidone have been shown to modify gut flora and it was further demonstrated that weight gain, often observed in patients during such treatment, was secondary to altered of gut microbiota by the antipsychotics (229–231). Moreover, olanzapine-induced metabolic dysfunction in rats was attenuated by antibiotic administration (232). Consequently, probiotics administration could provide a novel therapeutic strategy for the prevention or reversal of weight gain following antipsychotic treatment.
We are witnessing a truly interesting time for psychiatry and neuroscience. The last two decades of research in the field of psycho-neuro-immunology have provided us with an increased understanding of the role of immunity in psychiatric disorders. Now, with the involvement of the microbiota-gut-brain axis, the second stage of this inevitable paradigm shift in psychiatry has begun. In the old paradigm, psychiatry was “starting” when “all” physical abnormalities (besides clearly organic disorders) were excluded. Currently, the split between purely psychological vs. medical background of psychiatric disorders is dissolving in response to improved understanding of psycho-neuro-immune interactions between psyche and soma. Having recognized that vast amounts of psychiatric symptomatology may have an immune, autoimmune and/or gut-derived background, one wonders when we face the separate nosological entities and when we face various manifestations of underlying immune processes. Professor Ronald S. Smith, the precursor of the inflammatory hypothesis of depression, stated the following in his sadly unfinished book, “Cytokines & Depression. How your immune system causes depression.” While describing immune pathogenesis of depression, he referred to the First Edition of Encyclopaedia Britannica and challenged the current approach to major depression diagnosis. In this edition, published in 1771, 37 different subtypes of “fever disease” were described as separate disorders. Smith wrote, “Eventually it was understood that fever in not one disease nor 37 kinds of fever diseases, but rather it is a trustworthy universal sign of acute immune system activation. Fever is a sign of acute immune system activation, regardless of any other signs, symptoms or diseases that it may be associated with. After this realization, fever was no longer a bewildering and complex disease, but instead, a simple, direct and easily understood signal of acute immune activation” (233). On the other hand, Susannah Cahalan in her New York Times Bestselling autobiography, “Brain on Fire: My Month of Madness,” describes her own horrifying experience of anti-NMDA receptor encephalitis. Initially, due to various psychotic symptoms, a diagnosis of bipolar affective disorder, schizophrenia, or schizoaffective disorder was suggested, however after further investigation, she was diagnosed with aforementioned encephalitis. After all, Cahalan was diagnosed with and treated for a neurologic, autoimmune disorder, and a psychiatric diagnosis was rejected. However, this still provokes the question; how many psychiatric patients suffer from similar autoimmune conditions with lesser manifestations of neurologic symptoms? When are we dealing with a psychiatric manifestation of “organic” disorder and when is it a “purely” psychiatric one? Maybe, as Smith suggested, we are more commonly witnessing psychiatric manifestations of immune system activation. The GI tract is the largest immune organ in the human body and also the biggest surface area of interaction between the internal and external environment. Taking this into consideration and in light of the discussion included in this review; the GI tract will undoubtedly have a major impact on psychiatric symptoms and treatment.
Since changes in exposure to food antigens have been shown to modulate immune response, further research using elimination diets in a subgroup of patients expressing increased levels of food-specific antibodies would be of value. The benefits of dietary interventions in psychiatric patients have been described previously (234). Moreover, the therapeutic effects of elimination diet were described in neurologic and GI disorders such as irritable bowel syndrome, Crohn's disease and migraine, and improvement in symptoms was believed to be related to decreased levels to food-specific IgG as a result of decreased exposure to food antigens (235–240). Also, augmenting psychiatric pharmacotherapy with digestive enzymes and interventions known to positively influence the gut barrier may have a positive effect on the microbiota-gut-brain axis and psychiatric symptomatology. For instance, supplementation of curcumin and zinc, which are both excellent “tightners” of gut barrier, along with pre and probiotics might improve symptoms of psychiatric and co-morbid inflammatory and autoimmune disorders (241, 242). Both turmeric and zinc exhibit anti-inflammatory effects and their anti-depressant effects were previously demonstrated (243, 244). Curcumin has a positive effect on gut barrier due to its ability to decrease levels of TNFα, a pro-inflammatory cytokine which negatively influences tight junctions and increases intestinal and BBB permeability (35, 36, 60, 64–66, 245). This cytokine is involved also in activation of kynurenic enzyme indoleamine 2,3 dioxygenase (IDO) which diverts tryptophan from serotonin pathway toward detrimental kynurenines. Consequently, curcumin decreases IDO activity (246). Moreover this herb is a well-known inhibitor of NF-κβ signaling involved in inflammatory response and increased permeability of both intestinal and blood-brain barriers (247).
To conclude, the assessment of intestinal permeability, gut derived immunity, immunoglobulins against various food, microbial, viral and parasitic antigens, and assessment of intestinal flora composition may be valuable in psychiatric diagnosis and therapy. Also, modification of intestinal flora may prevent metabolic side effects related with antipsychotic treatment. Finally, supplementation of probiotics or other interventions to positively influence the intestinal barrier could be used as a preventive measure of exposure to stress and its detrimental consequences.
LR and AS designed and wrote the first version of manuscript and performed literature search. LR wrote the final version of manuscript, prepared figures and tables.
Publication of this manuscript was funded by Medical University of Warsaw.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523:337–41. doi: 10.1038/nature14432
3. Buscaino V. Patologia extraneurale della schizophrenia. Fegato, tubo digerente, sistema reticolo-endoteliale. Acta Neurol. (1953) 8:1–60.
5. Baruk H. Digestive and hepatointestinal etiology of the various mental diseases. Schweiz Med Wochenschr. (1953) 83 (Suppl. 38):1517–8.
6. Baruk H. Biliary & hepatic poisons in pathogenesis of schizophrenia; experimental study. Confin Neurol. (1958) 18:254–63.
7. Baruk H. Study of blood ammonia in periodic psychosis and in epileptic state. Psychotoxic valve of certain digestive disorders therapeutic trials. Ann Med Psychol. (1962) 120:721–6.
8. Baruk H. Psychoses from digestive origins. In: Hemmings G and Hemmings, W. A, editors. The Biological Basis of Schizophrenia. Dordrecht: Springer (1978). p. 37–44.
9. Dohan FC. Schizophrenia and neuroactive peptides from food. Lancet (1979) 1:1031. doi: 10.1016/S0140-6736(79)92780-6
10. Maes M. A review on the acute phase response in major depression. Rev Neurosci. (1993) 4:407–16. doi: 10.1515/REVNEURO.1993.4.4.407
11. Maes M. Evidence for an immune response in major depression: a review and hypothesis. Prog Neuro-psychopharmacol Biol Psychiatry (1995) 19:11–38. doi: 10.1016/0278-5846(94)00101-M
12. Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog Neuro-psychopharmacol Biol Psychiatry (2005) 29:201–17. doi: 10.1016/j.pnpbp.2004.11.003
13. Sluzewska A, Rybakowski J, Bosmans E, Sobieska M, Berghmans R, Maes M, et al. Indicators of immune activation in major depression. Psychiatry Res. (1996) 64:161–7. doi: 10.1016/S0165-1781(96)02783-7
14. Strous RD, Shoenfeld Y. Schizophrenia, autoimmunity and immune system dysregulation: a comprehensive model updated and revisited. J Autoimmun. (2006) 27:71–80. doi: 10.1016/j.jaut.2006.07.006
15. Maes M, Galecki P, Chang YS, Berk M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog Neuro-psychopharmacol Biol Psychiatry (2011) 35:676–92. doi: 10.1016/j.pnpbp.2010.05.004
16. Maes M. Depression is an inflammatory disease, but cell-mediated immune activation is the key component of depression. Prog Neuro-psychopharmacology Biol Psychiatry (2011) 35:664–75. doi: 10.1016/j.pnpbp.2010.06.014
17. Myint AM, Kim YK. Cytokine-serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med Hypotheses (2003) 61:519–25. doi: 10.1016/S0306-9877(03)00207-X
18. Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. (2012) 13:465–77. doi: 10.1038/nrn3257
19. Smith RS. The macrophage theory of depression. Med Hypotheses (1991) 35:298–306. doi: 10.1016/0306-9877(91)90272-Z
20. Stone TW, Darlington LG. The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders. Br J Pharmacol. (2013) 169:1211–27. doi: 10.1111/bph.12230
21. Wichers MC and Maes M. The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-α-induced depression. J Psychiatry Neurosci. (2004) 29:11–17.
22. Wonodi I, Schwarcz R. Cortical kynurenine pathway metabolism: a novel target for cognitive enhancement in Schizophrenia. Schizophr Bull. (2010) 36:211–8. doi: 10.1093/schbul/sbq002
23. Brattsand R, Linden M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment Pharmacol Ther. (1996) 10 (Suppl. 2):81–90: discussion 91–82.
24. Joyce DA, Steer JH, Abraham LJ. Glucocorticoid modulation of human monocyte/macrophage function: control of TNF-α secretion. Inflamm Res. (1997) 46:447–51. doi: 10.1007/s000110050222
25. Besedovsky HO, del Rey A. Regulating inflammation by glucocorticoids. Nat Immunol. (2006) 7:537. doi: 10.1038/ni0606-537
26. Soderholm JD, Perdue MH. Stress and gastrointestinal tract. IStress I, and intestinal barrier function. Am J Physiol Gastrointest Liver Physiol. (2001) 280:G7–13. doi: 10.1152/ajpgi.2001.280.1.G7
27. Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. (2009) 124:3–20; quiz: 21–2. doi: 10.1016/j.jaci.2009.05.038
28. Farhadi A, Banan A, Fields J, Keshavarzian A. Intestinal barrier: an interface between health and disease. J Gastroenterol Hepatol. (2003) 18:479–97. doi: 10.1046/j.1440-1746.2003.03032.x
29. Zareie M, Johnson-Henry K, Jury J, Yang PC, Ngan BY, McKay DM, et al. Probiotics prevent bacterial translocation and improve intestinal barrier function in rats following chronic psychological stress. Gut (2006) 55:1553–60. doi: 10.1136/gut.2005.080739
30. Yang PC, Jury J, Söderholm JD, Sherman PM, McKay DM, Perdue MH. Chronic psychological stress in rats induces intestinal sensitization to luminal antigens. Am J Pathol. (2006) 168:104–14. doi: 10.2353/ajpath.2006.050575
31. Ferrier L. Significance of increased human colonic permeability in response to corticotrophin-releasing hormone (CRH). Gut (2008) 57:7–9. doi: 10.1136/gut.2007.129841
32. Lambert GP. Stress-induced gastrointestinal barrier dysfunction and its inflammatory effects. J Anim Sci. (2009) 87:E101–8. doi: 10.2527/jas.2008-1339
33. Keita AV, Soderholm JD. The intestinal barrier and its regulation by neuroimmune factors. Neurogastroenterol Motil. (2010) 22:718–33. doi: 10.1111/j.1365-2982.2010.01498.x
34. Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol. (2007) 178:4641–49. doi: 10.4049/jimmunol.178.7.4641
35. Schmitz H, Fromm M, Bentzel CJ, Scholz P, Detjen K, Mankertz J, et al. Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci. (1999) 112(Pt 1):137–46.
36. Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. (2006) 290:G496–504. doi: 10.1152/ajpgi.00318.2005
37. Beaurepaire C, Smyth D, McKay DM. Interferon-gamma regulation of intestinal epithelial permeability. J Interferon Cytokine Res. (2009) 29:133–44. doi: 10.1089/jir.2008.0057
38. Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Starkel P, et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc Natl Acad Sci USA. (2014) 111:E4485–93. doi: 10.1073/pnas.1415174111
39. Fukui H. Increased intestinal permeability and decreased barrier function: does it really influence the risk of inflammation? Inflamm Intest Dis. (2016) 1:135–45. doi: 10.1159/000447252
40. Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut (2001) 48:206–11. doi: 10.1136/gut.48.2.206
41. Yamaguchi N, Sugita R, Miki A, Takemura N, Kawabata J, Watanabe J, et al. Gastrointestinal Candida colonisation promotes sensitisation against food antigens by affecting the mucosal barrier in mice. Gut (2006) 55:954–60. doi: 10.1136/gut.2005.084954
42. Severance EG, Kannan G, Gressitt KL, Dickerson FB, Pletnikov MV, Yolken RH. Antibodies to food antigens: translational research in psychiatric disorders. Neurol Psychiatry Brain Res. (2012) 18:87–8. doi: 10.1016/j.npbr.2012.02.040
43. Chavez AM, Menconi MJ, Hodin RA, Fink MP. Cytokine-induced intestinal epithelial hyperpermeability: role of nitric oxide. Crit Care Med. (1999) 27:2246–51. doi: 10.1097/00003246-199910000-00030
44. Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. (2004) 286:G367–76. doi: 10.1152/ajpgi.00173.2003
45. Lambert GP. Intestinal barrier dysfunction, endotoxemia, and gastrointestinal symptoms: the 'canary in the coal mine' during exercise-heat stress? Med Sport Sci. (2008) 53:61–73. doi: 10.1159/000151550
46. Pals KL, Chang RT, Ryan AJ, Gisolfi CV. Effect of running intensity on intestinal permeability. J Appl Physiol. (1997) 82:571–6.
47. Lambert GP, Gisolfi CV, Berg DJ, Moseley PL, Oberley LW, Kregel KC. Selected contribution: Hyperthermia-induced intestinal permeability and the role of oxidative and nitrosative stress. J Appl Physiol. (2002) 92:1750–61; discussion: 1749. doi: 10.1152/japplphysiol.00787.2001
48. Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet (1984) 1:179–82. doi: 10.1016/S0140-6736(84)92109-3
49. Bode C, Bode JC. Effect of alcohol consumption on the gut. Best Pract Res Clin Gastroenterol. (2003) 17:575–92. doi: 10.1016/S1521-6918(03)00034-9
50. Leclercq S, Cani PD, Neyrinck AM, Starkel P, Jamar F, Mikolajczak M, et al. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun. (2012) 26:911–8. doi: 10.1016/j.bbi.2012.04.001
51. Choi HJ, Kim J, Park SH, Do KH, Yang H, Moon Y. Pro-inflammatory NF-kappaB and early growth response gene 1 regulate epithelial barrier disruption by food additive carrageenan in human intestinal epithelial cells. Toxicol Lett. (2012) 211:289–95. doi: 10.1016/j.toxlet.2012.04.012
52. Bjarnason I, Williams P, Smethurst P, Peters TJ, Levi AJ. Effect of non-steroidal anti-inflammatory drugs and prostaglandins on the permeability of the human small intestine. Gut (1986) 27:1292–7. doi: 10.1136/gut.27.11.1292
53. Smetanka RD, Lambert GP, Murray R, Eddy D, Horn M, Gisolfi CV. Intestinal permeability in runners in the 1996 Chicago marathon. Int J Sport Nutr. (1999) 9:426–33. doi: 10.1123/ijsn.9.4.426
54. van Ampting MT, Schonewille AJ, Vink C, Brummer RJ, van der Meer R, Bovee-Oudenhoven IM. Damage to the intestinal epithelial barrier by antibiotic pretreatment of salmonella-infected rats is lessened by dietary calcium or tannic acid. J Nutr. (2010) 140:2167–72. doi: 10.3945/jn.110.124453
55. Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature (2013) 502:96–9. doi: 10.1038/nature12503
56. Tulstrup MVL, Christensen EG, Carvalho V, Linninge C, Ahrné S, Højberg O, et al. Antibiotic treatment affects intestinal permeability and gut microbial composition in wistar rats dependent on antibiotic class. PLoS ONE (2015) 10:e0144854. doi: 10.1371/journal.pone.0144854
57. Becattini S, Taur Y, Pamer EG. Antibiotic-induced changes in the intestinal microbiota and disease. Trends Mol Med. (2016) 22:458–78. doi: 10.1016/j.molmed.2016.04.003
58. Daneman R, Rescigno M. The gut immune barrier and the blood-brain barrier: are they so different? Immunity (2009) 31:722–35. doi: 10.1016/j.immuni.2009.09.012
59. Esposito P, Chandler N, Kandere K, Basu S, Jacobson S, Connolly R, et al. Corticotropin-releasing hormone and brain mast cells regulate blood-brain-barrier permeability induced by acute stress. J Pharmacol Exp Ther. (2002) 303:1061–6. doi: 10.1124/jpet.102.038497
60. Yang GY, Gong C, Qin Z, Liu XH, Lorris Betz A. Tumor necrosis factor alpha expression produces increased blood-brain barrier permeability following temporary focal cerebral ischemia in mice. Brain Res Mol Brain Res. (1999) 69:135–43. doi: 10.1016/S0169-328X(99)00007-8
61. Pan W, Kastin AJ. Tumor necrosis factor and stroke: role of the blood-brain barrier. Prog Neurobiol. (2007) 83:363–74. doi: 10.1016/j.pneurobio.2007.07.008
62. Blamire AM, Anthony DC, Rajagopalan B, Sibson NR, Perry VH, Styles P. Interleukin-1beta -induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: a magnetic resonance study. J Neurosci. (2000) 20:8153–9. doi: 10.1523/JNEUROSCI.20-21-08153.2000
63. Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. (2004) 190:446–55. doi: 10.1016/j.expneurol.2004.08.008
64. Tsao N, Hsu HP, Wu CM, Liu CC, Lei HY. Tumour necrosis factor-alpha causes an increase in blood-brain barrier permeability during sepsis. J Med Microbiol. (2001) 50:812–21. doi: 10.1099/0022-1317-50-9-812
65. Lv S, Song HL, Zhou Y, Li LX, Cui W, Wang W, Liu P. Tumour necrosis factor-alpha affects blood-brain barrier permeability and tight junction-associated occludin in acute liver failure. Liver Int. (2010) 30:1198–210. doi: 10.1111/j.1478-3231.2010.02211.x
66. Wang W, Lv S, Zhou Y, Fu J, Li C, Liu P. Tumor necrosis factor-alpha affects blood-brain barrier permeability in acetaminophen-induced acute liver failure. Eur J Gastroenterol Hepatol. (2011) 23:552–8. doi: 10.1097/MEG.0b013e3283470212
67. Chaitanya GV, Cromer WE, Wells SR, Jennings MH, Couraud PO, Romero IA, et al. Gliovascular and cytokine interactions modulate brain endothelial barrier in vitro. J Neuroinflammation (2011) 8:162. doi: 10.1186/1742-2094-8-162
68. Enciu AM, Gherghiceanu M, Popescu BO. Triggers and effectors of oxidative stress at blood-brain barrier level: relevance for brain ageing and neurodegeneration. Oxid Med Cell Longev. (2013) 2013:297512. doi: 10.1155/2013/297512
69. Garcia Bueno B, Caso JR, Madrigal JL, Leza JC. Innate immune receptor Toll-like receptor 4 signalling in neuropsychiatric diseases. Neurosci Biobehav Rev. (2016) 64:134–47. doi: 10.1016/j.neubiorev.2016.02.013
70. Pålsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology (2004) 113:153–62. doi: 10.1111/j.1365-2567.2004.01976.x
71. Grigoleit J-S, Engler H, Schedlowski M. Experimental human endotoxemia, sickness behavior, and neuropsychiatric diseases. In: Müller N, Myint AM, Schwarz, MJ, editors. Immunology and Psychiatry: From Basic Research to Therapeutic Interventions. Cham: Springer International Publishing (2015). p. 63–82.
72. Velin AK, Ericson AC, Braaf Y, Wallon C, Soderholm JD. Increased antigen and bacterial uptake in follicle associated epithelium induced by chronic psychological stress in rats. Gut (2004) 53:494–500. doi: 10.1136/gut.2003.028506
73. Parrott JM, Redus L, O'Connor JC. Kynurenine metabolic balance is disrupted in the hippocampus following peripheral lipopolysaccharide challenge. J Neuroinflammation (2016) 13:124. doi: 10.1186/s12974-016-0590-y
74. Reichenberg A, Yirmiya R, Schuld A, Kraus T, Haack M, Morag A, Pollmacher T. Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry (2001) 58:445–52. doi: 10.1001/archpsyc.58.5.445
75. Wright CE, Strike PC, Brydon L, Steptoe A. Acute inflammation and negative mood: mediation by cytokine activation. Brain Behav Immun. (2005) 19:345–50. doi: 10.1016/j.bbi.2004.10.003
76. Grigoleit JS, Kullmann JS, Wolf OT, Hammes F, Wegner A, Jablonowski S, et al. Dose-dependent effects of endotoxin on neurobehavioral functions in humans. PLoS ONE (2011) 6:e28330. doi: 10.1371/journal.pone.0028330
77. Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS ONE (2008) 3:e2516. doi: 10.1371/journal.pone.0002516
78. Maes M, Mihaylova I, Leunis JC. Increased serum IgA and IgM against LPS of enterobacteria in chronic fatigue syndrome (CFS): indication for the involvement of gram-negative enterobacteria in the etiology of CFS and for the presence of an increased gut-intestinal permeability. J Affect Disord. (2007) 99:237–40. doi: 10.1016/j.jad.2006.08.021
79. Giloteaux L, Goodrich JK, Walters WA, Levine SM, Ley RE, Hanson MR. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome (2016) 4:30. doi: 10.1186/s40168-016-0171-4
80. Maes M, Kubera M, Leunis JC. The gut-brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol Lett. (2008) 29:117–24.
81. Maes M, Kubera M, Leunis JC, Berk M. Increased IgA and IgM responses against gut commensals in chronic depression: further evidence for increased bacterial translocation or leaky gut. J Affect Disord. (2012) 141:55–62. doi: 10.1016/j.jad.2012.02.023
82. Severance EG, Gressitt KL, Stallings CR, Origoni AE, Khushalani S, Leweke FM, et al. Discordant patterns of bacterial translocation markers and implications for innate immune imbalances in schizophrenia. Schizophr Res. (2013) 148:130–7. doi: 10.1016/j.schres.2013.05.018
83. Reichelt KL, Landmark J. Specific IgA antibody increases in schizophrenia. Biol Psychiatry (1995) 37:410–3. doi: 10.1016/0006-3223(94)00176-4
84. Cascella NG, Kryszak D, Bhatti B, Gregory P, Kelly DL, McEvoy JP, et al. Prevalence of celiac disease and gluten sensitivity in the United States clinical antipsychotic trials of intervention effectiveness study population. Schizophr Bull. (2011) 37:94–100. doi: 10.1093/schbul/sbp055
85. Lindstrom LH, Besev G, Gunne LM, Terenius L. CSF levels of receptor-active endorphins in schizophrenic patients: correlations with symptomatology and monoamine metabolites. Psychiatry Res. (1986) 19:93–100. doi: 10.1016/0165-1781(86)90001-6
86. Lindström LH, Nyberg F, Terenius L, Bauer K, Besev G, Gunne LM, et al. CSF and plasma beta-casomorphin-like opioid peptides in postpartum psychosis. Am J Psychiatry (1984) 141:1059–66. doi: 10.1176/ajp.141.9.1059
87. Dickerson F, Stallings C, Origoni A, Vaughan C, Khushalani S, Leister F, et al. Markers of gluten sensitivity and celiac disease in recent-onset psychosis and multi-episode schizophrenia. Biol Psychiatry (2010) 68:100–4. doi: 10.1016/j.biopsych.2010.03.021
88. Severance EG, Dickerson FB, Halling M, Krivogorsky B, Haile L, Yang S, et al. Subunit and whole molecule specificity of the anti-bovine casein immune response in recent onset psychosis and schizophrenia. Schizophr Res. (2010) 118:240–7. doi: 10.1016/j.schres.2009.12.030
89. Severance EG, Alaedini A, Yang S, Halling M, Gressitt KL, Stallings CR, et al. Gastrointestinal inflammation and associated immune activation in schizophrenia, Schizophr Res. (2012) 138:48–53. doi: 10.1016/j.schres.2012.02.025
90. Severance EG, Gressitt KL, Halling M, Stallings CR, Origoni AE, Vaughan C, et al. Complement C1q formation of immune complexes with milk caseins and wheat glutens in schizophrenia. Neurobiol Dis. (2012) 48:447–53. doi: 10.1016/j.nbd.2012.07.005
91. Severance EG, Gressitt KL, Alaedini A, Rohleder C, Enning F, Bumb JM, et al. IgG dynamics of dietary antigens point to cerebrospinal fluid barrier or flow dysfunction in first-episode schizophrenia. Brain Behav Immun. (2015) 44:148–58. doi: 10.1016/j.bbi.2014.09.009
92. Dickerson F, Stallings C, Origoni A, Vaughan C, Khushalani S, Alaedini A, et al. Markers of gluten sensitivity and celiac disease in bipolar disorder. Bipolar Disord. (2011) 13:52–8. doi: 10.1111/j.1399-5618.2011.00894.x
93. Dickerson F, Stallings C, Origoni A, Vaughan C, Khushalani S, Yolken R. Markers of gluten sensitivity in acute mania: a longitudinal study, Psychiatry Res. (2012) 196:68–71. doi: 10.1016/j.psychres.2011.11.007
94. Severance EG, Gressitt KL, Yang S, Stallings CR, Origoni AE, Vaughan C, et al. Seroreactive marker for inflammatory bowel disease and associations with antibodies to dietary proteins in bipolar disorder. Bipolar Disord. (2014) 16:230–40. doi: 10.1111/bdi.12159
95. Rudzki L, Pawlak D, Pawlak K, Waszkiewicz N, Malus A, Konarzewska B, et al. Immune suppression of IgG response against dairy proteins in major depression. BMC Psychiatry (2017) 17:268. doi: 10.1186/s12888-017-1431-y
96. Dickerson F, Adamos M, Katsafanas E, Khushalani S, Origoni A, Savage C, et al. The association between immune markers and recent suicide attempts in patients with serious mental illness: a pilot study. Psychiatry Res. (2017) 255:8–12. doi: 10.1016/j.psychres.2017.05.005
97. de Magistris L, Picardi A, Siniscalco D, Riccio MP, Sapone A, Cariello R, et al. Antibodies against food antigens in patients with autistic spectrum disorders. Biomed Res Int. (2013) 2013:729349. doi: 10.1155/2013/729349
98. Lucarelli S, Frediani T, Zingoni AM, Ferruzzi F, Giardini O, Quintieri F, et al. Food allergy and infantile autism. Panminerva Med. (1995) 37:137–41.
99. Esnafoglu E, Cirrik S, Ayyildiz SN, Erdil A, Erturk EY, Dagli A, et al. Increased serum zonulin levels as an intestinal permeability marker in autistic subjects. J Pediatr. (2017) 188:240–4. doi: 10.1016/j.jpeds.2017.04.004
100. Fiorentino M, Sapone A, Senger S, Camhi SS, Kadzielski SM, Buie TM, et al. Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol Autism (2016) 7:49. doi: 10.1186/s13229-016-0110-z
101. Vojdani A, O'Bryan T, Green JA, McCandless J, Woeller KN, Vojdani E, et al. Immune response to dietary proteins, gliadin and cerebellar peptides in children with autism. Nutr Neurosci. (2004) 7:151–61. doi: 10.1080/10284150400004155
102. Vojdani A, Campbell AW, Anyanwu E, Kashanian A, Bock K, Vojdani E. Antibodies to neuron-specific antigens in children with autism: possible cross-reaction with encephalitogenic proteins from milk, Chlamydia pneumoniae and Streptococcus group A. J Neuroimmunol. (2002) 129:168–77. doi: 10.1016/S0165-5728(02)00180-7
103. Vojdani A, Pangborn JB, Vojdani E, Cooper EL. Infections, toxic chemicals and dietary peptides binding to lymphocyte receptors and tissue enzymes are major instigators of autoimmunity in autism. Int J Immunopathol Pharmacol. (2003) 16:189–99. doi: 10.1177/039463200301600302
104. Thurman RG, Bradford BU, Iimuro Y, Knecht KT, Arteel GE, Yin M, et al. The role of gut-derived bacterial toxins and free radicals in alcohol-induced liver injury. J Gastroenterol Hepatol. (1998) 13(Suppl.):S39–50. doi: 10.1111/jgh.1998.13.s1.39
105. Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology (1995) 108:218–24. doi: 10.1016/0016-5085(95)90027-6
106. Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology (2000) 32:1008–17. doi: 10.1053/jhep.2000.19621
107. Wieser H. Chemistry of gluten proteins. Food Microbiol. (2007) 24:115–9. doi: 10.1016/j.fm.2006.07.004
108. Kaminski S, Cieslinska A, Kostyra E. Polymorphism of bovine beta-casein and its potential effect on human health. J Appl Genet. (2007) 48:189–98. doi: 10.1007/BF03195213
109. Teschemacher H. Opioid receptor ligands derived from food proteins. Curr Pharm Design (2003) 9:1331–44. doi: 10.2174/1381612033454856
110. Haq MRU, Kapila R, Saliganti V. Consumption of β-casomorphins-7/5 induce inflammatory immune response in mice gut through Th2 pathway. J Funct Foods (2014) 8:150–60. doi: 10.1016/j.jff.2014.03.018
111. Tveiten D, Reichelt KL. Exorphins in urine from schizoaffective psychotics. Open J Psychiatry. (2012) 2:220–7. doi: 10.4236/ojpsych.2012.23029
112. Bushara KO. Neurologic presentation of celiac disease. Gastroenterology (2005) 128:S92–7. doi: 10.1053/j.gastro.2005.02.018
113. Jackson JR, Eaton WW, Cascella NG, Fasano A, Kelly DL. Neurologic and psychiatric manifestations of celiac disease and gluten sensitivity. Psychiatr Q. (2012) 83:91–102. doi: 10.1007/s11126-011-9186-y
114. Baldwin JA. Schizophrenia and physical disease: a preliminary analysis of the data from the Oxford Record Linkage Study. In: Hemmings G, Editor. Biochemistry of Schizophrenia and Addiction: In Search of a Common Factor. Dordrecht: Springer (1980). p. 297–318.
115. Makikyro T, Karvonen JT, Hakko H, Nieminen P, Joukamaa M, Isohanni M, et al. Comorbidity of hospital-treated psychiatric and physical disorders with special reference to schizophrenia: a 28 year follow-up of the 1966 northern Finland general population birth cohort. Public Health (1998) 112:221–8.
116. Eaton W, Mortensen PB, Agerbo E, Byrne M, Mors O, Ewald H. Coeliac disease and schizophrenia: population based case control study with linkage of Danish national registers. BMJ (2004) 328:438–9. doi: 10.1136/bmj.328.7437.438
117. Briani C, Samaroo D, Alaedini A. Celiac disease: from gluten to autoimmunity. Autoimmun Rev. (2008) 7:644–50. doi: 10.1016/j.autrev.2008.05.006
118. Czaja-Bulsa G. Non coeliac gluten sensitivity - A new disease with gluten intolerance. Clin Nutr. (2015) 34:189–94. doi: 10.1016/j.clnu.2014.08.012
119. Sapone A, Bai JC, Ciacci C, Dolinsek J, Green PH, Hadjivassiliou M, et al. Spectrum of gluten-related disorders: consensus on new nomenclature and classification, BMC Med. (2012) 10:13. doi: 10.1186/1741-7015-10-13
120. Fourgeaud L, Boulanger LM, Synapse remodeling, compliments of the complement system. Cell (2007) 131:1034–6. doi: 10.1016/j.cell.2007.11.031
121. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
122. Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J. Neurosci. (2011) 31:3459–69. doi: 10.1523/JNEUROSCI.3932-10.2011
123. Shen Y, Meri S. Yin and Yang: complement activation and regulation in Alzheimer's disease. Prog Neurobiol. (2003) 70:463–72. doi: 10.1016/j.pneurobio.2003.08.001
124. Thambisetty M, Simmons A, Hye A, Campbell J, Westman E, Zhang Y, et al. Plasma biomarkers of brain atrophy in Alzheimer's disease. PLoS ONE (2011) 6:e28527. doi: 10.1371/journal.pone.0028527
125. Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp Neurol. (1999) 159:362–76. doi: 10.1006/exnr.1999.7170
126. Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson's disease are recognized by antibodies to complement proteins. Acta Neuropathol. (1992) 84:100–4. doi: 10.1007/BF00427222
127. Singhrao SK, Neal JW, Gasque P, Morgan BP, Newman GR. Role of complement in the aetiology of Pick's disease? J Neuropathol Exp Neurol. (1996) 55:578–93. doi: 10.1097/00005072-199605000-00010
128. Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD, et al. Dysregulation of the complement cascade in the hSOD1G93Atransgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation (2013) 10:119. doi: 10.1186/1742-2094-10-119
129. Hakobyan S, Boyajyan A, Sim RB. Classical pathway complement activity in schizophrenia. Neurosci Lett. (2005) 374:35–7. doi: 10.1016/j.neulet.2004.10.024
130. Mayilyan KR, Arnold JN, Presanis JS, Soghoyan AF, Sim RB. Increased complement classical and mannan-binding lectin pathway activities in schizophrenia. Neurosci Lett. (2006) 404:336–41. doi: 10.1016/j.neulet.2006.06.051
131. Zakharyan R, Khoyetsyan A, Arakelyan A, Boyajyan A, Gevorgyan A, Stahelova A, et al. Association of C1QB gene polymorphism with schizophrenia in Armenian population. BMC Med Genet. (2011) 12:126. doi: 10.1186/1471-2350-12-126
132. Sória SLd, Gubert MCd, Ceresér KM, Gama CS, Kapczinski F. Increased serum levels of C3 and C4 in patients with schizophrenia compared to eutymic patients with bipolar disorder and healthy. Rev Bras Psiquiat. (2012) 34:119–20. doi: 10.1590/S1516-44462012000100022
133. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature (2016) 530:177–83. doi: 10.1038/nature16549
134. Ashwood P, Wills S, Van de Water J. The immune response in autism: a new frontier for autism research. J Leukocyte Biol. (2006) 80:1–15. doi: 10.1189/jlb.1205707
135. Fang C, Garbuzova-Davis S, Tan J, Obregon D. C1q as a regulator of brain development: implications for autism spectrum disorders. Brain Disord Ther. (2015) 4:152. doi: 10.4172/2168-975X.1000152
136. Torrente F, Ashwood P, Day R, Machado N, Furlano RI, Anthony A, et al. Small intestinal enteropathy with epithelial IgG and complement deposition in children with regressive autism. Mol Psychiatry (2002) 7:375–82, 334. doi: 10.1038/sj.mp.4001077
137. Xiao J, Li Y, Gressitt KL, He H, Kannan G, Schultz TL, et al. Cerebral complement C1q activation in chronic Toxoplasma infection. Brain Behav. Immun. (2016) 58:52–6. doi: 10.1016/j.bbi.2016.04.009
138. Reichelt KL, Seim AR, Reichelt WH. Could schizophrenia be reasonably explained by Dohan's hypothesis on genetic interaction with a dietary peptide overload? Prog Neuropsychopharmacol Biol Psychiatry (1996) 20:1083–114. doi: 10.1016/S0278-5846(96)00099-1
139. Bauer K, Kornhuber J. Blood-cerebrospinal fluid barrier in schizophrenic patients. Eur Arch Psychiatry Neurol Sci. (1987) 236:257–9. doi: 10.1007/BF00380949
140. Bechter K, Reiber H, Herzog S, Fuchs D, Tumani H, Maxeiner HG. Cerebrospinal fluid analysis in affective and schizophrenic spectrum disorders: identification of subgroups with immune responses and blood-CSF barrier dysfunction. J Psychiatr Res. (2010) 44:321–30. doi: 10.1016/j.jpsychires.2009.08.008
141. Tarlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, Scalise M, et al. Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell (2016) 167:1481–94.e18. doi: 10.1016/j.cell.2016.11.013
142. Dickerson F, Wilcox HC, Adamos M, Katsafanas E, Khushalani S, Origoni A, et al. Suicide attempts and markers of immune response in individuals with serious mental illness. J Psychiatr Res. (2017) 87:37–43. doi: 10.1016/j.jpsychires.2016.11.011
143. Santhanam B, Barry Kendler F. Nutritional factors in autism: an overview of nutritional factors in the etiology and management of autism. Integr Med. (2012) 11:46. Availabe Online at: http://www.imjournal.com/openaccess/autism.pdf
144. Campbell-McBride N. Gut and Psychology Syndrome: Natural Treatment for Autism, Dyspraxia, A.D.D., Dyslexia, A.D.H.D., Depression, Schizophrenia. Cambridge, UK: Medinform (2010).
145. Wang L, Yu YM, Zhang YQ, Zhang J, Lu N, Liu N. Hydrogen breath test to detect small intestinal bacterial overgrowth: a prevalence case-control study in autism. Eur Child Adolesc Psychiatry (2017) 27:233–40. doi: 10.1007/s00787-017-1039-2.
146. Horvath K, Papadimitriou JC, Rabsztyn A, Drachenberg C, Tildon JT. Gastrointestinal abnormalities in children with autistic disorder. J Pediatr. (1999) 135:559–63. doi: 10.1016/S0022-3476(99)70052-1
147. Daileda T, Baek P, Sutter ME, Thakkar K. Disaccharidase activity in children undergoing esophagogastroduodenoscopy: a systematic review. World J Gastrointes Pharmacol Ther. (2016) 7:283–93. doi: 10.4292/wjgpt.v7.i2.283
148. Vojdani A, Bazargan M, Vojdani E, Samadi J, Nourian AA, Eghbalieh N, et al. Heat shock protein and gliadin peptide promote development of peptidase antibodies in children with autism and patients with autoimmune disease. Clin Diagn Lab Immunol. (2004) 11:515–24. doi: 10.1128/CDLI.11.3.515-524.2004
149. Bashir S, Al-Ayadhi L. Alterations in plasma dipeptidyl peptidase IV in autism: a pilot study. Neurol Psychiatry Brain Res. (2014) 20:41–44. doi: 10.1016/j.npbr.2014.03.001
150. Kostyra E, Sienkiewicz-Szłapka E, Jarmołowska B, Krawczuk S, Kostyra H. Opioid peptides derived from milk proteins. Polish J Food Nutr Sci. (2004) 13:25–35. Available Online at: http://journal.pan.olsztyn.pl//pdfy/2004/1s/rozdzial2.pdf
151. Maes M, De M, Scharpé S, Desnyder R, Ranjan R, Meltzer HY. Alterations in plasma dipeptidyl peptidase IV enzyme activity in depression and schizophrenia: effects of antidepressants and antipsychotic drugs. Acta Psychiatr Scand. (1996) 93:1–8. doi: 10.1111/j.1600-0447.1996.tb10612.x
152. Maes M, De Meester I, Verkerk R, De Medts P, Wauters A, Vanhoof G, et al. Lower serum dipeptidyl peptidase IV activity in treatment resistant major depression: relationships with immune-inflammatory markers. Psychoneuroendocrinology (1997) 22:65–78. doi: 10.1016/S0306-4530(96)00040-6
153. Brudnak MA, Rimland B, Kerry RE, Dailey M, Taylor R, Stayton B, et al. Enzyme-based therapy for autism spectrum disorders – is it worth another look? Med Hypotheses (2002) 58:422–8. doi: 10.1054/mehy.2001.1513
154. Saad K, Eltayeb AA, Mohamad IL, Al-Atram AA, Elserogy Y, Bjorklund G, et al. A Randomized, placebo-controlled trial of digestive enzymes in children with autism spectrum disorders. Clin Psychopharmacol Neurosci. (2015) 13:188–93. doi: 10.9758/cpn.2015.13.2.188
155. Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol. (2005) 2:416–22. doi: 10.1038/ncpgasthep0259
156. Visser J, Rozing J, Sapone A, Lammers K, Fasano A. Tight junctions, intestinal permeability, and autoimmunity: celiac disease and type 1 diabetes paradigms. Ann NY Acad Sci. (2009) 1165:195–205. doi: 10.1111/j.1749-6632.2009.04037.x
157. Fasano A. Leaky gut and autoimmune diseases. Clin Rev Allergy Immunol. (2012) 42:71–8. doi: 10.1007/s12016-011-8291-x
158. Nielsen PR, Benros ME, Dalsgaard S. Associations between autoimmune diseases and attention-deficit/hyperactivity disorder: a nationwide study. J Am Acad Child Adolesc Psychiatry (2017) 56:234–40.e1. doi: 10.1016/j.jaac.2016.12.010
159. Benros ME, Eaton WW, Mortensen PB. The epidemiologic evidence linking autoimmune diseases and psychosis. Biol psychiatry (2014) 75:300–6. doi: 10.1016/j.biopsych.2013.09.023
160. Eaton WW, Byrne M, Ewald H, Mors O, Chen CY, Agerbo E, Mortensen PB. Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am J Psychiatry (2006) 163:521–8. doi: 10.1176/appi.ajp.163.3.521
161. Chen P, Jiang T, Ouyang J, Chen Y. Depression, another autoimmune disease from the view of autoantibodies. Med Hypotheses (2009) 73:508–9. doi: 10.1016/j.mehy.2009.06.018
162. Gleicher N. Postpartum depression, an autoimmune disease? Autoimmun Rev. (2007) 6:572–76. doi: 10.1016/j.autrev.2007.04.002
163. Strous RD, Shoenfeld Y. Behavioral changes in systemic lupus erythematosus are of an autoimmune nature. Nat Clin Pract Rheumatol. (2007) 3:592–3. doi: 10.1038/ncprheum0622
164. Chen Y, Jiang T, Chen P, Ouyang J, Xu G, Zeng Z, Sun Y. Emerging tendency towards autoimmune process in major depressive patients: a novel insight from Th17 cells. Psychiatry Res. (2011) 188:224–30. doi: 10.1016/j.psychres.2010.10.029
165. Benros ME, Waltoft BL, Nordentoft M, Krogh J, Mortensen PB. Autoimmunity and infections as risk factors for depression and other severe mental illnesses. Neurol Psychiatry Brain Res. (2012) 18:40–1. doi: 10.1016/j.npbr.2012.02.005
166. Iseme RA, McEvoy M, Kelly B, Agnew L, Attia J, Walker FR. Autoantibodies and depression: evidence for a causal link? Neurosci Biobehav Rev. (2014) 40:62–79. doi: 10.1016/j.neubiorev.2014.01.008
167. Katzav A, Solodeev I, Brodsky O, Chapman J, Pick CG, Blank M, et al. Induction of autoimmune depression in mice by anti-ribosomal P antibodies via the limbic system. Arthritis Rheum. (2007) 56:938–48. doi: 10.1002/art.22419
168. Chandley MJ, Miller MN, Kwasigroch CN, Wilson TD, Miller BE. Increased antibodies for the alpha7 subunit of the nicotinic receptor in schizophrenia. Schizophr Res. (2009) 109:98–101. doi: 10.1016/j.schres.2009.01.023
169. Chang SH, Chiang SY, Chiu CC, Tsai CC, Tsai HH, Huang CY, et al. Expression of anti-cardiolipin antibodies and inflammatory associated factors in patients with schizophrenia. Psychiatry Res. (2011) 187:341–6. doi: 10.1016/j.psychres.2010.04.049
170. Laske C, Zank M, Klein R, Stransky E, Batra A, Buchkremer G, et al. Autoantibody reactivity in serum of patients with major depression, schizophrenia and healthy controls. Psychiatry Res. (2008) 158:83–6. doi: 10.1016/j.psychres.2006.04.023
171. Schott K J, Schaefer E, Richartz E, Batra A, Eusterschulte B, Klein R, et al. Autoantibodies to serotonin in serum of patients with psychiatric disorders. Psychiatry Res. (2003) 121:51–7. doi: 10.1016/S0165-1781(03)00137-9
172. Margari F, Petruzzelli MG, Mianulli R, Campa MG, Pastore A, Tampoia M. Circulating anti-brain autoantibodies in schizophrenia and mood disorders. Psychiatry Res. (2015) 230:704–8. doi: 10.1016/j.psychres.2015.10.029
173. Ezeoke A, Mellor A, Buckley P, Miller B. A systematic, quantitative review of blood autoantibodies in schizophrenia. Schizophr Res. (2013) 150:245–51. doi: 10.1016/j.schres.2013.07.029
174. Pathmanandavel K, Starling J, Dale RC, Brilot F. Autoantibodies and the immune hypothesis in psychotic brain diseases: challenges and perspectives. Clin Dev Immunol. (2013) 2013:257184. doi: 10.1155/2013/257184
175. Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. (2011) 10:759–72. doi: 10.1016/S1474-4422(11)70096-5
176. Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry (2012) 83:638–45. doi: 10.1136/jnnp-2011-301237
177. Zandi MS, Irani SR, Lang B, Waters P, Jones PB, McKenna P, et al. Disease-relevant autoantibodies in first episode schizophrenia. J Neurol. (2011) 258:686–8. doi: 10.1007/s00415-010-5788-9
178. Tsutsui K, Kanbayashi T, Tanaka K, Boku S, Ito W, Tokunaga J, et al. Anti-NMDA-receptor antibody detected in encephalitis, schizophrenia, and narcolepsy with psychotic features. BMC Psychiatry (2012) 12:37. doi: 10.1186/1471-244X-12-37
179. Steiner J, Walter M, Glanz W, Sarnyai Z, Bernstein HG, Vielhaber S, et al. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-D-aspartate glutamate receptor encephalitis. JAMA Psychiatry (2013) 70:271–8. doi: 10.1001/2013.jamapsychiatry.86
180. Lennox BR, Coles AJ, Vincent A. Antibody-mediated encephalitis: a treatable cause of schizophrenia. Brit J Psychiatry (2012) 200:92–4. doi: 10.1192/bjp.bp.111.095042
181. Lambert J, Vojdani A. Correlation of tissue antibodies and food immune reactivity in randomly selected patient specimens. J Clin Cell Immunol. (2017) 8b. doi: 10.4172/2155-9899.1000521
182. Stefferl A, Schubart A, Storch M, Amini A, Mather I, Lassmann H, Linington C. Butyrophilin, a milk protein, modulates the encephalitogenic T cell response to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis. J Immunol. (2000) 165:2859–65. doi: 10.4049/jimmunol.165.5.2859
183. Guggenmos J, Schubart AS, Ogg S,ersson M, Olsson T, Mather IH, et al. Antibody cross-reactivity between myelin oligodendrocyte glycoprotein and the milk protein butyrophilin in multiple sclerosis. J Immunol. (2003) 172:661–8. doi: 10.4049/jimmunol.172.1.661
184. Vojdani A, Tarash I. Cross-reaction between gliadin and different food and tissue antigens. Food Nutr Sci. (2013) 4:20–32. doi: 10.4236/fns.2013.41005
185. Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol. (2012) 10:735–42. doi: 10.1038/nrmicro2876
186. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. (2012) 13:701–12. doi: 10.1038/nrn3346
187. Gareau MG. Microbiota-gut-brain axis and cognitive function. Adv Exp Med Biol. (2014) 817:357–71. doi: 10.1007/978-1-4939-0897-4_16
188. Rea K, Dinan TG, Cryan JF. The microbiome: a key regulator of stress and neuroinflammation. Neurobiol Stress (2016) 4:23–33. doi: 10.1016/j.ynstr.2016.03.001
189. Kelly JR, Kennedy PJ, Cryan JF, Dinan TG, Clarke G, Hyland NP. Breaking down the barriers: the gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front Cell Neurosci. (2015) 9:392. doi: 10.3389/fncel.2015.00392
190. Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
191. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Trans Med. (2014) 6:263ra158. doi: 10.1126/scitranslmed.3009759
192. Donato KA, Gareau MG, Wang YJ, Sherman PM. Lactobacillus rhamnosus GG attenuates interferon-{gamma} and tumour necrosis factor-alpha-induced barrier dysfunction and pro-inflammatory signalling. Microbiology (2010) 156:3288–97. doi: 10.1099/mic.0.040139-0
193. Yan F, Polk DB. Disruption of NF-kappaB signalling by ancient microbial molecules: novel therapies of the future? Gut (2010) 59:421–6. doi: 10.1136/gut.2009.179614
194. Hegazy SK, El-Bedewy MM. Effect of probiotics on pro-inflammatory cytokines and NF-kappaB activation in ulcerative colitis. World J Gastroenterol. (2010) 16:4145–51. doi: 10.3748/wjg.v16.i33.4145
195. Desbonnet L, Garrett L, Clarke G, Bienenstock J, Dinan TG. The probiotic Bifidobacteria infantis: an assessment of potential antidepressant properties in the rat. J Psychiatr Res. (2008) 43:164–74. doi: 10.1016/j.jpsychires.2008.03.009
196. Desbonnet L, Garrett L, Clarke G, Kiely B, Cryan JF, Dinan TG. Effects of the probiotic Bifidobacterium infantis in the maternal separation model of depression. Neuroscience (2010) 170:1179–88. doi: 10.1016/j.neuroscience.2010.08.005
197. Lamprecht M, Bogner S, Schippinger G, Steinbauer K, Fankhauser F, Hallstroem S, et al. Probiotic supplementation affects markers of intestinal barrier, oxidation, and inflammation in trained men; a randomized, double-blinded, placebo-controlled trial. J Int Soc Sports Nutr. (2012) 9:45. doi: 10.1186/1550-2783-9-45
198. Ohland CL, MacNaughton WK. Probiotic bacteria and intestinal epithelial barrier function. Am J Physiol Gastrointest Liver Physiol. (2010) 298:G807–19. doi: 10.1152/ajpgi.00243.2009
199. Rao AV, Bested AC, Beaulne TM, Katzman MA, Iorio C, Berardi JM, et al. A randomized, double-blind, placebo-controlled pilot study of a probiotic in emotional symptoms of chronic fatigue syndrome. Gut Pathog. (2009) 1:6. doi: 10.1186/1757-4749-1-6
200. Messaoudi M, Lalonde R, Violle N, Javelot H, Desor D, Nejdi A, et al. Assessment of psychotropic-like properties of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in rats and human subjects. Brit J Nutr. (2011) 105:755–64. doi: 10.1017/S0007114510004319
201. Steenbergen L, Sellaro R, van Hemert S, Bosch JA, Colzato LS. A randomized controlled trial to test the effect of multispecies probiotics on cognitive reactivity to sad mood. Brain Behav Immun. (2015) 48:258–64. doi: 10.1016/j.bbi.2015.04.003
202. Akkasheh G, Kashani-Poor Z, Tajabadi-Ebrahimi M, Jafari P, Akbari H, Taghizadeh M, et al. Clinical and metabolic response to probiotic administration in patients with major depressive disorder: A randomized, double-blind, placebo-controlled trial. Nutrition (2016) 32:315–20. doi: 10.1016/j.nut.2015.09.003
203. Mohammadi AA, Jazayeri S, Khosravi-Darani K, Solati Z, Mohammadpour N, Asemi Z, et al. The effects of probiotics on mental health and hypothalamic-pituitary-adrenal axis: a randomized, double-blind, placebo-controlled trial in petrochemical workers. Nutr Neurosc. (2016) 19:387–95. doi: 10.1179/1476830515Y.0000000023
204. McKean J, Naug H, Nikbakht E, Amiet B, Colson N. Probiotics and subclinical psychological symptoms in healthy participants: a systematic review and meta-analysis. J Altern Complement. Med. (2016) 23:249–58. doi: 10.1089/acm.2016.0023
205. Benton D, Williams C, Brown A. Impact of consuming a milk drink containing a probiotic on mood and cognition. Eur J Clin Nutr. (2007) 61:355–361. doi: 10.1038/sj.ejcn.1602546
206. Akbari E, Asemi Z, Daneshvar Kakhaki R, Bahmani F, Kouchaki E, Tamtaji OR, et al. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer's disease: a randomized, double-blind and controlled trial. Front Aging Neurosci. (2016) 8:256. doi: 10.3389/fnagi.2016.00256
207. Huang R, Wang K, Hu J. Effect of probiotics on depression: a systematic review and meta-analysis of randomized controlled trials. Nutrients (2016) 8:E483. doi: 10.3390/nu8080483
208. Kim YK, Shin C. The microbiota-gut-brain axis in neuropsychiatric disorders: pathophysiological mechanisms and novel treatments. Curr Neuropharmacol. (2017) 16:559–73. doi: 10.2174/1570159X15666170915141036
209. Lawrence K, Hyde J. Microbiome restoration diet improves digestion, cognition and physical and emotional wellbeing. PLoS ONE (2017) 12:e0179017. doi: 10.1371/journal.pone.0179017
210. Misra S, Mohanty D. Psychobiotics: a new approach for treating mental illness? Crit Rev Food Sci Nutr. (2017). doi: 10.1080/10408398.2017.1399860. [Epub ahead of print].
211. Ng QX, Peters C, Ho CYX, Lim DY, Yeo WS. A meta-analysis of the use of probiotics to alleviate depressive symptoms. J Affect Disord. (2017) 228:13–9. doi: 10.1016/j.jad.2017.11.063
212. Rios AC, Maurya PK, Pedrini M, Zeni-Graiff M, Asevedo E, Mansur RB, et al. Microbiota abnormalities and the therapeutic potential of probiotics in the treatment of mood disorders. Rev Neurosci. (2017) 28:739–49. doi: 10.1515/revneuro-2017-0001
213. Romijn AR, Rucklidge JJ, Kuijer RG, Frampton C. A double-blind, randomized, placebo-controlled trial of Lactobacillus helveticus and Bifidobacterium longum for the symptoms of depression. Austral NZ J Psychiatry (2017) 51:810–21. doi: 10.1177/0004867416686694
214. Slykerman RF, Hood F, Wickens K, Thompson J, Barthow C, Murphy R, et al. Probiotic in pregnancy study, effect of Lactobacillus rhamnosus HN001 in pregnancy on postpartum symptoms of depression and anxiety: a randomised double-blind placebo-controlled trial. Ebiomedicine (2017) 24:159–65. doi: 10.1016/j.ebiom.2017.09.013
215. Wallace CJK, Milev R. The effects of probiotics on depressive symptoms in humans: a systematic review. Ann Gen Psychiatry (2017) 16:14. doi: 10.1186/s12991-017-0138-2
216. Dinan TG, Stanton C, Cryan JF. Psychobiotics: a novel class of psychotropic. Biol Psychiatry (2013) 74:720–6. doi: 10.1016/j.biopsych.2013.05.001
217. Naseribafrouei A, Hestad K, Avershina E, Sekelja M, Linlokken A, Wilson R, Rudi K. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil. (2014) 26:1155–62. doi: 10.1111/nmo.12378
218. Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun. (2015) 48:186–94. doi: 10.1016/j.bbi.2015.03.016
219. Aizawa E, Tsuji H, Asahara T, Takahashi T, Teraishi T, Yoshida S, et al. Possible association of Bifidobacterium and Lactobacillus in the gut microbiota of patients with major depressive disorder. J Affect Disord. (2016) 202:254–7. doi: 10.1016/j.jad.2016.05.038
220. Lin P, Ding B, Feng C, Yin S, Zhang T, Qi X, et al. Prevotella and Klebsiella proportions in fecal microbial communities are potential characteristic parameters for patients with major depressive disorder. J Affect Disord. (2017) 207:300–4. doi: 10.1016/j.jad.2016.09.051
221. Adams JB, Johansen LJ, Powell LD, Quig D, Rubin RA. Gastrointestinal flora and gastrointestinal status in children with autism–comparisons to typical children and correlation with autism severity. BMC Gastroenterol. (2011) 11:22. doi: 10.1186/1471-230X-11-22
222. De Angelis M, Francavilla R, Piccolo M, De Giacomo A, Gobbetti M. Autism spectrum disorders and intestinal microbiota. Gut Microbes (2015) 6:207–13. doi: 10.1080/19490976.2015.1035855
223. Tomova A, Husarova V, Lakatosova S, Bakos J, Vlkova B, Babinska K, et al. Gastrointestinal microbiota in children with autism in Slovakia. Physiol Behav. (2015) 138:179–87. doi: 10.1016/j.physbeh.2014.10.033
224. Strati F, Cavalieri D, Albanese D, De Felice C, Donati C, Hayek J, et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome (2017) 5:24. doi: 10.1186/s40168-017-0242-1
225. Williams BL, Hornig M, Buie T, Bauman ML, Cho Paik M, Wick I, et al. Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS ONE (2011) 6:e24585. doi: 10.1371/journal.pone.0024585
226. Nguyen TT, Kosciolek T, Eyler LT, Knight R, Jeste DV. Overview and systematic review of studies of microbiome in schizophrenia and bipolar disorder. J Psychiatr Res. (2018) 99:50–61. doi: 10.1016/j.jpsychires.2018.01.013
227. Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host's metabolism. Mol Psychiatry (2016) 21:786–96. doi: 10.1038/mp.2016.44
228. Gabbard SL, Lacy BE, Levine GM, Crowell MD. The impact of alcohol consumption and cholecystectomy on small intestinal bacterial overgrowth. Digest Dis Sci. (2014) 59:638–44. doi: 10.1007/s10620-013-2960-y
229. Bahr SM, Weidemann BJ, Castro AN, Walsh JW, deLeon O, Burnett CML, et al. Risperidone-induced weight gain is mediated through shifts in the gut microbiome and suppression of energy expenditure. EBioMedicine (2015) 2:1725–34. doi: 10.1016/j.ebiom.2015.10.018
230. Morgan AP, Crowley JJ, Nonneman RJ, Quackenbush CR, Miller CN, Ryan AK, et al. The antipsychotic olanzapine interacts with the gut microbiome to cause weight gain in mouse. PLoS ONE (2014) 9:e115225. doi: 10.1371/journal.pone.0115225
231. Bahr SM, Tyler BC, Wooldridge N, Butcher BD, Burns TL, Teesch LM, et al. Use of the second-generation antipsychotic, risperidone, and secondary weight gain are associated with an altered gut microbiota in children. Trans Psychiatry (2015) 5:e652. doi: 10.1038/tp.2015.135
232. Davey KJ, Cotter PD, O'Sullivan O, Crispie F, Dinan TG, Cryan JF, et al. Antipsychotics and the gut microbiome: olanzapine-induced metabolic dysfunction is attenuated by antibiotic administration in the rat. Trans Psychiatry (2013) 3:e309. doi: 10.1038/tp.2013.83
233. Smithh RS. Cytokines & Depression. How Your Immune System Causes Depression (1997). Available online at: http://www.cytokines-and-depression.com/.
234. Severance EG, Yolken RH, Eaton WW. Autoimmune diseases, gastrointestinal disorders and the microbiome in schizophrenia: more than a gut feeling. Schizophr Res. (2016) 176:23–35. doi: 10.1016/j.schres.2014.06.027
235. Alpay K, Ertas M, Orhan EK, Ustay DK, Lieners C, Baykan B. Diet restriction in migraine, based on IgG against foods: a clinical double-blind, randomised, cross-over trial. Cephalalgia (2010) 30:829–37. doi: 10.1177/0333102410361404
236. Drisko J, Bischoff B, Hall M, McCallum R. Treating irritable bowel syndrome with a food elimination diet followed by food challenge and probiotics. J Am Coll Nutr. (2006) 25:514–22. doi: 10.1080/07315724.2006.10719567
237. Mitchell N, Hewitt CE, Jayakody S, Islam M, Adamson J, Watt I, Torgerson DJ. Randomised controlled trial of food elimination diet based on IgG antibodies for the prevention of migraine like headaches. Nutr J. (2011) 10:85. doi: 10.1186/1475-2891-10-85
238. Aydinlar EI, Dikmen PY, Tiftikci A, Saruc M, Aksu M, Gunsoy HG, et al. IgG-based elimination diet in migraine plus irritable bowel syndrome. Headache (2013) 53:514–25. doi: 10.1111/j.1526-4610.2012.02296.x
239. Atkinson W, Sheldon TA, Shaath N, Whorwell PJ. Food elimination based on IgG antibodies in irritable bowel syndrome: a randomised controlled trial. Gut (2004) 53:1459–64. doi: 10.1136/gut.2003.037697
240. Bentz S, Hausmann M, Piberger H, Kellermeier S, Paul S, Held L, et al. Clinical relevance of IgG antibodies against food antigens in Crohn's disease: a double-blind cross-over diet intervention study. Digestion (2010) 81:252–64. doi: 10.1159/000264649
241. Sturniolo GC, Di Leo V, Ferronato A, D'Odorico A, D'Inca R. Zinc supplementation tightens “leaky gut” in Crohn's disease. Inflammatory Bowel Dis. (2001) 7:94–8. doi: 10.1097/00054725-200105000-00003
242. Zhang B, Guo Y. Supplemental zinc reduced intestinal permeability by enhancing occludin and zonula occludens protein-1 (ZO-1) expression in weaning piglets. Brit J Nutr. (2009) 102:687–93. doi: 10.1017/S0007114509289033
243. Lopresti AL, Maes M, Maker GL, Hood SD, Drummond PD. Curcumin for the treatment of major depression: a randomised, double-blind, placebo controlled study. J Affect Disord. (2014) 167:368–75. doi: 10.1016/j.jad.2014.06.001
244. Ranjbar E, Kasaei MS, Mohammad-Shirazi M, Nasrollahzadeh J, Rashidkhani B, Shams J, et al. Effects of zinc supplementation in patients with major depression: a randomized clinical trial. Iran J Psychiatry (2013) 8:73–9.
245. Aggarwal BB, Gupta SC, Sung B. Curcumin: an orally bioavailable blocker of TNF and other pro-inflammatory biomarkers. Brit J Pharmacol. (2013) 169:1672–92. doi: 10.1111/bph.12131
246. Jeong YI, Kim SW, Jung ID, Lee JS, Chang JH, Lee CM, et al. Curcumin suppresses the induction of indoleamine 2,3-dioxygenase by blocking the Janus-activated kinase-protein kinase Cdelta-STAT1 signaling pathway in interferon-gamma-stimulated murine dendritic cells. J Biol Chem. (2009) 284:3700–8. doi: 10.1074/jbc.M807328200
247. Buhrmann C, Mobasheri A, Busch F, Aldinger C, Stahlmann R, Montaseri A, et al. Curcumin modulates nuclear factor kappaB (NF-kappaB)-mediated inflammation in human tenocytes in vitro: role of the phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem. (2011) 286:28556–66. doi: 10.1074/jbc.M111.256180
Keywords: microbiota-gut-brain axis, intestinal permeability, autoimmunity, psychiatric disorders, food antigens, gluten, exorphins, immunoglobulins
Citation: Rudzki L and Szulc A (2018) “Immune Gate” of Psychopathology—The Role of Gut Derived Immune Activation in Major Psychiatric Disorders. Front. Psychiatry 9:205. doi: 10.3389/fpsyt.2018.00205
Received: 15 February 2018; Accepted: 03 May 2018;
Published: 29 May 2018.
Edited by:
Błazej Misiak, Wroclaw Medical University, PolandReviewed by:
Chuanjun Zhuo, Tianjin Medical University General Hospital, ChinaCopyright © 2018 Rudzki and Szulc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leszek Rudzki, cnVkemtpLmxAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.