
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 22 July 2020
Sec. Plant Development and EvoDevo
Volume 11 - 2020 | https://doi.org/10.3389/fpls.2020.01000
 Xiaoxue Zeng1,2
Xiaoxue Zeng1,2 Kohei Mishina1
Kohei Mishina1 Juqing Jia1
Juqing Jia1 Assaf Distelfeld3
Assaf Distelfeld3 Peter Jeff Maughan4Shinji Kikuchi2Hidenori Sassa2
Peter Jeff Maughan4Shinji Kikuchi2Hidenori Sassa2 Takao Komatsuda1,2*
Takao Komatsuda1,2*In many non-cultivated angiosperm species, seed dispersal is facilitated by the shattering of the seed head at maturity; in the Triticeae tribe, to which several of the world's most important cereals belong, shattering takes the form of a disarticulation of the rachis. The products of the genes Btr1 and Btr2 are both required for disarticulation to occur above the rachis nodes within the genera Hordeum (barley) and Triticum/Aegilops (wheat). Here, it has been shown that both Btr1 and Btr2 are specific to the Triticeae tribe, although likely paralogs (Btr1-like and Btr2-like) are carried by the family Poaceae including Triticeae. Aegilops tauschii (the donor of the bread wheat D genome) lacks a copy of Btr1 and disarticulation in this species occurs below, rather than above the rachis node; thus, the product of Btr1 appears to be required for disarticulation to occur above the rachis node.
Shattering of the seed head at maturity has evolved as an effective means of seed dispersal in many angiosperms and represents one of the most conspicuous differences between a wild species and its related domesticate(s) (Zohary et al., 2012; Olsen and Wendel, 2013; Dong and Wang, 2015). Since shattering does not allow harvesting to be carried out after physiological maturity, the selection of non-shattering types is considered to be a key crop domestication event.
The Triticeae tribe harbors a number of the most important cereal crop species, including barley (Hordeum vulgare), cereal rye (Secale cereale), and the various forms of wheat, including diploid einkorn (Triticum monococcum), tetraploid emmer (T. turgidum ssp. dicoccum), and durum (T. turgidum ssp. durum) and hexaploid bread (T. aestivum). Seed dispersal in wild Triticeae species is achieved by a process of disarticulation affecting various parts of the mature inflorescence (Frederiksen and Seberg, 1992; Sakuma et al., 2011). In “brittle rachis” types, the flower stalk disarticulates at a number of sites, while in “brittle rachilla” types, it occurs instead along the axis of the spikelet (Figure 1, Table 1). Both types are represented within each of the 32 genera belonging to the Triticeae tribe (Sakuma et al., 2011). The site of rachis disarticulation varies from species to species (Figure 1A): when it occurs above a rachis node, “wedge-type” dispersal units are formed (Figure 1B): these feature commonly among the wild relatives of the cereals, such as in H. vulgare ssp. spontaneum (the ancestor of domesticated barley), in T. dicoccoides (the ancestor of cultivated polyploidy wheats), in S. vavilovii (the ancestor of cereal rye) and in both T. monococcum ssp. boeoticum (the ancestor of einkorn wheat) and T. urartu (the donor of the A genome present in both durum and bread wheat) (Harlan and Zohary, 1966; Zohary et al., 2012). Breakage at a single site above the lowermost rachis node results in a “whole spike-type” or “umbrella-type” dispersal unit, while its occurrence below a rachis node results in the “barrel-type” dispersal units produced by Ae. tauschii, the donor of bread wheat's D genome (Figure 1C). Rachilla disarticulation can occur at two sites (Figure 1A): the most common phenotype involves breakage below every rachilla node, resulting in dispersal units similar to those produced by Ae. tauschii. Disarticulation below the glumes is rare in the Triticeae: the only known example is in Elytrigia repens (Sakuma et al., 2011); on the other hand, it is frequent among species belonging to the tribes Oryzeae, Paniceae and Andropogoneae (Table 1).
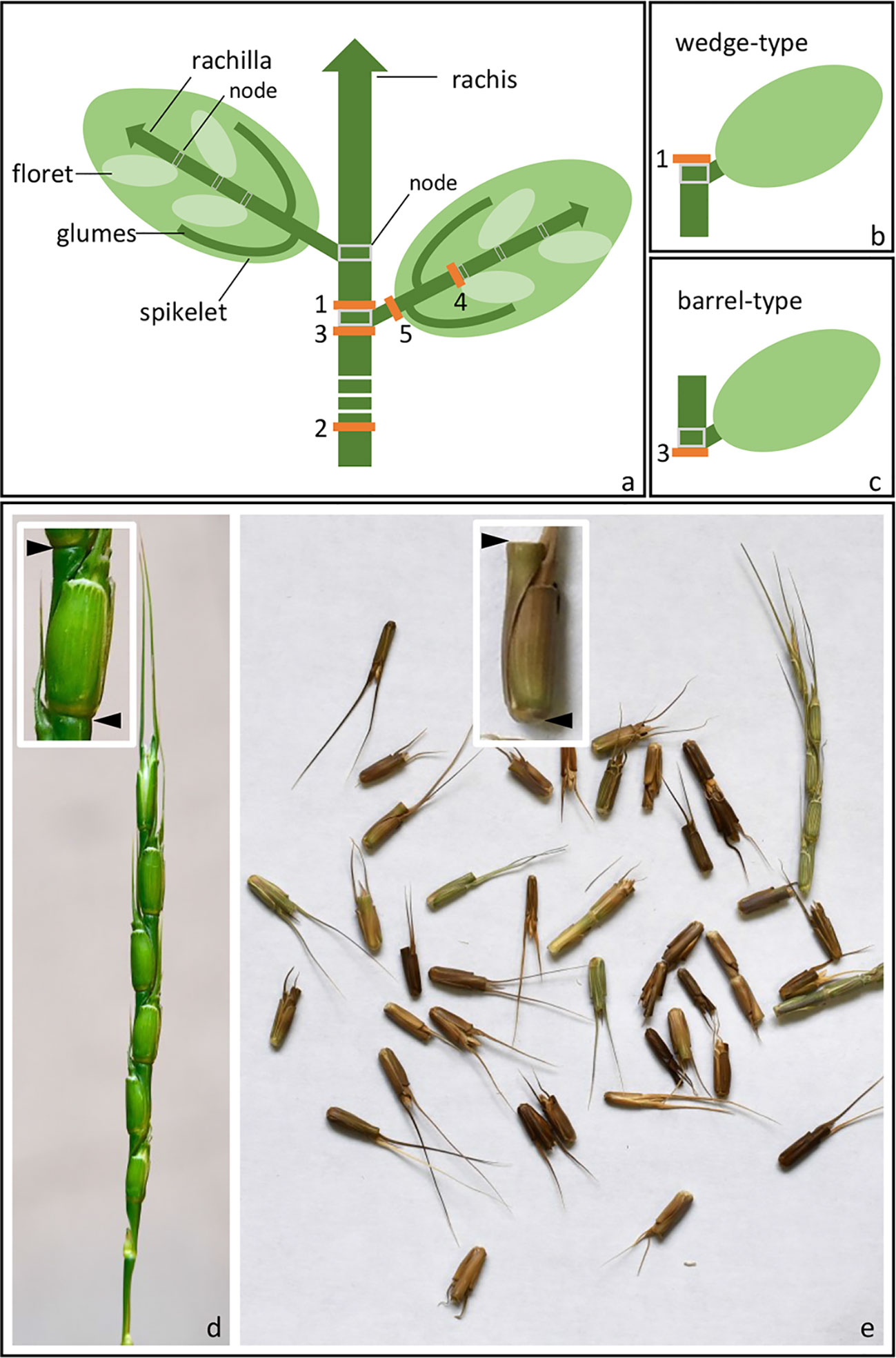
Figure 1 Spike disarticulation in Triticeae species. (A) In the mature spike, disarticulation can occur either (1) above the rachis nodes, (2) above the lowest rachis node, (3) below the rachis nodes, (4) below the rachilla nodes, or (5) below the glumes. (B) Disarticulation above the rachis node produces a wedge-type dispersal unit, while (C) disarticulation below the rachis node produces a barrel-type dispersal unit. (D, E) Barrel-type brittle rachis generated from Ae. tauschii (accession: AL8/78). Spike before (D) and upon (E) maturity was shown. Arrows indicate the disarticulation point.
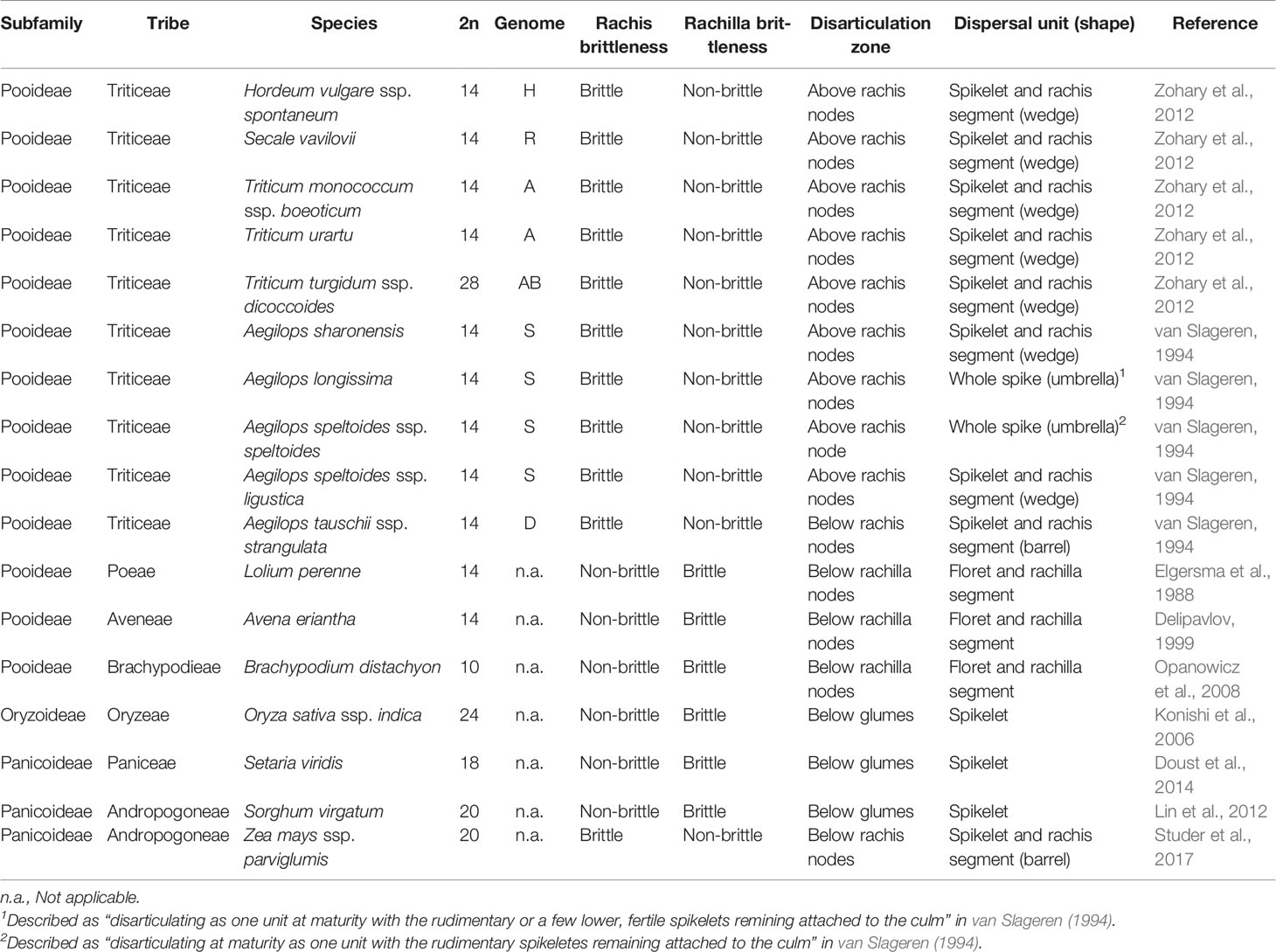
Table 1 Seed shattering characterized in the set of wild Poaceae species.
The formation of wedge-type dispersal units is genetically determined in barley by the genes Btr1 and Btr2, a pair of dominant, complementary, linked genes mapping to the short arm of chromosome 3H. The products of Btr1 and Btr2 are, respectively, a 196 and a 202 residue protein, the function of neither of which is currently known. A 1 bp deletion in the Btr1 coding sequence, and one of 11 bp in the Btr2 coding sequence are sufficient to convert a shattering to a non-shattering (“non-brittle”) phenotype (Pourkheirandish et al., 2015). In T. monococcum ssp. boeoticum, the substitution of a single residue in the Btr1 product converts a brittle to a non-brittle rachis (Pourkheirandish et al., 2018; Zhao et al., 2019). In the polyploid wheat species, mutations at both the A and B genome copies of Btr1 are required for the formation of a non-brittle rachis (Avni et al., 2017). Btr homoeoloci have been mapped to regions syntenous with the 3H region in a number of species (Urbano et al., 1988; King et al., 1997; Chen et al., 1998; Li and Gill, 2006; Jiang et al., 2014). Of the 22 species belonging to the genus Aegilops (van Slageren, 1994), Ae. speltoides is of particular interest in connection with spike disarticulation, because it features two morphological forms of rachis brittleness: while ssp. speltoides forms whole spike-type disarticulation units, ssp. ligustica disarticulates at each rachis node to produce wedge-type ones (Li and Gill, 2006). The whole spike-type disarticulation trait is recessive to the wedge-type one, assumed to reflect an allelic interaction at Brt1 (Li and Gill, 2006). Ae. tauschii exceptionally produces barrel-type dispersal units (van Slageren, 1994). Rather than mapping to the short arm of 3D, however, the locus responsible for this trait maps to the long arm of the chromosome as shown in three independent crosses (Amagai et al., 2015; Katkout et al., 2015; Zhang et al., 2015).
The evolutionary history of Btr1 and Btr2 remains obscure. The barley genome harbors a paralog of both Btr1 and Btr2 (respectively, Btr1-like and Btr2-like); while all four of these genes map to 3HS, Btr1-like and Btr2-like are separated from one another by just 4.2 kbp, but Btr2 maps 100 kbp away from this locus and Btr1 200 kbp away (Pourkheirandish et al., 2015). A similar situation pertains in T. turgidum (Avni et al., 2017). The relevant duplication events are known to have occurred post the divergence of the Hordeum and Brachypodium lineages (Pourkheirandish et al., 2015). The aim of the present study was to shed more light on the evolutionary events surrounding the acquisition in the Triticeae of Btr1 and Btr2.
A stock of T. monococcum ssp. boeoticum (accession KT1-1) was obtained from the National BioResource Project (NBRP)/KOMUGI, Kyoto University, Kyoto, Japan and used for PCR-cloning. Grains of S. vavilovii were kindly provided by Prof. Eva Bauer, Technische Universität München, Munich, Germany and used for phenotype observation and PCR-cloning. All the materials were grown in a greenhouse at NARO (Tsukuba, Japan).
The coding sequences of Btr1 (591 nt), Btr1-like (597 nt), Btr2 (609 nt) and Btr2-like (579 nt), all housed on the H. vulgare ssp. spontaneum (accession OUH602) BAC clone KR813335.1 (Pourkheirandish et al., 2015), were used as query sequences for a BLASTn search (E-value threshold: 1E−20) of the genomic sequences of S. vavilovii (Bauer et al., 2017), T. urartu (Ling et al., 2018), Ae. sharonensis, T. aestivum (Alaux et al., 2018), and Ae. speltoides ssp. speltoides (unpublished data provided by A Distelfeld [Tel Aviv University, Israel]), Ae. longissima (unpublished data provided by A Sharon [Tel Aviv University]), Ae. tauschii (Luo et al., 2017), T. dicoccoides (Avni et al., 2017), Lolium perenne (Byrne et al., 2015), Avena eriantha (provided by Maughan et al., 2019, [Brigham Young University, Provo, UT, USA]), and Brachypodium distachyon (International Brachypodium Initiative, 2010). A BLASTp search (E-value threshold: 1E-2 and identity threshold: 30%) was used to identify homologs present in rice (Kawahara et al., 2013), foxtail millet (Setaria italica) (Bennetzen et al., 2012), sorghum (Paterson et al., 2009), maize (Jiao et al., 2017), and Arabidopsis thaliana (Swarbreck et al., 2008).
Genomic DNA was extracted from fresh leaves of S. vavilovii and T. monococcum, as described by Komatsuda et al. (1998), to provide the template for PCRs driven by primers designed using Primer 3 software (bioinfo.ut.ee/primer3) from their respective genomic DNA contigs (Table S1). Each 10 µl PCR contained 0.25 U ExTaq polymerase (Takara, Tokyo, Japan), 1× ExTaq polymerase buffer, 0.3 μM of each primer, 200 μM dNTP, 2 mM MgCl2, 2.5% v/v DMSO, and 20 ng genomic DNA. The amplification regime comprised an initial denaturation step (94°C/5 min), followed by 30 cycles of 94°C/30 s, 57°C–62°C (primer pair dependent, see Table S1)/30 s, 72°C/90 s, and a final extension step of 72°C/10 min. The resulting amplicons were purified using a QIAquick PCR purification kit (Qiagen, Germantown, MD, USA), and sequenced using a reaction based on Big Dye Terminator v3.1 (Applied Biosystems, Foster City, CA, USA). The Agencourt CleanSeq system (Beckman Coulter Inc., Brea, CA, USA) was used to remove salts, non-incorporated dNTPs and dye terminator, and the sequence data acquired using an ABI Prism 3130/3730xL sequencer (Applied Biosystems).
The coding region of the S. vavilovii and T. monococcum Btr homologs was identified using the FGENESH program (linux1.softberry.com/berry.phtml) in conjunction with codon usage in a selection of monocotyledonous species (Solovyev et al., 2006). Multiple alignments of nucleotide and peptide sequences were obtained using the CLC Sequences Viewer v7.8.1 (www.qiagenbioinformatics.com). Predictions of polypeptide secondary structure were made using the SOSUI program (Hirokawa et al., 1998).
Nucleotide sequences were aligned using the appropriate algorithm provided by the MUSCLE program (Edgar, 2004), as implemented in the Mega v6 software package (Tamura et al., 2013) and the alignments used to conduct a phylogenetic analysis based on the neighbor-joining algorithm (Saitou and Nei, 1987) supported by 1,000 bootstrap replicates (Felsenstein, 1985). Polypeptide sequences were also aligned using the relevant MUSCLE algorithm and once the optimal model had been selected (Nei, 2000), the alignments were used to generate a phylogeny based on the maximum likelihood method; again, the analysis was supported by 1,000 bootstrap replicates.
Archival RNA-seq data capturing the transcriptome of the root, sheath, leaf, spike, stamen, pistil, stem and caryopsis of Ae. tauschii ssp. strangulata (Jia et al., 2013), were filtered to remove low quality reads using the FASTP program (Chen et al., 2018). Bowtie2 software (Langmead and Salzberg, 2012) was used to align and count the reads. Transcript abundances were expressed in the form reads per kilobases per million reads (RKPM) (Mortazavi et al., 2008), and were averaged where replicated samples were available.
The relevant statistical parameters associated with the genome assemblies are given in Table S2. A BLASTn search successfully retrieved Btr homologs from a number of members of the Triticeae, Poeae, Aveneae and Brachypodieae tribes (Supplemental Fasta File: Btr1_Btr1-like_DNA.fasta and Btr2_Btr2-like_DNA.fasta), as well as from a number of non-Pooideae species (identified using a BLASTp search), but not from A. thaliana. Species not belonging to the Triticeae tribe lacked a copy of Btr1, but harbored one or more Btr1-like genes (one in each of rice, foxtail millet and sorghum, two in L. perenne, three in Av. eriantha and four in B. distachyon). No Btr1-like sequence was recovered from maize (Table S3). Most of the Triticeae wild species harbored Btr1-like genes, with a copy number varying from one to three; none of these genes was interrupted by an intron (Figure S1, Table S3). Two Btr1-like copies were recovered from T. monococcum ssp. boeoticum: these were Btr1-like-A-1, predicted to encode a 195 residue polypeptide and Btr1-like-A-2 (a 194 residue polypeptide) (Table S3). The two sequences were 96% identical at the nucleotide level, including 22 polymorphic sites (Figure S2A), while their predicted polypeptide sequences shared 91% identity, differing at 17 sites (Figure S2B). Given that the species' inbreeding habit ensures a high level of homozygosity, the possibility that these two genes represent alleles is unlikely. The set of Btr1 sequences clustered within a single phylogenetic clade (the “Btr1 clade”), which was supported by a bootstrap probability of 99% (Figure S1). The remaining related sequences, referred to as ‘Btr1-like', also fell into this clade, implying that Btr1 and Btr1-like genes evolved from a single Triticeae sequence, which was later duplicated. The Btr1 open reading frame was in most cases a 591 bp sequence uninterrupted by introns (Table S3), as previously described for the copies present in barley (Pourkheirandish et al., 2015), T. monococcum (Pourkheirandish et al., 2018) and T. turgidum (Avni et al., 2017).
S. vavilovii contig 160742 harbored a sequence homologous to Btr1 (Table S3). Its coding sequence differed from the barley copy with respect to a small deletion involving nucleotides 532 through 535 (Figure S3A). In addition, the coding sequence was split into two exons. Its predicted product was somewhat shorter (186 residues) and included a frame shift in its N terminal region (Figure S3B). This S. vavilovii accession used to generate the whole genome sequence (Bauer et al., 2017), exhibited a brittle rachis (Figure S4). To confirm the presence of the 4 nt deletion, its Btr1 content was PCR-amplified from a template of the relevant S. vavilovii accession and the resulting amplicon sequenced. Two distinct Btr1 copies were recovered: one included the 4 nt deletion, while the other was a complete 591 nt sequence, predicted to encode the same 196 residues as the barley Btr1 gene does (Table S3). The two copies are referred to here as, respectively, Btr1-R-2 and -R-1. The two sequences differed at 18 sites in the coding region (Figure S3A); it remains unclear whether -R-2 and -R-1 are allelic, or whether they reside at independent loci, but the former is more likely, given that S. vavilovii is an out-pollinator.
The Ae. sharonensis genome harbored three copies of Btr1: one of these (TSL_WGS_sharonensis_v1_contig_98068 [3458-2918]) was a complete 591 nt sequence, predicted to encode 196 residues; the second (TSL_WGS_sharonensis_v1_contig_341236 [279-739]) was interrupted by one intron and was predicted by the FGENESH program to encode a 130 residue protein, lacking the codons lying between nucleotides 172 and 307 (Figure S5A, B); the third (TSL_WGS_sharonensis_v1_contig_1151931 [2022-1843]) was also a truncated sequence (Figure S6A), predicted to encode a 59 residue protein (lacking the codons beyond position 167) (Figure S6B).
The Ae. longissima genome harbored four copies of Btr1: three of these mapped to sites on chromosome 3S (nucleotides 86738095-86738685, 85162031-85162621 and 85013005-85012418) and are predicted to encode a polypeptide of length, respectively, 196, 196, and 195 residues; the fourth, which was contained within a scaffold not assignable to a specific chromosome, was a truncated sequence similar to the one present on the Ae. sharonensis TSL_WGS_sharonensis_v1_contig_1151931 (Figure S6A, B).
The Ae. tauschii genome lacked an intact copy of Btr1. The length of the homologous sequence was only 166 nt, mapping to a site on chromosome 3D (59424083-59424248) close to the position occupied by Btr1 copies in the other Triticeae species. The sequence aligned with the first 167 nucleotides of barley Btr1, and was identical with the truncated copies present in both Ae. longissima and Ae. sharonensis, except for the absence of the thymine base in the start codon (Figure S6A). The sequence was genuine, since a search of three independent genome sequence databases reporting the sequence of the AL8/78 accession (Marcussen et al., 2014; Xie et al., 2017; Zimin et al., 2017) retrieved the same sequence in each case (Table S4). An identical sequence was also represented in the assembly of bread wheat chromosome 3D (Table S4 and Table S5).
The predicted lengths of the products of the Btr1 and Btr1-like genes harbored by Poaceae species ranged from 170 to 212 residues, while among the Triticeae species, the range was 170–199 (most lay in the range 192–198) (Table S3). The majority of Btr1 genes encoded a 196 residue protein. Consistent with the nucleotide sequence-based phylogeny, two protein clades were recognized in the Triticeae (Figure 2). Most of the Triticeae species were found to encode one or more sequences in the BTR1 clade plus one or more in the BTR1-LIKE clade (Figure 2). Exceptionally, the Ae. tauschii genome encodes no BTR1 proteins, rather harboring three Btr1-like genes.
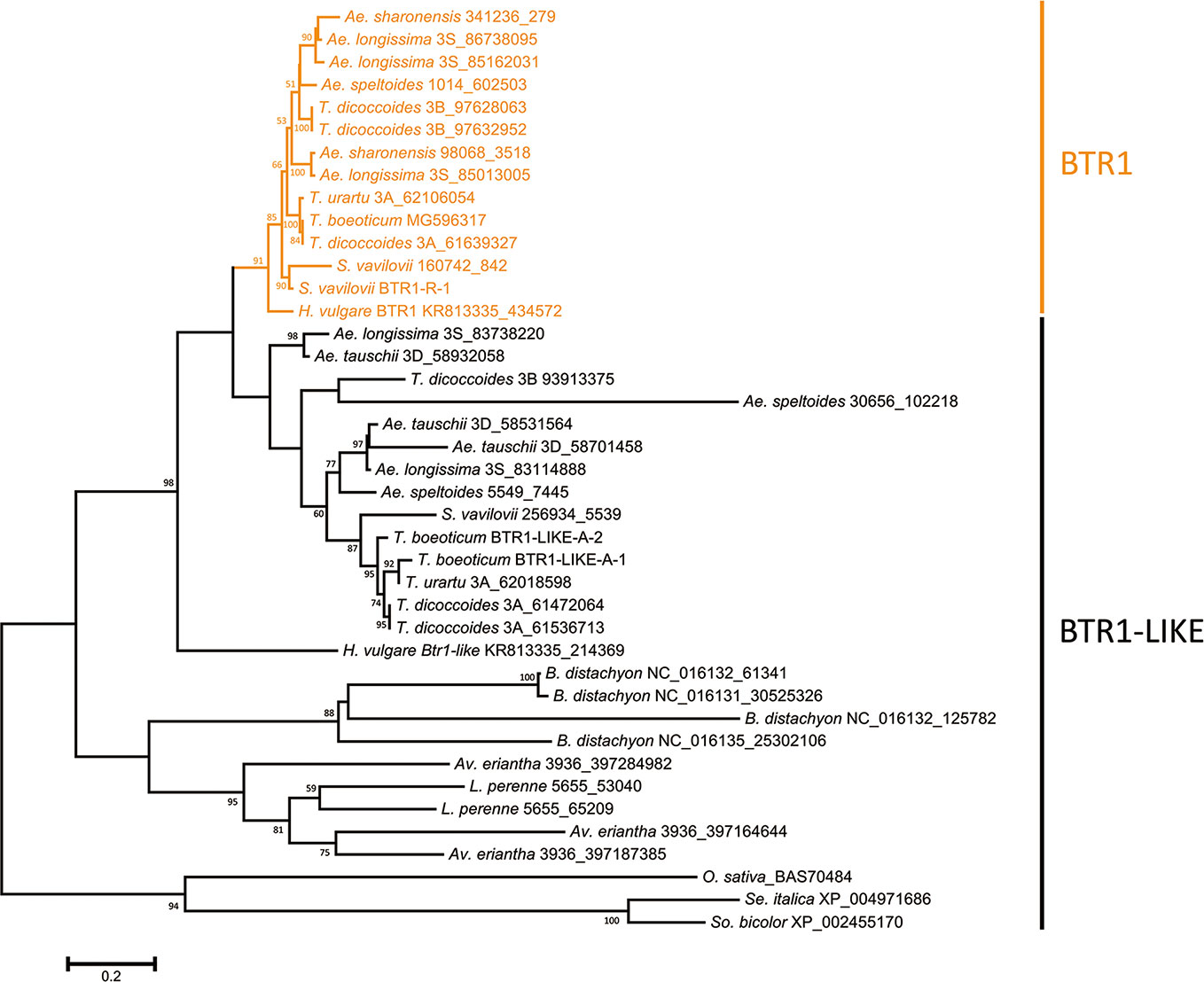
Figure 2 Phylogeny of the Poaceae BTR1 and BTR1-like proteins. The tree was constructed using the maximum likelihood method based on an alignment of deduced polypeptide sequences. The products of the Btr1 genes are shown in orange. Numbers given at a branch node indicate bootstrap probabilities (only those > 50% are shown). Accession/scaffold/contig numbers and the start location of each gene is given in Table S3.
The nucleotide sequences classified as either Btr2 or Btr2-like also formed two clades: the “Btr2 clade” was centered on the barley Btr2 gene, and was supported by a bootstrap probability of 97% (Figure S7). Grass species harbored a variable number of Btr2-like sequences. According to a BLASTn search, there was one copy in L. perenne, two in Av. eriantha and three in B. distachyon; while based on a BLASTp search, two copies were located in rice, and one each in foxtail millet, sorghum and maize (Table S6). Among the wild Triticeae species, most of the Btr2 and Btr2-like sequences were free of introns, as is also the case for barley Btr2. The exceptional case was the Ae. tauschii sequence mapping from nucleotides 58720400-58720945 on chromosome 3D, which was split into two exons as predicted by the FGENESH program. S. vavilovii, T. urartu, T. turdigum ssp. dicoccoides, Ae. sharonensis, Ae. longissima, Ae. speltoides ssp. speltoides and Ae. tauschii each harbored at least three Btr2 or Btr2-like sequences per diploid genome (the tetraploid species T. turgidum harbored six copies). The predicted length of the set of BTR2 and BTR2-LIKE proteins ranged from 129–426 residues, while among the Triticeae species, the range was much narrower (192–204) (Table S6). Two distinct clades were recognized among the Triticeae proteins (Figure 3): the one including barley BTR2 (the BTR2 clade) was supported by a bootstrap probability of 75%.
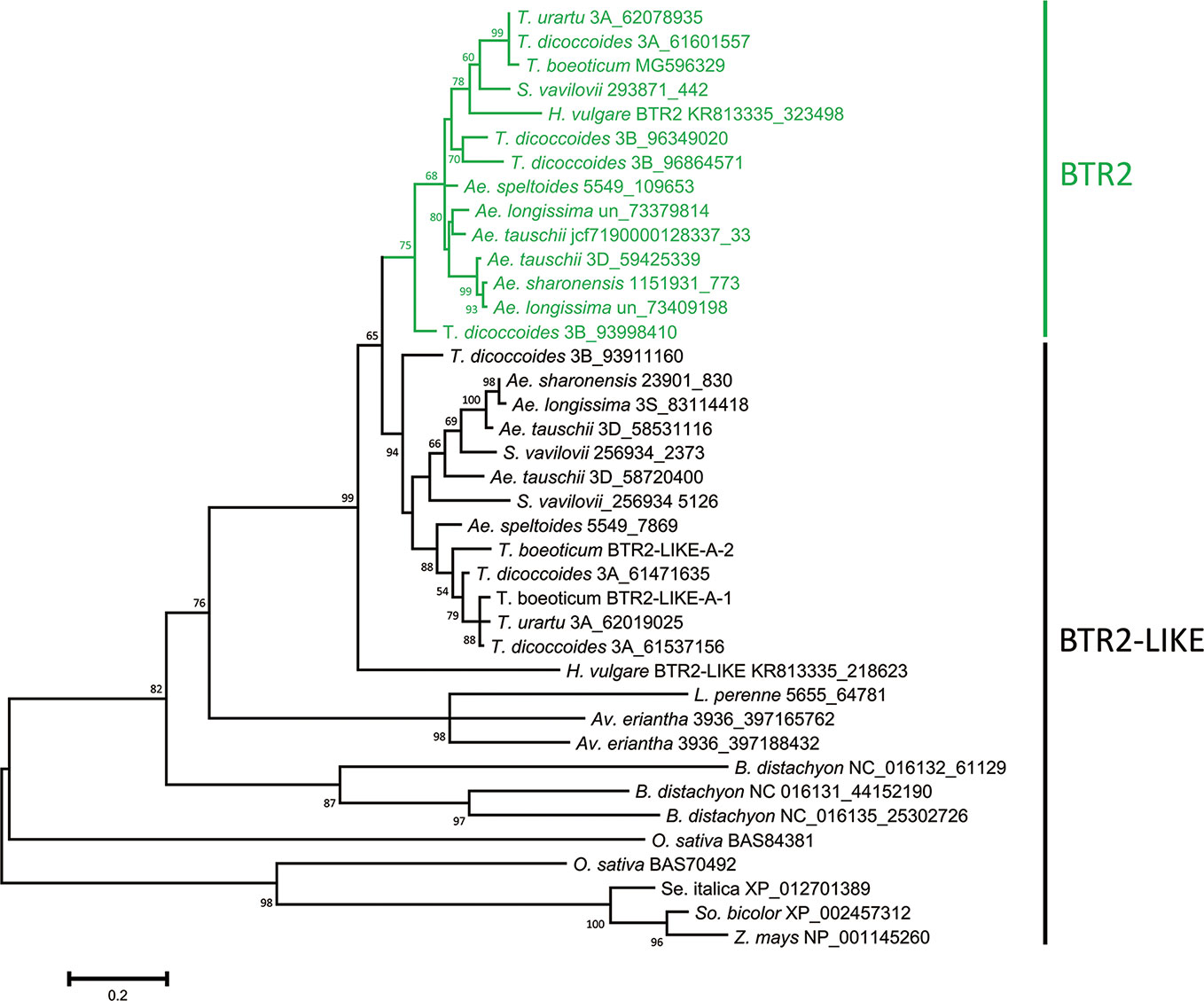
Figure 3 Phylogeny of the Poaceae BTR2 and BTR2-like proteins. The tree was constructed using the maximum likelihood method based on an alignment of deduced polypeptide sequences. The products of the Btr2 genes are shown in green. Numbers given at a branch node indicate bootstrap probabilities (only those > 50% are shown). Accession/scaffold/contig numbers and the start location of each gene is given in Table S6.
An alignment of BTR1 and BTR1-LIKE proteins encoded by the Triticeae species is shown in Figure 4. One of the two Ae. speltoides Btr1-like sequences, namely scaffold_30656 [102218-101631], was excluded because its sequence has clearly experienced a number of deletions. The Btr1-encoded proteins produced by T. urartu, T. monococcum ssp. boeoticum, T. turdigum ssp. dicoccoides (the copy on chromosome 3A), and S. vavilovii each featured two transmembrane helices (from residues 66–88 and 166–188), as is also the case for barley BTR1 (Pourkheirandish et al., 2015). The two proteins produced by T. turdigum ssp. dicoccoides (the copy on chromosome 3B) lacked a second transmembrane helix as a result of polymorphisms at three key residue sites (“G”, “A”, and “M” marked in Figure 4). The Btr1-encoded proteins produced by Ae. speltoides, Ae. sharonensis and Ae. longissima lacked any transmembrane helices due to polymorphisms in both domains: one or two in the 66–88 region and two or three in the 166–188 region. The latter polymorphisms in Ae. speltoides and Ae. longissima were identical to those present on the chromosome 3B-encoded T. turdigum ssp. dicoccoides BTR1. Like the barley BTR2 protein (Pourkheirandish et al., 2015), those proteins lacking transmembrane helices were predicted to be soluble, as were all of the Triticeae BTR2 proteins. An alignment of the relevant Triticeae sequences is shown in Figure 5. None of these proteins include the CAR-PIP motif present in barley BTR2 (Pourkheirandish et al., 2015), as a result of sequence polymorphisms (Figure 5).

Figure 4 Alignment of deduced Triticeae BTR1 and BTR1-like polypeptides. The two predicted transmembrane helix regions are shown boxed, and the transmembrane helices are colored orange. Variable residues in the transmembrane helices are colored gray.

Figure 5 Alignment of deduced Triticeae BTR2 and BTR2-like polypeptides. The CAR-PIP motif region is shown boxed and the motif present uniquely in barley is colored green. Variable residues in the motif region are colored gray.
In species harboring both Btr1-like and Btr2-like genes, in most cases, the loci lie close to one another: in foxtail millet, the separation is as little as 314 nt. The major exception is in rice, where the genes are separated by 18,000 nt (Table 2, Figure 6). In each case, the pair of genes is oriented head-to-head. The separation between the Btr1 and Btr2 loci is much greater: 111,000 nt in barley, 27,000 nt in T. urartu, 37,000 nt in T. dicoccoides (3A copy), and 763,000 nt in T. dicoccoides (3B copy); again, though, their orientation is consistently head-to-head. The separation between the Btr1-like and Btr2-like sequences is consistently smaller than between Btr1 and Btr2, because even where the relative location of the latter remains uncertain, at least the two genes each mapped onto a different contig or scaffold (Tables S3 and S6).
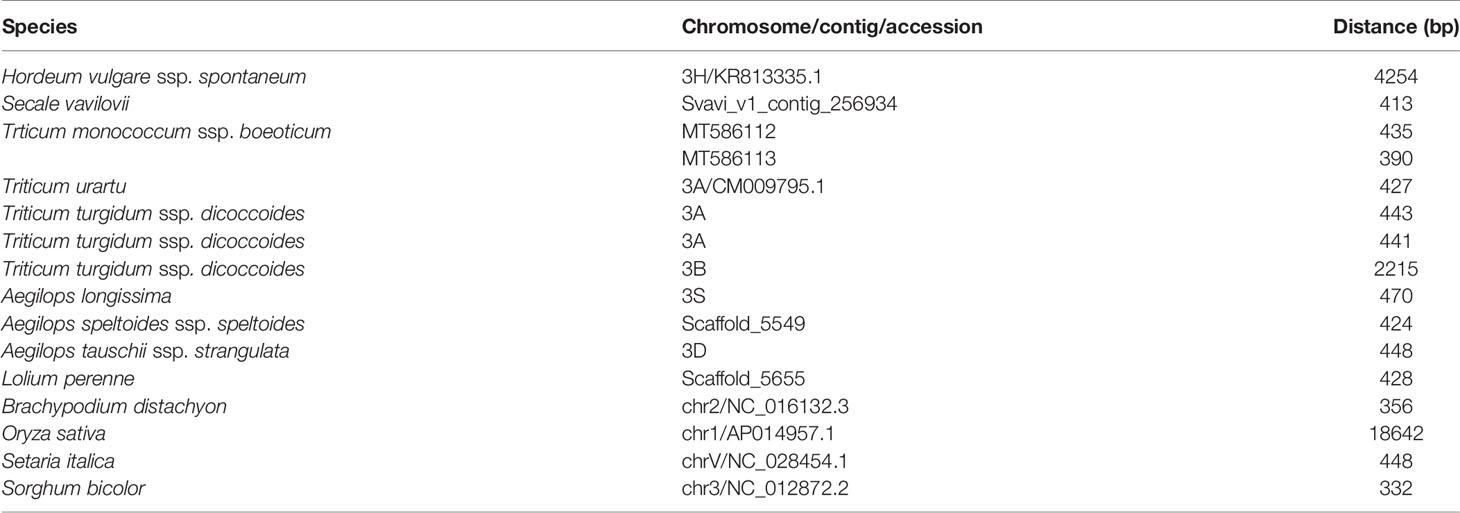
Table 2 Separation between Poaceae Btr1-like and Btr2-like genes.
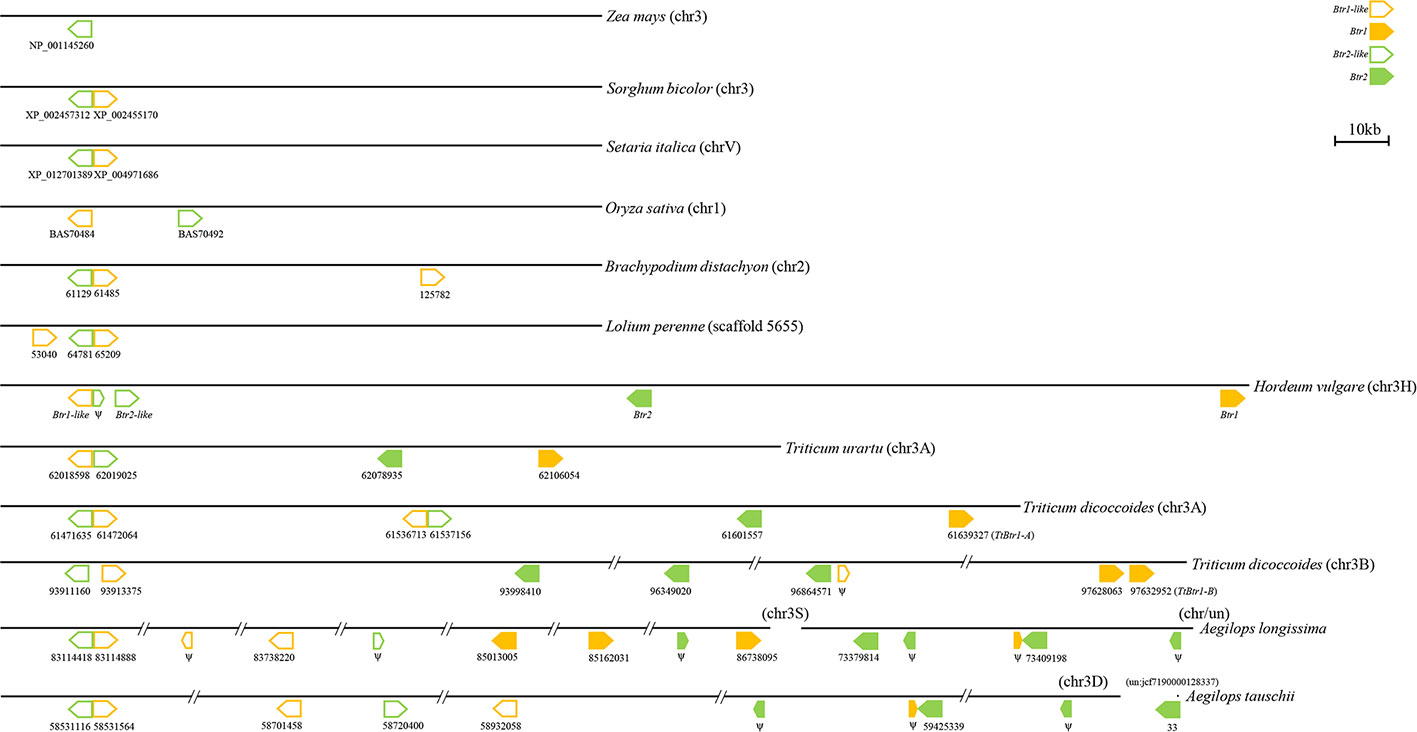
Figure 6 Organization of Btr1, Btr1-like, Btr2, and Btr2-like genes in the grasses. The direction of translation is arrowed. Numbers indicate either the gene's ID or the nucleotide positions of the gene's translation start point. Only the chromosome 2 genes of B. distachyon are shown.
The RPKM value was used to calculate the abundance of Btr transcript in Ae. tauschii. As shown in Figure S8, the two Btr2 genes are transcribed specifically in young spikes. Meanwhile, the linked pair Btr1-like (3D [58531564-58532142]) and Btr2-like (3D [58531116-58530508]) are less abundantly transcribed in young spike than the other copy of Btr1-like (3D [58932058-58931459]), whereas both were more abundantly transcribed in the stamen. Transcript of each of the orthologs of rice seed shattering genes (with the exception of the sh4 ortholog) is more abundant than Btr2, not only in young spikes but also elsewhere in the plant (Table S5, Figure S9).
The present analysis of a sample of Poaceae species showed that copies of both Btr1-like and Btr2-like have been retained in their expected genomic region, i.e., in the region sharing synteny with barley chromosome 3H (Salse, 2016). This level of conservation implies that their products likely perform an indispensable function. Given that the loci housing the Btr1-like and Btr2-like sequences lie close to one another (except in rice), and that the orientation of the two genes is invariably head-to-head. One hypothesis is that they are co-regulated, as is the case in Ae. tauschii (Figure S9), but further experiments are needed to verify it.
It has been demonstrated previously that Btr1 and Btr1-like evolved as a result of a duplication event (Pourkheirandish et al., 2015). The Btr1-like sequence is well conserved in the Poaceae, while Btr1 is only found in species belonging to the Triticeae tribe. The relationship between Btr2 and Btr2-like is similar. Species within the genera Lolium and Avena lack both Btr1 and Btr2, even though the Poeae and Aveneae tribes are considered to be closely related to the Triticeae (Kellogg, 1998). The suggestion is therefore that the duplication event(s) which led to the appearance of Btr1 and Btr2 occurred after the divergence of the Triticeae tribe from the Poeae and the Aveneae tribes. Whether all members of the Triticeae – which harbors some 30 genera (Barkworth and von Bothmer, 2009) - retain both Btr1 and Btr2 has yet to be determined.
Btr1 orthologs are required for disarticulation above the rachis nodes, since the loss-of-function btr1 mutant forms a non-brittle rachis in both Hordeum and Triticum spp. (Pourkheirandish et al., 2015; Avni et al., 2017; Pourkheirandish et al., 2018). The present analysis has established that all of the wild Triticeae species which exhibit disarticulation above the rachis nodes carry a copy of Btr1. Ae. speltoides ssp. speltoides is unique in disarticulating only above the lowest rachis node; this species harbors a copy of Btr1, so one hypothesis, plausibly testable using in situ RNA hybridization and/or transcriptomic profiling of micro-dissected rachis nodes, is that the gene is regulated differently in Ae. speltoides ssp. speltoides than in species which disarticulate above each rachis node. Ae. tauschii lacks an intact copy of Btr1 and disarticulates below the rachis nodes; the inference is that BTR1 is not required to effect disarticulation below the rachis nodes.
A Btr2 gene was harbored by each of the Triticeae species examined, implying that its product is involved in the determination of the brittle rachis trait; in particular, the gene was present in species which disarticulate above the rachis nodes. In barley, the finding that Btr2 expression occurs in a thin cell layer above the rachis node has been taken to imply that BTR2 contributes to the formation of the disarticulation zone (Pourkheirandish et al., 2015). Whether BTR2 in Ae. tauschii is involved in the same way below the rachis node remains an open question. However, it is clear that Btr2 transcript is generated in immature Ae. tauschii spikes, although at a rather low abundance (Figure S8). Note that Ae. tauschii harbors two copies of Btr2, so it is possible that one of these is expressed above the rachis nodes, but is inactive since Ae. tauschii lacks an intact copy of Btr1 to induce disarticulation there; meanwhile the second copy is perhaps expressed below the rachis nodes.
Ae. tauschii does not harbor an intact copy of Btr1, but this gene is not essential for this species, because its rachis disarticulates below the node. It is arguable that Ae. tauschii could be an evolutionary intermediate between the Poeae/Aveneae and the Triticeae tribes, since members of the former two tribes also lack Btr1. However, unlike members of the Poeae/Aveneae, Ae. tauschii does harbor an intact copy of Btr2. The argument would require that Btr2, and later Btr1, were acquired independently, which appears to be less plausible than the suggestion that the Btr1-like and Btr2-like pair was duplicated, allowing for a later divergence from Btr1-like to Btr1 and Btr2-like to Btr2, as suggested by Pourkheirandish et al. (2015). An alternative evolutionary pathway can be based on the assumption that the truncated Btr1 sequences present in Ae. tauschii (166 bp), Ae. sharonensis (167 bp) and Ae. longissima (167 bp) share a common origin. Ae. tauschii and Ae. sharonensis diverged some 2 Mya (Marcussen et al., 2014), after which Ae. tauschii lost its intact copy of Btr1, but retained the truncated one; meanwhile both Ae. sharonensis and Ae. longissima retained both the intact and the truncated Btr1 sequences. Disarticulation below the rachis nodes could have evolved in Ae. tauschii following the de novo recruitment (or perhaps neofunctionalization) of a co-operating gene(s). The latter may include orthologs of genes known to be responsible for shattering in rice, such as qSH1, sh4, SH5, SHAT1, CPL1 and OSH15 (Konishi et al., 2006; Li et al., 2006; Ji et al., 2010; Zhou et al., 2012; Yoon et al., 2014; Yoon et al., 2017), since orthologs of these genes are present in Ae. tauschii (Table S5), and are transcribed in the immature spike (Figure S9). Especially, the sh4 and OsCPL1 orthologs showed higher expression than the other ones in the immature spikes of Ae. tauschii.
All datasets presented in this study are included in the article/Supplementary Material.
XZ and TK planned and designed the research. TK, HS, SK, and JJ supervised the experiments. XZ performed the experiments. KM participated in the phylogenetic analysis and genome informatics; AD and PM analyzed the data and provided advices. XZ and TK wrote the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported in part by a grant-in-aid from the Japan Society for the Promotion of Science (JSPS) to TK (18H02184). XZ appreciates the Monbukagakusho scholarship from Japanese Government (MEXT).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SS declared a past collaboration with one of the authors TK to the handling Editor.
We thank Prof. A Sharon (Tel Aviv University) to provide the genomic data of Ae. longissima, Dr. V Korzun (KWS SAAT SE) to provide the data of S. vavilovii, Prof. MC Luo (University of California, Davis) to provide the photos of Ae. tauschii. We thank Prof. T Koba, Dr. T Oikawa, and Dr. M Ohta for their kind help and advice. We thank Dr. R Koebner for careful revision of the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.01000/full#supplementary-material
Data Sheet 1 | Btr1_Btr1-like_DNA.fasta.
Data Sheet 2 | Btr1_Btr1-like_Protein.fasta.
Data Sheet 3 | Btr2_Btr2-like_DNA.fasta.
Data Sheet 4 | Btr2_Btr2-like_Protein.fasta.
Alaux, M., Rogers, J., Letellier, T., Flores, R., Alfama, F., Pommier, C., et al. (2018). Linking the International Wheat Genome Sequencing Consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol. 19 (1), 111. doi: 10.1186/s13059-018-1491-4
Amagai, Y., Watanabe, N., Kuboyama, T. (2015). Genetic mapping and development of near-isogenic lines with genes conferring mutant phenotypes in Aegilops tauschii and synthetic hexaploid wheat. Euphytica 205 (3), 859–868. doi: 10.1007/s10681-015-1424-1
Avni, R., Nave, M., Barad, O., Baruch, K., Twardziok, S. O., Gundlach, H., et al. (2017). Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357 (6346), 93–97. doi: 10.1126/science.aan0032
Barkworth, M. E., von Bothmer, R. (2009). “Scientific Names in the Triticeae,” in Genetics and Genomics of the Triticeae. Eds. Feuillet, C., Muehlbauer, G. J. (New York: Springer), 3–30.
Bauer, E., Schmutzer, T., Barilar, I., Mascher, M., Gundlach, H., Martis, M. M., et al. (2017). Towards a whole-genome sequence for rye (Secale cereale L.). Plant J. 89 (5), 853–869. doi: 10.1111/tpj.13436
Bennetzen, J. L., Schmutz, J., Wang, H., Percifield, R., Hawkins, J., Pontaroli, A. C., et al. (2012). Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 30 (6), 555–561. doi: 10.1038/nbt.2196
Byrne, S. L., Nagy, I., Pfeifer, M., Armstead, I., Swain, S., Studer, B., et al. (2015). A synteny-based draft genome sequence of the forage grass Lolium perenne. Plant J. 84 (4), 816–826. doi: 10.1111/tpj.13037
Chen, Q. F., Yen, C., Yang, J. L. (1998). Chromosome location of the gene for brittle rachis in the Tibetan weedrace of common wheat. Genet. Resour. Crop Evol. 45 (5), 407–410. doi: 10.1023/a:1008635208146
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. doi: 10.1093/bioinformatics/bty560
Dong, Y., Wang, Y. Z. (2015). Seed shattering: from models to crops. Front. Plant Sci. 6, 476. doi: 10.3389/fpls.2015.00476
Doust, A. N., Mauro-Herrera, M., Francis, A. D., Shand, L. C. (2014). Morphological diversity and genetic regulation of inflorescence abscission zones in grasses. Am. J. Bot. 101 (10), 1759–1769. doi: 10.3732/ajb.1400186
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi: 10.1093/nar/gkh340
Elgersma, A., Leeuwangh, J. E., Wilms, H. J. (1988). Abscission and seed shattering in perennial ryegrass (Lolium perenne L.). Euphytica 39, 51–57. doi: 10.1007/bf00043367
Felsenstein, J. (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39 (4), 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Frederiksen, S., Seberg, O. L. E. (1992). Phylogenetic analysis of the Triticeae (Poaceae). Hereditas 116 (1-2), 15–19. doi: 10.1111/j.1601-5223.1992.tb00198.x
Harlan, J. R., Zohary, D. (1966). Distribution of wild wheats and barley. Science 153 (3740), 1074–1080. doi: 10.1126/science.153.3740.1074
Hirokawa, T., Boon-Chieng, S., Mitaku, S. (1998). SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14 (4), 378–379. doi: 10.1093/bioinformatics/14.4.378
International Brachypodium Initiative (2010). Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463 (7282), 763–768. doi: 10.1038/nature08747
Ji, H., Kim, S. R., Kim, Y. H., Kim, H., Eun, M. Y., Jin, I. D., et al. (2010). Inactivation of the CTD phosphatase-like gene OsCPL1 enhances the development of the abscission layer and seed shattering in rice. Plant J. 61 (1), 96–106. doi: 10.1111/j.1365-313X.2009.04039.x
Jia, J., Zhao, S., Kong, X., Li, Y., Zhao, G., He, W., et al. (2013). Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496 (7443), 91–95. doi: 10.1038/nature12028
Jiang, Y. F., Lan, X. J., Luo, W., Kong, X. C., Qi, P. F., Wang, J. R., et al. (2014). Genome-wide quantitative trait locus mapping identifies multiple major loci for brittle rachis and threshability in Tibetan semi-wild wheat (Triticum aestivum ssp. tibetanum Shao). PloS One 9 (12), e114066. doi: 10.1371/journal.pone.0114066
Jiao, Y., Peluso, P., Shi, J., Liang, T., Stitzer, M. C., Wang, B., et al. (2017). Improved maize reference genome with single-molecule technologies. Nature 546 (7659), 524–527. doi: 10.1038/nature22971
Katkout, M., Sakuma, S., Kawaura, K., Ogihara, Y. (2015). TaqSH1-D, wheat ortholog of rice seed shattering gene qSH1, maps to the interval of a rachis fragility QTL on chromosome 3DL of common wheat (Triticum aestivum). Genet. Resour. Crop Evol. 62 (7), 979–984. doi: 10.1007/s10722-015-0301-z
Kawahara, Y., de la Bastide, M., Hamilton, J. P., Kanamori, H., McCombie, W. R., Ouyang, S., et al. (2013). Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6 (1), 4. doi: 10.1186/1939-8433-6-4
Kellogg, E. A. (1998). Relationships of cereal crops and other grasses. Proc. Natl. Acad. Sci. U. S. A. 95 (5), 2005–2010. doi: 10.1073/pnas.95.5.2005
King, I. P., Law, C. N., Cant, K. A., Orford, S. E., Reader, S. M., Miller, T. E. (1997). Tritipyrum, a potential new salt-tolerant cereal. Plant Breed. 116 (2), 127–132. doi: 10.1111/j.1439-0523.1997.tb02166.x
Komatsuda, T., Nakamura, I., Takaiwa, F., Oka, S. (1998). Development of STS markers closely linked to the vrs1 locus in barley, Hordeum vulgare. Genome 41 (5), 680–685. doi: 10.1139/g98-069
Konishi, S., Izawa, T., Lin, S. Y., Ebana, K., Fukuta, Y., Sasaki, T., et al. (2006). An SNP caused loss of seed shattering during rice domestication. Science 312 (5778), 1392–1396. doi: 10.1126/science.1126410
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Li, W., Gill, B. S. (2006). Multiple genetic pathways for seed shattering in the grasses. Funct. Integr. Genomics 6 (4), 300–309. doi: 10.1007/s10142-005-0015-y
Li, C., Zhou, A., Sang, T. (2006). Rice domestication by reducing shattering. Science 311 (5769), 1936–1939. doi: 10.1126/science.1123604
Lin, Z., Li, X., Shannon, L. M., Yeh, C. T., Wang, M. L., Bai, G., et al. (2012). Parallel domestication of the Shattering1 genes in cereals. Nat. Genet. 44 (6), 720–724. doi: 10.1038/ng.2281
Ling, H. Q., Ma, B., Shi, X., Liu, H., Dong, L., Sun, H., et al. (2018). Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557 (7705), 424–428. doi: 10.1038/s41586-018-0108-0
Luo, M. C., Gu, Y. Q., Puiu, D., Wang, H., Twardziok, S. O., Deal, K. R., et al. (2017). Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551 (7681), 498–502. doi: 10.1038/nature24486
Marcussen, T., Sandve, S. R., Heier, L., Spannagl, M., Pfeifer, M., International Wheat Genome Sequencing, C, et al. (2014). Ancient hybridizations among the ancestral genomes of bread wheat. Science 345 (6194), 1250092. doi: 10.1126/science.1250092
Maughan, P. J., Lee, R., Walstead, R., Vickerstaff, R. J., Fogarty, M. C., Brouwer, C. R., et al. (2019). Genomic insights from the first chromosome-scale assemblies of oat (Avena spp.) diploid species. BMC Biol. 17 (1), 92. doi: 10.1186/s12915-019-0712-y
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5 (7), 621–628. doi: 10.1038/nmeth.1226
Olsen, K. M., Wendel, J. F. (2013). A bountiful harvest: genomic insights into crop domestication phenotypes. Annu. Rev. Plant Biol. 64, 47–70. doi: 10.1146/annurev-arplant-050312-120048
Opanowicz, M., Vain, P., Draper, J., Parker, D., Doonan, J. H. (2008). Brachypodium distachyon: making hay with a wild grass. Trends Plant Sci. 13 (4), 172–177. doi: 10.1016/j.tplants.2008.01.007
Paterson, A. H., Bowers, J. E., Bruggmann, R., Dubchak, I., Grimwood, J., Gundlach, H., et al. (2009). The Sorghum bicolor genome and the diversification of grasses. Nature 457 (7229), 551–556. doi: 10.1038/nature07723
Pourkheirandish, M., Hensel, G., Kilian, B., Senthil, N., Chen, G., Sameri, M., et al. (2015). Evolution of the grain dispersal system in barley. Cell 162 (3), 527–539. doi: 10.1016/j.cell.2015.07.002
Pourkheirandish, M., Dai, F., Sakuma, S., Kanamori, H., Distelfeld, A., Willcox, G., et al. (2018). On the origin of the non-brittle rachis trait of domesticated einkorn wheat. Front. Plant Sci. 8, 2031. doi: 10.3389/fpls.2017.02031
Saitou, N., Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4 (4), 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sakuma, S., Salomon, B., Komatsuda, T. (2011). The domestication syndrome genes responsible for the major changes in plant form in the Triticeae crops. Plant Cell Physiol. 52 (5), 738–749. doi: 10.1093/pcp/pcr025
Salse, J. (2016). Ancestors of modern plant crops. Curr. Opin. Plant Biol. 30, 134–142. doi: 10.1016/j.pbi.2016.02.005
Solovyev, V., Kosarev, P., Seledsov, I., Vorobyev, D. (2006). Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7 (Suppl 1), 11–12. doi: 10.1186/gb-2006-7-s1-s10
Studer, A. J., Wang, H., Doebley, J. F. (2017). Selection during maize domestication targeted a gene network controlling plant and inflorescence architecture. Genetics 207 (2), 755–765. doi: 10.1534/genetics.117.300071
Swarbreck, D., Wilks, C., Lamesch, P., Berardini, T. Z., Garcia-Hernandez, M., Foerster, H., et al. (2008). The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res. 36, D1009–D1014. doi: 10.1093/nar/gkm965
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30 (12), 2725–2729. doi: 10.1093/molbev/mst197
Urbano, M., Resta, P., Benedettelli, S., Blanco, A. (1988). “A Dasypyrum villosum L. Candargy chromosome related to homoeologous group 3 of wheat,” in Proceedings 7th International Wheat Genet Symposium, Eds. Miller, T. E., Koebner, R. M. D. (Cambridge Laboratory, Trumpington, Cambridge, UK: Institute of Plant Science Research), 169–173.
van Slageren, M. W. (1994). Wild wheats: a monograph of Aegilops L. and Amblyopyrum (Jaub. & Spach) Eig (Poaceae) (Wageningen: Wageningen Agricultural University).
Xie, J., Huo, N., Zhou, S., Wang, Y., Guo, G., Deal, K. R., et al. (2017). Sequencing and comparative analyses of Aegilops tauschii chromosome arm 3DS reveal rapid evolution of Triticeae genomes. J. Genet. Genomics 44 (1), 51–61. doi: 10.1016/j.jgg.2016.09.005
Yoon, J., Cho, L. H., Kim, S. L., Choi, H., Koh, H. J., An, G. (2014). The BEL1-type homeobox gene SH5 induces seed shattering by enhancing abscission-zone development and inhibiting lignin biosynthesis. Plant J. 79 (5), 717–728. doi: 10.1111/tpj.12581
Yoon, J., Cho, L. H., Antt, H. W., Koh, H. J., An, G. (2017). KNOX protein OSH15 induces grain shattering by repressing lignin biosynthesis genes. Plant Physiol. 174 (1), 312–325. doi: 10.1104/pp.17.00298
Zhang, Z., Zhu, H., Gill, B. S., Li, W. (2015). Fine mapping of shattering locus Br2 reveals a putative chromosomal inversion polymorphism between the two lineages of Aegilops tauschii. Theor. Appl. Genet. 128 (4), 745–755. doi: 10.1007/s00122-015-2469-1
Zhao, Y., Xie, P., Guan, P., Wang, Y., Li, Y., Yu, K., et al. (2019). Btr1-A induces grain shattering and affects spike morphology and yield-related traits in wheat. Plant Cell Physiol. 60 (6), 1342–1353. doi: 10.1093/pcp/pcz050
Zhou, Y., Lu, D., Li, C., Luo, J., Zhu, B. F., Zhu, J., et al. (2012). Genetic control of seed shattering in rice by the APETALA2 transcription factor shattering abortion1. Plant Cell 24 (3), 1034–1048. doi: 10.1105/tpc.111.094383
Zimin, A. V., Puiu, D., Luo, M. C., Zhu, T., Koren, S., Marçais, G., et al. (2017). Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 27 (5), 787–792. doi: 10.1101/gr.213405.116
Keywords: seed dispersal, disarticulation, phylogeny, duplication, Triticeae
Citation: Zeng X, Mishina K, Jia J, Distelfeld A, Maughan PJ, Kikuchi S, Sassa H and Komatsuda T (2020) The Brittle Rachis Trait in Species Belonging to the Triticeae and Its Controlling Genes Btr1 and Btr2. Front. Plant Sci. 11:1000. doi: 10.3389/fpls.2020.01000
Received: 13 May 2020; Accepted: 18 June 2020;
Published: 22 July 2020.
Edited by:
Laura Rossini, University of Milan, ItalyReviewed by:
Shun Sakuma, Tottori University, JapanCopyright © 2020 Zeng, Mishina, Jia, Distelfeld, Maughan, Kikuchi, Sassa and Komatsuda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takao Komatsuda, dGFrYW9AYWZmcmMuZ28uanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.