 R. Shawn Abrahams
R. Shawn Abrahams J. Chris Pires
J. Chris Pires M. Eric Schranz
M. Eric Schranz- 1Division of Biological Sciences, University of Missouri, Columbia, MO, United States
- 2Biosystematics Group, Wageningen University, Wageningen, Netherlands
Glucosinolates are a diverse group of plant metabolites that characterize the order Brassicales. The MAM locus is one of the most significant QTLs for glucosinolate diversity. However, most of what we understand about evolution at the locus is focused on only a few species and not within a phylogenetic context. In this study, we utilize a micro-synteny network and phylogenetic inference to investigate the origin and diversification of the MAM/IPMS gene family. We uncover unique MAM-like genes found at the orthologous locus in the Cleomaceae that shed light on the transition from IPMS to MAM. In the Brassicaceae, we identify six distinct MAM clades across Lineages I, II, and III. We characterize the evolutionary impact and consequences of local duplications, transpositions, whole genome duplications, and gene fusion events, generating several new hypothesizes on the function and diversity of the MAM locus.
Introduction
Glucosinolates (GSL) are a diverse class of amino-acid derived sulfur containing metabolites characteristic of plants of the order Brassicales (Rodman et al., 1998; Borpatragohain et al., 2016; Kliebenstein and Cacho, 2016; Olsen et al., 2016; Chhajed et al., 2019; Blazevic et al., 2020). When the plant experiences physical damage, such as chewing by herbivores, compartments of the cell rupture and release myrosinase enzymes that hydrolyze the GSLs to create an isothiocyanate anion, damaging the attacker (Rodman et al., 1998). Besides their roles in direct defense, GSLs have also been shown to play important roles such as nutrient transport and physiological signaling (del Carmen et al., 2013). They are considered a key innovation of the Brassicales, as adaptations in the biosynthesis pathway have been shown to correlate with increased rates of speciation (Edger et al., 2015). The GSL pathway is a model for investigating processes underlying natural variation within and among species; including the roles of genome and gene duplication (Kliebenstein, 2008; Bekaert et al., 2012; Hofberger et al., 2013; Edger et al., 2015; van den Bergh et al., 2016; Wisecaver et al., 2017). Aliphatic GSLs, the largest sub-group of compounds, are especially implicated in this rate of speciation as they are only found in the most species-rich groups such as the family Brassicaceae.
The often multi-gene methylthioalkylmalate (MAM) locus, also called the Elong locus, accounts for much of the natural variation observed in aliphatic GSLs (Kliebenstein et al., 2001a, b; Textor et al., 2004, 2007; Kroymann and Mitchell-Olds, 2005; Benderoth et al., 2006, 2009; Keurentjes et al., 2006; de Kraker et al., 2007; Wentzell et al., 2007; de Kraker and Gershenzon, 2011; Zhang et al., 2015; Kliebenstein and Cacho, 2016; Kumar et al., 2019; Petersen et al., 2019). MAM enzymes catalyze the condensation reaction that extends the carbon chain in amino acid derived GSL precursors (Benderoth et al., 2006). The extended amino acid expands the types (Kliebenstein and Cacho, 2016). Most of what we understand about the evolution of MAM has been learned from studying just a handful of species, without a broad phylogenetic context (Kliebenstein and Cacho, 2016). MAM diversification in the Brassicaceae is thought to have occurred independently in separate lineages. Specifically, MAM diversity has been largely examined in Lineage I of the family (Arabidopsis and relatives) and to a lesser extent in Lineage II (Brassica and relatives). This work has been supported by large gene datasets, though with differing gene tree topologies (Zhang et al., 2015; Supplementary Figure S1).
In Arabidopsis thaliana, phenotypic variation of the MAM locus is characterized by the accumulation of different majority carbon chain-length GSL profiles (Kliebenstein and Cacho, 2016). The most common profiles have majority three carbon (3C) or four carbon (4C) molecules, but can extend up to 8C majority profiles, with variability at the population level (Benderoth et al., 2009; Kliebenstein and Cacho, 2016). Copy number variation and allelic diversity/presence-absence drive these differences, as one MAM gene may mask the phenotype of another at the same locus (Benderoth et al., 2006, 2009). This plays out in the interactions between MAM1 and MAM2 in A. thaliana populations, where variation is well understood. The 4C majority phenotype is seen in populations where MAM1 and MAM2 are both present and intact or when MAM2 is absent. In populations lacking a MAM1 gene, the GSL profile exhibits a 3C majority phenotype. In some cases, MAM1 and MAM2 genes have been fused (e.g., gene chimerism) wherein they are reformed into a MAM1-like functional gene with partial MAM2 sequences, or vice versa (Benderoth et al., 2006). Crop Brassicas most commonly accumulate 3C, 4C, or a mix of 3C and 4C majority profiles, the latter displaying a seemingly unmasked phenotype, unlike what we see in A. thaliana (Benderoth et al., 2009; Kliebenstein and Cacho, 2016).
Naming conventions for MAM orthologs are either directly based on A. thaliana (MAM1, MAM2, and MAM3) or based on A. lyrata MAM (MAMa, MAMb, and MAMc) (Benderoth et al., 2009). The Arabidopsis centered model of MAM diversity is vulnerable to miss-characterization as Arabidopsis genes may be highly derived, and thus not generalizable. We also see that the number of genes at the MAM locus can vary between populations as well as species, potentially misleading ancestral state estimations with poor sampling. To accurately understand MAM diversification, it is necessary for gene selection across a broader species phylogeny with comparisons to their primary metabolic ancestor, isopropylmalate synthase (IPMS).
Though diverged, IPMS and MAM share a high sequence similarity and similar enzymatic function (Moghe and Last, 2015). IPMS contains two conserved protein domains: a pyruvate carboxylase (HMGL-like), that is involved in the carbon condensation reaction, and a leucine allosteric domain (LeuA), that commits the protein to the leucine biosynthesis pathway forming a homodimer (Koon et al., 2004). MAM genes only retain the HMGL-like domain, the loss of LeuA being considered a key step in the transition of MAM from an IPMS-like gene (de Kraker et al., 2007). To our knowledge, no previous work has investigated when the loss of this domain occurred in the evolution of the locus.
In this study, we examine the evolutionary history and diversity of the MAM/IPMS gene family, uncovering critical steps in the origin of MAM and identifying patterns of domain-specific diversity across the Brassicaceae and its sister-family the Cleomaceae. We utilize a genomic networking methodology to analyze the wealth of newly available genome sequences (Zhao et al., 2017; Zhao and Schranz, 2019). The method analyses the conserved physical location of gene family members across queried genomes, known as synteny, to characterize the impact of different gene duplication types in the expansion of the MAM/IPMS gene family (Zhao et al., 2017; Zhao and Schranz, 2019). Ultimately we show that a mix of gene duplication types and domain changes played important roles in the evolution and innovation of the MAM locus.
Materials and Methods
Genomic Network Construction
The genomic network analysis included 40 complete plant genomes representing 38 different species. This included 34 Brassicaceae species from Lineages I, II, III, and Aethionema arabicum as sister to the rest of the family, three genomes from the sister-family Cleomaceae, and three outgroup species (Theobroma cacao, Citrus sinensis, and Vitis vinifera) (Supplementary Table S1). For each genome, we utilized protein sequences in FASTA format and a BED/GFF file. One of two Capsella rubella genomes was excluded from downstream analysis due to insufficient quality. The Thellungiella halophila and Thellungiella salsuginea are two different sequencing efforts of the same species, now under the name Eutrema salsugineum. The genome sequenced as Alyssum linifolium has since been identified as Descurainia pinnata. Network analyses were performed as described in Zhao et al. (2017). Reciprocal all-against-all whole genome protein sequence comparison were made using RAPSeach2 (Zhao et al., 2012). MCScanX (Tang et al., 2008; Wang et al., 2012) was used to calculate generic collinearity between genomes and all comparisons were saved to generate the full genomic network.
Gene Family Network
We identified candidate IPMS/MAM genes using HMMER (Finn et al., 2011), cross-referencing the Pfam, PDBe, and GO databases with domain signature HMGL-like PF00682, and filtered by an inclusion threshold e-value of 0.007. Selected genes were later filtered by relative branch lengths as compared to known IPMS and MAM genes and then queried against the overall syntenic network with a 25 gene window to extract the gene family network. We visualized the resulting network in Cytoscape version 3.3.0 (Shannon et al., 2003). We then pruned the network of gene nodes that did not contain an HMGL-like domain but were dragged in by potential domain fusions. Clique percolation, as implemented in CFindier (Derényi et al., 2005; Palla et al., 2005; Fortunato, 2010), was used to locate all K-clique comments to identify communities or clusters of gene nodes.
Phylogenetic Inference
Full amino acid sequences for all gene family members were aligned using MAFFT (Kuraku et al., 2013; Katoh et al., 2017) and cleaned using Phyutility at a 50% occupancy threshold (Smith and Dunn, 2008). We used RAxML (Stamatakis, 2014) for phylogenetic inference with the GTRCAT model (Boostrap = 1000). The same procedure was repeated for the HMGL-like domain region of each gene FASTA file as estimated by HMMER. Supplemental sequence comparisons were made using MView (Madeira et al., 2019) and analyzed using R.
Results
Synteny and Domain Analysis
Micro-synteny network analysis identified three major syntenic clusters (Figure 1), two of which encompass many genes of the known MAM gene clade (orange and green clusters) and one encompassing the known IPMS gene clade (blue cluster). Of the syntenic clusters found in the MAM clade, the green cluster identifies the ancestral MAM position, what we will call the MAM-Ancestral locus, and is equivalent to the Elong locus. The orange cluster represents a transposed and retained MAM locus-specific to Lineage II of the Brassicaceae, which we will call the MAM-Transposed locus. The analysis also recovered the 4th cluster of an unnamed lineage of genes that have retained only a single HMGL-like domain and are found in both our outgroup and in-group genomes. The A. thaliana representative gene of this clade (AT2G26800) has been shown to play a role in seed amino acid concentration (Peng et al., 2015). Relative branch lengths showed this gene clade as highly diverged from both MAM and IPMS sequences. Because of this, all genes of this clade were filtered from downstream analyses.
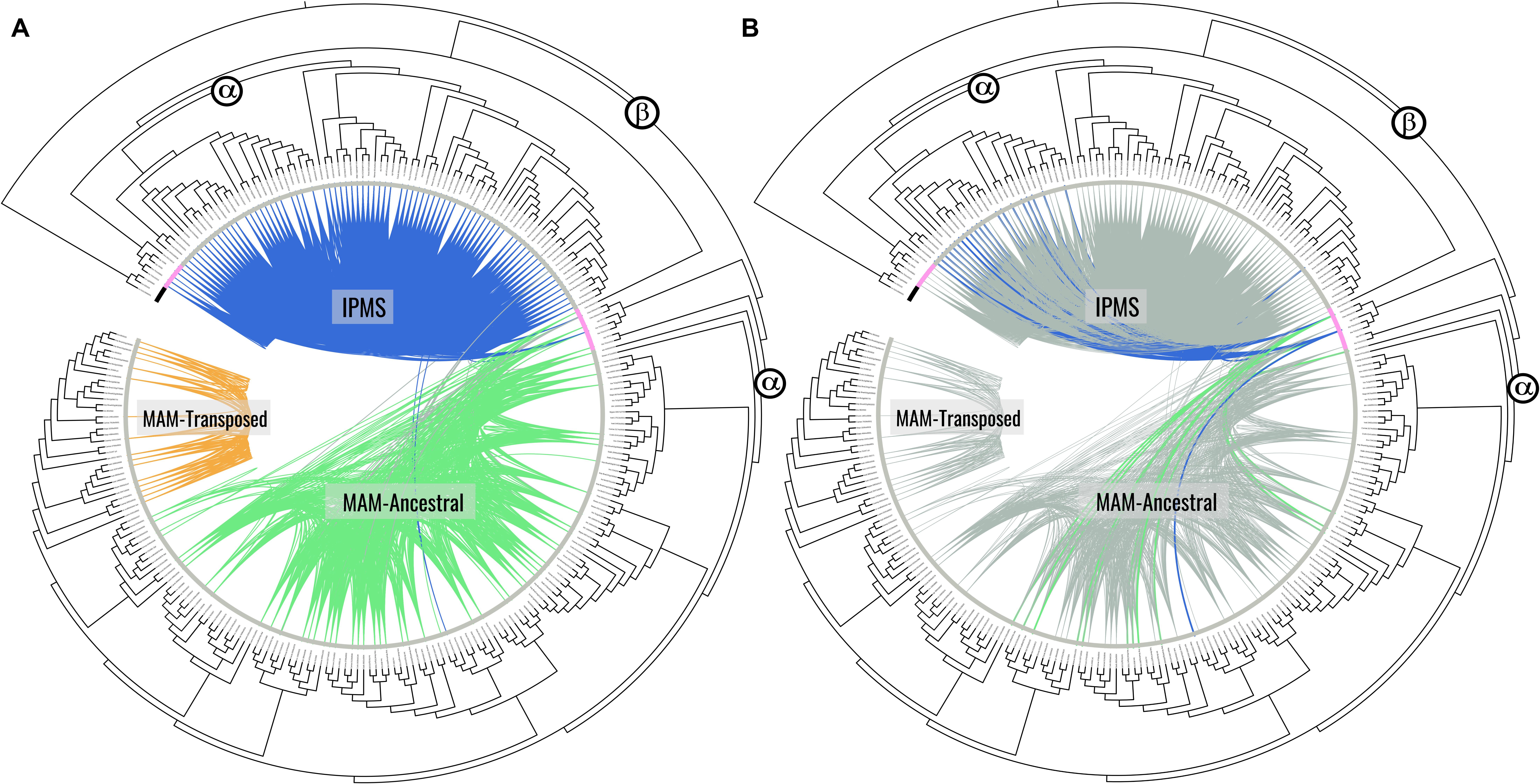
Figure 1. Synteny clusters and gene tree phylogeny of identified IPMS and MAM genes consisting of 262 total. For (A,B) the bar along the tips represent species lineage where black bars indicate genes from out group genomes, the pink bar indicates genes from Cleomaceae genomes, and the gray bar indicates genes from Brassicaceae genomes. (A) Syntenic cluster analysis identified three distinct gene clusters, each representing a different conserved genomic location. The IPMS cluster in blue, the MAM-Ancestral cluster in green, and the novel lineage specific MAM-Transposed cluster in orange. Gray lines here indicate connections between the IPMS and MAM clusters. (B) Emphasizes those connections between MAM-like genes in the Cleomaceae that exhibit both IPMS & MAM cluster membership (Clevi.0004s0713 and tha_Th2v24105) despite being physically located at the MAM-Ancestral locus in their respective genomes. [For Bootstrap scores: Supplementary Figure S6; Online interactive trees: (A) http://bit.ly/2tHVgYK; (B) http://bit.ly/2Svu8Vf].
95.7% of IPMS genes identified by sequence were also found in the IPMS syntenic cluster. 39.6% of MAM genes, not associated with the conserved Lineage II transposition, were found in the MAM-Ancestral syntenic cluster. 51.6% of genes found in the Lineage II transposed sub-clade were found in the syntenic cluster. Differences in percent synteny are tied to increased rates of tandem duplications, as the local duplicate syntenic signal was often masked, and transposed duplication events, which remove syntenic context. It is expected that many new transposed duplicates are in the process of pseudogenization and are not active MAM genes.
All genes at the Cleomaceae MAM-Ancestral locus have retained their LeuA domain from their time as IPMS duplicates, with some showing syntenic connections to both the MAM-Ancestral and IPMS syntenic cluster (Figure 1). For example, Th2v2405 from Tarenaya hassleriana has more syntenic connections with IPMS cluster members than with genes of the MAM-Ancestral locus, despite belonging to the direct orthologous chromosomal region of the MAM-Ancestral locus in the Brassicaceae (Figures 1, 2). Genes of the Cleomaceae MAM-Ancestral locus and the IPMS locus also appear to have a shared pattern of gene dosage. A duplication of the IPMS locus following WGD, brings the total IPMS gene number to two, followed by a compensatory reduction in MAM gene number at the MAM-Ancestral locus (Figure 2C). An exception to this is found in the Tarenaya hassleriana genome, where a novel transposed MAM-like gene has lost the LeuA domain. This allows for three MAM-like genes to co-occur with two IPMS genes (Supplementary Figure S3).
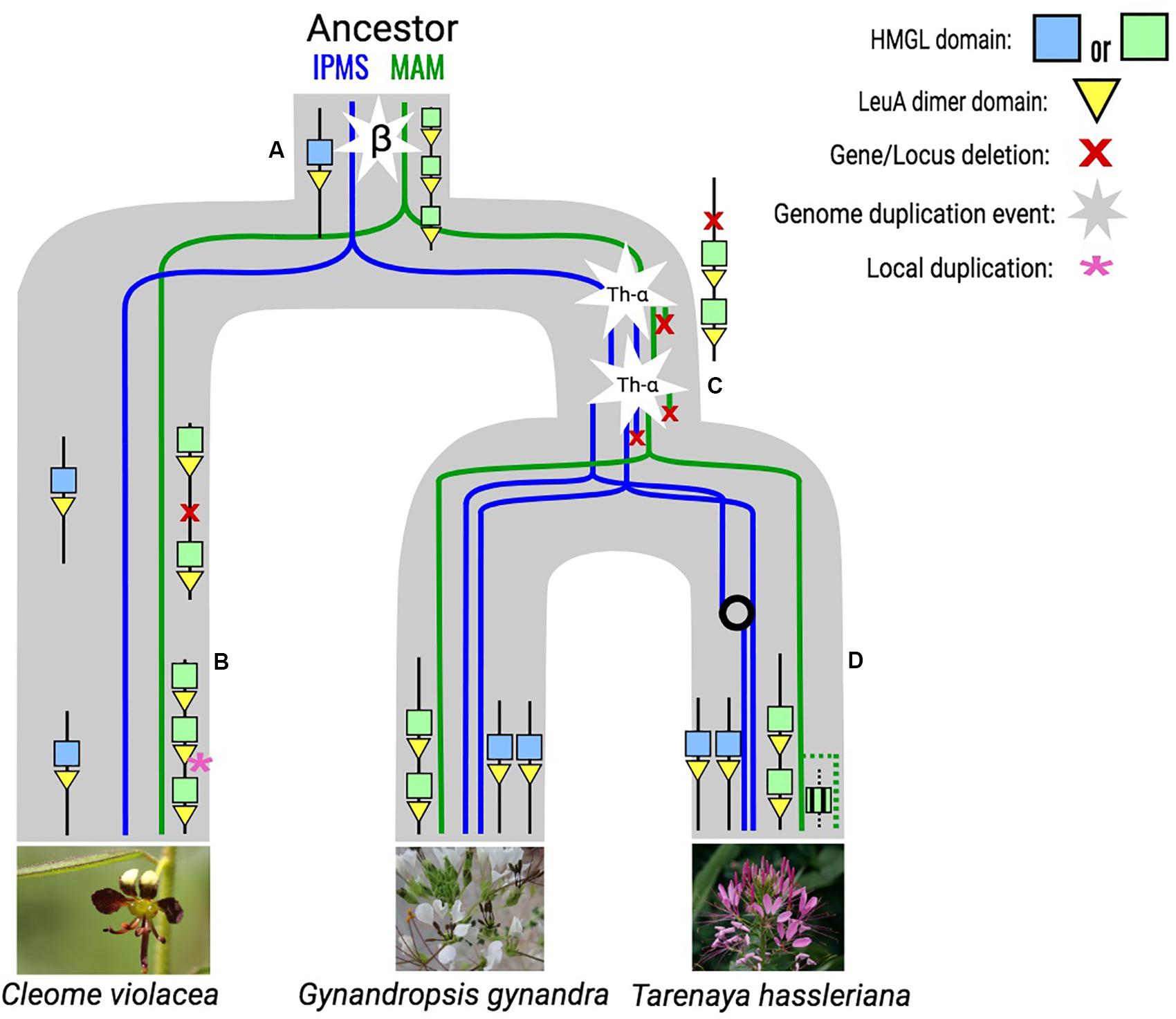
Figure 2. Inferred evolutionary trajectory of MAM and IPMS loci in the cleomaceae based on genomic synteny and phylogenetic information. (A) The MAM-Ancestral locus originated from the β whole genome duplication event and is characterized by MAM–like genes that experience local duplication and have retained their LeuA domain. (B) In the genome of Cleome violacea there is a gene deletion followed by a novel tandem duplication at the MAM locus. (C) Following the Th-α whole genome triplication the IPMS locus is duplicated and the MAM locus experiences compensatory gene loss and is reflected in the Gyanandropsis gynandra genome. (D) In the Tarenaya hassleriana genome, the IPMS locus experiences a gene conversion event that maintains sequence similarity between the two copies. There is also a novel transposition of the MAM-like gene from the MAM-Ancestral locus that does not maintain the LeuA domain. As the placement of the Th-α whole genome duplication event is not confirmed to be fully shared by both lineages, an alternative reconstruction is also possible.
Gene Family Relationships
The HMGL-like domain and full protein sequence gene trees identified distinct IPMS and MAM clades (Figure 1). In both cases, Cleomaceae genes are sister to a larger Brassicaceae clade, and Aethionema arabicum is sister to the rest of the Brassicaceae, which agrees with the species tree topology. Within the core Brassicaceae, the domain and full sequence trees display topological incongruence to each other (Figure 3) and neither perfectly match the species tree.
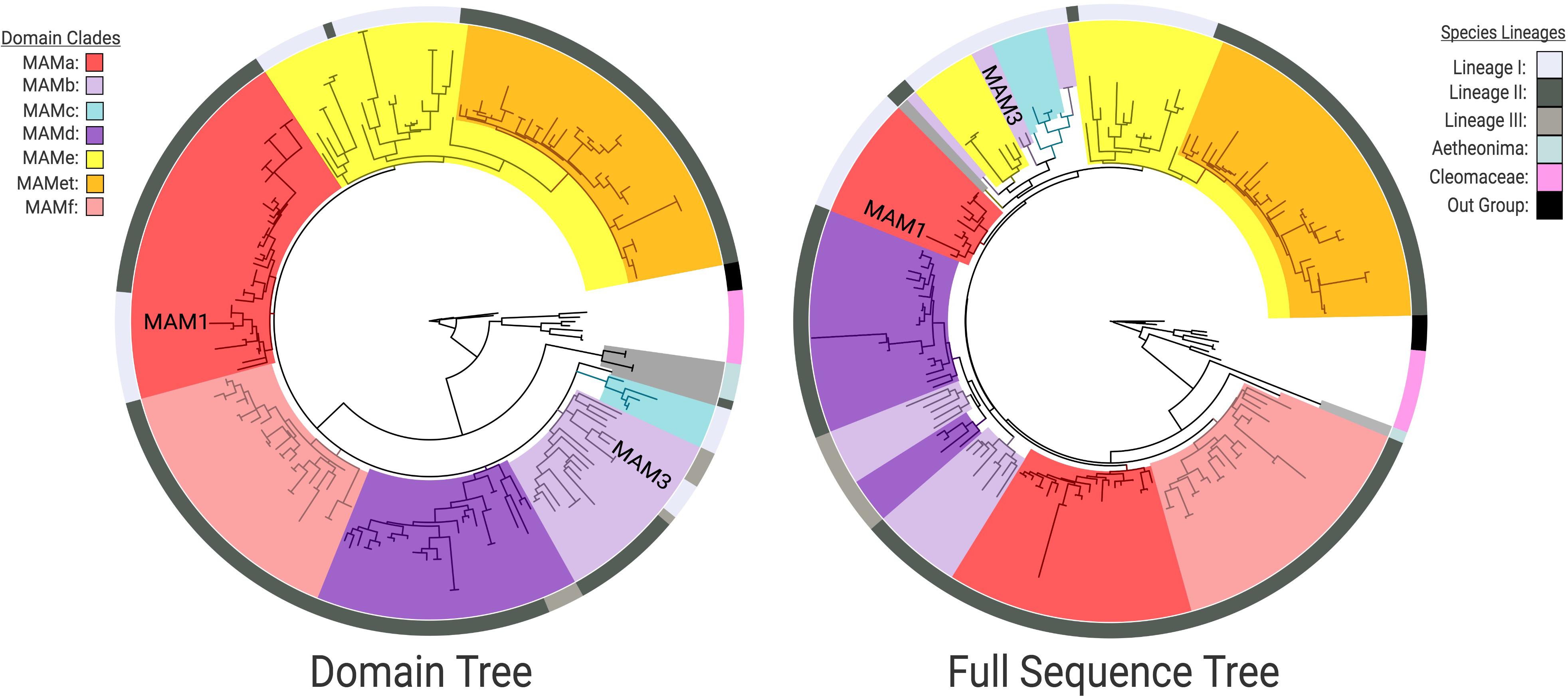
Figure 3. Clade comparison between Brassicaceae MAM domain and full sequence gene trees highlighting incongruence. HMGL-like sequences were used for the domain tree and resolved six clades of MAM (MAMa-f) but could not infer branching order. In the full sequence tree there is a breakdown of MAMa and MAMb that is correlated with species lineage. In both trees, the placement of MAM1 and MAM3 from the Arabidopsis thaliana Col genome are indicated. [For Bootstrap scores: Supplementary Figure S7 – Domain Tree; Supplementary Figure S8 – Full Sequence; Online interactive trees: Domain – http://bit.ly/2Hb5jIS; Full Sequence – http://bit.ly/37btHEZ].
The domain tree divides MAM into six supported clades (Figure 3). Though the branching order could not be determined, the supported clades were assigned MAMa-f. These domain clade designations are based on the Arabidopsis lyrata MAM gene-tree clades. Given the branch length, a measure of sequence divergence, of the genes found at the MAM-Transposed locus (Figure 3), the sub-clade of MAMe was designated MAMet. The closest non-MAMet domain sequence to the group was a MAMe sequence from the Lunaria annua genome.
Summary amino acid comparison at 80% similarity threshold shows MAMa is the most conserved domain, MAMe is the most variable domain, and MAMet and MAMc are the most diverged (Supplementary Figure S5). Exon/Intron comparisons of full MAMet genes show the expected number of domains for a functional MAM gene but with differences in exon size. When plotted on the species tree, MAMa-b and MAMe are ancestral to Lineage I, MAMa-b and MAMd-f are ancestral to Lineage II, and MAMb and MAMd are ancestral to Lineage III (Figure 4 and Supplementary Figure S3).
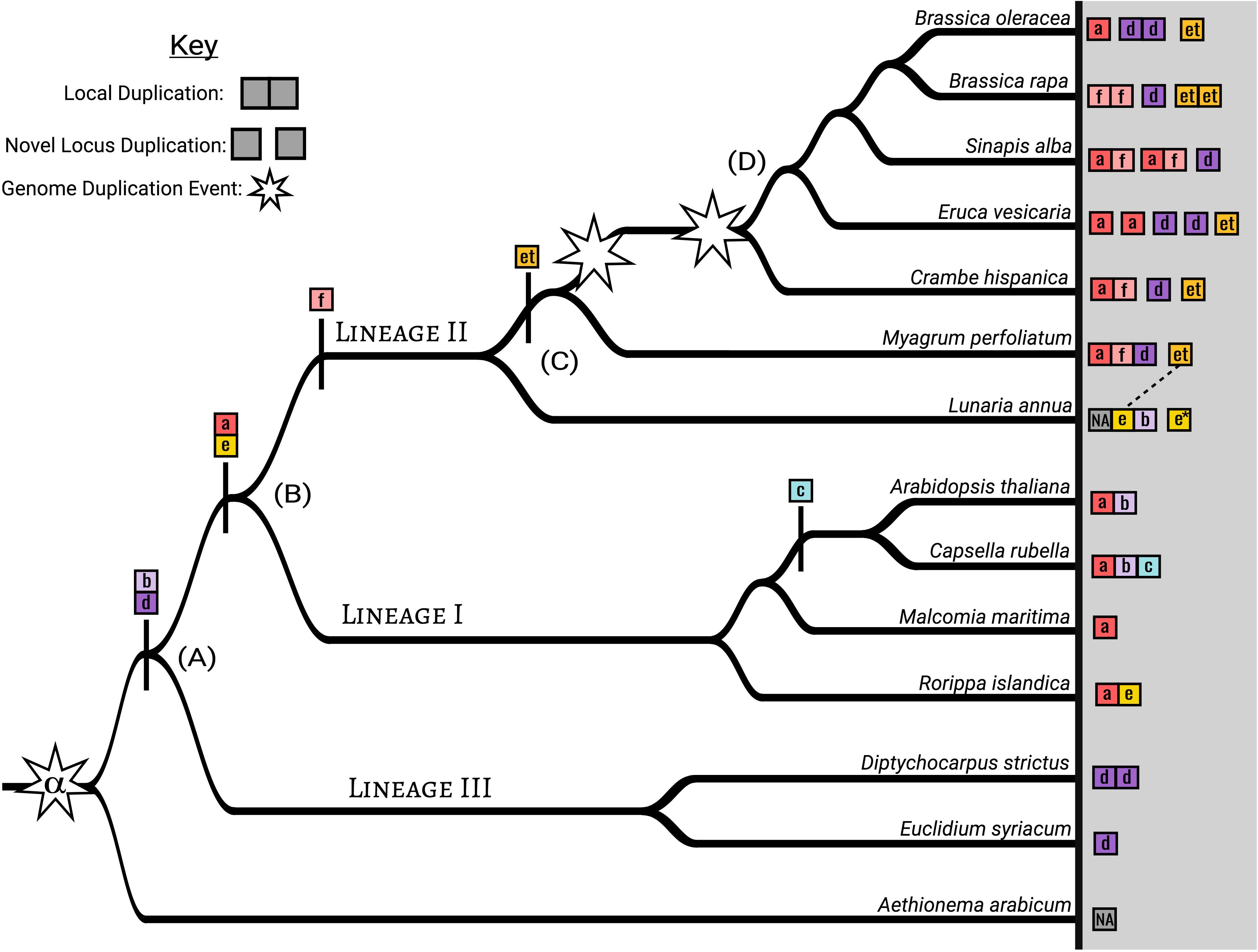
Figure 4. MAM clade and genomic context diversity within the Brassicaceae based on a subsample of analyzed genomes. Each square represents a MAM gene with an indicated HMGL-like domain type. Connected squares are found at the same physical location in the genome and not connected squares represent separate MAM loci (i.e., the MAM-Ancestral locus, a syntenic duplicate of the of the MAM-Ancestral locus, or MAM-Transposed). Non-syntenic gene transpositions were not included. (A) We estimate that the shared ancestor of Lineages I, II, and III maintained both MAMb and MAMd domain types. In the Lineage III genomes sampled MAMb genes were not located at the MAM-Ancestral locus, but at transposed loci. (B) At the ancestor of Lineage I and II, MAMa and MAMe appear, while the MAMc innovation occurs within a sub clade of Lineage I. (C) MAMf originates at the ancestor of Lineage II. The MAMet transposition that creates the MAM-Transposed locus occurs following the split from Lunaria annua, with all MAMet genes being closely related to a MAMe gene at the MAM-Ancestral locus. Lunaria annua also contains a context duplication of the MAM-Ancestral locus* that does not appear to be associated with whole genome duplication. (D) The unnamed whole genome duplication found in the tribe Brassiceae of Lineage II has resulted in multiple context duplications of the MAM-Ancestral locus. Full comparison is found in Supplementary Figure S3.
The MAM full-sequence tree shows bootstrap support between clades, but also a breakdown of some domain clades as well as clade nesting (Figure 3). MAMa and MAMb separate by species lineage, while MAMc is unique to a small subset of Lineage I species and appears closely related to MAMb and MAMe. MAMd, and MAMe are primarily the same as in the domain tree, but with other domains nested within. MAMf is consistent with the domain tree and sister to Lineage II MAMa.
To test for potential gene fusion events, full sequences of MAMa and MAMb Lineage I genes were broken up into “before the domain,” “domain,” and “after domain” sequences (Supplementary Figure S4). Pairwise sequence comparisons were made between the Lineage I gene segments and corresponding segments of Lineage I MAMe genes, and Lineage II genes for MAMa or Lineage III genes for MAMb. In both cases, the domain portion best matches the corresponding domain regardless of Lineage. For Lineage I MAMb, the region before the domain is more similar to Lineage I MAMe than it is to Lineage II MAMb. For Lineage I MAMa, the region before the domain is more similar on average to Lineage I MAMe but was not significantly different from Lineage II MAMa.
Discussion
The origin of all specialized metabolic pathways is primary metabolic genes, often with similar enzymatic chemistry (Moghe and Last, 2015). This transition is mediated by the process of gene duplication and subsequent drift and neo/subfunctionalization (Conant and Wolfe, 2008; Moghe and Last, 2015). For the MAM locus of the glucosinolate (GSL) biosynthesis pathway, the role of tandem duplication events in the evolution of the locus has been well characterized at the population level. The majority of work has only looked at Arabidopsis and its close relatives, and to a lesser extent, in the crop Brassicas (Kliebenstein and Cacho, 2016). Much of what we understand about the MAM locus function has not been understood in the context of phylogeny, except to say that based on gene tree relationships, Lineage II and Lineage II have independently diversified from some initial gene substrate (Benderoth et al., 2009; Zhang et al., 2015). In this study, we utilized a micro-synteny network of genomes and phylogenetic inference to elucidate the evolutionary history of the MAM locus.
MAM in the Cleomaceae
The inclusion of Cleomaceae genomes in our analysis has provided novel insight into the origin of the MAM locus, following the whole genome duplication (WGD) event β, the hypothesized origin of MAM from IPMS (van den Bergh et al., 2016). We estimate through micro-synteny and gene tree information that the Ancestral-MAM locus at the formation of the Cleomaceae was characterized by multiple MAM-like gene duplicates, the result of tandem duplications or local transposition (Figure 2). These genes are different from what has been characterized in the Brassicaceae orthologous Ancestral-MAM locus, the Elong locus. They have retained their LeuA domain, the loss of which has been considered a critical step in the evolution of Brassicaceae MAM (de Kraker et al., 2007). Within the Cleomaceae, some genes of the Ancestral-MAM locus exhibit both Ancestral-MAM and IPMS syntenic cluster identity (Figure 1). The syntenic window for these intermediates is shifted in comparison to other analyzed neighboring MAM-like genes. This allows for the inclusion of neighboring non-MAM genes that are more characteristic of the IPMS genomic context. This evidence supports the hypothesis that the Ancestral-MAM locus was once a full context duplicate of the IPMS locus, and in the process of specialization over millions of years, degraded in collinearity.
How these MAM-like genes interact with GSL biosynthesis is unknown, but they have shown levels of expression in the leaf, seed, and roots in Tarenaya hassleriana (van den Bergh et al., 2016). The retention of the LeuA domain suggests that MAM-like proteins may have some continued interaction with IPMS or leucine biosynthesis. The ways in which genes respond to duplication events are constrained by their biochemical interactions, and therefore may shed insight into enzyme behavior (Bekaert et al., 2012; Birchler and Veitia, 2012; Conant et al., 2014; McLysaght et al., 2014). For example, given that IPMS experiences purifying selection of local gene duplicates and that MAM-like Cleomaceae genes found at the MAM-Ancestral locus do exhibit some local duplication, it is likely that these MAM-like genes have significantly sub- or neofunctionalized from their IPMS ancestor in terms of biochemical role. With that said, the dosage effects of IPMS are broader than only limiting local duplication, and through stoichiometric effects constrain most duplication types. Only after the β WGD event, is IPMS able to be retained and reduced in multiples of two. A pattern we see recapitulated after subsequent WGD events, with a few potential exceptions (Supplementary Figure S2). Following Th-α, the Cleomaceae whole-genome triplication (WGT) or hexaploidy, there is an expected full context duplication of the IPMS locus, but with no context duplication of the Ancestral-MAM locus (Figure 2C). In fact, we see a compensatory loss of a MAM-like gene following the increase in IPMS copy number. The presence of stoichiometric conflict between IPMS and these MAM-like genes would support the hypothesis that they have retained some IPMS role and constraint. Further sampling across the Cleomaceae will be necessary to see if these patterns hold.
In the Tarenaya hassleriana genome, there is a novel a transposition of MAM (Figure 2D). This transposed gene does not have a LeuA domain, bringing the overall MAM/IPMS gene number beyond what would be expected under an IPMS dosage constraint (Supplementary Figure S2). This transposed locus has been shown to express in several tissues and to a greater extent in the leaf when compared to MAM-like counterparts at the Ancestral-MAM locus (van den Bergh et al., 2016). Increased species sampling, as well as an understanding of population-level variation in Cleomaceae MAM, is necessary for any conclusions on the dosage to be explored further using these methods. Direct biochemical assays of these MAM-like proteins will also be critical for characterizing any role they may play in glucosinolate biosynthesis and how that may differ from what is seen in the Brassicaceae. The Cleomaceae, and potentially the Capparaceae, which also shares the β duplication event (Edger et al., 2015), could serve as a powerful window into the evolution of early Brassicaceae MAM and a model for how gene families transition from primary to specialized metabolism.
MAM in the Brassicaceae
Between Lineages I, II, and III of the Brassicaceae, we have identified six distinct clades of MAM, MAMa-f, based on conserved HMGL-like domain sequences (Figure 3 and Supplementary Figure S3). Based on occurrence patterns across the family, we can say that MAMb and MAMd clades are ancestral to all three lineages, and MAMa and MAMe may be ancestral to only Linages I and Lineage II. The latter conclusion could not be confirmed by gene tree information and may be vulnerable to sampling bias. The dispute between the chloroplast and nuclear species tree topologies could also affect the evolutionary relationships between the MAM clades and hamper our ability to predict (Nikolov et al., 2019). Improved sampling across the Brassicaceae is necessary before a robust estimation of the ancestral type can be made. That said, we are confident that MAMc, MAMet, and MAMf domain types are more recent innovations occurring in Lineage I and Lineage II, with specific branch placements (Figure 4 and Supplementary Figure S3).
Given the functional role this domain plays in MAM biochemistry, we expect amino acid differences between domain types to be associated with generalizable patterns in MAM function. MAMa is the most conserved of the domains (Supplementary Figure S5B), suggesting that MAMa genes may contribute a necessary function to GSL biosynthesis, as compared to other MAM types. MAMc and MAMet are the most diverged, each having several unique amino acid substitutions when compared to other domain types (Supplementary Figure S5B). Across all the domains, some sites were characterized by amino acid variability within and between domain types. Based on the characterization of MAM proteins in Brassica juncea (Kumar et al., 2019), we identified that oxo-acid binding sites were most often found at flexible amino acid positions followed by COA binding sites (Supplementary Figure S5A). A better understanding of these patterns can give us insight into the forces driving the adaptation of MAM.
The domain and full-sequence gene trees conflict most significantly within the core Brassicaceae (Figure 3). In the full-sequence tree Lineage I MAMa and MAMb genes appear more closely related to MAMe genes than to other genes of their shared domain. Sequence comparison reveals split-sequence similarities in both MAMa and MAMb domain clade groups. This pattern suggests two possibilities: (1) MAM genes experienced convergent evolution of their amino acid sequences, or (2) a gene fusion event of separate MAM types occurred sometime during the divergence of Lineage I MAM. The latter scenario is both the more parsimonious conclusion, and it is supported by the previous characterization of population-level gene fusion events at the Ancestral-MAM locus (Benderoth et al., 2009). Given that Lineage I MAMa and MAMb genes show a close phylogenetic relationship to Lineage I MAMe, in conflict with the domain tree, it is the most likely donor gene. Both fusion events would have occurred at separate nodes of the Lineage I species tree, MAMa/MAMe fusion happening earlier than the MAMb/MAMe event. Improved sampling of Lineage I is necessary to identify the specific species branch points at which the events occurred. The fusion of MAM genes at the MAM-Ancestral locus, though largely studied from only a population level, may have been a critical driver of MAM diversity and innovation within Lineage I in the Brassicaceae.
Most of the genes in each domain clade exist at the MAM-Ancestral locus. This is true for genes of the MAMe group except for a nested clade of transposed genes, MAMet, that form the unique syntenic cluster MAM-Transposed (Figures 1, 4). There are subsequent transpositions from the MAM-Transposed locus, many of which show signs of degradation. The initial transposition occurred sometime following the split from the ancestor of Lunaria annua to the common ancestor of Thellungiella (Eutrema) and the rest of Lineage II (Supplementary Figure S3). Following the transposition event, there is a loss of all MAMe domain type genes. Of our dataset, L. annua is the only member of Lineage II to retain any copies of MAMe. Of those MAMe genes, most appear closely related to Lineage I MAMe genes, while one copy is most closely related to MAMet in both the domain and full sequence trees (Figure 3). This transposition event is the earliest conserved instance of a novel MAM context, which allows for an escape from cis-regulatory effects that may be experienced at the MAM-Ancestral locus (Chen and Ni, 2006; Conant and Wolfe, 2008). The possibilities exist that these genes are performing some yet to be characterized function or potentially may represent the GSL-PRO locus characterized in Brassica species. With this current analysis, we cannot further speculate on the role MAMet genes may be playing in GSL biosynthesis, except to say that experimental analysis of these genes will be necessary to understand their place in metabolic innovation.
Polyploidy offers another mechanism for MAM diversification, by escaping potential cis-regulatory effects of other MAM genes or sub- and neofunctionalization of resulting duplicates. In the Cleomaceae, the MAM-Ancestral locus duplicates are not retained following genome doubling, putatively due to the presence of their LeuA domain and restrictions under gene dosage. Without such dosage constraints in Brassicaceae MAM, most genomes sampled show retention of a duplicated MAM-Ancestral locus following known WGD events. For example, the WGT event in the tribe Brassiceae of Lineage II resulted in three homoeologous MAM-Ancestral loci in subsequently diploidized genomes (Figure 4 and Supplementary Figure S2). In Brassica rapa, Brassica oleracea, and Eruca vesicaria, the MAM-Ancestral loci maintain a single MAM domain type (MAMa, MAMd, or MAMf) at each. Whereas in other genomes, like Sinapis alba, MAMa and MAMf genes remain paired although duplicated at separate loci. We propose that phenotypic differences between Brassica and Arabidopsis, such as the ability to co-synthesize different carbon chain majority phenotypes, are facilitated by the physical separation of MAM genes within the genome. By influencing the rate of diversification for MAM genes at the different MAM-Ancestral loci and allowing for novel genomic interactions, the WGT may have been a critical step in driving the specialized metabolic innovation we see in this dynamic crop lineage.
Conclusion
The MAM/IPMS gene family serves as an excellent example of how a primary metabolic gene can, over millions of years and leveraging any source of novelty, give rise to a diverse lineage of highly adaptive specialized metabolic genes. Utilizing micro-synteny gene networks and broad phylogenetic sampling, we find that multiple modes of gene duplication have significantly influenced the evolutionary trajectory of the MAM locus and thereby diversity of aliphatic GSL profiles. By exploring some of the evolutionary consequences of whole-genome duplication, gene transposition, local duplication, and gene fusion, we have generated several new testable hypotheses as to the nature of MAM and GSL diversity. In the future, new experimental approaches and broad phylogenetically informed sampling will be critical to continue developing a robust understanding of this important gene family.
Data Availability Statement
The datasets generated for this study can be found in the information provided in Supplementary Material.
Author Contributions
RSA performed research and wrote the manuscript. JCP helped design the study and edited the manuscript. MES designed the study and helped write and edit the manuscript.
Funding
RSA was supported by a NSF GROW fellowship which allowed him to travel and work in Netherlands.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank J. Wiscaver, D. Kleibenstein, and the two reviewers for insights and critical feedback. We also thank Tao Zhao with help with the synteny network analysis pipeline.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00711/full#supplementary-material
FIGURE S1 | (A) Benderoth et al., 2009 describes the MAM lineage in terms of orthology to Arabidopsis lyrata gene tree clades. While the topology generally agrees with our tree, the emphasis on Arabidopsis and close relatives gives a limited picture of MAM diversity. This tree also supported the hypothesis that MAM has evolved separately in the Lineage I and II. (B) Zhang et al., 2015 generally agrees with this hypothesis though they do show shared clades not solely informed by the species tree. Some of their topology conflicts with our full sequence tree and yet agrees with the domain specific tree. This may be due to how their alignment was cleaned and their species sampling.
FIGURE S2 | The overall gene counts per genome for the MAM/IPMS gene family. Gene numbers, especially in IPMS, are correlated with recent polyploidy. Three genomes conflict with the expected IPMS dosage expectation of multiples of two. The Raphanus raphanistrum and Stanleya pinnata IPMS deviations may be an artifact of lower quality genomes, but the Eruca vesicaria retention appears to be a newly sub-functionalized IPMS copy, exhibiting an intermediate syntenic relationships to that of some MAM-Ancestral genes in the Cleomaceae. For MAM, the number of Loci indicates whether MAM-Ancestral or MAM-Transposed has experienced a context duplication. The number of genes at that locus is the overall total of genes across all syntenic loci of that type.
FIGURE S3 | Here we show the full domain clade distribution of MAM genes across the genomes, regardless of synteny or genomic position. This data was used ultimately to place the points of innovation for different MAM types in Figure 4.
FIGURE S4 | MAM protein sequences were divided into before domain, domain, and after domain segments and each significantly different section of the MAMa or MAMb genes from lineage I were compared to corresponding MAMe sections.
FIGURE S5 | Amino acid sequence comparisons at 80% sequence similarity. (A) Colored rectangles indicate specific biochemical functions as described by Kumar et al. (2019) in Brassica juncea. Green - metal binding sites; Yellow - catalytic sites; Red - 2- oxo acid binding sites; Blue - CoA binding sites. (B) Summarizes all sites with a uniquely divergent amino acid to quantify the significance of domain divergence.
FIGURE S6 | Full gene family phylogeny with bootstrap scores at 1000 bootstraps with syntenic clusters mapped. Used in Figure 1. May also be accessed via: http://bit.ly/2tHVgYK.
FIGURE S7 | Domain tree phylogeny with clades colored and bootstrap scores at 1000 bootstraps. Used in Figure 3. May also be accessed via: http://bit.ly/2Hb5jIS.
FIGURE S8 | Full gene family phylogeny with bootstrap scores at 1000 bootstraps with clades colored. Used in Figure 3. May also be accessed via: http://bit.ly/37btHEZ.
References
Bekaert, M., Edger, P. P., Hudson, C. M., Pires, J. C., and Conant, G. C. (2012). Metabolic and evolutionary costs of herbivory defense: systems biology of glucosinolate synthesis. New Phytol. 196, 596–605. doi: 10.1111/j.1469-8137.2012.04302.x
Benderoth, M., Pfalz, M., and Kroymann, J. (2009). Methylthioalkylmalate synthases: genetics, ecology and evolution. Phytochem. Rev. 8, 255–268. doi: 10.1007/s11101-008-9097-1
Benderoth, M., Textor, S., Windsor, A. J., Mitchell-Olds, T., Gershenzon, J., and Kroymann, J. (2006). Positive selection driving diversification in plant secondary metabolism. Proc. Natl. Acad. Sci. U.S.A. 103, 9118–9123. doi: 10.1073/pnas.0601738103
Birchler, J. A., and Veitia, R. A. (2012). Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. U.S.A. 109, 14746–14753. doi: 10.1073/pnas.1207726109
Blazevic, I., Montaut, S., Burcul, F., Olsen, C. E., Burow, M., Rollin, P., et al. (2020). Glucosinolate structural diversity, identification, chemical synthesis and metabolism in plants. Phytochemistry 169:112100. doi: 10.1016/j.phytochem.2019.112100
Borpatragohain, P., Rose, T. J., and King, G. J. (2016). Fire and Brimstone: Molecular interactions between sulfur and glucosinolate biosynthesis in model and crop Brassicaceae. Front. Plant Sci. 7:1735. doi: 10.3389/fpls.2016.01735
Chen, Z. J., and Ni, Z. (2006). Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. BioEssays 28, 240–252. doi: 10.1002/bies.20374
Chhajed, S., Misra, B. B., Tello, N., and Chen, X. (2019). Chemodiveristy of the glucosinolate-myrosinate system at the single cell type resolution. Front. Plant Sci. 10:618.
Conant, G. C., and Wolfe, K. H. (2008). Probabilistic cross-species inference of orthologous genomic regions created by whole-genome duplication in yeast. Genetics 179, 1681–1692. doi: 10.1534/genetics.107.074450
Conant, G. C., Birchler, J. A., and Pires, J. C. (2014). Dosage, duplication, and diploidization: clarifying the interplay of multiple models for duplicate gene evolution over time. Curr. Opin. Plant Biol. 19, 91–98. doi: 10.1016/j.pbi.2014.05.008
de Kraker, J.-W., and Gershenzon, J. (2011). From amino acid to glucosinolate biosynthesis: protein sequence changes in the evolution of methylthioalkylmalate synthase in Arabidopsis. Plant Cell 23, 38–53. doi: 10.1105/tpc.110.079269
de Kraker, J. W., Luck, K., Textor, S., Tokuhisa, J. G., and Gershenzon, J. (2007). Two Arabidopsis genes (IPMS1 and IPMS2) encode isopropylmalate synthase, the branchpoint step in the biosynthesis of leucine. Plant Physiol. 143, 970–986. doi: 10.1104/pp.106.085555
del Carmen, M., Moreno, D. A., and Carvajal, M. (2013). The physiological importance of glucosinolates on plant response to abiotic stress in Brassica. Int. J. Mol. Sci. 14, 11607–11625. doi: 10.3390/ijms140611607
Derényi, I., Palla, G., and Vicsek, T. (2005). Clique percolation in random networks. Phys. Rev. Lett. 94:160202.
Edger, P. P., Heidel-Fischer, H. M., Bekaert, M., Rota, J., Glockner, G., Platts, A. E., et al. (2015). The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. U.S.A. 112, 8362–8366. doi: 10.1073/pnas.1503926112
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Fortunato, S. (2010). Community detection in graphs. Phy. Rep. 486, 75–174. doi: 10.1016/j.physrep.2009.11.002
Hofberger, J. A., Lyons, E., Edger, P. P., Pires, J. C., and Schranz, M. E. (2013). Whole genome and tandem duplicate retention facilitated glucosinolate pathway diversification in the mustard family. Genome Biol. Evol. 5, 2155–2173. doi: 10.1093/gbe/evt162
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Keurentjes, J. J., Fu, J., de Vos, C. H., Lommen, A., Hall, R. D., Bino, R. J., et al. (2006). The genetics of plant metabolism. Nat. Genet. 38, 842–849. doi: 10.1038/ng1815
Kliebenstein, D. J. (2008). A role for gene duplication and natural variation of gene expression in the evolution of metabolism. PLoS One 3:e1838. doi: 10.1371/journal.pone.0001838
Kliebenstein, D. J., and Cacho, N. I. (2016). Nonlinear selection and a blend of convergent, divergent and parallel evolution shapes natural variation in glucosinolates. Adv. Bot. Res. 80, 31–55. doi: 10.1016/bs.abr.2016.06.002
Kliebenstein, D. J., Gershenzon, J., and Mitchell-Olds, T. (2001a). Comparative quantitative trait loci mapping of aliphatic, indolic and benzylic glucosinolate production in Arabidopsis thaliana leaves and seeds. Genetics 159, 359–370.
Kliebenstein, D. J., Lambrix, V. M., Reichelt, M., Gershenzon, J., and Mitchell-Olds, T. (2001b). Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell 13, 681–693. doi: 10.1105/tpc.13.3.681
Koon, N., Squire, C. J., and Baker, E. N. (2004). Crystal structure of LeuA from Mycobacterium tuberculosis, a key enzyme in leucine biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 101, 8295–8300. doi: 10.1073/pnas.0400820101
Kroymann, J., and Mitchell-Olds, T. (2005). Epistasis and balanced polymorphism influencing complex trait variation. Nature 435, 95–98. doi: 10.1038/nature03480
Kumar, R., Lee, S. G., Augustine, R., Reichelt, M., Vassao, D. G., Palavalli, M. H., et al. (2019). Molecular basis of the evolution of methylthioalkylmalate synthase and the diversity of methionine-derived glucosinolates. Plant Cell 31, 1633–1647. doi: 10.1105/tpc.19.00046
Kuraku, S., Zmasek, C. M., Nishimura, O., and Katoh, K. (2013). aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 41, W22–W28. doi: 10.1093/nar/gkt389
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
McLysaght, A., Makino, T., Grayton, H. M., Tropeano, M., Mitchell, K. J., Vassos, E., et al. (2014). Ohnologs are overrepresented in pathogenic copy number mutations. Proc. Natl. Acad. Sci. U.S.A. 111, 361–366. doi: 10.1073/pnas.1309324111
Moghe, G. D., and Last, R. L. (2015). Something old, something new: conserved enzymes and the evolution of novelty in plant specialized metabolism. Plant Physiol. 169, 1512–1523. doi: 10.1104/pp.15.00994
Nikolov, L. A., Shushkov, P., Nevado, B., Gan, X., Al-Shehbaz, I. A., Filatov, D., et al. (2019). Resolving the backbone of the Brassicaceae phylogeny for investigating trait diversity. New Phytol. 222, 1638–1651. doi: 10.1111/nph.15732
Olsen, C. E., Huang, X. C., Hansen, C. I. C., Cipollini, D., Orgaard, M., Mathes, A., et al. (2016). Glucosinolate diversity within a phylogenetic framework of the tribe Cardamineae (Brassicaceae) unraveled with HPLC-MS/MS and NMR-based analytical distinction of 70 desulfoglucosinolates. Phytochemistry 132, 33–56. doi: 10.1016/j.phytochem.2016.09.013
Palla, G., Derényi, I., Farkas, I., and Vicsek, T. (2005). Uncovering the overlapping community structure of complex networks in nature and society. Nature 435, 814–818. doi: 10.1038/nature03607
Peng, C., Uygun, S., Shiu, S.-H., and Last, R. L. (2015). The Impact of the Branched-Chain Ketoacid Dehydrogenase Complex on Amino Acid Homeostasis in Arabidopsis. Plant Physiol. 169, 1807–1820. doi: 10.1104/pp.15.00461
Petersen, A., Hansen, L. G., Mirza, N., Crocoli, C., Mirza, O., and Halkier, B. A. (2019). Changing substrate specificity and iteration of amino acid chain elongation in glucosinolate biosynthesis through targeted mutagenesis of Arabidopsis methylthioalkylmalate synthase 1. Biosci. Rep. 39:BSR20190446.
Rodman, J. E., Soltis, P. S., Soltis, D. E., Sytsma, K. J., and Karol, K. G. (1998). Parallel evolution of glucosinolate biosynthesis inferred from congruent nuclear and plastid gene phylogenies. Am. J. Bot. 85, 997–1006. doi: 10.2307/2446366
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Smith, S. A., and Dunn, C. W. (2008). Phyutility: a phyloinformatics tool for trees, alignments, and molecular data. Bioinformatics 24, 715–716. doi: 10.1093/bioinformatics/btm619
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., and Paterson, A. H. (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Textor, S., Bartram, S., Kroymann, J., Falk, K. L., Hick, A., Pickett, J. A., et al. (2004). Biosynthesis of methionine-derived glucosinolates in Arabidopsis thaliana: recom- binant expression and characterization of methylthioalkylmalate synthase, the condensing enzyme of the chain-elongation cycle. Planta 218, 1026–1035. doi: 10.1007/s00425-003-1184-3
Textor, S., de Kraker, J. W., Hause, B., Gershenzon, J., and Tokuhisa, J. G. (2007). MAM3 catalyzes the formation of all aliphatic glucosinolate chain lengths in Arabidopsis. Plant Physiol. 144, 60–71. doi: 10.1104/pp.106.091579
van den Bergh, E., Hofberger, J. A., and Schranz, M. E. (2016). Flower power and the mustard bomb: comparative analysis of gene and genome duplications in glucosinolate biosynthetic pathway evolution in Cleomaceae and Brassicaceae. Am. J. Bot. 103, 1212–1222. doi: 10.3732/ajb.1500445
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wentzell, A. M., Rowe, H. C., Hansen, B. G., Ticconi, C., Halkier, B. A., and Kliebenstein, D. J. (2007). Linking metabolic QTLs with network and cis-eQTLs controlling biosynthetic pathways. PLoS Genet. 3, 1687–1701. doi: 10.1371/journal.pgen.0030162
Wisecaver, J. H., Borowsky, A. T., Tzin, V., Jander, G., Kliebenstein, D. J., and Rokas, A. (2017). A global co-expression network approach for connecting genes to specialized metabolic pathways in plants. Plant Cell 29, 944–959. doi: 10.1105/tpc.17.00009
Zhang, J., Wang, X., Cheng, F., Wu, J., Liang, J., Yang, W., et al. (2015). Lineage-specific evolution of Methylthioalkylmalate synthases (MAMs) involved in glucosinolates biosynthesis. Front. Plant Sci. 6:18. doi: 10.3389/fpls.2015.00018
Zhao, T., Holmer, R., de Bruijn, S., Angenent, G. C., van den Burg, H. A., and Schranz, M. E. (2017). Phylogenomic synteny network analysis of MADS-box transcription factor genes reveals lineage-specific transpositions, ancient tandem duplications, and deep positional conservation. Plant Cell 29, 1278–1292. doi: 10.1105/tpc.17.00312
Zhao, T., and Schranz, M. E. (2019). Network-based microsynteny analysis identifies major differences and genomic outliers in mammalian and angiosperm genomes. Proc. Natl. Acad. Sci. U.S.A. 116, 2165–2174. doi: 10.1073/pnas.1801757116
Keywords: glucosinolates, brassicaceae, gene family, polyploidy, gene duplication, gene fusion
Citation: Abrahams RS, Pires JC and Schranz ME (2020) Genomic Origin and Diversification of the Glucosinolate MAM Locus. Front. Plant Sci. 11:711. doi: 10.3389/fpls.2020.00711
Received: 15 October 2019; Accepted: 05 May 2020;
Published: 04 June 2020.
Edited by:
Ralph Kissen, Norwegian University of Science and Technology, NorwayReviewed by:
Priyakshee Borpatragohain, Southern Cross University, AustraliaJoshua Trujillo, Purdue University, United States
Copyright © 2020 Abrahams, Pires and Schranz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Eric Schranz, ZXJpYy5zY2hyYW56QHd1ci5ubA==