 Lu-Xian Liu
Lu-Xian Liu Ying-Xue Du1
Ying-Xue Du1 Fu-De Shang
Fu-De Shang Pan Li
Pan Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 24 April 2020
Sec. Plant Systematics and Evolution
Volume 11 - 2020 | https://doi.org/10.3389/fpls.2020.00361
Saxifragaceae, a family of over 600 species and approximately 30 genera of herbaceous perennials, is well-known for intergeneric hybridization. Of the main lineages in this family, the Heuchera group represents a valuable model for the analysis of plastid capture and its impact on phylogeny reconstruction. In this study, we investigated plastome evolution across the family, reconstructed the phylogeny of the Heuchera group and examined putative plastid capture between Heuchera and Tiarella. Seven species (11 individuals) representing Tiarella, as well as Mitella and Heuchera, were selected for genome skimming. We assembled the plastomes, and then compared these to six others published for Saxifragaceae; the plastomes were found to be highly similar in overall size, structure, gene order and content. Moreover, ycf15 was lost due to pseudogenization and rpl2 lost its only intron for all the analyzed plastomes. Comparative plastome analysis revealed that size variations of the plastomes are purely ascribed to the length differences of LSC, SSC, and IRs regions. Using nuclear ITS + ETS and the complete plastome, we fully resolved the species relationships of Tiarella, finding that the genus is monophyletic and the Asian species is most closely related to the western North American species. However, the position of the Heuchera species was highly incongruent between nuclear and plastid data. Comparisons of nuclear and plastid phylogenies revealed that multiple plastid capture events have occurred between Heuchera and Tiarella, through putative ancient hybridization. Moreover, we developed numerous molecular markers for Tiarella (e.g., plastid hotspot and polymorphic nuclear SSRs), which will be useful for future studies on the population genetics and phylogeography of this disjunct genus.
In the past, studies have relied heavily on organellar markers for inferring phylogenetic relationships. However, chloroplast and mitochondrial genes often show markedly different phylogenetic patterns from nuclear markers (Rieseberg and Soltis, 1991; Toews and Brelsford, 2012). Various factors, including convergent evolution, lineage sorting, hybridization and introgression, may cause phylogenetic incongruence between nuclear and plastid DNA (e.g., Soltis and Kuzoff, 1995; McKinnon et al., 2001; Acosta and Premoli, 2010). Incomplete lineage sorting and hybridization/introgression are particularly hard to distinguish due to their similar phylogenetic signatures (Wendel and Doyle, 1998). However, hybridization is now more readily detected due to advances in statistical methods and data collection strategies (Joly et al., 2009; Folk et al., 2018). One extreme result of hybridization is plastid capture, in which the cytoplasm of one species is replaced by that of another species through inter-species hybridization and subsequent backcrossing, yielding a plant with a novel combination of nuclear and plastid genomes (Rieseberg and Soltis, 1991). Plastid capture has been reported across numerous plant lineages (Okuyama et al., 2005; Fehrer et al., 2007; Acosta and Premoli, 2010; Gurushidze et al., 2010; Mir et al., 2010).
Saxifragaceae, a family of over 600 species and approximately 30 genera of herbaceous perennials, are found mostly in the Northern Hemisphere with centers of diversity in the Himalayas, East Asia, and Western North America (Deng et al., 2015). Due to factors such as morphological stasis, convergent morphological evolution, and disjunct distributions, relationships within Saxifragaceae have historically confused botanists (Engler, 1930; Morgan and Soltis, 1993). The family comprises two main lineages: Saxifragoids, which include Saxifraga L. and Saxifragella Engl., and Heucheroids, which include all other genera. The Heucheroids are noted for examples of intergeneric hybridization. For example, nine genera of herbaceous perennials (Bensoniella C.V. Morton, Conimitella Rydb., Elmera Rydb., Tellima R. Br., Tolmiea Torr. et A. Gray, Tiarella L., Lithophragma (Nutt.) Torr. et A. Gray, Mitella L., and Heuchera L.) comprise the Heuchera group, a particularly valuable model for the analysis of plastid capture and its impact on phylogeny reconstruction due to evidence of numerous hybridization events among and within those genera (Soltis et al., 1991; Soltis and Kuzoff, 1995).
Within the Heuchera group, Mitella and Heuchera are the two largest genera, consisting of 21 and 43 currently recognized species, respectively (Folk and Freudenstein, 2014; Okuyama, 2015). Lithophragma and Tiarella comprise nine and three species, respectively. Tolmiea (2 spp.) and the remaining four monotypic genera (Bensoniella, Conimitella, Elmera, and Tellima) are solely distributed in western North America. Six of these genera are known to hybridize (Soltis and Bohm, 1985; Soltis and Soltis, 1986; Soltis et al., 1991), with intergeneric hybrids being reported between Heuchera and Tiarella (Spongberg, 1972), Tellima and Tolmiea (Soltis and Bohm, 1985), and Mitella and Conimitella (Soltis and Soltis, 1986). Numerous interspecific hybrids have been reported within Heuchera (Rosendahl et al., 1936). Several studies have reported that plastid capture likely occurred between species of Heuchera, as well as between Tellima and Mitella and between Heuchera and Mitella. Likewise, there is evidence for gene flow between Tiarella and Heuchera (Soltis, 1991; Soltis et al., 1991; Folk et al., 2017).
Tiarella is a small genus within the Heuchera group and contains only three species: Tiarella polyphylla D. Don, Tiarella cordifolia L., and Tiarella trifoliata L. (Wu and Raven, 2001; Wells and Elvander, 2009). The genus shows an interesting eastern Asia (T. polyphylla)-western North America (T. trifoliata)-eastern North America (T. cordifolia) disjunct distribution pattern, with one species each in these three areas. Soltis (1991) resolved T. trifoliata and T. cordifolia as paraphyletic to Heuchera as well as some other members of the Heuchera group based on restriction site analysis of chloroplast (cp) DNA (T. polyphylla was not sampled in this study). Subsequently, Soltis and Kuzoff (1995) revealed a clade containing T. trifoliata and T. cordifolia based on nrITS-1 (nuclear internal transcribed spacer) and nrITS-2 sequences. The monophyly of Tiarella was confirmed using ITS data by Xiang et al. (1998), who resolved the Asian species as sister to the two North American species. However, Okuyama et al. (2008) revealed that T. polyphylla and T. cordifolia formed a clade with moderate support and was sister to T. trifoliata. Recently, the ancestral area of this genus was found to be western North America as inferred by S-DIVA based on cpDNA + nrDNA data (Deng et al., 2015). However, the phylogenetic relationships among T. trifoliata, T. cordifolia, and T. polyphylla are not yet well resolved and these previous studies have lacked the population-level sampling needed for robust inference of hybridization histories.
High-throughput sequencing technologies have revolutionized the ease with which genomic data can be acquired for any plant species, from angiosperms to bryophytes, regardless of model species or non-model species (Dodsworth, 2015). Advances in sequencing technology has impacted biodiversity science, including systematics, population genetics, DNA barcoding and ecological investigations. Genome skimming sequencing, which involves random sampling of a small percentage of total genomic DNA (gDNA), was first identified by Straub et al. (2012) as a straightforward way to obtain genome-scale data with minimal lab processing. It is a simpler method compared to RNA-seq and RAD-seq, and the high-copy fraction of the genome (plastome, mitogenome, and repetitive nuclear elements) can be deeply sequenced through shallow sequencing of total gDNA. The nrITS and nrETS, various plastid markers and polymorphic nuclear simple sequence repeats (nSSRs) are commonly acquired using this technique (Liu et al., 2018a). This approach has been widely used at different taxonomic levels, for intraspecific “ultra-barcoding” (Kane et al., 2012), intergeneric (McPherson et al., 2013; Li et al., 2017; Liu et al., 2018b), and family level or above phylogenomic analyses (Besnard et al., 2013; Malé et al., 2015; Liu et al., 2017; Xu et al., 2018).
Here, we used a genome skimming approach to investigate the phylogeny of Tiarella and test the hypothesis of plastid capture between Tiarella and Heuchera. Seven species, in total 11 individuals, were selected for genome skimming. We specifically aimed to: (1) assemble, characterize and compare the plastomes among representatives of Saxifragaceae to gain insights into evolutionary patterns; (2) acquire nrITS, nrETS, and plastome data to resolve the phylogeny of Tiarella and the direction of plastid capture between Tiarella and Heuchera; (3) develop and screen appropriate intrageneric markers for Tiarella, such as plastid hotspot regions and polymorphic nSSRs.
Seven species from the Heuchera group (11 individuals) including T. polyphylla (4), T. cordifolia (2), T. trifoliata (1), Heuchera richardsonii R. Br. (1), Heuchera villosa Michx. (1), Mitella diphylla L. (1), and Mitella formosana (Hayata) Masam. (1) were selected for genome skimming (Supplementary Table S1). Fresh healthy leaves were collected in the field and immediately dried with silica gel for later DNA isolation. Voucher specimens are deposited at the Herbarium of Zhejiang University (HZU).
For broader comparisons, the complete plastomes of Heuchera parviflora Bartl (GenBank accession number: KR478645; Folk et al., 2015), as well as the following Saxifragaceae not in the Heuchera group were downloaded from GenBank: Bergenia scopulosa T.P. Wang (KY412195; Bai et al., 2018), Mukdenia rossii (Oliv.) Koidz (MG470844; Liu et al., 2018a), Oresitrophe rupifraga Bunge (MF774190 and MG470845; Liu et al., 2018a), and Saxifraga stolonifera Curtis (MH191389; Dong et al., 2018).
Total DNA was extracted using Plant DNAzol Reagent (LifeFeng, Shanghai) according to the manufacturer’s protocol with ∼2 mg of the silica-dried leaf tissue. The high molecular weight DNA was sheared using a Covaris S220-DNA Sonicator (Covaris, Inc., Woburn, MA, United States), yielding fragments ≤800 bp. The fragmentation quality was checked on an Agilent Bioanalyzer 2100 (Agilent Technologies). Libraries with an insert size of ∼500 bp were sequenced on an Illumina HiSeq 2500 (paired-end, 150 bp reads) by Beijing Genomics Institute (Shenzhen, China).
The raw reads were first filtered by quality with average Phred score < 30 (0.001 error probability). Subsequently, the remaining high-quality sequences were assembled into contigs using the CLC Genomics Workbench v12.0.3 (CLC Inc., Aarhus, Denmark) with the following optimized parameters: deletion and insertion costs of 3, mismatch cost of 2, minimum contig length of 200, bubble size of 98, length fraction, and similarity fraction of 0.9. All resultant contigs were mapped to the reference plastome (H. parviflora) using BLAST (NCBI BLAST v2.2.31) and the mapped contigs were ordered and oriented according to the reference genome (Folk et al., 2015). The draft genome was constructed by connecting overlapping terminal sequences, and the plastome was finalized by re-mapping cleaned reads to the draft genome.
Complete plastome annotation was performed through the online program Dual Organellar GenoMe Annotator (DOGMA; Wyman et al., 2004). The initial annotation was subsequently inspected and adjusted manually comparing with the reference genome sequence in order to confirm the start and stop codons and the exon-intron boundaries of genes. The tRNA genes were verified using tRNAscan-SE v1.21 (Schattner et al., 2005) with default settings. All the plastome sequences were deposited in GenBank (Figure 1) and the circular gene maps were drawn by the OrganellarGenomeDRAW tool (OGDRAW v1.3.1; Lohse et al., 2013) followed by manual modification.
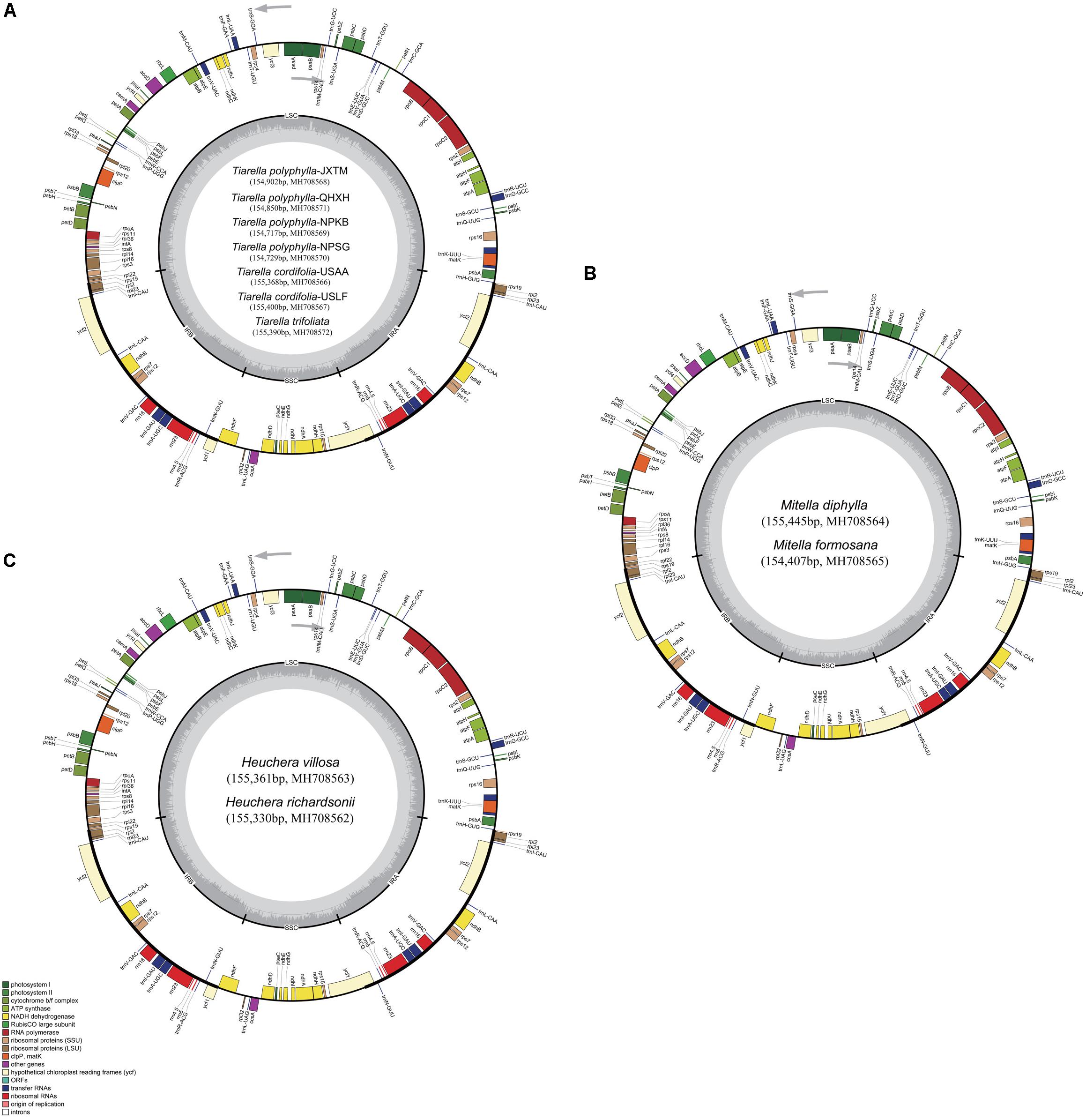
Figure 1. Plastome maps of Tiarella, Heuchera, and Mitella. (A) Seven Tiarella plastomes including four T. polyphylla individuals, two T. cordifolia, and one T. trifoliata, (B) Two Mitella plastomes including M. diphylla and M. formosana (C) Two Heuchera plastomes including H. villosa and H. richardsonii. Genes inside the circle are transcribed clockwise, gene outside are transcribed counter-clockwise. The light gray inner circle corresponds to the AT content, the dark gray to the GC content. Genes belonging to different functional groups are shown in different colors; see the legend for groups.
To compare the sequence variation within the Saxifragaceae, we combined publicly available plastomes of B. scopulosa, M. rossii, O. rupifraga (representing the Darmera group of genera; Soltis et al., 2001), and S. stolonifera. Multiple sequence alignments of the 12 Saxifragaceae species were performed in MAFFT v7.017 (Katoh and Standley, 2013) under standard parameters, and visually inspected and manually adjusted in GENEIOUS v8.1.7 (Kearse et al., 2012). The sequence identity of the 12 Saxifragaceae plastomes was plotted using the mVISTA program with LAGAN mode (Frazer et al., 2004). Plastid DNA rearrangement analyses of the 12 Saxifragaceae plastomes were performed via whole genome alignment in Mauve v2.3.1 (Darling et al., 2004).
To screen variable characters within the genus Tiarella, multiple alignments of the seven plastomes of Tiarella individuals were carried out using MAFFT v7.017. The average number of nucleotide different (K) and total number of mutations (Eta) was determined to analyze nucleotide diversity (π) using DnaSP v5.0 (Librado and Rozas, 2009).
In addition, the software CandiSSR v20170602 (Xia et al., 2015) was used to identify candidate polymorphic nSSRs within the genus Tiarella based on multiple assembled sequences. Plastid and mitochondrial contigs of the Tiarella species were removed from the assembled sequences using NCBI BLAST (v2.2.31) with the plastid (KR478645) and mitochondrial (KR559021) genomes of H. parviflora as references. The parameters implemented in CandiSSR are as follows: the flanking sequence length of 100, blast e-value cutoff of 1e-10, blast identity cutoff of 95, blast coverage cutoff of 95. Primers were automatically designed in the pipeline based on the Primer3 package (Untergasser et al., 2012) for each target SSRs.
To assemble the nrITS and nrETS sequences of all the accessions (including outgroup, in total nine species, 14 individuals), we downloaded the nrITS and nrETS sequences of H. parviflora (GenBank accession number: KM496213 for nrITS and KM496002 for nrETS) from GenBank as references. The assembled contigs were mapped to the reference sequences using BLAST search, and the mapped contigs with the highest score were aligned with the reference sequences again. Then, nrITS and nrETS sequences for all the accessions were extracted from the alignment files. To validate the accuracy of the nrITS and nrETS sequences we acquired, the ITS and ETS sequences from each Tiarella species were verified by PCR amplification and Sanger sequencing using the newly developed specific primers (nrITS F: CTCGCCGTT ACTAAGGGAATC, R: CGTAACAAGGTTTCCGTAGGTG; nrETS F: AGCCATTCGCAGTTTCACAG, R: TGGGACC TTGTGCTACACTTTG).
For phylogenetic inference, Maximum likelihood (ML) and Bayesian inference (BI) analyses were performed for the three different datasets: (1) complete plastome sequences; (2) a set of 77 protein coding sequences (CDS) shared by the plastomes (3) nrDNA sequence data including ITS and ETS. For plastid data, the ingroup included 81 accessions from the genera Tiarella, Heuchera, Bensoniella, and Mitella. These sequences included 11 accessions of seven taxa sequenced in this study, one accession downloaded from GenBank (H. parviflora, the GenBank accession number: KR478645), and the alignment data file containing 69 plastomes from Dryad (doi: https://doi.org/10.5061/dryad.cd546; published by Folk et al., 2017). For nrDNA data, the ingroup included 65 accessions from the same four genera, which consisted of 11 accessions sequenced in this study, as well as other 54 accessions downloaded from GenBank (Supplementary Table S2). The species used in the plastid and nrDNA datasets were kept largely consistent, except for some species for which nrDNA data were not available. One individual of M. rossii and two individuals of O. rupifraga were selected as outgroups based on Deng et al. (2015).
First, we used PARTITIONFINDER v2.1.1 (Lanfear et al., 2017) to determine the optimal data partition scheme and nucleotide substitution models for the two alignments according to the Akaike information criterion (AICc), resulting in three partitions for the 77 CDS regions (the first, second and third codon positions) and four partitions for nrDNA (ETS, ITS1, 5.8s rRNA and ITS2) (Table 1). The software jModelTest v2.1.4 (Posada, 2008) was used to determine the best-fit nucleotide substitution model (GTR + I + G) for the complete plastome sequences. ML analyses were implemented in RAxML-HPC v8.1.11 on the CIPRES cluster (Miller et al., 2010) using the optimal partitioning scheme and substitution model. 1000 bootstrap iterations were conducted with other parameters using the default settings. BI analyses were performed in MrBayes v3.2.3 (Ronquist and Huelsenbeck, 2003) using the same model selection criteria for two datasets. The Markov chain Monte Carlo (MCMC) algorithm was run with two independent chains with a random starting tree and default priors for 5,000,000 generations, with every 1000 generations for trees sampling. Convergence of the MCMC chains was assumed when the average standard deviation of split frequencies reached 0.01 or less.
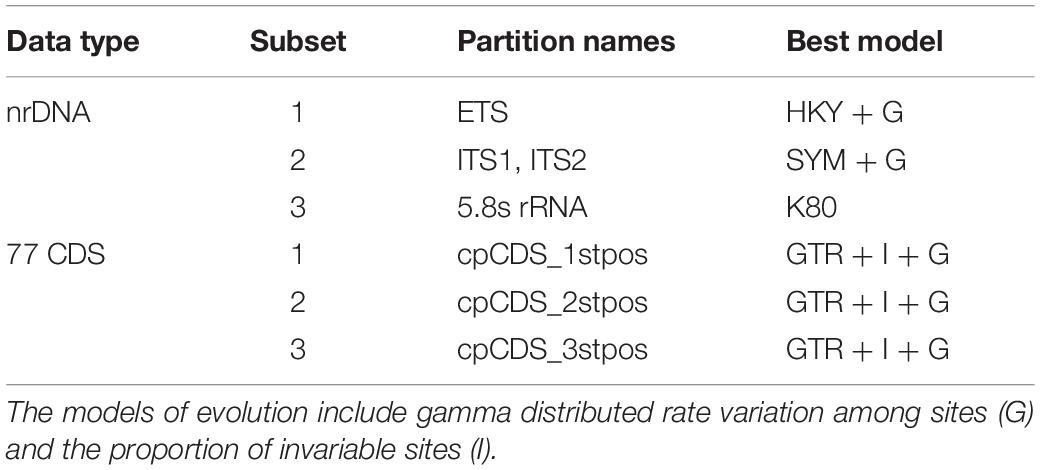
Table 1. Best partitioning scheme and nucleotide substitution models for nrDNA and 77 CDS data determined by PartitionFinder v2.1.1.
We generated paired-end clean reads ranging from 15,431,294 for T. polyphylla-NPSG to 49,735,470 for M. diphylla, respectively (Table 2). After filtering by quality with average Phred score < 30, the average length of remaining paired-end reads ranged from 100.43 bp (T. polyphylla-NPKB) to 135.53 bp (H. villosa), and the number of reads were between 13,331,090 (T. polyphylla-NPSG) and 44,127,123 (M. diphylla). The number of de novo assembled contigs from genome skimming data ranged from 315,998 (T. trifoliata) to 562,337 (M. diphylla). Each draft plastome of all the 11 accessions was generated from three initial contigs corresponding to the LSC, SSC and IRB/IRA regions, with no gaps or undetermined sites. The nucleotide coverage at each site for final plastome reconstruction was ranged from 11 (T. polyphylla-NPSG) to 5996 (H. richardsonii), and the average nucleotide coverage was ranged from 159 (T. polyphylla-NPSG) to 4682 (H. richardsonii), respectively (Table 2). Eventually, we found that each draft plastome sequence was absolutely identical with the corresponding final genome sequence.
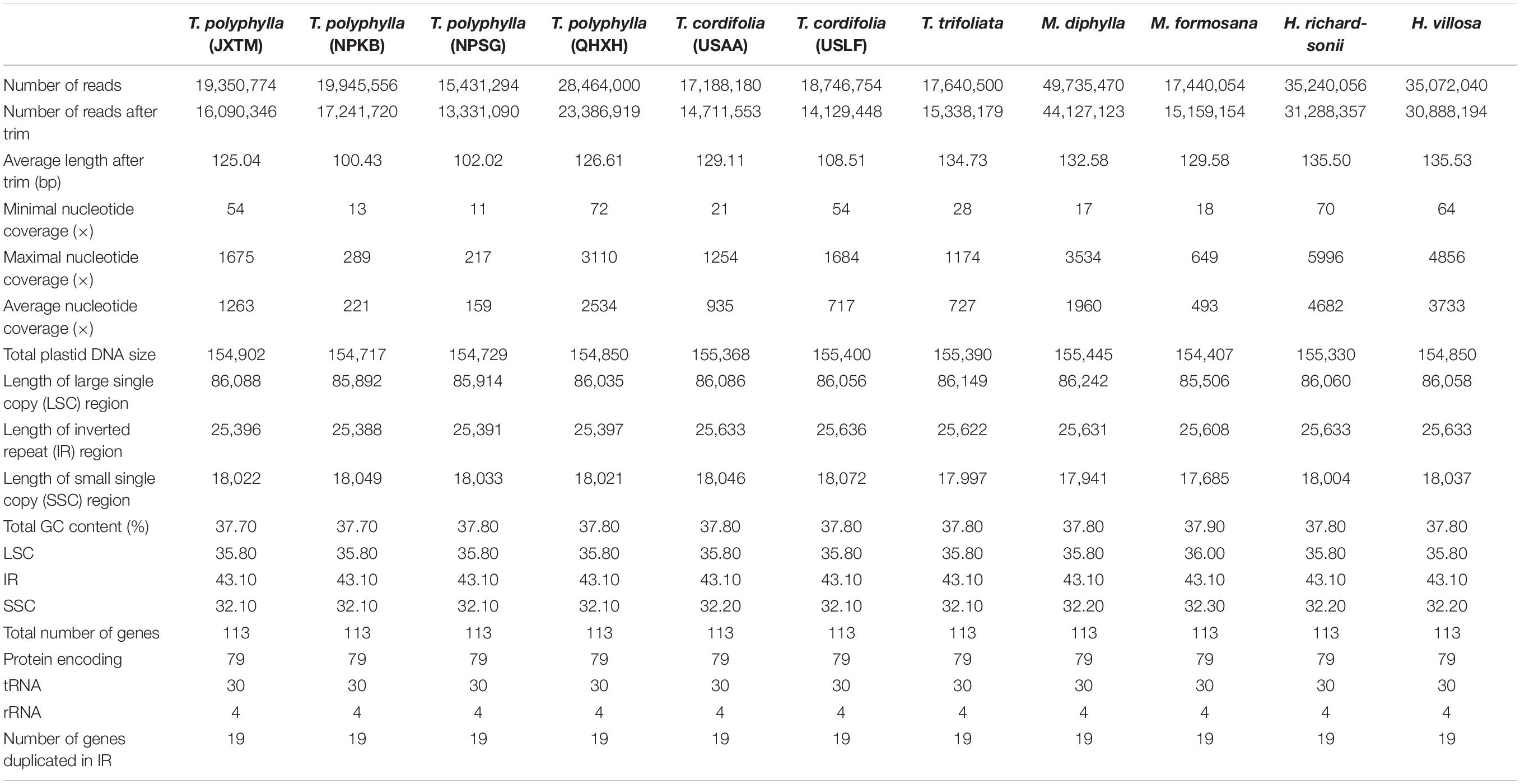
Table 2. Summary of the 11 individuals from the Heuchera group sequenced in this study.
All the plastomes sequenced in this study had a standard angiosperm structure comprising two copies of the IR region (25,388–25,636 bp) separated by the LSC region (85,506–86,242 bp) and SSC region (17,685–18,072 bp; Figure 1 and Table 2). For Tiarella species, the complete plastomes ranged from 154,717 bp in T. polyphylla-NPKB to 155,400 bp in T. cordifolia-USLF (Table 2). The overall GC content ranged narrowly from 37.70% to 37.80%, whereas the GC content in the LSC, SSC and IR regions were 35.80%, 32.10–32.20%, and 43.10%, respectively. For Heuchera and Mitella species, the complete plastomes ranged from 154,407 bp in M. formosana to 155,330 bp in H. richardsonii (Table 2). The overall GC content ranged from 37.80% to 37.90%, whereas the GC content in the LSC, SSC, and IR regions were 35.80–36.00%, 32.20–32.30%, and 43.10%, respectively.
The plastomes of all 11 accessions encoded an identical set of 132 genes, of which 113 were unique and 19 were duplicated in the IR regions (Table 2). Among the 113 unique genes, there were 79 protein-coding genes, 30 tRNA genes and four rRNA genes (Table 3). Six tRNA genes and eight protein-coding genes contained single intron, and three genes including rps12, clpP, and ycf3 contained two introns. The 5′-end exon of the rps12 gene was located in the LSC region, and the intron and 3′-end exon of the gene were situated in the IR region.
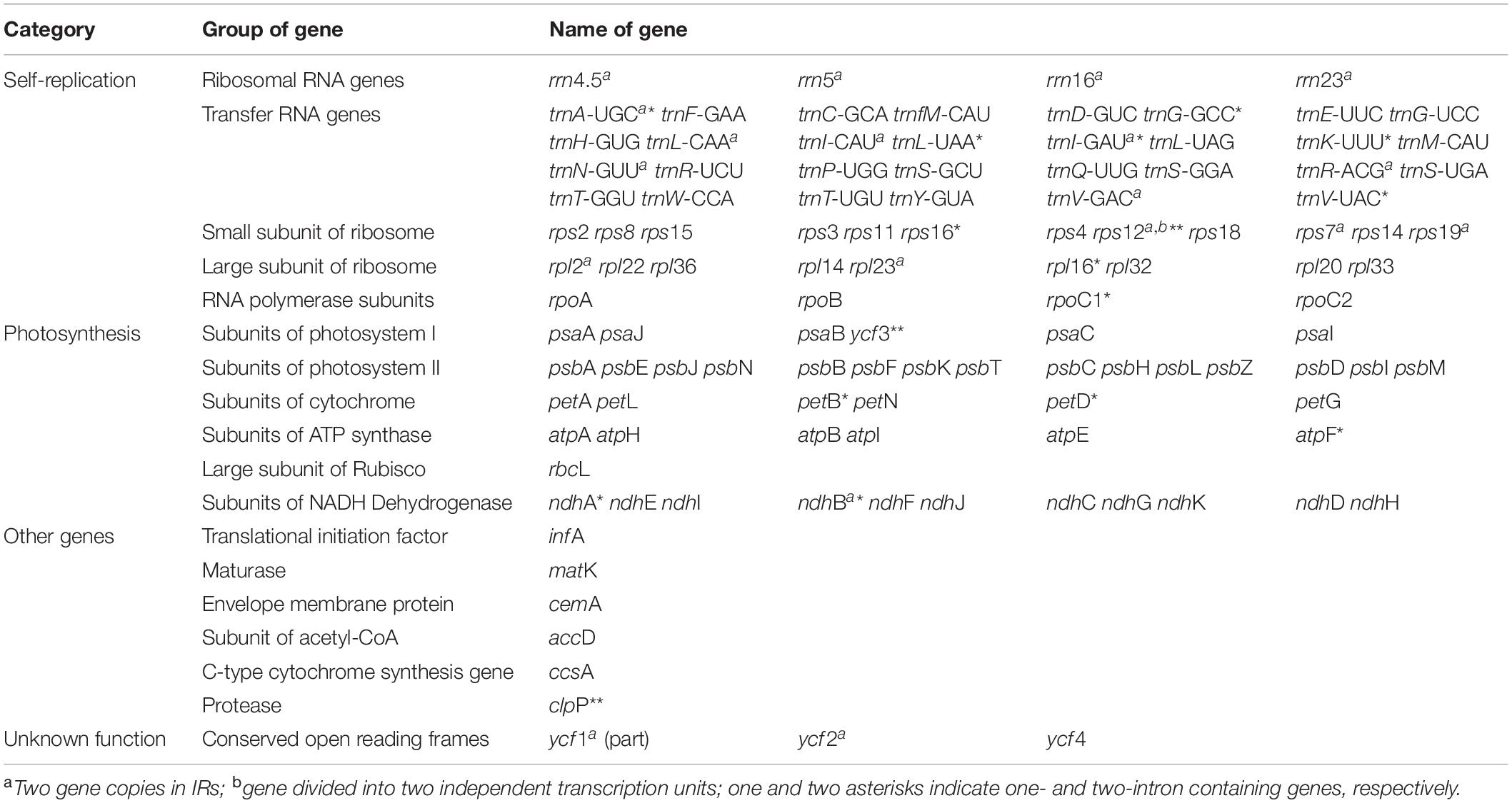
Table 3. Genes contained in Saxifragaceae plastomes (113 genes in total).
There were seventeen complete Saxifragaceae plastome sequences available in GenBank, including the 11 genomes sequenced in this study. We selected 12 of them representing different species to compare their structural organization. The 12 Saxifragaceae plastomes exhibited high levels of sequence similarity and structural conservation (Figure 2), and no rearrangement occurred in gene organization after verification. IR regions were more conservative than the LSC and SSC regions (Supplementary Figure S1).

Figure 2. Mauve alignment of 12 Saxifragaceae plastomes. Within each of the alignment, local collinear blocks are represented by blocks of the same color connected by lines.
Among the 12 Saxifragaceae species, the plastome size ranged from 150,752 to 156,960 bp in length. S. stolonifera exhibited the smallest genome size, while M. rossii had the largest genome size. The plastome size was typically between 154,000 bp and 157,000 bp. Size variation in Saxifragaceae plastomes is attributable to the length differences of LSC, SSC, and IRs regions (Figure 3). We compared the exact IR border positions and their adjacent genes between the Saxifragaceae plastomes (Figure 3). The genes rps19-rpl2-trnH and ycf1-ndhF were located in the junctions of LSC/IR and SSC/IR regions. The ycf1 gene spanned the SSC/IRA region and the pseudogene fragment of ψ ycf1 varies from 1156 to 1330 bp. The ndhF gene shared four nucleotides with the ψ ycf1 in T. polyphylla, T. cordifolia-USAA, three Heuchera and two Mitella species, but was separated from ψ ycf1 by a spacer with the length ranging from 12 to 57 bp in the rest species. The rps19 gene did not extend to the IRB region in M. rossii and O. rupifraga, but crossed the LSC/IRB region with 62 bp located at the IRB region in the other Saxifragaceae species. The rpl2 gene was separated from the LSC/IRB border by a spacer varied from 72 to 135 bp, as well as the trnH gene was separated from the IRA/LSC border by a spacer varied from 1 to 65 bp.
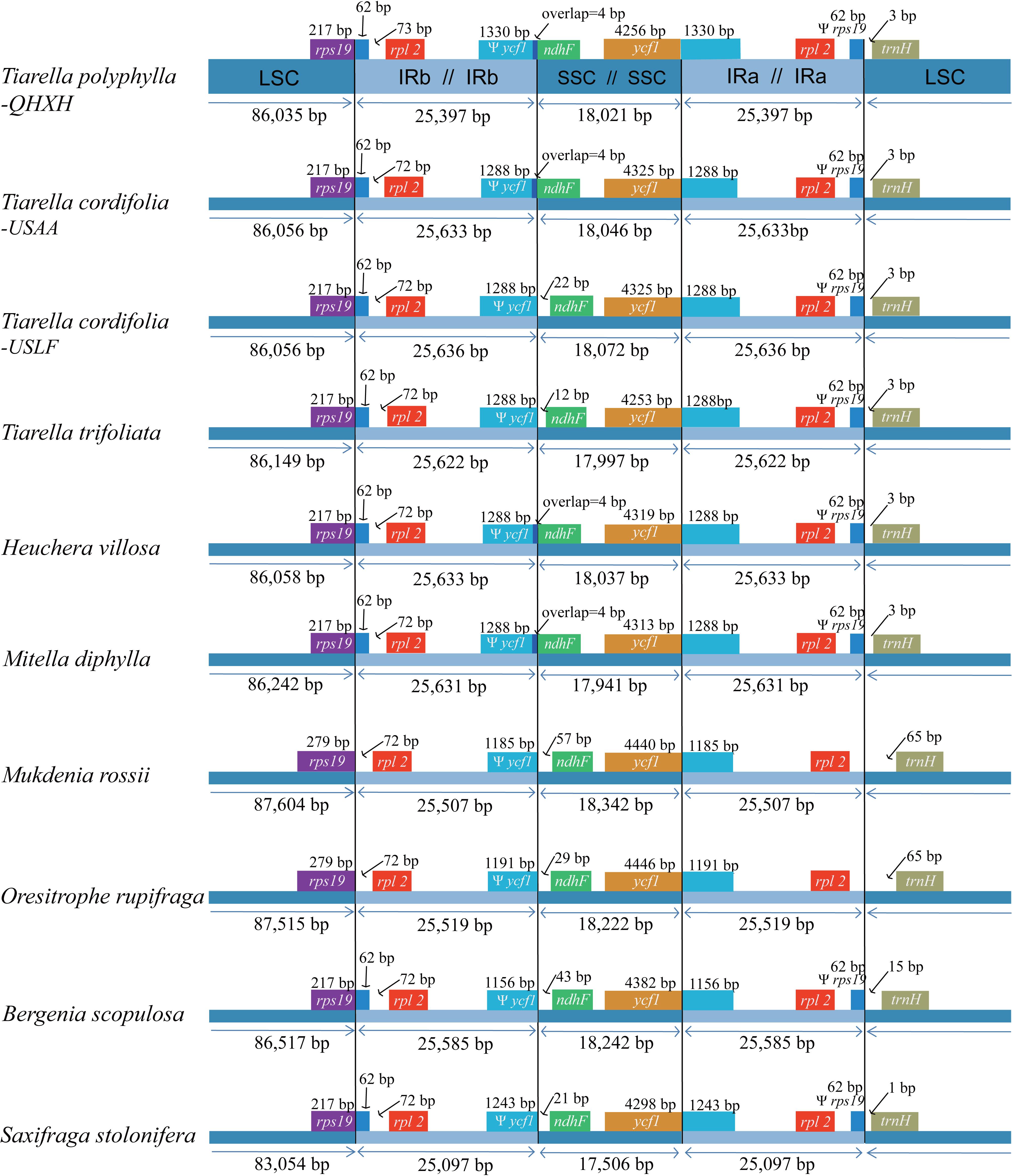
Figure 3. Comparison of boundaries of the large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions among the ten Saxifragaceae plastomes. We show only Heuchera villosa and Mitella diphylla as representative of identical boundary types in these genera. For the location of the two inverted repeat regions (IRA and IRB), refer to Figure 1.
The ycf15 gene, which was represented only by a small open reading frame (ORF), appeared to have been lost from all species in the family via pseudogenization, while the infA gene, commonly lost across the angiosperms, was always present. Likewise, the rpl2 gene was missing its single intron in all Saxifragaceae species investigated here. Therefore, the loss of ycf15 and the rpl2 intron may represent two synapomorphies for Saxifragaceae (Downie et al., 1991; Dong et al., 2013).
For the entire plastome, the IR regions of Saxifragaceae plastomes (nucleotide diversity, π = 0.00024) showed more conserved than either the LSC (π = 0.00237) or the SSC (π = 0.00387) regions. We compared coding genes, non-coding regions and intron regions among seven individuals of the three Tiarella species to find divergence hotspots, generating 119 loci (46 coding genes, 61 intergenic spacers, and 12 intron regions) (Figure 4). For the 119 regions, the π value for each locus ranged from 0.00042 (psbC) to 0.01667 (rpl32-trnL). We found nine highly divergent regions (π > 0.01): psbI-trnS (0.01524), trnR-atpA (0.01045), petN-psbM (0.0101), psbC-trnS (0.01306), trnT-trnL (0.01171), trnL-trnF (0.01157), petG-trnW (0.01633), rpl32-trnL (0.01667), and ndhD-psaC (0.01011). These levels of intrageneric variation suggested the suitability of these regions as markers for the genus Tiarella.
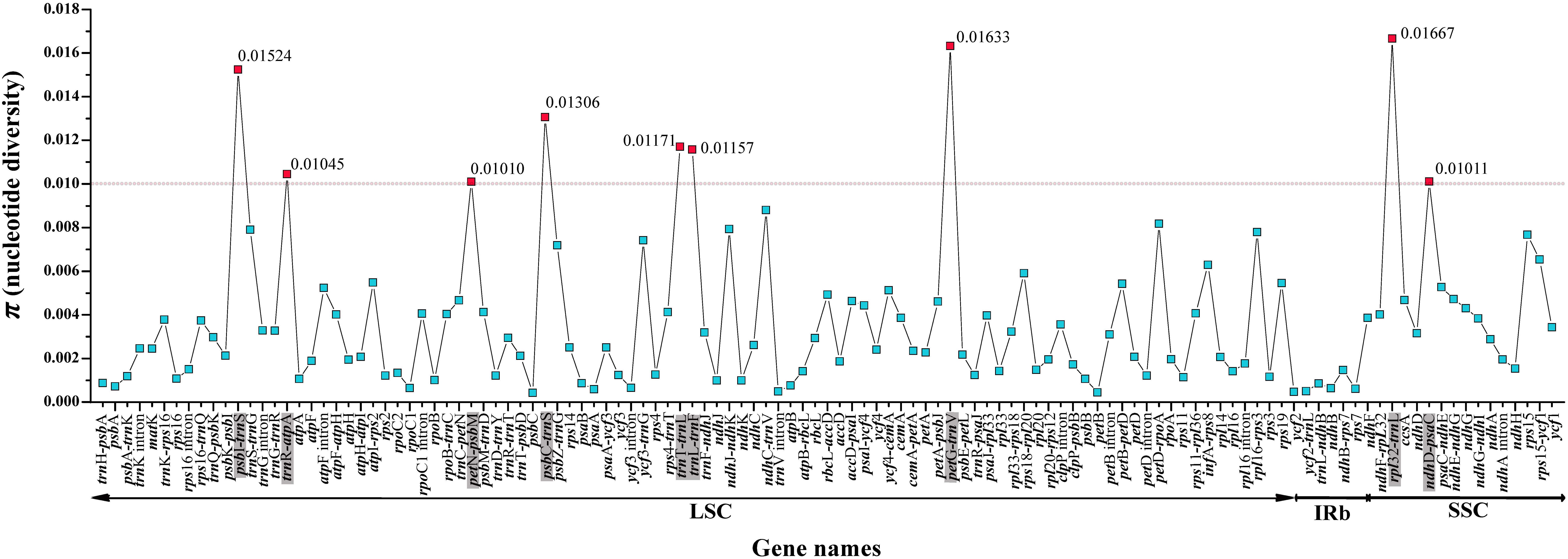
Figure 4. Comparative analysis of the nucleotide variability (π) values within the genus Tiarella. The dotted line represents π > 0.01; genes surpassing this threshold are highlighted in gray.
A total of 2,600 candidate polymorphic nSSRs were identified within the genus Tiarella. After removing the loci with the sequence similarity <90% (211) and no available primers designed (589), we obtained 1,800 polymorphic nSSRs with the standard deviation ranged from 0.40 to 4.97 for this genus (Supplementary Table S2). Among them, di- (1,009), tri- (722), tetra- (43), penta- (17), and hexanucleotides (9) accounted for 56.06%, 40.11%, 2.39%, 0.94%, and 0.50%, respectively (Figure 5). With regard to these nSSRs, GA (158) of di-, GAA (36) of tri-, TTTG (5) of tetra-, AAAAG (4) of penta-, and ATATAC (2) of hexanucleotides occupied the predominant proportion.
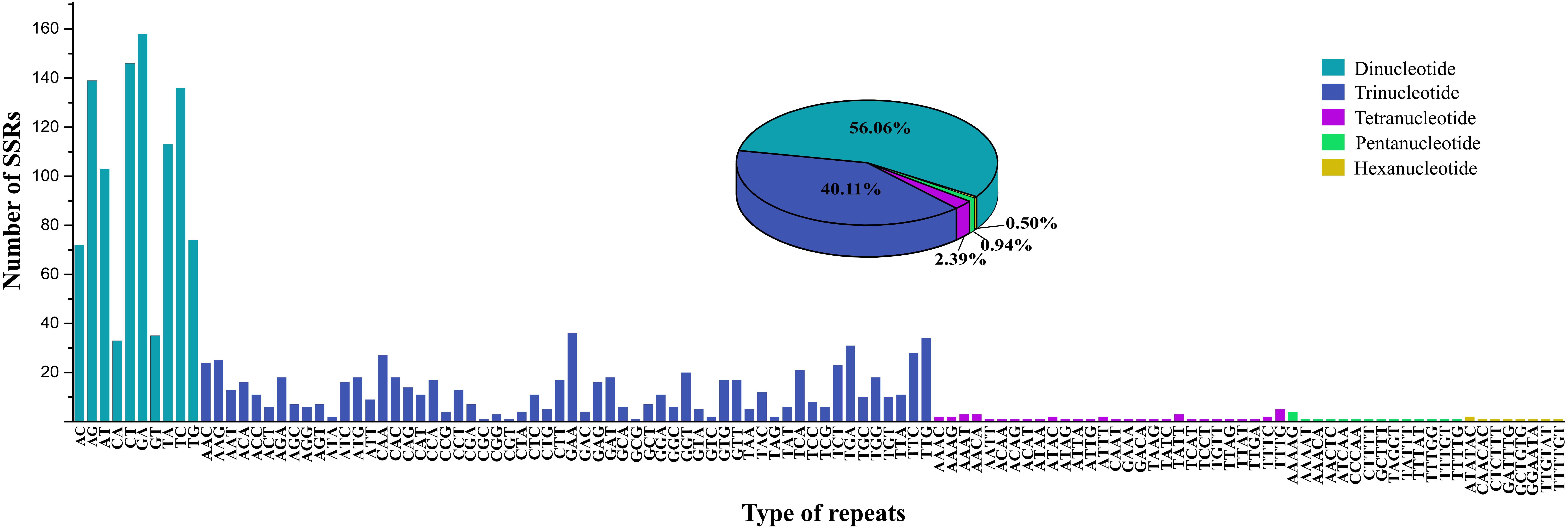
Figure 5. The distribution information of the polymorphic nuclear simple sequence repeats (nSSRs) within the genus Tiarella. The pie chart shows the overall prevalence of repeat lengths; the histogram shows the frequency distribution of prevalent repeat classes.
For the nuclear dataset, we obtained the nrITS and nrETS regions of the fourteen species sequenced here and supplemented this matrix with data from GenBank (primarily from Okuyama et al., 2008; Folk and Freudenstein, 2014). The nrITS regions newly sequenced in this study ranged from 631 bp in T. polyphylla-NPKB to 656 bp in M. diphylla. The length of the nrETS region varied slightly from 457 bp in O. rupifraga to 466 bp in H. richardsonii and M. diphylla. These sequences have been deposited in GenBank (Supplementary Table S3). Sequences for nrITS and nrETS regions of Tiarella obtained using Sanger sequencing are identical to those we obtained from genome skimming data. The concatenated matrix of nrITS and nrETS comprised 68 taxa with an aligned length of 1,078 bp. The aligned lengths of the complete plastome and 77-CDS datasets (in total 84 individuals) was 163,155 bp and 67,806 bp, respectively.
For the analyses with nrDNA data, the tree topologies recovered from ML and BI analyses were congruent with one another (Figure 6). The monophyly of Tiarella had maximal support (reported hereafter as ML bootstrap support (BS)/BI posterior probability (PP); BS/PP = 100/1). Within Tiarella, the four individuals of T. polyphylla (BS/PP = 100/1) and two individuals of T. cordifolia (BS/PP = 98/1) were resolved as reciprocally monophyletic, and we obtained decisive support for the topology [(T. polyphylla, T. trifoliata), T. cordifolia]. Heuchera was also monophyletic with full support in BI analysis (PP = 1) when including the monotypic genus Bensoniella (to date this placement of Bensoniella has been seen only in ribosomal DNA data; Okuyama et al., 2008; Folk and Freudenstein, 2014). In addition, the three Mitella species investigated were found to be polyphyletic. Of them, Mitella stauropetala Piper was sister to the Tiarella clade with high support (BS/PP = 98/0.97), while the other two Mitella species were clustered with the Heuchera clade. Mitella formosana was weakly supported to be sister to the Heuchera clade (BS/PP < 50/0.5), followed by M. diphylla, which is subsequently sister to the above clade (BS < 50%, PP = 0.99).
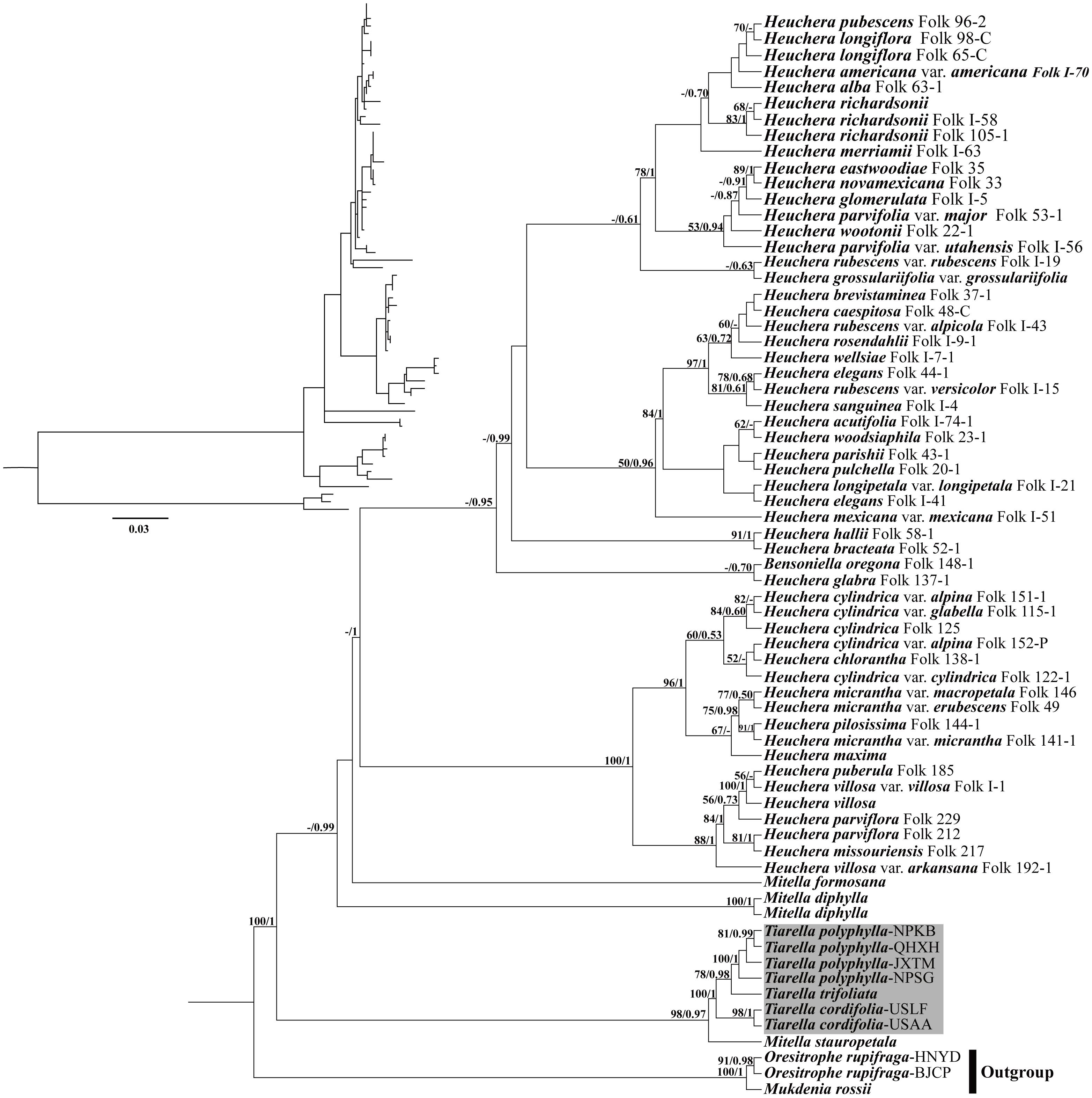
Figure 6. Phylogenetic tree reconstruction using maximum likelihood (ML) based on ITS + ETS sequences. The inset topology in the upper left shows the relative branch lengths in per-site substitutions. Numbers above the branches represent ML bootstrap/Bayesian posterior probability (BS/PP). Hyphens indicate a bootstrap value or posterior probability <50%.
For the analyses of plastid data, the ML and BI trees were identical in topology within each data matrix for the plastomes (Figure 7) and the 77 CDS regions (Supplementary Figure S2). Between datasets, the results were generally consistent; support values were generally greater in the plastid tree than for the 77-CDS tree, probably due to the inclusion of more informative loci. However, the results based on plastid data are strongly incongruent with those based on nrDNA data. The Heuchera species were divided into three major clades (labeled clades A, B, C in Figure 7), all with full support (BS/PP = 100/1). M. stauropetala and the two T. cordifolia individuals were nested deeply within Heuchera plastid Clade A. The other two Tiarella species formed a clade sister to Heuchera plastid Clade A; they subsequently formed a clade with M. pentandra Hook. and M. formosana. Clade B entirely comprised most of the remaining Heuchera species, including 26 accessions from 20 species. For the third Heuchera plastid clade (Clade C), four Heuchera species (five accessions) formed a strongly supported clade (BS/PP = 100/1) sister to M. diphylla. Hence, while monophyletic for nuclear data, plastid data suggest polyphyly of Tiarella as well as other genera in the Heuchera group.

Figure 7. Phylogenetic tree reconstruction using maximum likelihood (ML) based on whole plastome sequences. The inset topology in the upper left shows the relative branch lengths in per-site substitutions. Numbers above the branches represent ML bootstrap/Bayesian posterior probability (BS/PP). Hyphen indicate a bootstrap value or posterior probability <50%. Clade labels follow those for chloroplast clades recovered in Folk et al. (2017). Gray highlighted labels indicate the position of Tiarella plastomes.
We recovered multiple instances of strong incongruence between our nuclear and plastid phylogenies. The genera Tiarella and Heuchera together with Bensoniella were monophyletic in nrDNA data. However, both Heuchera and Tiarella were clearly polyphyletic in plastid data. Evolutionary processes including convergent evolution, lineage sorting and reticulate evolution could explain discordances between nuclear and plastid phylogenies (Acosta and Premoli, 2010). However, in this case, the probability of generating sequence convergence for an entire plastome is low given the size of the plastid genome and that the sampling locations of the taxa included here are widely geographically distant. Incomplete lineage sorting may likewise be excluded for the following reasons: (1) the plastid DNA in the Heuchera group is haploid and maternally inherited (Soltis et al., 1990), and hence the effective population size of plastid loci is generally one-quarter of that of nuclear loci (Birky et al., 1983), which means stochastic lineage sorting of an organelle locus is four times faster than that of a nuclear locus (Moore, 1995); and (2) previous application of a coalescent method on the Heuchera group demonstrated that incomplete lineage sorting alone does not adequately explain the plastid phylogeny of this group (Folk et al., 2017).
Plastid capture is an evolutionary process through which inter-species hybridization (commonly with subsequent backcrosses) yields a lineage with a novel genomic combination, where the plastome of one species occurs in the nuclear background of another species (Rieseberg and Soltis, 1991). Plastid introgression is generally thought to be more prevalent than nuclear introgression due to its characteristic inheritance, its usually complete lack of recombination, and the low influence of selection on its conserved housekeeping loci (Martinsen et al., 2001; Avise, 2004). Plastid capture, generally attributed to introgression, has been reported in many plant taxa (Tsitrone et al., 2003; Fehrer et al., 2007; Acosta and Premoli, 2010). These evolutionary events have often been accompanied by a clear absence of introgression of biparentally inherited nuclear genes (Soltis and Soltis, 1995; Fehrer et al., 2007), even in broad surveys of genome loci (Folk et al., 2017).
The Heuchera group has been cited frequently as a particularly valuable system for studying plastid capture (Folk et al., 2017 and citations therein). Several examples of plastid capture have been documented in this group using cpDNA restriction-site data together with allozyme data (Soltis, 1991), suggesting plastid capture between species of Heuchera, as well as between Tellima and Mitella (Soltis, 1991; Soltis et al., 1991). Significantly, Tiarella and some accessions of Heuchera are not only very similar in their cpDNAs, but they also hybridize frequently (Spongberg, 1972). One intergeneric hybrid, known in the nursery trade as “×Heucherella” (= Heuchera × Tiarella), is a common ornamental plant. Previous studies likewise revealed the possibility of ancient hybridization (that is, hybridization events ancestral to extant species) and subsequent plastid transfer between Heuchera and Tiarella (Soltis et al., 1991; Soltis and Kuzoff, 1995; Folk et al., 2017).
The most striking difference between our nuclear and plastid trees involves the phylogenetic position of Heuchera and Tiarella (Figures 6, 7). In our nrDNA phylogeny (Figure 6), Heuchera was monophyletic when including the monotypic genus Bensoniella (BS/PP = -/1). This is consistent with Okuyama et al. (2008) from which we got many of our ITS + ETS sequences. However, using sequences from six nuclear loci (including ITS, ETS and four single-copy nuclear genes, GBSSI-A, GBSSI-B, GS-II, PepCK) and 39 morphological characters, Folk and Freudenstein (2014) confidently supported the monophyly of Heuchera with concatenation and coalescent analyses. The monophyly of Heuchera was further confirmed by a subsequent study (Folk et al., 2017), which employed a targeted enrichment approach to generate a ∼400,000 bp dataset of 277 low-copy nuclear loci. Tiarella was well supported as monophyletic and a distant relative of Heuchera in our nuclear phylogeny (Figure 6), which is also proved by previous studies (Okuyama et al., 2008, 2012; Folk and Freudenstein, 2014; Folk et al., 2017). Our findings, in combination with previous work, strongly suggest that Heuchera and Tiarella are both monophyletic and not closely related based on evidence from the nuclear genome. Plastid DNA, by contrast, confidently resolves Tiarella species as polyphyletic with more derived positions (Figure 7 and Supplementary Figure S2). T. polyphylla and T. trifoliata formed a clade sister to Clade A, whereas the two T. cordifolia individuals were embedded within in Clade A. Reconciling the discordance between plastid and nuclear phylogenies, particularly among Tiarella and Heuchera species, requires at least two plastid capture events to explain the incongruences we observed, one to explain the more derived position of T. polyphylla and T. trifoliata in the plastid phylogeny as compared to nuclear data, and a second to explain the embedded position of T. cordifolia among Heuchera species. We hypothesize that the ancestor of the genus Tiarella initially captured the plastid of a Heuchera lineage (most likely an ancestral member of Clade A) through an ancient hybridization. Subsequently, T. cordifolia captured the plastid genome from a member, again likely ancestral, of Heuchera plastid Clade A. Although we have improved sampling at the population level and among outgroups, our results are congruent with previous work by Folk et al. (2017). Interestingly, a single species of Heuchera had two widely different plastid phylogenetic placements, indicating the prevalence of chloroplast capture in the group. Two accessions of H. richardsonii formed a clade embedded in the second Heuchera clade (Clade B, as seen previously), yet the third individual was embedded in the first Heuchera clade (Clade A), representing a putatively novel placement. However, this individual may correspond to a cryptic form of the naturally occurring hybrid H. richardsonii × H. americana L. examined in previous work and also found in chloroplast clade A (Folk et al., 2017; see also Rosendahl et al., 1936). Finally, the Mitella species we sampled in phylogenetic analysis were polyphyletic in both nuclear and chloroplast trees, in agreement with previous studies (Okuyama et al., 2008, 2012; Deng et al., 2015; Folk et al., 2017). However, M. stauropetala, was recovered as sister to Tiarella in nrDNA data, but was embedded in the first Heuchera clade (Clade A) in plastid phylogenies. This inconsistency, as observed in previous work, is also most likely due to the presence of plastid capture.
In this study, seven species of Saxifragaceae were selected for genome skimming and assembly of complete plastomes, nrITS and nrETS sequences. All the plastomes we sequenced ranged from 154,407 to 155,400 bp in length, encoding 113 identical genes including 79 protein-coding genes, 30 tRNA genes and four rRNA genes. Comparative analysis of plastomes with other Saxifragaceae species revealed that there no rearrangements occurred in gene organization, size variations of the plastomes are purely ascribed to the length differences of LSC, SSC, and IRs regions. Phylogenetic analyses inferred by nuclear and plastid data fully resolved the phylogeny of Tiarella, the topology of [(T. polyphylla, T. trifoliata), T. cordifolia] was favored with maximal support (BS = 100%, PP = 1.00). Comparisons of nuclear and plastid phylogenies revealed that plastid capture events have occurred multiple times among species and genera of the Heuchera group through ancient hybridization. Furthermore, molecular markers, including plastid hotspots and nuclear polymorphic nSSRs, were efficiently generated for the genus Tiarella, for future population genetics and phylogeographic studies.
The datasets generated for this study can be found in the all the plastome, ETS and ITS sequences sequenced in this study were deposited in GenBank, and the corresponding accession numbers are presented in the manuscript.
L-XL, F-DS, and PL conceived the ideas. L-XL and PL contributed to the sampling. L-XL and Y-XD performed the experiment. L-XL and S-YW analyzed the data. The manuscript was written and improved by L-XL, RF, DS, and PL.
This research was supported by the National Natural Science Foundation of China (Grant Nos. 31900188 and 31970225) and Natural Science Foundation of Zhejiang Province (Grant No. LY19C030007).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00361/full#supplementary-material
FIGURE S1 | Visualization of alignment of 17 Saxifragaceae chloroplast genome sequences with Saxifraga stolonifera as the reference. The horizontal axis indicates the coordinates within the chloroplast genome. The vertical scale indicates the percentage of identity, ranging from 50 to 100%. Genome regions are color codes as protein coding, intron, mRNA, and conserved non-coding sequences (CNS).
FIGURE S2 | Phylogenetic tree reconstruction using maximum likelihood (ML) based on 77 shared protein-coding genes. The inset topology in the upper left shows the relative branch lengths in per-site substitutions. Numbers above the branches represent ML bootstrap/Bayesian posterior probability (BS/PP). Hyphen indicate a bootstrap value or posterior probability <50%.
TABLE S1 | Locality and voucher information for seven Saxifrageae species including 11 individuals used in this study. Voucher specimens are deposited at the herbarium of Zhejiang University (HZU), Hangzhou, Zhejiang, China.
TABLE S2 | GenBank number for ETS and ITS sequences used in this study. Asterisk indicates the newly generated sequences.
TABLE S3 | The detail information of polymorphic nSSRs identified and primer pairs designed for each detected loci within Tiarella.
Acosta, M. C., and Premoli, A. C. (2010). Evidence of chloroplast capture in South American Nothofagus (subgenus Nothofagus, Nothofagaceae). Mol. Phylogenet. Evol. 54, 235–242. doi: 10.1016/j.ympev.2009.08.008
Avise, J. C. (2004). Molecular Markers, Natural History and Evolution, Second Edn. Sunderland, MA: Sinauer.
Bai, G. Q., Fang, L. Y., Li, S. F., and Cui, X. (2018). Characterization of the complete chloroplast genome sequence of Bergenia scopulosa (Saxifragales: Saxifragaceae). Conserv. Genet. Resour. 10, 363–366. doi: 10.1007/s12686-017-0825-y
Besnard, G., Christin, P. A., Malé, P. J. G., Coissac, E., Ralimanana, H., and Vorontsova, M. S. (2013). Phylogenomics and taxonomy of Lecomtelleae (Poaceae), an isolated panicoid lineage from Madagascar. Ann. Bot. 112, 1057–1066. doi: 10.1093/aob/mct174
Birky, C. W., Maruyama, T., and Fuerst, P. (1983). An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics 103, 513–527.
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Deng, J. B., Drew, B. T., Mavrodiev, E. V., Gitzendanner, M. A., Soltis, P. S., and Soltis, D. E. (2015). Phylogeny, divergence times, and historical biogeography of the angiosperm family Saxifragaceae. Mol. Phylogenet. Evol. 83, 86–98. doi: 10.1016/j.ympev.2014.11.011
Dodsworth, S. (2015). Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 20, 525–527. doi: 10.1016/j.tplants.2015.06.012
Dong, W., Xu, C., Cheng, T., and Zhou, S. (2013). Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS One 8:e77965. doi: 10.1371/journal.pone.0077965
Dong, W. P., Xu, C., Wu, P., Cheng, T., Yu, J., and Zhou, S. L. (2018). Resolving the systematic positions of enigmatic taxa: manipulating the chloroplast genome data of Saxifragales. Mol. Phylogenet. Evol. 126, 321–330. doi: 10.1016/j.ympev.2018.04.033
Downie, S. R., Olmstead, R. G., Zurawski, G., Soltis, D. E., and Soltis, P. S. (1991). Six independent losses of the chloroplast DNA rpl2 intron in dicotyledons: molecular and phylogenetic implications. Evolution 45, 1245–1259. doi: 10.1111/j.1558-5646.1991.tb04390.x
Engler, A. (1930). “Saxifragaceae,” in Die Naturlichen Flanzenfamilien, Second Edn, Vol. 18a, eds K. Engler and K. Prantl (Berlin: Wilhelm Engelmann), 74–226.
Fehrer, J., Gemeinholzer, B., Chrtek, J. Jr., and Bräutigam, S. (2007). Incongruent plastid and nuclear DNA phylogenies reveal ancient intergeneric hybridization in Pilosella hawkweeds (Hieracium, Cichorieae, Asteraceae). Mol. Phylogenet. Evol. 42, 347–361. doi: 10.1016/j.ympev.2006.07.004
Folk, R. A., and Freudenstein, J. V. (2014). Phylogenetic relationships and character evolution in Heuchera (Saxifragaceae) on the basis of multiple nuclear loci. Am. J. Bot. 101, 1532–1550. doi: 10.3732/ajb.1400290
Folk, R. A., Mandel, J. R., and Freudenstein, J. V. (2017). Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Syst. Biol. 66, 320–337. doi: 10.1093/sysbio/syw083
Folk, R. A., Mandel, J. R., and Freudenstein, J. V. A. (2015). A protocol for targeted enrichment of intron-containing sequence markers for recent radiations: a phylogenomics example from Heuchera (Saxifragaceae). Appl. Plant Sci. 3:1500039. doi: 10.3732/apps.1500039
Folk, R. A., Soltis, P. S., Soltis, D. E., and Guralnick, R. (2018). New prospects in the detection and comparative analysis of hybridization in the tree of life. Am. J. Bot. 105, 364–375. doi: 10.1002/ajb2.1018
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279.
Gurushidze, M., Fritsch, R. M., and Blattner, F. R. (2010). Species-level phylogeny of Allium subgenus Melanocrommyum: incomplete lineage sorting, hybridization and trnF gene duplication. Taxon 59, 829–840. doi: 10.1002/tax.593012
Joly, S., Mclenachan, P. A., and Lockhart, P. J. (2009). A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 174, E54–E70.
Kane, N., Sveinsson, S., Dempewolf, H., Yang, J. Y., Zhang, D., Engels, J. M., et al. (2012). Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 99, 320–329. doi: 10.3732/ajb.1100570
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stoneshavas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Li, P., Lu, R. S., Xu, W. Q., Ohitoma, T., Cai, M. Q., Qiu, Y. X., et al. (2017). Comparative genomics and phylogenomics of east asian tulips (Amana, Liliaceae). Front. Plant Sci. 8:451. doi: 10.3389/fpls.2017.00451
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, L.-X., Li, R., Worth, J. R. P., Li, X., Li, P., Cameron, K. M., et al. (2017). The complete chloroplast genome of Chinese bayberry (Morella rubra, Myricaceae): implications for understanding the evolution of fagales. Front. Plant Sci. 8:968. doi: 10.3389/fpls.2017.00968
Liu, L., Wang, Y., He, P., Li, P., Lee, J., Soltis, D. E., et al. (2018a). Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics 19:235. doi: 10.1186/s12864-018-4633-x
Liu, L., Zhang, C., Wang, Y., Dong, M., Shang, F., and Li, P. (2018b). The complete chloroplast genome of Caryopteris mongholica and phylogenetic implications in Lamiaceae. Conserv. Genet. Resour. 10, 281–285. doi: 10.1007/s12686-017-0802-5
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Malé, P. J., Bardon, L., Besnard, G., Coissac, E., Delsuc, F., Engel, J., et al. (2015). Genome skimming by shotgun sequencing helps resolve the phylogeny of a pantropical tree family. Mol. Ecol. Resour. 14, 966–975. doi: 10.1111/1755-0998.12246
Martinsen, G. D., Whitham, T. G., Turek, R. J., and Keim, P. (2001). Hybrid populations selectively filter gene introgression between species. Evolution 55, 1325–1335. doi: 10.1111/j.0014-3820.2001.tb00655.x
McKinnon, G. E., Vaillancourt, R. E., Jackson, H. D., and Potts, B. M. (2001). Chloroplast sharing in the Tasmanian eucalypts. Evolution 55, 703–711. doi: 10.1111/j.0014-3820.2001.tb00806.x
McPherson, H., Merwe, M. V. D., Delaney, S. K., Edwards, M. A., Henry, R. J., Mcintosh, E., et al. (2013). Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 13:8. doi: 10.1186/1472-6785-13-8
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), 2010, (New Orleans, LA: IEEE), 1–8.
Mir, C., Jarne, P., Sarda, V., Bonin, A., and Lumaret, R. (2010). Contrasting nuclear and cytoplasmic exchanges between phylogenetically distant oak species (Quercus suber L. and Q. ilex L.) in Southern France: inferring crosses and dynamics. Plant Biol. 11, 213–226. doi: 10.1111/j.1438-8677.2008.00106.x
Moore, W. S. (1995). Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution 49, 718–726. doi: 10.1111/j.1558-5646.1995.tb02308.x
Morgan, D. R., and Soltis, D. E. (1993). Phylogenetic relationships among members of Saxifragaceae sensu lato based on rbcL sequence data. Ann. Mol. Bot. Gard. 80, 631–660.
Okuyama, Y. (2015). Mitella amamiana sp. nov., the first discovery of the genus Mitella (Saxifragaceae) in the Central Ryukyus. Acta Phytotax. Geobot. 67, 17–27.
Okuyama, Y., Fujii, N., Wakabayashi, M., Kawakita, A., Ito, M., Watanabe, M., et al. (2005). Nonuniform concerted evolution and chloroplast capture: heterogeneity of observed introgression patterns in three molecular data partition phylogenies of Asian Mitella (Saxifragaceae). Mol. Biol. Evol. 22, 285–296. doi: 10.1093/molbev/msi016
Okuyama, Y., Pellmyr, O., and Kato, M. (2008). Parallel floral adaptations to pollination by fungus gnats within the genus Mitella (Saxifragaceae). Mol. Phylogenet. Evol. 46, 560–575. doi: 10.1016/j.ympev.2007.09.020
Okuyama, Y., Tanabe, A. S., and Kato, M. (2012). Entangling ancient allotetraploidization in Asian Mitella: an integrated approach for multilocus combinations. Mol. Biol. Evol. 29, 429–439. doi: 10.1093/molbev/msr236
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Rieseberg, L. H., and Soltis, D. E. (1991). Phylogenetic consequences of cytoplasmic gene flow in plants. Am. J. Bot. 5, 65–84.
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rosendahl, C. O., Butters, F. K., and Lakela, O. (1936). A Monograph on the Genus Heuchera. Minneapolis, MN: University of Minnesota Press.
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689.
Soltis, D. E. (1991). Chloroplast DNA variation within and among genera of the Heuchera group: evidence for extensive chloroplast capture and the paraphyly of Heuchera and Mitella. Am. J. Bot. 78, 1091–1112. doi: 10.1002/j.1537-2197.1991.tb14517.x
Soltis, D. E., and Bohm, B. A. (1985). Chromosomal and flavonoid chemical confirmation of intergeneric hybridization between Tolmiea and Tellima (Saxifragaceae). Can. J. Bot. 63, 1309–1312. doi: 10.1139/b85-182
Soltis, D. E., and Kuzoff, R. K. (1995). Discordance between nuclear and chloroplast phylogenies in the Heuchera group (Saxifragaceae). Evolution 49, 727–742. doi: 10.1111/j.1558-5646.1995.tb02309.x
Soltis, D. E., Kuzoff, R. K., Mort, M. E., Zanis, M., Fishbein, M., Hufford, L., et al. (2001). Elucidating deep-level phylogenetic relationships in Saxifragaceae using sequences for six chloroplastic and nuclear DNA regions. Ann. Mo. Bot. Gard. 88, 669–693.
Soltis, D. E., Mayer, M. S., Soltis, P. S., and Edgerton, M. (1991). Chloroplast-DNA variation in Tellima grandiflora (Saxifragaceae). Am. J. Bot. 78, 1379–1390. doi: 10.1002/j.1537-2197.1991.tb12604.x
Soltis, D. E., and Soltis, P. S. (1986). Intergeneric hybridization between Conimitella williamsii and Mitella stauropetala (Saxifragaceae). Syst. Bot. 11, 293–297.
Soltis, D. E., Soltis, P. S., and Ness, B. D. (1990). Maternal inheritance of the chloroplast genome in Heuchera and Tolmiea (Saxifragaceae). J. Hered. 81, 168–170.
Spongberg, S. A. (1972). The genera of Saxifragaceae in the Southeastern United States. J. Arnold Arboretum 53, 409–498.
Straub, S. C., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., and Liston, A. (2012). Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am. J. Bot. 99, 349–364. doi: 10.3732/ajb.1100335
Toews, D. P. L., and Brelsford, A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21, 3907–3930. doi: 10.1111/j.1365-294X.2012.05664.x
Tsitrone, A., Kirkpatrick, M., and Levin, D. A. (2003). A model for chloroplast capture. Evolution 57, 1776–1782. doi: 10.1111/j.0014-3820.2003.tb00585.x
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3-new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Wendel, J. F., and Doyle, J. J. (1998). “Phylogenetic incongruence: window into genome history and molecular evolution,” in Molecular Systematics of Plants II, eds D. E. Soltis, P. S. Soltis, and J. J. Doyle (Dordrecht: Kluwer Academic Publishers), 265–296. doi: 10.1007/978-1-4615-5419-6_10
Wu, Z., and Raven, P. (2001). Flora of China, Vol. 8: Brassicaceae Through Saxifragaceae. St. Louis, MI: Missouri Botanical Garden Press, 506.
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xia, E. H., Yao, Q. Y., Zhang, H. B., Jiang, J. J., Zhang, L. P., and Gao, L. Z. (2015). CandiSSR: an efficient pipeline used for identifying candidate polymorphic SSRs based on multiple assembled sequences. Front. Plant Sci. 6:1171. doi: 10.3389/fpls.2015.01171
Xiang, Q. Y., Soltis, D. E., and Soltis, P. S. (1998). The eastern Asian and eastern and western North American floristic disjunction: congruent phylogenetic patterns in seven diverse genera. Mol. Phylogenet. Evol. 10, 178–190. doi: 10.1006/mpev.1998.0524
Keywords: the Heuchera group, Saxifragaceae, plastome, phylogenomics, Heucherella
Citation: Liu L-X, Du Y-X, Folk RA, Wang S-Y, Soltis DE, Shang F-D and Li P (2020) Plastome Evolution in Saxifragaceae and Multiple Plastid Capture Events Involving Heuchera and Tiarella. Front. Plant Sci. 11:361. doi: 10.3389/fpls.2020.00361
Received: 20 September 2019; Accepted: 12 March 2020;
Published: 24 April 2020.
Edited by:
Nina Rønsted, National Tropical Botanical Garden, United StatesReviewed by:
Angela Jean McDonnell, Chicago Botanic Garden, United StatesCopyright © 2020 Liu, Du, Folk, Wang, Soltis, Shang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fu-De Shang, ZnVkZXNoYW5nQGhlbnUuZWR1LmNu; Pan Li, cGFubGlfemp1QDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.