 Jongrae Kim†
Jongrae Kim† EonSeon Jin
EonSeon Jin- Department of Life Science, Research Institute for Natural Science, Hanyang University, Seoul, South Korea
Chlamydomonas reinhardtii is being transformed from a model organism to an industrial organism for the production of pigments, fatty acids, and pharmaceuticals. Genetic modification has been used to increase the economic value of C. reinhardtii. However, low gene-editing efficiency and position-effects hinder the genetic improvement of this microorganism. Recently, site-specific double-stranded DNA cleavage using CRISPR-Cas9 system has been applied to regulate a metabolic pathway in C. reinhardtii. In this study, we proved that site-specific gene expression can be induced by CRISPR-Cas9-mediated double-strand cleavage and non-homologous end joining (NHEJ) mechanism. The CRISPR-Cas9-mediated knock-in method was adopted to improve gene-editing efficiency and express the reporter gene on the intended site. Knock-in was performed using a combination of ribonucleoprotein (RNP) complex and DNA fragment (antibiotics resistance gene). Gene-editing efficiency was improved via optimization of a component of RNP complex. We found that when the gene CrFTSY was targeted, the efficiency of obtaining the desired mutant by the knock-in method combined with antibiotic resistance was nearly 37%; 2.5 times higher than the previous reports. Additionally, insertion of a long DNA fragment (3.2 and 6.4 kb) and site-specific gene expression were analyzed. We demonstrated the knock-out phenotype of CrFTSY and on-site inserted gene expression of luciferase and mVenus at the same time. This result showed that CRISPR-Cas9-mediated knock-in can be used to express the gene of interest avoiding position-effects in C. reinhardtii. This report could provide a new perspective to the use of gene-editing. Furthermore, the technical improvements in genetic modification may accelerate the commercialization of C. reinhardtii.
Introduction
Chlamydomonas reinhardtii is widely used as a model organism and considered to be a potential cell factory to produce value-added compounds (Khan et al., 2018; Salomé and Merchant, 2019). Production of compounds such as zeaxanthin, sesquiterpene, bio-hydrogen, and human epidermal growth factor, have been reported in C. reinhardtii (Torzillo and Seibert, 2013; Lauersen et al., 2016; Baek et al., 2018; Baier et al., 2018a). These reports have increased the attention on the commercial use of C. reinhardtii. Moreover, the availability of C. reinhardtii as a biotechnological platform has been maximized through easy genetic modification techniques (Scaife et al., 2015).
Genetic modifications have been used to enhance the production of value-added compounds and produce fine chemicals in C. reinhardtii (Fu et al., 2019; Salomé and Merchant, 2019). Although gene transformation in C. reinhardtii is well-developed and easy, gene overexpression in C. reinhardtii is considered to be a huge obstacle for the advancement of the industry (Doron et al., 2016). Strategies such as developing a strong promoter (Kumar et al., 2013; Scranton et al., 2016; López-Paz et al., 2017), increasing translation using introns (Baier et al., 2018b), increasing gene expression stability using self-cleaving peptides (Rasala et al., 2012; Plucinak et al., 2015), developing a new reporter system using ferredoxin fused hydrogenase for effective screening (Eilenberg et al., 2016), and sequence-based optimization of RNA transcription (Weiner et al., 2018) have been developed for enhancing gene overexpression. The gene expression system is gradually being optimized by improving the technique. Though gene expression is notably advanced in C. reinhardtii, random insertion of the transformed gene is still problematic (Weiner et al., 2018; Jia et al., 2019). The random insertion leads to different levels of protein expression of the same gene called position-effect and also causes unexpected mutations. Therefore, the present study aimed to improve the heterologous gene expression technique by avoiding position-effects and inserting genes effectively at the desired site.
For the specific gene knock-out, gene-editing techniques like zinc-finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN) are used to create specific double-stranded DNA cleavages (Gaj et al., 2013). However, these techniques have high off-target mutation tendency and low feasibility (Gupta and Musunuru, 2014). Using the clustered regularly interspaced short palindromic repeats-associated protein 9 (CRISPR-Cas9) in eukaryotes, researchers can edit (cleave and knock-out) specific locations on genome more easily (Jinek et al., 2012; Cho et al., 2013; Cong et al., 2013; Mali et al., 2013). CRISPR-Cas9 system requires three basic components: the nuclease CRISPR-associated protein 9 (Cas9) cleaving the RuvC site which is 3-nucleotide far from PAM, a CRISPR RNA (crRNA) containing a 20-base pair sequence complementary to the target DNA, and a trans-activating crRNA (tracrRNA). crRNA and tracrRNA can be physically linked to form single guide RNA (gRNA) (Jinek et al., 2012). In C. reinhardtii, the CRISPR-Cas9 was first applied via DNA vector system in 2014 and recently ribonucleoprotein (RNP) system has been developed (Jiang et al., 2014; Baek et al., 2016; Guzmán-Zapata et al., 2019). CRISPR-Cas9 system is the ideal tool for gene-editing; however, it requires efficient selective markers for reducing the time and labor. Presently, phenotypic changes such as visual changes are used to improve the selection efficiency, however, the method is less efficient, requires a lot of labor (Baek et al., 2016; Shin et al., 2016; Greiner et al., 2017; Jeong et al., 2017b), and is not suitable for most genes. Therefore, gene-editing methods using counter-selective markers such as adenine phosphoribosyl transferase (APT), nitrate reductase (NR), peptidylprolyl isomerase (FKB12), tryptophan synthase beta subunit (TSB), and orotidine 5′-phosphate decarboxylase (UMP) have been recently proposed (Shin et al., 2016; Wang et al., 2016; Jiang and Weeks, 2017; Serif et al., 2018; Guzmán-Zapata et al., 2019). Counter selection can be efficient; however, they are eventually dependent on specific phenotypes. To overcome this limitation, pre-selection using antibiotic resistance for enhancing the efficiency of gene-editing has been reported (Shin et al., 2016; Jiang and Weeks, 2017). However, the conditions have not been optimized and the gene-editing efficiency reported has not exceeded >15% till date (Greiner et al., 2017).
As mentioned above, advanced gene expression tools and gene-editing techniques have played a major role in increasing the commercial use of C. reinhardtii. However, the efficiency of these methods is low and the rate of unintended mutations is high. In this study, to develop the new technique of genetic modification in C. reinhardtii, we investigated the knock-in method to improve gene-editing efficiency while inducing gene expression at the desired location. Phenotypic studies indicated that the two desired characteristics were obtained simultaneously. We optimized the technique that increased gene-editing efficiency to >30% and successfully demonstrated site-specific gene expression by avoiding random mutations.
Materials and Methods
Culture Condition
Chlamydomonas reinhardtii CC4349, CC124, and CC503 (purchased from Chlamydomonas Resource Center, University of Minnesota, United States) were maintained photoheterotrophically in Tris-Acetate-Phosphate (TAP) medium at 25°C with continuous light (80 μmol photons m–2 s–1) on an orbital shaker (100 rpm). The cells were cultivated till the log phase in liquid TAP medium under the same conditions for all the experiments. For the selection and maintenance of transformant lines, solid TAP medium fortified with 30 μg/mL hygromycin-B (Thermo Fisher, MA, United States) for ΔCrFTSY-Ga and with 30 μg/mL hygromycin-B, 30 μg/mL paromomycin (Sigma-Aldrich, MO, United States) for ΔCrFTSY-mV, were used.
Knock-in Mutant Generation by CRISPR-Cas9
To optimize the gene-specific knock-out efficiency, RNP method was used and the optimal conditions were determined with slight modification according to the method described previously (Baek et al., 2016). The target sequence of CrFTSY and the gRNA sequence, 5′-CGATCTTCAGAGCAGTGCGG-3′, that was the same as that of the previous study (Baek et al., 2016), were used to avoid the off-target effect. Cas9 protein was purchased from ToolGen Inc. (Seoul, South Korea) and gRNA was synthesized in vitro using GeneArtTM Precision gRNA Synthesis Kit (Thermo Fisher, MA, United States) following the manufacturer’s protocol. The inserted DNA fragments, aph7 gene (aminoglycoside phosphotransferase7, resistance against hygromycin B) (1.6 kb) (Figure 1 left) and aph7-GLuc (Gaussia Luciferase) (3.2 kb) (Figure 2A) DNA cassettes, were amplified by polymerase chain reaction using amplifying primers (sense: 5′-CGGTTCCTGGCCTTTTGCTGG-3′ and antisense: 5′-CAAGTACCATCAACTGACGTTACATTCTG-3′). The inserted DNA fragment, aph8 (aminoglycoside phosphotransferase8, resistance against paromomycin) – mVenus (yellow fluorescence protein) – aph7 (6.4 kb) (Figure 6A) DNA cassette, was linearized by restriction enzyme (ScaI, SpeI). All the inserted DNA fragments were purified by agarose gel extraction.
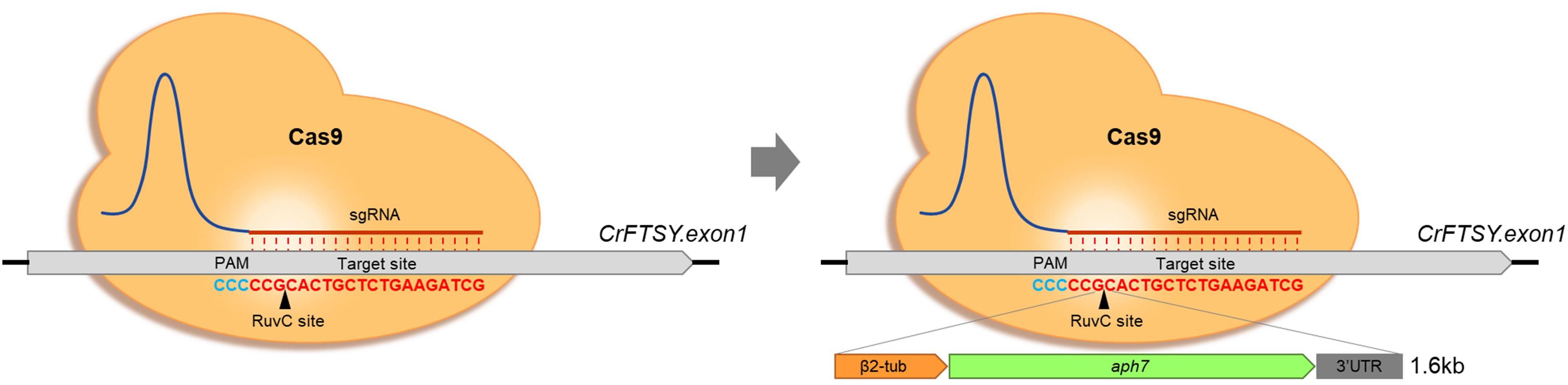
Figure 1. Optimization of gene-editing condition based on CRISPR-Cas9-mediated knock-in strategy. Formal knock-out method by CRISPR-Cas9 (Left) and knock-in with donor DNA (Right). The inserted DNA fragment consisted of β2-tubulin promoter, aph7 (hygromycin-B resistance), rbcs2 3′UTR.
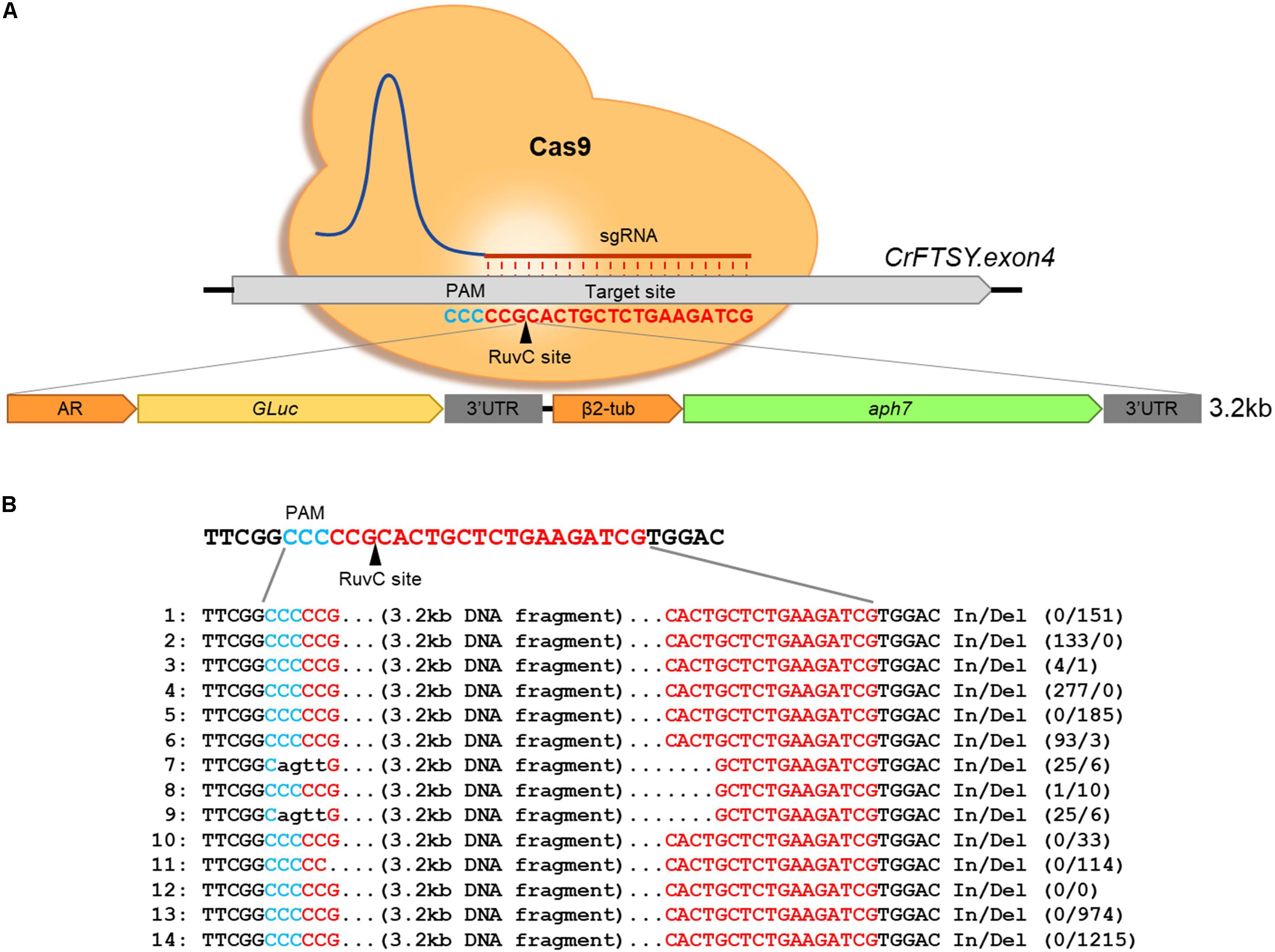
Figure 2. Target-specific long length DNA insertion via CRISPR-Cas9-mediated knock-in method. (A) Schematic diagram of large DNA fragment insertion in CrFTSY. Target-specific DNA cleavage via CRISPR-Cas9 and insertion of the large gene of interest (3.2 kb, GLuc-aph7 DNA cassette) simultaneously. (B) Sequence analysis done by the Sanger sequencing method to identify the inserted DNA fragment on ΔCrFTSY-Ga.
To generate the target-specific knock-in mutant using RNP in Chlamydomonas, 2 × 107 cells were transformed with Cas9 protein premixed with gRNA (RNP complex). Lyophilized Cas9 protein was dissolved in gRNA solution prepared with nuclease-free water and the mixture was incubated for 10 min at room temperature (20–25°C) to make a complex. Cells mixed with the RNP complex were transferred into 4 mm cuvettes and transformed by Gene Pulser XcellTM Electroporation Systems (Bio-Rad, CA, United States) set at 600 V, 50 μF, and 200 Ω. After transformation, the cells were incubated overnight (16–20 h) and spread on the selective medium. Colonies appeared within 2 weeks. The antibiotic resistance colonies were transferred to liquid medium for the sequence analysis. This pre-selected colony number is classified as “candidate” for the calculation of knock-in efficiency.
Identification of DNA Sequence of CrFTSY and Inserted DNA Sequence
The genomic DNA was prepared from the colonies grown on selective medium using the Chelex-100 method (Wan et al., 2011). Targeted region (around 200 bp from gRNA sequence) was amplified by PCR with specific primers (sense: 5′-GGTGTCCCCGCAATCACCAAC-3′, antisense: 5′-CACCCACACCCACCTTGAGCGAG-3′). The PCR product was purified by agarose gel electrophoresis and sequenced using Sanger sequencing by Macrogen Inc. (Seoul, South Korea).
Measurement of Chlorophyll Content
Wild-type and mutant cells were cultured in liquid TAP medium untill the mid-log phase. To make comparison of colorimetric analysis, the cultured cells were collected from the liquid media, and 2.5 × 106 cells were deposited on solid TAP medium. The cells were incubated for 24 h and the difference in color characteristics was compared. For measurement of the chlorophyll contents, 1 × 107 cells were suspended in 80% (v/v) acetone, and the absorbance was measured by spectrophotometer at wavelength of 663 and 647 nm. The formula used for calculating the content is given by Melis et al. (1987).
Measurement of Gaussia Luciferase Activity
Luciferase activity was measured by a previously reported protocol (Kim et al., 2018) with slight modifications to confirm GLuc expression. GLuc is secreted in the medium hence, its activity was measured using the complete cell culture (Ruecker et al., 2008) and it is more sensitive than using only the cells. The mutant cells were cultured till the absorbance at 750 nm reached 1.0 indicating mid-log phase. A small volume (200 μL) of culture was used for the luciferase assay using the Renilla Luciferase Assay Kit (Promega, WI, United States). The cells were suspended in 40 μL of lysis buffer and lysed by vigorous vortexing. The lysed cells were pelleted by centrifugation at 13,000 rpm for 5 min. Then, 80 μL supernatant was mixed with equal volume of substrate solution. The chemiluminescence was measured immediately after mixing the two solutions in a Glo MaxTM 20/20 luminometer (Promega, WI, United States). The statistical difference was analyzed by Student’s t-test (p-value < 0.05).
Fluorescence Microscopic Analysis
ΔCrFTSY-mV was cultivated in selective medium as mentioned above. Five microliters of cultured cells were dropped on a glass slide and covered with a coverslip. Fluorescence was detected by a fluorescence microscope (Eclipse Ni, Nikon, Tokyo, Japan). Fluorescence detection wavelengths were 540 ± 20 nm with YFP filter for mVenus and 630 ± 30 nm with Texas RED filter for auto-fluorescence of chlorophyll. The magnification was 400×.
Southern Blot Analysis
Southern blot was used to confirm single DNA insertion in mutants. Genomic DNA was extracted from cells in the log phase by the CTAB method (Porebski et al., 1997). The genomic DNA was isolated (20 μg) and digested by PvuII or SmaI restriction enzymes. Digested genomic DNA was separated using 0.8% agarose gel and transferred to a nylon membrane by capillary transfer. Membrane attached DNA was developed through hybridization with an aph7 probe and amplified by PCR with specific primers (sense: 5′-ATGATTCCTACGCGAGCCTG-3′, antisense: 5′-ATCCGGCTCATCACCAGGTA-3′) using the Gene Images AlkPhos Direct Labeling and Detection System (Amersham, Little Chalfont, Bucks, United Kingdom). The process was carried out according to the manufacturer’s protocol.
Western Blot Analysis
Cultivated cells (1 × 106) were boiled in SDS-PAGE loading buffer, electrophoresed on 15% SDS-polyacrylamide gels, and transferred to a PVDF membrane using Xcell II blot module (Thermo Fisher, MA, United States). GLuc antibody (NEB, MA, United States) was used to detect GLuc protein. ATP-β antibody (Agrisera, Vännäs, Sweden) was used as a reference. HRP-conjugated goat anti-rabbit IgG (H + L) antibody (Life Technologies, CA, United States) was used as a secondary antibody. GLuc and ATP-β were visualized on an X-ray film by chemiluminescence using EPD Western Reagent (ELPIS-BIOTECH, Daejeon, South Korea).
Results
Improvement of Gene-Editing Efficiency by CRISPR-Cas9-Mediated Knock-in
In this study, the selective marker system aph7 DNA cassette was used to optimize the knock-in method in C. reinhardtii. This strategic knock-in method was induced by CRISPR-Cas9-mediated knock-out, and the selective marker gene was inserted into the cleaved site by NHEJ (Figure 1). The mutants were screened more effectively by the expression of the antibiotic selective marker in the inserted DNA fragment.
Although the knock-in method has been reported previously, the gene-editing efficiency was very low (under 15%). Therefore, in this study gene-editing efficiency was analyzed by using different concentrations of RNP complex required for transformation (Table 1). Every single experiment resulted in a different number of colonies but a similar percentage of positive colonies (Supplementary Figure 1). The gene-editing efficiency of CrFTSY (ΔCrFTSY) was observed to be maximum at 36.8%, when transformed with 100 μg Cas9 and 70 μg gRNA. Even on transformation with 10 μg Cas9 and 7 μg gRNA, ΔCrFTSY efficiency was >16.5%, which was higher than previously reported (Baek et al., 2016; Shin et al., 2016; Greiner et al., 2017). Thus, we optimized the concentration of RNP complex for maximum gene-editing efficiency. Additionally, we investigated the gene-editing efficiency depending on the presence or absence of a cell wall. The gene-editing efficiency in C. reinhardtii strains CC503 (no cell wall) and CC124 (with cell wall) were observed under the same method and it was found to be similar to that of C. reinhardtii CC4349 (Supplementary Figure 2). This result indicated that the knock-in method is universally applicable independent of the cell type.
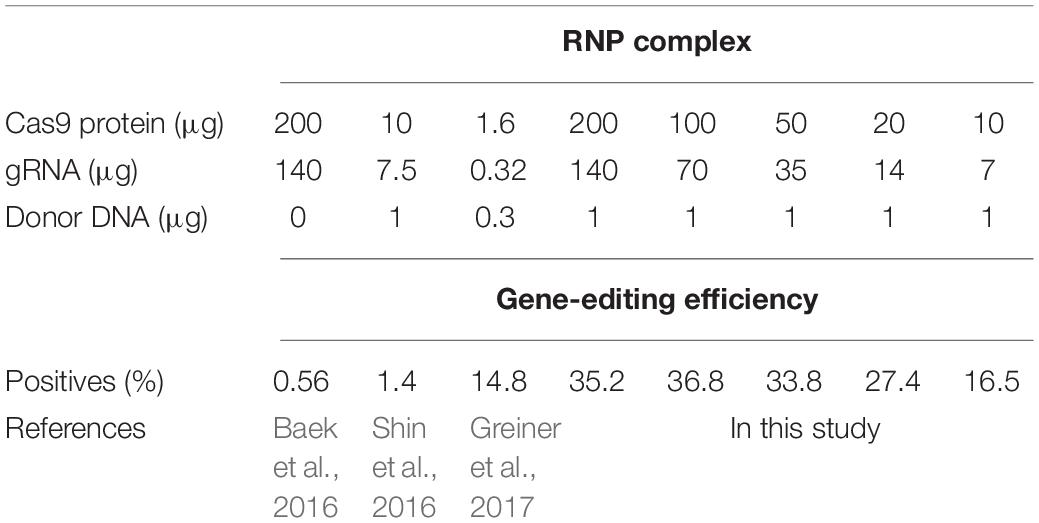
Table 1. Gene-editing efficiency obtained via the knock-in method.
Insertion of Long-Length DNA Fragment on Intended Site
In the gene expression using foreign DNA, transformed foreign DNA integrates in the nuclear genome of C. reinhardtii. This DNA integration in the genome predominantly leads to unexpected mutations and position-effects (Leon and Fernandez, 2007). Therefore, we investigated the possibility of on-site foreign gene expression through the knock-in method to reduce position effects. Firstly, a 3.2 kb long DNA fragment was transformed into the target site of by the knock-in method described above (Figure 2A). Large DNA insertion in the target site was confirmed by genomic PCR in 14 positives among 39 candidates (36% gene-editing efficiency) of ΔCrFTSY_Ga colonies in which GLuc-aph7 DNA cassette was inserted into CrFTSY (Supplementary Figure 3). ΔCrFTSY-Ga DNA was sequenced and compared with the expected sequence after integration (Figure 2B and Supplementary Data Sheet 2). Targeted DNA sequences on CrFTSY were neatly cleaved by Cas9 in ΔCrFTSY-Ga mutants except mutants 7 and 9. Among the 14 positive mutants, clean insertion without any In/Del was detected only in mutant 12. As evident in this result, the inserted DNA sequence resulted in mutations during integration into the genome. Thus, it is important to identify the exact sequence of the insertion site. Although the problems related to the mutations of inserted DNA remain unsolved, however, this result showed that the insertion of DNA longer than 3 kb is possible at the desired site.
Expression of the Foreign Gene at the Desired Site by Knock-in
We also analyzed the expression of the foreign genes at the desired site. To confirm the normal expression of GLuc inserted at the target site, we confirmed the copy number of DNA insert in ΔCrFTSY-Ga mutants. Mutant 2 was excluded in the further analysis as it was mixed with non-mutant cells. Mutants 13 and 14 were also excluded as the promoter sequence of GLuc got deleted. Southern blot analysis was performed to determine the copy number of DNA insert. Genomic DNA was digested by restriction enzymes and hybridized with the specific probe. All ΔCrFTSY-Ga mutants had a single copy of the DNA insert (Figure 3 and Supplementary Figure 4). This result suggests that luciferase activity observed in the next experiment was due to a single Gaussia luciferase gene inserted on CrFTSY.
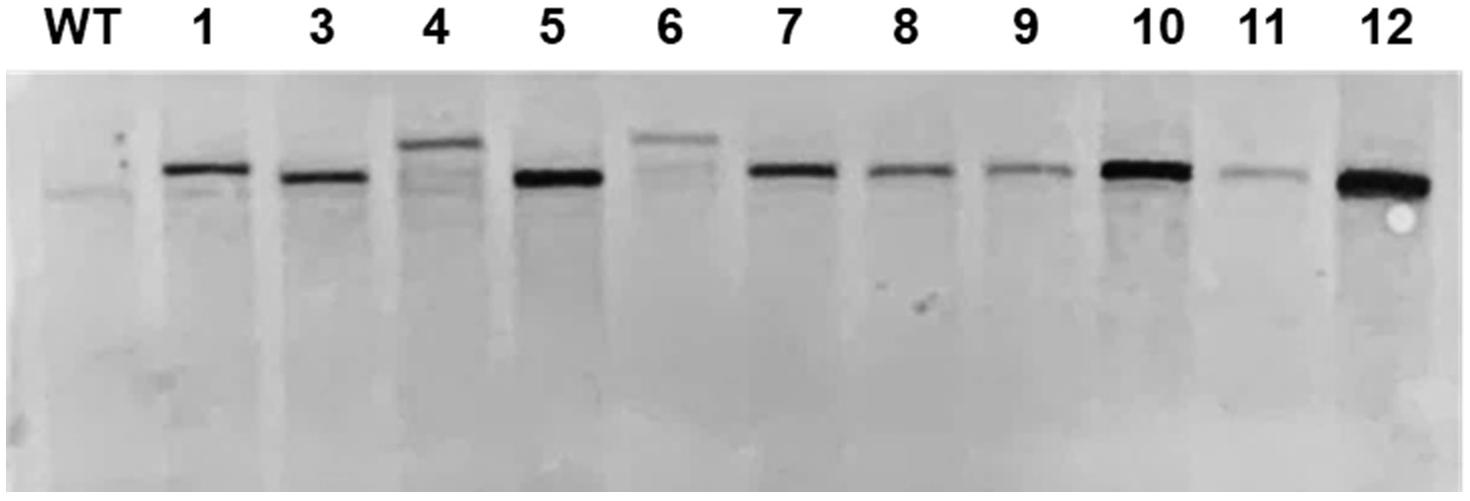
Figure 3. Southern blot for validation of copy number of an inserted DNA fragment. Extracted genomic DNA was digested by SmaI and blotted on a nylon membrane. The aph7 probe was used for detection of DNA integration.
Chlamydomonas reinhardtii with mutation in CrFTSY appeared to be pale green in color compared to the color of the wild type due to the reduction in chlorophyll content on a theoretical basis (Kirst et al., 2012). We observed that 11 ΔCrFTSY-Ga mutants obtained were pale green in color compared to that of the wild type on solid TAP medium (Figure 4A). Moreover, to validate this visual difference between WT and mutants, we measured the chlorophyll content of all the samples, including WT (Figure 4B). The levels of chlorophyll-a (4.80 ± 0.76 nmol mL–1) and -b (1.34 ± 0.53 nmol mL–1) in ΔCrFTSY-Ga mutants were reduced to 63 and 38% of wild type chlorophyll-a and chlorophyll-b (7.63 ± 0.43 and 3.53 ± 0.83), respectively. Therefore, the chlorophyll a/b ratio was increased by 1.8 ± 0.2-fold in ΔCrFTSY-Ga mutants compared to that of wild type, as also shown in the previous report (Baek et al., 2016). The results clearly reflected the phenotypic differences when the FTSY was knocked out (Figure 4).
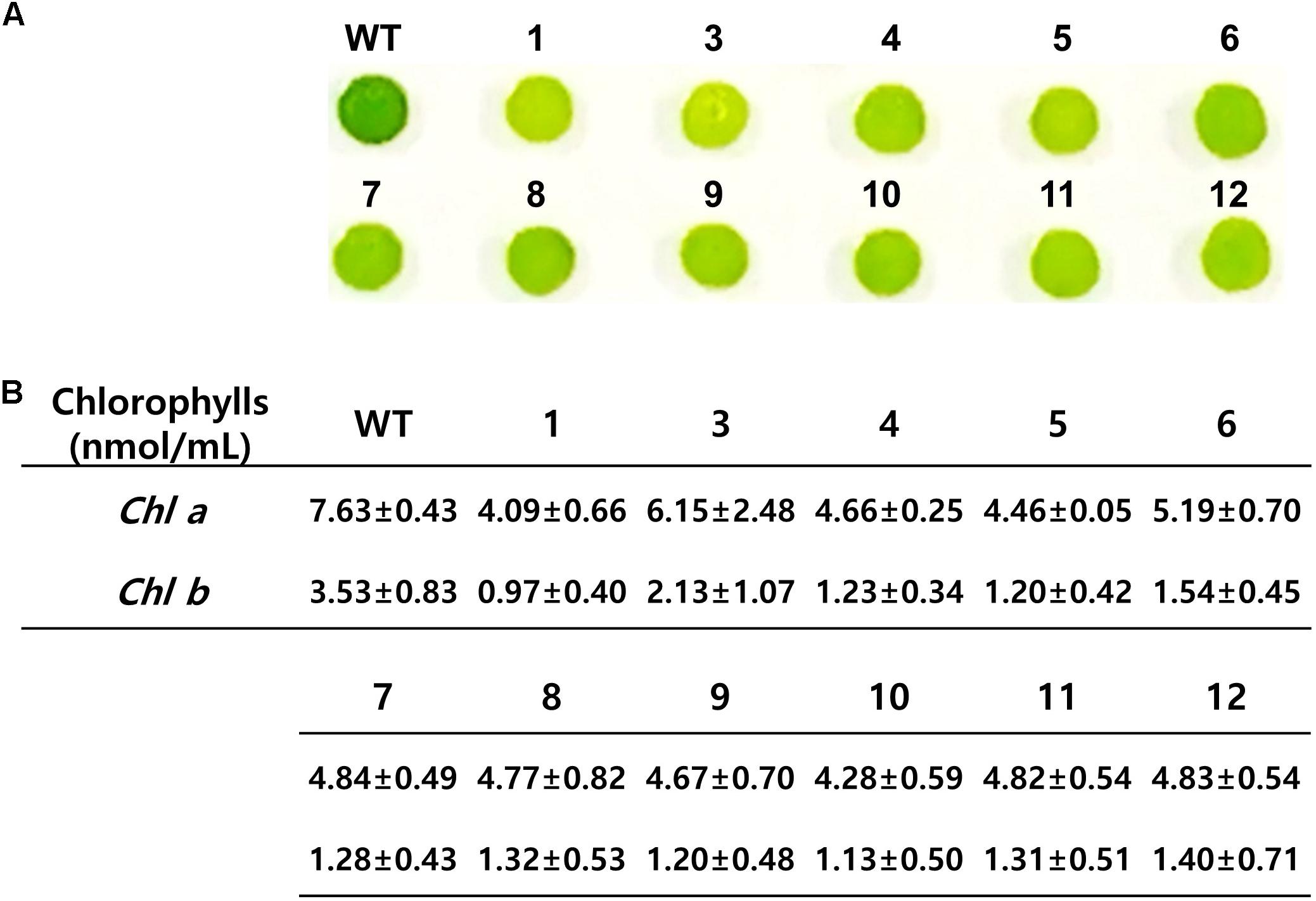
Figure 4. Phenotypic characteristics of CrFTSY knock-out. Wild-type and ΔCrFTSY-Ga mutants grew on TAP agar medium under 80 μmol photon m–2 s–1 light. (A) Pale green color was detected in ΔCrFTSY-Ga mutants compared to dark green color exhibited by wild type. (B) Amount (nmol mL–1) of Chlorophyll-a and -b of ΔCrFTSY-Ga mutants (n = 3; the values shown are means ± SD).
The foreign gene expression on the intended site was verified by confirming the protein expression of GLuc (Supplementary Figure 5) and measuring the luciferase activity (Figure 5). We used the cultured cells and medium together for the luciferase analysis as mentioned before. As shown in Figure 5, all ΔCrFTSY-Ga mutants successfully expressed GLuc while luciferase activity was negligible in the wild type. The results of this experiment demonstrated that a gene of interest can be expressed on the desired site without random insertional mutation. Mean difference between GLuc activity in 11 different mutants was 20%, which was significantly lower than the mean difference (75%) found in random integration mutants (Kim et al., 2018). This constant expression level in 11 mutants was possible due to the insertion of a single copy of DNA fragment at the intended site on the genome.
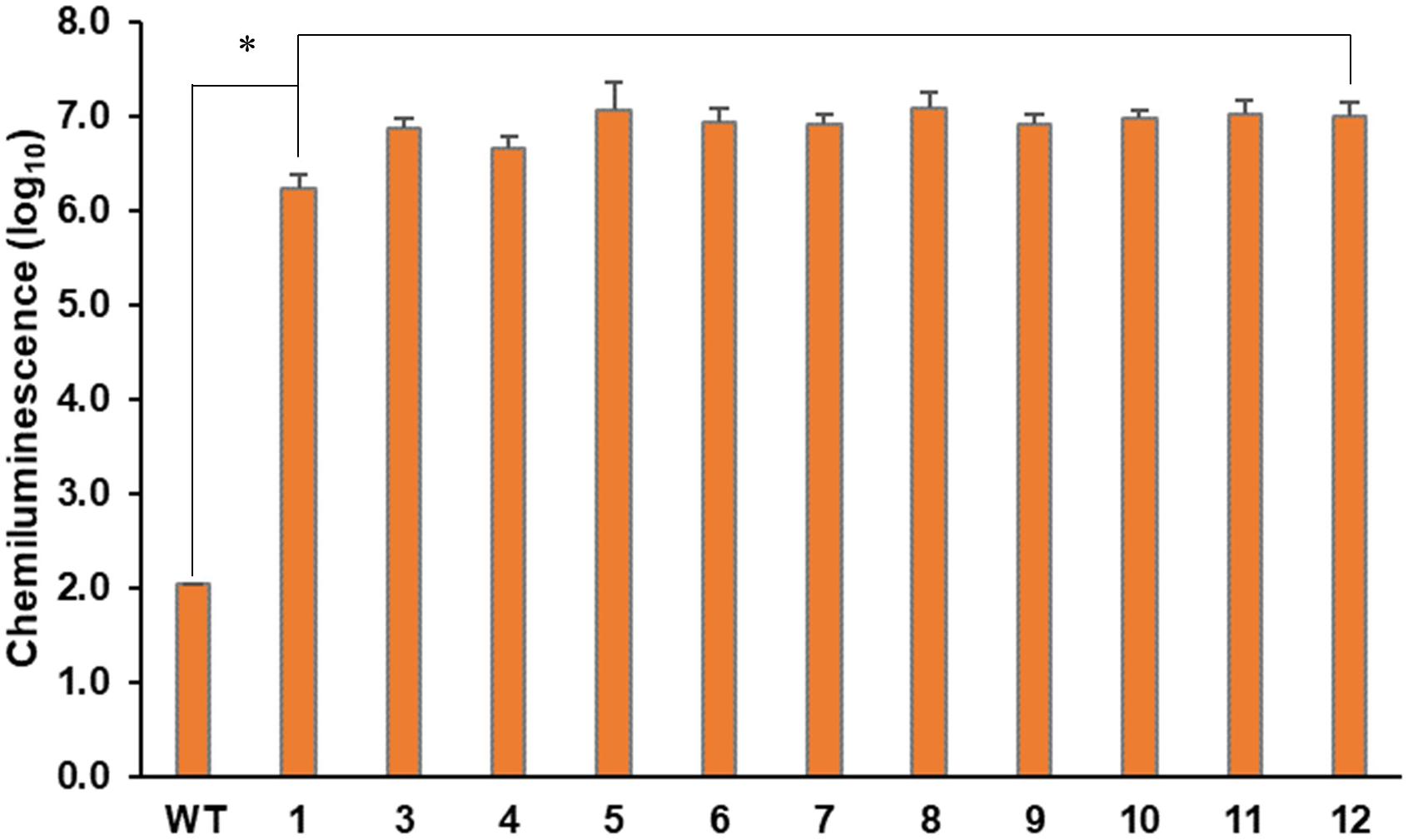
Figure 5. Phenotypic characteristics of expression of the inserted DNA fragment. Gaussia luciferase assays from ΔCrFTSY-Ga mutants were performed in triplicate independently. The asterisk indicates the statistical difference determined by Student’s t-test (n = 3; the values shown are means ± SD; p < 0.05).
Determination of the Maximum Size of DNA Fragment Inserted on Target by Knock-in
We tested the possibility of insertion of a DNA fragment larger than 3.2 kb through the knock-in method, which confirmed the insertion of a 6.4 kb long DNA fragment in the target site. For this additional experiment, we used the same methodology as described in the section “Expression of the Foreign Gene at the Desired Site by Knock-in.” The DNA insertion in the target site was confirmed by genomic PCR. Four positives among 12 candidates (33% gene-editing efficiency) of ΔCrFTSY_mV in which aph8-mVenus-aph7 DNA cassette was inserted into CrFTSY were obtained (Supplementary Figure 6). From the sequence analysis at the insertion site, we found long length In/Del mutations (Figure 6A and Supplementary Data Sheet 2). Two mutants (mutants 1 and 2) had 126 bp insertion and two different mutants (mutants 3 and 4) had around 700 bp deletion. As in the above result (Figure 2), In/Del occurred non-specifically in this experiment. Nevertheless, the knock-in method deleted the desired genes and reconfirmed that the introduction of DNA fragment for overexpression was effective. mVenus expression in the selected mutant was visualized by fluorescence microscopy (Figure 6B). Hence, we confirmed that a DNA fragment of up to 6.4 kb long could be inserted at the desired location and overexpressed. From these results, we demonstrated that the CRISPR-Cas9-mediated knock-in method was an effective method that allowed the gene deletion and overexpression of foreign genes in a single experiment.
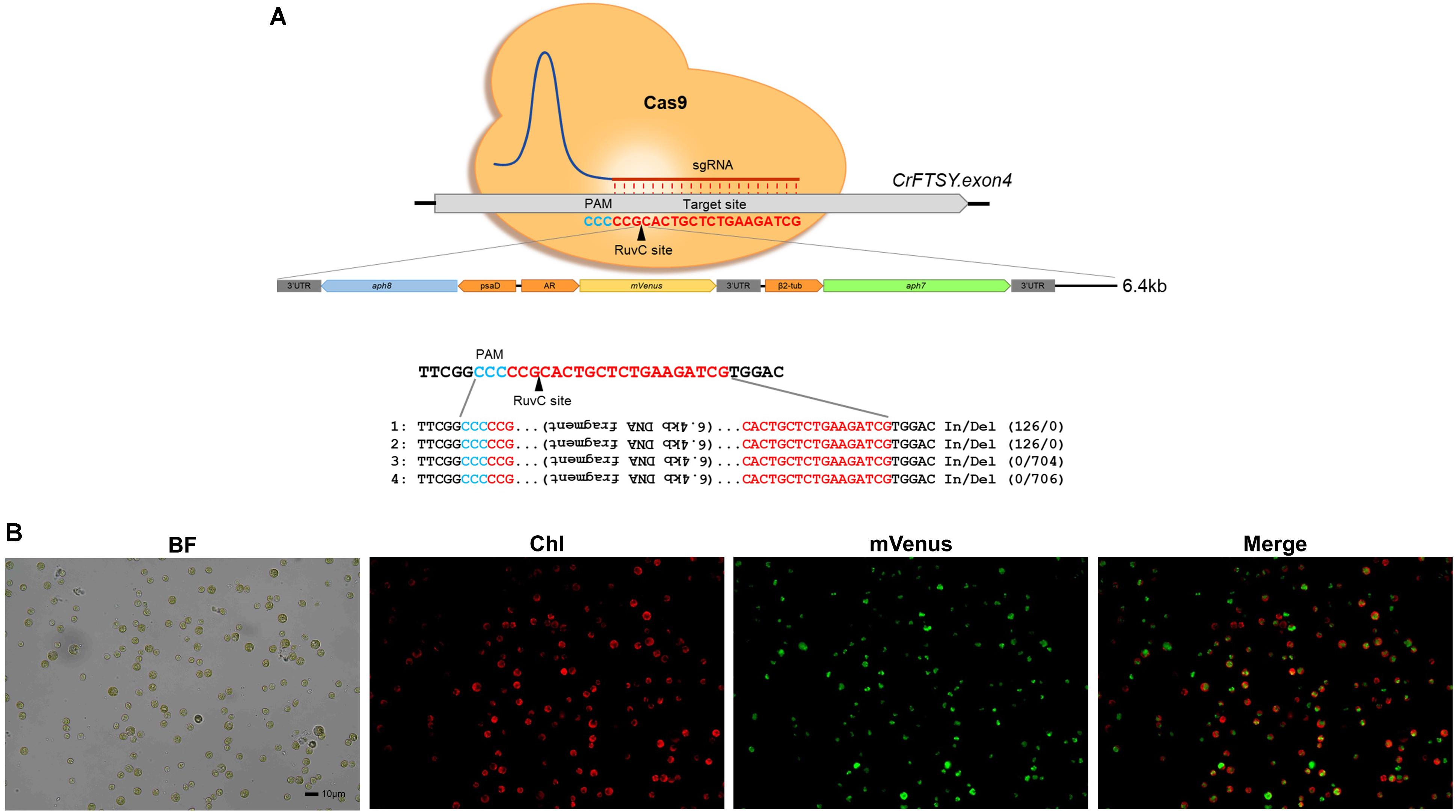
Figure 6. Target specific 6.4 kb long DNA (aph8–mVenus–aph7 DNA cassette) insertion via CRISPR-Cas9-mediated knock-in. (A) Sequence analysis of target site and inserted DNA fragment. (B) Visualization of mVenus expression in ΔCrFTSY_mV at insertion site by fluorescence microscopy (red, chlorophyll; green, mVenus). Scale bar indicates 10 μm.
Discussion
Increasing the commercial use of C. reinhardtii requires improvement of the strain by genetic modification for the production of high-value compounds. C. reinhardtii has been extensively studied for fundamental research and industrial use based on its genome sequence data and well-developed molecular tool kit (Scaife et al., 2015; Crozet et al., 2018; Salomé and Merchant, 2019). Moreover, the genetic modification techniques are highly developed and the engineering strategies of metabolic pathways are well established (Plucinak et al., 2015; Baier et al., 2018b; Fu et al., 2019; Kong et al., 2019).
Despite several refined concepts, the improvement of C. reinhardtii is still not sufficient and simple. Due to the non-specific disruption of genes generating the desired mutants has been difficult, hampering reverse-genetic studies (Fu et al., 2019; Park et al., 2019). The existing transformation methods of C. reinhardtii cannot target a specific gene, thus researchers cannot regulate precisely the desired genes (Leon and Fernandez, 2007; Jia et al., 2019; Kim et al., 2019). These problems can be overcome by the recently developed gene-editing techniques. Gene-editing techniques based on RNP using Cas9 proteins are being recognized as the most effective gene specific knock-out methods to date (Patel et al., 2019). Cas9-mediated gene knock-out has been reported for several genes and the use of donor DNA with RNP, called knock-in, has emerged recently. However, the gene-editing efficiency reported in previous studies was not satisfactory (Shin et al., 2016; Greiner et al., 2017; Jeong et al., 2017a).
Recently, some reports suggested that gene selection can be achieved with high yields (up to 30%) through counter selection without the use of antibiotic genes (Jiang and Weeks, 2017; Serif et al., 2018; Guzmán-Zapata et al., 2019). However, these methods are mostly functional for specific genes and cannot be applied universally.
In this study, we used the antibiotic gene (aph7) as donor DNA to ensure high selection efficiency for the optimization of the Cas9-mediated knock-in method. In contrast to the previously reported CRISPR-Cas9-mediated knock-out methods, the use of a selective marker in the knock-in method of our study enhanced the knock-out efficiency by inserting of an external DNA into the cleaved site (Figure 1). The method of using the antibiotic gene employed in this study has been proven to be generally applicable while effectively performing the gene-editing of other genes (AGP and LCYE) (unpublished data). Therefore, we suggest that the use of antibiotic genes as donor DNA is an efficient method when using the RNP-based gene-editing. In addition, through the optimization of RNP complex used in this study, the gene-editing efficiency was increased up to 37% (Table 1 and Supplementary Figure 1). These results indicate that the RNP complex of 100 μg:70 μg (Protein:gRNA) is optimal for the target specific gene editing in C. reinhardtii.
An additional benefit of establishing the knock-in method is that it avoids the position-effects of random mutations that occur during the transformation process. We strategically utilized this methodology to validate the expression of the gene of interest at the desired position (Figures 2–5). The successful expression of the report gene (GLuc and mVenus) and the fact that the single copy of DNA insert was integrated into genome showed that our strategy of avoiding position-effect and expressing the gene of interest at the target location successful (Figures 3–6). The success of our target-oriented gene insertion strategy could provide a new strategic perspective for future C. reinhardtii improvement studies.
In this paper, we reported an improved gene-editing technique. Despite several improvements in different techniques, researchers still face the problem of tedious processes for species improvement. Even though the target-oriented gene insertion based on knock-in method was performed, validation for the precise knock-in was still required. Besides, due to the lack of information on the sites effectively targeted by Cas9, the location of gene insertion is limiting, which unavoidably involves the deletion of one gene. Therefore, it is important to discover target locations with high gene-editing efficiency without affecting the biological function of the cell. As our results show in Figures 2, 6, the inserted gene was integrated into genomic DNA by NHEJ, therefore it is necessary to develop a technique to prevent the mutation in sequences in the integration process.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
EJ and JK conceived and designed the experiments. KB confirmed the optimal condition of RNP. JK and SL performed the knock-in study and analyzed the gene expression. JK, SL, and EJ wrote the manuscript. All authors reviewed the manuscript.
Funding
This study was supported by the Korea Carbon Capture & Sequestration R&D Center (KCRC) (NRF-2014M1A8A1049273), and the Basic Core Technology Development Program for the Oceans and the Polar Regions (NRF-2015M1A5A1037053) of the National Research Foundation of Korea, Ministry of Science and ICT, Government of Korea.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00306/full#supplementary-material
References
Baek, K., Kim, D. H., Jeong, J., Sim, S. J., Melis, A., Kim, J. S., et al. (2016). DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9 ribonucleoproteins. Sci. Rep. 6:30620. doi: 10.1038/srep30620
Baek, K., Yu, J., Jeong, J., Sim, S. J., Bae, S., and Jin, E. (2018). Photoautotrophic production of macular pigment in a Chlamydomonas reinhardtii strain generated by using DNA−free CRISPR−Cas9 RNP−mediated mutagenesis. Biotechnol. Bioeng. 115, 719–728. doi: 10.1002/bit.26499
Baier, T., Kros, D., Feiner, R. C., Lauersen, K. J., MüLler, K. M., and Kruse, O. (2018a). Engineered fusion proteins for efficient protein secretion and purification of a human growth factor from the green microalga Chlamydomonas reinhardtii. ACS Synth. Biol. 7, 2547–2557. doi: 10.1021/acssynbio.8b00226
Baier, T., Wichmann, J., Kruse, O., and Lauersen, K. J. (2018b). Intron-containing algal transgenes mediate efficient recombinant gene expression in the green microalga Chlamydomonas reinhardtii. Nucleic Acids Res. 46, 6909–6919. doi: 10.1093/nar/gky532
Cho, S. W., Kim, S., Kim, J. M., and Kim, J.-S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232. doi: 10.1038/nbt.2507
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Crozet, P., Navarro, F. J., Willmund, F., Mehrshahi, P., Bakowski, K., Lauersen, K. J., et al. (2018). Birth of a photosynthetic chassis: a MoClo toolkit enabling synthetic biology in the microalga Chlamydomonas reinhardtii. ACS Synth. Biol. 7, 2074–2086. doi: 10.1021/acssynbio.8b00251
Doron, L., Segal, N. A., and Shapira, M. (2016). Transgene expression in microalgae—from tools to applications. Front. Plant Sci. 7:505. doi: 10.3389/fpls.2016.00505
Eilenberg, H., Weiner, I., Ben-Zvi, O., Pundak, C., Marmari, A., Liran, O., et al. (2016). The dual effect of a ferredoxin-hydrogenase fusion protein in vivo: successful divergence of the photosynthetic electron flux towards hydrogen production and elevated oxygen tolerance. Biotechnol. Biof. 9:182. doi: 10.1186/s13068-016-0601-3
Fu, W., Nelson, D. R., Mystikou, A., Daakour, S., and Salehi-Ashtiani, K. (2019). Advances in microalgal research and engineering development. Curr. Opin. Biotechnol. 59, 157–164. doi: 10.1016/j.copbio.2019.05.013
Gaj, T., Gersbach, C. A., and Barbas Iii, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405. doi: 10.1016/j.tibtech.2013.04.004
Greiner, A., Kelterborn, S., Evers, H., Kreimer, G., Sizova, I., and Hegemann, P. (2017). Targeting of photoreceptor genes in Chlamydomonas reinhardtii via zinc-finger nucleases and CRISPR/Cas9. Plant Cell 29, 2498–2518. doi: 10.1105/tpc.17.00659
Gupta, R. M., and Musunuru, K. (2014). Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Invest. 124, 4154–4161. doi: 10.1172/JCI72992
Guzmán-Zapata, D., Sandoval-Vargas, J. M., Macedo-Osorio, K. S., Salgado-Manjarrez, E., Castrejón-Flores, J. L., Oliver-Salvador, M. D. C., et al. (2019). Efficient editing of the nuclear APT reporter gene in Chlamydomonas reinhardtii via expression of a CRISPR-Cas9 module. Int. J. Mol. Sci. 20:1247. doi: 10.3390/ijms20051247
Jeong, J., Baek, K., Kirst, H., Melis, A., and Jin, E. (2017a). Loss of RP54 function leads to a truncated light-harvesting antenna size in Chlamydomonas reinhardtii. Biochim. Biophys. Acta Bioenerg. 1858, 45–55. doi: 10.1016/j.bbabio.2016.10.007
Jeong, J., Baek, K., Yu, J., Kirst, H., Betterle, N., Shin, W., et al. (2017b). Deletion of the chloroplast LTD protein impedes LHCI import and PSI–LHCI assembly in Chlamydomonas reinhardtii. J. Exp. Bot. 69, 1147–1158. doi: 10.1093/jxb/erx457
Jia, B., Xie, X., Wu, M., Lin, Z., Yin, J., Huang, Y., et al. (2019). Understanding the functions of endogenous DOF transcript factor in Chlamydomonas reinhardtii. Biotechnol. Biof. 12:67. doi: 10.1186/s13068-019-1403-1
Jiang, W., Brueggeman, A. J., Horken, K. M., Plucinak, T. M., and Weeks, D. P. (2014). Successful transient expression of Cas9 and single guide RNA genes in Chlamydomonas reinhardtii. Eukaryot. Cell 13, 1465–1469. doi: 10.1128/EC.00213-14
Jiang, W. Z., and Weeks, D. P. (2017). A gene-within-a-gene Cas9/sgRNA hybrid construct enables gene editing and gene replacement strategies in Chlamydomonas reinhardtii. Algal Res. 26, 474–480. doi: 10.1016/j.algal.2017.04.001
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Khan, M. I., Shin, J. H., and Kim, J. D. (2018). The promising future of microalgae: current status, challenges, and optimization of a sustainable and renewable industry for biofuels, feed, and other products. Microb. Cell Fact. 17:36. doi: 10.1186/s12934-018-0879-x
Kim, J., Kwak, H. S., Sim, S. J., and Jin, E. (2019). Overexpression of malic enzyme isoform 2 in Chlamydomonas reinhardtii PTS42 increases lipid production. Bioresour. Technol. Rep. 7:100239. doi: 10.1016/j.biteb.2019.100239
Kim, J., Liu, L., Hu, Z., and Jin, E. (2018). Identification and functional analysis of the psaD promoter of Chlorella vulgaris using heterologous model strains. Int. J. Mol. Sci. 19:1969. doi: 10.3390/ijms19071969
Kirst, H., García-Cerdán, J. G., Zurbriggen, A., and Melis, A. (2012). Assembly of the light-harvesting chlorophyll antenna in the green alga Chlamydomonas reinhardtii requires expression of the TLA2-CpFTSY gene. Plant Physiol. 158, 930–945. doi: 10.1104/pp.111.189910
Kong, F., Yamaoka, Y., Ohama, T., Lee, Y., and Li-Beisson, Y. (2019). Molecular genetic tools and emerging synthetic biology strategies to increase cellular oil content in Chlamydomonas reinhardtii. Plant Cell Physiol. 60, 1184–1196. doi: 10.1093/pcp/pcz022
Kumar, A., Falcao, V. R., and Sayre, R. T. (2013). Evaluating nuclear transgene expression systems in Chlamydomonas reinhardtii. Algal Res. 2, 321–332. doi: 10.1016/j.algal.2013.09.002
Lauersen, K. J., Baier, T., Wichmann, J., Wördenweber, R., Mussgnug, J. H., Hübner, W., et al. (2016). Efficient phototrophic production of a high-value sesquiterpenoid from the eukaryotic microalga Chlamydomonas reinhardtii. Metab. Eng. 38, 331–343. doi: 10.1016/j.ymben.2016.07.013
Leon, R., and Fernandez, E. (2007). “Nuclear transformation of eukaryotic microalgae,” in Transgenic Microalgae as Green Cell Factories. Advances in Experimental Medicine and Biology, Vol. 616, eds R. León, A. Galván, and E. Fernández (New York, NY: Springer), 1–11. doi: 10.1007/978-0-387-75532-8_1
López-Paz, C., Liu, D., Geng, S., and Umen, J. G. (2017). Identification of Chlamydomonas reinhardtii endogenous genic flanking sequences for improved transgene expression. Plant J. 92, 1232–1244. doi: 10.1111/tpj.13731
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., Dicarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi: 10.1126/science.1232033
Melis, A., Spangfort, M., and Andersson, B. (1987). Light−absorption and electron−transport balance between photosystem II and photosystem I in spinach chloroplasts. Photochem. Photobiol. 45, 129–136. doi: 10.1111/j.1751-1097.1987.tb08413.x
Park, S., Nguyen, T. H. T., and Jin, E. (2019). Improving lipid production by strain development in microalgae: strategies, challenges and perspectives. Bioresour. Technol. 262:121953. doi: 10.1016/j.biortech.2019.121953
Patel, V. K., Soni, N., Prasad, V., Sapre, A., Dasgupta, S., and Bhadra, B. (2019). CRISPR–Cas9 system for genome engineering of photosynthetic microalgae. Mol. Biotechnol. 61, 541–561. doi: 10.1007/s12033-019-00185-3
Plucinak, T. M., Horken, K. M., Jiang, W., Fostvedt, J., Nguyen, S. T., and Weeks, D. P. (2015). Improved and versatile viral 2 A platforms for dependable and inducible high−level expression of dicistronic nuclear genes in Chlamydomonas reinhardtii. Plant J. 82, 717–729. doi: 10.1111/tpj.12844
Porebski, S., Bailey, L. G., and Baum, B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Report. 15, 8–15. doi: 10.1007/bf02772108
Rasala, B. A., Lee, P. A., Shen, Z., Briggs, S. P., Mendez, M., and Mayfield, S. P. (2012). Robust expression and secretion of Xylanase1 in Chlamydomonas reinhardtii by fusion to a selection gene and processing with the FMDV 2A peptide. PLoS One 7:e43349. doi: 10.1371/journal.pone.0043349
Ruecker, O., Zillner, K., Groebner-Ferreira, R., and Heitzer, M. (2008). Gaussia-luciferase as a sensitive reporter gene for monitoring promoter activity in the nucleus of the green alga Chlamydomonas reinhardtii. Mol. Genet. Genom. 280, 153–162. doi: 10.1007/s00438-008-0352-3
Salomé, P. A., and Merchant, S. S. (2019). A series of fortunate events: introducing Chlamydomonas as a reference organism. Plant Cell 31, 1682–1707. doi: 10.1105/tpc.18.00952
Scaife, M. A., Nguyen, G. T., Rico, J., Lambert, D., Helliwell, K. E., and Smith, A. G. (2015). Establishing Chlamydomonas reinhardtii as an industrial biotechnology host. Plant J. 82, 532–546. doi: 10.1111/tpj.12781
Scranton, M. A., Ostrand, J. T., Georgianna, D. R., Lofgren, S. M., Li, D., Ellis, R. C., et al. (2016). Synthetic promoters capable of driving robust nuclear gene expression in the green alga Chlamydomonas reinhardtii. Algal Res. 15, 135–142. doi: 10.1016/j.algal.2016.02.011
Serif, M., Dubois, G., Finoux, A.-L., Teste, M.-A., Jallet, D., and Daboussi, F. (2018). One-step generation of multiple gene knock-outs in the diatom Phaeodactylum tricornutum by DNA-free genome editing. Nat. Commun. 9:3924. doi: 10.1038/s41467-018-06378-9
Shin, S.-E., Lim, J.-M., Koh, H. G., Kim, E. K., Kang, N. K., Jeon, S., et al. (2016). CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci. Rep. 6:27810. doi: 10.1038/srep27810
Torzillo, G., and Seibert, M. (2013). “Hydrogen production by Chlamydomonas reinhardtii,” in Handbook of Microalgal Culture: Applied Phycology and Biotechnology, 2nd Edn, eds A. Richmond and Q. Hu (Oxford: Blackwell Publishing Ltd.), 417–432.
Wan, M., Rosenberg, J. N., Faruq, J., Betenbaugh, M. J., and Xia, J. (2011). An improved colony PCR procedure for genetic screening of Chlorella and related microalgae. Biotechnol. Lett. 33, 1615–1619. doi: 10.1007/s10529-011-0596-6
Wang, Q., Lu, Y., Xin, Y., Wei, L., Huang, S., and Xu, J. (2016). Genome editing of model oleaginous microalgae Nannochloropsis spp. by CRISPR/Cas9. Plant J. 88, 1071–1081. doi: 10.1111/tpj.13307
Keywords: Chlamydomonas reinhardtii, genetic modification, CRISPR-Cas9, knock-in, on-site gene overexpression
Citation: Kim J, Lee S, Baek K and Jin E (2020) Site-Specific Gene Knock-Out and On-Site Heterologous Gene Overexpression in Chlamydomonas reinhardtii via a CRISPR-Cas9-Mediated Knock-in Method. Front. Plant Sci. 11:306. doi: 10.3389/fpls.2020.00306
Received: 04 November 2019; Accepted: 03 March 2020;
Published: 20 March 2020.
Edited by:
Goetz Hensel, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), GermanyReviewed by:
Fantao Kong, Dalian University of Technology, ChinaSang Jun Sim, Korea University, South Korea
Zhangli Hu, Shenzhen University, China
Copyright © 2020 Kim, Lee, Baek and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: EonSeon Jin, ZXNqaW5AaGFueWFuZy5hYy5rcg==
†These authors have contributed equally to this work