
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 31 January 2020
Sec. Plant Biotechnology
Volume 10 - 2019 | https://doi.org/10.3389/fpls.2019.01720
This article is part of the Research Topic Proceedings of ICPSBBB 2018 - 2nd International Conference on Plant Synthetic Biology, Bioengineering and Biotechnology View all 9 articles
 Jun Hyung Lee1,2
Jun Hyung Lee1,2 Mitra Mazarei1
Mitra Mazarei1 Alexander C. Pfotenhauer2,3Aubrey B. Dorrough1
Alexander C. Pfotenhauer2,3Aubrey B. Dorrough1 Magen R. Poindexter1,2
Magen R. Poindexter1,2 Tarek Hewezi1Scott C. Lenaghan2,3
Tarek Hewezi1Scott C. Lenaghan2,3 David E. Graham4
David E. Graham4 C. Neal Stewart Jr.1,2*
C. Neal Stewart Jr.1,2*CRISPR/Cas9 has been widely applied to various plant species accelerating the pace of plant genome editing and precision breeding in crops. Unintended effects beyond off-target nucleotide mutations are still somewhat unexplored. We investigated the degree and patterns of epigenetic changes after gene editing. We examined changes in DNA methylation in genome-edited promoters of naturally hypermethylated genes (AT1G72350 and AT1G09970) and hypomethylated genes (AT3G17320 and AT5G28770) from Arabidopsis. Transgenic plants were developed via Agrobacterium-mediated floral dip transformation. Homozygous edited lines were selected from segregated T2 plants by an in vitro digestion assay using ribonucleoprotein complex. Bisulfite sequencing comparisons were made between paired groups of edited and non-edited plants to identify changes in DNA methylation of the targeted loci. We found that directed mutagenesis via CRISPR/Cas9 resulted in no unintended morphological or epigenetic alterations. Phenotypes of wild-type, transgenic empty vector, and transgenic edited plants were similar. Epigenetic profiles revealed that methylation patterns of promoter regions flanking target sequences were identical among wild-type, transgenic empty vector, and transgenic edited plants. There was no effect of mutation type on epigenetic status. We also evaluated off-target mutagenesis effects in the edited plants. Potential off-target sites containing up to 4-bp mismatch of each target were sequenced. No off-target mutations were detected in candidate sites. Our results showed that CRISPR/Cas9 did not leave an epigenetic footprint on either the immediate gene-edited DNA and flanking DNA or introduce off-target mutations.
As the global population continues to rapidly expand, food scarcity becomes a major issue. In recent decades, biotechnological advancements in genetic engineering have led to a great impact on modern agriculture through crop improvement. More recently, several precise genome editing tools have been developed using customizable, sequence-specific nucleases such as zinc-finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) (Gaj et al., 2013), fueling advances in the field of crop improvement. Among these genome editing approaches, CRISPR/Cas9 has rapidly become the best choice for gene editing in various plant species (reviewed by Jaganathan et al., 2018) because of its simplicity, efficiency, and design flexibility.
Despite its wide application for crop improvement, the risks of gene-editing technology need thorough examination. Off-target effects are one of the major concerns of using CRISPR/Cas9 because Cas9 is tolerant to some mismatched sequences distal from the protospacer adjacent motif (PAM). Although the off-target effects are unlikely to occur in plants (Bao et al., 2019), high-frequency off-target mutagenesis was reported in human cells (Fu et al., 2013) and represents a potential limitation for biomedical and clinical applications (Zhang et al., 2015). Therefore, many studies have been conducted to minimize off-target effects and to improve specificity of CRISPR/Cas9 (reviewed by Moon et al., 2019). Unintended effects beyond off-target mutations, however, are still somewhat unexplored. We know of no published studies that have explored epigenetic changes in genome-edited organisms.
Epigenetic changes, such as DNA methylation, histone modifications, and non-coding RNA changes, can affect genome stability and gene expression. DNA methylation is the most common epigenetic ‘footprint’ in plants and plays important roles in various biological processes such as transposon silencing, plant development, and plant responses to biotic and abiotic environmental stimuli (reviewed by Zhang et al., 2018). The patterns of methylation are important in various regions of DNA, including promoters. For example, genome-wide mapping of DNA methylation in Arabidopsis showed that, in general, methylation in transcribed regions tends to promote higher and constitutive gene expression, while genes methylated in promoter regions show tissue-specific expression (Zhang et al. 2006b). Promoter DNA methylation is usually associated with gene repression by direct inhibition of the binding of transcription factors (Domcke et al., 2015; Zhu et al., 2016), but in some cases it promotes gene expression by a still unknown mechanism (Song et al., 2013; Lang et al., 2017).
In the present study, we aim to assess epigenetic changes attributable to CRISPR/Cas9-mediated genome editing in plants. To evaluate epigenetic profiles, especially with regard to promoter methylation, we selected four genes in which a differentially methylated region was located on their promoter: naturally hypermethylated genes (AT1G72350 and AT1G09970) and hypomethylated genes (AT3G17320 and AT5G28770) from Arabidopsis. To our knowledge, this is the first study about potential effects of genome editing on DNA methylation status in plants.
Four Arabidopsis genes (AT1G72350, AT1G09970, AT3G17320, and AT5G28770) containing differentially methylated regions (200-bp long) in their promoters (Hewezi et al., 2017) were selected for CRISPR/Cas9-mediated genome editing. The 200-bp sequence was entered in the web-based tool CRISPOR (http://crispor.tefor.net/crispor.py; Haeussler et al., 2016) to design optimal gRNAs for each target in consideration of their GC content and the presence of potential off-target sites (Table 1). Polymerase chain reaction (PCR) products flanking the target sites were sequenced to evaluate any possible allelic variation or single-nucleotide polymorphisms. Unless noted otherwise, Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA) was used for all PCR. The primer sequences used in this study are listed in Supplementary Table S1.

Table 1 Target genes containing differentially methylated regions (200-bp) in their respective promoters, selected gRNA sequences within these regions, and gRNA GC-contents.
The binary vector pKSE401 (Addgene plasmid # 62202; Xing et al., 2014) was used in this study to generate sgRNA/Cas9 constructs. The synthesized oligonucleotides of gRNAs (Integrated DNA Technologies, Coralville, IA) specific to the target sequence with appropriate overhangs were annealed and then ligated into the BsaI site in the pKSE401 vector. In brief, two complementary single stranded oligonucleotides (one strand consisting of a 5ˊ-ATTG overhang plus 20-nt gRNA sequence, and the other strand consisting of a 5ˊ-AAAC overhang and the complementary sequence) were mixed together and incubated at 95°C for 2 min followed by cooling to 4°C using a thermal cycler. The pKSE401 vector was digested by BsaI, then the annealed double strand fragments were ligated into the vector.
The pKSE401 vectors harboring the sgRNA/Cas9 expression cassette were transformed into Agrobacterium tumefaciens strain EHA105 using the freeze-thaw method. For an empty vector control, pKSE401 vector without any gRNA sequence was transformed. The binary vectors were then introduced into Arabidopsis thaliana Col-0 plants via the floral dip method (Zhang et al. 2006a). T1 seedlings were selected on Murashige and Skoog plates containing kanamycin (50 mg/l) for 2 weeks, and then transplanted to soil for further growth for genotyping and selfing to the next generation. For bisulfite sequencing, homozygous edited plants were selected in the T2 generation. Experimental plants were grown in a growth room kept at 23°C under a 16 h photoperiod.
Genomic DNA from T1 plants was extracted using the CTAB method and the presence of the Cas9 gene was confirmed by PCR. PCR amplifications were carried out with an initial denaturation step at 98°C for 30 s, followed by 35 cycles comprising of 98°C for 10 s, 60°C for 30 s, and 72°C for 20 s, and a final extension for 10 min at 72°C. From the PCR-confirmed transgenic lines, DNA fragments flanking the target sites were amplified using specific primers and then directly sequenced by Sanger method to find edited lines. To select homozygous edited plants in the T2 generation, a genotyping method using PCR/RNP complex was adopted (Liang et al., 2018). In brief, template DNAs were amplified using specific primers and then used for in vitro sgRNA synthesis using HiScribeTM T7 Quick High Yield RNA Synthesis Kit (New England Biolabs) according to the manufacturer’s instructions. sgRNAs were then cleaned by Monarch® RNA Cleanup Kit (New England Biolabs) before adding to the Cas9 digestion mixture. For each digestion reaction, RNP complex in Cas9 reaction buffer (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 100 µg/ml BSA) consisted of 0.5 µg sgRNA, 0.5 µg Cas9 protein, and ddH2O up to 10 µl. The mixture was pre-incubated at 25°C for 10 min before adding 1 µl of PCR amplicon flanking the target site. The PCR/RNP mixture was then incubated at 37°C for 3 h for digestion, followed by incubation at 65°C for 10 min to stop the reaction. The reaction products were then analyzed immediately on 2% agarose gel. Only the PCR products undigested by RNP, which were assumed as homologous or biallelic mutants, were selected for direct sequencing to determine mutations.
To avoid variability caused by inbreeding, T2 homozygous plants from segregating populations were used for DNA methylation experiments. Experiments consisted of up to four independent biological replicates for each construct. Genomic DNA was extracted from leaves of 4-week-old plants using the Plant DNeasy Mini Kit (Qiagen, Germantown, MD). Aliquots of 500 ng of each DNA sample were subjected to bisulfite treatment using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. PCR was performed by using bisulfite-treated DNA as templates. The sequences of interest were amplified using polymerase Takara Ex Taq Hot Start (Clontech, Mountain View, CA) and methylation-neutral primers. PCR amplifications were carried out with an initial denaturation step at 94°C for 5 min, followed by 40 cycles comprising of 94°C for 45 s, 55°C for 45 s, and 72°C for 1 min, and a final extension for 10 min at 72°C. The PCR-amplified fragment of each bisulfite-treated DNA sample was gel-purified using the Zymoclean Gel DNA Recovery Kit (Zymo Research) and cloned into the pGEM-T Easy vector (Promega, Madison, WI). For each PCR amplicon, at least seven independent colonies were analyzed by Sanger sequencing. Unmethylated lambda phage DNA (Promega) was used as a control for bisulfite conversion efficiency. The primers used in this study are listed in Supplementary Table S1.
The potential off-target sites containing up to 4-bp mismatches were predicted by the web-based tool CRISPOR. Genomic DNA was extracted from selected T2 plants and PCR was performed using primer sets designed to flank the potential off-target sites. PCR products were then gel-purified using Zymoclean Gel DNA Recovery Kit (Zymo Research) and directly sequenced to evaluate off-target mutagenesis.
Four Arabidopsis genes containing differentially methylated regions (DMRs) in their promoters (Hewezi et al., 2017) were selected. Two hundred-bp promoter regions of two genes (AT1G72350 and AT1G09970) are naturally hypermethylated, whereas promoters of the other two genes (AT3G17320 and AT5G28770) are hypomethylated (Hewezi et al., 2017). CRISPR/Cas9 target sites were selected within the DMRs. To evaluate any possible allelic variation or single-nucleotide polymorphisms in the DMRs, genomic DNA was extracted from randomly selected wild-type plants, and the PCR amplicons flanking the regions were directly sequenced. The results confirmed that there were no variations within the selected target sites (data not shown). To make CRISPR/Cas9 vectors, two complementary oligonucleotides of gRNA sequence were annealed together and then placed between AtU6-26 promoter and gRNA scaffold sequence in pKSE401 vector in which Zea mays codon-optimized Cas9 was expressed by a CaMV 35S promoter (Figure 1). A total of four vectors were constructed to target the four genes to study the effects of genome editing on DNA methylation status.
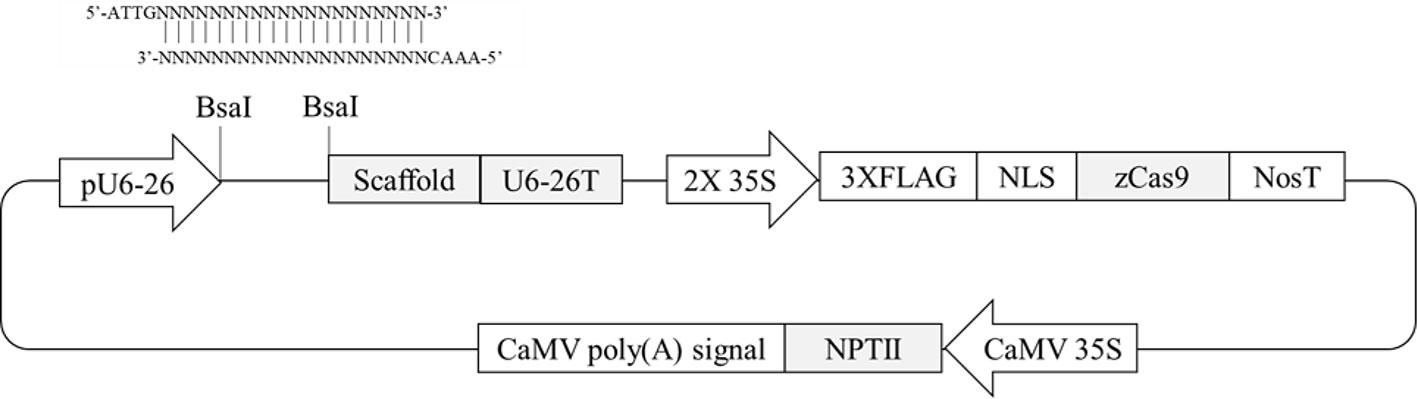
Figure 1 Schematic diagram of CRISPR/Cas9 plant transformation vector pKSE401. Oligonucleotides of gRNA sequence were annealed and then inserted at the BsaI-digestion site between the pU6-26 promoter and gRNA scaffold.
CRISPR/Cas9 vectors were transformed into A. thaliana Col-0 plants via the floral dip method, and putative transgenic plants were screened by kanamycin selection. Six to fourteen T1 plants per construct, in which the presence of Cas9 vector was confirmed by PCR, were used for genotyping to detect mutation and selfing to the next generation. PCR amplicons flanking the target sites were directly sequenced to check for mutations. The mutagenesis rates in T1 plants varied with a range of 30% to 100% depending on the target genes, but all the edited plants were heterozygous or biallelic mutants with multiple peaks on the sequence chromatogram downstream from the expected Cas9 cleavage sites (Table 2; Supplementary Figures S1–S4).
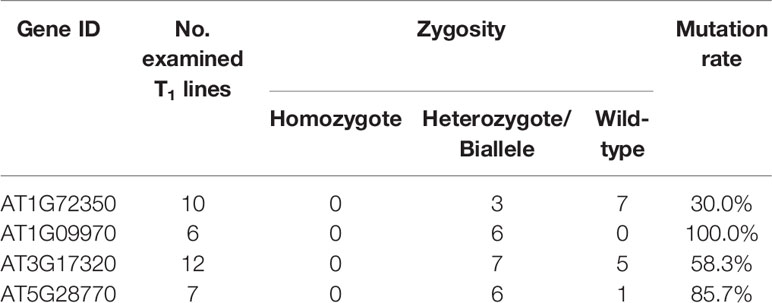
Table 2 Mutation events in T1 transgenic lines of the target genes.
In order to obtain homozygous edited plants for bisulfite sequencing, T2 plants were generated. To minimize the number of plants required for sequence confirmation from approximately twenty T2 seedlings per edited line, a recently developed genotyping method using PCR/RNP complex was applied (Liang et al., 2018). Because homozygous or biallelic mutants have lost their complementary sequence to the gRNA, PCR amplicons of the target site are not digested by Cas9, resulting in a single uncut band only (Figure 2; lanes 1, 3, 12, and 14). PCR amplicons from heterozygous mutants, however, contained both edited and original gRNA sequences that were partially digested by Cas9, which resulted in two bands (Figure 2; lanes 2, 7, 8 10, and 13). Unedited T2 plants were also generated by segregation from heterozygous T1 mutants, and their PCR amplicons were completely digested like the wild-type control (Figure 2; lanes 4, 5, 6, 9, and 11). Based on the digestion pattern from the PCR/RNP assay, we selected the lines with exclusively uncut bands to be used for further sequence analysis. Direct sequencing of the PCR amplicons detected mutations for all four target genes: 1-nt deletion (C) in AT1G72350; 1-nt insertion (+G or +T) in AT1G09970; 1-nt insertion (+T) and 5-nt deletion (GCTGG) in AT3G17320; and 1-nt insertion (+A) and 4-nt deletion (GACG) in AT5G28770 (Figure 3). No phenotypic differences were apparent (Figure 4), and we used these plants for allele-specific bisulfite sequencing analysis.
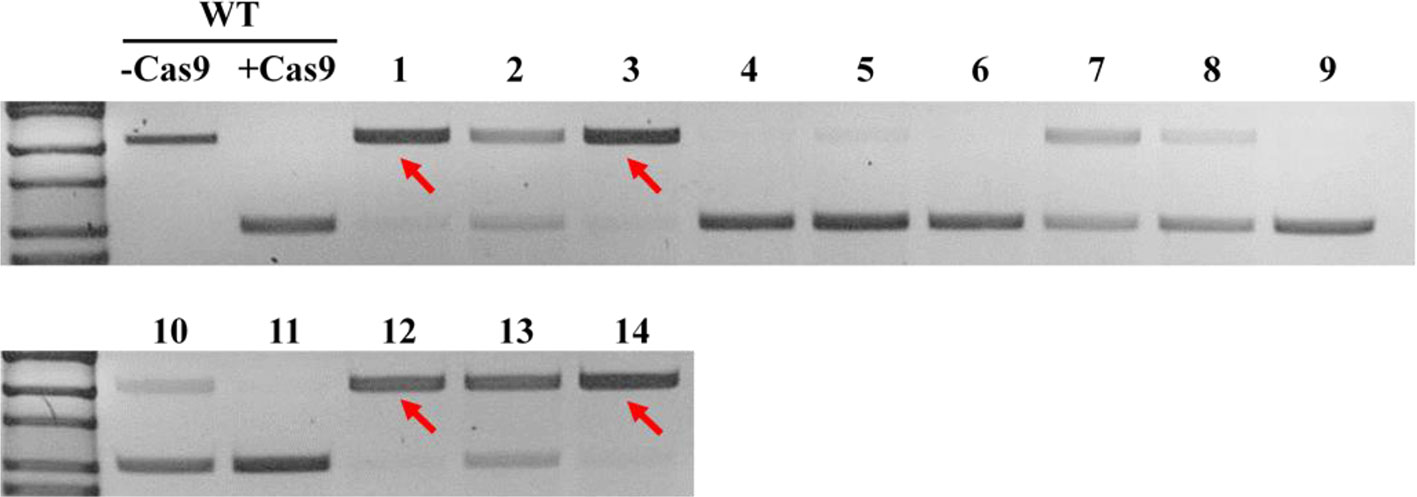
Figure 2 Representative genotyping result using PCR/ribonucleoprotein (RNP) complex to select potential homozygous lines. Independent edited plants in the T2 generation targeting AT1G72350 were analyzed using PCR/RNP complex, a mixture of in vitro synthesized sgRNA, Cas9 enzyme, and PCR products flanking target site. PCR amplicons identical to the sgRNA were completely digested by Cas9 (lanes 4, 5, 6, 9, and 11), while amplicons of homozygous or biallelic-mutants (red arrows) were not digested. Partially digested amplicons showing both cut- and uncut-bands indicated heterozygous mutants (lanes 2, 7, 8, 10, and 13).
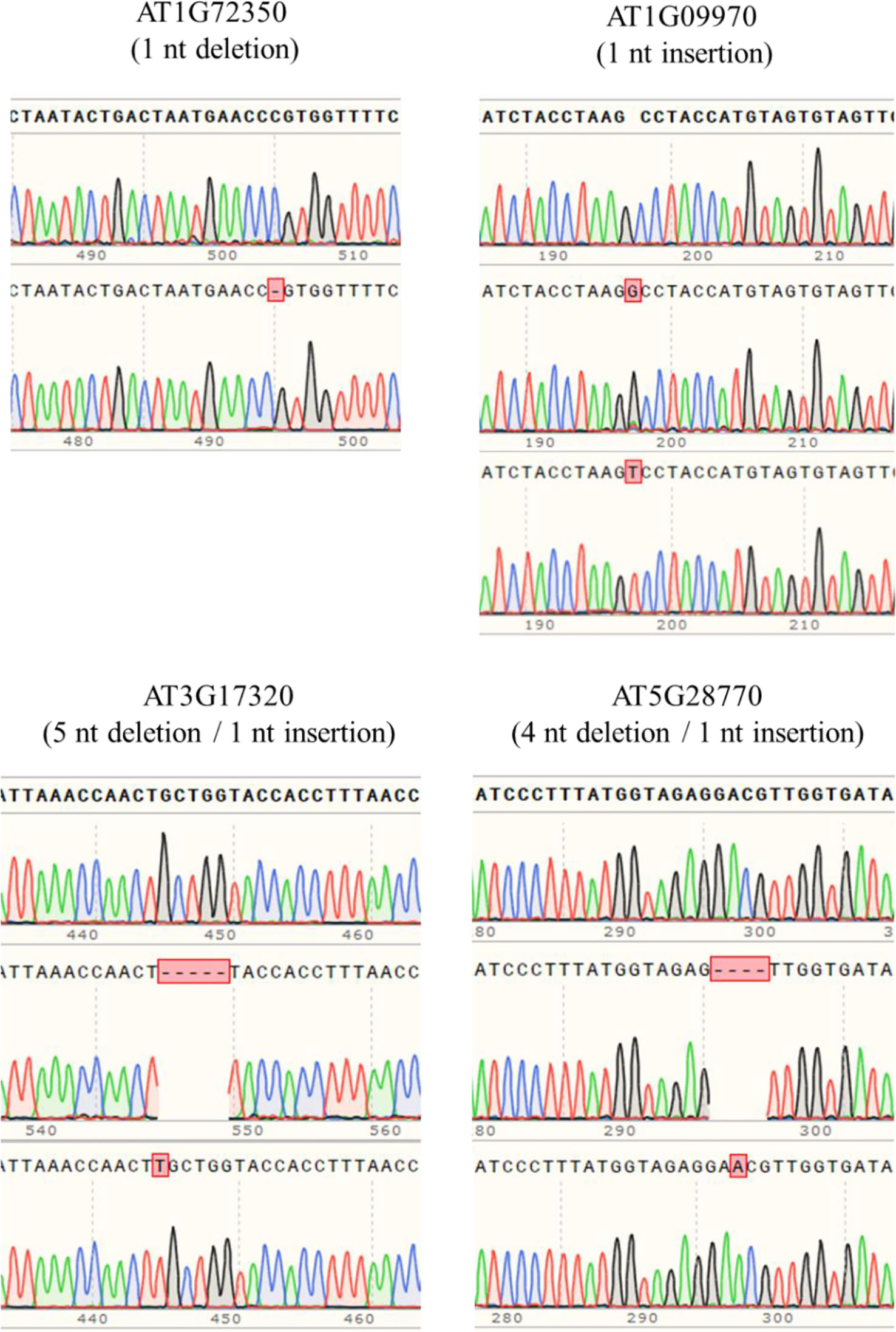
Figure 3 Detection of mutations by the Sanger sequencing method. PCR products flanking target sites were amplified from putative homozygous edited T2 plants, and then directly sequenced. Sequence alignment showed that 1-nt insertion/deletion was the major mutation type, but up to 5-nt deletion occurred.

Figure 4 Representative wild type, transgenic empty vector, and transgenic edited T2 plants at 4 weeks old.
According to analysis using the web-based tool CRISPOR, there were no homologous sequences to the four gRNAs flanking a PAM sequence, and no loci containing even up to 3-nt mismatches were found. Potential off-target sites with 4-nt mismatches were predicted for three target genes: 8 loci for AT1G72350; 2 loci for AT1G09970; and 6 loci for AT3G17320 (Table 3). The potential off-target sites were amplified from selected T2 plants using the specific primers and then directly sequenced. Sequence alignment analysis revealed that no off-target mutagenesis occurred. We also checked the presence of CRISPR/Cas9 construct in the selected T2 plants. Most of the plants still contained the construct (Supplementary Figure S5) with the exception of three lines targeting AT3G7320, indicating off-target mutagenesis on loci with 4-nt mismatches may not be likely to occur in plants by CRISPR/Cas9 editing.

Table 3 Potential off-target analysis of edited T2 plants.
To identify changes in DNA methylation patterns associated with CRISPR/Cas9-mediated genome editing, we used the bisulfite sequencing approach for analyzing bisulfite-converted DNA providing single-base resolution across the entire amplicon. Experiments were performed with wild-type, transgenic empty vector, and transgenic edited plants (Figure 4). Cloning of bisulfite PCR products followed by sequencing with vector-specific primers was performed to obtain the best sequencing results for quantification of methylation. The degree of bisulfite-conversion was determined by sequencing. Bisulfite conversion efficiency was up to 97%, as determined using unmethylated lambda phage DNA. DNA methylation of the edited plants in the locus-specific gene-edited promoters of hypermethylated (AT1G72350 and AT1G09970) and hypomethylated (AT3G17320 and AT5G28770) genes was compared to that of the control plants. These analyses showed no alterations in methylation patterns of the corresponding locus in wild-type, transgenic empty vector, and transgenic edited plants (Figures 5–8). Also, there was no apparent association between types of mutation and methylation patterns (Figures 7C, D and 8C, D). We also performed cross-check analysis to see if there were methylation pattern changes in other areas of the genome that was not targeted: (AT1G72350) region in AT1G09970-edited plant; (AT1G09970) region in AT1G72350-edited plant; (AT3G17320) region in AT5G28770-edited plant; and (AT5G28770) region in AT3G17320-edited plant. We selected these genes since in the previous study by Hewezi et al., 2017, these genes showed differentially methylated patterns by changing the methylation status in response to stimuli. We wanted to examine whether Cas9-associated biological activities affect DNA methylation status not just on the target sites where Cas9 binding followed by DNA repair processes, but also on other sites in the genome. No differences in methylation patterns of the target regions were detected between the wild type and the edited plants (Supplementary Figures S6–S9). Altogether, our results suggest that CRISPR/Cas9-mediated genome editing did not cause unintended effects and did not leave epigenetic artifacts on target sequences.
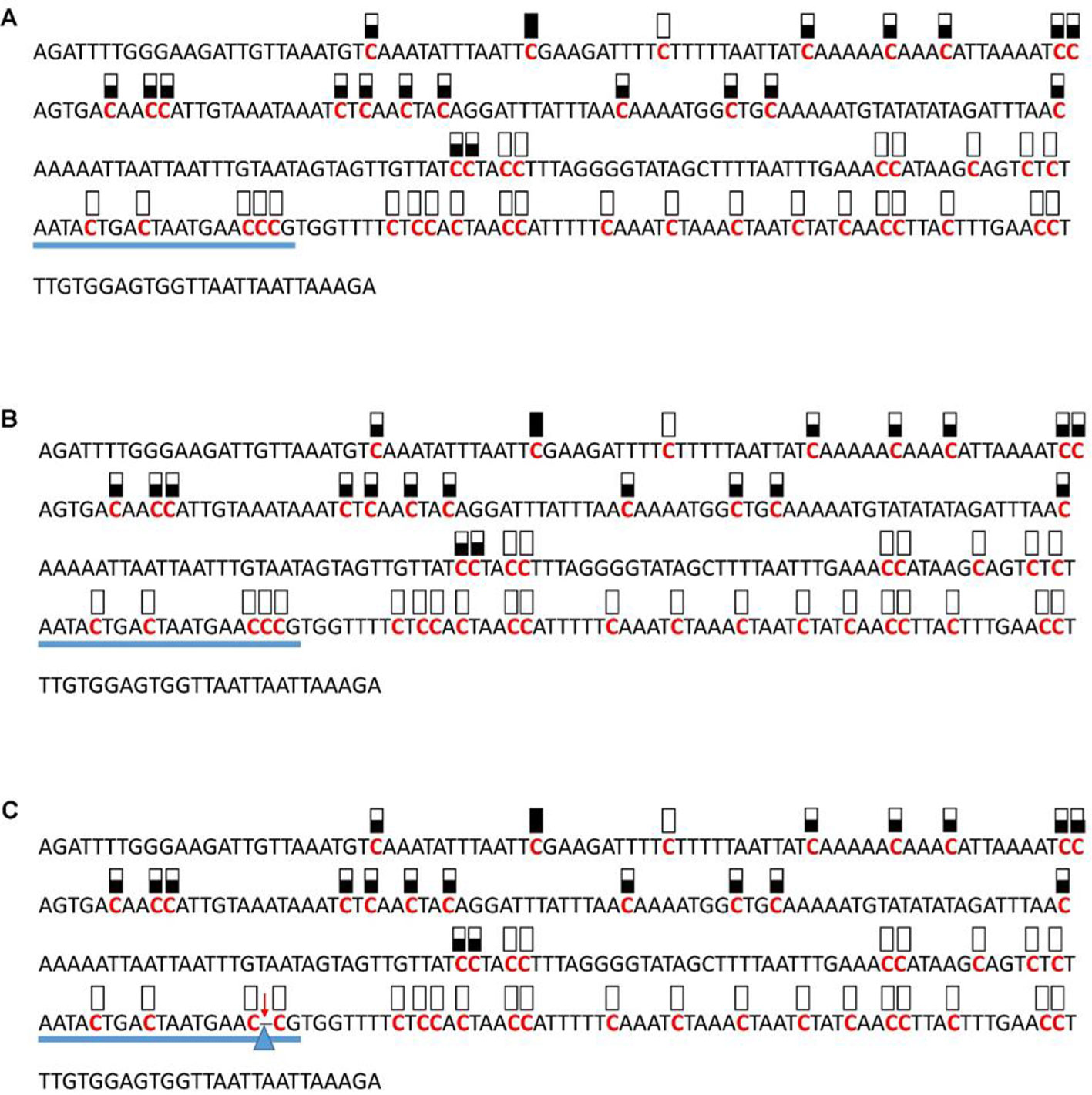
Figure 5 Pattern of methylation of the AT1G72350 promoter in wild-type (A), transgenic empty vector (B), and transgenic edited (C) plants. Because the data were obtained by sequencing of independent pGEM-T colonies, an average level of methylation was determined for each cytosine. Solid boxes indicate that the cytosine at this position were methylated (>90%), open boxes indicate that cytosine methylation was not detected (<10%), and half-shaded boxes indicate that the cytosine was methylated at a range of 40-60%. Underlines indicate the 20-bp target sequences. The arrow and triangle in (C) indicate the one nucleotide “C” deletion. This figure presents results of 4 independent biological replicates.
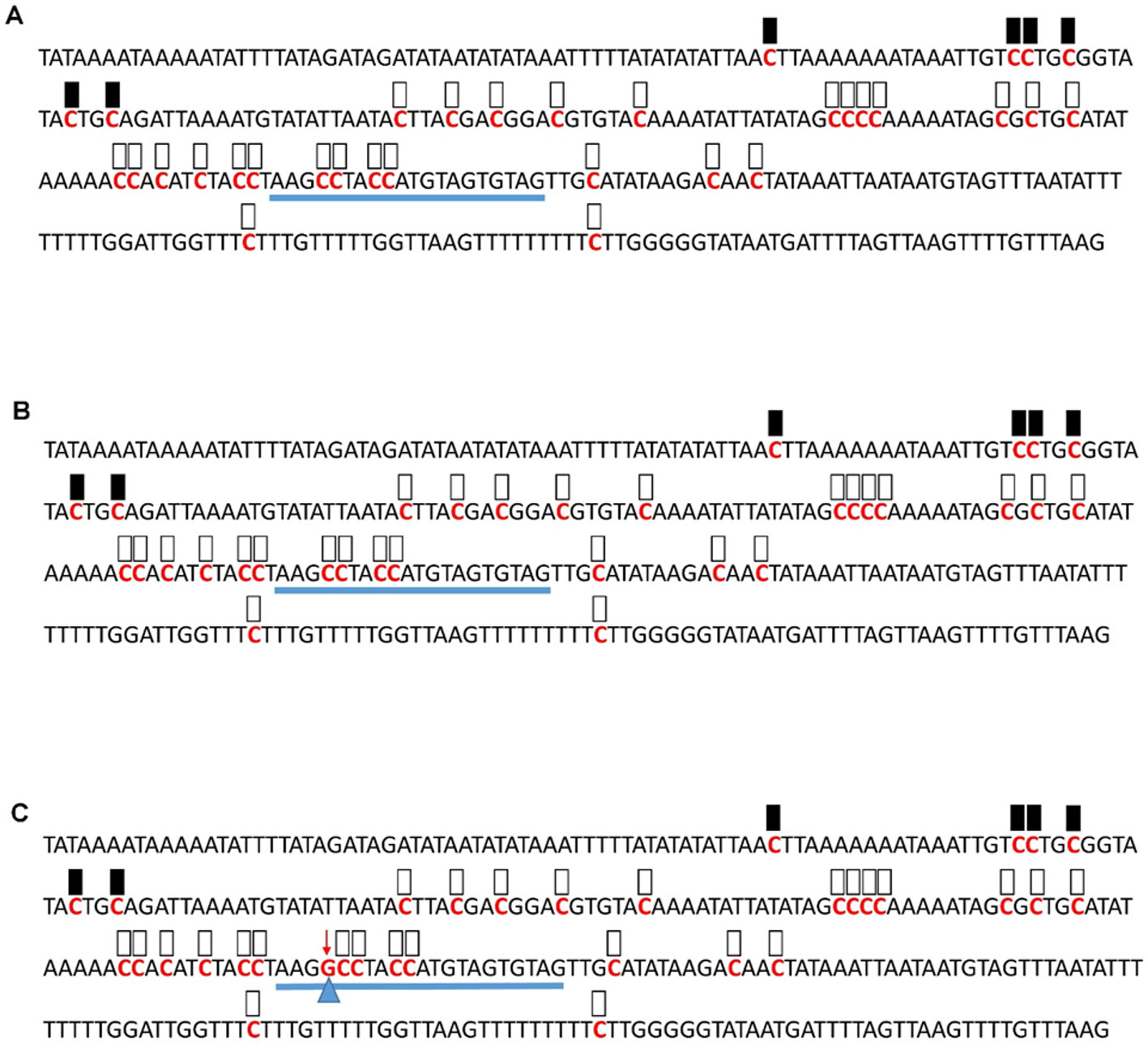
Figure 6 Pattern of methylation of the AT1G09970 promoter in wild-type (A), transgenic empty vector (B), and transgenic edited (C) plants. Because the data were obtained by sequencing of independent pGEM-T colonies, an average level of methylation was determined for each cytosine. Solid boxes indicate that the cytosine at this position were methylated (>90%), open boxes indicate that cytosine methylation was not detected (<10%), and half-shaded boxes indicate that the cytosine was methylated at a range of 40-60%. Underlines indicate the 20-bp target sequences. The arrow and triangle in (C) indicate the one nucleotide “G” addition. This figure presents results of 4 independent biological replicates.
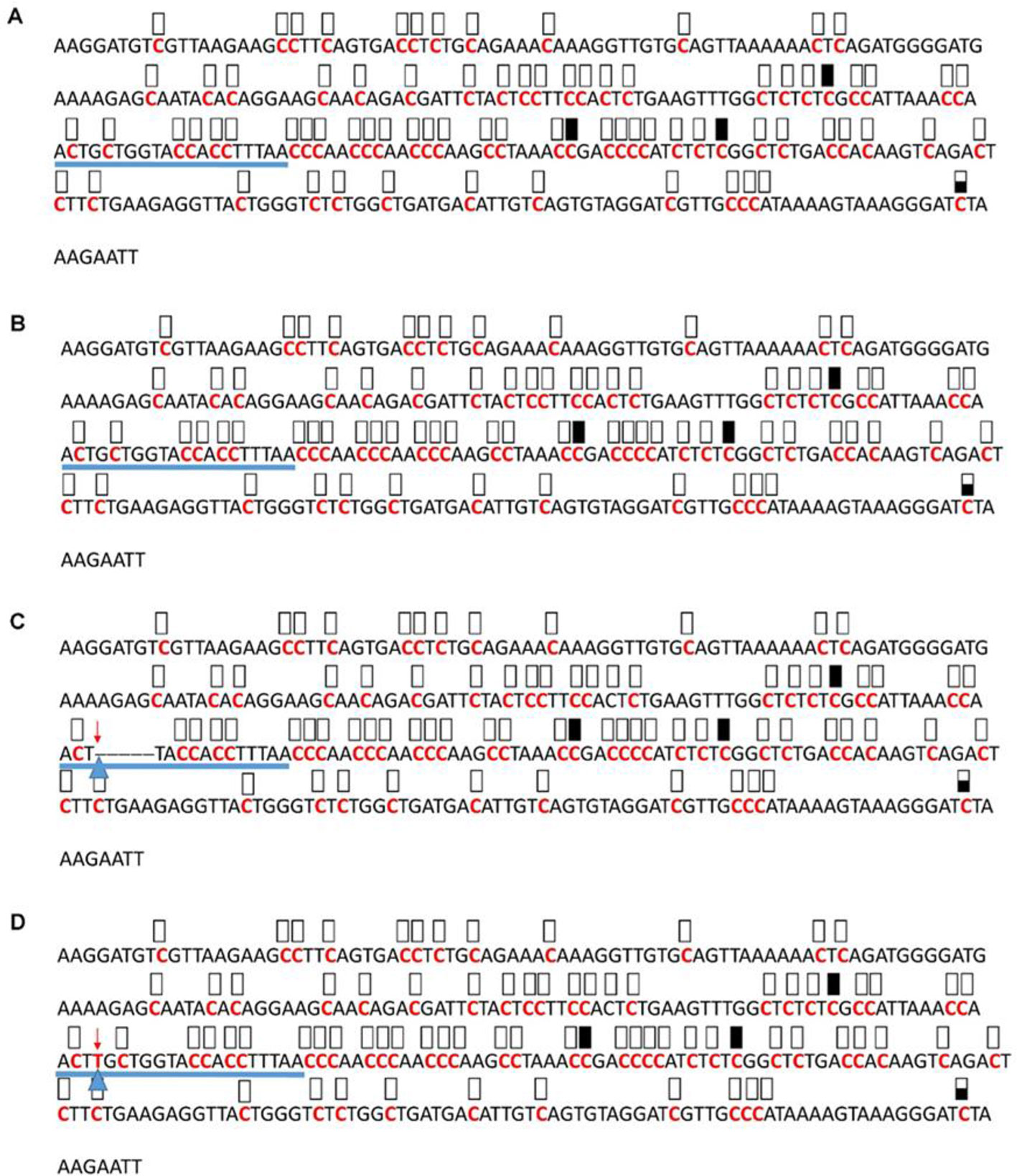
Figure 7 Pattern of methylation of the AT3G17320 promoter in wild-type (A), transgenic empty vector (B), and transgenic edited (C, D) plants. Because the data were obtained by sequencing of independent pGEM-T colonies, an average level of methylation was determined for each cytosine. Solid boxes indicate that the cytosine at this position were methylated (>90%), open boxes indicate that cytosine methylation was not detected (<10%), and half-shaded boxes indicate that the cytosine was methylated at a range of 40-60%. Underlines indicate the 20-bp target sequences. The arrow and triangle in (C) indicate the five nucleotide “GCTGG” deletion. The arrow and triangle in (D) indicate the one nucleotide “T” addition. This figure presents results of 4 independent biological replicates.
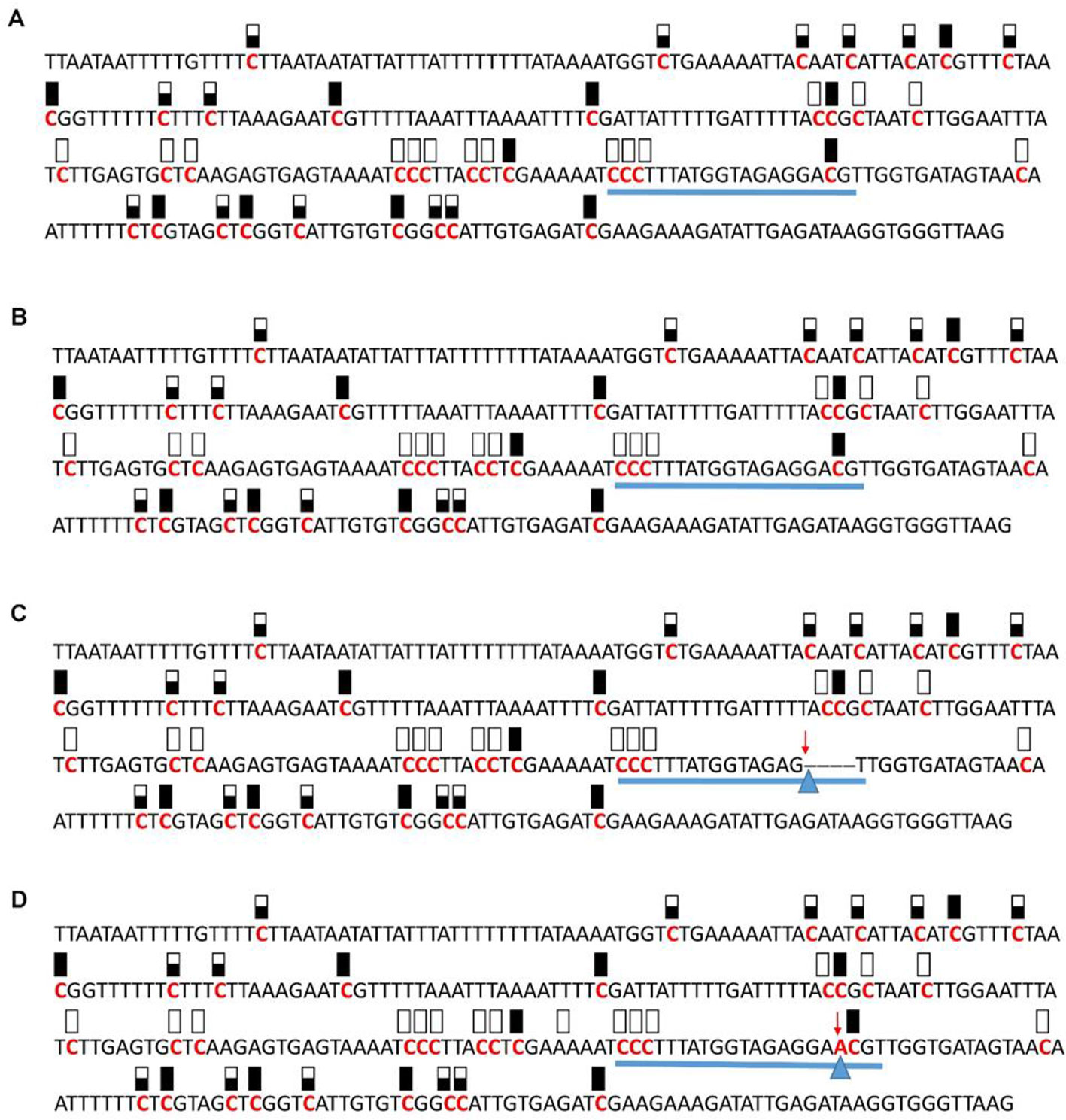
Figure 8 Pattern of methylation of the AT5G28770 promoter in wild-type (A), transgenic empty vector (B), and transgenic edited (C, D) plants. Because the data were obtained by sequencing of independent pGEM-T colonies, an average level of methylation was determined for each cytosine. Solid boxes indicate that the cytosine at this position were methylated (>90%), open boxes indicate that cytosine methylation was not detected (<10%), and half-shaded boxes indicate that the cytosine was methylated at a range of 40-60%. Underlines indicate the 20-bp target sequences. The arrow and triangle in (C) indicate the four nucleotide “GACG” deletion. The arrow and triangle in (D) indicate the one nucleotide “A” addition. This figure presents results of 4 independent biological replicates.
CRISPR/Cas9 approaches are powerful tools for crop improvement and are largely considered to be viable precision-breeding methods (Scheben et al., 2017). For example, major traits associated with productivity in ‘groundcherry’ (Physalis pruinsa) were improved by CRISPR/Cas9-mediated mutation on orthologues of tomato domestication and improvement genes, SELF-PRUNING genes, and CLAVATA1 gene that control plant architecture, flower production, and fruit size (Lemmon et al., 2018). Although some CRISPR/Cas9-edited crops are not currently regulated as genetically modified organisms (GMO) by the USDA in the United States (Waltz, 2018), the controversy surrounding the regulation of genome-edited crops is still in flux. The Court of Justice of the European Union (ECJ) recently ruled that CRISPR/Cas9-edited plants should be subject to the same draconian GM regulations in the EU (Callaway, 2018). In order to answer regulatory questions and engender public acceptance, potential adverse effects in genome-edited plants should be thoroughly assessed. In the present study, we provided locus-specific epigenetic profiles of edited plants to assess potential adverse effects of plant genome editing by CRISPR/Cas9. We generated transgenic Arabidopsis plants to edit four target genes that have differentially methylated regions in their promoters using CRISPR/Cas9. In T1 plants, we obtained 30—100% mutation frequencies (Table 2), which were similar to published studies. In dicot plants, a wide range of mutation frequency has been reported in CRISPR/Cas9-mediated genome editing, ranging from 30% to 92% in Arabidopsis (Feng et al., 2014); from 81.8% to 87.5% in tobacco (Gao et al., 2015); from 84% to 100% in tomato (Pan et al., 2016); and from 20% to 100% in soybean (Cai et al., 2018). Our previous study suggested that gRNA GC-content may play a role in sgRNA efficacy; no edited plants were generated when gRNAs with less than 40% GC-content were used, and a higher mutation frequency was obtained when a higher GC-content was used (unpublished data). Similar results were reported in other studies (Zhang et al., 2014; Pan et al., 2016), but very high GC-content was also less effective and may increase the risk of off-target cleavage (Wang et al., 2014; Tsai et al., 2015).
Off-target mutation is a common and critical problem associated with CRISPR/Cas9-mediated genome editing in human cells, but it has rarely been reported in gene-edited plants. Several studies have reported that there was no off-target mutation, not only in Arabidopsis, but also various crop species (Feng et al., 2014; Pan et al., 2016; Braatz et al., 2017; Jia et al., 2017; Tang et al., 2017; Cai et al., 2018). We analyzed the potential off-target mutagenesis on loci highly similar to the target sequence. No mutations were detected at any of these candidate off-target sites (Table 3), demonstrating that we designed highly specific gRNAs. It has been proposed that the risk of off-target mutations in plants, unlike that in therapeutic applications, may not be a critical issue because any unwanted phenotypic effects caused by off-target mutations or somatic mutations can be eliminated by the subsequent selection process of plant tissue culture-based transformation after editing (Zhao and Wolt, 2017).
DNA methylation is one of the most extensively studied epigenetic modifications of genomic DNA. Numerous DNA methylation studies have demonstrated that DNA methylation is critical in many regulatory processes such as silencing of gene expression, cellular differentiation, transposon mobility, genome stability, and genomic imprinting (Bewick and Schmitz, 2017; Elhamamsy, 2017; Niederhuth and Schmitz, 2017; Yaish, 2017; Bartels et al., 2018; Bräutigam and Cronk, 2018; Zhang et al., 2018). Much effort has been paid to characterize variation of DNA methylation across different biological samples, developmental stages, and disease status (Zhang et al. 2006b; Dinh et al., 2012; Breuil-Broyer et al., 2016; Hewezi et al., 2018; Xu et al., 2018), however, changes in DNA methylation patterns associated with CRISPR/Cas9-mediated genome editing have not been explored yet. We examined the impact of CRISPR/Cas9-mediated genome editing on the DNA methylation patterns of four gene promoters (hypermethylated genes AT1G72350 and AT1G09970; and hypomethylated genes AT3G17320 and AT5G28770) in which differentially methylated region was located on their promoter (Hewezi et al., 2017). We conducted targeted DNA methylation analysis by treating genomic DNA with bisulfite, amplifying and sequencing targeted regions of interest. Bisulfite genomic sequencing is regarded as a gold-standard technology for detection of DNA methylation over the genomes of interest because it provides a qualitative, quantitative, and efficient approach to identify methylcytosine at single base-pair resolution (Li and Tollefsbol, 2011; Wreczycka et al., 2017). Bisulfite-mediated cytosine conversion paired with subsequent PCR amplification and sequencing represents a highly promising approach (Henderson et al, 2010; Masser et al., 2015). We performed Sanger sequencing of bisulfite converted DNA, which is the most used methods for analysis of targeted regions (i.e., a promoter region of a single gene). Under these experimental conditions, our results showed that CRISPR/Cas9-mediated genome editing did not change DNA methylation patterns among wild-type, transgenic empty vector, and transgenic edited plants, regardless of the types of mutation caused by CRISPR/Cas9-mediated gene editing in the same target gene promoter and/or between the gene promoters by cross-check analysis. Our results suggest that there was not an association between CRISPR/Cas9-mediated genome editing and DNA methylation. In this context, given the increased knowledge about highly efficient CRISPR/Cas9 as the most-used genome editing tool, determining unintended effects beyond off-target mutations such as methylation status presented here provides further insights into the application of this precise genome editing platform.
All datasets generated for this study are included in the article/Supplementary Material.
JL and MM designed and performed the experiments, analyzed the data, and wrote the manuscript. AP designed gRNAs and performed cloning of CRISPR/Cas9 constructs. AD conducted off-target analysis. MP performed genomic DNA extractions, PCR, cloning of bisulfite converted fragments, and assisted with plant care. TH, SL, DG, and CS conceived of the study and its design and coordination, and assisted with interpretation of results and revisions to the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors, NS.
We would like to thank Ralph Laurel for his help with plant care and Sarbottam Piya for useful discussion. This research was sponsored in part by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed by UT-Battelle, LLC, for the US Department of Energy (DOE) under contract DE-AC05-00OR22725. This work was partially funded by the Center for Bioenergy Innovation supported by the Office of Biological and Environmental Research in the DOE Office of Science. This manuscript has been co-authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the DOE. The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan). We also acknowledge funding from Hatch grants to CNS, SCL, and TH, as well as funding from UT AgResearch to support the Center for Agricultural Synthetic Biology.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01720/full#supplementary-material
Bao, A., Burritt, D. J., Chen, H., Zhou, X., Cao, D., Tran, L.-S. P. (2019). The CRISPR/Cas9 system and its applications in crop genome editing. Crit. Rev. Biotechnol. 39, 321–336. doi: 10.1080/07388551.2018.1554621
Bartels, A., Han, Q., Nair, P., Stacey, L., Gaynier, H., Mosley, M., et al. (2018). Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 19, 2144. doi: 10.3390/ijms19072144
Bewick, A. J., Schmitz, R. J. (2017). Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 36, 103–110. doi: 10.1016/j.pbi.2016.12.007
Braatz, J., Harloff, H.-J., Mascher, M., Stein, N., Himmelbach, A., Jung, C. (2017). CRISPR-Cas9 targeted mutagenesis leads to simultaneous modification of different homoeologous gene copies in polyploid oilseed rape (Brassica napus). Plant Physiol. 174, 935–942. doi: 10.1104/pp.17.00426
Bräutigam, K., Cronk, Q. (2018). DNA methylation and the evolution of developmental complexity in plants. Front. Plant Sci. 9, 1447. doi: 10.3389/fpls.2018.01447
Breuil-Broyer, S., Trehin, C., Morel, P., Boltz, V., Sun, B., Chambrier, P., et al. (2016). Analysis of the Arabidopsis superman allelic series and the interactions with other genes demonstrate developmental robustness and joint specification of male–female boundary, flower meristem termination and carpel compartmentalization. Ann. Bot. 117, 905–923. doi: 10.1093/aob/mcw023
Cai, Y., Chen, L., Liu, X., Guo, C., Sun, S., Wu, C., et al. (2018). CRISPR/Cas9-mediated targeted mutagenesis of GmFT2a delays flowering time in soya bean. Plant Biotechnol. J. 16, 176–185. doi: 10.1111/pbi.12758
Callaway, E. (2018). CRISPR plants now subject to tough GM laws in European Union. Nature 560, 16. doi: 10.1038/d41586-018-05814-6
Dinh, H. Q., Dubin, M., Sedlazeck, F. J., Lettner, N., Scheid, O. M., von Haeseler, A. (2012). Advanced methylome analysis after bisulfite deep sequencing: an example in Arabidopsis. PloS One 7, e41528. doi: 10.1371/journal.pone.0041528
Domcke, S., Bardet, A. F., Ginno, P. A., Hartl, D., Burger, L., Schübeler, D. (2015). Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528, 575–579. doi: 10.1038/nature16462
Elhamamsy, A. R. (2017). Role of DNA methylation in imprinting disorders: an updated review. J. Assist. Reprod. Gen. 34, 549–562. doi: 10.1007/s10815-017-0895-5
Feng, Z., Mao, Y., Xu, N., Zhang, B., Wei, P., Yang, D.-L., et al. (2014). Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 111, 4632–4637. doi: 10.1073/pnas.1400822111
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826. doi: 10.1038/nbt.2623
Gaj, T., Gersbach, C. A., Barbas, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405. doi: 10.1016/j.tibtech.2013.04.004
Gao, J., Wang, G., Ma, S., Xie, X., Wu, X., Zhang, X., et al. (2015). CRISPR/Cas9-mediated targeted mutagenesis in Nicotiana tabacum. Plant Mol. Biol. 87, 99–110. doi: 10.1007/s11103-014-0263-0
Haeussler, M., Schönig, K., Eckert, H., Eschstruth, A., Mianné, J., Renaud, J.-B., et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148. doi: 10.1186/s13059-016-1012-2
Henderson, I. R., Chan, S. R., Cao, X., Johnson, L., Jacobsen, S. E. (2010). Accurate sodium bisulfite sequencing in plants. Epigenetics 5, 47–49. doi: 10.4161/epi.5.1.10560
Hewezi, T., Lane, T., Piya, S., Rambani, A., Rice, J. H., Staton, M. (2017). Cyst nematode parasitism induces dynamic changes in the root epigenome. Plant Physiol. 174, 405–420. doi: 10.1104/pp.16.01948
Hewezi, T., Pantalone, V., Bennett, M., Stewart, C. N., Jr., Burch-Smith, T. M. (2018). Phytopathogen-induced changes to plant methylomes. Plant Cell Rep. 37, 17–23. doi: 10.1007/s00299-017-2188-y
Jaganathan, D., Ramasamy, K., Sellamuthu, G., Jayabalan, S., Venkataraman, G. (2018). CRISPR for crop improvement: an update review. Front. Plant Sci. 9, 985. doi: 10.3389/fpls.2018.00985
Jia, H., Zhang, Y., Orbović, V., Xu, J., White, F. F., Jones, J. B., et al. (2017). Genome editing of the disease susceptibility gene CsLOB1 in citrus confers resistance to citrus canker. Plant Biotechnol. J. 15, 817–823. doi: 10.1111/pbi.12677
Lang, Z., Wang, Y., Tang, K., Tang, D., Datsenka, T., Cheng, J., et al. (2017). Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. U.S.A. 114, E4511–E4519. doi: 10.1073/pnas.1705233114
Lemmon, Z. H., Reem, N. T., Dalrymple, J., Soyk, S., Swartwood, K. E., Rodriguez-Leal, D., et al. (2018). Rapid improvement of domestication traits in an orphan crop by genome editing. Nat. Plants 4, 766–770. doi: 10.1038/s41477-018-0259-x
Li, Y., Tollefsbol, T. O. (2011). DNA methylation detection: bisulfite genomic sequencing analysis. Methods Mol. Biol. 791, 11–21. doi: 10.1007/978-1-61779-316-5_2
Liang, Z., Chen, K., Yan, Y., Zhang, Y., Gao, C. (2018). Genotyping genome-edited mutations in plants using CRISPR ribonucleoprotein complexes. Plant Biotechnol. J. 16, 2053–2062. doi: 10.1111/pbi.12938
Masser, D. R., Stanford, D. R., Freeman, W. M. (2015). Targeted DNA methylation analysis by next-generation sequencing. J. Vis. Exp. 96, e52488. doi: 10.3791/52488
Moon, S. B., Ko, J.-H., Kim, J.-S., Kim, Y.-S. (2019). Improving CRISPR genome editing by engineering guide RNAs. Trends Biotechnol. 37, 870–881. doi: 10.1016/j.tibtech.2019.01.009
Niederhuth, C. E., Schmitz, R. J. (2017). Putting DNA methylation in context: from genomes to gene expression in plants. Biochim. Biophys. Acta 1860, 149–156. doi: 10.1016/j.bbagrm.2016.08.009
Pan, C., Ye, L., Qin, L., Liu, X., He, Y., Wang, J., et al. (2016). CRISPR/Cas9-mediated efficient and heritable targeted mutagenesis in tomato plants in the first and later generations. Sci. Rep. 6, 24765. doi: 10.1038/srep46916
Scheben, A., Wolter, F., Batley, J., Puchta, H., Edwards, D. (2017). Towards CRISPR/Cas crops – bringing together genomics and genome editing. New Phytol. 216, 682–698. doi: 10.1111/nph.14702
Song, Q.-X., Lu, X., Li, Q.-T., Chen, H., Hu, X.-Y., Ma, B., et al. (2013). Genome-wide analysis of DNA methylation in soybean. Mol. Plant 6, 1961–1974. doi: 10.1093/mp/sst123
Tang, L., Mao, B., Li, Y., Lv, Q., Zhang, L., Chen, C., et al. (2017). Knockout of OsNramp5 using the CRISPR/Cas9 system produces low Cd-accumulating indica rice without compromising yield. Sci. Rep. 7, 1–12. doi: 10.1038/s41598-017-14832-9
Tsai, S. Q., Zheng, Z., Nguyen, N. T., Liebers, M., Topkar, V. V., Thapar, V., et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197. doi: 10.1038/nbt.3117
Waltz, E. (2018). With a free pass, CRISPR-edited plants reach market in record time. Nat. Biotechnol. 36, 6–7. doi: 10.1038/nbt0118-6b
Wang, T., Wei, J. J., Sabatini, D. M., Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. doi: 10.1126/science.1246981
Wreczycka, K., Gosdschan, A., Yusuf, D., Grüning, B., Assenov, Y., Akalin, A. (2017). Strategies for analyzing bisulfite sequencing data. J. Biotechnol. 261, 105–115. doi: 10.1016/j.jbiotec.2017.08.007
Xing, H.-L., Dong, L., Wang, Z.-P., Zhang, H.-Y., Han, C.-Y., Liu, B., et al. (2014). A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 14, 327. doi: 10.1186/s12870-014-0327-y
Xu, Y., Zhang, L., Wu, G. (2018). Epigenetic regulation of juvenile-to-adult transition in plants. Front. Plant Sci. 9, 1048. doi: 10.3389/fpls.2018.01048
Yaish, M. W. (2017). Epigenetic modifications associated with abiotic and biotic stresses in plants: an implication for understanding plant evolution. Front. Plant Sci. 8, 1983. doi: 10.3389/fpls.2017.01983
Zhang, H., Lang, Z., Zhu, J.-K. (2018). Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506. doi: 10.1038/s41580-018-0016-z
Zhang, H., Zhang, J., Wei, P., Zhang, B., Gou, F., Feng, Z., et al. (2014). The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol. J. 12, 797–807. doi: 10.1111/pbi.12200
Zhang, X., Henriques, R., Lin, S.-S., Niu, Q.-W., Chua, N.-H. (2006a). Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 1, 641–646. doi: 10.1038/nprot.2006.97
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S. W., Chen, H., et al. (2006b). Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126, 1189–1201. doi: 10.1016/j.cell.2006.08.003
Zhang, X.-H., Tee, L. Y., Wang, X.-G., Huang, Q.-S., Yang, S.-H. (2015). Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids 4, e264. doi: 10.1038/mtna.2015.37
Zhao, H., Wolt, J. D. (2017). Risk associated with off-target plant genome editing and methods for its limitation. Emerg. Top. Life Sci. 1, 231–240. doi: 10.1042/ETLS20170037
Keywords: CRISPR/Cas9, genome editing, bisulfite sequencing, DNA methylation, epigenetic change
Citation: Lee JH, Mazarei M, Pfotenhauer AC, Dorrough AB, Poindexter MR, Hewezi T, Lenaghan SC, Graham DE and Stewart CN Jr. (2020) Epigenetic Footprints of CRISPR/Cas9-Mediated Genome Editing in Plants. Front. Plant Sci. 10:1720. doi: 10.3389/fpls.2019.01720
Received: 29 October 2019; Accepted: 06 December 2019;
Published: 31 January 2020.
Edited by:
Nicola Joan Patron, Earlham Institute, United KingdomReviewed by:
Holger Puchta, Karlsruhe Institute of Technology (KIT), GermanyCopyright © 2020 Lee, Mazarei, Pfotenhauer, Dorrough, Poindexter, Hewezi, Lenaghan, Graham and Stewart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Neal Stewart Jr., bmVhbHN0ZXdhcnRAdXRrLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.