 Jieping Li
Jieping Li Fanjun Chen
Fanjun Chen Yanqing Li1
Yanqing Li1 Pengcheng Li
Pengcheng Li Guohua Mi
Guohua Mi Lixing Yuan
Lixing Yuan- 1Key Laboratory of Plant-Soil Interaction, MOE, Department of Plant Nutrition, College Resources and Environmental Sciences, China Agricultural University, Beijing, China
- 2Key Laboratory of Plant Stress Biology, State Key Laboratory of Cotton Biology, Department of Plant Science, School of Life Sciences, Henan University, Kaifeng, China
- 3Jiangsu Key Laboratory of Crop Genetics and Physiology, Key Laboratory of Plant Functional Genomics, Co-Innovation Center for Modern Production Technology of Grain Crops, MOE, College of Agriculture, Yangzhou University, Yangzhou, China
In maize, shoot-borne roots dominate the whole root system and play essential roles in water and nutrient acquisition and lodging tolerance. Shoot-borne roots initiate at shoot nodes, including crown roots from the belowground nodes and brace roots from aboveground nodes. In contrast to crown roots, few genes for brace roots development have been identified. Here, we characterized a maize AP2/ERF transcription factor, ZmRAP2.7, to be involved in brace roots development. ZmRAP2.7 expressed in all types of roots, and the encoded protein localized in the nucleus with transcriptional activation activity. A maize transposon insert mutant RAP2.7-Mu defective in ZmRAP2.7 expression revealed a decreased number of brace roots but not crown roots. Maize Corngrass1 mutant, which showed an elevated expression of ZmRAP2.7, however, revealed an increased number of brace roots. The ZmRAP2.7-based association analysis in a maize panel further identified a SNP marker at the fifth exon of gene to be associated with number of brace roots. These results uncovered a function of ZmRAP2.7 in brace roots development and provided the valuable gene and allele for genetic improvement of maize root systems.
Introduction
Roots are essential organs for exploring and exploiting soil resources, such as water and mineral nutrients, and providing anchorage (Lynch, 1995, 2007; Hodge et al., 2009). Root system architecture (RSA) is fundamental for crop growth and productivity, particularly under abiotic stress condition (de Dorlodot et al., 2007). Maize (Zea mays L.) is one of the most important food and feed crop, and the productivity is sensitive to nutrient deficiency, drought stress, and root lodging in high-density populations (Mueller et al., 2012; Lobell et al., 2014; Xue et al., 2017). A simulation study in the USA Corn Belt suggested that the historical maize yield trend can be explained by the improvement of root systems (Hammer et al., 2009). The deep root system is proposed to be optimal for water and nitrogen acquisition, while the shallow root system is suitable for phosphorus absorption (Lynch, 2011, 2013). However, since it is difficult to evaluate roots traits under field conditions, plant breeders are seldom considered root traits as a selection criterion (Cai et al., 2012). Therefore, identification of QTLs or genes for RSA is required to provide the valuable targets for marker-assisted selection or genetic modification to promote root-based approach on crop yield improvement.
In maize, the root systems are formed during the embryogenesis and post-embryonic development (Hochholdinger and Tuberosa, 2009). The post-embryonic shoot-borne roots dominate the whole root system, representing the major components for resource acquisition and lodging tolerance for the adult plants. Shoot-borne roots are initiated at the consecutive shoot nodes and consist of crown roots (CR) from belowground nodes and brace roots (BR) from aboveground nodes (Hochholdinger, 2009). Using maize rootless concerning crown and seminal roots (rtcs) mutant, ZmRTCS gene have been identified for the involvement in developing shoot-borne roots (Taramino et al., 2007). ZmRTCS and its close homolog gene ZmRTCL both encoded LOB domain proteins that regulated shoot-borne roots initiation and elongation, respectively (Taramino et al., 2007; Xu et al., 2015). Both genes act as the downstream of auxin response factor ZmARF35, suggesting a role of auxin pathway in shoot-borne roots development (Xu et al., 2015). Using a teosinte-maize population, a flowering-time gene ZmCCT was also found to control number of shoot-borne roots (Zhang et al., 2018). For adult maize plants, the developed brace roots grown into the soil are mainly responsible for nutrients and water absorption and against lodging. The anatomical, morphological, and transcriptomic pattern of brace roots significantly differed from those of crown roots, suggesting the genetic differences between these two types of roots in maize (Li et al., 2011; Yu et al., 2015, 2016). Some QTLs for brace roots traits have been mapped (Cai et al., 2012; Ku et al., 2012; Gu et al., 2016), but the underlying genes have not yet been identified. A multidrug-and-toxin-extrusion (MATE) transporter gene may confer to brace roots development as the corresponding mutant big embryo1 revealed more number of brace roots than the wild-type plants (Suzuki et al., 2015). Nevertheless, the molecular mechanism of brace roots development is still poorly understood.
Some AP2 transcription factors were found to be involved in regulating crown roots development in rice (Zhao et al., 2009; Kitomi et al., 2011). For example, CROWNROOTLESS5 (CRL5) is involved in crown roots initiation (Kitomi et al., 2011), and another AP2 transcription factor, ERF3 can interact with WOX11 to control crown roots initiation and development (Zhao et al., 2009, 2015). The AP2 transcription factors belong to a large gene family of plant transcription factors, which contain the highly conserved AP2/ERF DNA-binding domain. The AP2 members were assigned into four subfamilies: AP2, RAV (related to ABI3/VP1), dehydration-responsive element-binding protein (DREB), and ERF (Yamaguchi-Shinozaki and Shinozaki, 2006; Dietz et al., 2010; Sharoni et al., 2011; Mizoi et al., 2012). Phylogenetic analysis predicted a total of 184 AP2-like genes in maize genome (Du et al., 2014), and none of them was reported to regulate root development.
Maize Corngrass1 mutant with elevated expression of miR156 exhibited more brace roots (Chuck et al., 2007a). As the downstream of miR156, miR172 was depressed in Corngrass1 (Chuck et al., 2007b). Since some member of AP2/ERF (AP2) transcription factors act as the targets of miR172, the miR156-miR172-AP2 pathway was proposed to be a regulatory pathway that controlled the timing of the juvenile-to-adult phase transition and subsequently influenced flowering time and floral development (Irish, 1997; Chuck et al., 2007a,b; Buckler et al., 2009; Castelletti et al., 2014). As the elevated expression of an AP2 gene, ZmRAP2.7, as the target of miR172, was observed in the node with brace roots of Corngrass1, we then suspected the role of ZmRAP2.7 in maize brace roots development. The ZmRAP2.7 encoded protein was localized in the nucleus with the activity of transcriptional activation. The maize mutant RAP2.7-Mu defective in ZmRAP2.7 expression revealed a decreased number of brace roots. Thus, this finding suggested the function of ZmRAP2.7 in maize brace roots development.
Materials and Methods
Plant Stocks and Growth Conditions
The ZmRAP2.7 Mu-transposon insertion mutant (Stock ID: UFMu-00629; Locus ID: mu1019979), named RAP2.7-Mu, was obtained from the Uniform-Mu project in the Maize Genetics Cooperation Stock Center1 (McCarty et al., 2005). The corresponding wild-type inbred line W22 was grown in a climate-controlled greenhouse (16/8 h light-dark cycle at temperatures of 29/24°C). For RNA extraction, different organs were collected at the silking stage, including root, the node with or without brace roots. The Corngrass1 mutant was deposited in the Maize Genetics Cooperation Stock Center as CG1 (Stock ID: 310D; Chuck et al., 2007a).
The RAP2.7-Mu and W22 plants were grown in the field at Shangzhuang (SZ) Experimental Station (Beijing, N40°08′12.15″, E116°10′44.83″) during the summers in 2016 and 2017 and at Sanya (SY) Experiment Station (Hainan, N18°22′55.83″, E109°11′43.94″) during the winter in 2016. Leaf samples were collected at the seedling stage, and the genomic DNA was then extracted for the genotyping. A BCF1 segregation population from the cross of Corngrass1 and W22 line was planted, and the node tissue with the primordia of brace roots was then sampled for RNA extraction.
Cloning and Sequence Analysis of ZmRAP2.7
The reference sequence of ZmRAP2.7 (GRMZM2G700665) was identified from Phytozome database2. Full length of ZmRAP2.7 cDNA was amplified using the specific primers located at 5′-UTR and 3′-UTR regions (Supplementary Table S1). The PCR product was then ligated into CloneSmarter-TOPO vector (TaiHe Biotechnology, Beijing, China) and sequenced. The structure of ZmRAP2.7 encoded protein was predicted by SMART3. The secondary structure was predicted by PSIPRED4 and the tertiary structure by SWISS-MODEL5.
Subcellular Localization of ZmRAP2.7 Protein
The coding sequence of ZmRAP2.7 was cloned into the BamHI and EcoRI sites of the pEZS-NL transient expression vector under the control of the 35S promoter to generate pEZS-35S:ZmRAP2.7-EGFP construct (Zuo et al., 2015). Maize protoplasts were isolated from etiolated maize seedlings of inbred B73 for transformation as described by Yoo et al. (2007). After the incubation at 24°C for 12 h in the dark, GFP fluorescence in the transformed protoplasts was visualized using a LSM510 META confocal scanning laser inverted microscope (Carl Zeiss, Jena, Germany).
Transactivation Activity Assay of ZmRAP2.7 Protein
ZmRAP2.7 cDNA was cloned into the EcoRI and BamHI sites of pGBKT7 vector to generate pGBKT7-ZmRAP2.7 construct. This plasmid with empty vector control was then transformed into yeast strain AH109 to analysis the transactivation activity. Yeast transformants with OD600 of 0.1 were plated on various selective media, SD/-Trp and SD/-Trp-His, and incubated at 30°C for 3 days.
Phylogenetic Analysis of ZmRAP2.7
To analysis phylogenetic tree of ZmRAP2.7, the peptide sequences of selected members of AP2 transcription factor subfamily of Oryza sativa, Sorghum bicolor, Populus trichocarpa, and Arabidopsis thaliana were obtained from the database. The peptide sequences of 28 maize AP2 members were obtained from PlantTFDB database6. The sequence alignment was performed by the software DNAMAN, MEGA (version 6) (Tamura et al., 2007) and ClustalX2.0 (Larkin et al., 2007). Phylogenetic tree was constructed using the neighbor-joining method (Saitou and Nei, 1987).
Gene Expression Analysis
Total RNA was isolated using Trizol reagent (Takara, Dalian, China) and treated with DNaseI to eliminate genomic DNA contamination. The cDNA was amplified using a PrimeScript™ RT reagent Kit with gDNA Eraser kits (Takara, Dalian, China). Expression of ZmRAP2.7 was analyzed by qRT-PCR method (Bio-Rad, Hercules, CA, United States) using the fluorescent DNA intercalating dye SYBR Green I Master Mix (TAKARA, Dalian, China). The gene-specific oligonucleotide primers for the gene expression analysis were designed, and the efficiency and specificity of the candidate primers were examined by a melting curve analysis from 55 to 99°C. All primer sequences are listed in Supplementary Table S1. Three biological replicates and three technical repetition were performed. The thermal cycling program was as follows: 40 cycles at 95°C for 3 min, 95°C for 10 s, and 58°C for 30 s. Expression levels were normalized to the maize ubiquilin-1 (ZmUBQ1) gene as an internal control, and the data were analyzed based on the comparative 2-ΔΔCT formula (Livak and Schmittgen, 2001).
Identification of Mu Transposon Insertion Line RAP2.7-Mu
The maize genotype was identified by the method as described by Settles et al. (2007). Two gene-specific primers AP2SF and AP2SR were designed beside the predicted insertion position sequence in the chromosome 8 (131,577,941~131,577,949) (B73 RefGen_v3). Combined with a Mu-TIR-specific primer TIR6 (McCarty et al., 2005), the transposon fragments were amplified and the corresponding insertion site was verified. Plant genotypes were analyzed using primers sets AP2SF + AP2SR and AP2SR + TIR6, and the homozygous for either wild type or mutant and the heterozygote were then identified (Supplementary Figure S1).
Root Phenotyping
The number of shoot-borne roots, including brace roots and crown roots, was evaluated for the different maize genotypes (RAP2.7-Mu, Corngrass1, and the corresponding wild types) by a modified “shovelomics” method (Trachsel et al., 2011). Roots of field-grown maize plants were excavated and then cleaned by removing the soil. Each brace roots and crown roots were cut following the order of node, and the number of roots was counted. The order of whorl was recorded from the bottom to upper nodes (Supplementary Figure S2). Significant difference between different genotypes was determined by Student’s t-tests (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
Candidate Gene Association Analysis of ZmRAP2.7 With Root Traits in the AM508 Panel
A maize association panel composed of 508 diverse inbred lines (AM508) was used to analyze the traits of number of brace and crown roots under the field conditions (Li et al., 2013). Root phenotype of each inbred lines was evaluated at Shangzhuang (SZ) Experimental Station (Beijing, N40°08′12.15″, E116°10′44.83″) and Quzhou (QZ) Experimental Station (Hebei, N36°51′48.55″, E115°00′58.62″) during the summer in 2012. At each location, all lines were planted in one-row plots with an incompletely random design. Each row was 4 m long, 0.5 m wide, and contained 17 plants. Root traits of five randomly selected plants per row were evaluated. The best linear unbiased predictor (BLUP) values from two locations were used for the association analysis. Polymorphisms (single-nucleotide polymorphism, SNPs) of ZmRAP2.7 were obtained from database7, and their association to the investigated traits was calculated by TASSEL5.0, under the standard MLM, with MAF ≥ 0.05.
Results
Elevated Expression of ZmRAP2.7 in Corngrass1 Revealed More Brace Roots
The maize Corngrass1 mutant developed more brace roots than its corresponding wild type (Figure 1A). In the field, the number of shoot-borne roots averaged 122.3 in Corngrass1, about six fold more than that wild-type plants (Figure 1B). Increased number of shoot-borne roots in Corngrass1 was mainly explained by brace roots, while the number of crown roots was similar between two genotypes. The previous study showed that in Corngrass1 expression of miR156 was elevated and subsequently repressed miR172 (Navarro et al., 2017). ZmRAP2.7, one of the putative target genes of miR172, was then supposed to be upregulated in Corngrass1 and probably responded to the brace roots phenotype.
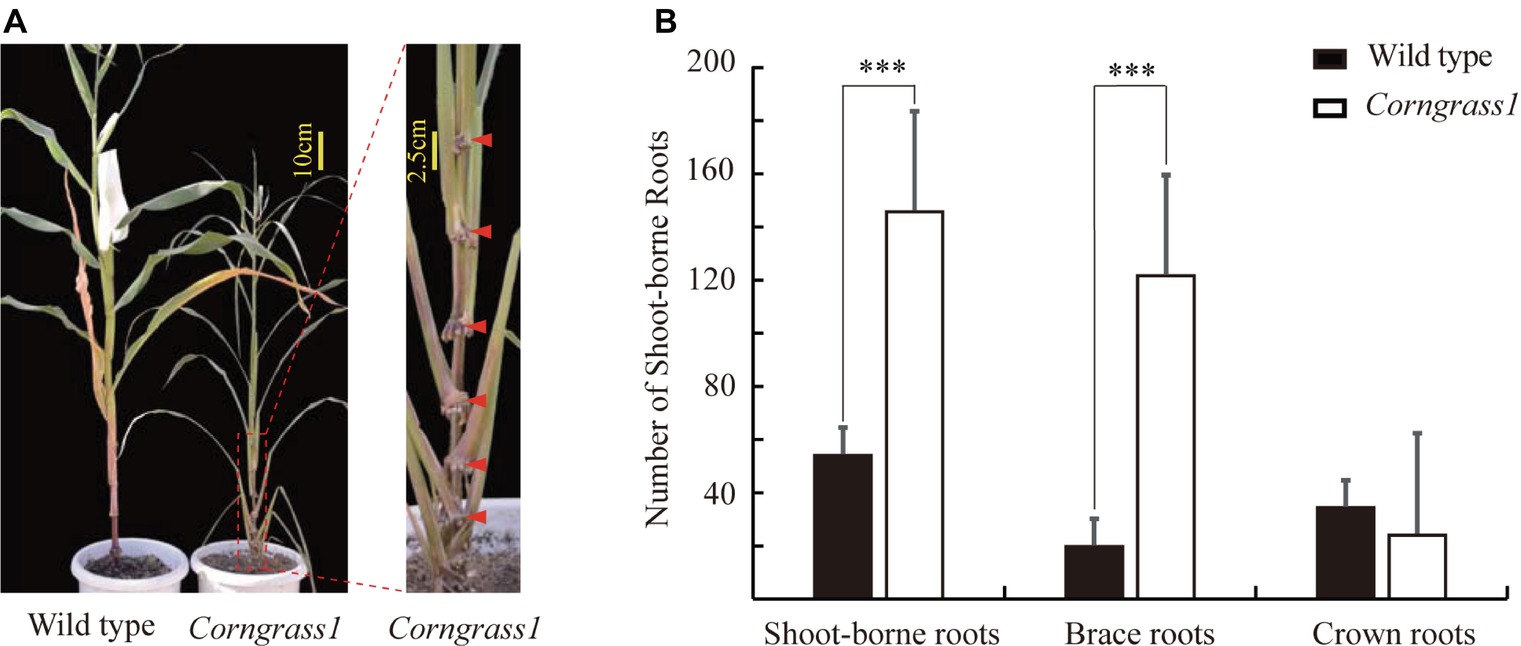
Figure 1. Increased number of brace roots in Corngrass1 mutant. (A) Corngrass1 mutant and wild-type plants were grown in greenhouse for 68 days. Upper nodes in Corngrass1 still maintained at juvenile phase and initiated shoot-borne roots. (B) Number of shoot-borne roots, which consist of brace and crown roots, in field-grown Corngrass1 mutant and wild-type plants at mature stage. Bars indicate mean (±SD) (n = 6). Significant difference within each group was indicated by an asterisk (***p < 0.001) according to Student’s t-tests.
To define the function of ZmRAP2.7 in maize brace roots development, the expression pattern of ZmRAP2.7 in different organs was surveyed from database (B73 genome V3, https://www.maizegdb.org/). ZmRAP2.7 expression levels were abundant in all types of roots, including primary, seminal, and shoot-borne roots (Supplementary Figure S3). We further examined ZmRAP2.7 expression in root (R), node with brace roots (NBR), and node without brace roots (N) of plants at the silking stage (Supplementary Figure S4). Again, ZmRAP2.7 showed the highest expression levels in roots. Remarkably, ZmRAP2.7 expression in the node with the primordia of brace roots was about 10 times higher than that in the node without the primordia of brace roots (Figure 2A), suggesting that ZmRAP2.7 may function in brace roots development. Furthermore, in the developed nodes at the silking stage, ZmRAP2.7 expression in Corngrass1 was about 30-fold higher than that in wild type (Figure 2B). Thus, the elevated expression of ZmRAP2.7 in the node may explain the more number of brace roots in Corngrass1.
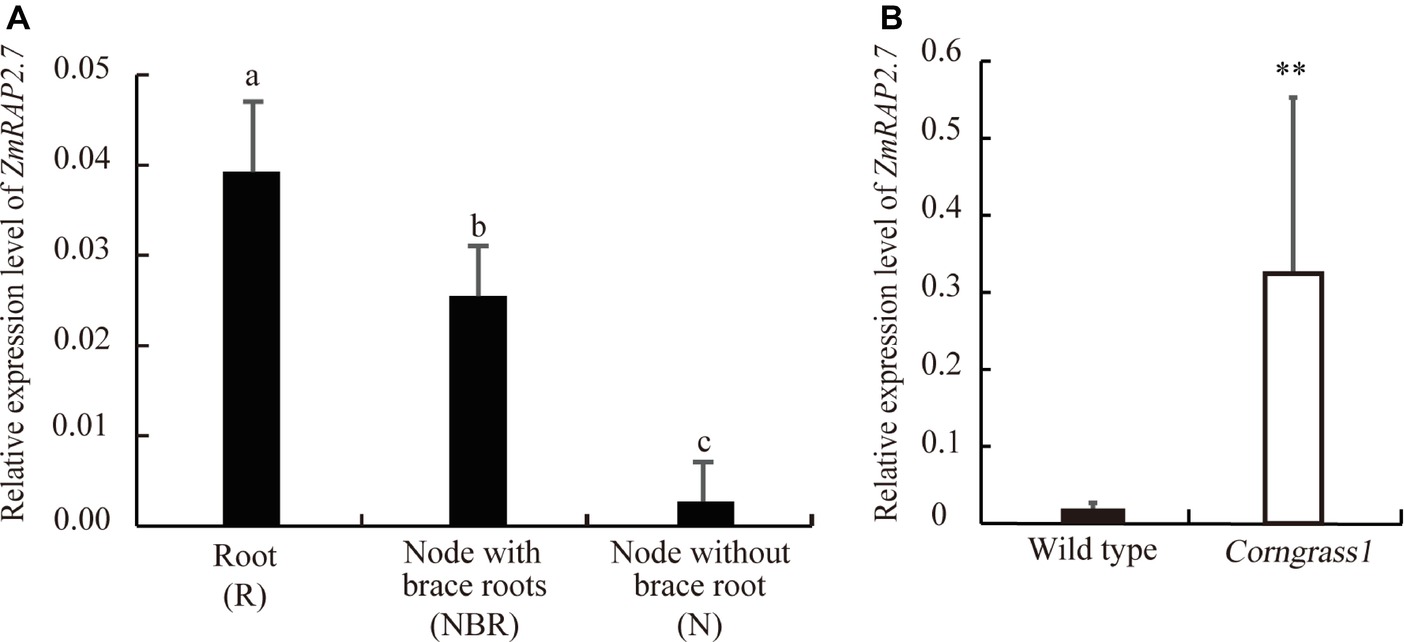
Figure 2. Preferential expression of ZmRAP2.7 in nodes initiated brace roots and upregulation in Corngrass1 mutant. (A) Relative expression levels of ZmRAP2.7 in root (R), nodes with brace roots (NBR), and nodes without brace roots (N) in field-grown wild-type plants at flowering stage. Bars indicate mean (±SD) (n = 3). Significant difference was indicated by different letters according to Tukey’s test. (B) Relative expression levels of ZmRAP2.7 in nodes of field-grown Corngrass1 mutant and wild-type plants at flowering stage. Bars indicate mean (±SD) (n = 3). Significant difference was indicated by an asterisk (**p < 0.01) according to Student’s t-tests.
By the phylogenetic analysis, ZmRAP2.7 (GRMZM2M2G700665) and the close homolog ZmEREB81 (GRMZM2G416701) were assigned into the Cluster I and ts6 (tassel seed6, GRMZM5G862109) and sid1 (sister of indeterminate spikelet 1, GRMZM5G176175) into the Cluster II (Figure 3A). The clusters I and II were probably caused by duplicated segments of the Poaceae genome originating from the ancestral whole genome duplication (Figure 3B). A pair of two genes in each cluster was resulted from the genome allotetraploidization. In the Cluster II, both ts6 and sid1 were the target genes of miR172 and function in floral organ identity (http://www.mirbase.org/; Chuck et al., 2007b). The ts6 and sid1 were constitutively expressed in most of organs as revealed by database (http://www.maizegdb.org/; Stelpflug et al., 2016) (Supplementary Figure S3). In the Cluster I, both ZmRAP2.7 and ZmEREB81 were highly expressed in roots. Since ZmEREB81 lost the miR172 target site, ZmRAP2.7 was supposed to be the candidate gene for regulating root development.
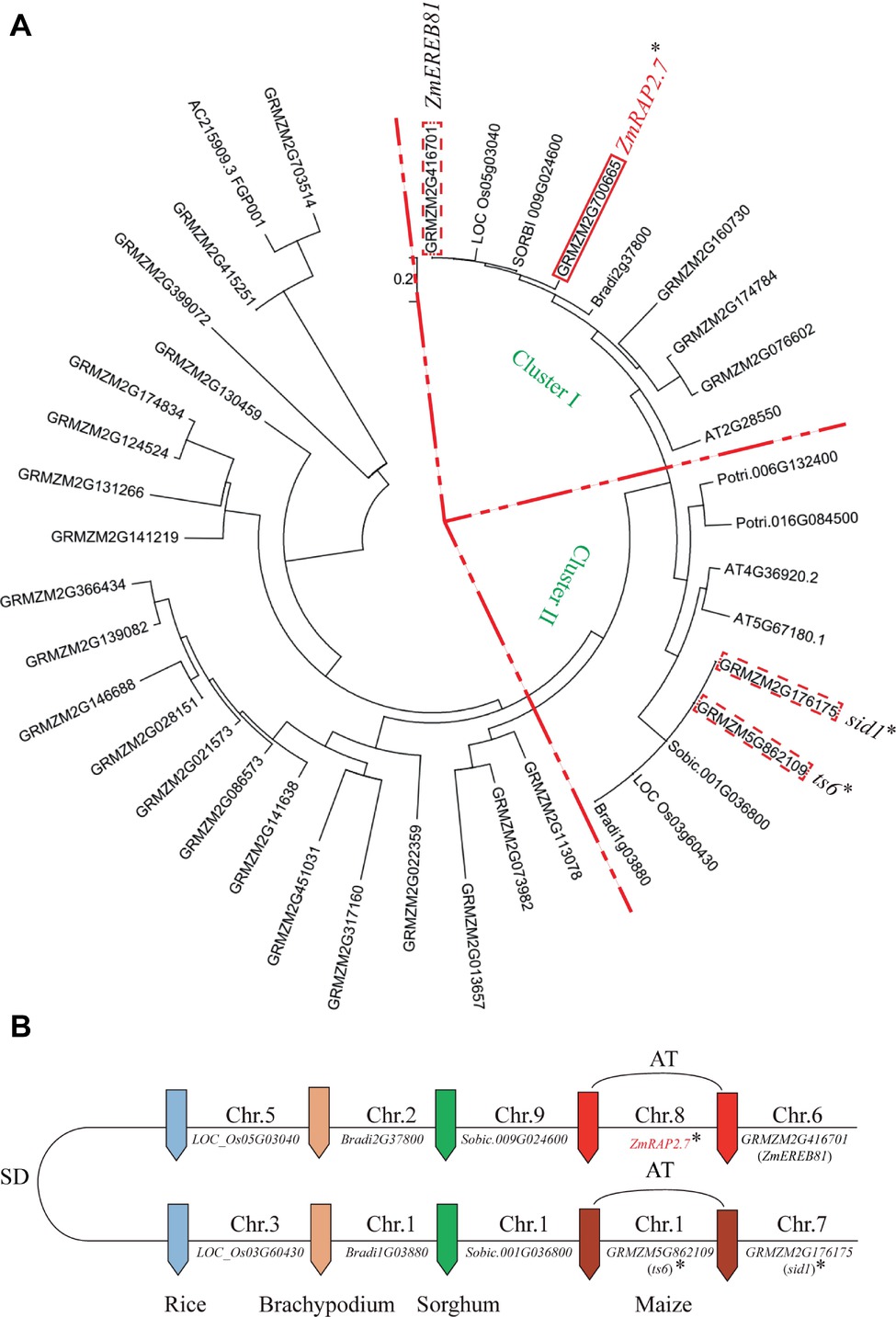
Figure 3. Phylogenetic tree and comparative genome analysis of ZmRAP2.7 and its homologs in different grass species. (A) The phylogenetic tree was constructed using MEGA software. GRMZM2M2G700665 (ZmRAP2.7), Sobic.009G024600, GRMZM2G416701 (ZmEREB81), Os05G03040, and Bradi2G37800 were assigned into Cluster I. GRMZM5G862109 (ts6), GRMZM5G176175 (sid1), Sobic.001G036800, Os03G60430, and Bradi1G03880 were assigned into Cluster II. (B) Schematic representation of the chromosome locations of ZmRAP2.7 and homolog genes within the framework of Poaceae inter- and intra-species collinearity. The clusters I and II resulted from duplicated segments of the Poaceae genome originating from the ancestral whole genome duplication (SD) and a pair of two genes within each cluster from the genome allotetraploidization (AT). Genes contained the targeted sites of miR172 were indicated with an asterisk.
Number of Brace Roots Decreased in ZmRAP2.7 Transposon Insertion Mutant
To verify the role of ZmRAP2.7 in brace roots development, we characterized the Mu-transposon insertion mutant (RAP2.7-Mu) in which ZmRAP2.7 expression was disrupted. By sequencing the PCR products amplified from Mu-specific and ZmRAP2.7 gene-specific primers, the Mu transposon was found to be inserted into the second exon of ZmRAP2.7 gene at +626 to +634 bp region with a repeat sequence 5′-GCGGCAAGC-3′ (Figure 4A). By the semi-quantitative RT-PCR analysis of seedlings root sample, ZmRAP2.7 expression in wild-type plants was revealed as an apparent band while no signal was detected in RAP2.7-Mu (Figure 4B). Thus, this result indicated that ZmRAP2.7 expression was completely disrupted in RAP2.7-Mu by a transposon insertion into the coding sequence.

Figure 4. Isolation of RAP2.7-Mu transposon insertion mutant defective in ZmRAP2.7 expression. (A) A Mu transposon was inserted in the second exon of ZmRAP2.7 gene at the position between +626 and +634 bp with the two 9 bp repeats 5′-GCGGCAAGC-3′. The miR172 target sequence was located in the ninth exon at the position between +1,292 and +1,312 bp. Genotypes were analyzed using primers sets AP2SF + AP2SR and AP2SR + TIR6. (B) Expression levels of ZmRAP2.7 in roots of the RAP2.7-Mu and the corresponding wild-type plants grown in pots.
The number of shoot-borne roots, including brace and crown roots, was then compared between RAP2.7-Mu and the corresponding wild-type plants under the field conditions. To rule out the possible influence of other insertions or genomic modifications on the investigated traits in RAP2.7-Mu, we generated a F2 segregating populations from the cross of ZmRAP2.7-Mu and wild-type W22, and the association between ZmRAP2.7-Mu allele and root phenotype was then analyzed. In the field trials at SZ2016, by genotyping of each individual within the F2 population, we obtained 28 plants with homozygous ZmRAP2.7-Mu allele (−/−), 60 plants with heterozygous ZmRAP2.7-Mu allele (−/+), and 23 plants with homozygous wild-type allele (+/+), presenting a segregation ratio of 1:2:1. On average, the homozygous RAP2.7-Mu plants had significantly 8.6% less number of shoot-borne roots (89.4) than those with wild-type allele (97.8) (Figures 5A,B), while no difference was observed between heterozygous and wild-type allele. Thus, this indicated that the decrease number of brace roots in ZmRAP2.7-Mu resulted from a recessive mutation in ZmRAP2.7. Number of crown roots in each whorl (W1–W8) was similar between both genotypes. By contrast, less brace roots observed in RAP2.7-Mu plants were mainly explained by one whorl less (W9, W10) than that of wild type (W9–W11). Therefore, the disruption of ZmRAP2.7 expression significantly inhibited the development of brace roots rather than crown roots. In addition, the homozygous RAP2.7-Mu also showed an early pollen shed time than the homozygous wild type (8 days earlier in SZ2016 and 4 days in SZ2017) (Supplementary Table S3). Besides brace roots development, the role of ZmRAP2.7 in flowering time was also confirmed as described by Salvi et al. (2007).
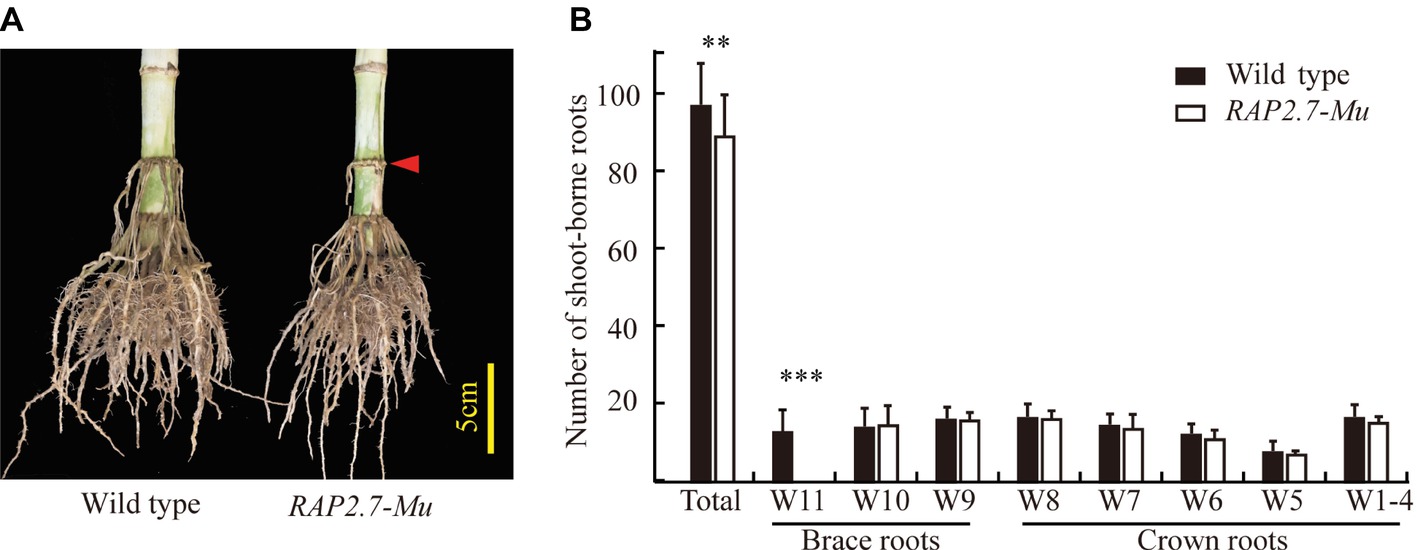
Figure 5. Decreased number of brace roots in RAP2.7-Mu mutant as revealed by F2 segregating population. (A) Phenotype of brace roots development in RAP2.7-Mu mutant and wild-type W22 plants grown in the field at mature stage (75 days after sowing). The difference in brace roots between two genotypes was signed with a red arrow. (B) Number of brace and crown roots of maize plants grown in Shangzhuang (2016) at mature stage (75 days after sowing). The RAP2.7-Mu mutant and wild-type W22 plants were genotypically selected from a F2 segregating population. The whorl corresponding to node numbers in orders was indicated as W1–W11. Bars indicate mean (±SD) (n = 23–28). Significant difference within each group was indicated with an asterisk (**p < 0.01; ***p < 0.001) according to Student’s t-tests.
To confirm the root phenotype of RAP2.7-Mu, we further generated the F2:3 families from either homozygous RAP2.7-Mu or homozygous wild-type plants. The root phenotypes were evaluated in SY2016 and SZ2017. In SY2016, the RAP2.7-Mu showed about 42.0 shoot-borne roots on average, about 14% significant reduction compared with wild type (Figure 6). The wild-type plants developed the brace roots in the eighth whorl (W8), while RAP2.7-Mu failed to initiate the brace roots at the same whorl (Figure 6). The number of crown roots (W1–W7) was similar between both genotypes. Compared to plants in SY2016, number of shoot-borne roots, mainly for brace roots, significantly increased in SZ2017, indicating the brace roots development also influenced by the environments. Nevertheless, number of shoot-borne roots in RAP2.7-Mu (72.5) also decreased up to 12% than that in wild type (82.7) (Figure 6). Again, the decrease was mainly explained by the less number of brace roots in the 10th whorl (W10). In addition, the root dry weight showed about 70% reduction in RAP2.7-Mu plants, while the shoot dry weight remained similar between two genotypes (Supplementary Figure S5). Collectively, these results indicated that ZmRAP2.7 was involved in the development of brace roots rather than crown roots.
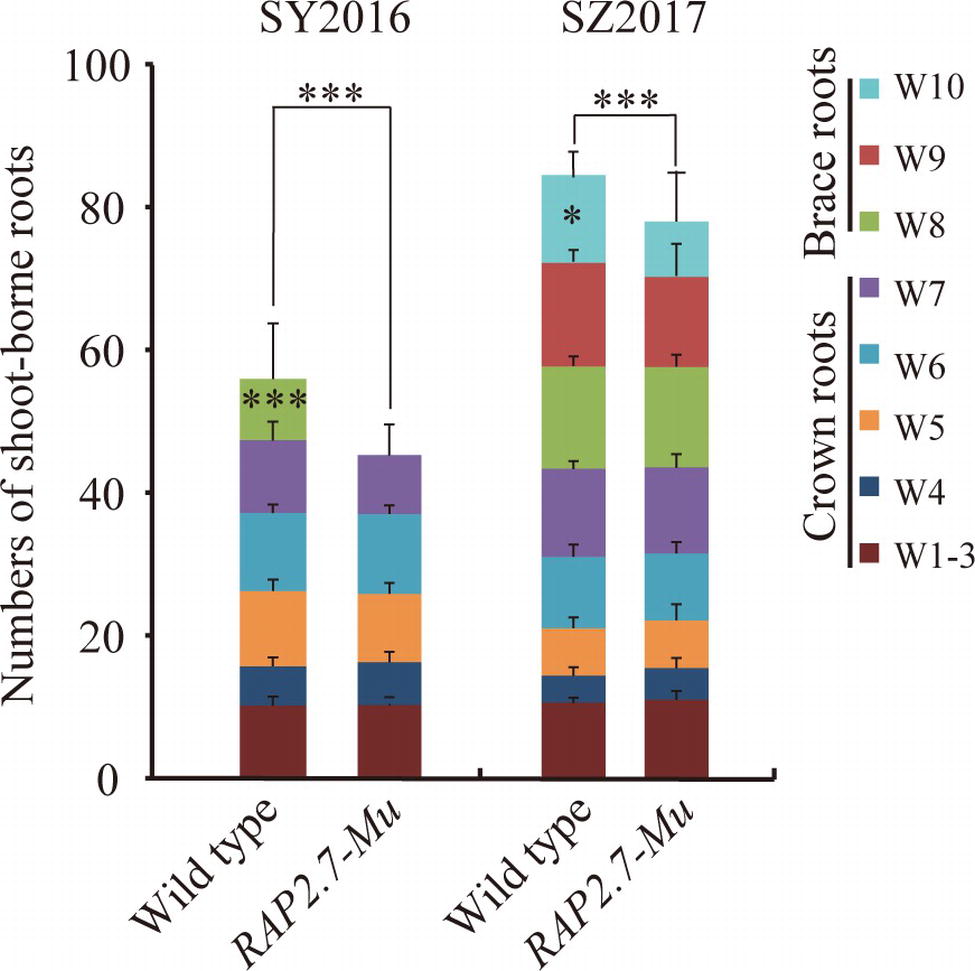
Figure 6. Decreased number of brace roots in RAP2.7-Mu mutant as revealed by F2:3 families. Number of brace and crown roots of maize plants grown in Sanya (SY2016) and Shangzhuang (SZ2017) at mature stage (about 78 days after sowing). The F2:3 families’ plants of RAP2.7-Mu mutant and wild-type W22 were compared. The whorl corresponding to node numbers in orders was indicated as W1–W10. Bars indicate mean (±SD) (n = 36). Significant difference within each group was indicated by an asterisk (***p < 0.001) according to Student’s t-tests.
The candidate gene association analysis for ZmRAP2.7 was then conducted in a maize panel (AM508) consisted of 508 diverse lines (Figure 7). The allele variation of ZmRAP2.7 among these genotypes was extracted according to the corresponding SNPs from the database8 (Yang et al., 2011). A total of 49 SNPs, spanning 5′- and 3′-untranslation region (UTR) and 9 exons of ZmRAP2.7 were identified among these lines. Using the mixed linear model, the SNP1499, which located at the fifth exon of ZmRAP2.7 gene, revealed a significant association with number of brace roots (Figure 7). This SNP1499 could contribute to 12.5% of the phenotypic variation within the population. By contrast, SNP1499 was not associated with number of crown roots, in agreement with the role of ZmRAP2.7 in brace roots development as revealed by mutant analysis. Additionally, the favorite alleles variation identified in ZmRAP2.7 could contribute to generate molecular marker for selecting the traits for brace roots.
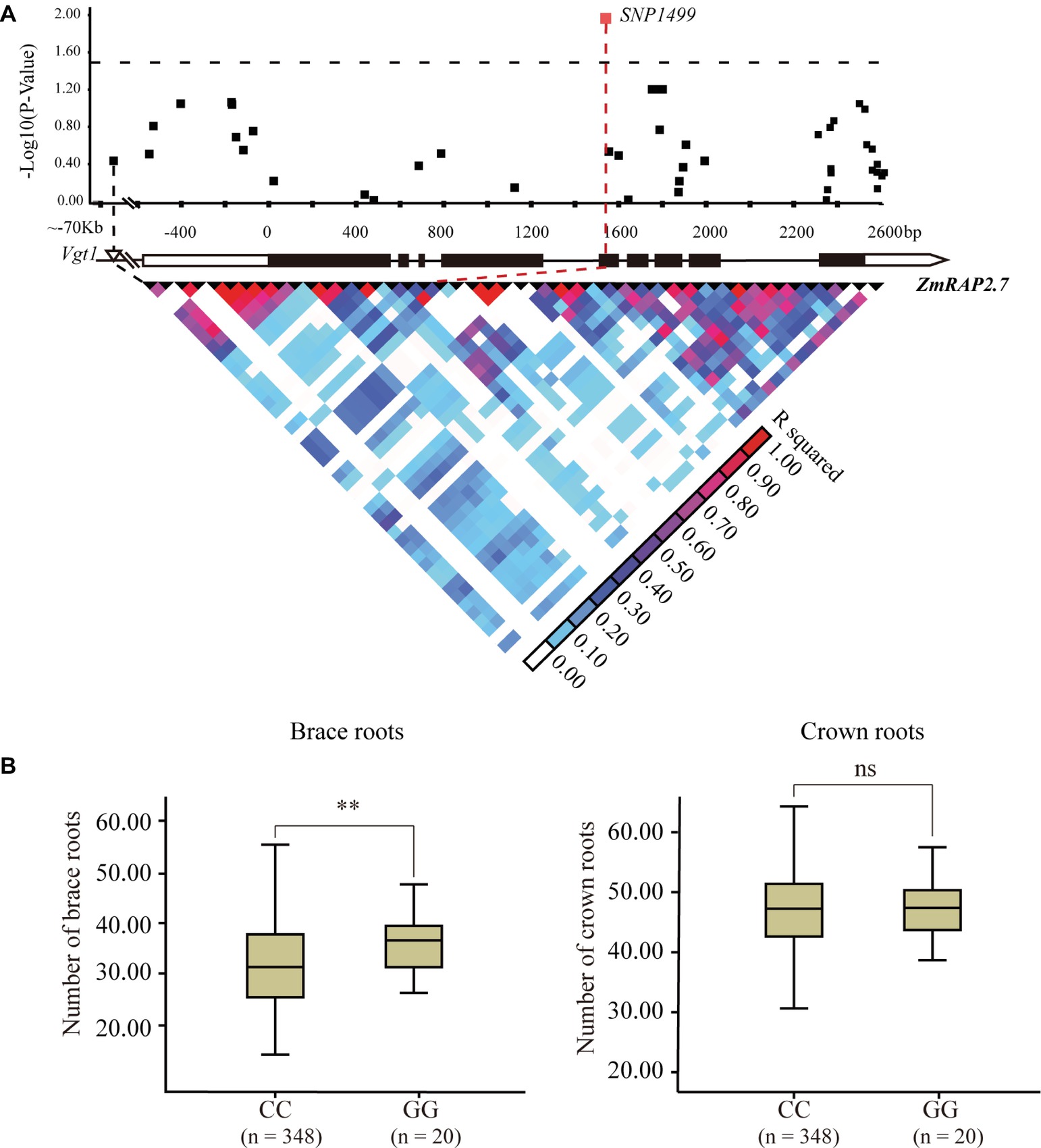
Figure 7. Natural variation in ZmRAP2.7 gene was significantly associated with number of brace roots within a maize association panel (AM508). (A) ZmRAP2.7-based association mapping and pairwise linkage disequilibrium (LD) analysis. The dots represent SNPs. A SNP variant at fifth exon (SNP1499) associated with number of brace roots was highlighted in red. (B) Number of crown roots and brace roots in haplotypes of ZmRAP2.7 (GG and CC alleles at SNP1499). n denotes the number of genotypes belonging to each haplotype group. Significant difference within each group was indicated by an asterisk (**p < 0.01) according to Student’s t-tests, and no significant was indicated as n.s.
ZmRAP2.7 Localized in the Nucleus and Revealed Transcription Activity
The ZmRAP2.7 gene contained an open reading frame of 1,413 bp and encodes a predicted 470 amino acids protein with a predicted molecular weight of 51.66 kDa. ZmRAP2.7 protein was predicted to contain two α-helices (Supplementary Figures S6A,B) with the two conserved AP2/ERF domains (153–215 aa and 245–308 aa) (Supplementary Figure S6C). Thus, the ZmRAP2.7 protein was predicted as one of the members in AP2 transcription factor family.
To investigate the subcellular localization of ZmRAP2.7 protein, the full-length ORFs of ZmRAP2.7 was fused to EGFP and transiently expressed in maize leaf protoplasts (Figure 8A). In contrast to GFP fluorescence throughout the whole cell, the ZmRAP2.7-dependent green fluorescence was mainly localized in the nucleus (Figure 8A), indicating a nuclear localization of ZmRAP2.7 protein. The yeast strains transformed with the pGBKT7-ZmRAP2.7 were able to grow on the selected medium SD/-Trp/-His in which those stains with empty vector pGBDKT7 could not grow (Figure 8B). This result indicated that ZmRAP2.7 revealed the transcriptional activity, suggesting a role of ZmRAP2.7 as a transcription activator.
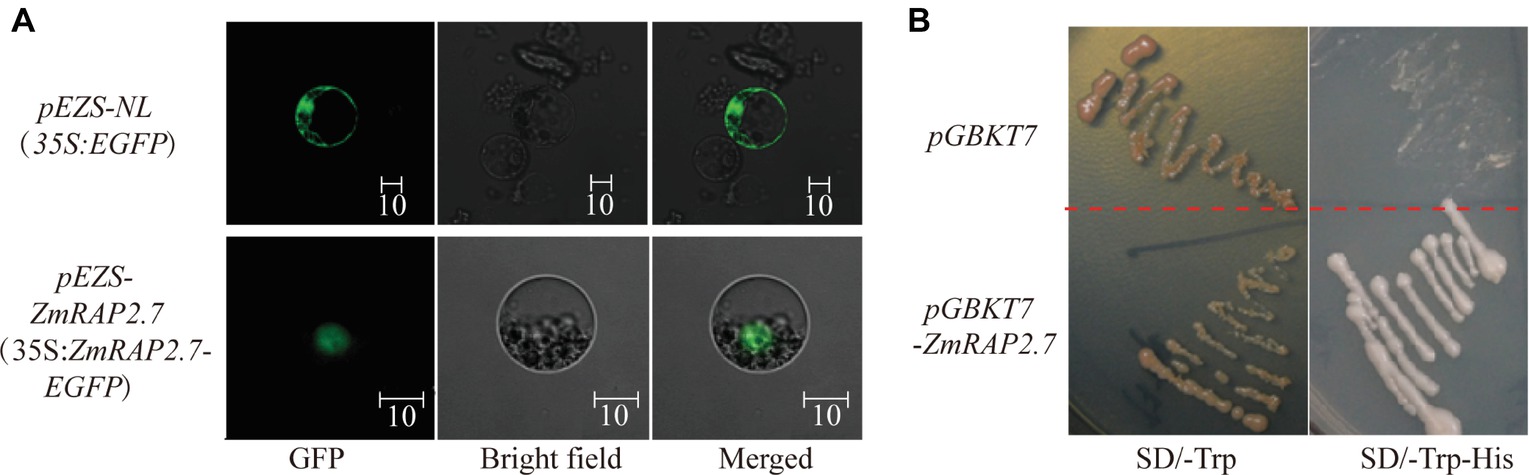
Figure 8. ZmRAP2.7 localized in the nuclear with transcription activity. (A) Nuclear localization of ZmRAP2.7 protein in maize. The constructs pEZS-35S:EGFP and pEZS-35S:ZmRAP2.7-EGFP were transiently expressed into maize mesophyll protoplasts. ZmRAP2.7-dependent GFP fluorescence was observed in the nuclear. The scale bar represents 10 μm. (B) Transcription activation of ZmRAP2.7 in yeasts. The constructs pGKBT7 and pGKBT7-ZmRAP2.7 were transformed in yeasts and grew for 3 days on the selected medium as indicated.
Discussion
The brace roots are important component for the whole root system of maize. By growing into the soils, the brace roots contribute significantly to lodging tolerance and water and nutrient uptake efficiency. However, few genes have been cloned for maize brace roots development, and the underlying mechanism remains to be elucidated. In this study, we characterized a maize AP2 transcription factor gene, ZmRAP2.7, with an essential function in the brace roots development. In addition, the identified allele variation of ZmRAP2.7 could be used to generate valuable markers for genetic improvement of root traits.
Several evidences indicated that ZmRAP2.7 gene was involved in brace roots development in maize. First, ZmRAP2.7 was mainly expressed in roots (Supplementary Figure S3) and the nodes with initiated brace roots (Figure 2A). Second, the alteration of ZmRAP2.7 expression resulted in the changes of number of braces roots (Figures 1, 2, 4–6). In contrast to wild type, RAP2.7-Mu plants defective in ZmRAP2.7 expression showed lower number of brace roots (Figures 4–6), while Corngrass1 plants with the elevated ZmRAP2.7 expression showed higher number of brace roots (Figures 1A, 2B). Third, ZmRAP2.7 protein was localized in the nuclear and revealed transcriptional activity. However, the downstream genes of ZmRAP2.7 and how they regulate brace roots development remain largely unknown. As maize ZmRTCS and ZmRTCL and their upstream regulator ZmARF35 control shoot-borne roots development (Taramino et al., 2007; Xu et al., 2015), it would be worthy to investigate whether this pathway might be regulated by ZmRAP2.7. In addition, two rice AP2 transcription factor genes (OsERF3 and OsCRL5) were found to control shoot-borne crown root initiation and elongation by regulating the response regulators (RRs) of cytokinin signaling (Zhao et al., 2009, 2015; Kitomi et al., 2011). Thus, ZmRAP2.7 may also regulate cytokinin pathway for controlling shoot-borne roots development in maize.
Many previous studies in maize demonstrated that ZmRAP2.7 gene was a negative regulator for the flowering time as revealed by genome-wide association analysis (Buckler et al., 2009), QTL analysis (Castelletti et al., 2014) and transgenic analysis (Salvi et al., 2007). The transgenic maize lines overexpressing ZmRAP2.7 revealed a delayed flowering time and also an increase of node number (Salvi et al., 2007). Meanwhile, the SNPs located in the genomic region of ZmRAP2.7 were found to be significantly associated with the node number within a nested association mapping (NAM) population (Wallace et al., 2014). Thus, a role of ZmRAP2.7 in regulating the node number could be supposed besides the flowering time. The alternations of ZmRAP2.7 expression in Corngrass1 or RAP2.7-Mu plants resulted in changes of the number of brace roots associated with the node numbers (Figures 1, 5, 6). Because miR156 and miR172 pathways control the transition between juvenile and adult stage (Lauter et al., 2005; Chuck et al., 2007b; Yang et al., 2011; Navarro et al., 2017), the downstream gene ZmRAP2.7 might function in keeping the stem nodes at juvenile stage to maintain their meristematic ability for developing brace roots.
As ZmRAP2.7 was involved in controls of both brace roots development and flowering time, a pleiotropy effect of ZmRAP2.7 could be expected. Using a maize association panel (AM508), number of brace roots was found to be significantly associated with several developmental traits including the date for heading, silking, and pollen shed (Supplementary Table S2; Li et al., 2013; Yang et al., 2014). This phenotypic correlation suggested a genetic relationship between brace roots and flowers development in maize, which could be explained by the pleiotropy function of ZmRAP2.7 in both developmental processes. Indeed, RAP2.7-Mu plants showed an early flowering time and less number of brace roots (Figures 5, 6; Supplementary Table S3). QTL analysis of a teosinte-maize population also established a genetic association between number of shoot-borne roots and flowering time and identified ZmCCT gene co-regulating both traits (Zhang et al., 2018). A similar case was also found in wheat and barley that loss function of VERNALIZATION1 (VRN1) leads to promote flowering time and root phenotypes (Deng et al., 2015; Voss-Fels et al., 2017).
However, the functions of ZmRAP2.7 in brace roots and flower development might be regulated independently. Within AM508, the SNP1499 was significantly associated with number of brace roots, but not associated with the date of heading, silking, and pollen shed (Supplementary Figure S7). Previous studies showed that Vegetative to generative transition 1 (Vgt1), a 2-Kb noncoding region positioned 70 Kb upstream of ZmRAP2.7, could function as a cis-acting regulatory element to repress the ZmRAP2.7 expression level and resulted in an earlier flowering time (Salvi et al., 2007; Castelletti et al., 2014; Navarro et al., 2017). Indeed, within AM508, the Vgt1 allelic variation could also contribute to the phenotypic variation of flowering time. However, Vgt1 allelic variation was not associated with the number of brace roots (Supplementary Figure S8). Thus, the distinct allelic variation of ZmRAP2.7 could be supposed to regulate brace roots or flowers development independently, but the underlying mechanism remains to be elucidated.
Taken together, this study characterized a maize AP2 transcription factor, ZmRAP2.7, with essential functions in brace roots development. ZmRAP2.7 could stimulate brace roots development and also repress the flowering time. In addition, natural variations of ZmRAP2.7 associated with root traits allow to develop the molecular markers for improving root systems in maize.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
JL and LY conceived and designed the experiments. JL conducted most of the experiments. FC performed the field trials. YL and YW genotyped and phenotyped the RAP2.7-Mu mutant. PL collected the root phenotypes in AM508 maize panel. JL and LY analyzed the data and wrote the manuscript. GM helped to revise the manuscript. All authors read and proved the manuscript.
Funding
This work was financially supported by the National Key Research and Development Program of China (No. 2016YFD0100700) and the National Natural Science Foundation of China (Nos. 31430095, 31471934, and 31572186).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Xiaohong Yang, China Agricultural University, for kindly support on the data of Vgt1 allelic variation in AM508 panel. We thank the reviewers for their critical readings and constructive comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00820/full#supplementary-material
Abbreviations
AT, Allotetraploidization; BR, Brace roots; CG1, Corngrass1; CR, Crown roots; CRL5, CROWNROOTLESS5; LD, Linkage disequilibrium; QTL, Quantitative trait locus; RR, Response regulators of cytokinin signaling; RSA, Root system architecture; rtcs, Rootless concerning crown and seminal roots; SD, Segments duplicated; SNP, Single-nucleotide polymorphism; Tp1, Teopod1; Tp2, Teopod2; UTR, Untranslated region; Vgt1, Vegetative to generative transition 1; VRN1, VERNALIZATION1.
Footnotes
1. http://www.maizegdb.org/documentation/uniformmu/
2. https://phytozome.jgi.doe.gov/
3. http://smart.embl-heidelberg.de/
4. http://bioinf.cs.ucl.ac.uk/psipred/
References
Buckler, E. S., Holland, J. B., Bradbury, P. J., Acharya, C. B., Brown, P. J., Browne, C., et al. (2009). The genetic architecture of maize flowering time. Science 325, 714–718. doi: 10.1126/science.1174276
Cai, H. G., Chen, F. G., Mi, G. H., Zhang, F. S., Maurer, H. P., Liu, W. X., et al. (2012). Mapping QTLs for root system architecture of maize (Zea mays L.) in the field at different developmental stages. Theor. Appl. Genet. 125, 1313–1324. doi: 10.1007/s00122-012-1915-6
Castelletti, S., Tuberosa, R., Pindo, M., and Salvi, S. (2014). A MITE transposon insertion is associated with differential methylation at the maize flowering time QTL Vgt1. G3 4, 805–812. doi: 10.1534/g3.114.010686
Chuck, G., Cigan, A. M., Saeteurn, K., and Hake, S. (2007a). The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat. Genet. 39, 544–549. doi: 10.1038/ng2001
Chuck, G., Meeley, R., Irish, E., Sakai, H., and Hake, S. (2007b). The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1. Nat. Genet. 39, 1517–1521. doi: 10.1038/ng.2007.20
De Dorlodot, S., Forster, B., Pagès, L., Price, A., Tuberosa, R., and Draye, X. (2007). Root system architecture: opportunities and constraints for genetic improvement of crops. Trends Plant Sci. 12, 474–481. doi: 10.1016/j.tplants.2007.08.012
Deng, W., Casao, M. C., Wang, P., Sato, K., Hayes, P. M., Finnegan, E. J., et al. (2015). Direct links between the vernalization response and other key traits of cereal crops. Nat. Commun. 6:5882. doi: 10.1038/ncomms6882
Dietz, K. J., Vogel, M. O., and Viehhauser, A. (2010). AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma 245, 3–14. doi: 10.1007/s00709-010-0142-8
Du, H. W., Huang, M., Zhang, Z. X., and Cheng, S. Y. (2014). Genome-wide analysis of the AP2/ERF gene family in maize waterlogging stress response. Euphytica 198, 115–126. doi: 10.1007/s10681-014-1088-2
Gu, D. D., Mei, X. P., Yu, T. T., Sun, N. N., Xu, D., Liu, C., et al. (2016). QTL identification for brace-root traits of maize in different generations and environments. Crop Sci. 57, 13–21. doi: 10.2135/cropsci2016.01.0031
Hammer, G. L., Dong, Z. S., McLean, G., Doherty, A., Messina, C., Schussler, J., et al. (2009). Can changes in canopy and/or root system architecture explain historical maize yield trends in the U.S. Corn Belt? Crop Sci. 49, 299–312. doi: 10.2135/cropsci2008.03.0152
Hochholdinger, F. (2009). “The maize root system: morphology, anatomy, and genetics” in Handbook of maize: Its biology. eds. J. L. Bennetzen and S. C. Hake (New York: Springer Science & Business Media), 145–159.
Hochholdinger, F., and Tuberosa, R. (2009). Genetic and genomic dissection of maize root development and architecture. Curr. Opin. Plant Biol. 12, 172–177. doi: 10.1016/j.pbi.2008.12.002
Hodge, A., Berta, G., Doussan, C., Merchan, F., and Crespi, M. (2009). Plant root growth, architecture and function. Plant Soil 321, 153–187. doi: 10.1007/s11104-009-9929-9
Irish, E. E. (1997). Experimental analysis of tassel development in the maize mutant tasselseed6. Plant Physiol. 114, 817–825. doi: 10.1104/pp.114.3.817
Kitomi, Y., Ito, H., Hobo, T., Aya, K., Kitano, H., and Inukai, Y. (2011). The auxin responsive AP2/ERF transcription factor CROWN ROOTLESS5 is involved in crown root initiation in rice through the induction of OsRR1, a type-A response regulator of cytokinin signaling. Plant J. 67, 472–484. doi: 10.1111/j.1365-313X.2011.04610.x
Ku, L. X., Sun, Z. H., Wang, C. L., Zhang, J., Zhao, R. F., Liu, H. Y., et al. (2012). QTL mapping and epistasis analysis of brace root traits in maize. Mol. Breed. 30, 697–708. doi: 10.1007/s11032-011-9655-x
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lauter, N., Kampani, A., Carlson, S., Goebel, M., and Moose, S. P. (2005). microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc. Natl. Acad. Sci. U. S. A. 102, 9412–9417. doi: 10.1073/pnas.0503927102
Li, Y., Fu, Y., Huang, J., Wu, C., and Zheng, C. (2011). Transcript profiling during the early development of the maize brace root via solexa sequencing. FEBS J. 278, 156–166. doi: 10.1111/j.1742-4658.2010.07941.x
Li, H., Peng, Z. Y., Yang, X. H., Wang, W. D., Fu, J. J., Wang, J. H., et al. (2013). Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–U72. doi: 10.1038/ng.2484
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lobell, D. B., Roberts, M. J., Schlenker, W., Braun, N., Little, B. B., Rejesus, R. M., et al. (2014). Greater sensitivity to drought accompanies maize yield increase in the U.S. midwest. Science 344, 516–519. doi: 10.1126/science.1251423
Lynch, J. (1995). Root architecture and plant productivity. Plant Physiol. 109, 7–13. doi: 10.1104/pp.109.1.7
Lynch, J. P. (2007). Roots of the second green revolution. Aust. J. Bot. 55, 493–512. doi: 10.1071/BT06118
Lynch, J. P. (2011). Root phenes for enhanced soil exploration and phosphorus acquisition: tools for future crops. Plant Physiol. 156, 1041–1049. doi: 10.1104/pp.111.175414
Lynch, J. P. (2013). Steep, cheap and deep: an ideotype to optimize water and N acquisition by maize root systems. Ann. Bot. 112, 347–357. doi: 10.1093/aob/mcs293
McCarty, D. R., Settles, A. M., Suzuki, M., Tan, B. C., Latshaw, S., Porch, T., et al. (2005). Steady-state transposon mutagenesis in inbred maize. Plant J. 44, 52–61. doi: 10.1111/j.1365-313X.2005.02509.x
Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta Gene Regul. Mech. 1819, 86–96. doi: 10.1016/j.bbagrm.2011.08.004
Mueller, N. D., Gerber, J. S., Matt, J., Ray, D. K., Navin, R., and Foley, J. A. (2012). Closing yield gaps through nutrient and water management. Nature 494, 390–390. doi: 10.1038/nature11907
Navarro, J. A. R., Willcox, M., Burgueno, J., Romay, C., Swarts, K., Trachsel, S., et al. (2017). A study of allelic diversity underlying flowering-time adaptation in maize landraces. Nat. Genet. 49, 476–480. doi: 10.1038/ng.3784
Saitou, N., and Nei, M. (1987). The neighbor-joining method - a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Salvi, S., Sponza, G., Morgante, M., Tomes, D., Niu, X., Fengler, K. A., et al. (2007). Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc. Natl. Acad. Sci. U. S. A. 104, 11376–11381. doi: 10.1073/pnas.0704145104
Settles, A. M., Holding, D. R., Tan, B. C., Latshaw, S. P., Liu, J., Suzuki, M., et al. (2007). Sequence-indexed mutations in maize using the UniformMu transposon-tagging population. BMC Genomics 8:116 doi: 10.1186/1471-2164-8-116
Sharoni, A. M., Nuruzzaman, M., Satoh, K., Shimizu, T., Kondoh, H., Sasaya, T., et al. (2011). Gene structures, classification and expression models of the AP2/EREBP transcription factor family in rice. Plant Cell Physiol. 52, 344–360. doi: 10.1093/pcp/pcq196
Stelpflug, S. C., Sekhon, R. S., Vaillancourt, B., Hirsch, C. N., Buell, C. R., De Leon, N., et al. (2016). An expanded maize gene expression atlas based on RNA sequencing and its use to explore root development. Plant Genome 9, 1–16. doi: 10.3835/plantgenome2015.04.0025
Suzuki, M., Sato, Y., Wu, S., Kang, B. H., and McCarty, D. R. (2015). Conserved functions of the mate transporter big embryo1 in regulation of lateral organ size and initiation rate. Plant Cell 27, 2288–2300. doi: 10.1105/tpc.15.00290
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Taramino, G., Sauer, M., Stauffer Jay, L., Multani, D., Niu, X., Sakai, H., et al. (2007). The maize (Zea mays L.) RTCS gene encodes a LOB domain protein that is a key regulator of embryonic seminal and post-embryonic shoot-borne root initiation. Plant J. 50, 649–659. doi: 10.1111/j.1365-313X.2007.03075.x
Trachsel, S., Kaeppler, S. M., Brown, K. M., and Lynch, J. P. (2011). Shovelomics: high throughput phenotyping of maize (Zea mays L.) root architecture in the field. Plant Soil 341, 75–87. doi: 10.1007/s11104-010-0623-8
Voss-Fels, K. P., Robinson, H., Mudge, S. R., Richard, C., Newman, S., Wittkop, B., et al. (2017). VERNALIZATION1 modulates root system architecture in wheat and barley. Mol. Plant. 11, 226–229. doi: 10.1016/j.molp.2017.10.005
Wallace, J. G., Bradbury, P. J., Zhang, N. Y., Gibon, Y., Stitt, M., and Buckler, E. S. (2014). Association mapping across numerous traits reveals patterns of functional variation in maize. PLoS Genet. 10, 1–10. doi: 10.1371/journal.pgen.1004845
Xu, C. Z., Tai, H. H., Saleem, M., Ludwig, Y., Majer, C., Berendzen, K. W., et al. (2015). Cooperative action of the paralogous maize lateral organ boundaries (LOB) domain proteins RTCS and RTCL in shoot-borne root formation. New Phytol. 207, 1123–1133. doi: 10.1111/nph.13420
Xue, J., Xie, R. Z., Zhang, W. F., Wang, K. R., Hou, P., Ming, B., et al. (2017). Research progress on reduced lodging of high-yield and -density maize. J. Integr. Agric. 16, 2717–2725. doi: 10.1016/S2095-3119(17)61785-4
Yamaguchi-Shinozaki, K., and Shinozaki, K. (2006). Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 57, 781–803. doi: 10.1146/annurev.arplant.57.032905.105444
Yang, X. H., Gao, S. B., Xu, S. T., Zhang, Z. X., Prasanna, B. M., Li, L., et al. (2011). Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol. Breed. 28, 511–526. doi: 10.1007/s11032-010-9500-7
Yang, N., Lu, Y. L., Yang, X. H., Huang, J., Zhou, Y., Ali, F., et al. (2014). Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet. 10:e1004573. doi: 10.1371/journal.pgen.1004573
Yoo, S. D., Cho, Y. H., and Sheen, J. (2007). Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2, 1565–1572. doi: 10.1038/nprot.2007.199
Yu, P., Baldauf, J., Lithio, A., Marcon, C., Nettleton, D., Li, C., et al. (2016). Root type specific reprogramming of maize pericycle transcriptomes by local high nitrate results in disparate lateral root branching patterns. Plant Physiol. 170, 1783–1798. doi: 10.1104/pp.15.01885
Yu, P., Hochholdinger, F., and Li, C. (2015). Root-type-specific plasticity in response to localized high nitrate supply in maize (Zea mays). Ann. Bot. 116, 751–762. doi: 10.1093/aob/mcv127
Zhang, Z., Zhang, X., Lin, Z., Wang, J., Xu, M., Lai, J., et al. (2018). The genetic architecture of nodal root number in maize. Plant J. 93, 1032–1044. doi: 10.1111/tpj.13828
Zhao, Y., Cheng, S. F., Song, Y. L., Huang, Y. L., Zhou, S. L., Liu, X. Y., et al. (2015). The interaction between rice ERF3 and WOX11 promotes crown root development by regulating gene expression involved in cytokinin signaling. Plant Cell 27, 2469–2483. doi: 10.1105/tpc.15.00227
Zhao, Y., Hu, Y. F., Dai, M. Q., Huang, L. M., and Zhou, D. X. (2009). The WUSCHEL-related Homeobox gene WOX11 is required to activate shoot-borne crown root development in rice. Plant Cell 21, 736–748. doi: 10.1105/tpc.108.061655
Keywords: AP2 transcription factor, brace roots, candidate gene association, shoot-borne roots, root development, Zea mays L.
Citation: Li J, Chen F, Li Y, Li P, Wang Y, Mi G and Yuan L (2019) ZmRAP2.7, an AP2 Transcription Factor, Is Involved in Maize Brace Roots Development. Front. Plant Sci. 10:820. doi: 10.3389/fpls.2019.00820
Edited by:
Stefan de Folter, Center for Research and Advanced Studies (CINVESTAV), MexicoCopyright © 2019 Li, Chen, Li, Li, Wang, Mi and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lixing Yuan, eXVhbmxpeGluZ0BjYXUuZWR1LmNu