 Marco Kreuzer
Marco Kreuzer Caroline Howard
Caroline Howard Bhaskar Adhikari
Bhaskar Adhikari Colin A. Pendry
Colin A. Pendry Julie A. Hawkins
Julie A. Hawkins
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 14 May 2019
Sec. Computational Genomics
Volume 10 - 2019 | https://doi.org/10.3389/fpls.2019.00586
This article is part of the Research Topic Advanced Technologies for the Quality Control and Standardization of Plant Based Medicines View all 20 articles
DNA barcoding of herbal medicines has been mainly concerned with authentication of products in trade and has raised awareness of species substitution and adulteration. More recently DNA barcodes have been included in pharmacopoeias, providing tools for regulatory purposes. The commonly used DNA barcoding regions in plants often fail to resolve identification to species level. This can be especially challenging in evolutionarily complex groups where incipient or reticulate speciation is ongoing. In this study, we take a phylogenomic approach, analyzing whole plastid sequences from the evolutionarily complex genus Berberis in order to develop DNA barcodes for the medicinally important species Berberis aristata. The phylogeny reconstructed from an alignment of ∼160 kbp of chloroplast DNA for 57 species reveals that the pharmacopoeial species in question is polyphyletic, complicating development of a species-specific DNA barcode. Instead we propose a DNA barcode that is clade specific, using our phylogeny to define Operational Phylogenetic Units (OPUs). The plastid alignment is then reduced to small, informative DNA regions including nucleotides diagnostic for these OPUs. These DNA barcodes were tested on commercial samples, and shown to discriminate plants in trade and therefore to meet the requirement of a pharmacopoeial standard. The proposed method provides an innovative approach for inferring DNA barcodes for evolutionarily complex groups for regulatory purposes and quality control.
DNA barcoding has two major objectives: specimen identification, where an unknown sequence is matched to a sequence of a known species, and species discovery, which is equivalent to species delimitation and species description (DeSalle, 2006). DNA barcoding of herbal medicines is mainly concerned with authentication, the identification of specimens for quality assurance (Sgamma et al., 2017). In the last decade, DNA barcoding of herbal medicines has raised awareness of species substitution and adulteration, highlighting issues surrounding the quality of herbal medicines in the global market (Newmaster et al., 2013; Srirama et al., 2017). Regulation of herbal medicines is a pressing issue for regulatory agencies (Directive 2001/83/Ec, 2001; Directive 2004/83/EC, 2004; Vlietinck et al., 2009). Published pharmacopoeial standards for authentication predominantly rely on chemical and anatomical methods (e.g., British Pharmacopoeia, 2016), but DNA barcoding offers new tools for regulatory purposes (de Boer et al., 2015) and DNA barcodes have recently been incorporated into the British Pharmacopoeia for the first time (British Pharmacopoeia Commission, 2017). Here we investigate opportunities and limitations of DNA barcoding using next-generation sequence data of an evolutionarily complex genus. The aim is to design new methodological approaches for producing DNA barcodes for regulatory purposes, pharmacovigilance and quality assurance.
To date, the British Pharmacopoeia has approved 6 annotated DNA barcodes for the individual identification of the following species: Anethum graveolens Sowa (ITS2); Glehnia littoralis (ITS2); Ocimum tenuiflorum (trnH-psbA); Myristica fragrans (trnH-psbA); Phellodendron amurense (trnH-psbA); and Phellodendron chinense (trnH-psbA). The British Pharmacopoeia Commission (2017) have also published guidelines for the use of these barcodes, guiding users through the extraction of DNA, amplification of barcode markers, sequencing and comparison to pharmacopoeial standards. This development of bespoke barcode markers for different species is an approach likely to continue since there is no single, universal DNA barcode for land plants (Hollingsworth et al., 2011). For taxonomic purposes, several propositions have been made (e.g., Kress et al., 2005; Chase et al., 2007; CBOL Plant Working Group et al., 2009). Following Hollingsworth et al. (2011), most studies use a combination of the plastid regions matK, rbcL, the intergenic spacer trnH-psbA and the nuclear ITS2. Advances in sequencing technology have encouraged the barcoding community to augment the standard barcoding approach (Kane et al., 2012; Vaughn et al., 2014; Coissac et al., 2016; Zhang et al., 2017). In the era of next-generation sequencing, some researchers have even argued for the use of whole plastid genomes as barcodes (Kane et al., 2012; Vaughn et al., 2014; Coissac et al., 2016; Zhang et al., 2017; Manzanilla et al., 2018). How whole plastid genomes might be best deployed for pharmacopoeial purposes has hardly been explored yet.
Methodological approaches for specimen identification using DNA barcodes commonly rely on either distance-based measures or phylogenetic methods (Austerlitz et al., 2009). The former are based on the assumption that intra- and interspecific variation do not overlap (e.g., Hebert et al., 2004), also referred to as the barcoding gap (Meyer and Paulay, 2005). Accurate specimen identification using distance-based approaches such as BLAST are highly dependent on a well-curated database in which all members of a group are ideally represented by several individuals (Meyer and Paulay, 2005). The drawbacks of using distance-based approaches are that there is no objective distance threshold criterion and that the nearest neighbor is not always the closest relative (Moritz and Cicero, 2004). Specimen identification using phylogenetic methods is based on membership of a query sequence to a specific clade (Casiraghi et al., 2010). One difficulty associated with using tree-based barcoding methods is that phylogenies inferred from the barcode sequence might not be resolved sufficiently for an individual to be allocated to a clade, and that clades may exhibit poor support, questioning the robustness of any phylogenetic hypothesis (Moritz and Cicero, 2004). The use of concatenated DNA sequences for species tree inference has been shown to produce more robust phylogenetic hypotheses (Rokas et al., 2003). However, phylogenetic methods of DNA barcoding are not suitable when the underlying system is not based on strictly hierarchical ancestor-descendant relations structures, such as in nested structures (Goldstein and DeSalle, 2005).
Whether specimens of different species can be differentiated depends on the choice of the DNA barcode and the reproductive isolation and evolutionary history of the species under investigation. Although relatively high success rates for the identification of genera has been reported when using common barcodes in plants, limited sequence variation is often the cause of the failure to distinguish between closely related species (Seberg and Petersen, 2009; Parmentier et al., 2013; Braukmann et al., 2017). One incentive for employing genomic approaches for barcoding is that broader genome coverage increases the variation in the barcoding data set (Coissac et al., 2016). However, closely related species may not exhibit a DNA barcoding gap even when the most variable regions are employed. In the case of incipient speciation where lineage sorting is incomplete, species are likely to be paraphyletic (Rieseberg and Brouillet, 1994; Fazekas et al., 2009). Furthermore, cytoplasmic genomes can have different evolutionary histories compared with nuclear genomes because of processes such as chloroplast capture (Rieseberg and Soltis, 1991), and specimens may group geographically rather than taxonomically (Acosta and Premoli, 2010). The success of DNA barcoding may therefore be limited in some plant groups because of their biology and evolutionary history (Percy et al., 2014).
The genus Berberis is a case in which DNA barcoding using only a few regions has had limited success (Roy et al., 2010). Similarly, a phylogeny of Berberis based on ndhF and ITS loci failed to resolve boundaries of several species (Adhikari et al., 2015). Berberis aristata is a medicinal plant that has been in traditional use in India for centuries and is nowadays traded throughout the world (Srirama et al., 2017). Local market studies suggest that several species are traded under the same vernacular name (Srivastava and Rawat, 2013), including B. aristata and B. asiatica. B. aristata is described in several pharmacopoeias (Ayurvedic Pharmacopoeia of India, 2001; British Pharmacopoeia, 2016). Chemical and anatomical tests are deficient and conventional macro-morphological and microscopic examination do not distinguish the traded materials (Chandra and Purohit, 1980; Srivastava et al., 2004) therefore there is a strong incentive for the development of a DNA barcoding method for their identification.
The aim of this study is to investigate whole plastid sequences of the genus Berberis as a resource for barcode design, utilizing a whole plastid phylogeny of the species in order to better understand the difficulties of using barcoding for pharmacopoeial purposes. In light of the challenges of this complex group, we develop a method for identifying short, informative plastid barcode regions based on diagnostic nucleotides. These barcodes, which are informative of clade membership in a phylogenetic context, are tested on commercial samples, and their utility for regulatory purposes and quality control outlined.
This study includes 85 specimens from 57 species (Table 1). The dataset includes sequences from two putative new species (named in this study as B_newsppA and B_newsppB) and one unidentified species (B_spp).
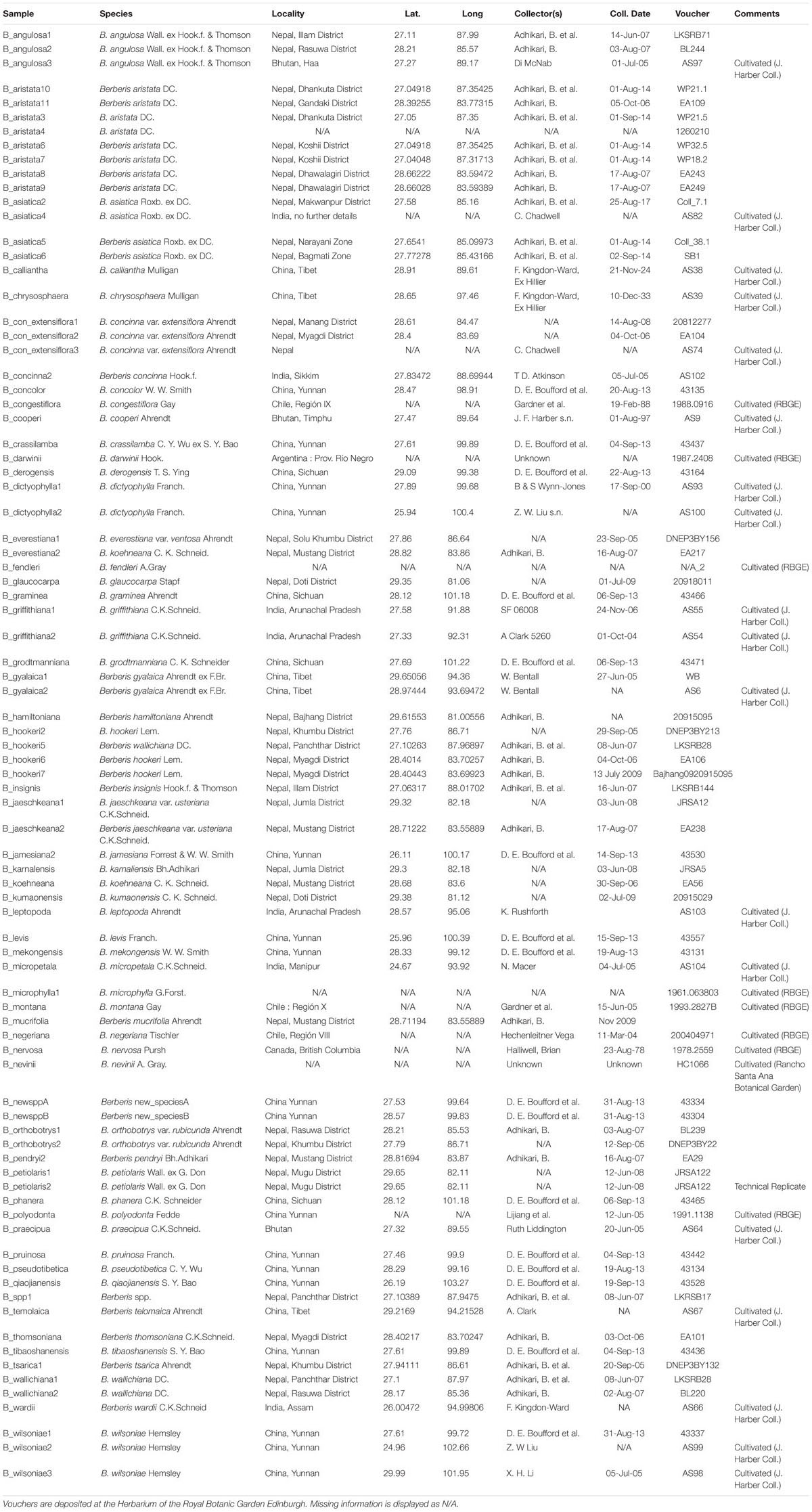
Table 1. Specimen information.
DNA was extracted using either the Qiagen DNeasy Plant Kit following the manufacturer’s protocol or the CTAB method (Doyle and Doyle, 1987). The quality of the extractions was checked for the degree of degradation on 1 or 1.5% agarose gels. Furthermore, we performed PCR amplifications of the rbcL gene in different dilutions (1:1, 1:10 and 1:100) and finally we measured the DNA concentration on a Qubit® Fluorometer (Life Technologies, Carlsbad, CA, United States), using the dsDNA High Sensitivity kit. The concentrations after extraction ranged from 1.5 to 34.8 ng/μl.
The library preparation for the shotgun sequencing was performed according to Meyer and Kircher (2010). The libraries were sequenced in two runs on a MiSeq® and a NextSeq®. Depending on their integrity, the DNA samples were sheared mechanically to a fragment size of approximately 400 bp using a Covaris© sonicator with peak incident power of 75; duty factor of 10%, and 200 cycles per burst. The duration of treatment was chosen according to the observed fragment size on agarose gels and ranged between 30s (medium degradation) and 40s (genomic DNA).
We followed the protocol for blunt-end repair, adapter ligation and adapter fill-in. After each of these steps, the DNA was cleaned-up with AMPure® XP beads (Agencourt®). Before the indexing PCR, the DNA quantity was measured on a Qubit©. Depending on the concentration of adapter-ligated libraries, we aimed to use between 50 and 100 ng of DNA as input for the indexing PCR where possible. Higher concentrations may impair the PCR reaction. In order to avoid high duplication levels, a minimal number of PCR cycles were applied. Libraries with concentrations lower than 40 ng were amplified with 16 PCR cycles. If more than 40 ng of library was used for the PCR, 12 cycles were applied. We used the index sequences (“barcodes”) as suggested by the protocol. The final libraries were washed using AMPure® XP beads (Agencourt®). We then measured for concentration with Qubit© and assessed the fragment size using Bioanalyzer® (Agilent). The libraries were diluted to 10 mM and pooled together. The libraries were sequenced in two runs on either an Illumina MiSeq® using the MiSeq v2 reagent kit with the 250 bp paired-end option or a NextSeq® with the NextSeq 500 High Output kit performing 150 bp paired-end sequencing.
The adapters of the raw reads were removed either with the built-in Illumina software on sequencers or using cutadapt v. 1.10 (Martin, 2011). Raw reads were trimmed using Trimmomatic v.0.33 (Bolger et al., 2014) with the options LEADING:3, TRAILING:3, SLIDINGWINDOW:4:20. Reads from Illumina NextSeq were discarded when shorter than 30 bp and from MiSeq when shorter than 50 bp. The read quality was checked with FastQC (Andrews, 2010).
The reference genome for B. aristata7 was reconstructed using a hybrid strategy of read mapping and de novo assembly. All reads were mapped to the reference plastid genome of Berberis bealei (Ma et al., 2013 GenBank reference KF176554), using the Geneious medium-low sensitivity “Map to Reference” function with five iterations. The resulting contig was then checked manually for low coverage and low pairwise identity regions. One read from each of these regions was extracted and all reads were then mapped against these individual reads as a new reference sequence using the same settings as above. The iterations lead to an extension of the read to a contig (typically up to 2,500 bp). The consensus sequences were then mapped to the reference obtained from the first read mapping. This method allowed large indels in the B. aristata reference that were not detected by the read mapping algorithm to be identified. The built-in de novo algorithm in Geneious 7.1.7 was used for the de novo assembly of the plastid genome. We performed the assembly only with reads that matched to the reference sequence of B. bealei. The ten largest contigs, ranging in length from 1,132 to 29,132 bp, were then mapped to the B. aristata reference and checked for ambiguities. All reads were then mapped again to the new consensus sequence.
We made our plastid genome reconstructions by mapping to a reference genome, having verified that the levels of variation between B. aristata, our reference, and the chloroplast genome of a member of the distantly related congeneric (B. bealei; Ma et al., 2013 GenBank reference KF176554), were structurally congruent. Reconstructions to a reference permitted a more rapid and cost-effective generation of high quality data than de novo assembly. The quality filtered paired-end reads were mapped to a reference genome of B. aristata7 with Burrows-Wheeler Alignment tool (BWA, ver. 0.7.12, Li and Durbin, 2009). The reference genome was indexed using option “bwa index.” Read pairs that survived the quality check were mapped with default options of the command “bwa mem.” The resulting SAM file was converted to BAM format with “samtools view” and sorted with “samtools sort” in SAMtools v. 1.2 (Li et al., 2009). Optical read duplicates were removed with Picard tools1. We used the single nucleotide polymorphism (SNP) calling workflow in GATK (McKenna et al., 2010; Van der Auwera et al., 2013). Regions that contain insertions and deletions are often badly aligned. Therefore, a local realignment process was applied with the command “–T IndelRealigner” in GATK. Variant calling was performed on the realigned BAM files with the “–T HaploTypeCaller” module with haploid settings (“-ploidy 1”). The output is a genomic variant call file (GVCF) that contains base call information for all sites of the markers. The variant calls were then exported with “–T GenotypeGVCFs” to the standard variant call format (VCF). SNP and indel variants were then filtered separately. The first SNP filter applied is quality by depth (QD), which can be considered as the quality of the variant call standardized by the depth of coverage. QD avoids inflation of the Phred quality score for the variant call caused by deep coverage. Variants that had a QD < 2 were filtered out as recommended by Van der Auwera et al. (2013). The FisherStrand (FS) quality filter is a Phred-scaled probability that strand bias exists at a specific site. Specifically, the score is a measure for whether an alternate allele was seen more or less often on either forward or reverse reads. The mapping quality (MQ) in GATK is calculated as the root mean square quality over all reads at a given site. The sites where variance resulted in an MQ score < M 40 were treated as missing data in order to avoid carry-over of reference- specific base pairs. The final sequence was reconstructed with the command “–T FastaAlternateReferenceMaker” in GATK. We checked our pipeline by visual comparison of the final plastid sequence with the BAM file for selected samples.
The plastids were aligned using the MAFFT v7.215 aligner (Katoh and Standley, 2013) with default options. The alignment of repetitive regions such as poly A sequences was not straight-forward, therefore two alignment files were created: the first alignment was used for phylogenetic inference, and blocks where no unambiguous alignment could be constructed were removed. Furthermore, the inverted repeats were removed, since SNP calling on these repeats was difficult to address. Reads with polymorphisms in only one region will map to the other repeat as well. Random mapping to inverted repeat regions often results in apparently heterozygous read alignments, precluding unique assignments of SNPs to a specific inverted repeat. The second alignment was used for the barcoding analysis. Regions were masked (coded as “N”) where no unambiguous alignment was possible.
The online platforms DOGMA (Wyman et al., 2004) and CpGAVAS (Liu et al., 2012) were used for the annotation of the genome of B. aristata7. The full genome sequences were imported into Apollo (Lee et al., 2009). The annotation of B. aristata was compared with the previously published annotation of B. bealei (Ma et al., 2013). Start and stop codons were checked manually. The annotation was visualized using OGdraw.
The sequences of matK, rbcL, and trnH-psbA of B. aristata were extracted from the annotated reference B. aristata7. The sequences were then aligned to the plastid genomes using BLAT (Kent, 2002). The output was parsed to produce a BED file, which denotes the start and end position of an alignment. The respective sequence was then extracted with the “getfasta” option in BEDTools (Quinlan and Hall, 2010).
A two-step pipeline was devised to reconstruct the ITS2 from shotgun sequencing data. Firstly, reads that map to the ITS2 reference were filtered and then a de novo assembly was performed using these reads. Filtering prior to de novo assembly reduces computation time substantially. The reference sequence of ITS2 (Berberis repens, BOLD accession: HIMS1138-12) was indexed with BWA (Li and Durbin, 2009) using the command “bwa index.” Trimmed and filtered reads were mapped to the reference with “bwa mem.” Mapped reads were then separated from unmapped reads with SAMtools (Li et al., 2009) “samtools view –b –F 4,” resulting in a BAM file with only mapped reads. The mapped reads were then extracted to fastq format using Picard tools (see footnote 1) with the command “SamToFastq.” The reads were then used for de novo assembly using SPAdes v3.7.0 (Bankevich et al., 2012) and the longest contig extracted.
The phylogeny of the plastid alignment was estimated using RAxML v. 8.2.10 (Stamatakis, 2014). The best model of substitution was calculated under the Aikaike Information Criterion in jModeltest2. The ML phylogeny was estimated with 1,000 bootstrap replicates under the GTRGAMMA + I substitution model using the online CIPRES portal (Miller et al., 2010). The whole alignment was considered as a single partition. Members of the compound-leaved Berberis were set as outgroup (B. nervosa, B. polyodonta and B. nevinii).
Potential novel Berberis-specific barcodes were explored by extracting SNP positions of the multiple sequence alignment of whole plastid genomes with the program SNP-sites (Page et al., 2016). The SNPs were summarized in 500 bp windows and their distribution plotted with Circos (Krzywinski et al., 2009). Potential barcodes were selected spanning regions where a 500 bp window had a sequence variability of >5%, and a maximum amount of missing/masked data <3%. The 500 bp regions were then compared to the annotated plastid genome and the barcodes were constructed to correspond with genomic regions, such as intergenic spacers that are flanked by conservative regions suitable for primer design. These Berberis specific barcodes derived from the whole plastid alignment were evaluated, along with the commonly used barcodes ITS2, rbcL, matK, and trnH-psbA.
The individual barcode regions were aligned using MAFFT v7.215 (Katoh and Standley, 2013) with default options and were then manually trimmed. A first step was to infer a maximum likelihood tree of the barcode with RAxML v.8.2.9 (Stamatakis, 2014) with 1,000 rapid bootstrap replicates (“–f a”) under the GTRCAT model. The potential barcodes were sorted according to the percent variable sites, percent parsimony informative sites, recovery of B. aristata and B. asiatica groups and the recovery of groups present in the whole plastid phylogeny. The selected barcodes were concatenated and a maximum likelihood phylogeny was built with the same parameters as described above. Phylogenies of the selected barcodes were inferred under the GTRCAT model in RAxML v. 8.2.9 (Stamatakis, 2014). Additionally, haplotype networks were constructed with the function haploNet in the R package pegas (Paradis, 2010). Finally the alignment of each selected barcode was then reduced to SNP sites only and diagnostic polymorphisms were identified for each group in order to delimit a minimal barcode.
The first test data consisted of three commercial samples, supposedly of B. aristata (Table 2). Sequences for the commercial samples were generated and the sequence data used to make identifications according to the diagnostic loci in Table 4.

Table 2. Commercial samples analyzed in this study.
The whole plastid phylogeny is shown in Figure 1. Nine groups, eight of which are monophyletic, are identified and numbered 1 to 9. The aristata, asiatica and Mahonia clades (numbered 4, 5, and 9 in Figure 1) are of most importance in terms of authentication. The plastid phylogeny reveals that B. aristata is not monophyletic since B. jaeschkeana, B. karnaliensis and B. mucrifolia are nested amongst the specimens of this species in clade 4. The topology of the phylogeny is consistent with morphological and biogeographical characters, and with the topology based on nuclear sequence data (Kreuzer et al., in prep.). The annotated plastid sequence of B_aristata7 is shown in Supplementary Figure S1 and the corresponding sequence is found on Genbank with reference number MK714340.
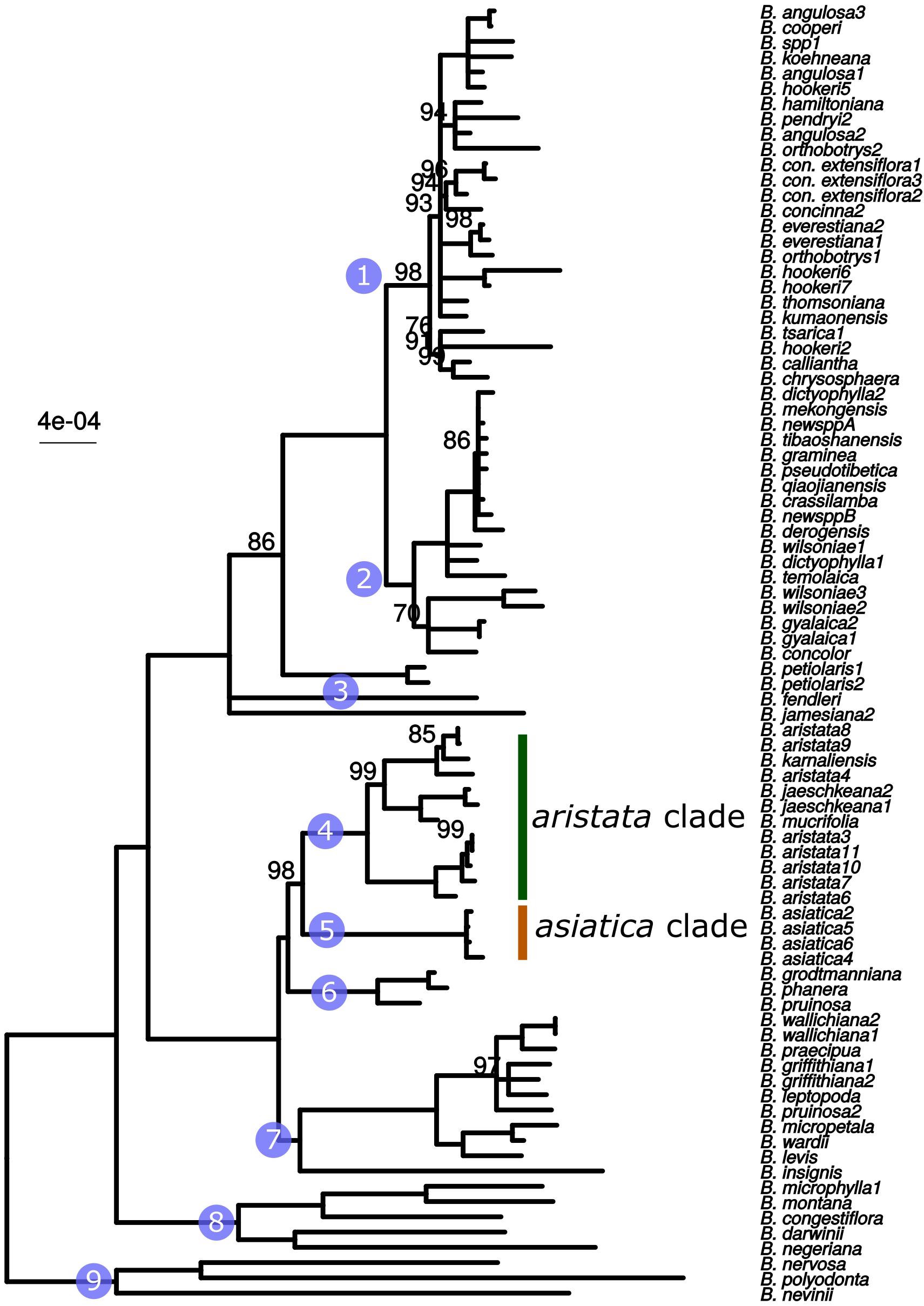
Figure 1. ML phylogeny based on whole plastid sequences. Note that B. aristata, in the aristata clade, is a polyphyletic species, but that B. asiatica samples in the asiatica group comprise a monophyletic group. Numbers above branches are bootstrap values between 51 and 99. Branches with support <50 were collapsed to polytomies, bootstrap values of 100 are not shown.
The barcoding analysis aimed to find a set of informative nucleotides that are unique to clades of interest. The topology of the whole plastid genome phylogeny was used to determine evolutionarily meaningful groups, termed Operational Phylogenetic Units (OPUs). Barcodes were then constructed for identifying these OPUs, rather than individual species. A barcoding method based on diagnostic characters was preferred over distance or purely phylogenetic approaches, because of its ease of application to regulatory purposes and to provide an alternative approach in an evolutionarily complex group. The density of SNPs in 500 bp windows along the whole plastid alignment is shown in Figure 2. The bins contained between 0 and 124 variable sites per 500 bp. The inspection of bins with >25 SNPs (5%) resulted in 21 potential barcode regions. Several of the highly variable bins fell into regions where the alignment was partly masked due to ambiguous alignment, leaving 13 bins for further inspection. Two neighboring bins were combined into a single potential barcode of 1,000 bp, and a set of four bins combined into a 2000 base pair barcode. The barcode of 2,000 bp (SSC_noncoding2) was further examined by partitioning the alignment into 50 bp windows and reducing the barcode size (SSC_noncoding2, Figure 3). The trnH-psbA intergenic spacer was identified among one of the seven highly variable regions, and together with the matK, rbcL and ITS2 barcodes, selected because they are commonly used barcode regions, eleven barcode candidates were investigated (Table 3). None of the individual barcodes retrieved phylogenies with the same topology as the whole plastid phylogeny. Although the matK phylogeny is not well resolved overall, species from the aristata and asiatica groups were recovered. B. asiatica is monophyletic in the non-coding SSC_noncoding2 phylogeny, but species from the aristata clade are separated into two groups. The percent variable sites varied between 2.2 in rbcL and 9.85 in the intergenic spacer ndhI-ndhG (Table 3) and the latter was chosen along with matK and SSC_noncoding2 as barcodes for phylogenetic and haplotype analysis (Figure 4).
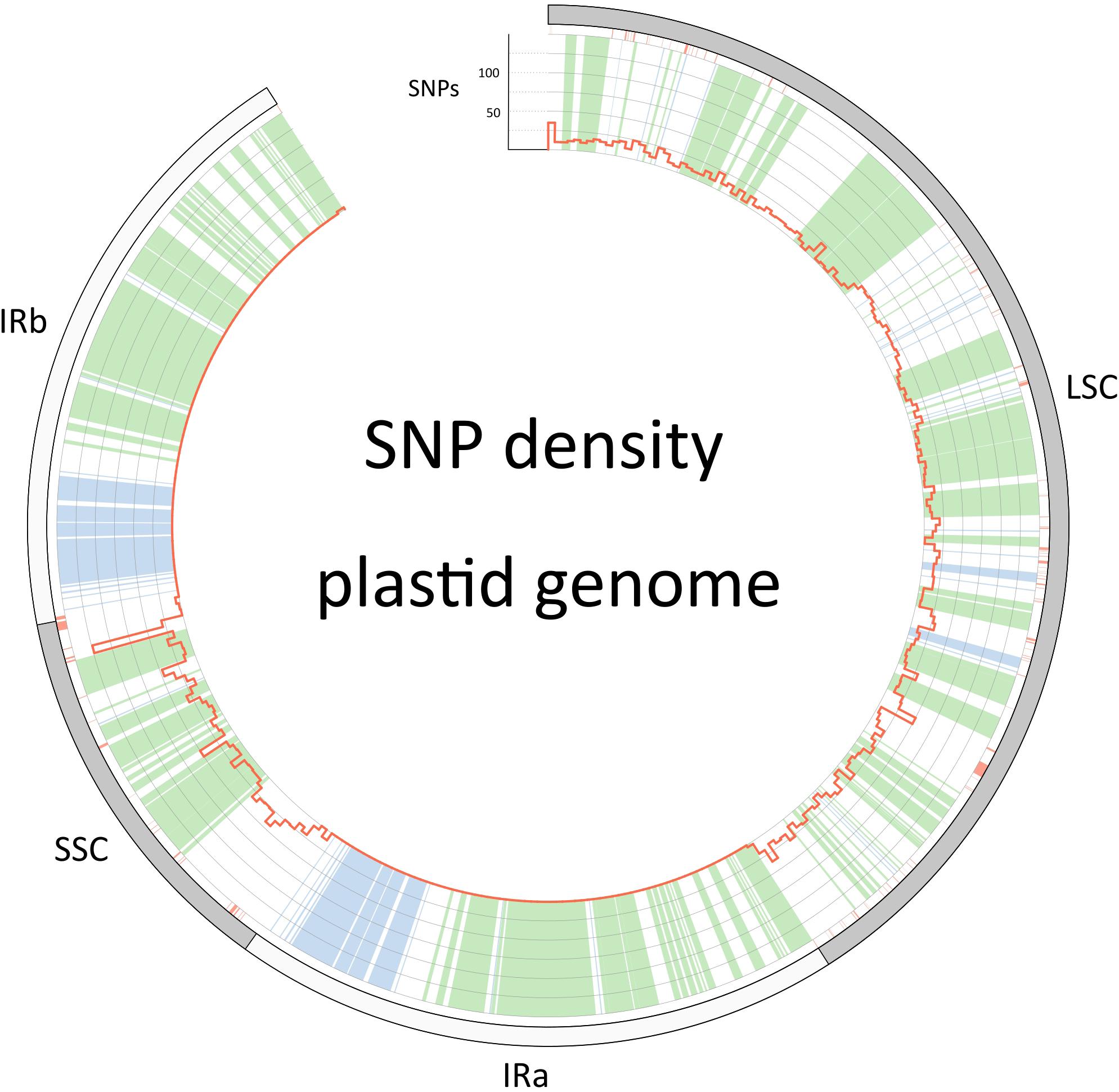
Figure 2. Single nucleotide polymorphism density along the plastid genome (red histograms). The outer circle describes the boundaries of the large single copy, the inverted repeats (IRa and IRb) and the small single copy. Regions that are colored green in the inner circle are coding regions, blue are RNA genes (rRNA and tRNA genes) and white is non-coding sequence. Red color below the outer circle shows regions that have been masked and are thus coded as “N”.
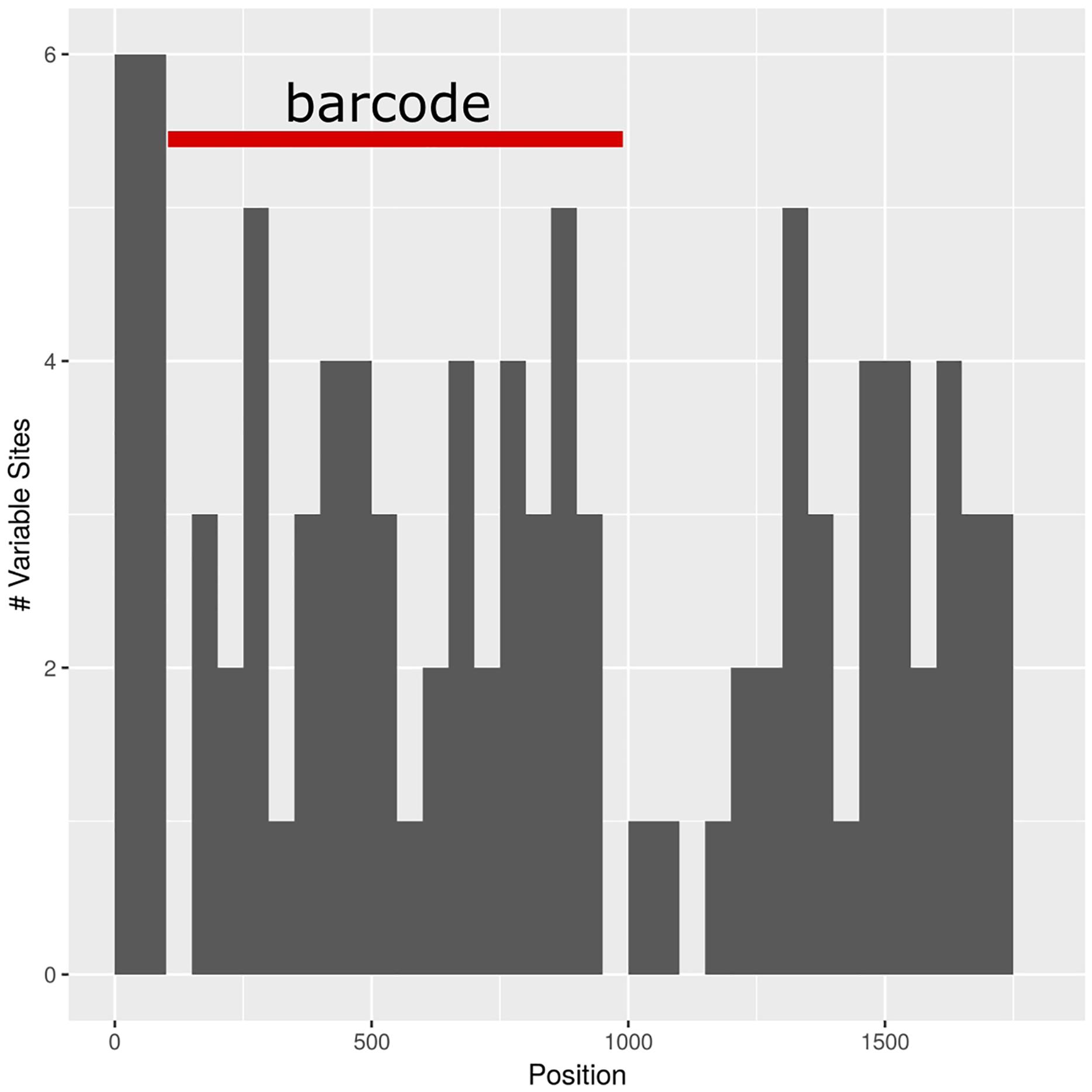
Figure 3. Subselection of barcode regions with the SSC_noncoding2 region. The newly determined barcode is marked in red.
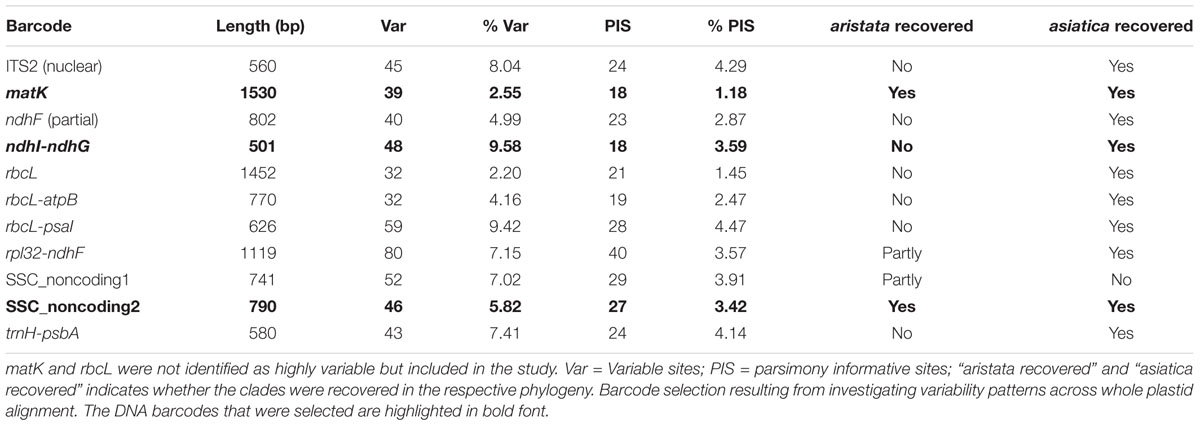
Table 3. Barcode selection resulting from investigating variability patterns across whole plastid alignment.
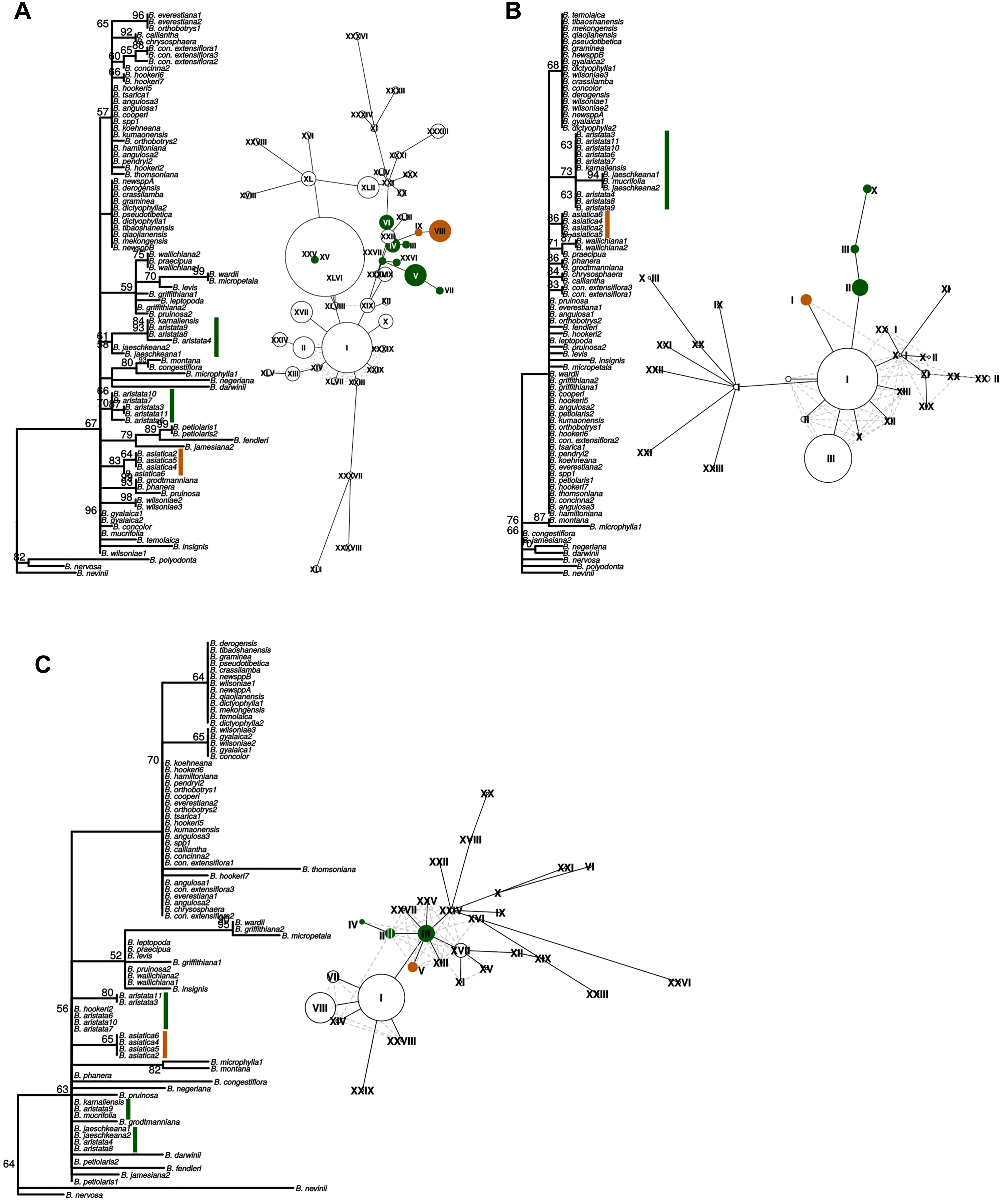
Figure 4. “Maximum likelihood phylogenies and haplotype networks of individual barcodes. (A) SSC_noncoding2, (B) matK, (C) ndhI-ndhG. Values on braches in the phylogeny are ML bootstraps. Species belonging to the Berberis aristata clade as recovered from the total plastid phylogeny are identified by green bars, and the B. asiatica clades by orange bars on the phylogeny. The same colors are used for the haplotype networks, where Roman numerals indicate different haplotypes and the size of the circles corresponds to the number of samples sharing this haplotype. Species contributing to B. aristata clade haplotypes are as follows: SSC_noncoding2 – XXV = B. mucrifolia (1 plant), VI = B. aristata (2 plants), IV = B. aristata (2 plants), III = B. aristata (1 plant), XXVI = B. jaeschkeana (1 plant), XXVII = B. jaeschkeana (1 plant), V = B. aristata (2 plants) and B. karnaliensis (1 plant), VII = B. aristata (1 plant); matK – X = B. jaeschkeana (2 plants) and B. mucrifolia (1 plant), III = B. aristata (3 plants) II = B. aristata (5 plants) and B. karnaliensis (1 plant); ndhI-ndhG – IV = B. aristata (2 plants), II = B. aristata (2 plants) and B. hookeri (1 plant), III = B. aristata (3 plants) and B. jaeschkeana (2 plants) and B. karnaliensis (1 plant) and B. mucrifolia (1 plant).”
These three barcodes yielded 133 variable positions in total. Nine positions were sufficient to identify seven of the nine groups with clade-specific nucleotide variants. Groups 3 and 8 (Figure 1) share a barcode, in other words their barcodes are identical. The phylogeny of the concatenated barcodes matK, SSC_noncoding2 and ndhI-ndhG barcodes is shown in Figure 5. The topology of the tree differs substantially from the total-evidence tree inferred from whole plastid sequences. However, four of the major clades are identified in both trees. Haplotype networks constructed for each of the separate data sets showed variation in the haplotype associated with the B. aristata clade (Figure 4). There was no haplotype unique to B. aristata: for the SSC_noncoding2 region one of the B. aristata haplotypes is found also in B. karnaliensis; for the matK region there is also a haplotype shared between B. aristata and B. karnaliensis; for ndhI-ndhG there is a haplotype found in B. aristata, B. jaeschkeana, B. karnaliensis and B. mucrifolia. The lack of species-specific haplotypes even in these most variable regions underlines the necessity of a clade-based approach. However, for pharmacopoeial purposes the haplotype networks reveal separation of the B. aristata clade haplotypes and B. asiatica haplotypes.
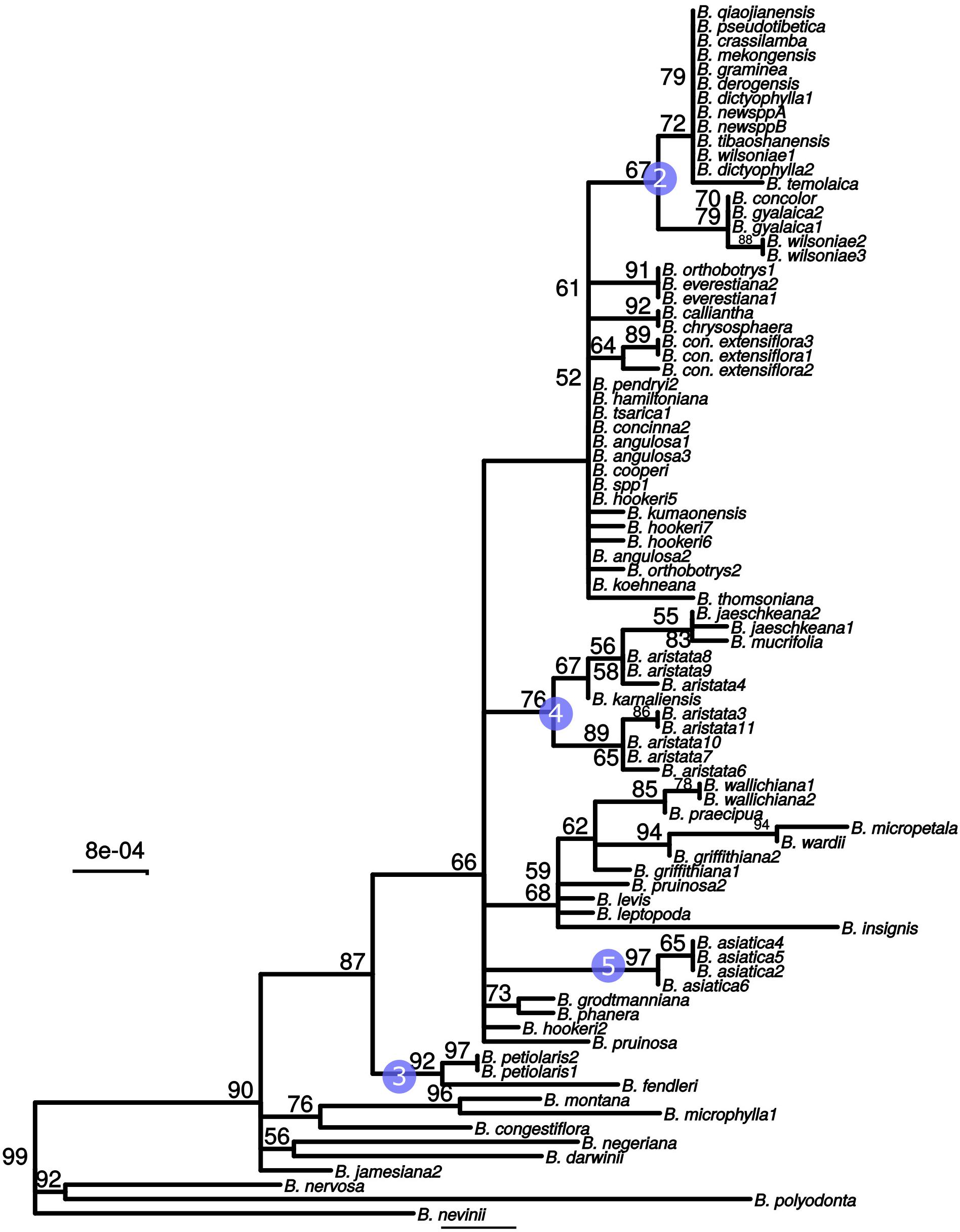
Figure 5. Maximum likelihood tree from the concatenated barcodes matK, SSC_noncoding2 and ndhI-ndhG. Nodes with bootstrap support <50 were collapsed to polytomies. Bootstrap values between 50 and 99 are shown above branches. No number indicates a bootstrap value of 100. Numbered circles indicate groups that were recovered in the whole plastid phylogeny (see Figure 1).
The minimal barcode consists of nine positions and includes barcodes unique to seven groups. No unique SNPs were identified for groups 3, 6, and 8. No individual barcode for groups 6 and 8 could be constructed (Table 4). The barcodes were evaluated with the test data set. The commercial samples Market1 and Market2 were identified as belonging to the Mahonia clade. The sample Market11 shared the barcode with B. asiatica samples.

Table 4. Top: Matrix of informative barcode positions.
DNA barcoding for quality assurance and pharmacovigilance has great potential and is likely to be implemented as a routine diagnostic method. In this study, we present an approach for barcoding of an evolutionarily complex group of species and demonstrate that these barcodes can identify the species in commercial samples. Our purpose was to provide a barcode for pharmacopoeial purposes that discriminates B. aristata and B. asiatica since these are the pharmacopoeial species and the main substitute, respectively. We present a solution for barcoding that meets regulatory needs.
With the emergence of new sequencing technologies, whole plastid sequencing has been proposed as an extension of the current barcoding concept (Coissac et al., 2016). It has been shown that whole plastid sequences increase phylogenetic resolution (Parks et al., 2009) and simultaneously increase the effectiveness of discriminating between species. In this study, we show how whole plastid next-generation sequencing can be used to investigate sequence variability patterns for the discovery of informative DNA barcodes. We confirm the difficulty of barcoding Berberis species as suggested by Roy et al. (2010), even when whole plastid sequences are used for comparison. Although the sampling was limited, with only a few of the species represented with multiple samples, the low resolution of the plastid phylogeny at shallow phylogenetic levels and the presence of polyphyletic species (e.g., B. aristata) indicates evolutionary reasons for the failure of barcoding this genus to species level (Mutanen et al., 2016). DNA barcoding is challenging in groups where frequent hybridization occurs in conjunction with plastid capture or where lineage sorting has not yet been completed (Fazekas et al., 2009). A salient point arising from our study is that the pharmacopoeial species, B. aristata, is polyphyletic. One explanation for this finding is hybridization, a phenomenon documented in Berberis (Adhikari et al., 2012). Low resolution among the closely related species of Berberis as reported in the whole plastid phylogeny, could point toward retention of ancestral polymorphism or incomplete lineage sorting (Naciri and Linder, 2015). Misidentification of B. jaeschkeana, B. karnaliensis and/or B. mucrifolia is unlikely, since these have been included in recent revisionary work (Adhikari et al., 2012). Polyphyletic species are likely to persist where they are morphologically robust entities, and the development of methods for their identification, in this case for pharmacopoeia, benefits from understanding of their evolutionary history. The case of barcoding medicinal Berberis species provides an example of how barcoding for regulatory purposes in an evolutionarily complex group can be approached. Phylogenies can be essential for formulating adequate barcoding hypotheses; the whole plastid phylogeny reveals that at least three species are nested in the clade with the main species. The polyphyly of B. aristata indicates that universal barcodes are unlikely to delineate these species, and haplotype analysis shows this is the case for three of the most variable regions. Furthermore, several clades show low resolution at terminal branches. We have therefore adapted our classification scheme and defined meaningful OPUs that do not correspond to existing species limits. OPUs are the entities that can be discriminated by the barcodes put forward. The OPUs in this study are delimited using an integrative approach based on the interpretation of a whole plastid phylogeny, coupled with the detection of diagnostic nucleotides in relatively short barcodes for well-supported groups. These DNA barcodes can be targeted by PCR and Sanger sequencing and therefore offer a simple and fast identification test for regulatory purposes and quality control. Appropriate OPUs would be identified on a case-by-case basis for other evolutionarily complex groups for regulatory purposes. This is because for evolutionarily complex groups barcodes do not confirm species identity. The novelty of our approach lies in using whole plastid phylogeny to identify of short, easily amplified markers that incorporate clade-specific SNPs, and although we expect it to be more widely applicable it is only appropriate when the non-pharmacopoeial species belonging to the OPU are neither candidate adulterants nor substitute species, as is the case here.
The barcode presented in this study is based on diagnostic nucleotides for groups of species, referred to here as OPUs. Like the morphological classification of species, diagnostic methods provide a set of unique characters to assign specimens to species or species groups (Little and Stevenson, 2007). Diagnostic methods are particularly well-suited to pharmacopoeial purposes because a sequence generated from test material can be compared to a published sequence in a way that is comparable to other pharmacopoeial standards. The barcode we propose would require the user to amplify and sequence three regions, whereas the barcodes included in the British Pharmacopoeia to date are single regions (British Pharmacopoeia, 2016). We have limited the number of loci that would be part of the test to three because incorporating more loci would make the test more unwieldy for users. Limiting the number of regions necessarily reduces the number of informative sites. Identifying the most informative regions, as we do here, is therefore important. A deficiency of the diagnostic method is that further samples might show variation that is not present amongst the samples used for barcode design. However, there is scope to modify the published barcodes, perhaps by using the IUPAC nucleotide codes, if novel variants are reported.
The diagnostic method has been implemented in various analysis tools (Sarkar et al., 2008; Weitschek et al., 2013), mainly for specimen identification. Some of the algorithms use logic mining techniques (Bertolazzi et al., 2009). Logic mining for DNA barcoding refers to a two-step process, in which the barcode is first reduced to a set of very informative nucleotides and thereafter a logic mining method is applied, to define a set of formulas for separating the species. More recent approaches, such as BLOG 2.0 (Weitschek et al., 2013), provide a diagnostic, character-based methodology to species identification that is based on supervised machine learning. Character-based approaches circumvents analytical issues such as the nearest-neighbor problem in distance-based methods (DeSalle et al., 2005). Although the in silico mixtures presented in this study were created from the samples that were used for producing the DNA barcode and are therefore not true test samples, the analysis demonstrates the utility of analyzing mixed samples based on diagnostic nucleotides when shotgun sequencing data is available.
We believe that the development of clade-specific DNA barcodes is the way forward when investigating evolutionarily complex species. The barcodes we present are readily understandable and easily applicable for large-scale and routine testing of samples using PCR and Sanger sequencing. DNA barcoding is beyond doubt a powerful method for specimen identification, but its implementation as a routine process for quality assurance (Sgamma et al., 2017) and pharmacovigilance (de Boer et al., 2015) will depend on the ease of application. Neither phylogenetic nor distance methods are appropriate, since they depend on large databases, sophisticated tools and lack objective criteria. For this reason, the British Pharmacopoeia (BP) approach is to present a sequence which samples must match for authentication. Pharmacopoeias ensure the safe use of pharmaceuticals by defining certain quality standards and DNA barcodes have recently been published in the BP for the first time (British Pharmacopoeia Commission, 2017). The question “does this sample correspond to the pharmacopoeial species?” is addressed by comparison to the pharmacopoeial sequence, since methods based on diagnostic nucleotides provide an easy and straight-forward way to answer the question. Identifying such sequences for inclusion in a pharmacopeia is the challenge addressed by this study. The whole plastid approach described here could become a model that can be applied to species that are difficult to resolve. Success depends on devising a sampling strategy that includes species that are closely related to the target species. Furthermore, the inclusion of distantly related, congeneric species increases the confidence in detected diagnostic nucleotide polymorphisms.
JH, CH, CP, and MK contributed to the conception and design of the study. BA and CP provided samples and made taxonomic identifications. CH and MK conducted the laboratory work. MK performed the data analysis and wrote the first draft of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
This work was conducted as part of the MedPlant ITN and received funding from the European Union’s Seventh Framework Program for research, technological development and demonstration under grant agreement no. 606895.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to acknowledge the herbal medicines research group, the NGS core facility at the National Institute for Biological Standards and Control (NIBSC) and Edward Mee for help in NGS sequencing. We also would like to thank the group of JH at the University of Reading for facilitating lab work and discussions of the manuscript. Julian Harber has contributed to this study by providing samples from his personal collection.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00586/full#supplementary-material
FIGURE S1 | Gene map of the plastid genome of Berberis aristata. Genes on the outside of the circle are transcribed clockwise and genes on the inside anti-clockwise. The dark gray histograms in the inner circle show the GC content.
Acosta, C. M., and Premoli, A. C. (2010). Evidence of chloroplast capture in south american nothofagus (subgenus nothofagus, nothofagaceae). Mol. Phylogenet. Evol. 54, 235–242. doi: 10.1016/j.ympev.2009.08.008
Adhikari, B., Milne, R., Pennington, R. T., Särkinen, T., and Pendry, C. A. (2015). Systematics and biogeography of Berberis s. l. inferred from nuclear ITS and chloroplast ndhF gene sequences. Taxon 64, 39–48. doi: 10.12705/641.21
Adhikari, B., Pendry, C. A., Pennington, R. T., and Milne, R. I. (2012). A revision of Berberis s.s. (berberidaceae) in nepal. Edinburgh J. Bot. 69, 447–522. doi: 10.1017/S0960428612000261
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ doi: 10.1017/s0960428612000261 (accessed May 2, 2019).
Austerlitz, F., David, O., Schaeffer, B., Bleakley, K., Olteanu, M., Leblois, R., et al. (2009). DNA barcode analysis: a comparison of phylogenetic and statistical classification methods. BMC Bioinformatics 10(Suppl. 1):S10. doi: 10.1186/1471-2105-10-S14-S10
Ayurvedic Pharmacopoeia of India (2001). Ayurvedic Pharmacopoeia of India. New Delhi: Government of India, Ministry of Health and Family Welfare.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bertolazzi, P., Felici, G., and Weitschek, E. (2009). Learning to classify species with barcodes. BMC Bioinformatics 10(Suppl. 14):S7. doi: 10.1186/1471-2105-10-S14-S7
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Braukmann, T. W. A., Kuzmina, M. L., Sills, J., Zakharov, E. V., and Hebert, P. D. N. (2017). Testing the efficacy of DNA barcodes for identifying the vascular plants of Canada. PLoS One 12:e0169515. doi: 10.1371/journal.pone.0169515
British Pharmacopoeia (2016). British Pharmacopoeia. London: Medicines and Healthcare Regulatory Agency (MHRA).
British Pharmacopoeia Commission (2017). British Pharmacopoeia Appendix XI V Deoxyribonucleic Acid (DNA) Based Identification Techniques for Herbal Drugs. London: TSO.
Casiraghi, M., Labra, M., Ferri, E., Galimberti, A., and de Mattia, F. (2010). DNA barcoding: a six-question tour to improve users’ awareness about the method. Brief. Bioinform. 11, 440–453. doi: 10.1093/bib/bbq003
Chandra, P., and Purohit, A. N. (1980). Berberine contents and alkaloid profile of Berberis species from different altitudes. Biochem. Syst. Ecol. 8, 379–380. doi: 10.1016/0305-1978(80)90040-X
Chase, M. W., Cowan, R. S., Hollingsworth, P. M., Berg, C., Van Den Madriñán, S., Petersen, G., et al. (2007). A proposal for a standardised protocol to barcode all land plants published by: international association for plant taxonomy ( iapt ) linked references are available on jstor for this article: new trends a proposal in plant to barcode all land plants fo. Taxon 56, 295–299. doi: 10.1002/tax.562004
Coissac, E., Hollingsworth, P. M., Lavergne, S., and Taberlet, P. (2016). From barcodes to genomes: extending the concept of DNA barcoding. Mol. Ecol. 25, 1423–1428. doi: 10.1111/mec.13549
de Boer, H. J., Ichim, M. C., and Newmaster, S. G. (2015). DNA barcoding and pharmacovigilance of herbal medicines. Drug Saf. 38, 611–620. doi: 10.1007/s40264-015-0306-8
DeSalle, R. (2006). Species discovery versus species identification in dna barcoding efforts: response to rubinoff. Conserv. Biol. 20, 1545–1547. doi: 10.1111/j.1523-1739.2006.00543.x
DeSalle, R., Egan, M. G., and Siddall, M. (2005). The unholy trinity: taxonomy, species delimitation and DNA barcoding. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 1905–1916. doi: 10.1098/rstb.2005.1722
Directive 2001/83/Ec (2001). On the Community code relating to medicinal products for human use. Off. J. Eur. Union L 311, 67–128.
Directive 2004/83/EC (2004). On minimum standards for the qualification and status of third country nationals or stateless persons as refugees or as persons who otherwise need international protection and the content of the protection granted. Off. J. Eur. Union L 136, 85–90.
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Fazekas, A. J., Kesanakurti, P. R., Burgess, K. S., Percy, D. M., Graham, S. W., Barrett, S. C. H., et al. (2009). Are plant species inherently harder to discriminate than animal species using DNA barcoding markers? Mol. Ecol. Resour. 9 (Suppl. s1), 130–139. doi: 10.1111/j.1755-0998.2009.02652.x
Goldstein, P. Z., and DeSalle, R. (2005). Phylogenetic species, nested hierarchies, and character fixation. Cladistics 16, 364–384. doi: 10.1111/j.1096-0031.2000.tb00356.x
Hebert, P. D. N., Stoeckle, M. Y., Zemlak, T. S., and Francis, C. M. (2004). Identification of birds through DNA barcodes. PLoS Biol. 2:e312. doi: 10.1371/journal.pbio.0020312
Hollingsworth, P. M., Graham, S. W., and Little, D. P. (2011). Choosing and using a plant DNA barcode. PLoS One 6:e19254. doi: 10.1371/journal.pone.0019254
Kane, N., Sveinsson, S., Dempewolf, H., Yang, J. Y., Zhang, D., Engels, J. M. M., et al. (2012). Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 99, 320–329. doi: 10.3732/ajb.1100570
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kent, W. J. (2002). BLAT—the blast-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A., and Janzen, D. H. (2005). Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. U.S.A. 102, 8369–8374. doi: 10.1073/pnas.0503123102
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lee, E., Harris, N., Gibson, M., Chetty, R., and Lewis, S. (2009). Apollo: a community resource for genome annotation editing. Bioinformatics 25, 1836–1837. doi: 10.1093/bioinformatics/btp314
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Little, D. P., and Stevenson, D. W. (2007). A comparison of algorithms for the identification of specimens using DNA barcodes: examples from gymnosperms. Cladistics 23, 1–21. doi: 10.1111/j.1096-0031.2006.00126.x
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and genbank submission of completely sequenced chloroplast genome sequences. BMC Genomics 13:715. doi: 10.1186/1471-2164-13-715
Ma, J., Yang, B., Zhu, W., Sun, L., Tian, J., and Wang, X. (2013). The complete chloroplast genome sequence of mahonia bealei (Berberidaceae) reveals a significant expansion of the inverted repeat and phylogenetic relationship with other angiosperms. Gene 528, 120–131. doi: 10.1016/j.gene.2013.07.037
Manzanilla, V., Kool, A., Nguyen Nhat, L., Nong Van, H., Le Thi Thu, H., and De Boer, H. J. (2018). Phylogenomics and barcoding of panax: toward the identification of ginseng species. BMC Evol. Biol. 18:44. doi: 10.1186/s12862-018-1160-y
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meyer, C. P., and Paulay, G. (2005). DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 3:e422. doi: 10.1371/journal.pbio.0030422
Meyer, M., and Kircher, M. (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010:pdb.prot5448. doi: 10.1101/pdb.prot5448
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA.
Moritz, C., and Cicero, C. (2004). DNA barcoding: promise and pitfalls. PLoS Biol. 2:e354. doi: 10.1371/journal.pbio.0020354
Mutanen, M., Kivelä, S. M., Vos, R. A., Doorenweerd, C., Ratnasingham, S., Hausmann, A., et al. (2016). Species-level para- and polyphyly in DNA barcode gene trees: strong operational bias in european lepidoptera. Syst. Biol. 65, 1024–1040. doi: 10.1093/sysbio/syw044
Naciri, Y., and Linder, H. P. (2015). Species delimitation and relationships: the dance of the seven veils. Taxon 64, 3–16. doi: 10.12705/641.24
Newmaster, S. G., Grguric, M., Shanmughanandhan, D., Ramalingam, S., and Ragupathy, S. (2013). DNA barcoding detects contamination and substitution in north american herbal products. BMC Med. 11:222. doi: 10.1186/1741-7015-11-222
Page, A. J., Taylor, B., Delaney, A. J., Soares, J., Seemann, T., Keane, J. A., et al. (2016). SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genomics 2:e000056. doi: 10.1099/mgen.0.000056
Paradis, E. (2010). Pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420. doi: 10.1093/bioinformatics/btp696
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Parmentier, I., Duminil, J., Kuzmina, M., Philippe, M., Thomas, D. W., Kenfack, D., et al. (2013). How effective are DNA barcodes in the identification of african rainforest trees? PLoS One 8:e54921. doi: 10.1371/journal.pone.0054921
Percy, D. M., Argus, G. W., Cronk, Q. C., Fazekas, A. J., Kesanakurti, P. R., Burgess, K. S., et al. (2014). Understanding the spectacular failure of DNA barcoding in willows (Salix): does this result from a trans-specific selective sweep? Mol. Ecol. 23, 4737–4756. doi: 10.1111/mec.12837
Plant Working Group, C. B. O. L., Hollingsworth, P. M., Forrest, L. L., Spouge, J. L., Hajibabaei, M., Ratnasingham, S., et al. (2009). A DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 106, 12794–12797. doi: 10.1073/pnas.0905845106
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rieseberg, L. H., and Soltis, D. E. (1991). Phylogenetic consequences of cytoplasmic gene flow in plants. Evol. Trends Plants 5, 65–84. doi: 10.1007/s00606-006-0485-y
Rokas, A., Williams, B. L., King, N., and Carroll, S. B. (2003). Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425, 798–804. doi: 10.1038/nature02053
Roy, S., Tyagi, A., Shukla, V., Kumar, A., Singh, U. M., Chaudhary, L. B., et al. (2010). Universal plant DNA barcode loci may not work in complex groups: a case study with Indian Berberis species. PLoS One 5:e13674. doi: 10.1371/journal.pone.0013674
Sarkar, I. N., Planet, P. J., and Desalle, R. (2008). CAOS software for use in character-based DNA barcoding. Mol. Ecol. Resour. 8, 1256–1259. doi: 10.1111/j.1755-0998.2008.02235.x
Seberg, O., and Petersen, G. (2009). How many loci does it take to DNA barcode a crocus? PLoS One 4:e4598. doi: 10.1371/journal.pone.0004598
Sgamma, T., Lockie-williams, C., Kreuzer, M., Williams, S., Scheyhing, U., Koch, E., et al. (2017). DNA barcoding for industrial quality assurance. Planta Med. 83, 1117–1129. doi: 10.1055/s-0043-113448
Srirama, R., Santhosh Kumar, J. U., Seethapathy, G. S., Newmaster, S. G., Ragupathy, S., Ganeshaiah, K. N., et al. (2017). Species adulteration in the herbal trade: causes, consequences and mitigation. Drug Saf. 40, 651–661. doi: 10.1007/s40264-017-0527-0
Srivastava, S., and Rawat, A. K. S. (2013). Quality evaluation of ayurvedic crude drug daruharidra, its allied species, and commercial samples from herbal drug markets of India. Evid. Based. Complement. Alternat. Med. 2013:472973. doi: 10.1155/2013/472973
Srivastava, S. K., Rawat, A. K. S., and Mehrotra, S. (2004). Pharmacognostic evaluation of the root of Berberis asiatica. Pharm. Biol. 42, 467–473. doi: 10.1080/13880200490886256
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From fastq data to high confidence variant calls: the genome analysis toolkit best practices pipeline.[GATK]. Curr. Protoc. Bioinforma. 43:11.10.1-33
Vaughn, J. N., Chaluvadi, S. R., Tushar Rangan, L., and Bennetzen, J. L. (2014). Whole plastome sequences from five ginger species facilitate marker development and define limits to barcode methodology. PLoS One 9:e108581. doi: 10.1371/journal.pone.0108581
Vlietinck, A., Pieters, L., and Apers, S. (2009). Legal requirements for the quality of herbal substances and herbal preparations for the manufacturing of herbal medicinal products in the European union. Planta Med. 75, 683–688. doi: 10.1055/s-0029-1185307
Weitschek, E., Van Velzen, R., Felici, G., and Bertolazzi, P. (2013). BLOG 2.0: a software system for character-based species classification with DNA barcode sequences. What it does, how to use it. Mol. Ecol. Resour. 13, 1043–1046. doi: 10.1111/1755-0998.12073
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Keywords: DNA barcoding, next-generation sequencing, operational phylogenetic units, herbal medicines, Berberis, pharmacopoeia, pharmacopoeial standards, plastome
Citation: Kreuzer M, Howard C, Adhikari B, Pendry CA and Hawkins JA (2019) Phylogenomic Approaches to DNA Barcoding of Herbal Medicines: Developing Clade-Specific Diagnostic Characters for Berberis. Front. Plant Sci. 10:586. doi: 10.3389/fpls.2019.00586
Received: 29 November 2018; Accepted: 18 April 2019;
Published: 14 May 2019.
Edited by:
Nunzio D’Agostino, University of Naples Federico II, ItalyReviewed by:
Michael R. McKain, The University of Alabama, United StatesCopyright © 2019 Kreuzer, Howard, Adhikari, Pendry and Hawkins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Kreuzer, bWFyY28uYy5rcmV1emVyQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.