
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 07 May 2019
Sec. Plant Breeding
Volume 10 - 2019 | https://doi.org/10.3389/fpls.2019.00513
 V. B. Reddy Lachagari1*Ravi Gupta2,3Sivarama Prasad Lekkala1Lakshmi Mahadevan2,3Boney Kuriakose4,5Navajeet Chakravartty1A. V. S. K. Mohan Katta1Sam Santhosh4Arjula R. Reddy6George Thomas4,5
V. B. Reddy Lachagari1*Ravi Gupta2,3Sivarama Prasad Lekkala1Lakshmi Mahadevan2,3Boney Kuriakose4,5Navajeet Chakravartty1A. V. S. K. Mohan Katta1Sam Santhosh4Arjula R. Reddy6George Thomas4,5Purpleputtu (Oryza sativa ssp. indica cv. Purpleputtu) is a unique rice landrace from southern India that exhibits predominantly purple color. This study reports the underlying genetic complexity of the trait, associated domestication and de-domestication processes during its coevolution with present day cultivars. Along-with genome level allelic variations in the entire gene repertoire associated with the purple, red coloration of grain and other plant parts. Comparative genomic analysis using ‘a panel of 108 rice lines’ revealed a total of 3,200,951 variants including 67,774 unique variations in Purpleputtu (PP) genome. Multiple sequence alignment uncovered a 14 bp deletion in Rc (Red colored, a transcription factor of bHLH class) locus of PP, a key regulatory gene of anthocyanin biosynthetic pathway. Interestingly, this deletion in Rc gene is a characteristic feature of the present-day white pericarped rice cultivars. Phylogenetic analysis of Rc locus revealed a distinct clade showing proximity to the progenitor species Oryza rufipogon and O. nivara. In addition, PP genome exhibits a well conserved 4.5 Mbp region on chromosome 5 that harbors several loci associated with domestication of rice. Further, PP showed 1,387 unique when SNPs compared to 3,023 lines of rice (SNP-Seek database). The results indicate that PP genome is rich in allelic diversity and can serve as an excellent resource for rice breeding for a variety of agronomically important traits such as disease resistance, enhanced nutritional values, stress tolerance, and protection from harmful UV-B rays.
Rice is the staple food for more than half of the world’s population and substantially meets both food and calorie requirements. Rice cultivation covers about 165 million hectares globally with an annual production of 758.8 million MT (Food and Agriculture Organization of the United Nations [FAOUN], 2017) and is a critical component of the global food security system. Diverse Asian population rely on rice to cover 35–80% of their calorie needs, while global reliance is about 21%. The two subspecies of cultivated rice, O. sativa, namely indica and japonica, occupy more than 90% of the Asian rice crop acreage (Muthayya et al., 2014). Evolution of rice from its progenitors is marked by great complexity as it appears to have involved diverse lineages, domestication/de-domestication processes and selection, both natural and artificial. Domestication and selection of different populations of Asian wild progenitor rices, O. rufipogon and O. nivara might have begun more than 10,000 years ago giving rise to the present day Asian cultivated rices (Yang et al., 2015; Choi et al., 2017; Qiu et al., 2017). Wild rices predominantly exhibit varying grain colors and this trait is known to be associated with domestication (Civáň and Brown, 2017). Rice germplasm collections comprise various colored rice lines, though these are neither cultivated widely nor used extensively in crop improvement programs. Colored rices have been widely used as entries in trials for the discovery of genes that confer resistance to bacteria, fungi and insects (Ahuja et al., 2010). Colored rices of various hues were described as red, brown, purple, and black, based largely on pericarp and/or hull coloration due to accumulation of anthocyanins, their precursors, flavonoids or their combinations, called co-pigmentation, besides other polyphenolic derivatives. Anthocyanins, the end products of anthocyanin pathway, are ubiquitous pigments known to be present in flowering plants. Naturally occurring rice landraces that accumulate anthocyanins, proanthocyanidins, and anthocyanin derivatives have been widely described (Reddy et al., 1995; Oh et al., 2018). Historically, colored rices have been deemed specialty rices by various ancient Asian cultures. For example, black rice has been described as forbidden rice or Emperor’s rice in China and red rices have been used in some religious celebrations in south and southeast Asia. However, due to changed consumer preference for white grained rices, they were not exploited in the breeding programs despite their special features such as enhanced levels of antioxidant compounds and biotic and abiotic stress tolerance (Reddy et al., 2007). In addition, red/purple rices exhibit some well described domestication related traits, though in varying intensity, such as seed dormancy, grain shattering, photo-period sensitivity, long duration, tillering, and lodging.
Purpleputtu (PP) is a colored landrace that exhibits purple color in all aerial parts including seeds except in nodes and pollen (Reddy et al., 1995). It is an indica landrace cultivated in small restricted areas in farmer fields in southern India, often used as border lines to demarcate test plots in experimental fields, primarily serving as a pollen barrier due to its height (Rangaswamy et al., 1988). The genetic control of pericarp color in PP has been described and molecular biological basis of the control of the underlying anthocyanin pathway has been elucidated (Reddy et al., 1994, 1995, 2007; Oh et al., 2018). Earlier studies on color in japonica rices revealed the contours of the genetic circuitry that govern color pathway (Furukawa et al., 2007). Regulation of the anthocyanin pathway, both in indica and japonica subspecies, by different classes of transcription factors and repressors have been identified and tissue specific expression of some of these genes deciphered (Reddy et al., 1995; Sweeney et al., 2006; Rahman et al., 2013).
Allelic variations at certain target loci of the anthocyanin pathway that lead to the formation of many diverse flavonoids and anthocyanins have been described (Reddy et al., 1995; Kim et al., 2011, 2015; Maeda et al., 2014; Chin et al., 2016). However, not much is known about allelic variations at loci associated with the pathway in terms of mutations, deletions and rearrangements. Scant information exists on differences at the genomic level between colored and white grained rices. Advancement of next generation sequencing (NGS) technologies along with the availability of the reference genome sequences for both japonica and indica rices provided an unprecedented opportunity to investigate the genome wide distribution of allelic variations that control complex pathways such as those that differentiate colored rices from white rice. Deep sequencing coupled with comparative genomic analysis using extensively sequenced diverse rice lines and SNP-Seek database provide a great opportunity to gain incisive insights into the genetic and molecular basis of a diverse array of traits. Further, genomic analysis of diverse genotypes such as wild progenitors, land races, cultivars and modern rices is expected to throw new light on domestication, selection sweeps of specific genomic regions and evolution of colorless grain phenotype. Present day colored rices, i.e., PP, had evolved from their colored progenitors such as O. rufipogon over thousands of years of cultivation, domestication and natural selection. It is interesting to investigate as to how such a complex trait governed by many genes across the genome has evolved and maintained even when selection is biased toward white grained rices in modern time. Interestingly, colored rices are known for traits associated with disease resistance and abiotic stress tolerance as demonstrated by numerous reports of introgression breeding via wide hybridization to essentially transfer useful genes from progenitors and wild relatives to present day cultivars, a form of de-domestication.
The present study is aimed at understanding the basis of existence and maintenance of PP, a fully colored rice, by whole genome deep sequencing and comparative genomics using a global collection of thousands of rice lines including progenitors such as O. rufipogon, O. nivara; and the reference genome of Nipponbare rice (Kawahara et al., 2013; Lachagari et al., 2019). We uncovered a significant number of genome-wide allelic variations in PP including those in genes associated with anthocyanin biosynthesis and genomic regions associated with the domestication-related genes controlling dormancy, seed shattering and diseases response. Additionally, we report here the discovery of unique alleles in genomic regions showing extreme conservation through evolution and thus representing selection sweeps. Besides, we identified numerous unique alleles at loci associated with major structural and regulatory genes of the pathways determining purple phenotype.
Whole genome shotgun sequencing of PP genomic DNA (80× coverage) on Illumina HiSeq 2000 platform yielded 43.47 GB of raw data that include a total of 430,403,016 paired end reads of 100 bp. More than 81.7% of the data exceeded Q30 Phred quality score. The quality score of read 1 (forward) and read 2 (reverse) are shown in Supplementary Figures S1A,B, respectively, while position-based quality of each nucleotide for read 1 and 2 is shown in Supplementary Figures S1C,D, respectively. Read based GC content estimates show that more than 36% of reads have less than 30% of GC content (Supplementary Figure S1E). The data was deposited in NCBI SRA database with an accession number PRJNA309223. The reads were aligned to the reference genome (Os-Nipponbare-Reference-IRGSP-1.0, MSU release 7) using BWA. Overall, 95.7% of the total reads were mapped covering 94% of the reference genome. The reads with mapping quality value ≥30 were retained for further analysis after removing duplicates. Out of total generated reads, 263,679,866 were aligned to the reference genome with an average of 49.5× read depth and 80.61% genome-wide coverage (Table 1 and Supplementary Figure S2A). Supplementary Figures S2B–D indicate percentage of aligned reads, chromosome-wise average read depth and coverage, respectively.
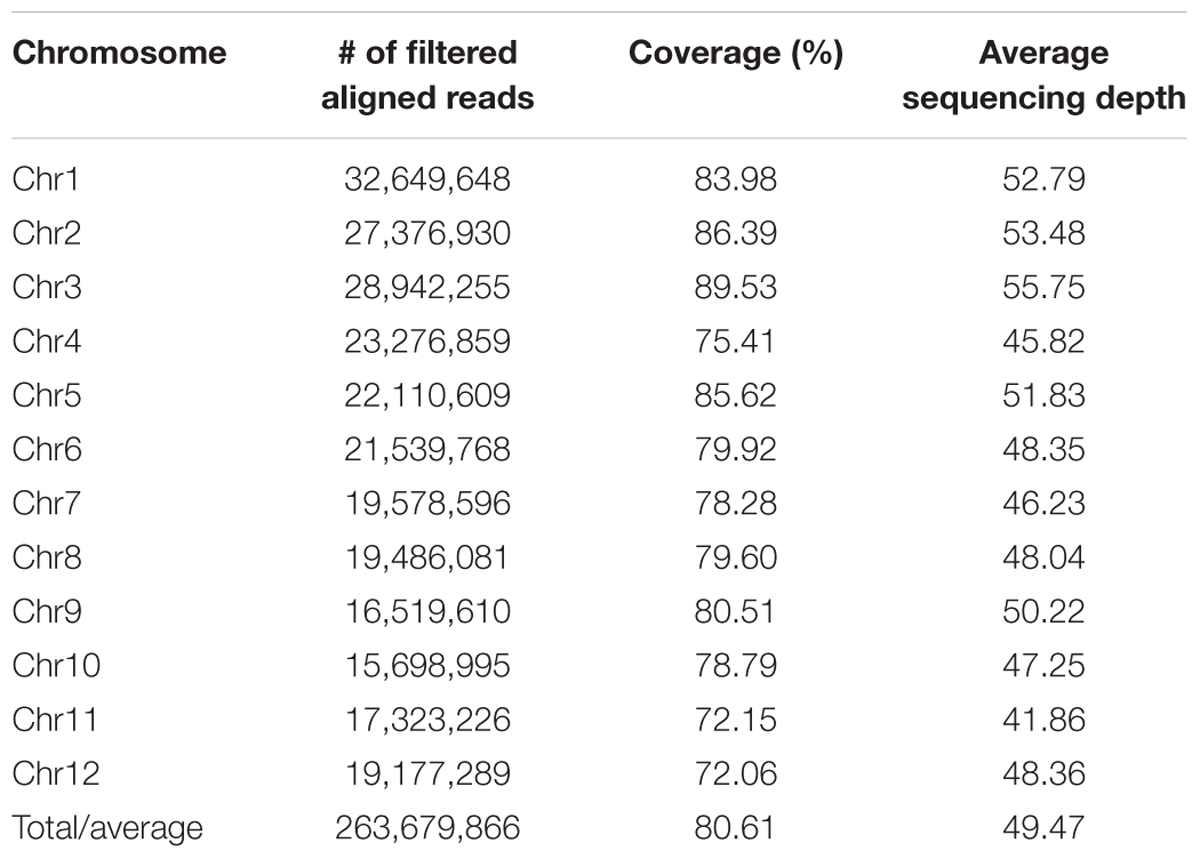
Table 1. Chromosome wise distribution and read statistics of Purpleputtu (PP) genome.
Assembled unmapped reads of PP was used for repeats masking using rice as reference model. A total of 2,124 genes were predicted, out of which 1,239 gene sequences were annotated based on UniProt, NCBI NR and Phytozome database (Supplementary Table S1). Of the 1,239 genes sequences, set of 70 and 1,069 have characterized and uncharacterized gene information respectively from homologous rice cultivar sequences. The remaining 76 sequences were annotated to the orthologous sequences of maize, wheat, purple false brome, cutgrasses, and sorghum. A set of 885 sequences did not find suitable match in any of the above-said databases, indicating that they are unique in PP genome and their functional domain information was extracted to find out the functions of gene which are unique to PP (Supplementary Table S1).
Comprehensive genome-wide mapping diagram indicates the read depth, gene density, insertion density, deletion density, and SNP density (Figure 1). A total of 3,200,951 variants (2,824,513 SNPs and 376,445 INDELs) were identified in PP (Table 2) with read depth ≥5 and variant quality score ≥50 against Nipponbare reference genome (Supplementary Table S2). A majority of the variants (88.24% of SNPs and 94.44% of INDELs) were found to be homozygous. Most of the SNP changes observed were of transition type: A>G (18.92%), C>T (16.47%), G>A (16.43%), and T>C (18.89%) with a Ts/Tv ratio of 2.41 (Figure 2A). A majority (70%) of the identified changes are short INDELs of length 1–2 bp (Figure 2B). Of the total variants 1,058,815 (33.8%) map to the repeat region of the genome (Supplementary Table S3). The variant density was estimated to be 756 SNPs and 100 INDELs per 100 Kb in PP in comparison with MSU release 7 assembly. Chromosome (Chr) 10 shows the highest SNP density (889/100 Kb), while the lowest SNP density (624/100 Kb) was found in Chr5. Chr1 shows the highest INDEL density (109/100 Kb) whereas Chr4 has the lowest (85/100 Kb) (Supplementary Table S2). We observed that 67% of the variant desert region (Chr5) falls into repeat regions with a majority of the repeat class being putative retrotransposons as compared with a few other indica lines (Wang et al., 2009). The average read depth and coverage in this variant desert region was observed to be 43× and 81%. A total of 652 genes overlap were found in the variant desert region, half of which do not have a single variant. Of these genes 368 and 43 genes belong to the retrotransposon and transposon proteins, respectively (Supplementary Tables S4, S5).
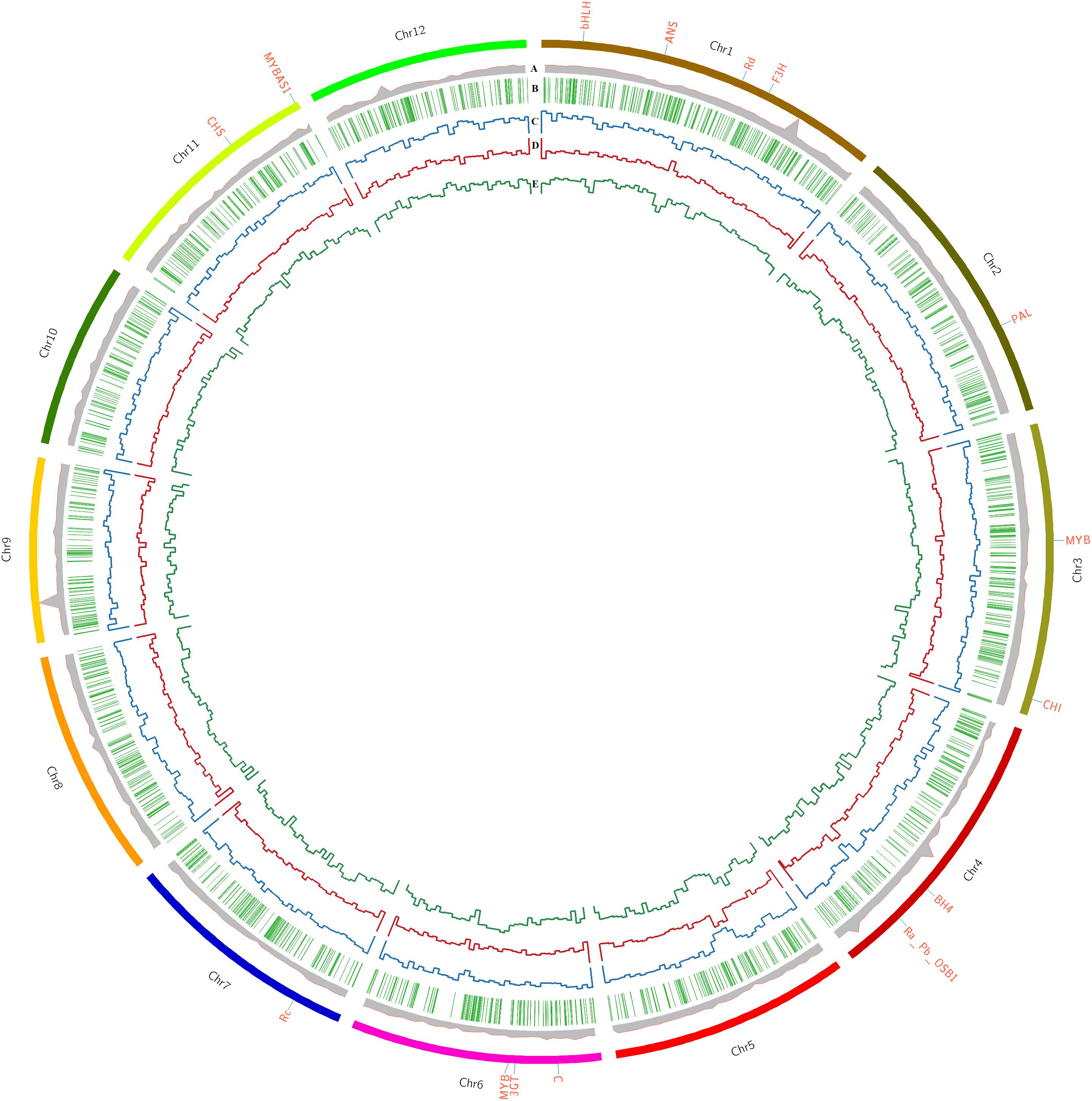
Figure 1. Comprehensive genome-wide analysis of Purpleputtu (PP) characteristics with Nipponbare reference genome. Circle diagram represents (A) read depth, (B) gene density, (C) insertion density, (D) deletion density, (E) SNP density. Outer circle of diagram represents the chromosomes of PP genome, each one labeled with important genes.

Table 2. Summary statistics of various identified variants.
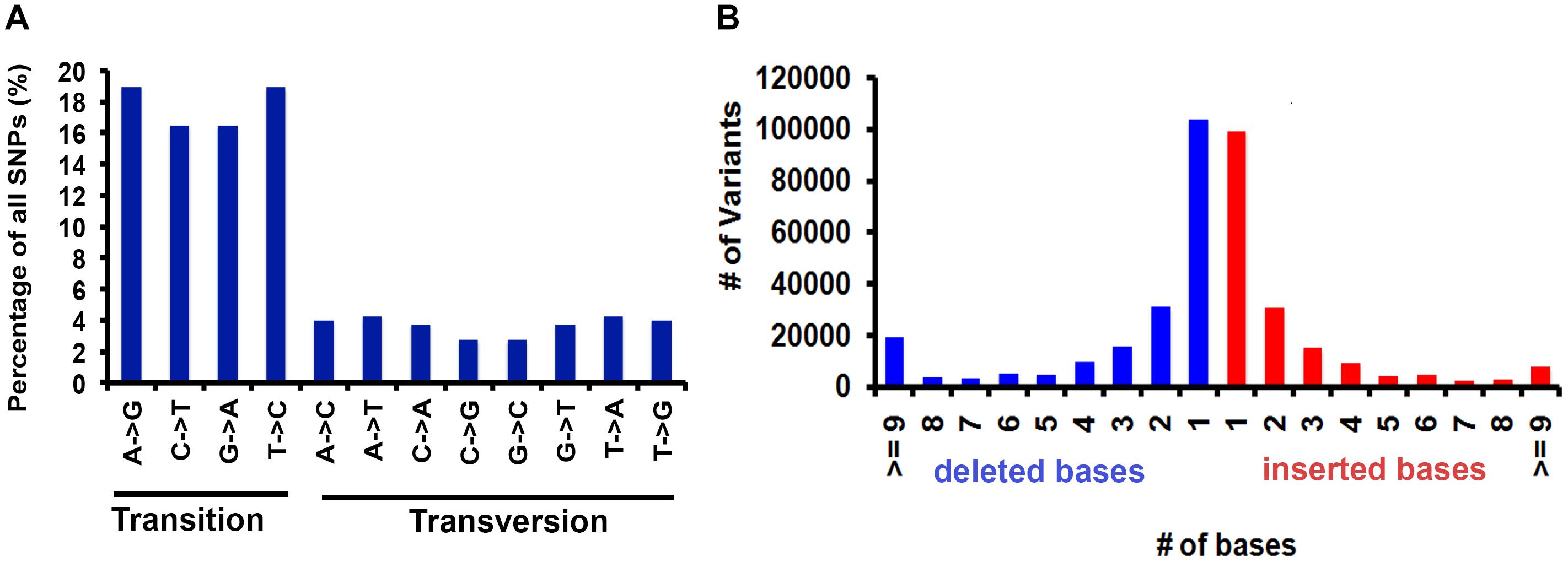
Figure 2. Base distribution statistics of variants identified in PP genome. (A) Percentage of transition and transversions. (B) Distribution of deleted and inserted bases.
The variants were annotated using an in-house pipeline against the gene model provided by MSU release 7. Overall, ∼32% of the variants span the genic region and the remaining 68% fall in the non-genic regions (Figure 3A). Out of the total genic variants, 50% overlap exonic region and of these 79.6% falls in the coding regions. Of the total variants in the coding regions, 37.6% were synonymous and the remaining 63.4% were non-synonymous type. About half (55.8%) of the non-synonymous type variants belong to missense class. We found that 33% of the total variants are in the repeat regions. Of these, 23.3% were genic and the remaining 76.7% were inter-genic. Further, a breakup of repeat class variants revealed that many of them belong to retrotransposons and few to miniature inverted-repeat transposable element (MITE) repeat class and cacta-like transposons (Figure 3B). These may serve as a valuable resource for selecting functional markers in genetic mapping programs. We further investigated the variant density around the transcription start sites (TSS) and found that it peaks at around 420 bp upstream and dips at ∼165 bp downstream (Figure 3C). The variant density gradually decreases toward zero on both sides of the TSS. Deep analysis of variants present in 1.5 Kb upstream shows that maximum variations were present in the genes for pyrrolidone-carboxylate peptidase, WD domain/G-beta repeat domain containing protein, actin, dehydrogenase E1 component domain containing protein and CBL-interacting protein kinase 1 (Figure 3D), all variants and associated genes are depicted in Supplementary Table S6.
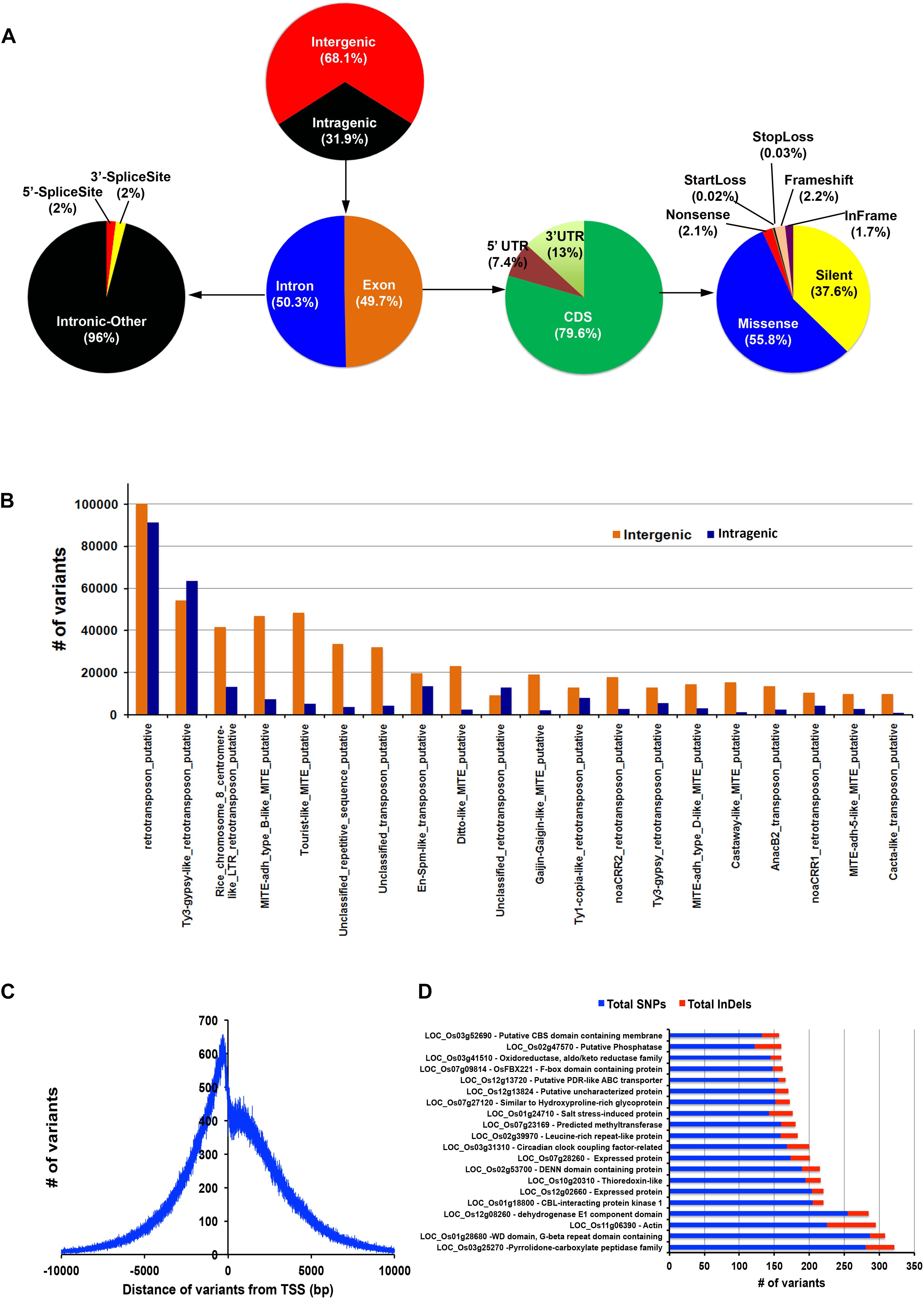
Figure 3. Comprehensive variant detection and annotation. Panel (A) indicates the distribution of variants in different regions of genome. Panel (B) represents the number of variant in different types of repeats. (C) Variant density graph from transcription start site (TSS). (D) Genes containing highest SNPs/INDELs variations.
To capture unique variants of PP, we compared its genome with a panel of 108 rice lines (Supplementary Table S7) covering different red rices, progenitors, landraces, modern cultivars spanning across sativa group (indica, temperate and tropical japonica) and Australian, aromatic and wild rices. This panel also includes other wild/progenitor species such as O. longistaminata, O. brachyantha, O. barthii, O. meridionalis, O. nivara, O. glaberrima, and O. rufipogon. A deeper analysis of the genome sequences with this panel allowed us to identify a set of unique alleles in PP genome. Out of 3,200,951 variants, 67,774 were found to be unique to PP (Supplementary Table S8). Of these, 64,394 are SNPs and 3,380 are INDELs, which include 390 INDELs that span exonic regions. Zygosity analysis of unique variants revealed that 49,025 (72.74%) are homozygous and 18,749 (27.66%) are heterozygous. Among these unique variants, 24,576 were mapped to genic regions and 43,198 to intergenic regions (Supplementary Table S8). The genic regions (12,831) span across 9,370 genes indicating that unique variations occur in almost one fifth of the total genes in rice. Further classification of unique variants in 5′ UTRs, 3′ UTRs, intronic regions and splice junction sequences shows 7,087 missense SNPs, 4,942 silent variations, 371 non-sense mutations, and 23 start-loss and 18 stop-loss variants. Analysis of SNP desert region of chromosome 5 revealed 224 unique variants of which 13 are INDELs and 211 SNPs (Supplementary Table S9). A set of 63 variants are uncovered in genic region of which 34 are silent mutations, 26 are missense mutations and one is a non-sense mutation. A majority of these variations are localized in Ty3/Gypsy class of retrotransposons. Variant analysis also identified 96 unique variants of PP genome associated with morphological traits, physiological traits and resistance or tolerance to biotic and abiotic stresses (Supplementary Table S10).
Purpleputtu genome was subjected to a deep comparative analysis by aligning with International Rice Genome Sequencing Project (IRGSP v1.0) assembly1 having 3K SNP-Seek data set. The resultant 1,962,843 SNPs in PP were compared with 5,854,680 SNPs of 3K global rice collection to identify rare variants with high effect. PP variants were merged with those of 3K SNP-Seek dataset and rare variants were obtained with minor allele frequency (MAF) cutoff of <0.01. The dendrogram made with 3K dataset comparison shows that PP is close to IRIS-313-8921, IRIS-313-8498 (Supplementary Figure S5). A total of 481,205 rare SNP loci were found amongst the combined dataset; which has 5,323,594 SNPs in common variant loci. Of all rare variants, 479,818 loci were only called in PP and not called in 3K dataset; therefore, they were denoted as unique. The remaining 1,387 SNP loci/SNPs were found to be unique to PP and identified one each of stop loss, stop gained and splice site accepter variants having high effects (Supplementary Table S11). The Armadillo-like helical domain-containing protein has a stop gained mutation in exon 2 leading to a truncated protein. However, the other two mutations were observed in two different conserved hypothetical proteins. The chromosome-wise distribution of these unique SNPs are as follows: Chr1 (141), Chr2 (100), Chr3 (100), Chr4 (129), Chr5 (108), Chr6 (124), Chr7 (114), Chr8 (132), Chr9 (102), Chr10 (101), Chr11 (128), Chr12 (108). The localization of these SNPs in their respective loci is listed in Supplementary Table S11.
The functional classification of the variants was performed at different levels: pathway, ontology and traits. For the pathway study we compared the variants against rice metabolic pathway RiceCyc v3.3 database (Dharmawardhana et al., 2013). The pathways with the highest number of variants include cytokinins glucoside biosynthesis, betanidin degradation, sucrose degradation to ethanol and lactate, and cellulose biosynthesis (Supplementary Table S12) (Gupta et al., 2017a). Betanidin degradation eliminates betacyanin pigment pathway (which leads to production of red, purple, and violet betacyanin pigments which are predominant in Caryophyllaceae); here it is worth noting that anthocyanins and betacyanins are mutually exclusive in flowering plants (Rodriguez-Amaya, 2018). Similarly, sucrose degradation is a prerequisite for preventing root hypoxia. There is some information on the role of this process in salinity tolerance (Behr et al., 2017). Cytokinin glucoside biosynthesis is reported to be associated with indeterminate growth. Of the total unique variants in PP, 25,447 variants mapped to genes associated with 338 pathways (Figure 4). Of these, 13,439 are silent, 10,871 are missense, 66 are non-sense, 26 are start-loss, and 43 are stop-loss mutations. Interestingly, PP exhibits many unique variations at diverse loci controlling the highly conserved ubiquitous flavonoid biosynthetic pathway. These variants span across genes controlling sub-pathways such as flavonoid biosynthesis (PWY1F-FLAVSYN), flavonol biosynthesis (PWY-3101), anthocyanin biosynthesis [pelargonidin 3-O-glucoside, cyanidin 3-O-glucoside] (PWY-5125), anthocyanin biosynthesis [delphinidin 3-O-glucoside] (PWY-5153), proanthocyanidin biosynthesis from flavanols (PWY-641). Besides this, other biotic and abiotic stress responsive pathways were also found to harbor unique variants in PP (Supplementary Table S11) (Gupta et al., 2017a,b, 2018a). Specifically, 122 variations were identified in flavonoid biosynthesis pathway (PWY1F-FLAVSYN) genes of which 62 are silent and 57 are missense variants, 1 each of non-sense, frame-shift and in-frame variations. As many as 40 variants were observed in the flavonol biosynthetic pathway (PWY-3101). Considering the pathway-based analysis, it is inferred that pelargonidin/cyanidin 3-O-glucoside sub-pathway (PWY-5125) consists of 34 variations in which 9 are silent, 23 are missense, 1 each of frame-shift and in frame variations. In contrast, 24 variations were observed in an evolutionarily silenced delphinidin route of anthocyanin biosynthesis pathway (PWY-5153) of rice. Only one missense mutation was observed in enzymes responsible for proanthocyanidin pathway from flavanols (PWY-641) in rice. Trait ontology analysis for unique variants in PP was performed using Q-TARO database (Yonemaru et al., 2010). Out of this 67,774 variants, 6,283 were mapped to the regions associated with morphological (2,287), physiological (1,361), resistance or tolerance traits (2,436) and other traits (199) (Supplementary Table S13). Among these 2,812, 2, and 8 variants were found to be missense, start-loss and stop-loss, respectively. Interestingly, the start-loss variations are observed only in flowering and cold tolerance traits whereas stop-loss variants were observed in genomic locations associated with sterility, and various biotic and abiotic stress traits such as blast resistance, soil stress tolerance, drought tolerance and cold tolerance (Gupta et al., 2017b). Of all the variants that mapped to the trait-associated loci, the highest number of variants mapped to dwarf, sterility and blast resistance categories (Figure 5 and Supplementary Table S13). In addition, variants were observed in traits related to seed, leaf, flowering, germination, drought tolerance, stress tolerance, and lodging resistance which reflect the typical phenotype of PP.
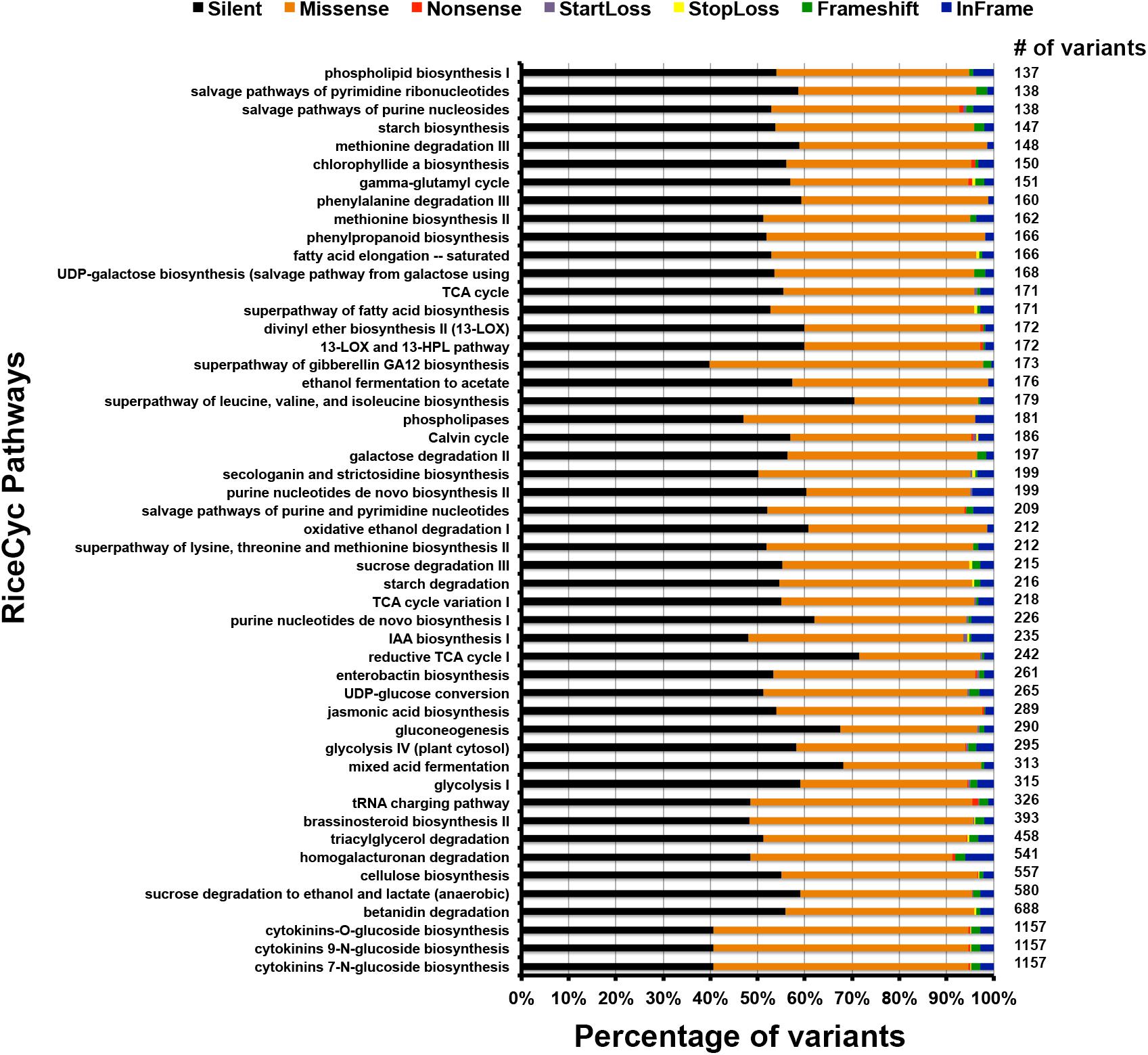
Figure 4. Bar diagram showing total number of different variants and their association in various metabolic pathways.
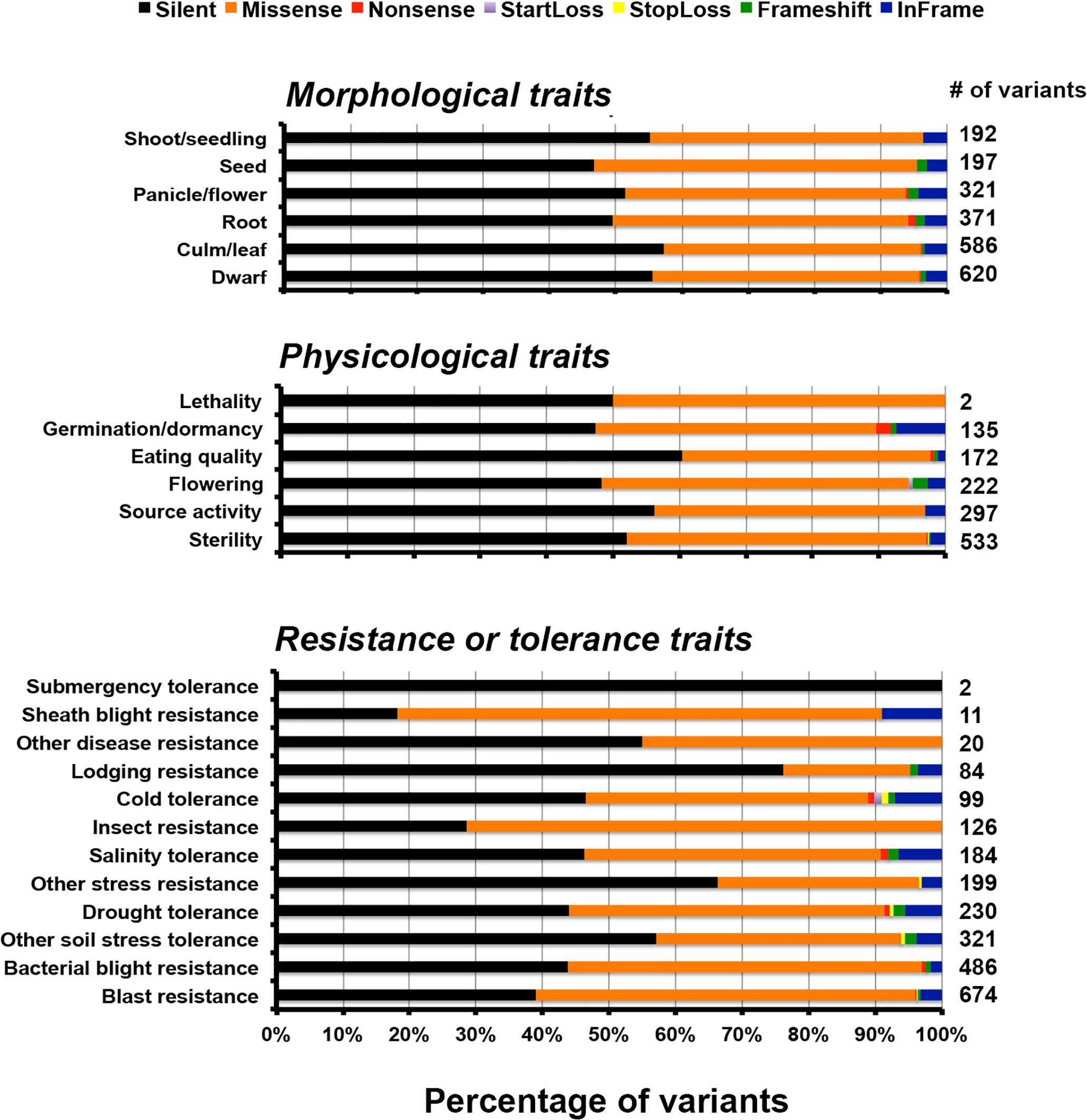
Figure 5. Bar diagram showing total number of different variants involved in morphological, physiological traits, and resistance to biotic and abiotic stress.
The unique variants of PP mapped to different transcription factors (TFs) in rice including MYB, bHLH, FAR1, NAC, WRKY, ERF, bZIP, and AP2 (Supplementary Table S14). The highest number of variants (1,752) was localized in Far-Red Impaired Response 1 (FAR1) family transcription factors which controls the far-red light signaling pathway by modulating phyA expression by activating FHY1 and FHL, thus maintaining homeostasis (Lin et al., 2007; Gupta et al., 2019). In contrast, whirly family of transcription factors involved in defense response show low number of variants, viz., three, indicating high level of conservation. NAC transcription factor family, which is one of the largest families of plant-specific transcription factors and play an important role in response to various plant stresses show 682 variants (Nakashima et al., 2012). The bHLH class of transcription factors, including the Rc (red color), which are involved in flavonoid/anthocyanin biosynthesis besides other biological processes such as wound and drought stress, light signaling and hormone signaling shows 672 variants (Carretero-Paulet et al., 2010). Interestingly, 319 of them are missense, 3 non-sense, 15 frame-shift and 79 in-frame mutations and the remaining are silent; all of these changes put together indicate significant variation in this locus. MYB genes and related genes of rice, known to be involved in the regulation of anthocyanin biosynthetic pathways as well as in other different biological processes in rice were found to have 898 variants of which 433 are missense variants (Ambawat et al., 2013; Liu et al., 2015). The WRKY class of transcriptional factors show 483 variants of which 157 are silent and 272 are missense mutations, which are involved in several biotic and abiotic stress responses (Phukan et al., 2016; Gupta et al., 2018b). A set of 47 missense variations and one stop-loss mutation is observed in AP2 transcription factor family genes which are involved in abiotic stress response in rice (Fu and Xue, 2010).
We studied independent events of domestication in the PP genome, which can offer an extremely useful system for studying the genetic basis of parallel evolution with other rice genomes. A significant trait altered by rice domestication and de-domestication is hull color. Wild progenitors of two cultivated rice species have predominantly black-colored hulls (Bh4) and straw colored hulls; these are the phenotypic effects of Bh4 and straw hull 4 (Sh4) candidate gene expression (Zhu et al., 2011; Vigueira et al., 2013). Examination of evolutionary relationship of Bh4 genes of PP with other 70 rice cultivar genes is shown in Supplementary Figure S3. The phylogenetic tree shows the Bh4 DNA sequence variations, that provides clues to the parallel evolution of hull color variation in PP and other rice cultivars. Supplementary Figure S3 shows that out of 70 rice cultivars, 19 have variations suggesting that the same gene is responsible for parallel trait evolution.
The variants that mapped to the anthocyanin pathway genes were analyzed for the resultant changes in the encoded proteins and these include both regulatory and structural genes such as C (Chr6), Ra (Chr4), Rc (Chr7), bHLH (Chr1), Rd (Chr1), MYB (Chr3), MYB (Chr6), MYBAS1 (Chr11), chalcone synthase (CHS) (Chr11), chalcone isomerase (CHI) (Chr3), leucoanthocyanidin dioxygenase (ANS) (Chr1), flavanone 3-hydroxylase (F3H) (Chr1), 3-O-glucosyltransferase (3GT) (Chr6), Bh4 (Chr4) and phenylalanine ammonia-lyase (PAL) (Chr2) (Figure 1). The variants of anthocyanin pathway genes were explored to understand the key differences that may explain the uniqueness of PP genome in showing distinct color phenotype (Cingolani et al., 2012). Of the 585 variants observed, 388 were identified as upstream variants followed by intronic (99), missense (45), synonymous (37) and 1 stop-gain variant (Supplementary Table S15). Comparative analysis of 5 cM Rc locus (Chr7) spanning 42.6 cM to 47.7 cM in three BAC clones (AP003748, AP005098, AP005779) revealed unique variations in PP placing it as a distinct clade within the indica group, separated from all other rice lines (Sweeney et al., 2006). Contrary to expectations, PP with its colored pericarp phenotype shows the 14 bp deletion signature in Rc gene which is a characteristic feature of the present white pericarped rice lines. Further evolutionary relationship of PP Rc gene to other Rc gene homologs of rice cultivars (includes Australian, Elite, Indica, Tropical Japonica, Aromatic, O. barthii, O. glaberrima, O. rufipogon, O. meridionalis, O. longistaminata, O. brachyantha, and wild species, i.e., BHA-Redrice, CA97_053, CA97_053, Daldhal, HK47, MV98_80, Nivara-IRGC106154, SHA-Redrice, IRGC105327, P46, VOC4, Yuan3_4) indicate divergence from other rice cultivars (Supplementary Figure S4). MYBAS1 gene (LOC_Os11g47460) on Chr11 was found to have 56 missense SNPs responsible for changes in 42 amino acids, which could be a possible paralog of the Rc gene on Chr7. In addition to that, the Rc gene (LOC_Os07g11020) on Chr7 exhibits an intronic variation responsible for deletion of ‘GAGA’ at position 138 that does not affect the functions of the gene. In addition, homology search for Rc genes against other rice genomes, did not provide any significant hit for any alternate functional gene. Notably, anthocyanin regulatory gene Ra (Pb/OSB1/LOC_Os04g47080) on Chr4 which is reported to be associated with purple color pericarp (Wang and Shu, 2007) shows one missense variation leading to M64T, and two frame shift variations causing T575fs and V545fs. Both of these frameshift mutations in this gene lead to alternate chain of 30 amino acids (PLGAGINIGWSPWTDTS QVCLICCRRTWE∗) in the C terminus when compared to Nipponbare reference genome. bHLH (LOC_Os01g09900) on Chr1 shows interesting variations leading to amino acid changes (E304K, P148L, V76A and disruptive in-frame insertion at A98AA). MYB gene (LOC_Os06g19980) on Chr3 shows a total of 14 variations in which 7, 3, 2, and 1 are downstream gene variant, in-frame deletion, intron variant and synonymous variant, respectively; remaining 1 variant belongs to missense variation and the changes in amino acid A263G possibly does not alter the secondary structure of the protein. MYB genes (LOC_Os06g19980) on Chr6 has 23 missense variations causing changes in amino acids, viz. S27R, M47L, R63L, R66L, D72G, L80N, I82S, A83P, I86V, Q112E, S116I, E147G, E148D, I151V, L163V, T202A, I232T, L296R, R298G, S446L, Y607H, and A639P; these changes possibly alter the structure and function of this MYB protein. The gene encoding PAL (LOC_Os02g41630) enzyme on Chr2 catalyzing the formation of 4-coumaroyl-CoA from phenylalanine has one synonymous, one intronic variation and only one missense variation changing amino acid A621V of the protein. CHS (Os11g0530600) on Chr11 has 1 SNP leading to a single amino acid change, N158S. F3H (LOC_Os01g50490) on Chr1, the gene encoding the enzyme involved in conversion of flavonone to dihydroflavonones has 49 unique variations in which 22 SNPs lead to protein structure variations. We found four SNPs each causing amino acid changes to proline and glutamic acid, viz. T3P, A79P, L99P, L474P, and Q80E, K407E, D410E, D414E, respectively. In addition, the remaining SNPs cause mutations of L6V, T15M, V34A, S55G, N77T, D103N, A110V, C389S, L392F, I393V, R418G, G420D, G424W, and T488A. Possibly, these mutations contribute toward hyper-accumulation of anthocyanins in PP. One silent variation was identified in ANS1 (LOC_Os01g27490) gene on Chr1, encoding the key enzyme involved in the conversion of leucoanthocyanidins to anthocyanidins, the penultimate step in anthocyanin biosynthesis. The last enzyme 3GT (LOC_Os06g18790) of this pathway was found on Chr6 of PP genome, converting anthocyanidins to anthocyanins and had three unique SNPs (M186V, K190Q, K190R) that may have a role in accumulation of pigments in PP (Brazier-Hicks et al., 2007).
Oryza sativa, an independently domesticated rice that has been in cultivation for more than 12,000 years, has become the predominant cereal staple for most of the ∼3 billion plus Asians. With an estimated 30% rise in population by 2050 and unpredictable climate changes, rice breeders must gear up for substantial increase in rice productivity across various agroclimatic regions. Modern rice breeding technologies are increasingly utilizing the genetic resources of progenitors and wild relatives by introgression of new genes associated with important agronomical traits into cultivars. These mainly include genes for biotic and abiotic stress tolerance, growth, and maturity traits. Though colored rices constitute a significant proportion of germplasm collections, they were not extensively used as genetic resources in breeding. Progenitors and domesticated colored rices were reported to be potential source of genes for resistance to diseases and pest, and tolerance to abiotic stresses. Besides, diverse colored pigments are recognized for their nutritional quality and anti-oxidant properties. Further, colored rices were found to be good subject material for understanding domestication and de-domestication processes in grasses in general and rice in particular (Choi et al., 2017; Qiu et al., 2017). We set out to uncover novel and unique alleles in one such fully colored rice line PP, that is cultivated sporadically with no evidence of any directional selection or crossing in its long history of domestication and cultivation.
Whole genome sequencing of PP rice and mapping with Nipponbare genome was performed to discover genome-wide DNA variations. A total of 263,679,866 reads were mapped to the reference genome. The assembly and annotation of unmapped reads indicates the presence of many uncharacterized genes having homology with the other rice species such as O. nivara, O. meridionalis, O. glumipatula, O. glaberrima, O. brachyantha, O. barthii, O. punctata, O. rufipogon, and O. alta (Supplementary Table S1). Interestingly, 56 gene models showed homology with a red rice line (O. punctata) and all these genes are uncharacterized indicating the presence of novel genes associated with red/purple pigmentation. Further, comparative genomic analysis using ‘a panel of 108 rice lines’ spanning both indica and japonica, progenitors, land races uncovered 64,349 SNPs and 3,380 INDELs unique to PP genome. In all, we captured unique variants in one fifth of the total gene models of rice. Of the 3,200,951 polymorphic SNPs identified, about a third span across exonic regions and three fourth of them fall in coding sequences. Further, about 33% of the variants are mapped to repeat regions, retrotransposons and transposable elements, a finding that falls within the range reported for Oryza (Stein et al., 2018). The present data show that Chr1 has the highest number of INDELs and Chr10 has the highest number of SNPs per 100 kb. Similarly, the lowest INDEL density was on Chr4 and lowest SNP density on Chr5. In addition, distribution of SNP-rich and SNP-poor regions in each chromosome of PP was identified, which also corroborates with earlier findings in rice, Arabidopsis, and wheat (Nordborg et al., 2005; Ravel et al., 2006; Subbaiyan et al., 2012).
The mapped genome of PP revealed a clear bias toward transitions (almost twice that of transversions) deviating from the expected ratio of 0.5. Higher Ts/Tv ratios were reported in rice, maize, otus, medicago, diploid wheat, Triticum monococcum and barley (Batley et al., 2003a; Vitte and Bennetzen, 2006; Subbaiyan et al., 2012; Bindusree et al., 2017). Due to wobble effect, transitions manifest mostly into silent mutations that do not alter the amino acid and thus conserves the amino acid chain (Wakeley, 1996). Among transitions, the C/T transitions were more in number, presumably due to a simple methylation being the cause of this mutation (Coulondre et al., 1978). Higher frequency of C/T mutations has been reported in other crops such as common bean, maize, grape, and citrus (Batley et al., 2003b; Lijavetzky et al., 2007; Terol et al., 2008). Generally, genomic segments with higher SNP frequency were shorter than the lower SNP frequency segments. The SNP-poor region having 4.3 Mb between 9.3 Mb and 13.6 Mb on Chr5 was identified in PP genome that is popularly described as ‘SNP desert’ and described in detail (Supplementary Tables S4, S5, S9). This conserved region in rice has been earlier reported in certain indica and japonica lines (Wang et al., 2009; Yonemaru et al., 2010; Subbaiyan et al., 2012). It is well known that selective sweeps during the long process of domestication in both indica and japonica rice are responsible for lower SNP frequencies across some regions in the genome (Subbaiyan et al., 2012).
The progenitors of the present-day rice lines and most of the wild rices exhibit red color in the pericarp and some other plant parts. However, the present-day modern rice cultivars predominantly have white pericarp. The disappearance of purple–red–black–brown color in grain in modern cultivars is a great example of agronomical spreading of a single variation at the Rc locus through the course of domestication and natural and artificial selection (Furukawa et al., 2007; Sweeney et al., 2006; Gross and Olsen, 2010). The Rc locus of PP shows the well characterized 14 bp deletion in the fifth exon; this deletion has been reported to be the hallmark of white rices, yet the pericarp of PP that bears this signature, is colored. In contrast, almost all white pericarped lines, including all modern rice cultivars, have the same deletion at Rc locus. The Rc gene encodes a bHLH TF that regulates DFR activity. We predict an alternate TF that may be regulating DFR in PP grain. It is to be noted that the Rc locus in the colored progenitor rufipogon lacks this 14 bp deletion. It may be the result of gene flow from cultivars to PP which indicates de-domestication. Introduction of many disease resistance genes, such as Xa21, from wild progenitors by introgression breeding and directed selection is a routine approach to exploit novel genes. We conclude that the Rc locus of PP is conserved due to simple cultivation and non-directional selection for color. The white pericarp phenotype controlled by the deletion-containing Rc locus in modern rice cultivars is due to selection pressure for white grain. Our results challenge the notion that the 14 bp deletion is an invariant signature of white rices only.
In depth analysis of variants revealed several unique SNPs/INDELs in structural and regulatory genes of various pathways responsible for stress response, genotypic and phenotypic effects on different traits of PP rice (Supplementary Tables S12, S13 and Figure 4). Loci associated with cytokinin glucoside biosynthesis, betanidin degradation, sucrose degradation to ethanol and lactate, and cellulose showed the highest number of variants. Furthermore, our results revealed unique variations at loci associated with biosynthesis of betacyanin, anthocyanins, and flavonoids. It is well known that betanidin degradation is a prerequisite step in eliminating betacyanin production in plants. It is worth noting that most flowering plants do not accumulate betacyanins and thus these two different color pigments are mutually exclusive (Shoeva et al., 2016; Khoo et al., 2017; Rodriguez-Amaya, 2018). Sucrose degradation is a prerequisite for preventing root hypoxia and salinity tolerance in some plants. Cytokinin glucoside biosynthesis is reported to be associated with indeterminate growth. The unique variant analysis uncovered 25,447 variants mapped to different genes associated with 338 pathways (Figure 4). Many structural, regulatory and inhibitory genes (Table 3) dispersed across the genome are involved in the anthocyanin pathway that leads to purple color formation in various tissues in PP (Reddy et al., 1995). The production of anthocyanins such as cyanidin, pelargonidin, and delphinidin derivatives through a multistep anthocyanins pathway is mediated by several enzymes PAL, cinnamate 4-hydroxylase (C4H), 4-coumarate CoA ligase (4CL), CHS, CHI, F3H, dihydroxyflavonol reductase (DFR); leucoanthocyanidin dioxygenase (LDOX)/anthocyanidin synthase (ANS), GT/3GT, acyltransferase (AT), methyltransferase (MT) (Aza-González et al., 2012). We found several unique variations in genes encoding PAL, CHS, F3H, ANS, and 3GT enzymes of this pathway in PP when compared with ‘a panel of 108 rice lines’ and ‘SNP-Seek dataset.’ Variation (A621V) was found in alpha helical region of the PAL enzyme that is positioned far away from the catalytic site, indicating unlikely effect on structural and functional role of this enzyme (Bata et al., 2018). Changes of N158S in CHS enzyme in PP when compared with CHS crystal structure of Freesia hybrida (PDB ID:4WUM) indicated change of charged side change amino acid N to uncharged side chain amino acid S at a location near the catalytic residues in PP CHS. This change may be enhancing the catalytic activity of the enzyme (Sun et al., 2015). The hydroxylation pattern of flavonoids controls their color, stability, and antioxidant capacity. We identified the gene encoding F3H in PP having 22 unique mutations, when compared with 3K dataset of rice, which may be relevant in the maintenance of purple color through generations. These may also be relevant for stability, antioxidant activity and stress defense capacity (Liu et al., 2014). Furthermore, analysis of structural and functional differences of PP F3H enzyme with that of white rice and other related cultivars indicates that it belongs to the plant Cytochrome P450 gene family (Schoenbohm et al., 2000). Similarly, several silent mutations were identified in the Ans gene encoding ANS1 enzyme that catalyzes the penultimate step of the anthocyanin pathway, namely conversion of leucoanthocyanidin to anthocyanidin (Reddy et al., 1996, 2007).
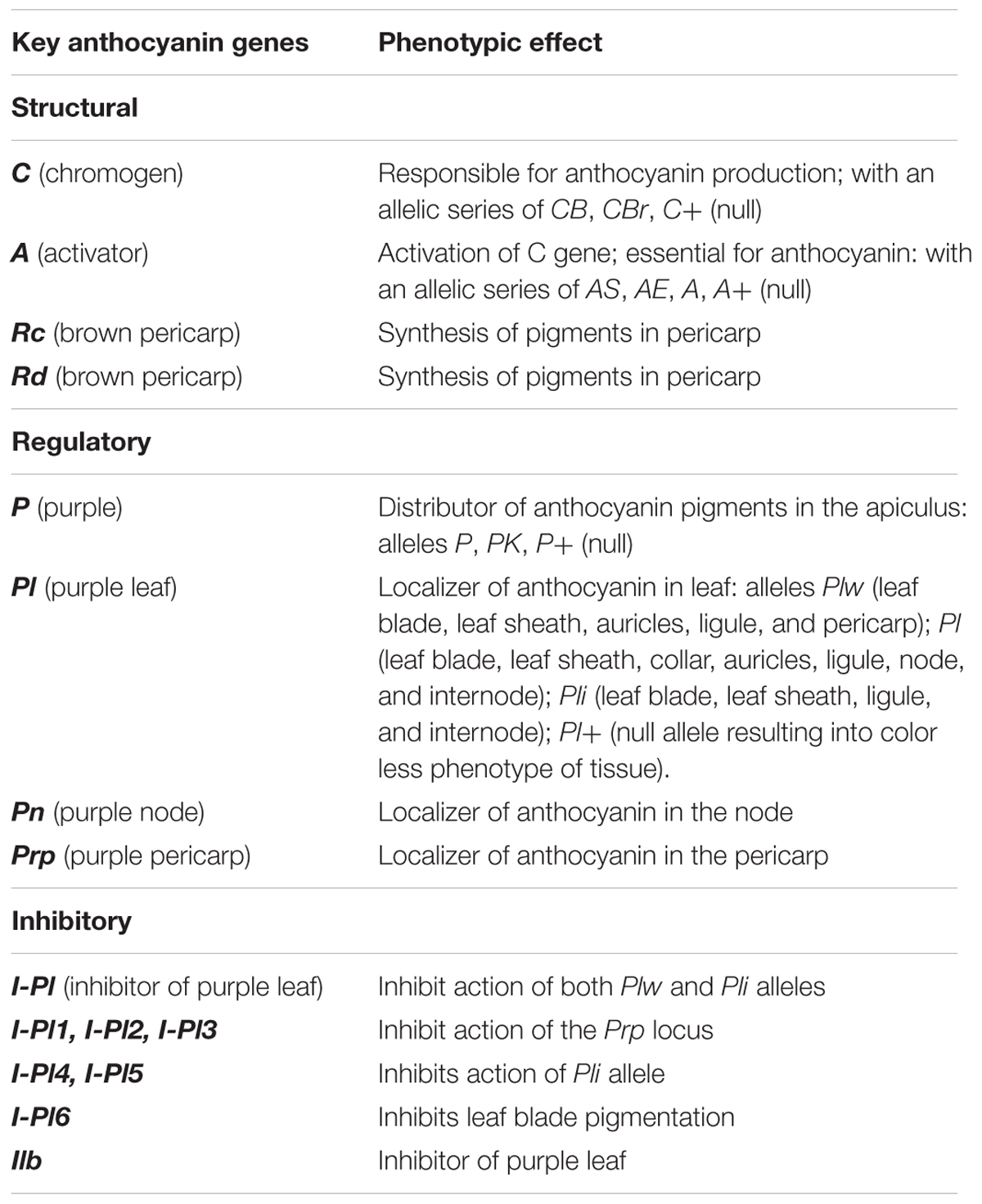
Table 3. List of structural, regulatory, and inhibitory genes involved anthocyanin pathway with their phenotypic effects.
Recently, Sun et al. (2018) proposed a C-S-A gene model for rice hull pigmentation: C1 that encodes a MYB transcription factor and acts as a color-producing gene, and S1 that encodes a bHLH protein that functions in a tissue-specific manner. C1 interacts with S1 and activates expression of A1, which encodes a dihydroflavonol reductase (Sun et al., 2018). We uncovered unique variants of various TFs such as MYB, bHLH, FAR1, NAC, WRKY, ERF, bZIP, bHLH, AP2 and others (Supplementary Table S14) responsible for different biological, molecular and cellular processes in PP. The role of bHLH, MYB, MYBAS1 TFs and other genes such F3H, Ra, and Rc in regulating anthocyanins and other associated pathways responsible for PP color and texture (Supplementary Table S15) (Pireyre and Burow, 2015) has been described here. In addition, as many as 96 novel variations in PP rice with known effects on morphological, physiological effects and stress response phenotypes are also reported.
Overall present study deals with whole-genome variations in PP rice examined by identifying SNP and INDEL polymorphisms using ‘a panel of 108 rice lines’ and ‘SNP-Seek dataset.’ Base substitutions and distribution of DNA polymorphism over PP genome provides important insights into the molecular basis underlying phenotypic traits exhibited by the genotype. Further, the unique allele variations in different genes participating in flavonoid biosynthesis and anthocyanin pathways responsible for purple color in PP genome were revealed by comparative analysis. Unique SNPs identified in anthocyanin pathway occurring in both structural genes and regulatory transcription factors, will help to breed rice with high nutraceutical content, particularly, flavonoids that have antioxidant activity.
Seeds of PP was germinated, and 15-day-old seedlings were used for genomic DNA extraction using Qiagen DNeasy kit (Qiagen). Qubit 2.0 fluorometer (Thermo Fisher) was used to quantify and NanoDrop 2000 (Thermo Fisher) for quality check of the isolated DNA. The DNA was fragmented to 300 bp size using a Covaris M220 focused ultrasonicator. The fragmented DNA was purified, and sequencing libraries were prepared using Illumina TruSeq DNA sample prep kit (Illumina Inc., United States) as per manufacturer’s specifications. The quantity and size distribution of the libraries were carried out using a Bioanalyzer 2100 (Agilent Technologies). The quantified libraries were subjected for whole genome sequencing on Illumina HiSeq-2000 platform (Illumina Technologies) by paired-end sequencing to generate 90-base pair long, small reads with an insert size of 200–350 bp (Supplementary Figure S2). Standard Illumina pipeline was used to filter the whole genome sequencing data. To remove low-quality reads and reads containing adaptor/primer contamination, FASTQ files were further subjected to stringent quality control using NGS QC Toolkit (v2.3) (Patel and Jain, 2012).
BWA software (v0.7) was used for the mapping of high-quality filtered reads against reference genome reference genome (Os-Nipponbare-Reference-IRGSP-1.0, MSU release 7) download from Rice Genome Annotation Project Database2 (Li and Durbin, 2009; Kawahara et al., 2013). Further, only uniquely aligned reads (with mapping quality ≥30 and minimum read depth 10) were considered in this analysis. Base quality score re-calibration and INDEL realignment were performed using Genome Analysis Toolkit (GATK, v2.1.13) and genome-wide coverage was estimated by Samtools (v0.1.16) (Li et al., 2009; McKenna et al., 2010).
Unmapped reads of PP genome were assembled using MaSuRCA (v3.2.1) assembler with default options (Zimin et al., 2013). The assembled genome was masked using RepeatMasker (v4.0.6) with default parameters, using rice as a model (Smit et al., 2015). Subsequently the masked genome was used in Augustus v3.2.1 using rice as model organism for gene prediction (Stanke et al., 2008). The annotations of identified genes were done using the Diamond program against NCBI NR, UniProt and Phytozome (v11.0) databases (Buchfink et al., 2015). Also, domain based functional annotation of all genes was performed using InterProScan v5.33.72 (Mitchell et al., 2018).
Minimum variant frequency of ≥90%, average base quality of the SNP ≥30 and minimum read depth of 10 were the stringent criteria followed to filter the identified SNPs and INDELs. If three or more SNPs were present in any 10-bp window, the SNPs and INDELs were filtered. Genome wide distribution of DNA polymorphisms was analyzed by calculating their frequency at every 100 Kb interval on each rice chromosome. Circos was used to visualize the distribution of the SNPs and INDELs on rice chromosomes (Krzywinski et al., 2009). Such distribution is assessed by integrating the position of DNA polymorphisms with GFF file containing rice genomic annotation. Customized Perl scripts were used to perform the genomic distribution and annotation of SNPs and INDELs. SnpEff (v3.1) tool was used to identify synonymous and non-synonymous SNPs, and large-effect SNPs and INDELs (Cingolani et al., 2012). Cut-off for number of non-synonymous SNPs per Kb gene length was determined using Box and Whisker plot to identify the outlier genes.
We downloaded raw data of 108 selected rice cultivars (Supplementary Table S7) and alignment of this data was performed with reference genome (Os-Nipponbare-Reference-IRGSP-1.0, MSU release 7) using BWA software (v0.7). The combined vcf file was generated using similar parameter and tools. Further, comparative variants analysis was performed to identify the unique variants in PP rice using in-house PERL program.
SNP dataset derived from alignment to IRGSP v1.0 was downloaded from SNP-seek database3. Corresponding SNPs of PP were filtered as per SNP-seek database norms and compared using Bcftools to determine unique alleles. Identification of different transcripts associated with these variants were also performed. Variations in transcription factor genes were detected using PlantTFDB (Jin et al., 2016). Moreover, comparative analysis of PP genome variations with 3,023 rice genome collections available at Rice SNP-Seek Database was performed (Mansueto et al., 2016).
All pathways associated with different variants have been annotated using rice metabolic pathway database genes (RiceCyc v3.3) using default parameters (Dharmawardhana et al., 2013). QTLs/Genes morphological, physiological and resistance/tolerance traits were downloaded from Q-TARO database4. SNPs in each QTLs/Genes were identified through co-localization of the coordinates in Q-TARO (Yonemaru et al., 2010; Yamamoto et al., 2012).
Search of Bh4 gene for selected 108 rice lines against various public databases provides only 70 lines with bh4 genes sequences, while Rc gene was present for selected cultivars. Further multiple sequence alignments using ClustalW and phylogenetic tree construction using the Neighbor-Joining method and branch length is estimated using the bootstrap in MEGAv6 (Tamura et al., 2013).
Whole genome data of PP rice submitted in SRA database with BioProject ID: PRJNA309223.
The datasets generated for this study can be found in data generated in this work was related to rice plant and submitted in SRA database (Acc No. PRJNA309223).
GT, SS, and VL conceived the work and designed the experiments. RG, VL, BK, SL, AMK, NC, and LM performed in silico and in vitro experiments. RG, VL, GT, and AR analyzed the results. All authors contributed to writing the manuscript, discussed the results, and commented on the manuscript.
This study was supported by SciGenom Research Foundation (SGRF).
VL, SL, BK, NC, AMK, AR, and GT were employed by AgriGenome Labs Pvt. Ltd., India, SS was employed by SciGenom Labs Pvt. Ltd., India, and RG and LM were employed by MedGenome Labs Ltd., India.
We would like to express our gratitude to Dr. Saurabh Gupta, Scientist, AgriGenome Labs Pvt. Ltd., Hyderabad, India, for his help in the manuscript writing. We also acknowledge Tamil Nadu Agricultural University for providing PP seeds. This manuscript has been released as a Pre-Print at BioRxive https://www.biorxiv.org/content/10.1101/536326v2.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00513/full#supplementary-material
Ahuja, I., de Vos, R. C., Bones, A. M., and Hall, R. D. (2010). Plant molecular stress responses face climate change. Trends Plant Sci. 15, 664–674. doi: 10.1016/j.tplants.2010.08.002
Ambawat, S., Sharma, P., Yadav, N. R., and Yadav, R. C. (2013). MYB transcription factor genes as regulators for plant responses: an overview. Physiol. Mol. Biolo. Plants 19, 307–321. doi: 10.1007/s12298-013-0179-1
Aza-González, C., Núñez-Palenius, H. G., and Ochoa-Alejo, N. (2012). Molecular biology of chili pepper anthocyanin biosynthesis. J. Mex. Chem. Soc. 56, 93–98.
Bata, Z., Madaras, E., Leveles, I., Hammerschmidt, F., Paizs, C., Poppe, L., et al. (2018). Bioactive 3D structure of phenylalanine ammonia-lyase reveal key insights into ligand binding dynamics. Biophys. J. 114:406a. doi: 10.1016/j.bpj.2017.11.2248
Batley, J., Barker, G., O’Sullivan, H., Edwards, K. J., and Edwards, D. (2003a). Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data. Plant Physiol. 132, 84–91. doi: 10.1104/pp.102.019422
Batley, J., Mogg, R., Edwards, D., O’sullivan, H., and Edwards, K. J. (2003b). A high-throughput SNuPE assay for genotyping SNPs in the flanking regions of Zea mays sequence tagged simple sequence repeats. Mol. Breed. 11, 111–120.
Behr, J. H., Bouchereau, A., Berardocco, S., Seal, C. E., Flowers, T. J., and Zörb, C. (2017). Metabolic and physiological adjustment of Suaeda maritima to combined salinity and hypoxia. Ann. Bot. 119, 965–976.
Bindusree, G., Natarajan, P., Kalva, S., and Madasamy, P. (2017). Whole genome sequencing of Oryza sativa L. cv. Seeragasamba identifies a new fragrance allele in rice. PLoS One 12:e0188920. doi: 10.1371/journal.pone.0188920
Brazier-Hicks, M., Offen, W. A., Gershater, M. C., Revett, T. J., Lim, E. K., Bowles, D. J., et al. (2007). Characterization and engineering of the bifunctional N-and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc. Natl. Acad. Sci. U.S.A. 104, 20238–20243. doi: 10.1073/pnas.0706421104
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12:59. doi: 10.1038/nmeth.3176
Carretero-Paulet, L., Galstyan, A., Roig-Villanova, I., Martínez-García, J. F., Bilbao-Castro, J. R., and Robertson, D. L. (2010). Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 153, 1398–1412. doi: 10.1104/pp.110.153593
Chin, H. S., Wu, Y. P., Hour, A. L., Hong, C. Y., and Lin, Y. R. (2016). Genetic and evolutionary analysis of purple leaf sheath in rice. Rice 9:8.
Choi, J. Y., Platts, A. E., Fuller, D. Q., Wing, R. A., and Purugganan, M. D. (2017). The rice paradox: multiple origins but single domestication in Asian rice. Mol. Biol. Evol. 34, 969–979.
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Civáň, P., and Brown, T. A. (2017). Origin of rice (Oryza sativa L.) domestication genes. Genet. Resour. Crop Evol. 64, 1125–1132. doi: 10.1007/s10722-017-0518-0
Coulondre, C., Miller, J. H., Farabaugh, P. J., and Gilbert, W. (1978). Molecular basis of base substitution hotspots in Escherichia coli. Nature 274:775. doi: 10.1038/274775a0
Dharmawardhana, P., Ren, L., Amarasinghe, V., Monaco, M., Thomason, J., Ravenscroft, D., et al. (2013). A genome scale metabolic network for rice and accompanying analysis of tryptophan, auxin and serotonin biosynthesis regulation under biotic stress. Rice 6:15. doi: 10.1186/1939-8433-6-15
Food and Agriculture Organization of the United Nations [FAOUN] (2017). Rice Market Monitor, Vol. 21. Rome: FAOUN. doi: 10.1186/1939-8433-6-15
Fu, F. F., and Xue, H. W. (2010). Coexpression analysis identifies Rice Starch Regulator1, a rice AP2/EREBP family transcription factor, as a novel rice starch biosynthesis regulator. Plant Physiol. 154, 927–938. doi: 10.1104/pp.110.159517
Furukawa, T., Maekawa, M., Oki, T., Suda, I., Iida, S., Shimada, H., et al. (2007). The Rc and Rd genes are involved in proanthocyanidin synthesis in rice pericarp. Plant J. 49, 91–102. doi: 10.1111/j.1365-313x.2006.02958.x
Gross, B. L., and Olsen, K. M. (2010). Genetic perspectives on crop domestication. Trends Plant Sci. 15, 529–537. doi: 10.1016/j.tplants.2010.05.008
Gupta, S., Gupta, V., Singh, V., and Varadwaj, P. K. (2018a). Extrapolation of significant genes and transcriptional regulatory networks involved in Zea mays in response in UV-B stress. Genes Genom. 40, 973–990. doi: 10.1007/s13258-018-0705-1
Gupta, S., Singh, Y., Kumar, H., Raj, U., Rao, A. R., and Varadwaj, P. K. (2018b). Identification of novel abiotic stress proteins in Triticum aestivum through functional annotation of hypothetical proteins. Interdiscipl. Sci. Computat. Life Sci. 10, 205–220. doi: 10.1007/s12539-016-0178-3
Gupta, S., Kumari, M., Kumar, H., and Varadwaj, P. K. (2017a). Genome-wide analysis of miRNAs and Tasi-RNAs in Zea mays in response to phosphate deficiency. Funct. Integr. Genom. 17, 335–351. doi: 10.1007/s10142-016-0538-4
Gupta, S., Yadav, B. S., Raj, U., Freilich, S., and Varadwaj, P. K. (2017b). Transcriptomic analysis of soil grown T. aestivum cv. Root to reveal the changes in expression of genes in response to multiple nutrients deficiency. Front. Plant Sci. 8:1025. doi: 10.3389/fpls.2017.01025
Gupta, S., Mishra, V. K., Kumari, S., Chand, R., and Varadwaj, P. K. (2019). Deciphering genome-wide WRKY gene family of Triticum aestivum L. and their functional role in response to abiotic stress. Genes Genom. 41, 79–94. doi: 10.1007/s13258-018-0742-9
Jin, J., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J., et al. (2016). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Research 45, D1040–D1045.
Kawahara, Y., de la Bastide, M., Hamilton, J. P., Kanamori, H., McCombie, W. R., Ouyang, S., et al. (2013). Improvement of the Oryza sativa nipponbare reference genome using next generation sequence and optical map data. Rice 6:4. doi: 10.1186/1939-8433-6-4
Khoo, H. E., Azlan, A., Tang, S. T., and Lim, S. M. (2017). Anthocyanidins and anthocyanins: colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr. Res. 61:1361779. doi: 10.1080/16546628.2017.1361779
Kim, C. K., Cho, M. A., Choi, Y. H., Kim, J. A., Kim, Y. H., Kim, Y. K., et al. (2011). Identification and characterization of seed-specific transcription factors regulating anthocyanin biosynthesis in black rice. J. Appl. Genet. 52, 161–169. doi: 10.1007/s13353-011-0027-3
Kim, C. K., Seol, Y. J., Shin, Y., Lim, H. M., Lee, G. S., Kim, A. R., et al. (2015). Whole-genome resequencing and transcriptomic analysis to identify genes involved in leaf-color diversity in ornamental rice plants. PLoS One 10:e0124071. doi: 10.1371/journal.pone.0124071
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645.
Lachagari, V. R., Gupta, R., Lekkala, S. P., Mahadevan, L., Kuriakose, B., Chakravartty, N., et al. (2019). Whole genome sequencing and comparative genomic analysis reveal novel allelic variations unique to a purple colored rice landrace (Oryza sativa ssp. indica cv. Purpleputtu). bioRxiv [Preprint]. doi: 10.1101/536326
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lijavetzky, D., Cabezas, J. A., Ibáñez, A., Rodríguez, V., and Martínez-Zapater, J. M. (2007). High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genomics 8:424. doi: 10.1186/1471-2164-8-424
Lin, R., Ding, L., Casola, C., Ripoll, D. R., Feschotte, C., and Wang, H. (2007). Transposase-derived transcription factors regulate light signaling in Arabidopsis. Science 318, 1302–1305. doi: 10.1126/science.1146281
Liu, J., Osbourn, A., and Ma, P. (2015). MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol. Plant 8, 689–708. doi: 10.1016/j.molp.2015.03.012
Liu, S., Ju, J., and Xia, G. (2014). Identification of the flavonoid 3’-hydroxylase and flavonoid 3’,5’-hydroxylase genes from Antarctic moss and their regulation during abiotic stress. Gene 543, 145–152. doi: 10.1016/j.gene.2014.03.026
Maeda, H., Yamaguchi, T., Omoteno, M., Takarada, T., Fujita, K., Murata, K., et al. (2014). Genetic dissection of black grain rice by the development of a near isogenic line. Breed. Sci. 64, 134–141. doi: 10.1270/jsbbs.64.134
Mansueto, L., Fuentes, R. R., Chebotarov, D., Borja, F. N., Detras, J., Abriol-Santos, J. M., et al. (2016). SNP-Seek II: a resource for allele mining and analysis of big genomic data in Oryza sativa. Curr. Plant Biol. 7, 16–25. doi: 10.1016/j.cpb.2016.12.003
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., et al. (2018). InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360.
Muthayya, S., Sugimoto, J. D., Montgomery, S., and Maberly, G. F. (2014). An overview of global rice production, supply, trade, and consumption. Ann. N. Y. Acad. Sci. 1324, 7–14. doi: 10.1111/nyas.12540
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 1819, 97–103.
Nordborg, M., Hu, T. T., Ishino, Y., Jhaveri, J., Toomajian, C., Zheng, H., et al. (2005). The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 3:e196. doi: 10.1371/journal.pbio.0030196
Oh, J. H., Lee, Y. J., Byeon, E. J., Kang, B. C., Kyeoung, D. S., and Kim, C. K. (2018). Whole-genome resequencing and transcriptomic analysis of genes regulating anthocyanin biosynthesis in black rice plants. 3 Biotech 8:115.
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Phukan, U. J., Jeena, G. S., and Shukla, R. K. (2016). WRKY transcription factors: molecular regulation and stress responses in plants. Front. Plant Sci. 7:760. doi: 10.3389/fpls.2016.00760
Pireyre, M., and Burow, M. (2015). Regulation of MYB and bHLH transcription factors: a glance at the protein level. Mol. Plant 8, 378–388. doi: 10.1016/j.molp.2014.11.022
Qiu, J., Zhou, Y., Mao, L., Ye, C., Wang, W., Zhang, J., et al. (2017). Genomic variation associated with local adaptation of weedy rice during de-domestication. Nat. Commun. 8:15323. doi: 10.1038/ncomms15323
Rahman, M. M., Lee, K. E., Lee, E. S., Matin, M. N., Lee, D. S., Yun, J. S., et al. (2013). The genetic constitutions of complementary genes Pp and Pb determine the purple color variation in pericarps with cyanidin-3-O-glucoside depositions in black rice. J. Plant Biol. 56, 24–31. doi: 10.1007/s12374-012-0043-9
Rangaswamy, M., Natarajamoorthy, K., and Rangasamy, S. R. S. (1988). Use of purple puttu rice variety as a pollen barrier in CMS line seed production. Int. Rice Res. Newsl. 13, 4–28.
Ravel, C., Praud, S., Murigneux, A., Canaguier, A., Sapet, F., Samson, D., et al. (2006). Single-nucleotide polymorphism frequency in a set of selected lines of bread wheat (Triticum aestivum L.). Genome 49, 1131–1139. doi: 10.1139/g06-067
Reddy, A. M., Reddy, V. S., Scheffler, B. E., Wienand, U., and Reddy, A. R. (2007). Novel transgenic rice overexpressing anthocyanidin synthase accumulates a mixture of flavonoids leading to an increased antioxidant potential. Metab. Eng. 9, 95–111. doi: 10.1016/j.ymben.2006.09.003
Reddy, A. R., Scheffler, B., Madhuri, G., Srivastava, M. N., Kumar, A., Sathyanarayanan, P. V., et al. (1996). Chalcone synthase in rice (Oryza sativa L.): detection of the CHS protein in seedlings and molecular mapping of the chs locus. Plant Mol. Biol. 32, 735–743 . doi: 10.1007/bf00020214
Reddy, V. S., Dash, S., and Reddy, A. R. (1995). Anthocyanin pathway in rice (Oryza sativa L): identification of a mutant showing dominant inhibition of anthocyanins in leaf and accumulation of proanthocyanidins in pericarp. Theor. Appl. Genet. 91, 301–312. doi: 10.1007/bf00220892
Reddy, V. S., Goud, K. V., Sharma, R., and Reddy, A. R. (1994). Ultraviolet-B-responsive anthocyanin production in a rice cultivar is associated with a specific phase of phenylalanine ammonia lyase biosynthesis. Plant Physiol. 105, 1059–1066. doi: 10.1104/pp.105.4.1059
Rodriguez-Amaya, D. B. (2018). Update on natural food pigments-A mini-review on carotenoids, anthocyanins, and betalains. Food Res. Int. doi: 10.1016/j.foodres.2018.05.028
Schoenbohm, C., Martens, S., Eder, C., Forkmann, G., and Weisshaar, B. (2000). Identification of the Arabidopsis thaliana flavonoid 3’-hydroxylase gene and functional expression of the encoded P450 enzyme. Biol. Chem. 381, 749–753.
Shoeva, O. Y., Mock, H. P., Kukoeva, T. V., Börner, A., and Khlestkina, E. K. (2016). Regulation of the flavonoid biosynthesis pathway genes in purple and black grains of Hordeum vulgare. PLoS One 11:e0163782. doi: 10.1371/journal.pone.0163782
Smit, A. F. A., Hubley, R., and Green, P. (2015). RepeatMasker Open-4.0. Available at: http://www.repeatmasker.org/ (accessed June 20, 2018).
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Stein, J. C., Yu, Y., Copetti, D., Zwickl, D. J., Zhang, L., Zhang, C., et al. (2018). Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 50, 285–296. doi: 10.1038/s41588-018-0040-0
Subbaiyan, G. K., Waters, D. L., Katiyar, S. K., Sadananda, A. R., Vaddadi, S., and Henry, R. J. (2012). Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 10, 623–634. doi: 10.1111/j.1467-7652.2011.00676.x
Sun, W., Meng, X., Liang, L., Jiang, W., Huang, Y., He, J., et al. (2015). Molecular and biochemical analysis of chalcone synthase from Freesia hybrid in flavonoid biosynthetic pathway. PLoS One 10:e0119054. doi: 10.1371/journal.pone.0119054
Sun, X., Zhang, Z., Chen, C., Wu, W., Ren, N., Jiang, C., et al. (2018). The C-S-A gene system regulates hull pigmentation and reveals evolution of anthocyanin biosynthesis pathway in rice. J. Exp. Bot. 69, 1485–1498. doi: 10.1093/jxb/ery001
Sweeney, M. T., Thomson, M. J., Pfeil, B. E., and McCouch, S. (2006). Caught red-handed: Rc encodes a Basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 18, 283–294. doi: 10.1105/tpc.105.038430
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Terol, J., Naranjo, M. A., Ollitrault, P., and Talon, M. (2008). Development of genomic resources for Citrus clementina: characterization of three deep-coverage BAC libraries and analysis of 46,000 BAC end sequences. BMC Genomics 9:423. doi: 10.1186/1471-2164-9-423
Vigueira, C. C., Li, W., and Olsen, K. M. (2013). The role of Bh4 in parallel evolution of hull colour in domesticated and weedy rice. J. Evol. Biol. 26, 1738–1749. doi: 10.1111/jeb.12171
Vitte, C., and Bennetzen, J. L. (2006). Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc. Natl. Acad. Sci. U.S.A. 103, 17638–17643. doi: 10.1073/pnas.0605618103
Wakeley, J. (1996). The excess of transitions among nucleotide substitutions: new methods of estimating transition bias underscore its significance. Trends Ecol. Evol. 11, 158–162. doi: 10.1016/0169-5347(96)10009-4
Wang, C., and Shu, Q. (2007). Fine mapping and candidate gene analysis of purple pericarp gene Pb in rice (Oryza sativa L.). Chin. Sci. Bull. 52, 3097–3104. doi: 10.1007/s11434-007-0472-x
Wang, L., Hao, L., Li, X., Hu, S., Ge, S., and Yu, J. (2009). SNP deserts of Asian cultivated rice: genomic regions under domestication. J. Evol. Biol. 22, 751–761. doi: 10.1111/j.1420-9101.2009.01698.x
Yamamoto, E., Yonemaru, J. I., Yamamoto, T., and Yano, M. (2012). OGRO: the overview of functionally characterized genes in rice online database. Rice 5:26. doi: 10.1186/1939-8433-5-26
Yang, X., Fuller, D. Q., Huan, X., Perry, L., Li, Q., Li, Z., et al. (2015). Barnyard grasses were processed with rice around 10000 years ago. Sci. Rep. 5:16251.
Yonemaru, J. I., Yamamoto, T., Fukuoka, S., Uga, Y., Hori, K., and Yano, M. (2010). Q-TARO: QTL annotation rice online database. Rice 3, 194–203. doi: 10.1007/s12284-010-9041-z
Zhu, B. F., Si, L., Wang, Z., Zhu, Y. Z. J., Shangguan, Y., Lu, D., et al. (2011). Genetic control of a transition from black to straw-white seed hull in rice domestication. Plant Physiol. 155, 1301–1311. doi: 10.1104/pp.110.168500
Keywords: Purpleputtu, rice, WGS, SNPs/INDELs, variant calling, anthocyanin pathway
Citation: Lachagari VBR, Gupta R, Lekkala SP, Mahadevan L, Kuriakose B, Chakravartty N, Mohan Katta AVSK, Santhosh S, Reddy AR and Thomas G (2019) Whole Genome Sequencing and Comparative Genomic Analysis Reveal Allelic Variations Unique to a Purple Colored Rice Landrace (Oryza sativa ssp. indica cv. Purpleputtu). Front. Plant Sci. 10:513. doi: 10.3389/fpls.2019.00513
Received: 27 February 2019; Accepted: 03 April 2019;
Published: 07 May 2019.
Edited by:
Luigi Cattivelli, Council for Agricultural Research and Economics, ItalyReviewed by:
Fengxia Liu, China Agricultural University, ChinaCopyright © 2019 Lachagari, Gupta, Lekkala, Mahadevan, Kuriakose, Chakravartty, Mohan Katta, Santhosh, Reddy and Thomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: V. B. Reddy Lachagari, vb.reddy@aggenome.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.