 Wei Zhao
Wei Zhao Shanshan Chu
Shanshan Chu Yongqing Jiao
Yongqing Jiao- 1Collaborative Innovation Center of Henan Grain Crops, College of Agronomy, Henan Agricultural University, Zhengzhou, China
- 2Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture, Oil Crops Research Institute, Chinese Academy of Agricultural Sciences, Wuhan, China
Circular RNAs (circRNAs) are new endogenous non-coding RNA family members that arise during pre-mRNA splicing in a reversed order in which the 3′ and 5′ ends are covalently closed. Compared to the comprehensive investigation of circRNAs in animals, circRNA research in plants is still in its infancy. Genome-wide identification and characterization of circRNAs have recently been performed in several plant species. CircRNAs are ubiquitously expressed and abundant in plants. The expression of circRNAs is often dependent on cell-type, tissue, and developmental stage, and it is particularly stress-inducible in plants. CircRNAs might play important roles in various biological processes in plants, including development and the response to biotic and abiotic stresses. Here, we review the current literature and provide a brief overview of circRNAs and their research status in plants, as well as the bioinformatic tools and database resources for circRNA analysis.
Introduction
Unlike the better-known linear mRNAs formed by linear splicing, circular RNAs (circRNAs) are novel members of the non-coding RNA family and are generated during post-transcriptional processes via backsplicing of precursor messenger RNAs (pre-mRNAs) (Figures 1A,B) (Jeck et al., 2013). CircRNAs could be derived from any genomic location, such as exonic, intronic, and intergenic regions (Chen, 2016). In animals, circRNAs are widespread and are the predominant isoform of exons originating from protein-coding genes spliced in a non-canonical order (Salzman et al., 2012; Westholm et al., 2014; Rybak-Wolf et al., 2015). Unlike normal linear RNAs, circRNAs form covalently closed loop structures with neither 5′–3′ polarities nor polyadenylated tails. Thus, circRNAs are resistant to RNase R, which is a strong 3′–5′ exoribonuclease that is able to efficiently degrade linear RNAs. Therefore, circRNAs can be segregated and enriched from eukaryotic total RNAs by RNase R digestion (Suzuki and Tsukahara, 2014; Chen and Yang, 2015).
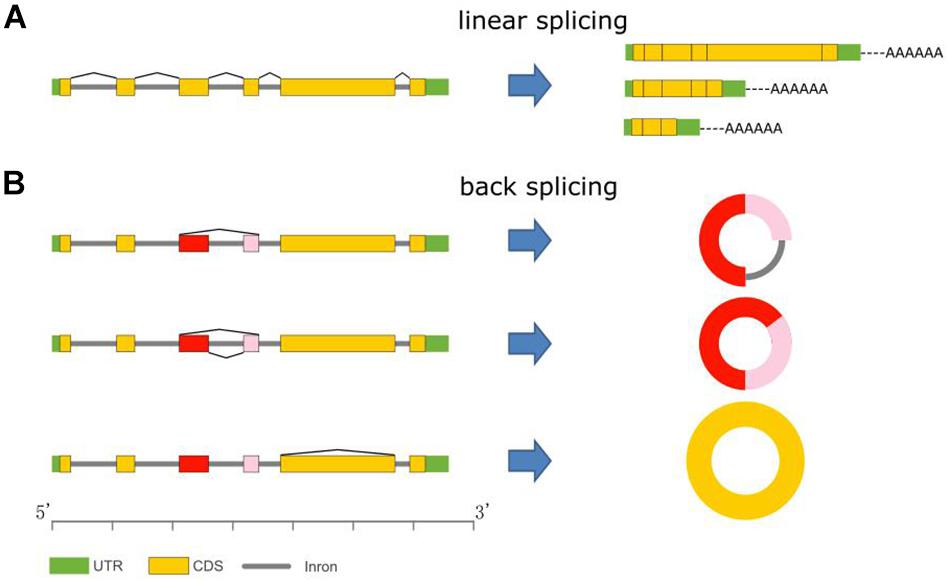
Figure 1. CircRNA biogenesis. (A) mRNA variants via alternative linear splicing. All mRNAs have polyA tails. (B) CircRNA variants formation via backsplicing.
Although the existence of circular transcripts has been observed for decades, such as the hepatitis δ virus and some plant viroids, circRNAs were typically regarded as rare byproducts of errant splicing during mRNA processing in eukaryotic cells (Sanger et al., 1976; Kos et al., 1986; Memczak et al., 2013). Due to the recent developments in high-throughput deep sequencing technology, exonuclease-based enrichment strategies, and novel bioinformatic tools, numerous circRNAs have been discovered and identified in many organisms, such as humans (Jeck et al., 2013), mouse (Fan et al., 2015), zebrafish (Shen et al., 2017), archaea (Danan et al., 2012), rice (Lu et al., 2015), and Arabidopsis (Ye et al., 2015), leading to the consensus that circRNAs are ubiquitous and abundant in eukaryotes.
Plant circRNAs possess features that differ from animal circRNAs. For example, reverse complementary elements, which are important for circularization, are enriched in the flanking introns of circRNAs in animals (Jeck and Sharpless, 2014). In contrast, in plants, most of the identified circRNAs contain comparatively fewer repetitive and reverse complementary sequences in the flanking introns that bracket the circRNAs (Lu et al., 2015; Ye et al., 2015). Additionally, in animals, certain circRNAs have been reported to act as miRNA sponges to regulate the expression of target genes. However, studies of circRNAs in plants have not implied the potential suitability of circRNAs as miRNA sponges (Hansen et al., 2013; Memczak et al., 2013; Westholm et al., 2014). Thus, plant circRNAs might possess different mechanisms of biogenesis and have different functional roles from animal circRNAs.
In this review, we present a concise and up-to-date overview of circRNAs in plants. Particularly, we focus on the abundance and expression patterns of circRNAs in various plant species and then discuss the available bioinformatic resources that can be used to characterize circRNAs based on high-throughput sequencing data. Finally, the potential functionality of circRNAs in plants is explored.
CircRNA Abundance in Plants
It is challenging to separate circRNAs from other RNAs, such as miRNA and mRNA, based on size or electrophoretic mobility. Due to the lack of a free polyadenylated tail, circRNAs have evaded recognition by poly (A) enrichment approaches. Thus, although circRNAs have been observed in eukaryotic cells for decades, it has not been possible to comprehensively evaluate them. Recent developments in high-throughput deep sequencing coupled with exonuclease-based enrichment strategies and computational approaches have resulted in the identification of thousands of circRNAs in animals, including in Drosophila (Westholm et al., 2014), humans (Salzman et al., 2012), mouse (Fan et al., 2015), and zebrafish (Shen et al., 2017).
Similarly, limited studies on higher plants have revealed that circRNAs are also widespread and abundant in plant species (Table 1). The genome-wide identification of plant circRNAs was first performed in Oryza sativa and Arabidopsis thaliana. Ye et al. (2015) identified 12,037 circRNAs from the root and shoot tissues of O. sativa and 6,012 circRNAs from the leaves of A. thaliana. Furthermore, Ye et al. (2017) also identified 2,806 circRNAs in the root tissues of rice seedlings. Lu et al. (2015) reported 2,354 circRNAs in the panicles and mature leaves of rice at different flowering time stages. In addition to the two well-known model plants, circRNAs have also been identified from the other monocotyledon and dicotyledon species, such as barley, tomato, wheat, soybean, and kiwifruit (Table 1). Darbani et al. (2016) reported 62 circRNAs in the leaves and immature seeds of barley, while Zuo et al. (2016) identified 854 circRNAs from mesocarp samples at the mature green stage of tomato. Additionally, Tan et al. (2017) also identified 796 circRNAs from the fruits of tomato. Using an RNase R-treated enrichment approach, Wang et al. (2017) detected 88 circRNAs in the leaves of wheat seedlings. Zhao et al. (2017a) identified 5,372 circRNAs in the leaves, stems, and roots of soybean seedlings, and another study also identified 5,367 circRNAs associated with resistance to defoliating insects in soybean leaves (Zhao et al., 2017b). Wang et al. (2017) identified 3,582 circRNAs from the leaf, root, and stem tissues of four kiwifruit materials originating from three species belonging to the genus Actinidia. Chen et al. (2018) detected 2,804 circRNAs in the seedling leaves of maize. In summary, an abundance (95,143) of circRNAs has been identified from various plant species (Chu et al., 2018); however, regrettably, only a few circRNAs have been verified by experimental approaches. With the development of high-throughput technology and new bioinformatic tools, we anticipate that more circRNAs will be identified in plants in the future.
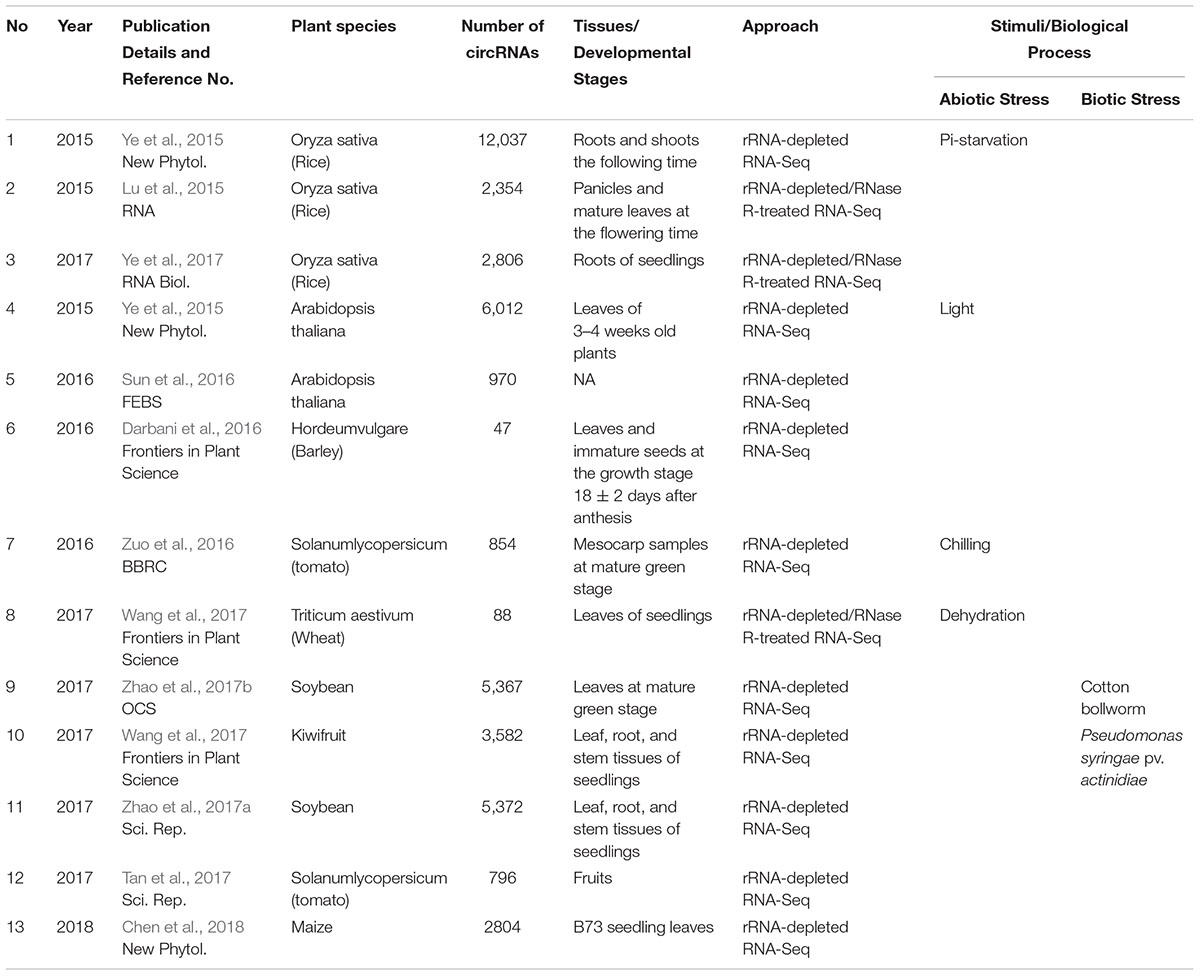
Table 1. Studies showing the genome-wide identification of circRNAs in plants.
Features of Plant circRNAs
The size of circRNAs ranges from smaller than 100 nt to larger than 4 kb. For example, the majority of human circRNAs are a few hundred nucleotides in length, while soybean circRNAs are mainly between 150 and 600 bp, and only a few are greater than 2 kb (Jeck et al., 2013; Zhang et al., 2014; Zhao et al., 2017a).
A conserved feature in animals and plants is that the genesis of circRNAs depends on RNA polymerase II-mediated transcription and backsplicing reactions of pre-mRNAs (Sun et al., 2016). Previous studies have demonstrated that the reverse complementary sequences were significantly enriched in the flanking introns bracketing circRNAs and that these short intronic repeat sequences could facilitate circRNA production in animals (Jeck et al., 2013; Chen and Yang, 2015; Chen, 2016). For example, in humans, circularized exons are typically bracketed by long introns, which contain abundant Arthrobacter luteu (Alu) elements (Jeck et al., 2013). However, there are comparatively fewer of these repetitive elements in plant circRNAs. For example, the proportion of reverse complementary sequences was only 6.2, 2.7, and 0.3% in the intronic sequences flanking exonic circRNAs in rice, soybean, and A. thaliana, respectively (Lu et al., 2015; Ye et al., 2015; Zhao et al., 2017a). In maize, sequences related to LINE1-like elements (LLEs) and their Reverse Complementary Pairs (LLERCPs) are significantly enriched in the flanking regions of circRNAs, which indicates that transposons may be involved in the formation of circRNAs in plants (Wang et al., 2017).
CircRNAs are generated when the pre-mRNA splicing machinery backsplices to join a down-stream splice donor to an upstream splice acceptor. The 3′ and 5′ ends normally present in a linear mRNA molecule have been joined together covalently in circRNAs. The U2-dependent spliceosome is responsible for the splicing of the vast majority of introns in both plants and animals, with GT and AG terminal dinucleotides at their 5′ and 3′ termini, respectively (Szczesniak et al., 2013). However, the mechanisms of selection for certain sequences to circularize by spliceosomes are poorly characterized. The analysis of splice signals of circRNAs in humans has revealed that most of the exonic circRNAs contain canonical GT/AG splicing signals, while some also harbor non-GT/AG splicing signals (Ye et al., 2017). In plants, the splice signals of circRNAs differ between monocot and dicot species. In rice, only a small portion (7.3%) of circRNAs possess canonical GT/AG (CT/AC) splicing signals, and a large number of circRNAs share diverse non-GT/AG splicing signals, such as GC/GG, CA/GC, GG/AG, GC/CG, and CT/CC (Ye et al., 2017). In A. thaliana, among the 803 identified circRNAs with the GT-AG signal, the majority showed the canonical splicing signal GT/AG, while only 9 circRNAs were generated from non-canonical splicing signals (Sun et al., 2016). However, these diverse splice signal patterns should be further verified in more plant species.
CircRNAs are conserved and have various isoforms that are generated by alternative circularization in plants (Figure 1B). In O. sativa and A. thaliana, 6,074 and 5,152 circRNA-host genes could generate exonic circRNAs, of which 12.2 and 14.5% constitute orthologous genes. Furthermore, over 300 orthologous circRNA-host genes could generate circRNAs from a similar position (Ye et al., 2015).
Alternative circularization constitutes another circRNA formation mechanism that can generate a variety of additional circRNAs from one gene (Zhang et al., 2014). A previous study demonstrated that over 50% of gene loci produced circRNAs through alternative circularization in human and mouse cells (Zhang et al., 2014). Similarly, in rice, a large number of circRNA isoforms are produced by the same locus, and over half of the circRNAs in rice were generated by alternative circularization (Ye et al., 2015). For example, the gene LOC_Os11g02080 was predicted to generate 41 isoforms, while the gene LOC_Os12g02040 was predicted to generate 38 isoforms, which were further validated by the successful sequencing of reverse transcription (RT)-PCR products (Ye et al., 2015).
Bioinformatic Resources for Plant circRNAs
The advances in high-throughput deep sequencing technology have enabled scientists to generate millions of sequencing reads in a short time period. In response to the mass generation of RNA sequencing (RNA-Seq) data, new computational algorithms for the precise and efficient identification of circRNAs have been developed (Szabo et al., 2015). Several different bioinformatic tools, such as circRNA finder (Westholm et al., 2014), CIRCexplorer (Zhang et al., 2014), CIRI (Gao et al., 2015), find circ (Memczak et al., 2013), Mapsplice (Wang et al., 2010), PcircRNA_finder (Chen L. et al., 2016), and circseq-cup (Ye et al., 2017), have been developed specifically for this purpose (Table 2). However, these bioinformatic tools perform differently in terms of precision and sensitivity when identifying circRNAs from RNA-Seq data and also differ in computational costs. PcircRNA_finder was specially developed for plant circRNA identification and provided a more comprehensive, sensitive, and precise prediction method for plant circRNAs, while the other algorithms were established for animals (Chen L. et al., 2016). Hansen et al. (2016) comparatively analyzed circRNA prediction tools. They found that different tools could produce highly divergent results with high false positive ratios; however, combining the output of the different tools could reduce the false positive fraction significantly (Hansen et al., 2016). Zeng et al. (2017) also comprehensively evaluated different circRNA detection tools using four different datasets, including a positive dataset, background dataset, mixed dataset, and real datasets. Generally, CIRI, CIRCexplorer, and KNIFE perform better in terms of achieving a balance between precision and sensitivity. These comparison results proved valuable for improving algorithms and have provided useful guidelines for current algorithms used for data interpretation by researchers.

Table 2. An overview of the bioinformatics tools available for the prediction of circRNAs.
Increasing numbers of circRNA datasets have been produced, which has urgently necessitated the efficient organization and management of these datasets. Currently, several animal circRNA databases have been established, such as circ2Traits (Ghosal et al., 2013), nc2Cancer (Chen et al., 2015), circBase (Glažar et al., 2014), starBase v2.0 (Li et al., 2014), CircNet (Liu Y.C. et al., 2016), deepBase v2.0 (Zheng et al., 2016), CircInteractome (Dudekula et al., 2016), and circRNADb (Chen X. et al., 2016) (Table 3). Comparatively, only one plant-specific circRNA database, PlantcircBase1, has been created recently (Chu et al., 2017). PlantcircBase records published and unpublished circRNAs with universal identifiers from different plants species, including O. sativa, A. thaliana, Zea mays (Z. mays), Solanum lycopersicum (S. lycopersicum), Glycine max (G. max), Camellia sinensis (C. sinensis), Gossypium arboreum (G. arboreum), Gossypium hirsutum (G. hirsutum), Gossypium raimondii (G. raimondii), Ptelea trifoliata (P. trifoliata), Symphytum tuberosum (S. tuberosum), Triticum aestivum (T. aestivum), and Hordeum vulgare (H. vulgare). PlantcircBase also provides a visualization of specific circRNA structures, potential interaction networks involving circRNA-miRNA-mRNA in the corresponding species, and validation information by Sanger sequencing. However, more significant information on functional annotation, tissue expression, interaction with other molecules, and phylogenetic conservation are still needed, which would make PlantcircBase a more comprehensive resource for research into plant circRNAs. The development of high-throughput sequencing technologies and the growing availability of various bioinformatic resources will greatly promote circRNA research.
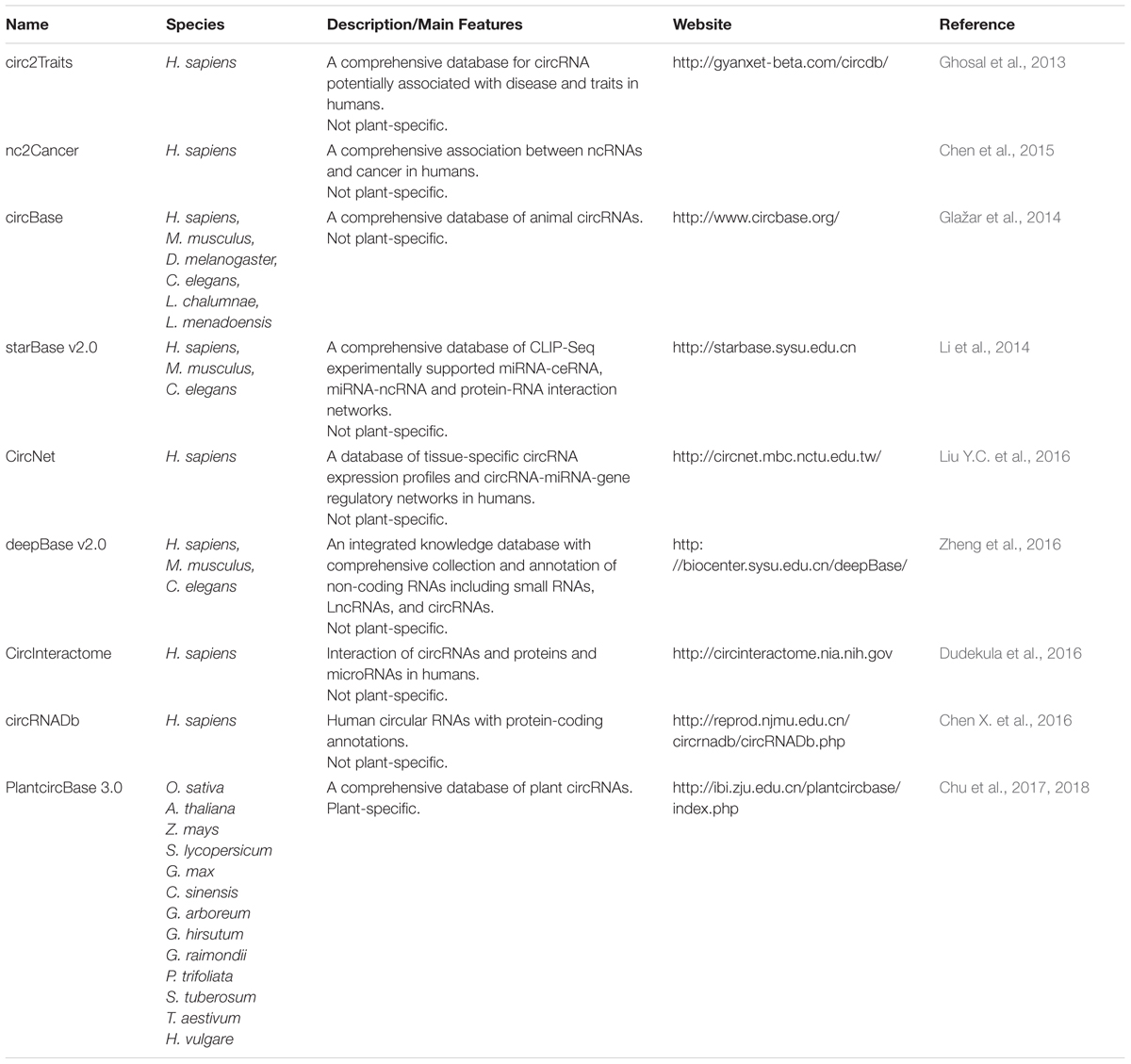
Table 3. Public circRNA databases.
Expression Patterns of Plant circRNAs
CircRNAs usually exhibit specific cell-type, tissue, and developmental stage expression patterns in animals (Westholm et al., 2014; Fan et al., 2015). For example, in Drosophila, circRNAs originate from neural genes and exhibit enhanced accumulation in neural tissues (Westholm et al., 2014). Using single-cell universal poly(A)-independent RNA sequencing technology, Fan et al. (2015) discovered 2,891 circRNAs in mouse preimplantation embryos, of which a large proportion showed developmental-specific expression patterns.
In plants, circRNAs also exhibit specific expression patterns, as observed in animals (Lu et al., 2015; Darbani et al., 2016). Darbani et al. (2016) treated field-grown barley plants with a foliar application of iron or zinc solution and collected the seed transfer cells using laser capture micro-dissection for RNA-Seq analysis. They ultimately identified 62 transfer cell-specific circRNAs and demonstrated that these circRNAs could respond to the foliar application of micronutrients in barley (Darbani et al., 2016). Lu et al. (2015) validated 30 rice circRNAs experimentally, of which three and four circRNAs showed panicle-specific and leaf-specific expression, respectively.
Plant circRNAs also exhibit stress-inducible expression patterns. Ye et al. (2015) found that 27 circRNAs were differentially expressed under phosphate-sufficient -sufficient and -starvation conditions in rice. Additionally, by comparing the expressions of the circRNAs between low- and high-light stress conditions, they found that many circRNAs were specifically expressed following high-light treatment in Arabidopsis (Ye et al., 2015). In tomato, approximately 19% (163/854) of circRNAs exhibited chilling responsive expression patterns (Zuo et al., 2016). In wheat, 62 circRNAs were differentially expressed in dehydration-stressed seedling leaves (Wang Y. et al., 2016).
In addition to abiotic stress, circRNAs were also reported to be responsive to biotic stresses. For example, in soybean, 199 circRNAs were found to be differentially expressed between resistant and susceptible samples under defoliation damage by cotton bollworm feeding (Zhao et al., 2017b). Another study in kiwifruit revealed that 584 circRNAs were differentially expressed during Pseudomonas syringae pv. actinidiae (Psa) infection (Wang et al., 2017). These results suggest that circRNAs might play important and diverse functional roles in response to biotic and abiotic stresses in plants.
Putative Functions of Plant circRNAs
Although the abundance of circRNAs has been recognized, their functions have remained largely unclear. However, accumulating evidence suggests that circRNAs could play important functional roles in various biological processes, such as miRNA binding, protein binding, and transcriptional regulation (Hansen et al., 2013; Memczak et al., 2013; Chen, 2016).
Previous studies in mammals demonstrated that circRNAs can function as miRNA sponges or potent ceRNA molecules to bind specific miRNAs to prohibit them from regulating their target genes (Memczak et al., 2013). For example, in mouse, the sex-determining region Y (Sry) is a highly expressed, testis-specific circRNA that harbors 16 putative binding sites for miR-138 and serves as a miR-138 sponge (Rybak-Wolf et al., 2015). In humans, ciRS-7 (also termed CDR1as), as a circular miR-7 inhibitor, functions as an efficient microRNA sponge. It harbors more than 70 conventional miR-7 binding sites and strongly suppresses miR-7 activity, resulting in increased levels of miR-7 targets (Memczak et al., 2013). However, there is no further evidence to support that miRNA sponge function has been ascribed to the majority of circRNAs in animals. Nevertheless, only 6.6 and 5.0% of circRNAs were predicted to potentially target mimics of miRNAs in rice and Arabidopsis, respectively (Ye et al., 2015). Recently, 24 tomato circRNAs and 6 wheat circRNAs were thought to act as miRNA sponges, which requires further validation (Zuo et al., 2016; Wang et al., 2017). Os08circ16564, a rice circRNA, was predicted to harbor the target sites of miR172 and miR810 (Lu et al., 2015). Transgenic analysis for the overexpression of Os08circ16564 showed that the expression level of miR170 did not differ between Os08circ16564-transgenic rice and the control (Lu et al., 2015). It is thus unclear if miRNA inhibition is a general functional aspect of circRNAs in plants and animals—a topic that requires further experimental investigation.
It was previously thought that circRNAs could not produce a natural protein since most of them were not associated with polysomes (Guo et al., 2014). However, some engineered circRNAs with an internal ribosome entry site (IRES) could be translated in vivo (Wang and Wang, 2015). Recent research findings in mammals and flies have revealed that endogenous circRNAs containing IRES sequences and ATG could produce proteins. For example, circMbl, a circRNA that exists in the heads of flies, could generate a 37.04 kDa protein (Pamudurti et al., 2017). The Circ-ZNF609 circRNA in humans contains an intact open reading frame (ORF) with start and stop codons that could be translated into a protein in a splicing-dependent and cap-independent manner (Legnini et al., 2017). Moreover, the circ-ZNF609-derived protein plays important functional roles in myogenesis (Legnini et al., 2017). To date, no plant circRNAs have been reported to generate proteins. As research progresses, we expect that the protein coding circRNAs and their potential functions will be revealed in plants.
Previous studies revealed that circRNAs are associated with various physiological and pathophysiological processes, such as insulin biosynthesis and secretion (Xu et al., 2015), neurological diseases (Satoh and Yamamura, 2004; Junn et al., 2009; Lukiw, 2013), degenerative diseases (Ashwal-Fluss et al., 2014; Liu Q. et al., 2016), cardiovascular diseases (Burd et al., 2010; Wang K. et al., 2016; Du et al., 2017), and cancers (Qu et al., 2015; Sand et al., 2016; Shang et al., 2016; Xuan et al., 2016), which indicates that circRNAs might be novel biomarkers for disease diagnosis and therapy in humans. CircRNAs were also reported to serve as a class of aging biomarkers in the central nervous system (CNS) (Westholm et al., 2014). In plants, biomarkers have been widely used for both molecular fundamental research and applied practices in crop breeding (Yang et al., 2011). CircRNAs could also be potential biomarkers in plants due to their unique characteristics, including resistance to degradation, long half-lives, and the ease and specificity of detection (Lai et al., 2018). A recent study revealed that circRNAs can function as bona fide biomarkers of functional exon-skipped AS variants, including in the homeotic MADS-box transcription factor family in Arabidopsis (Conn et al., 2017). CircRNAs usually exhibit specific cell-type, tissue, and developmental stage expression patterns, and furthermore, the expression of circRNAs and circRNA isoforms is often induced under diverse environmental stresses, such as low- and high-light stresses, Pi-starvation conditions, low temperature stress, dehydration stress, and chewing injury stress by insects, which suggests that circRNAs might play important roles in plant development or in the response to biotic and abiotic stresses. One subclass of circRNAs in human cells, EIciRNAs (exon–intron circRNAs), was shown to enhance the transcription of the gene from which they were derived through interaction with U1 snRNP and RNA Polymerase II in the promoter region of the circRNA-host gene (Li Z. et al., 2015). Thus, EIciRNAs have the potential to act as transcriptional regulators to induce the expression of circRNA-host genes. In plants, Zhao et al. (2017b) discovered 293 EIcircRNAs, including 183 and 175 in resistant and susceptible samples, under defoliation damage stress by cotton bollworm feeding in soybean, which indicated that EIcircRNAs might participate in the response to chewing injury resistance processes in plants. In addition, some barley circRNAs that are highly expressed in the mitochondria might be involved in micronutrient homeostasis. The overexpression of PSY1-circ1, a circRNA derived from Phytoene Synthase 1 (PSY1) in tomato, resulted in a significant decrease in lycopene and β-carotene accumulation in transgenic tomato fruits, which suggests the involvement of circRNAs in plant development (Tan et al., 2017). However, the detailed mechanisms underlying their regulatory roles in response to biotic and abiotic stresses are still poorly understood.
Future Perspectives
Interest in the identification and characterization of plant circRNAs is growing. With abundance of circRNAs being identified and new insights into circRNAs generated rapidly, the biogenesis and functionality of circRNAs has become a pertinent research topic. The expression of circRNAs is usually specific to cell-type, tissue, and developmental stage, and is also stress-inducible in both animals and plants, which indicates that circRNAs may represent a new layer of post-transcriptional gene regulation. Evidence in animals indicates that circRNAs are potentially important regulators in various biological processes and are associated with human disorders, including cancers (Li Y. et al., 2015).
CircRNAs potentially represent another level of post-transcriptional gene regulators. Although circRNAs were previously regarded as a novel class of non-coding RNAs, several studies have provided initial evidence for the coding of proteins in animals by certain endogenous circRNAs (Legnini et al., 2017; Pamudurti et al., 2017; Yang et al., 2017). However, the coding potential of plant circRNAs has not been investigated thus far. In the future, elucidating and understanding the functional roles of circRNAs, such as in plant development, the response to biotic and abiotic stresses, and in translation, might constitute the primary research topic in plant circRNAs. Additionally, the differences in circRNA biogenesis between plants and animals would also be an interesting research avenue.
Data Availability
No datasets were generated or analyzed for this study.
Author Contributions
WZ and YJ wrote and revised the manuscript. SC helped to prepare the materials and revise the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2016YFD0100603), National Natural Science Foundation of China (31522042), National Transgenic Project (2016ZX08004003), Natural Science Foundation of Hubei (2016CFA049), Chinese Academy of Agricultural Sciences (Y2017JC14), and Henan Agricultural University.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnote
References
Ashwal-Fluss, R., Meyer, M., Pamudurti, N. R., Ivanov, A., Bartok, O., Hanan, M., et al. (2014). circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 56, 55–66. doi: 10.1016/j.molcel.2014.08.019
Burd, C. E., Jeck, W. R., Liu, Y., Sanoff, H. K., Wang, Z., and Sharpless, N. E. (2010). Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 6:e1001233. doi: 10.1371/journal.pgen.1001233
Chen, L., Yu, Y., Zhang, X., Liu, C., Ye, C., and Fan, L. (2016). PcircRNA_finder: a software for circRNA prediction in plants. Bioinformatics 32, 3528–3529. doi: 10.1093/bioinformatics/btw496
Chen, X., Han, P., Zhou, T., Guo, X., Song, X., and Li, Y. (2016). circRNADb: a comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 6:34985. doi: 10.1038/srep34985
Chen, L., Zhang, P., Fan, Y., Lu, Q., Li, Q., Yan, J., et al. (2018). Circular RNAs mediated by transposons are associated with transcriptomic and phenotypic variation in maize. New Phytol. 217, 1292–1306. doi: 10.1111/nph.14901
Chen, L. L. (2016). The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 17, 205–211. doi: 10.1038/nrm.2015.32
Chen, L. L., and Yang, L. (2015). Regulation of circRNA biogenesis. RNA Biol. 12, 381–388. doi: 10.1080/15476286.2015.1020271
Chen, Z., Liu, K., Yan, Z., Xiang, S., and Sun, Z. (2015). nc2Cancer: a database for cancer-associated human ncRNAs. Chin. J. Bioinformatics 13, 77–81.
Chu, Q., Bai, P., Zhu, X., Zhang, X., Mao, L., Zhu, Q. H., et al. (2018). Characteristics of plant circular RNAs. Brief Bioinform. doi: 10.1093/bib/bby111 [Epub ahead of print].
Chu, Q., Zhang, X., Zhu, X., Liu, C., Mao, L., Ye, C., et al. (2017). PlantcircBase: a database for plant circular RNAs. Mol. Plant 10, 1126–1128. doi: 10.1016/j.molp.2017.03.003
Conn, V. M., Hugouvieux, V., Nayak, A., Conos, S. A., Capovilla, G., Cildir, G., et al. (2017). A circRNA from SEPALLATA3 regulates splicing of its cognate mRNA through R-loop formation. Nat. Plants 3:17053. doi: 10.1038/Nplants.2017.53
Danan, M., Schwartz, S., Edelheit, S., and Sorek, R. (2012). Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 40, 3131–3142. doi: 10.1093/nar/gkr1009
Darbani, B., Noeparvar, S., and Borg, S. (2016). Identification of circular RNAs from the parental genes involved in multiple aspects of cellular metabolism in barley. Front. Plant Sci. 7:776. doi: 10.3389/fpls.2016.00776
Du, W. W., Yang, W., Chen, Y., Wu, Z. K., Foster, F. S., Yang, Z., et al. (2017). Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur. Heart J. 38, 1402–1412. doi: 10.1093/eurheartj/ehw001
Dudekula, D. B., Panda, A. C., Grammatikakis, I., De, S., Abdelmohsen, K., and Gorospe, M. (2016). CircInteractome: a web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 13, 34–42. doi: 10.1080/15476286.2015.1128065
Fan, X., Zhang, X., Wu, X., Guo, H., Hu, Y., Tang, F., et al. (2015). Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 16:148. doi: 10.1186/s13059-015-0706-1
Gao, Y., Wang, J., and Zhao, F. (2015). CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 16:4. doi: 10.1186/s13059-014-0571-3
Ghosal, S., Das, S., Sen, R., Basak, P., and Chakrabarti, J. (2013). Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 4:283. doi: 10.3389/fgene.2013.00283
Glažar, P., Papavasileiou, P., and Rajewsky, N. (2014). circBase: a database for circular RNAs. RNA 20, 1666–1670. doi: 10.1261/rna.043687.113
Guo, J. U., Agarwal, V., Guo, H., and Bartel, D. P. (2014). Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 15:409. doi: 10.1186/s13059-014-0409-z
Hansen, T. B., Jensen, T. I., Clausen, B. H., Bramsen, J. B., Finsen, B., Damgaard, C. K., et al. (2013). Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388. doi: 10.1038/nature11993
Hansen, T. B., Veno, M. T., Damgaard, C. K., and Kjems, J. (2016). Comparison of circular RNA prediction tools. Nucleic Acids Res. 44:e58. doi: 10.1093/nar/gkv1458
Hoffmann, S., Otto, C., Doose, G., Tanzer, A., Langenberger, D., Christ, S., et al. (2014). A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome Biol. 15:R34. doi: 10.1186/gb-2014-15-2-r34
Jeck, W. R., and Sharpless, N. E. (2014). Detecting and characterizing circular RNAs. Nat. Biotechnol. 32, 453–461. doi: 10.1038/nbt.2890
Jeck, W. R., Sorrentino, J. A., Wang, K., Slevin, M. K., Burd, C. E., Liu, J., et al. (2013). Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19, 141–157. doi: 10.1261/rna.035667.112
Junn, E., Lee, K. W., Jeong, B. S., Chan, T. W., Im, J. Y., and Mouradian, M. M. (2009). Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. U.S.A. 106, 13052–13057. doi: 10.1073/pnas.0906277106
Kos, A., Dijkema, R., Arnberg, A. C., van der Meide, P. H., and Schellekens, H. (1986). The hepatitis delta (delta) virus possesses a circular RNA. Nature 323, 558–560. doi: 10.1038/323558a0
Lai, X., Bazin, J., Webb, S., Crespi, M., Zubieta, C., and Conn, S. J. (2018). “CircRNAs in plants,” in Circular RNAs. Advances in Experimental Medicine and Biology, Vol. 1087, ed. J. Xiao (Singapore: Springer), 370.
Legnini, I., Di Timoteo, G., Rossi, F., Morlando, M., Briganti, F., Sthandier, O., et al. (2017). Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell 66, 22–37. doi: 10.1016/j.molcel.2017.02.017
Li, J. H., Liu, S., Zhou, H., Qu, L. H., and Yang, J. H. (2014). starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–D97. doi: 10.1093/nar/gkt1248
Li, Y., Zheng, Q. P., Bao, C. Y., Li, S. Y., Guo, W. J., Zhao, J., et al. (2015). Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 25, 981–984. doi: 10.1038/cr.2015.82
Li, Z., Huang, C., Bao, C., Chen, L., Lin, M., Wang, X., et al. (2015). Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 22, 256–264. doi: 10.1038/nsmb.2959
Liu, Q., Zhang, X., Hu, X., Dai, L., Fu, X., Zhang, J., et al. (2016). Circular RNA related to the chondrocyte ECM regulates MMP13 expression by functioning as a MiR-136 ‘Sponge’ in human cartilage degradation. Sci. Rep. 6:22572. doi: 10.1038/srep22572
Liu, Y. C., Li, J. R., Sun, C. H., Andrews, E., Chao, R. F., Lin, F. M., et al. (2016). CircNet: a database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res. 44, D209–D215. doi: 10.1093/nar/gkv940
Lu, T., Cui, L., Zhou, Y., Zhu, C., Fan, D., Gong, H., et al. (2015). Transcriptome-wide investigation of circular RNAs in rice. RNA 21, 2076–2087. doi: 10.1261/rna.052282.115
Lukiw, W. J. (2013). Circular RNA (circRNA) in Alzheimer’s disease (AD). Front. Genet. 4:307. doi: 10.3389/fgene.2013.00307
Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. doi: 10.1038/nature11928
Pamudurti, N. R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., et al. (2017). Translation of CircRNAs. Mol. Cell 66, 9.e7–21.e7. doi: 10.1016/j.molcel.2017.02.021
Qu, S., Song, W., Yang, X., Wang, J., Zhang, R., Zhang, Z., et al. (2015). Microarray expression profile of circular RNAs in human pancreatic ductal adenocarcinoma. Genom Data 5, 385–387. doi: 10.1016/j.gdata.2015.07.017
Rybak-Wolf, A., Stottmeister, C., Glazar, P., Jens, M., Pino, N., Giusti, S., et al. (2015). Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 58, 870–885. doi: 10.1016/j.molcel.2015.03.027
Salzman, J., Gawad, C., Wang, P. L., Lacayo, N., and Brown, P. O. (2012). Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7:e30733. doi: 10.1371/journal.pone.0030733
Sand, M., Bechara, F. G., Sand, D., Gambichler, T., Hahn, S. A., Bromba, M., et al. (2016). Circular RNA expression in basal cell carcinoma. Epigenomics 8, 619–632. doi: 10.2217/epi-2015-0019
Sanger, H. L., Klotz, G., Riesner, D., Gross, H. J., and Kleinschmidt, A. K. (1976). Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. U.S.A. 73, 3852–3856. doi: 10.1073/pnas.73.11.3852
Satoh, J., and Yamamura, T. (2004). Gene expression profile following stable expression of the cellular prion protein. Cell. Mol. Neurobiol. 24, 793–814. doi: 10.1007/s10571-004-6920-0
Shang, X., Li, G., Liu, H., Li, T., Liu, J., Zhao, Q., et al. (2016). Comprehensive circular RNA profiling reveals that hsa_circ_0005075, a new circular RNA biomarker, is involved in hepatocellular crcinoma development. Medicine 95:e3811. doi: 10.1097/MD.0000000000003811
Shen, Y., Guo, X., and Wang, W. (2017). Identification and characterization of circular RNAs in zebrafish. FEBS Lett. 591, 213–220. doi: 10.1002/1873-3468.12500
Song, X., Zhang, N., Han, P., Lai, R. K., Wang, K., and Lu, W. (2016). Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 44:e87. doi: 10.1093/nar/gkw075
Sun, X., Wang, L., Ding, J., Wang, Y., Wang, J., Zhang, X., et al. (2016). Integrative analysis of Arabidopsis thaliana transcriptomics reveals intuitive splicing mechanism for circular RNA. FEBS Lett. 590, 3510–3516. doi: 10.1002/1873-3468.12440
Suzuki, H., and Tsukahara, T. (2014). A view of pre-mRNA splicing from RNase R resistant RNAs. Int. J. Mol. Sci. 15, 9331–9342. doi: 10.3390/ijms15069331
Szabo, L., Morey, R., Palpant, N. J., Wang, P. L., Afari, N., Jiang, C., et al. (2015). Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 16:126. doi: 10.1186/s13059-015-0690-5
Szczesniak, M. W., Kabza, M., Pokrzywa, R., Gudys, A., and Makalowska, I. (2013). ERISdb: a database of plant splice sites and splicing signals. Plant Cell Physiol. 54:e10. doi: 10.1093/pcp/pct001
Tan, J., Zhou, Z., Niu, Y., Sun, X., and Deng, Z. (2017). Identification and functional characterization of tomato CircRNAs derived from genes involved in fruit pigment accumulation. Sci. Rep. 7:8594. doi: 10.1038/s41598-017-08806-0
Wang, K., Long, B., Liu, F., Wang, J. X., Liu, C. Y., Zhao, B., et al. (2016). A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur. Heart J. 37, 2602–2611. doi: 10.1093/eurheartj/ehv713
Wang, Y., Yang, M., Wei, S., Qin, F., Zhao, H., and Suo, B. (2016). Identification of circular RNAs and their targets in leaves of Triticum aestivum L. under dehydration stress. Front. Plant Sci. 7:2024. doi: 10.3389/fpls.2016.02024
Wang, K., Singh, D., Zeng, Z., Coleman, S. J., Huang, Y., Savich, G. L., et al. (2010). MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38:e178. doi: 10.1093/nar/gkq622
Wang, Y., and Wang, Z. (2015). Efficient backsplicing produces translatable circular mRNAs. RNA 21, 172–179. doi: 10.1261/rna.048272.114
Wang, Z., Liu, Y., Li, D., Li, L., Zhang, Q., Wang, S., et al. (2017). Identification of circular RNAs in kiwifruit and their species-specific response to bacterial canker pathogen invasion. Front. Plant Sci. 8:413. doi: 10.3389/fpls.2017.00413
Westholm, J. O., Miura, P., Olson, S., Shenker, S., Joseph, B., Sanfilippo, P., et al. (2014). Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 9, 1966–1980. doi: 10.1016/j.celrep.2014.10.062
Xu, H., Guo, S., Li, W., and Yu, P. (2015). The circular RNA Cdr1as, via miR-7 and its targets, regulates insulin transcription and secretion in islet cells. Sci. Rep. 5:12453. doi: 10.1038/srep12453
Xuan, L., Qu, L., Zhou, H., Wang, P., Yu, H., Wu, T., et al. (2016). Circular RNA: a novel biomarker for progressive laryngeal cancer. Am. J. Transl. Res. 8, 932–939.
Yang, X. S., Wu, J., Ziegler, T. E., Yang, X., Zayed, A., Rajani, M. S., et al. (2011). Gene expression biomarkers provide sensitive indicators of in planta nitrogen status in maize. Plant Physiol. 157, 1841–1852. doi: 10.1104/pp.111.187898
Yang, Y., Fan, X. J., Mao, M. W., Song, X. W., Wu, P., Zhang, Y., et al. (2017). Extensive translation of circular RNAs driven by N-6-methyladenosine. Cell Res. 27, 626–641. doi: 10.1038/cr.2017.31
Ye, C. Y., Chen, L., Liu, C., Zhu, Q. H., and Fan, L. (2015). Widespread noncoding circular RNAs in plants. New Phytol. 208, 88–95. doi: 10.1111/nph.13585
Ye, C. Y., Zhang, X., Chu, Q., Liu, C., Yu, Y., Jiang, W., et al. (2017). Full-length sequence assembly reveals circular RNAs with diverse non-GT/AG splicing signals in rice. RNA Biol. 14, 1055–1063. doi: 10.1080/15476286.2016.1245268
Zeng, X., Lin, W., Guo, M., and Zou, Q. (2017). A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 13:e1005420. doi: 10.1371/journal.pcbi.1005420
Zhang, X. O., Wang, H. B., Zhang, Y., Lu, X., Chen, L. L., and Yang, L. (2014). Complementary sequence-mediated exon circularization. Cell 159, 134–147. doi: 10.1016/j.cell.2014.09.001
Zhao, W., Cheng, Y., Zhang, C., You, Q., Shen, X., Guo, W., et al. (2017a). Genome-wide identification and characterization of circular RNAs by high throughput sequencing in soybean. Sci. Rep. 7:5636. doi: 10.1038/s41598-017-05922-9
Zhao, W., Zhang, C., Shen, X., Xiao, L., Lu, J., Zhang, Y., et al. (2017b). Characterization of circRNAs associated with resistance to defoliating insects in soybean. Oil Crop Sci. 2, 23–37.
Zheng, L. L., Li, J. H., Wu, J., Sun, W. J., Liu, S., Wang, Z. L., et al. (2016). deepBase v2.0: identification, expression, evolution and function of small RNAs, LncRNAs and circular RNAs from deep-sequencing data. Nucleic Acids Res. 44, D196–D202. doi: 10.1093/nar/gkv1273
Keywords: circular RNA, circRNA, non-coding RNA, bioinformatics, review
Citation: Zhao W, Chu S and Jiao Y (2019) Present Scenario of Circular RNAs (circRNAs) in Plants. Front. Plant Sci. 10:379. doi: 10.3389/fpls.2019.00379
Received: 05 January 2019; Accepted: 12 March 2019;
Published: 02 April 2019.
Edited by:
Mathew G. Lewsey, La Trobe University, AustraliaReviewed by:
Chloe Zubieta, Centre National de la Recherche Scientifique (CNRS), FranceMing-Bo Wang, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Australia
Copyright © 2019 Zhao, Chu and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongqing Jiao, amlhb3lvbmdxaW5nQGhlbmF1LmVkdS5jbg==