
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 22 January 2019
Sec. Plant Systematics and Evolution
Volume 10 - 2019 | https://doi.org/10.3389/fpls.2019.00005
 Dan Zong1,2
Dan Zong1,2 Peihua Gan1,2
Peihua Gan1,2 Anpei Zhou1,2
Anpei Zhou1,2 Yao Zhang1,2
Yao Zhang1,2 Xinlian Zou1,2
Xinlian Zou1,2 Anan Duan1,2,3
Anan Duan1,2,3 Yu Song4,5*
Yu Song4,5* Chengzhong He1,2,3*
Chengzhong He1,2,3*Populus, a core genus of Salicaceae, plays a significant ecological role as a source of pioneer species in boreal forests. However, interspecific hybridization and high levels of morphological variation among poplars have resulted in great difficulty in classifying species for systematic and comparative evolutionary studies. Here, we present phylogenetic analyses of 24 newly sequenced Populus plastomes and 36 plastomes from GenBank, which represent seven genera of Salicaceae, in combination with a matrix of eighteen morphological characters of 40 Populus taxa to reconstruct highly supported relationships of genus Populus. Relationships among the 60 taxa of Salicaceae strongly supported two monophyletic genera: Populus and Salix. Chosenia was nested within the genus Salix, and five clades within Populus were divided. Clade I included the three taxa P. euphratica, P. pruinosa, and P. ilicifolia. Clade II contained thirteen taxa [P. adenopoda, P. alba, P. bolleana, P. davidiana, P. hopeiensis, P. nigra, P. qiongdaoensis, P. rotundifolia, P. rotundifolia var. duclouxiana, P. tremula, P. tremula × alba, P. tomentosa, and P. tomentosa (NC)]. Clade III included the ten taxa P. haoana, P. kangdingensis, P. lasiocarpa, P. pseudoglauca, P. qamdoensis, P. schneideri, P. simonii, P. szechuanica, P. szechuanica var. tibetica, and P. yunnanensis. Clade IV included P. cathayana, P. gonggaensis, P. koreana, P. laurifolia, P. trinervis, P. wilsonii, and P. xiangchengensis. The last clade comprised P. angustifolia, P. balsamifera, P. deltoides, P. deltoides × nigra, P. fremontii, P. mexicana, and P. trichocarpa. This phylogeny is also supported by morphological traits, including bark smoothness, bud size, petiole shape, leaf inflorescence, male anther length and male anther tip.
The family Salicaceae is primarily distributed in cold, tropical and warm temperate regions worldwide and is a dioecious woody and shrub plant throughout the northern hemisphere (Leskinen and Alström-Rapaport, 1999; Wang et al., 2014). Salicaceae includes over 50 genera, which comprise nearly 1000 species (Stevens, 2001; Chase et al., 2002; Byng et al., 2016). Phylogenetic analyses of Salicaceae using DNA sequences from the chloroplast gene rbcL (Azuma et al., 2000; Chase et al., 2002) and ITS of nuclear rDNA (Leskinen and Alström-Rapaport, 1999) strongly suggest that Populus and Salix are both monophyletic.
The species of the genus Populus, commonly known as poplars, aspen and cottonwood, are widely distributed throughout the northern hemisphere from subtropical to boreal forests (Brawdshaw et al., 2000), and China is one of the most important poplar distribution areas (Wan et al., 2013). Populus species play important roles in ecosystems and serve as model organisms for basic research in molecular biology and genetics, as well as in plant domestication and conservation (Kuzovkina and Vietto, 2014), mainly due to their fast growth rates, easy vegetative propagation, numerous wood uses, and small genome sizes (Braatne et al., 1992; Stettler et al., 1996; Cronk, 2005; Hamzeh et al., 2006).
Eckenwalder (1996) classified the genus Populus into 29 species grouped into six sections based on important morphological characteristics: persistence of the floral disk, presence or absence of marked foliar heteroblasty, overall leaf shape, distribution and shape of foliar teeth, number of carpels and flattened vs. round petiole. Section Abaso comprises a single species, P. mexicana; section Aigeiros contains three species and widely distributed in Europe and North America; section Leucoides shows a limited geographic distribution, with two species in China and the remaining species in southeastern United States; section Turanga contains three species and is geographically restricted to Central and West Asia and North Africa; sections Tacamahaca and Populus are relatively large, comprising nine and ten species, respectively (Hamzeh et al., 2006). However, the phylogenetic affinities between sections and the position of several taxa within these sections remain controversial. Many of the Populus species within a section, as well as between sections, are cross-compatible with each other (Hamzeh et al., 2006; Fladung and Schroeder, 2010). For example, species of the sections Aigeiros and Tacamahaca are sexually compatible, and natural hybridization occurs among several species of these sections (Zsuffa, 1975; Rajora and Zsuffa, 1984). Because of interspecific hybridization and high levels of morphological variation among poplars, the number of Populus species currently described in the literature ranges from 22 to 85, and hundreds of Populus hybrids and cultivars exist (Eckenwalder, 1977a,b, 1996; Dickmann and Stuart, 1983; Wang and Gilbert, 2007; Wan et al., 2013). Discrepancies in the total number of Populus species could be attributed to the misinterpretation of some hybrids and difficulties involved in delineating species boundaries (Hamzeh and Dayanandan, 2004).
For the assessment of genetic relationships among Populus species, different methods have been applied. A phylogenetic analysis of poplars using 76 morphological traits of leaves, flowers, fruits, and inflorescences supported the monophyly of all sections except for Tacamahaca, which resolved into two paraphyletic groups (Eckenwalder, 1996). ITS sequences were used in a study of fifteen representative species from five sections to resolve the relationships among the species in the genus Populus (Shi et al., 2001). The results showed that Populus is a monophyletic group and can be divided into two main clades: section Leuce and the remaining sections. Another phylogenetic study, based on three non-coding regions of cpDNA (intron of trnL and intergenic regions of trnT-trnL and trnL-trnF) and two nuclear rDNAs (ITS1 and ITS2), showed polyphyletic relationships among species in the sections Tacamahaca and Aigeiros (Hamzeh and Dayanandan, 2004). The phylogenetic tree of seventeen species or hybrids from four sections in Populus based on the chloroplast marker trnL-F showed that the Populus section formed a separate clade, while sections Tacamahaca and Turanga could not be clearly separated, and P. szechuanica var. tibetica formed a weakly supported clade with the other species of section Tacamahaca (Wei et al., 2010). Moreover, based on the different combinations of plastid genomes (rbcL-a, psbI-psbK, psbA-trnH, and trnL-trnF), the phylogenetic relationship of 63 Populus individuals representing 32 species from five sections suggests that sections Populus and Leucoides formed a separate clade, while section Tacamahaca was divided into two groups, albeit with very weak bootstrap support (Yun et al., 2015). Although previous molecular systematic studies have been conducted, they were unable to solve the phylogenic relationships of Populus well. To reconstruct a more reliable phylogeny of Populus, it was necessary to sample more taxa, as well as to use more variable markers and genome sequences to resolve the phylogenetic problems at a lower taxonomic level. In addition, the most recent molecular studies of Populus have not explicitly analyzed morphology, therefore, a combined analysis including both molecular data and morphological data. Here, 24 newly sequenced Populus plastomes, sixteen Populus, fifteen Salix, and four previously reported plastomes and one partial Chosenia chloroplast genome sequence, all of which represent seven genera of the Salicaceae family, were used to reconstruct phylogenetic relationships.
Twenty-four taxa, P. adenopoda, P. alba, P. bolleana, P. cathayana, P. davidiana, P. deltoides, P. euphratica, P. gonggaensis, P. haoana, P. hopeiensis, P. kangdingensis, P. lasiocarpa, P. pseudoglauca, P. qamdoensis, P. rotundifolia var. duclouxiana, P. schneideri, P. tomentosa, P. tomentosa narrow crown (P. tomentosa (NC)), P. simonii, P. deltoides × nigra, P. szechuanica, P. szechuanica var. tibetica, P. trinervis, and P. yunnanensis, representing the genus Populus of the family Salicaceae, were sampled following the taxonomy system of Eckenwalder (1996) and the Flora of China (Fang et al., 1999; Wu, 1999). We collected healthy, tender and fresh leaves from adult plants of target species. Details of the samples are given in Table 1. The voucher herbarium specimens for the 24 sampled Populus taxa were deposited at Southwest Forestry University (SWFU), Kunming, China.
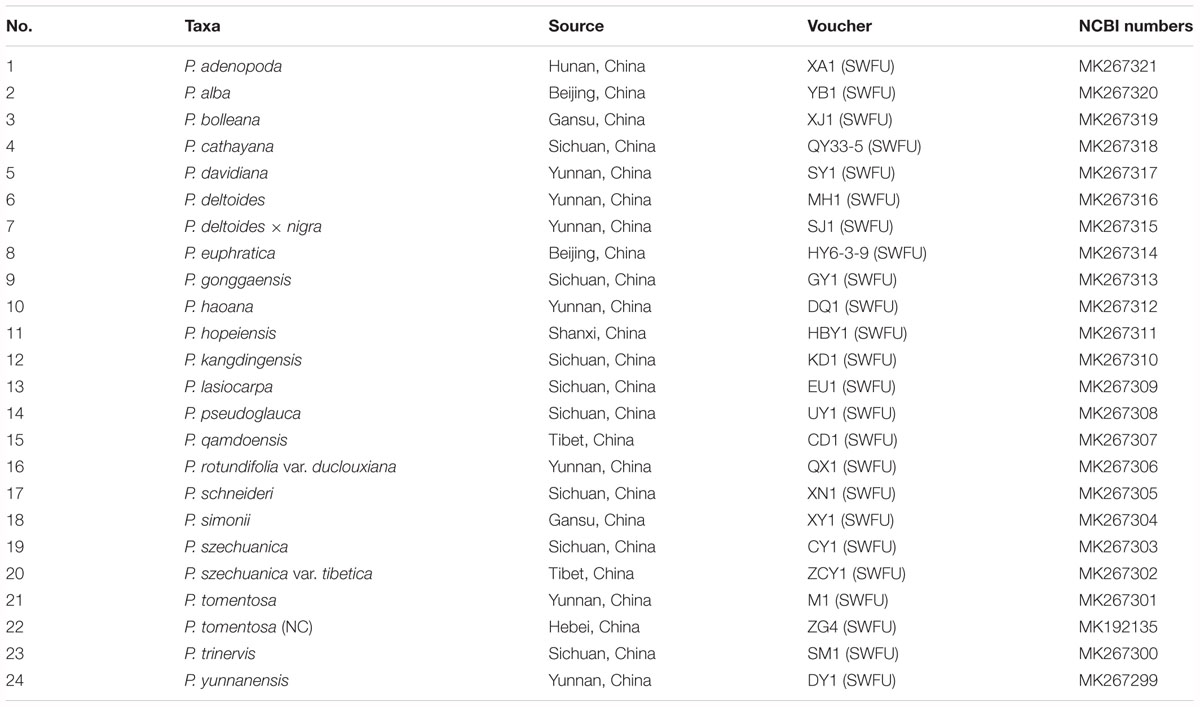
Table 1. List of samples, voucher collection information and GenBank accession numbers.
Total genome DNA was extracted with the Ezup plant genomic DNA prep Kit (Sangon Biotech, Shanghai, China). DNA samples were properly deposited in Key Laboratory of State Forestry Administration on Biodiversity Conservation in Southwest China, SWFU, Kunming. Total DNA was used to generate libraries with an average insert size of 400 bp and sequences using the Illumina HiSeqX platform. Approximately 15.0 GB of raw data were generated with 150 bp paired-end read lengths. Then, the raw data were used to assemble the complete chloroplast genome using GetOrganelle software (Jin et al., 2018) with P. trichocarpa as the reference. Genome annotation was performed with the program Geneious R8 (Biomatters Ltd., Auckland, New Zealand), and the start and stop codons were manually adjusted by comparison with the cp genome of P. trichocarpa. The tRNA genes were further confirmed using online tRNAscan-SE web servers (Schattner et al., 2005). The gene map of the annotated Populus chloroplast genome was drawn by OGdraw online (Lohse et al., 2007).
From NCBI GenBank, sixteen plastome sequences of Populus [P. angustifolia (GenBank accession number: MG262345), P. balsamifera (GenBank accession number: KJ664927), P. fremontii (GenBank accession number: KJ664926), P. ilicifolia (GenBank accession number: KX421095), P. koreana (GenBank accession number: MG262348), P. laurifolia (GenBank accession number: MG262350), P. mexicana (GenBank accession number: MG232351), P. nigra (GenBank accession number: KX377117), P. pruinosa (GenBank accession number: MG262355), P. qiongdaoensis (GenBank accession number: KX534066), P. rotundifolia (GenBank accession number: KX425853), P. tremula (GenBank accession number: KP861984), P. tremula × alba (GenBank accession number: KT780870), P. trichocarpa (GenBank accession number: EF489041), P. wilsonii (GenBank accession number: MG214781)], P. xiangchengensis (GenBank accession number: MH910611) reported in our previous study (Zong et al., 2019), and fifteen plastomes of Salix [S. arbutifolia (GenBank accession number: KX781246), S. babylonica (GenBank accession number: KT449800), S. chaenomeloides (GenBank accession number: MG262362), S. hypoleuca (GenBank accession number: MG262363), S. interior (GenBank accession number: KJ742926), S. magnifica (GenBank accession number: MG262364), S. minjiangensis (GenBank accession number: MG262365), S. oreinoma (GenBank accession number: MF189168), S. paraplegia (GenBank accession number: MG262366), S. purpurea (GenBank accession number: KP019639), S. rehderiana (GenBank accession number: MG262367), S. rorida (GenBank accession number: MG262368), S. suchowensis (GenBank accession number: KM983390), S. taoensis (GenBank accession number: MG262369) and S. tetrasperma (GenBank accession number: MF189169)] were collected. MAFFT version 7 software was used to align the 40 plastome sequences of Populus, fifteen plastomes of Salix, and all 55 plastomes, respectively (Katoh and Standley, 2013). After manual adjustment with MEGA 5 (Tamura et al., 2011), sliding window analysis was performed to assess the variability (Pi) of the whole plastomes in DnaSP version 5 software (Librado and Rozas, 2009). The window length was set to 600 bp, and the step size was set to 200 bp.
To estimate phylogenetic relationships within the Populus, 59 taxa with available complete plastid genomes and one taxa (Chosenia arbutifolia) with seven combined chloroplast fragments [matK (GenBank accession number: EU790701), ndhF (GenBank accession number: AY757181), rbcL-atpB (GenBank accession number: FJ788535), trnL (GenBank accession number: GQ244791), trnD-trnT (GenBank accession number: FJ788620), rbcL (GenBank accession number: AB012776), and trnL-F (GenBank accession number: AY757062)] were compared, with 40 taxa from Populus, fifteen taxa from Salix and four taxa from four genera that are members of the Salicaceae family, Flacourtia indica (GenBank accession number: MG262341), Idesia polycarpa (GenBank accession number: KX229742), Itoa orientalis (GenBank accession number: MG262342), and Poliothyrsis sinensis (GenBank accession number: MG262343) were sampled as outgroups. The 40 Populus genomes comprised the 24 new plastomes and sixteen previously published complete plastomes adopted from the NCBI (Chen et al., 2016; Wang et al., 2016; Zheng et al., 2016; Zong et al., 2019). The complete genome matrix was aligned using MAFFT version 7 software (Katoh and Standley, 2013) and then manually edited using MEGA 5 (Tamura et al., 2011). The matrix of 60 chloroplast genome sequences included 180,546 characters, 4,425 of which were parsimary informative sites. Based on these parsimary informative sites, we performed maximum likelihood and Bayesian inference analyses. A maximum likelihood method for phylogenetic analysis was performed based on the GTR+I+G model in the RAxML version 8 with 1000 bootstrap replicates (Stamatakis, 2014). Maximum likelihood (ML) bootstrap support values (BS) ≥70% were considered well supported, and ML BS <50% were considered poorly supported or unresolved. Bayesian inference (BI) was performed using MrBayes 3.1.2 (Ronquist and Huelsenbeck, 2003). jModelTest 2.0 (Darriba et al., 2012) was used to determine the best-fitting model for each dataset based on the Akaike information criterion (AIC), and the optimal model “GTR+F+R5” (freqA = 0.313, freqC = 0.187, freqG = 0.180, freqT = 0.320, R(a) [AC] = 0.949, R(b) [AG] = 1.621, R(c) [AT] = 0.916, R(d) [CG] = 0.552, R(e) [CT] = 1.621, R(f) [GT] = 1.000) was selected. The Markov chain Monte Carlo (MCMC) algorithm was run for 1000,000 generations, and a burn-in of 25% was used for the analysis. Internodes with posterior probability (PP) values ≥0.95 were considered statistically significant.
The classification of Populus was based on the Flora of China (Fang et al., 1999; Wu, 1999), and 40 taxa representing the genus of Populus were sampled. Morphological information was collected from personal observations of live plants, herbarium specimens, and the available literature (Fang et al., 1999; Argus, 2010), which were evaluated for eighteen morphological characters: (1) bark smoothness; (2) bark color; (3) branchlet; (4) bud; (5) bud size; (6) bud viscid; (7) petiole vs. leaf; (8) petiole shape; (9) petiole; (10) petiole glands; (11) leaf presence in short branchlet; (12) leaf apex; (13) leaf margin; (14) leaf base; (15) leaf; (16) leaf in inflorescence; (17) male anther length; and (18) male anther tip (Table 2), and a matrix of eighteen discrete morphological characters was constructed for all of the taxa investigated (Table 3).

Table 2. Character coding for morphological analysis.
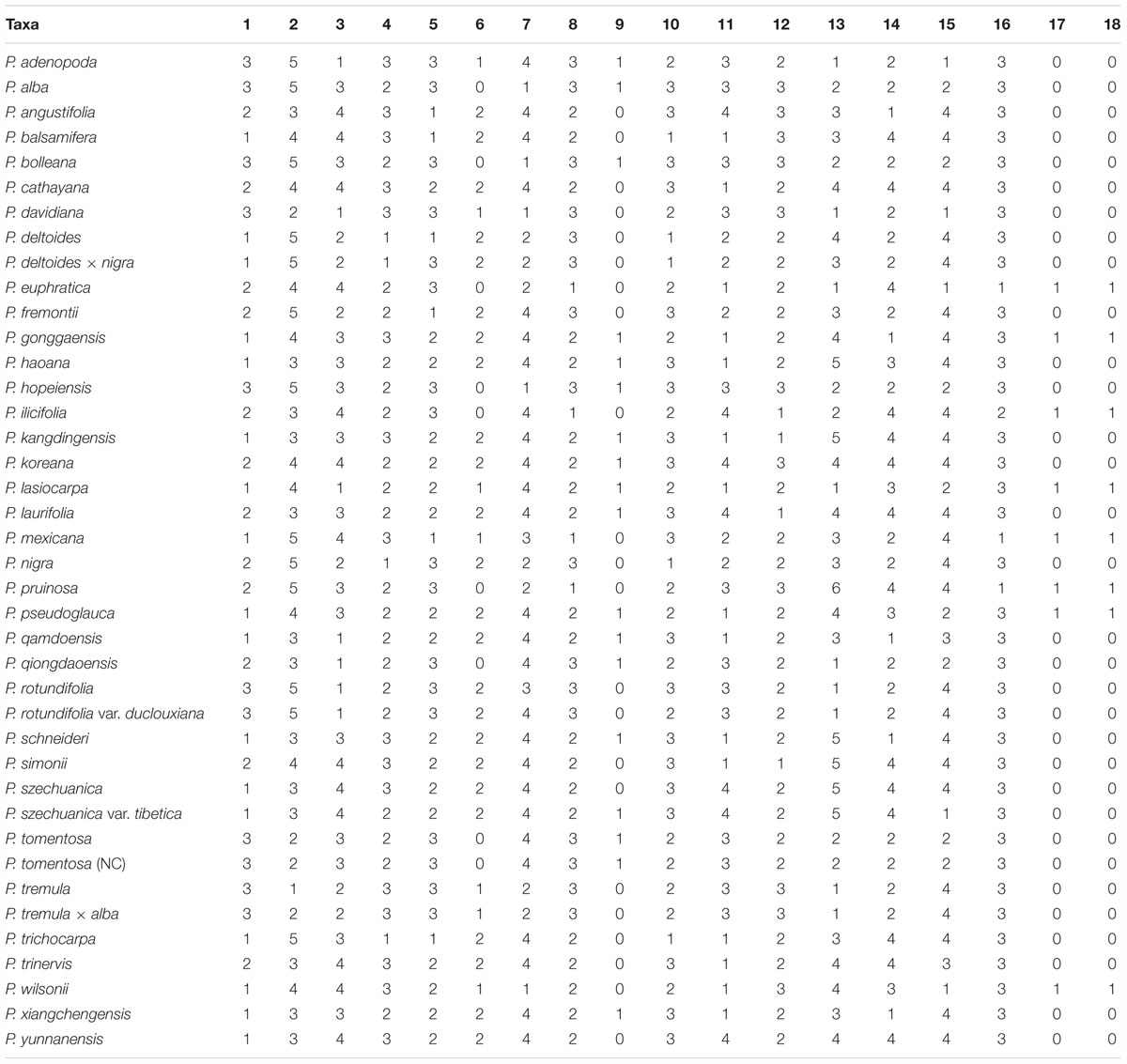
Table 3. Data matrix of morphological characters in the study.
To evaluate the morphological characters supporting relationships based on molecular data and to evaluate whether the investigated characters were phylogenetically conserved at the level of the whole phylogeny, we calculated mean Pagel’s lambda (λ) (Pagel, 1999) and Blomberg’s K values (Garland et al., 1992) at the species level for each character, thus obtaining phylogenetic information. Both indices assume the classic Brownian motion (BM) evolutionary model, with values varying from zero to one for λ and from zero to higher than one for K. λ values close to zero indicate there is no phylogenetic signal (the traits have evolved independently of phylogeny, and the traits of close relatives are not more similar than those of distant relatives), and λ values close to one indicate trait evolution according to BM. K-values close to zero indicate the phylogenetic signal is weaker than expected from the BM model of character evolution (low levels of phylogenetic character conservation). K-values close to or higher than one indicate a strong phylogenetic signal (Molina-Venegas and Rodríguez, 2017).
The significance of phylogenetic signals was determined by shuffling species’ character values (999 times) across the tips of the phylogenetic tree and comparing the resulting K-values to those computed from the observed character data (Eichenberg et al., 2015), whereas the statistical significance of λ was assessed based on a comparison with the likelihood of a model that assumes complete phylogenetic independence (Pagel, 1999). The Bayesian tree based on the complete chloroplast genome sequences provided the standard tree topology. Phylogenetic signal analyses were carried out using the routines provided in the picante package available for R (Kembel et al., 2010).
In this study, we determined the structure characteristics and gene contents of the complete plastid genomes of 24 Populus taxa. The cp genome lengths of the 24 Populus taxa ranged from 155,177 bp for P. rotundifolia var. duclouxiana to P. euphratica for 157,839 bp (Figure 1). All of these assembled into single circular, double-stranded DNA sequences, presenting a typical quadripartite structure, including one LSC with a length of 85,858 bp (P. euphratica) to 84,450 bp (P. rotundifolia var. duclouxiana), one SSC with a length of 16,421 bp (P. szechuanica var. tibetica) to 16,879 bp (P. hopeiensis), and a pair of IR with a length of 26,903 bp (P. hopeiensis) to 27,672 bp (P. cathayana) (Table 4). All 24 chloroplast genomes contained 130 genes, of which 112 were unique and eighteen were duplicated in IR regions. Among the 112 unique genes, 78 protein-coding genes, 30 tRNA genes and four rRNA genes were identified. Six protein-coding genes (atpF, ndhA, ndhB, petB, rpl2, and rpoC1) and six tRNA genes (trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC) had a single intron, while three protein-coding genes (ycf3, clpP, and rps12) possessed two introns (Supplementary Table S1). The GC content of these plastomes ranged from 36.5% in P. euphratica to 36.8% in P. tomentosa (NC), P. tomentosa, P. alba, P. bolleana, P. davidiana, P. rotundifolia var. duclouxiana, and P. hopeiensis. The GC content of the LSC region (34.2–34.6%) and SSC region (30.4–30.7%) was lower than that of the IR regions (41.9–42.3%). The high GC percentage of the IR regions was possibly due to the presence of four rRNA genes in these regions.
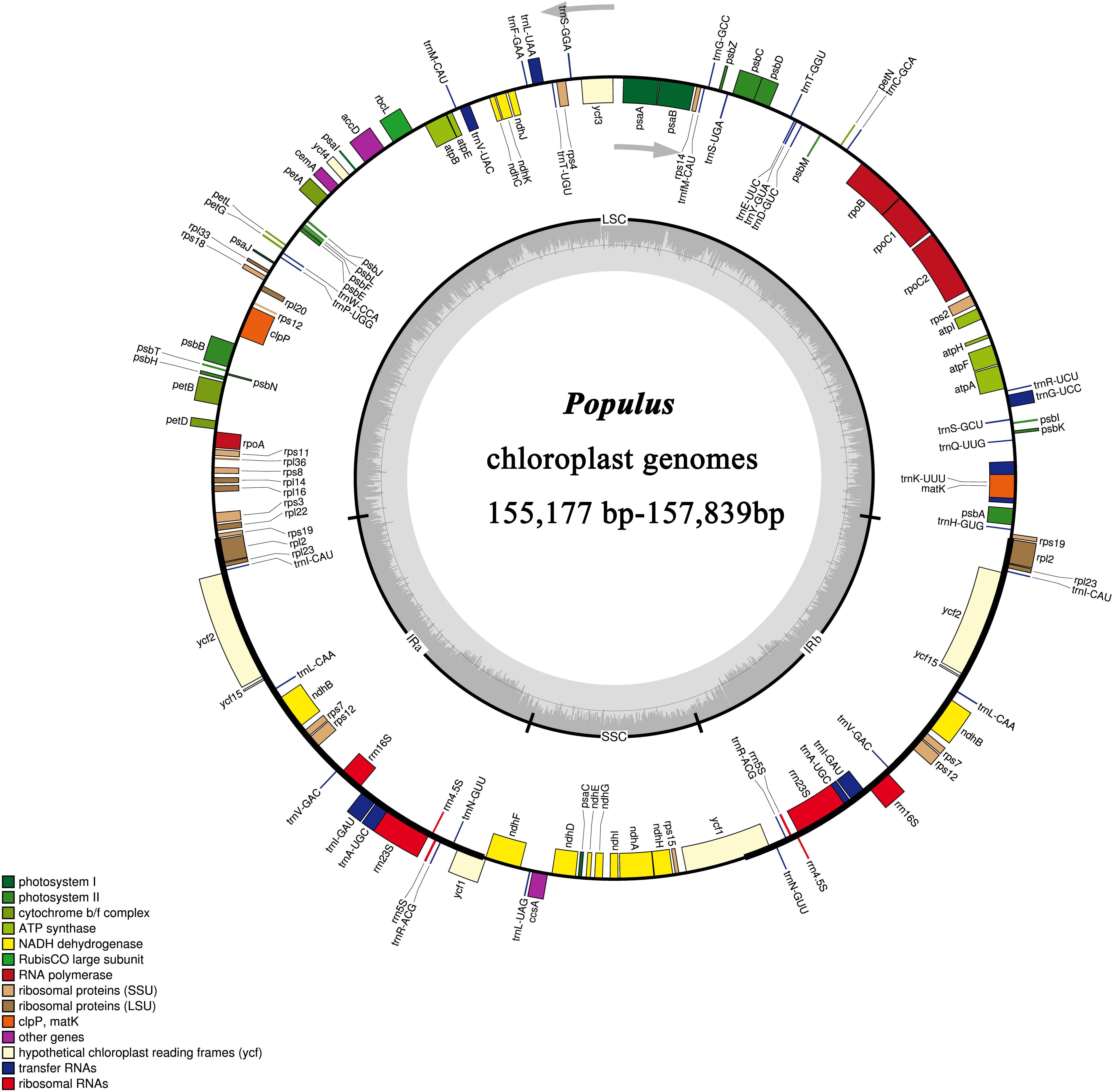
Figure 1. Gene map of 24 Populus chloroplast genomes. Genes inside the circle are transcribed clockwise and genes outside the circle are transcribed counter clockwise.
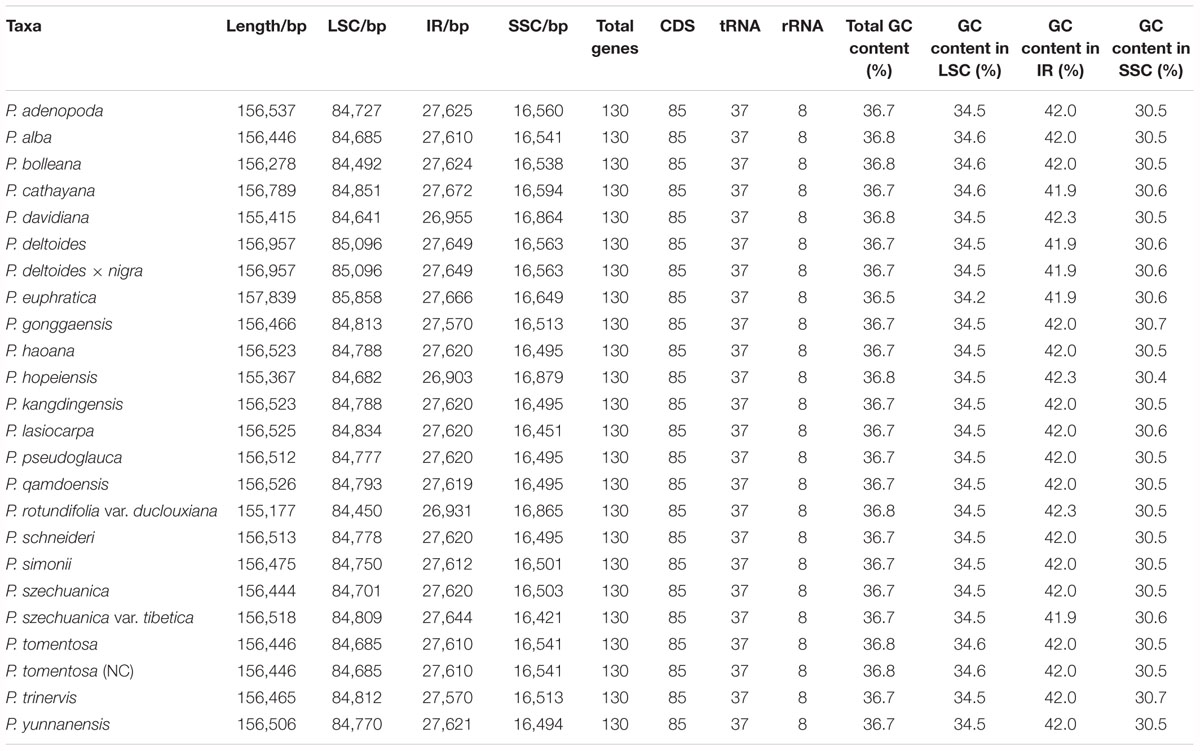
Table 4. Summary of complete chloroplast genomes for 24 Populus species.
The expansion and contraction of the IR region and the single copy (SC) boundary regions is considered a primary mechanism causing the length variation of angiosperm cp genomes (Kim and Lee, 2005). Although the overall genomic structure, including gene number and gene order, was well conserved among the Populus plastid genomes, these genomes exhibited obvious differences in the IR/SC boundary regions. The IR regions of the Populus plastid genomes ranged from 26,903 bp (P. hopeiensis) to 27,672 bp (P. cathayana) in size, and two complete or fragmented copies of rpl22 and ycf1 were located at the boundaries between the LSC or SSC regions and IRs regions in the 24 Populus plastomes. The full lengths of the rpl22 and ycf1 genes were 399 and 5442 to 5472 bp, respectively. The rpl22 gene crossed the IR-LSC boundary, with only 1 bp variation in sequence length among the Populus plastomes. The gene trnH in the LSC region contracted by 14 bp from the junction region of the IR-LSC boundary in four taxa, i.e., P. alba, P. bolleana, P. tomentosa, and P. tomentosa (NC), while the other taxa contracted by 3 bp. In addition, rps19 in the IRa region also contracted by a different number of bases (200–242 bp) among taxa. Gene ycf1 in the IRb region extended from 15 to 170 bp, whereas gene ycf1 in the IRa region extended 979 to 1725 bp (Figure 2).
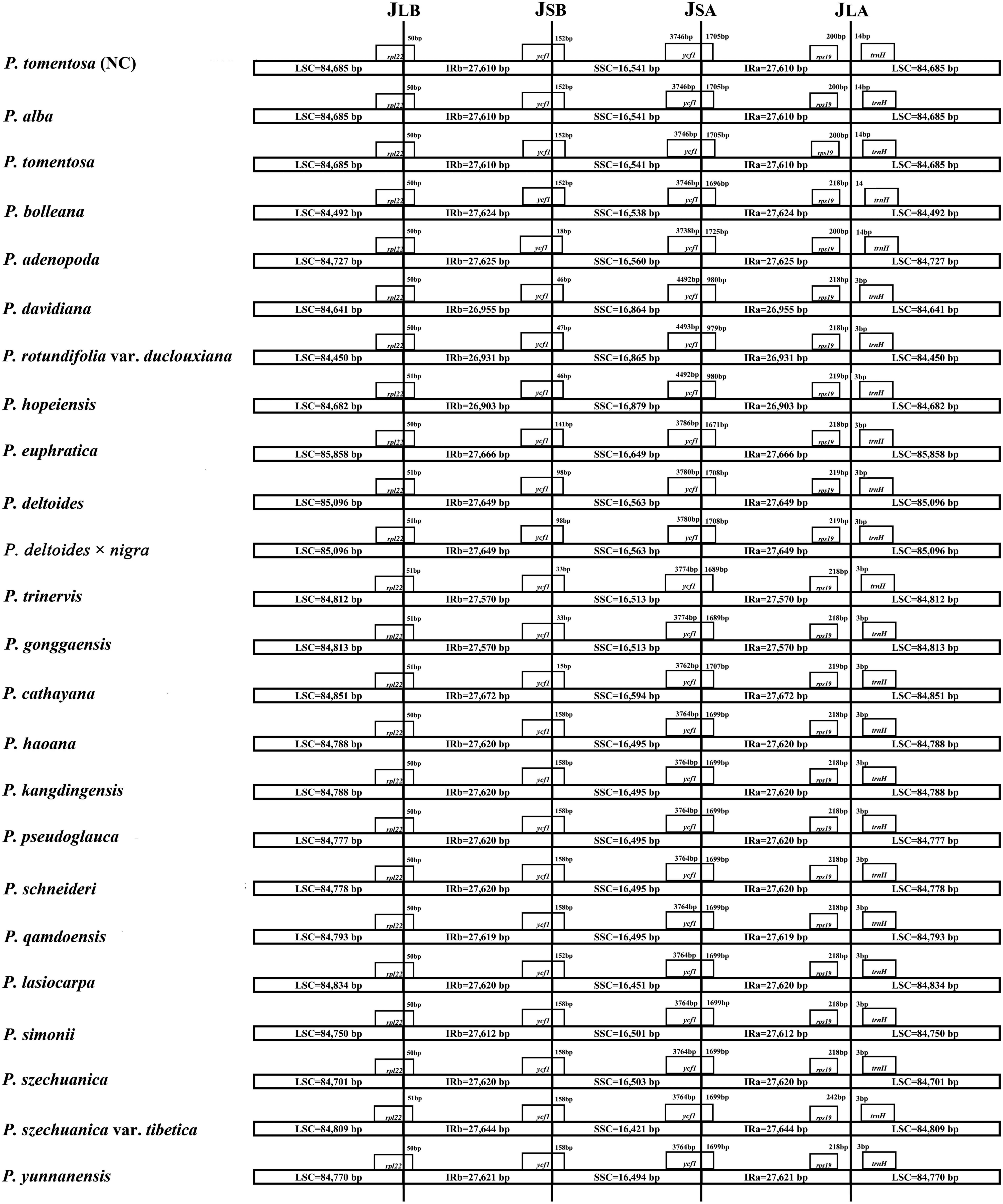
Figure 2. Comparison of the borders of the LSC, SSC, and IR regions of 24 Populus plastomes.
To investigate the levels of sequence divergence, the nucleotide variability (Pi) values within 600 bp in 40 Populus plastomes were calculated with DnaSP software. The nucleotide variability values within these 600 bp varied from 0 to 0.00713, with a mean of 0.00359, among the 40 plastomes (Supplementary Table S2). The pairwise nucleotide divergence between two of the 40 plastomes varied from 0.000 to 0.003 (Supplementary Table S3). Both results indicate high sequence similarity across the 40 chloroplast genomes, suggesting that the plastomes of Populus are highly conserved. All aligned sequences demonstrate surprising low divergences, with only ten regions displaying high variation (Pi > 0.008). The ten most dissimilar regions of the 40 plastomes were trnK intron + trnK-psbK, rpoB-petN, psbM-trnD, psbZ-trnfM, trnL-ndhJ, ndhC-trnV, ycf1, ndhF-ccsA, ccsA + ccsA-ndhD, and rps15-ycf1 (Figure 3A). Among these regions, six (trnK intron + trnK-psbK, rpoB-petN, psbM-trnD, psbZ-trnfM, trnL-ndhJ, and ndhC-trnV) were located in the LSC region, and four were located in the SSC region (ycf1, ndhF-ccsA, ccsA + ccsA-ndhD, and rps15-ycf1). In addition, eight hotspot regions were found (Pi > 0.012) in the comparison of fifteen Salix plastomes. These eight hotspot regions were trnK- trnQ, rpoB-petN, psbZ-trnfM, psaA-ycf3 + ycf3 intron, trnL intron + trnL-ndhJ, ndhC-trnV, ndhF, ccsA + ccsA-ndhD (Figure 3B). Twelve hotspot regions (trnH-trnK, psbK-atpA, atpH-atpI, rpoC1 intron, rpoB-psbM, psbM-trnE, trnE-psbD, trnL intron + trnL-ndhJ, ndhC-trnV, petA-psbF, and ycf1) were shared between Populus and Salix (Pi > 0.02) (Figure 3C).
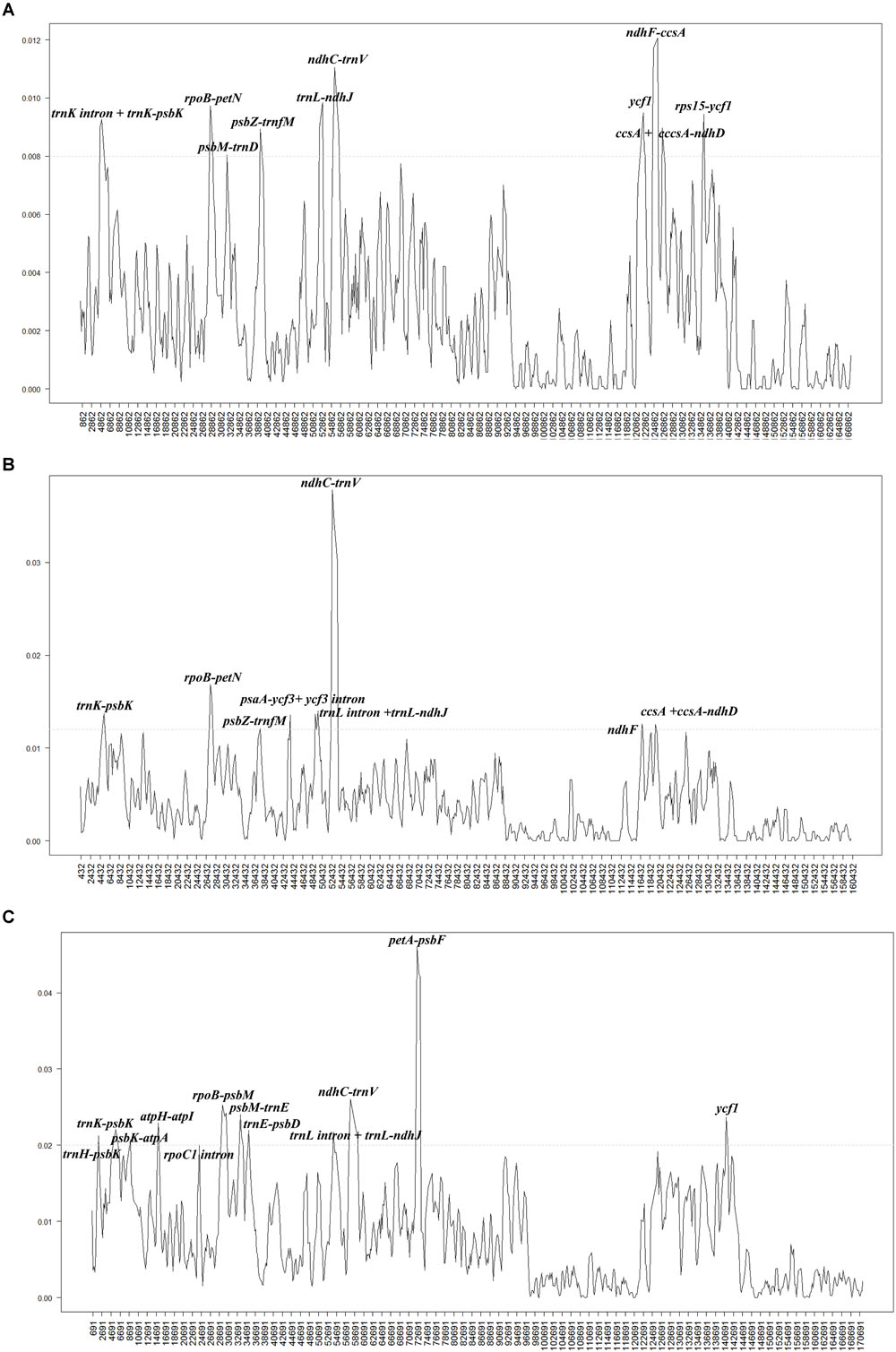
Figure 3. Comparison of the nucleotide variability (Pi) values of the plastomes: (A) 40 plastomes of Populus, (B) fifteen plastomes of Salix, and (C) 55 plastomes of Populus and Salix. The genetic divergence was calculated with DnaSP 5.0 software (window length: 600 bp and step size: 200 bp). X-axis: position of the midpoint of a window, Y-axis: nucleotide diversity of each window.
The matrix of the complete plastomes was used to reconstruct a phylogenetic tree of Populus and Salix (Figures 4, 5). Populus and Salix were two independent main clades (BS = 100% and PP = 1). C. arbutifolia was nested within the genus Salix, while the sisterhood of Populus and Salix was highly supported. Five well-supported clades were recovered within Populus (BS > 90% and PP = 1). Clade I (BS = 90% and PP = 1) comprised three taxa: P. euphratica, P. ilicifolia and P. pruinosa. Clade II (BS = 100% and PP = 1) contained thirteen plastomes for the taxa P. adenopoda, P. alba, P. bolleana, P. davidiana, P. hopeiensis, P. nigra, P. qiongdaoensis, P. rotundifolia, P. rotundifolia var. duclouxiana, P. tremula, P. tremula × alba, P. tomentosa, and P. tomentosa (NC). Clade III (BS = 100% and PP = 1) contained ten taxa: P. haoana, P. kangdingensis, P. lasiocarpa, P. pseudoglauca, P. qamdoensis, P. schneideri, P. simonii, P. szechuanica, P. szechuanica var. tibetica, and P. yunnanensis. Clade IV (BS = 97% and PP = 1) included the taxa P. cathayana, P. gonggaensis, P. koreana, P. laurifolia, P. trinervis, P. wilsonii, and P. xiangchengensis. The last clade (BS = 97% and PP = 1) was made up of P. angustifolia, P. balsamifera, P. deltoides, P. deltoides × nigra, P. fremontii, P. mexicana, and P. trichocarpa.
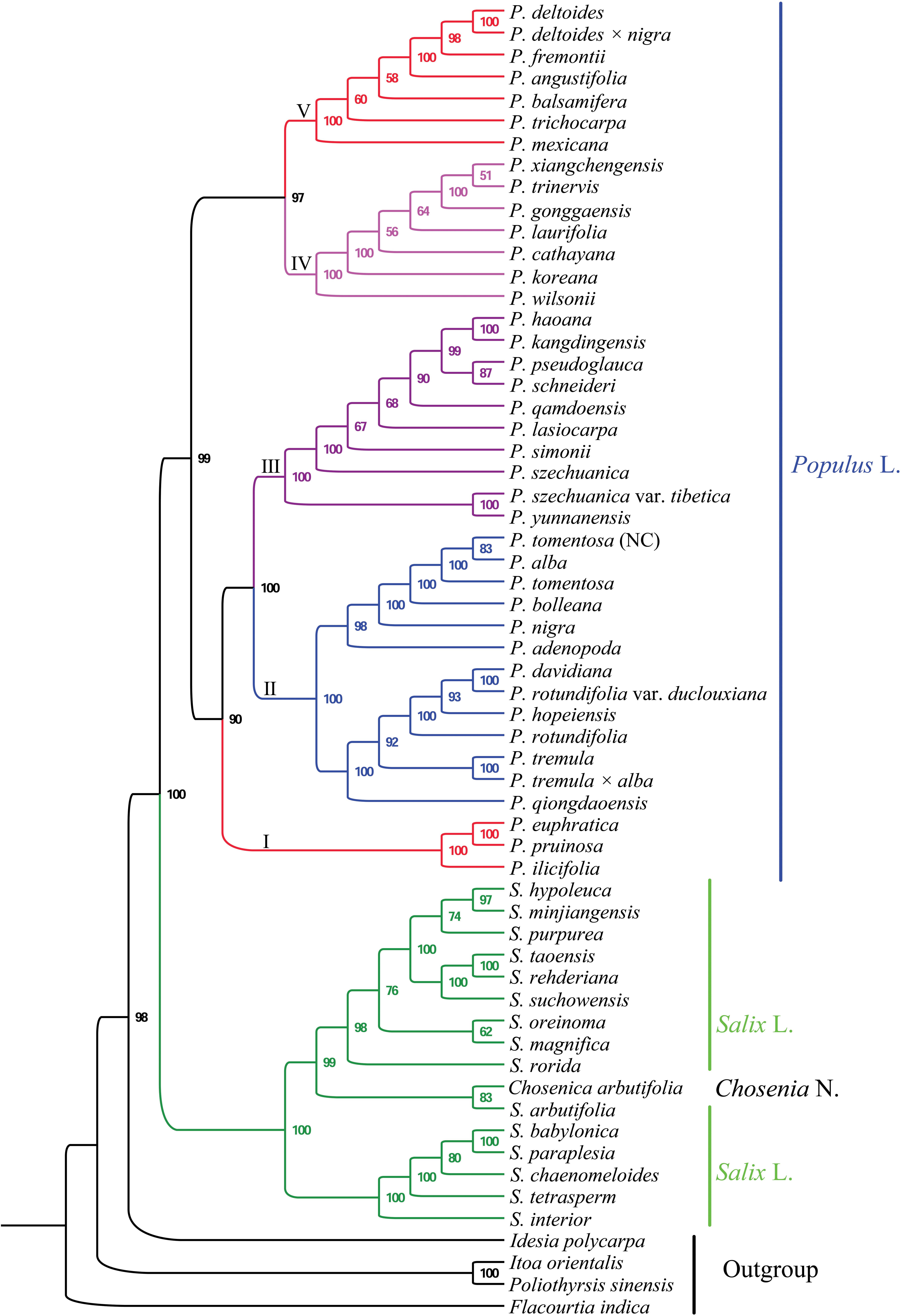
Figure 4. Phylogenetic tree reconstructions of Populus using maximum likelihood (ML) based on whole chloroplast genome sequences.
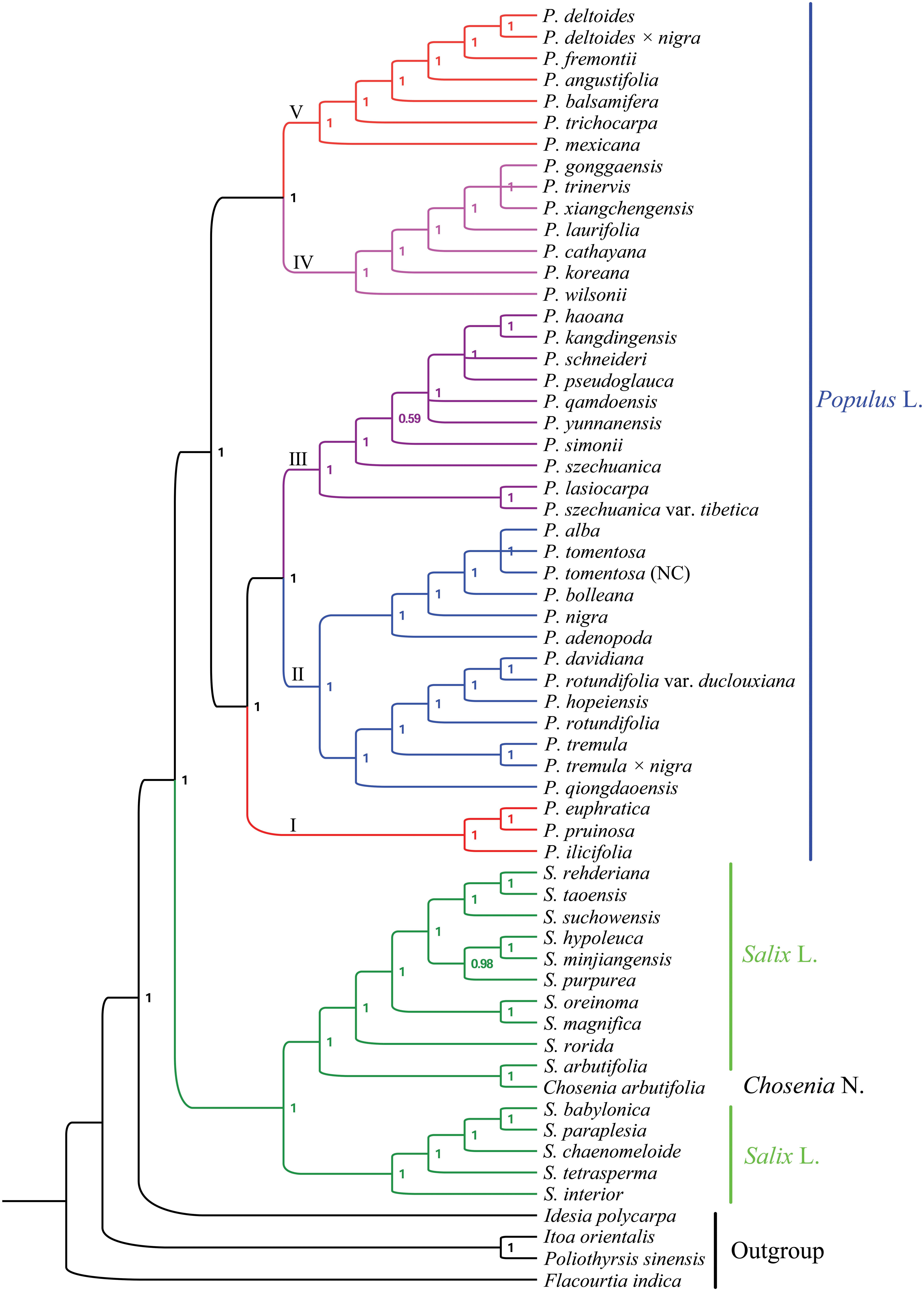
Figure 5. Phylogenetic tree reconstructions of Populus using Bayesian inference (BI) based on whole chloroplast genome sequences.
Analysis of morphological characters showed that six characters, i.e., bark smoothness (λ = 8.340867e-01, P < 0.001; K = 1.0480, P < 0.001), bud size (λ = 9.076553e-01, P < 0.001; K = 0.72461, P < 0.001), petiole shape (λ = 9.817749e-01, P < 0.001; K = 1.40960, P < 0.001), leaf in inflorescence (λ = 6.208698e-01, P < 0.001; K = 0.58131, P = 0.08759), anther length (λ = 4.917771e+00, P < 0.001; K = 1.50515, P < 0.001), and anther tip (λ = 4.917771e+00, P < 0.001; K = 1.50515, P < 0.001), were highly congruent with the molecular phylogenies (Table 5).
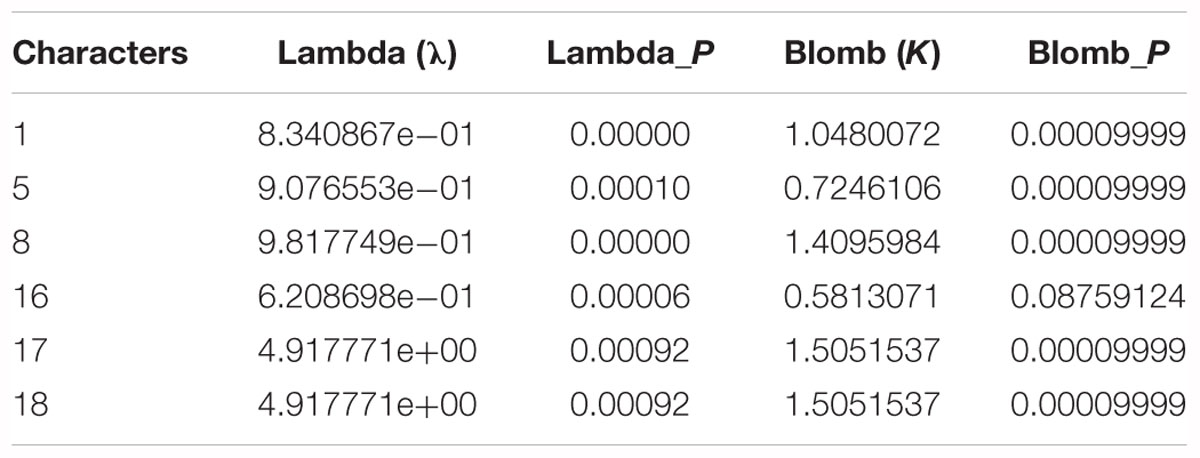
Table 5. Six characters test well congruent with molecular phylogenies.
Complete plastid genomes are valuable sources of genetic markers for phylogenetic analyses because of their highly conserved genome structure (Provan et al., 2001; Chaney et al., 2016). Although the plastid genome generally has a nearly collinear gene order, changes in the genome, such as sequence inversion and gene expansion or contraction at the boundary of the SC and IR regions, occur over the course of evolution (Cho et al., 2015; Choi et al., 2016). Our results showed that the genome size, gene order, and compositions of the 24 newly sequenced Populus genomes were very similar with each other and with previously sequenced Populus plastid genomes; the genome sizes ranged from 155,177 to 157,839 bp (Chen et al., 2016; Wang et al., 2016; Zheng et al., 2016; Han et al., 2017). Among the genomes, three plastomes of P. alba, P. tomentosa, and P. tomentosa (NC), two plastomes of P. haoana and P. kangdingensis, two plastomes of P. trinervis and P. xiangchengensis, and two plastomes of P. deltoides and P. deltoides × nigra, each had the same length. All of the newly examined chloroplast genomes of Populus contained more AT than GC contents, with values ranging from 36.5 to 39.8%, and the GC contents of the IR regions were higher than those of the SC regions, possibly due to the presence of rRNA genes. Furthermore, we found that all 24 Populus plastomes encoded 85 protein-coding genes, 37 tRNA genes and eight rRNA genes, and eighteen of these genes were duplicated within the IRs. Moreover, the total intron numbers in the plastome were the same among the 24 Populus genomes, and in each plastome, twelve genes contained one intron, and three genes contained two introns.
The IR/LS boundary regions of the 24 complete Populus cp genomes were compared and showed slight differences in the junction positions. The rpl22 gene crossed the LSC/IRb boundary with only one bp variation in sequence length among the Populus plastomes. The gene trnH was located in the LSC region of all genomes but contracted by 3 or 14 bp from the LSC/IRa junction region, with a contraction of 14 bp in the plastomes of P. alba, P. bolleana, P. tomentosa, and P. tomentosa (NC) and a contraction of 3 bp in the other taxa. Gene ycf1 in the IRb region extended from 15 to 170 bp, whereas gene ycf1 in the IRa region extended 979 to 1725 bp. The length changes in the truncated ycf1 gene directly drive the contraction of the IR regions in the plastome of Populus.
Although the length of the truncated ycf1 gene varied and the divergence of the unbroken ycf1 gene was high among the Populus taxa, the nucleotide variability of the whole plastome among these taxa was only 0.36%, which was approximately the same as the nucleotide variability of eight plastomes of Alseodaphne gracilis, A. huanglianshanensis, A. semecarpifolia, avocado, Cinnamomum micranthum, Machilus balansae, Phoebe sheareri, and P. omeiensis (0.30%) (Song et al., 2015, 2016, 2017, 2018) and was much lower than the sequence divergence among six Cymbidium species (3.70%) (Yang et al., 2013) and five Epimedium species (3.97%) (Zhang et al., 2016). In previous studies, trnL-trnF, rbcL-a, psbI-psbK, psbA-trnH, and ITS sequences did not completely resolve the phylogenetic relationships in the genus of Populus at the molecular level (Hamzeh and Dayanandan, 2004; Wei et al., 2010; Yun et al., 2015). Further work is still necessary to determine whether these ten variable loci (trnK intron + trnK-psbK, rpoB-petN, psbM-trnD, psbZ-trnfM, trnL-ndhJ, ndhC-trnV, ycf1, ndhF-ccsA, ccsA + ccsA-ndhD, and rps15-ycf1) could be used in Populus phylogenetic analyses or as excellent candidate markers for population genetic and phylogenetic analyses.
Most previous studies have used sequences from only one or more chloroplast loci in Salicaceae (Hamzeh and Dayanandan, 2004; Wei et al., 2010; Yun et al., 2015). However, the section delimitation and species relationships within Populus remain unclear. Our study included 60 plastid genomes for plants from seven genera of Salicaceae. All of these plastome sequences of Populus and related genera yielded a fully resolved tree, consistent with the Angiosperm Phylogeny Group’s most recent phylogeny, APG IV (Chase et al., 2002; Byng et al., 2016). In addition, two monophyletic genera: Populus and Salix, were strongly supported, and Chosenia taxa were nested within the genus Salix, which is consistent with previous studies (Leskinen and Alström-Rapaport, 1999; Chase et al., 2002). The results suggested that Populus is a monophyletic sister group to Salix.
Forty Populus taxa were separated into the following five clades in our study. Section Turanga in the study of Wu (1999) formed the first clade, including P. euphratica, P. ilicifolia, and P. pruinosa, in our phylogeny, which share the same character of micro flat petiole. Section Populus in the study of Wu (1999) formed the second clade, and the taxa in this clade shared the same character of flat petiole in contrast to other taxa (Figure 6). The phylogenetic placements of the first and second clades were consistent with previously published phylogenetic relationships (Li et al., 2007; Wei et al., 2010). The results of Li et al. (2007) showed that section Turanga and section Populus can be divided into two independent groups based on AFLP markers, and the trnL-F sequence analysis also supported section Populus as a separate clade (Wei et al., 2010). In our morphology analysis, sections Turanga and Populus shared the same characters of bud size, but the characters of bark smoothness, petiole shape and leaf in inflorescence were different (Figure 6). Therefore, we can distinguish the two sections according to the three characters. Interestingly, our phylogenomic analysis showed that P. nigra, which was classified in section Aigeiros in the study of Wu (1999) is nested among the members of section Populus. The character of bud size different between P. nigra and P. deltoides (section Aigeiros), but the characters of small bud, flat petiole, no leaf in inflorescence, short anther length and cuneate anther tip were similar in section Populus. Therefore, the plastid data and morphology characteristics both revealed that P. nigra had a close affinity to species of section Populus, which supported the possibility of ancient hybridization by which P. nigra appeared to be an introgressant of the P. alba lineage and some other presently unknown paternal lineage of section Populus (Smith, 1988; Hamzeh and Dayanandan, 2004).

Figure 6. Morphological character reconstruction of Populus on the Bayesian phylogenetic tree.
Section Tacamahaca in the study of Wu (1999) contained nearly 60 species and varieties that share the same characters of petiole eglandular, flower disk, and capsule usually glabrous or rarely pilose (Fang et al., 1999; Song, 2006). In our analysis, the members of section Tacamahaca were divided into clade III, clade IV and clade V, and the three clades shared the same character of a tetrete petiole. Clade III contained P. haoana, P. kangdingensis, P. qamdoensis, P. schneideri, P. simonii, P. szechuanica, P. szechuanica var. tibetica, P. yunnanensis, and the taxa of P. lasiocarpa and P. pseudoglauca, which had previously been retrieved as members of section Leucoides in the study of Wu (1999). Clade IV included the taxa P. cathayana, P. koreana, P. laurifolia, P. trinervis, P. xiangchengensis, P. gonggaensis and P. wilsonii, but the last two taxa were classified in section Leucoides. Clade V included P. angustifolia, P. balsamifera, P. trichocarpa, P. deltoides, P. deltoides × nigra, P. fremontii, and P. mexicana. All of the taxa in clade V had the character of a middle bud unlike the other two clades. Section Aigeiros in the study of Wu (1999), including P. deltoides, P. deltoides × nigra, and P. fremontii, was nested within clade V and shared the same characters of a middle bud, no leaf in inflorescence, short anther length and cuneate anther tip with the taxa of section Tacamahaca (Figure 6). Furthermore, the character of shallowly furrowed bark in P. fremontii was shared with P. cathayana, P. koreana, P. laurifolia, and P. trinervis which were members of section Tacamahaca. Therefore, our analysis did not support section Aigeiros as a sister section of sections Populus, Tacamahaca and Turanga. Curiously, P. deltoides × nigra was placed among the species of section Aigeiros, but the character of bud size was inherited from the parent P. nigra unlike the other members in clade V. Meanwhile, P. mexicana shared the four characters of petiole shape, leaf in inflorescence, male anther length and tip shape with section Turanga. Therefore, we speculated that P. mexicana may be caused by ancient hybridization or chloroplast capture from excluded samples or now-extinct North America poplar, similar to previous studies on two North America species P. trichocarpa and P. balsamifera (Huang et al., 2014; Liu et al., 2017).
Our analysis showed that clade V was a sister clade to clade IV. Clade V includes seven species (P. angustifolia, P. balsamifera, P. deltoides, P. deltoides × nigra, P. fremontii, P. mexicana, and P. trichocarpa) that are primarily distributed in North America. Clade IV comprises seven species that mainly exist within Eurasia. The phylogenetic relationships between clade IV and clade V matched well with the morphology data, such as bud size, although not well with smooth bark and petiole shape (Figure 6). The phylogenetic relationships between clade IV and clade V supported the hypothesis regarding the migration of species from Asia to North America (Tiffney and Manchester, 2001; Milne, 2006; Liu et al., 2017). The authors who proposed this hypothesis speculated that a common ancestor first appeared in North America and then dispersed to other continents via the North Atlantic Land Bridge and the Bering Land Bridge. Then, with the disappearance of the Bering Land Bridge, new species emerged due to the geographic isolation (Tiffney and Manchester, 2001; Milne, 2006; Liu et al., 2017).
The taxa in clade III and clade IV had an overlapping distribution, such as P. xiangchengensis and P. gonggaensis in clade IV, which were distributed in southwest China, while the taxa of P. schneideri and P. kangdingensis in clade III were also distributed in this area. Clade III and IV showed high similarity in morphology characters (Figure 6), thus complicating their distribution. Liu and Fu (2004) considered P. xiangchengensis a hybridization of P. schneideri and P. pseudoglauca based on morphological characteristics, while another study suggested that P. xiangchengensis was a likely hybrid species of P. kangdingensis and P. pseudoglauca (Wan et al., 2009). P. schneideri was also considered a natural hybrid formed by P. kangdingensis and P. cathayana (Chen et al., 2007; Wang, 2012). The uplift of the Qinghai-Tibet (Q-T) plateau began approximately 40 million years ago (Ma) following the collision of India and Asia (Chung et al., 1998). Some new niches were created by the uplifts, while other plants retreated in response to the climatic changes, thus providing strong opportunities for new hybrid species to emerge (Wang et al., 2005; Liu et al., 2006; Wang and Gilbert, 2007; Lu et al., 2014). Meanwhile, the uplift of the Q-T plateau also induced significant geographical effects (He et al., 2015). These complex factors account for section Tacamahaca’ s presence among the current phylogenetic relationships. Further research using more genetic data and more individuals per species is needed to deeply illuminate the complex relationships in section Tacamahaca in the future.
This study clarified the phylogenetically intergenetic and interspecific relationships of Populus. The genus was confirmed as monophyletic and subdivided into five well-resolved and strongly supported clades. Clade I included three taxa: P. euphratica, P. pruinosa and P. ilicifolia. Clade II contained thirteen plastomes of the taxa P. adenopoda, P. alba, P. bolleana, P. davidiana, P. hopeiensis, P. nigra, P. qiongdaoensis, P. rotundifolia, P. rotundifolia var. duclouxiana, P. tremula, P. tremula × alba, P. tomentosa and P. tomentosa (NC). Clade III included ten taxa: P. haoana, P. kangdingensis, P. lasiocarpa, P. pseudoglauca, P. qamdoensis, P. schneideri, P. simonii, P. szechuanica, P. szechuanica var. tibetica, and P. yunnanensis. Clade IV included P. cathayana, P. gonggaensis, P. koreana, P. laurifolia, P. trinervis, P. wilsonii and P. xiangchengensis. The last clade was made up of P. angustifolia, P. balsamifera, P. deltoides, P. deltoides × nigra, P. fremontii, P. mexicana, and P. trichocarpa. Six morphological characters, including bark smoothness, petiole shape, bud size, leaf in inflorescence, and anther length and tip, agreed well with the molecular phylogenies and can be used to subdivide the Populus genus.
DZ performed the experiments, analyzed the data, wrote the manuscript, and prepared the figures and/or tables. PG performed the experiments and prepared the figures and/or tables. AZ, YZ, and XZ analyzed the data and reviewed drafts of the manuscript. AD, YS, and CH conceived and designed the experiments, reviewed the drafts of the manuscript, and approved the final draft. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31460205, 31860219, and 31360184) and the Forestry Public Benefit Research Program (Grant No. 201104076).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Prof. Wuyuan Yin for his help with sample collection and species identification. We also thank the editor and the two reviewers for their constructive comments, which helped us, improve this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00005/full#supplementary-material
Argus, G. W. (2010). Salix in Flora of North America Editorial Committee. New York, NY: Oxford University Press.
Azuma, T., Kajita, T., Yokoyama, J., and Ohashi, H. (2000). Phylogenetic relationships of Salix (Salicaceae) based on rbcL sequence data. Am. J. Bot. 87, 67–75. doi: 10.2307/2656686
Braatne, J. H., Hinckly, T. M., and Stettler, R. F. (1992). Influence of soil water on the physiological and morphological components of plant water balance in Populus trichocarpa, Populus deltoides and their F1 hybrids. Tree Physiol. 11, 325–340. doi: 10.1093/treephys/11.4.325
Brawdshaw, H. D., Ceulemans, R., Davis, J., and Stettler, R. (2000). Emerging model systems in plant biology: poplar (Populus) as a model forest tree. J. Plant Growth Regul. 19, 306–313. doi: 10.1007/s003440000030
Byng, J. W., Chase, M. W., Christenhusz, M. J. M., Fay, M. F., Judd, W. S., Mabberley, D. J., et al. (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–21. doi: 10.1111/boj.12385
Chaney, L., Mangelson, R., Ramaraj, T., Jellen, E. N., and Maughan, P. J. (2016). The complete chloroplast genome sequences for four Amaranthus species (Amaranthaceae). Appl. Plant Sci. 4:1600063. doi: 10.3722/apps.1600063
Chase, M. K., Zmarzty, S., Lledo, M. D., Wurdack, K. J., Swensen, S. M., and Fay, M. F. (2002). When in doubt, put it in Flacourtiaceae: a molecular phylogenetic analysis based on plastid rbcL DNA sequence. Kew Bull. 57, 141–181. doi: 10.2307/4110825
Chen, K., Peng, Y. H., Wang, Y. H., Korpelainen, H., and Li, C. Y. (2007). Genetic relationships among poplar species in section Tacamahaca (Populus L.) from western Sichuan. China. Plant Sci. 172, 196–203. doi: 10.1016/j.plantsci.2006.08.009
Chen, Z. Y., Wang, W. W., Yang, W. L., and Ma, T. (2016). Characterization of the complete chloroplast genome of Populus ilicifolia. Conserv. Genet. Resour. 8, 1–3. doi: 10.1007/s12686-016-0566-3
Cho, K. S., Yun, B. K., Yoon, Y. H., Hong, S. Y., Mekapogu, M., Kim, K. H., et al. (2015). Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS One 10:e0125332. doi: 10.1371/journal.pone.0125332
Choi, K. S., Chung, M. G., and Park, S. (2016). The complete chloroplast genome sequences of three Veroniceae Species (Plantaginaceae): comparative analysis and highly divergent regions. Front. Plant Sci. 7:335. doi: 10.3389/fpls.2016.00355
Chung, S. L., Lo, C. H., Lee, T. Y., Zhang, Y. Q., Xie, Y. W., Li, X. H., et al. (1998). Diachronous uplift of the Tibetan plateau starting 40 Myr ago. Nature 394, 769–773. doi: 10.1038/29511
Cronk, Q. (2005). Plant eco-devo: the potential of poplar as a model organism. New Phytol. 166, 39–48. doi: 10.1111/j.1469-8137.2005.01369.x
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Dickmann, D. I., and Stuart, K. (1983). The culture of poplars in Eastern North America. Rhodora 19, 10–15.
Eckenwalder, J. E. (1977a). North American cottonwoods (Populus, Salicaceae) of sections Abaso, and Aigeiros. J. Arnold Arbor. 58, 193–208. doi: 10.5962/bhl.part.29239
Eckenwalder, J. E. (1977b). Systematics of Populus L. in southwestern North American with special reference to sect. Aigeiros Duby. Ph.D. thesis, University of California, Berkeley, CA.
Eckenwalder, J. E. (1996). “Systematics and evolution of Populus,” in Biology of Populus and its Implications for Management and Conservation, eds R. I. F. I. Stettler, H. D. Bradshaw, P. E. Heilman, and T. M. Hinckley (Ottawa: NRC Research Press), 7–32.
Eichenberg, D., Purschke, O., Ristok, C., Wessjohann, L., and Bruelheide, H. (2015). Trades-offs between physical and chemical carbon-based leaf defence: of intraspecific variation and trait evolution. J. Ecol. 103, 1667–1679. doi: 10.1111/1365-2745.12475
Fang, Z. F., Zhao, S. D., and Skvortsov, A. K. (1999). Flora of China (English Version). Beijing: Science press, 162–274.
Fladung, M., and Schroeder, H. (2010). SSR and SNP markers for the identification of clones, hybrids and species within the genus Populus. Silvae Genet. 59:6.
Garland, T., Harvey, P. H., and Ives, A. R. (1992). Procedures for the analysis of comparative data using phylogenetically independent contracts. Syst. Biol. 41, 18–32. doi: 10.1093/sysbio/41.1.18
Hamzeh, M., and Dayanandan, S. (2004). Phylogeny of Populus (Salicaceae) based on nucleotide sequences of chloroplast trnT-trnF region and nuclear rDNA. Am. J. Bot. 9, 1398–1408. doi: 10.3732/ajb.91.9.1398
Hamzeh, M., Périnet, P., and Dayanandan, S. (2006). Genetic relationships among species of Populus (Salicaceae) based on nuclear genomic data. J. Torrey Bot. Soc. 133, 519–527. doi: 10.3159/1095-5674(2006)133[519:GRASOP]2.0.CO;2
Han, X. M., Wang, Y. M., and Liu, Y. J. (2017). The complete chloroplast genome sequence of Populus wilsonii and its phylogenetic analysis. Mitochondrial DNA B 2, 932–933. doi: 10.1080/23802359.2017.1413291
He, C. Z., Li, J. M., Yun, T., Zong, D., Zhou, A. P., Ou, G. L., et al. (2015). SRAP analysis on the effect of geographic isolation on population genetic structure of Populus davidiana in Tibetan-inhabited regions in southwest China. For. Res. 28, 152–157.
Huang, D. I., Hefer, C. A., Kolosova, N., Douglas, C. J., and Cronk, Q. C. B. (2014). Whole plastome sequencing reveals deep plastid divergence and cytonuclear discordance between closely related balsam poplars, Populus balsamifera and P. trichocarpa (Salicaceae). New Phytol. 204, 693–703. doi: 10.1111/nph.12956
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., Yi, T. S., and Li, D. Z. (2018). GetOrganelle: a simple and fast pipeline for de novo assemble of a complete circular chloroplast genome using genome skimming data. bioRxiv [Preprint]. doi: 10.1101/256479
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kembel, S. W., Cowan, P. B., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: (R) tools for intergrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btg166
Kim, K. J., and Lee, H. L. (2005). Wide spread occurrence of small inversions in the chloroplast genomes of land plants. Mol. Cells 19, 104–113.
Kuzovkina, Y. A., and Vietto, L. (2014). An update on the cultivar registration of Populus and Salix (Salicaceae). Skvortsovia 1, 133–148.
Leskinen, E., and Alström-Rapaport, C. (1999). Molecular phylogeny of Salicaceae and closely related Flacourtiaceae: evidence from 5.8 S, ITS 1, and ITS 2 of the rDNA. Plant Syst. Evol. 215, 209–227. doi: 10.1007/BF00984656
Li, S. W., Zhang, Y. H., Zhang, Z. Y., An, X. M., He, C. Z., and Li, B. L. (2007). AFLP analysis of some species and hybrids in Populus. Sci. Silvae Sin. 43, 35–41.
Librado, P., and Rozas, J. (2009). Dnasp v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, J. Q., Wang, Y. J., Wang, A. L., Hideaki, O., and Abbott, R. J. (2006). Radiation and diversification within the Ligularia-Cremanthodium-Parasenecio complex (Asteraceae) triggered by uplift of the Qinghai-Tibetau. Mol. Phylogen. Evol. 38, 31–49. doi: 10.1016/j.ympev.2005.09.010
Liu, X., Wang, Z. S., Shao, W. H., Ye, Z. Y., and Zhang, J. G. (2017). Phylogenetic and taxonomic status analyses of the Abaso section from multiple nuclear genes and plastid fragments reveal new insights into the North America origin of Populus (Salicaceae). Front. Plant Sci. 7:2022. doi: 10.3389/fpls.2016.02022
Liu, Y. Q., and Fu, D. R. (2004). Development and utilization of section Tacamahaca gene resources on the Plateau of Western Sichuan. J. Cent. South For. Univ. 24, 129–131.
Lohse, M., Drechsel, O., and Bock, R. (2007). Organellar Genome DRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Lu, Z. Q., Tian, B., Liu, B. B., Yang, C., and Liu, J. Q. (2014). Origin of Ostryopsis intermedia (Betulaceae) in the southeast Qinghai-Tibet Plateau through hybrid speciation. J. Syst. Evol. 52, 250–259. doi: 10.1111/jse.12091
Milne, R. I. (2006). Northern hemisphere plant disjunctions: a window on tertiary land bridges and climate changes? Ann. Bot. 98, 465–472. doi: 10.1093/aob/mcl148
Molina-Venegas, R., and Rodríguez, M. Á (2017). Revisiting phylogenetic signal; strong or negligible impacts of polytomies and branch length information? BMC Evol. Biol. 17:113. doi: 10.1186/s12862-017-0898-y
Pagel, M. (1999). Inferring the historical patterns of biological evolution. Nature 401, 876–884. doi: 10.1038/44766
Provan, J., Powell, W., and Hollingsworth, P. M. (2001). Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol. Evol. 16, 142–147. doi: 10.1016/S0169-5347(00)02097-8
Rajora, O. P., and Zsuffa, L. (1984). “Interspecific crossability and its relation to the taxonomy of the genus Populus L,” in Proceedings of the Joint Meeting of the Working Parties S2-02-10 Poplar Provenances and S2-03-07 Breeding Poplar, XVII Session of the International Poplar Commission (Ottawa: National Research Council), 33–45.
Ronquist, F., and Huelsenbeck, J. P. (2003). Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, 686–689. doi: 10.1093/nar/gki366
Shi, Q. L., Zhuge, Q., Huang, M. R., and Wang, M. X. (2001). Phylogenetic relationship of Populus sections by ITS sequence analysis. Acta Bot. Sin. 43, 323–325.
Smith, R. L. (1988). Phylogenetics of Populus L. (Salicaceae) Based on Restriction Site Fragment Analysis of cpDNA. MS. thesis, University of Wisconsin, Madison, WI.
Song, H. Z. (2006). Genetic Diversity Evaluation and Cultivars Identification with Fingerprinting Based on AFLP in Five Sections of the Genus Populus. Ph.D. thesis, Chinese Academy of Sciences Forestry Biotechnology, Beijing.
Song, Y., Dong, W. P., Liu, B., Xu, C., Yao, X., Gao, J., et al. (2015). Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machuilus balansae in the family of Lauraceae. Front. Plant Sci. 6:662. doi: 10.3389/fpls.2015.00662
Song, Y., Yao, X., Liu, B., Tan, Y. H., and Corlett, R. T. (2018). Complete plastid genome sequences of three tropical Alseodaphne trees in the family Lauraceae. Holzforschung 72, 337–345. doi: 10.1515/hf-2017-0065
Song, Y., Yao, X., Tan, Y. H., Gan, Y., and Corlett, R. T. (2016). Complete chloroplast genome sequence of the avocado: gene organization, comparative analysis, and phylogenetic relationships with other Lauraceae. Can. J. For. Res. 46, 1293–1301. doi: 10.1139/cjfr-2016-0199
Song, Y., Yao, X., Tan, Y. H., Gan, Y., Yang, J. B., and Corlett, R. T. (2017). Comparative analysis of complete chloroplast genome sequences of two subtropical trees, Phoebe sheareri and Phoebe omeiensis (Lauraceae). Tree Genet. Genomes 13:120. doi: 10.1007/s11295-017-1196-y
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stettler, R. F., Bradshaw, H. D., Heilman, P. E., and Hinckley, T. M. (1996). Biology of Populus and its Implications for Management and Conservation. Ottawa: NRC research press.
Stevens, P. F. (2001). Angiosperm Phylogeny Website. Version 2017 (and More or Less Continuously Updated Since). Available at: http://www.mobot.org/MOBOT/research/APweb/
Tamura, K., Peterson, D., Peterson, N., Stecher, G., and Nei, M. (2011). Kumar, S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tiffney, B. H., and Manchester, S. R. (2001). The use of geological and paleontological evidence in evaluating plant phylogeographic hypotheses in the Northern Hemisphere Tertiary. Int. J. Plant Sci. 162, S3–S17. doi: 10.1086/323880
Wan, X., Zhang, F., Zhong, Y., Wang, C., Ding, Y., Hu, T., et al. (2009). Conservation and application of the genetic resource of native poplars in Southwest China. Sci. Silvae Sin. 45, 139–144. doi: 10.11707/j.1001-7488.20090423
Wan, X. Q., Zhang, F., Zhong, Y., Ding, Y. H., Wang, L. W., and Hu, T. X. (2013). Study of genetic relationships and phylogeny of the native Populus in southwest China based on nucleotide sequences of chloroplast trnT-trnF and nuclear DNA. Plant Syst. Evol. 299, 57–65. doi: 10.1007/s00606-012-0702-9
Wang, A. Z., Yang, M. H., and Liu, J. Q. (2005). Molecular phylogeny, recent radiation and evolution of gross morphology of the Rhubarb genus Rheum (Polygonaceae) inferred from chloroplast DNA trnL-F sequences. Ann. Bot. 96, 489–498. doi: 10.1093/aob/mci201
Wang, M. F. (2012). Analysis the Genetic Relationship of the Native Populus in Sichuan by ISSR and ITS Sequences. Ph.D. thesis, Department of Sichuan Agricultrual University, Sichuan.
Wang, T. J., Fan, L. Q., Guo, X. Y., Luo, X., and Wang, K. (2016). Characterization of the complete chloroplast genome of Populus qiongdaoensis T. Hong et P. Luo. Conserv. Genet. Resour. 8, 435–437. doi: 10.1007/s12686-016-0590-3
Wang, Y. Z., and Gilbert, M. G. (2007). “Stellera Linnaeus,” in Flora of China, eds C. Y. Wu and P. H. Raven (Beijing: Science Press).
Wang, Z. S., Du, S. H., Dayanandan, S., Wang, D. S., Zeng, Y. F., and Zhang, J. G. (2014). Phylogeny reconstruction and hybrid analysis of Populus (Salicaceae) based on nucleotide sequences of multiple single-copy nuclear genes and plastid fragments. PLoS One 9:e103645. doi: 10.1371/journal.pone.0103645
Wei, Z. Z., Guo, L. Q., Zhang, J. F., Li, B. L., Zhang, D. Q., and Guo, H. (2010). Phylogenetic relationship of Populus by trnL-F sequence analysis. J. Beijing For. Univ. 32, 27–33.
Yang, J. B., Tang, M., Li, H. T., Zhang, Z. R., and Li, D. Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Yun, T., Li, J. M., Zhou, A. P., Yan, L. X., Zong, D., Li, D., et al. (2015). Analysis of phylogenetic relationship of Populus based on sequence data of chloroplast regions. Plant Physiol. J. 51, 1339–1346.
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: light into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Zheng, H. L., Fan, L. Q., Wang, T. J., Zhang, L., Ma, T., and Mao, K. S. (2016). The complete chloroplast genome of Populus rotundifolia (Salicaceae). Conserv. Genet. Resour. 8, 339–401. doi: 10.1007/s12686-016-0568-1
Zong, D., Zhou, A. P., Li, D., and He, C. Z. (2019). The complete chloroplast genome of Populus xiangchengensis, an endemic species in southwest China. Mitochondrial DNA B 4, 70–71. doi: 10.1080/23802359.2018.1536463
Keywords: Populus, Salicaceae, chloroplast genome, phylogenetic, morphological traits
Citation: Zong D, Gan P, Zhou A, Zhang Y, Zou X, Duan A, Song Y and He C (2019) Plastome Sequences Help to Resolve Deep-Level Relationships of Populus in the Family Salicaceae. Front. Plant Sci. 10:5. doi: 10.3389/fpls.2019.00005
Received: 01 November 2018; Accepted: 07 January 2019;
Published: 22 January 2019.
Edited by:
Michael Eric Schranz, Wageningen University and Research, NetherlandsReviewed by:
Alejandra Moreno-Letelier, National Autonomous University of Mexico, MexicoCopyright © 2019 Zong, Gan, Zhou, Zhang, Zou, Duan, Song and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Song, c29uZ3l1QHh0YmcuYWMuY24= Chengzhong He, aGVjekBzd2Z1LmVkdS5jbg==; aGN6NzBAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.