 Qingying Zhang1*†
Qingying Zhang1*† Xuan Chen1†
Xuan Chen1† Hongyan Guo1
Hongyan Guo1 Luisa M. Trindade2
Luisa M. Trindade2 Elma M. J. Salentijn2Rong Guo1Mengbi Guo1Yanping Xu1Ming Yang1*
Elma M. J. Salentijn2Rong Guo1Mengbi Guo1Yanping Xu1Ming Yang1*- 1Industrial Crops Research Institute, Yunnan Academy of Agricultural Sciences, Kunming, China
- 2Wageningen University and Research Plant Breeding, Wageningen University and Research, Wageningen, Netherlands
Cannabis is one of the most important industrial crops distributed worldwide. However, the phylogeographic structure and domestication knowledge of this crop remains poorly understood. In this study, sequence variations of five chloroplast DNA (cpDNA) regions were investigated to address these questions. For the 645 individuals from 52 Cannabis accessions sampled (25 wild populations and 27 domesticated populations or cultivars), three haplogroups (Haplogroup H, M, L) were identified and these lineages exhibited distinct high-middle-low latitudinal gradients distribution pattern. This pattern can most likely be explained as a consequence of climatic heterogeneity and geographical isolation. Therefore, we examined the correlations between genetic distances and geographical distances, and tested whether the climatic factors are correlated with the cpDNA haplogroup frequencies of populations. The “isolation-by-distance” models were detected for the phylogeographic structure, and the day-length was found to be the most important factor (among 20 BioClim factors) that influenced the population structures. Considering the distinctive phylogeographic structures and no reproductive isolation among members of these lineages, we recommend that Cannabis be recognized as a monotypic genus typified by Cannabis sativa L., containing three subspecies: subsp. sativa, subsp. Indica, and subsp. ruderalis. Within each haplogroup which possesses a relatively independent distribution region, the wild and domesticated populations shared the most common haplotypes, indicating that there are multiregional origins for the domesticated crop. Contrast to the prevalent Central-Asia-Origin hypothesis of C. saltiva, molecular evidence reveals for the first time that the low latitude haplogroup (Haplogroup L) is the earliest divergent lineage, implying that Cannabis is probably originated in low latitude region.
Introduction
Cannabis is one of the oldest crops and has been distributed worldwide by humans. This plant may have been utilized for at least 10,000 years (Schultes et al., 1974; Long et al., 2016), and its cultivation in China can be traced back to around 6,000 years ago according to the archaeological findings and records of ancient literatures (Li, 1974; Yang, 1991). Cannabis has been developed as a multi-purpose crop, which is widely used for the production of biomaterials such as textile, paper, construction, and insulation materials, but also as functional foods, namely the oil and seeds, and for other applications including cosmetics and personal care products, and in the pharmaceutical industry. The global market for hemp has been estimated to consist of more than 25,000 products (Johnson, 2013; Salentijn et al., 2015). In recent years, the hemp industry has increasingly received attention and the development of high value products has been the main focus of various studies (Amaducci et al., 2015). Especially, cannabis plants can produce more than 100 pharmacologically active compounds (cannabinoids), with the most studied compounds being tetrahydrocannabinol (THC) and cannabidiol (CBD), and CBD has sparked an increasing interest for product development.
Based on the content of cannabinoids in this herbaceous annual crop, cannabis plants have been often classified as hemp, mostly referring to a fiber crop with low tetrahydrocannabinol (THC) and marijuana, the drug type with often high THC content. This plant comprises both wild and domesticated populations which can be either dioecious or monoecious cultivars. The flowering is very sensitive to photoperiod and cultivars can be early-, intermediate-, and late-ripening. Compared to the domesticated cannabis, the wild forms usually exhibit the following distinct morphological and physiological features: remarkably smaller seeds (mature achene, thousand seed weight < 10 g), easy seed shattering behavior (seeds readily disarticulate from the pedicel), long-term seed dormancy and the need for cold-moist stratification treatment to facilitate germination. For a long time, researchers have disputed the taxonomy of Cannabis regarding the definitions of species, subspecies, and/or varieties (McPartland and Guy, 2004, 2014; Hillig, 2005; Gilmore et al., 2007; Small, 2015). The issue of Cannabis taxonomy continues to puzzle botanical taxonomists (Piomelli and Russo, 2016; Welling et al., 2016; Mcpartland and Guy, 2017; Mcpartland and Hegman, 2018). Linnaeus named Cannabis sativa L. (hereafter as C. sativa) as a unique species. Later on, two species, C. indica Lam. (1785) and C. ruderalis Jan. (1924), were split from C. sativa based on certain distinct morphological Characteristics (Hillig and Mahlberg, 2004), while Small and coauthors recommended retaining only one species (C. sativa) but including two subspecies, subsp. sativa and subsp. indica, where each subspecies includes both domesticated and wild varieties (Small and Cronquist, 1976; Small, 2015). Recently, based on allozyme analysis results, Hillig (2005) suggested a taxonomic concept of three species (C. sativa, C. indica, and C. ruderalis) including seven putative taxa in the genus Cannabis.
Germplasm collections of Cannabis are the most valuable fundamental materials for breeding as they are a potential source of novel genes controlling important traits such as increased seed productivity, improved qualitative characteristics for example fiber quality, or resistance to adverse environmental factors such as cold, drought, strong wind, and pest/disease pressure. The native distribution range of Cannabis is commonly believed to be in Central Asia, Siberia, the Himalayas, and possibly extending into China (de Candolle, 1885; Vavilov, 1926; Li, 1974; Hillig, 2005; Small, 2015; Mcpartland and Hegman, 2018). Currently, the distribution of cannabis covers most of the Chinese territory, ranging from about 23 to 51° N, 80 to 125° E. China has been a major hemp growing country with the largest cultivation area and has developed many landraces and cultivars. China is part of the potential center of origin for cannabis, with abundant genetic resources in wild populations but also developed cultivars, thus provides a unique opportunity to investigate the domestication origin of cannabis plants. However, the wild populations of cannabis have been poorly studied, and the genetic diversity and structure of these populations, as well as the relationships among the wild populations and the domesticated cultivars remains largely unknown.
Chloroplast DNA (cpDNA) markers and phylogeographic methods have been proven to be very useful tools in investigating genetic diversity, population structure, domestication origin, and historical context of species (Avise, 2000, 2004, 2009). The cpDNA is a haploid (and thus are homoplasmic), non-recombining genome that is maternally inherited in most angiosperms (Schaal et al., 1998; Avise, 2009). However, like many other plant species, cannabis cpDNA displayed very low genetic diversity (Gilmore et al., 2007; Zhang et al., 2017; Mcpartland and Hegman, 2018). A key to successful utilization of cpDNA markers for estimating diversity and phylogenetic relationships among populations of Cannabis species requires obtaining sufficient genetic variation in cpDNA and developing suitable cpDNA markers. In this study, based on scrutinizing differences in the whole chloroplast genomes DNA sequences of four Cannabis accessions (Oh et al., 2016; Vergara et al., 2016), we developed five DNA markers for the most variable polymorphic regions and investigated the genetic diversity of an extensive set of Cannabis samples. These samples include wild populations, representative landraces and breeding cultivars from China, as well as some accessions from other countries (The Netherlands, France, Hungary, Italy, Russia, Nigeria, Korea, and USA). Our main objectives were: (1) to estimate the genetic diversity and elucidate the distribution patterns of the wild and domesticated cannabis from China; (2) to determine the main factors that affected the spatial distribution of cannabis and provide information on historical processes of this plant; (3) to infer the genetic relationships between the populations or lineages, as well as domestication origins of cannabis cultivars in China.
Materials and Methods
Plant Material
The studied material comprised 645 Cannabis individuals (derived from 52 accessions: 25 wild populations and 27 domesticated populations or cultivars), and four closely related out group species, Humulus scandens, Humulus yunnanensis, Humulus lupulus, and Aphananthe aspera. Information relevant to the samples is shown in Table 1.
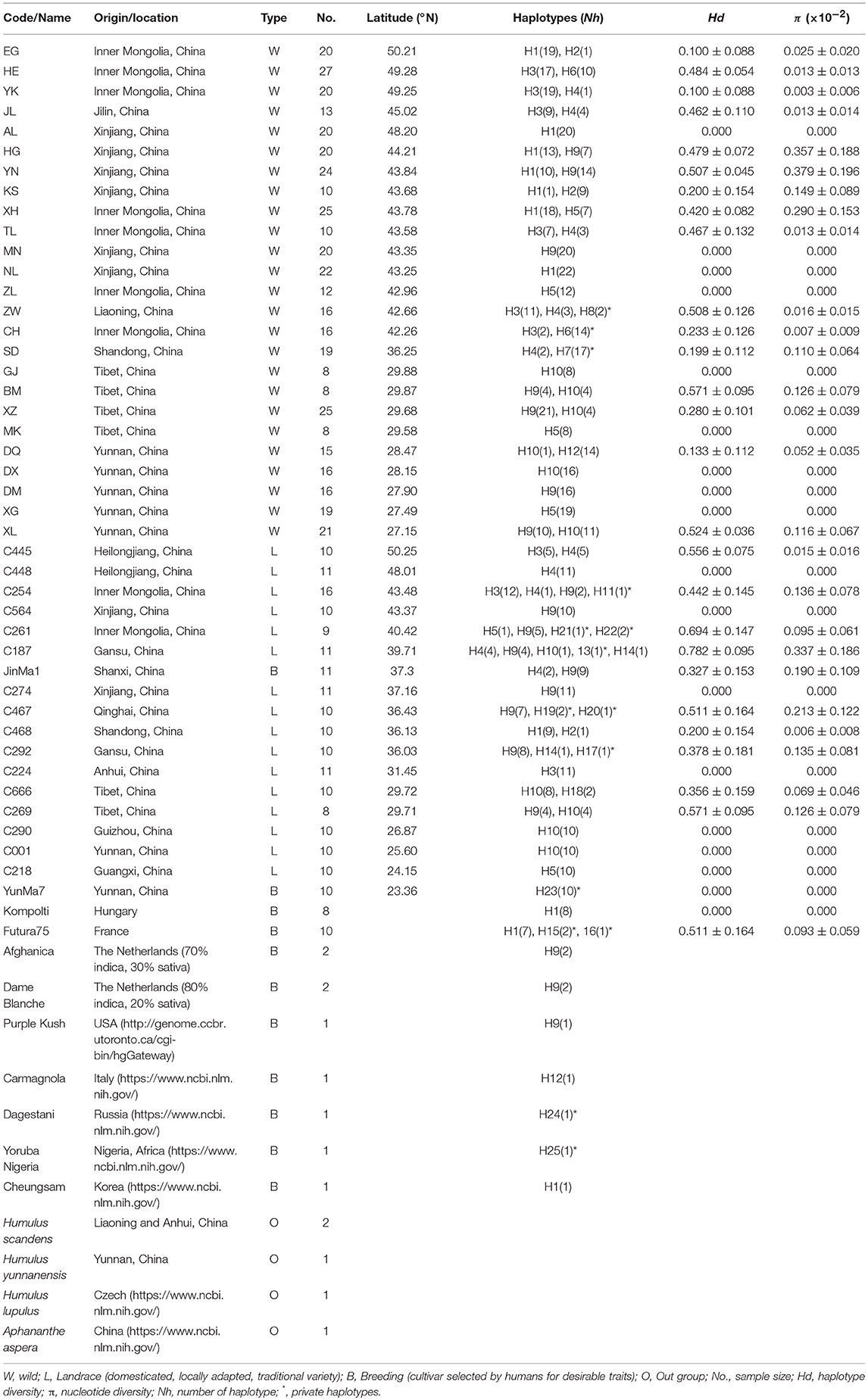
Table 1. Sample information and summary of haplotype distribution, genetic diversity for each population based on the combined five cpDNA regions.
Twenty-five wild populations represented by 430 individuals were collected from 2011 to 2016. These populations covered the only distribution ranges of extant wild Cannabis throughout China: Inner Mongolia, Jilin, Liaoning, Shandong, Xinjiang, Tibet, and Yunnan provinces or regions. The population size of wild Cannabis is generally ranging from hundreds to several thousand individuals. Healthy leaves were collected in the field and immediately dried with silica gel until DNA extractions. To increase the possibility of detecting variation within each population, individuals growing at least 10 m apart were randomly sampled and in addition, eight to thirty plants were sampled from the edges and the interior of populations, depending on the actual population size. For domesticated populations, 27 cultivars represented by 215 individuals were included. Eighteen cultivars (188 individuals) from China were obtained from the Industrial Crops Research Institute, Yunnan Academy of Agricultural Sciences, and two European hemp cultivars, Kompolti and Futura75, were obtained from Hungary and France, respectively. About 200 seeds from each cultivar were planted and during the flowering stage leaves were sampled for DNA extraction. Additionally, two marijuana materials (named Afghanica and Dame Blanche) from The Netherlands were used, whereas sequence data for another five cultivars (Purple Kush, Carmagnola, Dagestani, Yoruba Nigeria, and Cheungsam) were downloaded from GenBank and The Cannabis Genome Browser website (http://genome.ccbr.utoronto.ca/cgi-bin/hgGateway) (Table 1).
For the 43 hemp populations originating from China [25 wild populations (W) and 18 domesticated cultivars (L and B)] (Table 1, Figure 1), the sampled regions throughout China spanned an area from 50.25° to 23.36° N and from 79.44° to 126.08° E, with an altitude span from about 50 m above sea level in Anhui (C224) to 3,700 m in Tibet (MK).
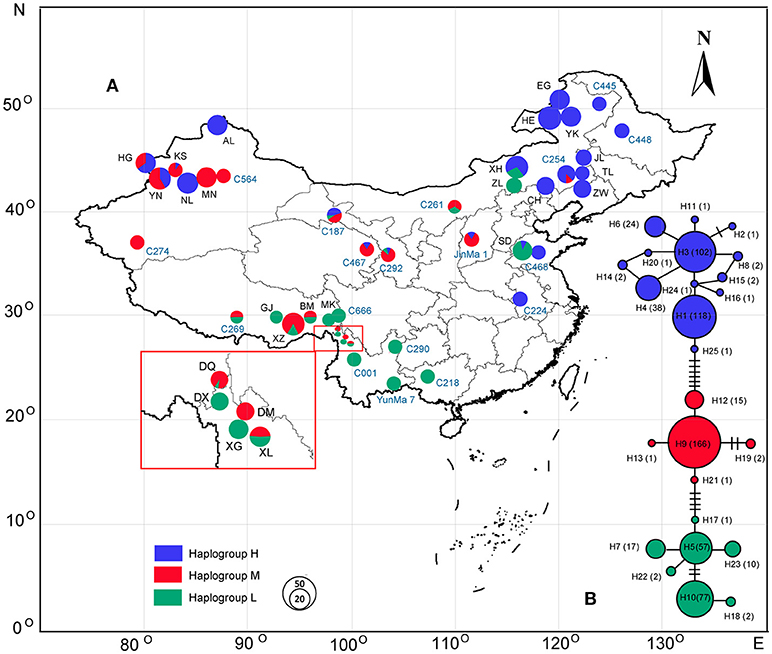
Figure 1. Geographic location of the 43 populations of Cannabis analyzed in the present study and haplogroup distribution patterns of Cannabis (see Table 1 for population codes); population codes in black represent the wild samples and blue ones are the domesticated accessions. (B) The haplotype network generated from the 25 haplotypes of Cannabis; pie chart size corresponds to the sample size of each population (A) or haplotype (B).
DNA Extraction, Primer Development, PCR Amplification, and Sequencing
Total DNA of each sample was extracted from leaf material according to the modified CTAB method (Doyle, 1991; Chen et al., 2015).
To develop genetic markers for population genetic analyses, we first tested 17 universal primer sets, developed for amplification of highly variable chloroplast DNA regions of angiosperms, on six individuals from different wild Cannabis populations. However, Cannabis individual sequences generated from these primers are too conserved to obtain variable sites suitable for population-level studies despite repeated tests (Zhang et al., 2017). Based on comparisons of the four available whole chloroplast genomes from cultivars of C. sativa (Oh et al., 2016; Vergara et al., 2016), we developed five pairs of PCR primers targeting several highly variable chloroplast regions (rps16; psaI-accD; rps11-rps8; rpl32-trnL; ndhF-rpl32). These new primers are suitable for the population genetic study of Cannabis and its closest relative Humulus (Table 2). Due to unsuccessful PCR amplification of the rps16 region of Humulus species, a specific forward primer for the genus Humulus was designed.
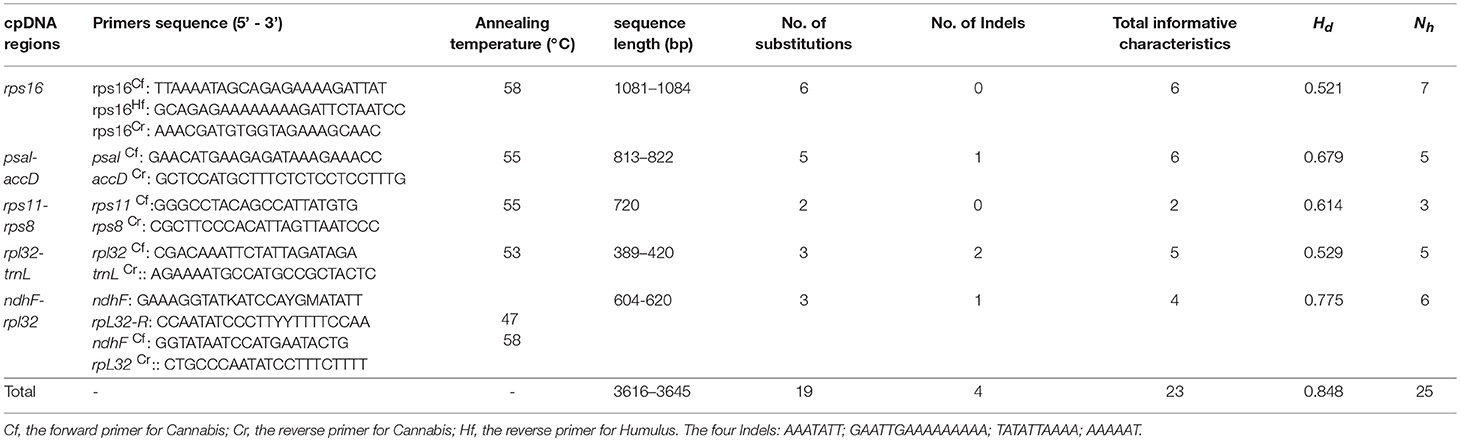
Table 2. Primer pairs of cpDNA regions used in this study and polymorphism on the 645 individuals of Cannabis.
PCR amplification reactions were carried out in a total volume of 25 μL, containing 2.0 μL DNA template (20–30 ng/μL), 2.5 μL 10 × PCR reaction Buffer (with Mg2+), 1.5 μL dNTPs mix (2.5 mmol/L), 0.5 μL each forward and reverse primers (10 μmol/L), 0.3 μL Taq DNA polymerase (5 U/μL, Beijing TransGen Biotech Co., Ltd., China), and 17.7 μL double-distilled water. Amplifications were conducted on an ABI Veriti Thermal Cycler (Applied Biosystems, Foster City, CA, USA) using the following program setting: an initial 4 min pre-denaturation at 94°C, followed by 30 cycles of 45 s at 94°C, 30 s at 47–58°C (Table 2), 45–90 s at 72°C, and a final 10 min at 72°C.
The obtained PCR products were purified with a Gel Extraction and PCR Purification Combo Kit (Beijing Tsingke BioTech Co., Ltd., China) and then bidirectional sequencing was performed on an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) employing the same primers used for PCR amplifications. All sequences of the rps16, psaI-accD, rps11-rps8, rpl32-trnL, and ndhF-rpl32 cpDNA regions have been deposited in GenBank under the accession numbers from MG731579 through MG731614.
Observation of Main Phenological and Morphological Traits
To test whether there are obvious differences among the 43 accessions (including both wild and domesticated germplasms) on phenotypic characteristics, we also carried out a Varieties Evaluation Field Trial in 2016 involving all 43 accessions. The trial site was located in Kunming, Yunnan province of China. This trial was set up as a randomized complete block design with three replicates and each plot was 6 m2, with a distance between rows of 40 cm (with a density about 50 plant individuals per square meter). The plots were directly seeded at a depth of 3–5 cm on May 28 in 2016 and all wild-type seeds were pretreated to facilitate germination before sowing about 10 days, and the whole trial was managed with normal management practices. Main phenological and morphological traits for each accession were investigated, including initiation of flowering, full flowering, seed full maturity time, stem diameter, plant height, and number of branches. These data were collected based on 20 individuals randomly selected for each plot (10 individuals for female and male respectively).
Data Analysis
Raw sequence data of the five amplified DNA fragments (amplicons) were assembled with SeqMan (DNAStar Inc., Madison, WI, USA) and carefully checked for genetic variation together with the chromatograms. Sequences were aligned using the CLUSTAL W (Thompson et al., 1994) followed by manual adjustment implemented in MEGA 6.0 (Tamura et al., 2013). Small insertion/deletion events (indels), excluding long mononucleotide repeats (poly A/T or poly G/C), were counted as single mutations. The haplotypes for each gene marker, and in the combined five-fragment dataset matrix, were identified using DNASP v5.10 (Librado and Rozas, 2009).
Based on the combined five-fragment dataset, the relationships among haplotypes were reconstructed by median-joining (MJ) network method (Bandelt et al., 1999) implemented in the software NETWORK v5.0.0.1 (available at http://www.fluxus-engineering.com) with the maximum parsimony (MP) post-processing option.
To detect genetic diversity and population structure, we carried out the following analyses. The distribution of three haplogroups (identified by phylogenetic tree and network) was plotted on maps of China using ArcGIS v 10.2 (ESRI Inc., Redlands, CA, USA). To define the most differentiated groups of populations we performed a spatial analysis of molecular variance (SAMOVA) using the software SAMOVA v 2.0 (available at http://cmpg.unibe.ch/software/samova2/) based on geographical coordinates and haplotype distribution data of Cannabis populations from China. Different hierarchical levels of genetic variation including within populations, among populations within groups and among groups were assessed by the analysis of molecular variance (AMOVA) implemented in Arlequin v 3.5.2.2 (Excoffier and Lischer, 2010), with significance assessed by 1,000 permutations on the 43 populations from China. The 43 populations were grouped into three population groups (Group H, Group M, and Group L) by SAMOVA based on variation in cpDNA or into two morphology groups by morphological and physiological features (the wild Group and domesticated Group) where the population genetic structure and the domestication pattern for Cannabis in China were assessed. Indices of nucleotide diversity (π) and haplotype diversity (Hd) were calculated for each population, population groups, and for all samples combined, using Arlequin v 3.5.2.2. Also, Tajima's D and Fu's Fs neutrality tests were conducted. Levels of gene flow (Fst and Nm) were measured using DNASP v5.10. Mantel tests were conducted to examine the correlation between two matrixes (genetic distances and pairwise geographical distances or latitude differences) with 9,999 permutations using GenALEx v 6.5 (Peakall and Smouse, 2012).
To identify the main climatic factors affecting the distribution of the Cannabis genetic lineages, we also tested correlations of 20 bioclimatic factors on a compilation of cpDNA haplogroup frequencies for 43 populations. The values of 19 BioClim variables were extracted by using DIVA-GIS v7.5 (http://www.diva-gis.org/) based on the global climate layer data (at 2.5 arc-min resolution) downloaded from the WorldClim v2.0 database (http://www.worldclim.org/), and the mean day length of cannabis growth season (from the Spring Equinox to Autumnal equinox) were calculated according to solar geometry (Spitters et al., 1986; Yuan et al., 2014) for 43 sampling sites. The correlation between environmental variables and haplogroup frequencies was analyzed by redundancy analysis (RDA). We first assessed the effects of all 20 climatic factors on haplogroup frequencies distribution. And then, to identify a minimum subset of climatic variables that significantly explain variation of genotype spatial distribution, we further tested the multicollinearity in the whole data set, and the redundant factors (variance inflation factors, VIF > 10) were excluded through stepwise regression. To explore the percent variance uniquely explained by each factor, the Analysis of Variance (ANOVA) was calculated. RDA and ANOVA analyses were performed using the vegan package in R version 3.3.1 (R Core Team, 2015).
Phylogenetic relationships based on the cpDNA haplotypes were deduced using MrBayes ver. 3.1.2 (Ronquist and Huelsenbeck, 2003). Using the sequences from Aphananthe and Humulus species as out groups, the divergence times for the major groups of these haplotypes were further estimated with BEAST v1.8.1 (Drummond and Rambaut, 2007) with GTR + G selected by MrModeltest 2.3 as the best substitution model for the data set (Nylander, 2004). The data was analyzed using a relaxed log-normal clock model and a Yule Process speciation model for the tree priors. As the earliest fossil species of Aphananthe was reported around 66–72.1 million years ago (Ma) from the Maastrichtian (66–72.1 Ma) in late Cretaceous (Ervín et al., 1986), the stem age of the Aphananthe was set to 66 Ma based on the low boundary age (node A in Figure 2B). Prior settings for calibrating node were: offset of 66 Ma, a log mean of 1.0 (log stdev of 0.5). Two independent runs were conducted for 10 million generations. Log files resulted from the two runs were combined using LogCombiner after the first 25% were discarded as burn-ins, and the convergence of the chains was checked in Tracer v1.6 (Rambaut et al., 2014). Similarly, the resulted trees were combined in LogCombiner, and the maximum clade credibility (MCC) tree was produced with Tree Annotator, and then viewed in FigTree v1.4.2.
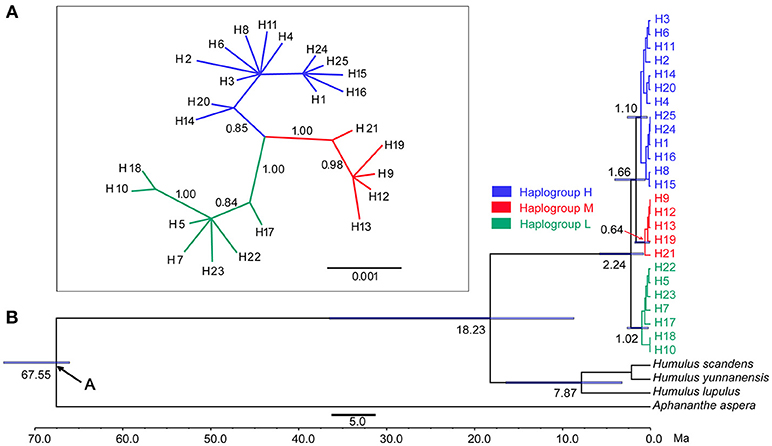
Figure 2. (A) Bayesian phylogenetic tree based on cpDNA data. (B) Divergence time estimated for the major clades of Cannabis by the BAEST analysis (Blue bars indicate the 95% highest posterior density credibility for node ages).
Results
Sequence Characteristics and Identification of cpDNA Haplotypes
We successfully obtained high quality sequences for all the five target cpDNA genes (rps16, psaI-accD, rps11-rps8, rpl32-trnL, ndhF-rpl32) for each of the 640 Cannabis individual plants. Five additional sequences of Cannabis lines were retrieved from the published chloroplast genomes (Table 1). In total, the combined alignment of the five cpDNA fragments (five-gene matrix) covered 3,635 base pairs in length, and harbored 19 single nucleotide polymorphisms (SNPs) and four indels varying up to 38 bp in length (Table 2), thus the proportion of variable sites was 1.57%. The indel mutations introduced by long mononucleotide repeats (poly A/T or poly G/C) were excluded from the analysis. The AT content was 70.5% and a total of 25 haplotypes (H1–H25) were identified based on the genetic variation found among the 645 samples. For the rps16 intron, and four intergenic spacers (psaI-accD, rps11-rps8, rpl32-trnL, and ndhF-rpl32), the sequence length and polymorphic informative characters are shown in Table 2.
Distribution of cpDNA Haplotypes, Phenotypic Characteristics and Genetic Diversity
In the haplotype network (Figure 1B), the 25 haplotypes were split into three distinct haplogroups: Haplogroup H (blue colored), Haplogroup M (red colored), and Haplogroup L (Green colored). Haplogroup H contained 13 haplotypes (H1, H2, H3, H4, H6, H8, H11, H14, H15, H16, H20, H24, H25), Haplogroup M contained 5 haplotypes (H9, H12, H13, H19, H21), and Haplogroup L contained 7 haplotypes (H5, H7, H10, H17, H18, H22, H23). The phylogenetic tree (Figure 2A) also exposed three well-supported lineages corresponding to the above-mentioned three haplogroups illustrated by the haplotype network. The haplotypes are not evenly distributed for each haplogroup (Figure 1B): In Haplogroup H, the two most common haplotypes, H1 (40.1%) and H3 (34.7%), were observed in 15 out of the 20 sampled populations north of 40° N. For Haplogroup M, the most common haplotype H9 (89.7%) and other 4 rare haplotypes were found in the area ranging from 27° to 43° N. In Haplogroup L, seven haplotypes, including the two major haplotypes H5 (34.3%) and H10 (46.4%), were mainly distributed throughout the area south of 30° N.
As the lineages displayed distinct structure by network analyses (Figure 1B) and structural phylogeographic distribution patterns (Figure 1A), SAMOVA analysis (based on a simulated annealing method) was performed to define groups of populations. The result showed that when k = 3 the differences between groups (FCT = 0.64) was the highest, and the 43 populations from China were divided into three groups: Group H, Group M, and Group L (Figure 1A). Group H included 16 populations mainly from the high latitude region: EG, HE, YK, JL, AL, HG, NL, XH, TL, ZW, CH, C445, C448, C254, C468, and C224. This group largely corresponds to haplogroup H. Group M also included 16 populations but from the middle latitude region: YN, KS, MN, BM, XZ, DQ, DM, XL, C564, C261, C187, JinMa 1, C274, C467, C269, and C292, corresponding to above haplogroup M. Group L included 11 populations mainly from low latitude region: GJ, MK, DX, XG, C290, C001, C218, C666, ZL, SD, and YunMa 7, corresponding to haplogroup L. Frequencies of the three lineages in each population and their geographical distribution are displayed in Figure 1.
Interestingly, we also noted that the main phenotypic traits of 43 wild or domesticated accessions originating from different latitudes shifted along latitudinal gradients (23.36–50.21° N), which matched the regular distribution of three lineages. Our phenotype data (Table S1) indicated there were very high variations among 43 accessions. The six measured traits involved three phenological Characteristics (initiation of flowering, full flowering, and seed full maturity time) and three morphological features (stem diameter, plant height, and number of branches). The correlations between the phenotypes and latitude were assessed, and all six traits had a negative, very strong, and significant (p < 0.001) relationship with latitude of origin, Pearson' correlation coefficients (r) respectively were 0.858, 0.949, 0.906, 0.911, 0.914, 0.815 for initiation of flowering, full flowering, seed full maturity, stem diameter, plant height, and number of branches, respectively. When the phenotype of three genetic groups (above mentioned SAMOVA grouping) were compared, group H had the shortest growth time (mean seed-maturity time, 77.2 ± 18.1 days), thinnest stem diameter (0.54 ± 0.22 cm), shortest plant height (99.2 ± 52.4 cm), and fewest branches (3.2 ± 1.7), while Group L had the longest growth time (mean seed-maturity time,133.6 ± 36.8 days), widest stem diameter (1.14 ± 0.40 cm), tallest height (238.0 ± 86.5 cm), and most branches (11.0 ± 3.9), and the traits data of Group M were in-between.
For genetic diversity features, our studies showed that the number of haplotypes is different among the 43 Chinese populations, plus the cultivars Futura75 and Kompolti, ranging from 1 to 5 haplotypes. We observed that out of the 25 haplotypes, 15 private haplotypes were exclusively found in three wild populations (ZW, CH, SD) and in nine cultivated accessions (C254, C261, C187, C292, C467, YunMa7, Futura75, Dagestani, Yoruba Nigeria). Haplotype diversity (Hd) and nucleotide diversity (Π) of each population are summarized in Table 1. The domesticated population C187 possessed the highest haplotype diversity (Hd = 0.782) and nucleotide diversity (Π = 0.00337), while the lowest number of haplotypes (Nh = 1; Hd = 0; Π = 0) were found in 18 other populations, including domesticated accessions and wild populations. Among the wild populations, the BM population had the highest haplotype diversity (Hd = 0.571), YN population had the highest nucleotide diversity (Π = 0.00379), and ZW had the highest number of haplotypes (Nh = 3, Hd = 0.508).
Isolation by Distance and Climatic Correlates of cpDNA Lineages Frequency
To examine whether the observed genetic distributions are correlated to geographical localization, Mantel tests were performed. Between Nei's pairwise genetic distances (Nei, 1978) and the two-dimensional geographical distances (based on longitudinal and latitudinal coordinates), and the results showed that there is a significant positive correlation among the 43 sampled populations from China (r = 0.379, p = 0.000) and the “isolation-by-distance” pattern was detected. Furthermore, the testing between Nei's pairwise genetic distances and the latitudinal differentiation also showed a significant positive correlation (r = 0.348, p = 0.000). Similarly, for the 25 wild populations alone, significant positive correlations were found between the genetic distances and pairwise geographical distances (r = 0.368, p = 0.000), as well as between the genetic distances and latitude differences (r = 0.416, p = 0.000).
Haplogroup distribution frequencies shifted smoothly along latitudinal gradients and the three lineages distinctively show a high-middle-low latitude distribution pattern (Figure 1). Based on the RDA analysis and ANOVA partition (Table 3), 15 out of the 20 tested BioClim variables had a significant (p < 0.05) relationship with haplogroup distribution frequencies for all the 43 populations (Table S2). This result indicated that climate obviously affected the genetic distribution of Cannabis populations. When the redundancy factors were removed, only MDL (Mean day length), Bio2 (Mean diurnal range), Bio8 (Mean temperature of wettest quarter), Bio13 (Precipitation of wettest month), Bio14 (Precipitation of driest month), Bio15 (Precipitation seasonality) formed a minimum subset of climatic variables. Based on the ANOVA analysis, MDL was the most significant factor influencing the haplogroup distribution frequencies (r2 = 0.6024, p < 0.001), and the subset of 6 climatic variables totally explained 74.2% of variation, and MDL accounted for the largest fraction of the total explained variation (20.8%).
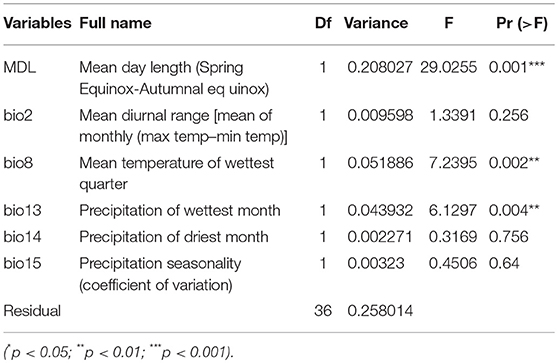
Table 3. ANOVA analyses between BioClim variables and the three cpDNA haplogroup frequencies for 43 Cannabis populations.
Genetic Structure and Gene Flow
Based on the groups defined by SAMOVA, the analysis of molecular variance (AMOVA) revealed that most variance (69.48%) of the total observed genetic variations was due to variations between-groups, 14.43% was attributed to variance among populations within groups, and 16.10% to variance within the same population (Table 4). F-statistics of all the three levels of hierarchy were highly significant (p < 0.001). Population genetic differences (Fst) within the High-latitude lineage (Group H) was higher than that of the lower-latitude lineages (Group M and Group L), while gene flow (Nm) within Group M and Group L was higher than in Group H (Table 5). For genetic diversity within each group, Group H had the highest haplotype diversity, and Group M had the highest nucleotide diversity and number of haplotypes.
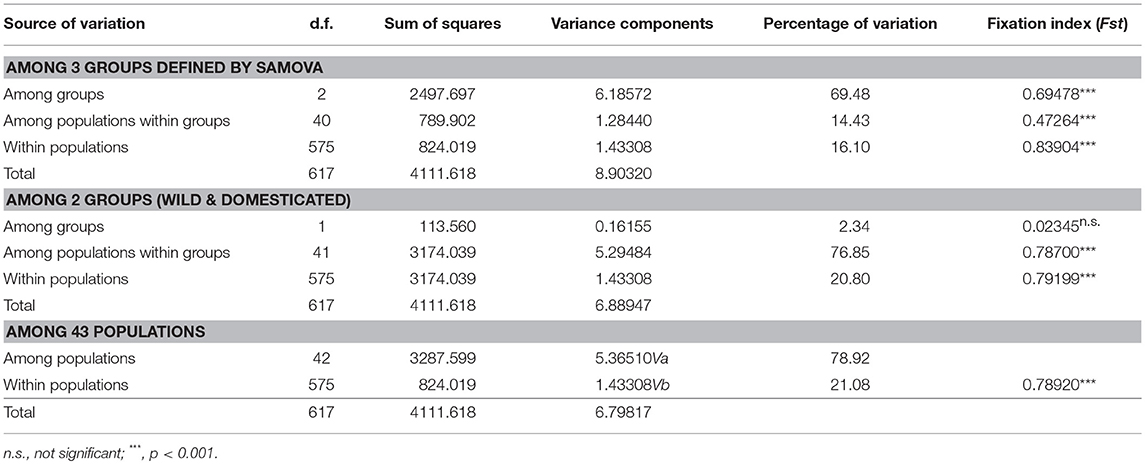
Table 4. Analysis of molecular variance (AMOVA) for on the Cannabis populations from China based on the five cpDNA regions.
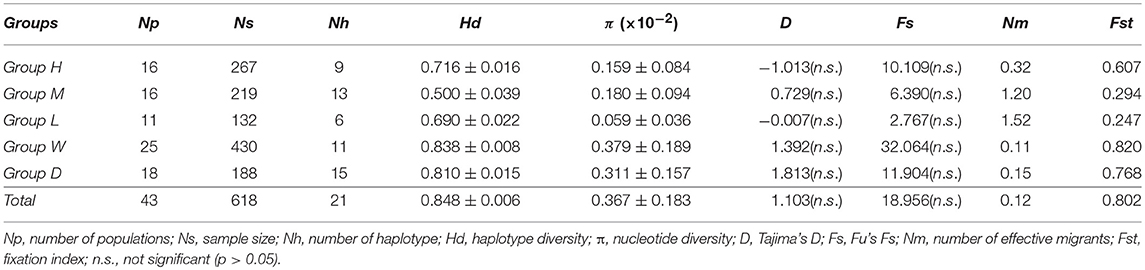
Table 5. Population genetic statistics among Cannabis population groups based on SAMOVA grouping, the two morphology groups (wild & domesticated) and all samples from China.
When two morphological groups (wild and domesticated) were considered for the same 43 populations, AMOVA analysis indicated low and non-significant (2.34% of molecular variance, Fst = 0.023, p = 0.19) genetic differentiation between the two groups. Most variance components were present among populations within groups. The degree of population differentiation was slightly higher in the wild group compared to the domesticated group. Results of the neutrality tests for each group and total sample set are shown in Table 5. All values of Fu's Fs and Tajima's D were statistically non-significance, suggesting stable populations on a different level.
Divergence Time Estimations
The phylogenetic tree (Figure 2) inferred from the five-gene matrix clustered the 25 haplotypes into a monophyletic clade, in which the haplotypes from the high, middle, and low latitude regions formed three monophyletic subclades, with strong statistical support. The stem age of Cannabis (Figure 2B) was estimated at 18.23 Ma with 95% highest posterior density (HPD) 8.83–36.56 Ma, and the crown age of this species was 2.24 Ma, with 95% HPD 0.81–5.81 Ma.
Discussion
Distinct Pattern of Lineage Distribution and Genetic Structure
One major finding of this study is that Cannabis can be divided into three distinct genetic lineages (Figure 1), namely the H, M, and L haplogroups. Interestingly the haplogroups exhibited latitudinal gradients distribution and this distinctive high-middle-low latitude pattern was supported by NETWORK, AMOVA, SAMOVA, and Mantel Tests based on cpDNA data. High-latitude group members (group H) were mainly distributed in regions north of about 40° N and Low-latitude group members (group L) were mainly distributed in areas south of about 30° N, while the middle-latitude group members (group M) were mainly distributed in the zone between about 30° N and 40° N. This current distribution pattern implies an adaptation to distinct latitudinal gradient climatic features. In the present study, the lineage distribution was significantly correlated with latitude and climatic factors. In particular, the day-length has a strong and significant (r2 = 0.6024, p < 0.001) influence on the haplogroup distribution frequencies in each population by RDA analysis and ANOVA partition (Table 3). Furthermore, our field phenotype trial results showed that phenological and morphological traits had a negative, very strong, and significant correlation with latitude of accession origin. For instance, Group H is characterized by short plant height, thin stem, fewer branches, and short life cycle. On the contrary, Group L demonstrates opposite characteristics compared with Group H. This is well-linked to the quantitative (facultative) short-day plant trait of Cannabis. The flowering of Cannabis is normally induced by a required duration of days with a minimum uninterrupted period of darkness (10–12 h for most cultivars) (Small, 2015). Due to the sensitivity to photoperiod, shortening day length can promote Cannabis plant pre-flowering. On the contrary, prolonged day length would delay this crop from shifting from a vegetative stage into a reproductive stage. Indeed, the northernmost distribution of group L is located at about 43° N, which is consistent with previous observations that cultivars from the southern (low latitude) areas have extended vegetative cycles and failed to produce seeds when grown in the North (High latitude areas) (Pahkala et al., 2008; Amaducci et al., 2012; Small, 2015). Our results suggest that photoperiod sensitivity is a potential factor that prevents group L from extending further north. In contrast, the southernmost boundary of group H is 31° N (landrace C224 in Figure 1A). It was surprising to observe that Cannabis lineages still present a distinctive high-middle-low latitude distribution pattern after several thousand years despite human activities. Nevertheless, each of the three haplogroups is not strictly limited to its main corresponding geographical locations: North of 40° N (Haplogroup H), 30 to 40° N (Haplogroup H), and South of 30° N (Haplogroup L). Some haplotypes of the haplogroups were aberrantly growing out of the main distribution latitude range (Figure 1A). For instance, haplotype H3 in cultivar C224, which belongs to Haplogroup H, was found in lower latitude areas around 31° N; while the haplotype H5 in wild population XH and ZL, which belongs to Haplogroup L, was found at a higher latitude area around 43° N. These exceptions may result from the influences of human agricultural activities. Clarke and Merlin (2016) have stated, “Humans and the Cannabis plant share an intimate history spanning millennia.” There might have been much more stringent distribution limits between haplogroups prior to human activities (see below).
The high genetic diversity of this crop has been reported based on nuclear genetic markers (Gao et al., 2014; Sawler et al., 2015; Soler et al., 2017), but this is the first report of genetic diversity from cpDNA markers. The rather low mutation rate among numerous organelle loci of Cannabis (Gilmore et al., 2007; Zhang et al., 2017), makes genetic analyses of populations based on single organelle sequence extremely difficult. Our results revealed a high level of haplotype diversity (Hd = 0.848) at the species level, a strong genetic differentiation among the three groups (Fst = 0.695), and the molecular variations observed are mostly between-cultivars (76.85%) or among groups (69.48%). It is worth noting that genetic variation at different levels of hierarchy contrasts to previous studies based on nuclear markers (Gilmore et al., 2003; Chen et al., 2015; Soler et al., 2017), where the largest molecular variation observed was due to differences within cultivars, instead of among cultivars. These contrasting results are probably due to the fact that the cpDNA markers are maternally inherited, and detect therefore variations only from the maternal parent, instead of an unspecified mixture of both parents, which occurs for nuclear markers. The haploid and non-recombining nature of the cpDNA makes it possible to better trace genealogical histories in plant populations (Avise, 2009).
Three Subspecies Classification
The genus Cannabis was previously placed in family Moraceae, then in its own family Cannabaceae together with Humulus (Rendle, 1925). This family contains ten genera based on molecular phylogenies (Sytsma et al., 2002; Mabberley, 2008; Yang et al., 2013). The cultivation and selection of hemp has been performed for several thousand years, and this has resulted in difficulty when classifying Cannabis accessions based only on morphological traits. In recent studies, three lineages have been identified in Cannabis by enzyme variants analysis (Hillig, 2005), 7 polymorphic sites of organelle DNA sequences (Gilmore et al., 2007), and EST-SSR markers (Gao et al., 2014) based on worldwide sampling. However, whether these three lineages should be treated as three distinct species, three varieties of a single species or other taxonomic treatments have been debated (Hillig, 2005; Gilmore et al., 2007; Piluzza et al., 2013; Small, 2015; Mcpartland and Hegman, 2018). In the present study, 645 Cannabis individuals (all 43 populations from China and 9 accessions from the other countries or regions) were split into three gene pools without exception. On the phylogenetic tree, all Cannabis haplotypes formed a monophyletic clade (Figure 2B) containing three distinct subclades, with each subclade significantly different from the others (Figure 2A). At first glance, the three distinct subclades could be treated as three different species corresponding to the three commonly recognized species C. sativa, C. indica, and C. ruderalis. However, there is no reproductive isolation that exists between these lineages in nature based on our observations as well as recognitions by most researchers (Beutler and Marderosian, 1978). Furthermore, few sequence variations have been detected in Cannabis chloroplast DNA: < 0.03% for the whole chloroplast genomes based on four Cannabis cultivars and < 0.24% for the 16 cpDNA non-coding regions based on six individuals of wild Cannabis (Zhang et al., 2017); < 0.1% for the 7 cpDNA regions (Gilmore et al., 2007). In addition, significantly lower divergence (0.41%) was observed between materials identified as C. sativa and C. indica based on DNA barcoding sequences (rbcL, matK, trnH-psbA, trnL-trnF, ITS), compared to the mean divergence of 3.0% that separated five pairs of plants considered as different species such as Humulus lupulus and H. japonicus in Canabaceae (McPartland and Guy (2014). These accumulating pieces of evidence also hint that a rank below that of species is more reasonable. Thus we suggest that Cannabis should be considered as a monotypic genus with only one species, Cannabis sativa L. Considering that the three distinctive lineages revealed by cpDNA molecular markers also clearly demonstrated obvious geographic regions as stated above, this species can be further divided into three subspecies. Meanwhile, based on nomenclature history of this species, original geographic range, and basic difference in phenotype, we recommend the naming of the three subspecies as: Cannabis sativa subsp. sativa, C. sativa subsp. indica, and C. sativa subsp. ruderalis, corresponding to the Haplogroup M, Haplogroup L, and Haplogroup H, respectively. Small and Cronquist (1976) also pointed out that C. sativa subsp. sativa is typically distributed at areas with latitudes north of 30° N. Our present results that the haplogroup M (i.e., subsp. sativa) is distributed in areas ranging from 27 to 43° N, is largely consistent with the observations by Small and Cronquist.
Divergence Time Inference and Evolutionary History
In the present study, we included Aphananthe aspera, the basal taxon of the family Cannabaceae, and all the three Humulus species (the sister group of Cannabis) as outgroups for the dating analysis based on cpDNA markers and large numbers of Cannabis individuals. The reconstructed phylogenetic tree (Figure 2) shows the stem age of C. sativa is at 18.23 Ma (95% HPD: 8.84–36.6 Ma), which means Humulus and Cannabis diverged from a common ancestor before 18.23 Ma. This time period is in agreement with the divergence time (about 14 Ma) inferred by Zhang et al. (2017). In fact, the history of Cannabis, Humulus and their extinct sister genus can be dated back to the Oligocene and Miocene Epoch (33.9–5.33 Ma) according to the fossil records (Tiffney, 1986; McPartland, 2018). The crown age of C. sativa is at 2.24 Ma (95% HPD: 0.81–5.81 Ma), which is also the stem age of the three lineages. This diversification time coincides with the Quaternary glaciation, the last of five known glaciations during Earth's history which is thought to have started at 2.58 Ma, indicating that the Quaternary glaciation could have played a major role in the evolutionary history of the three subspecies of C. sativa. The current distribution of the three subspecies could be explained as a consequence of secondary contact after historical divergence events.
The Central-Asia-Origin has been the prevalent opinion for C. saltiva (de Candolle, 1885; McPartland, 2018), although some botanists considered Europe as the center of origin (Thiébaut de Berneaud, 1835; Keppen, 1886), or a region spanning Asia and Europe (Herder, 1892; Vavilov, 1926). However, our molecular analyses revealed for the first time that the low latitude region distributed subsp. indica (Haplogroup L) possesses the basal group position within Cannabis, indicating that this species is possibly originated from low latitude areas in the evolutionary history of this plant. This finding does not support the hypothesis of the Central-Asia-origin of Cannabis, but is partly in agreement with the speculation of Linnaeus (1737) that the native range of C. saltiva was India Orientali (encompassing the Indian subcontinent, southeastern Asia, and the Malay Archipelago), Japonia (Japan), and Malabaria (the Malabar coast of southwest India). Indeed, the seeds from wild Cannabis populations in India are remarkably small, unlike those collected from any other area, also indicating that the wild Indian populations may be an ancient wild form (Small, 2015).
Multiregional Domestication Origin of Cannabis Plant
Each of the three haplogroups (M, L, and H) identified in this study contains haplotypes from both wild populations and cultivars. Within each haplogroup, the wild and domesticated populations shared the most common haplotypes. For instance, haplotype H1, H3, and H4 are the most common haplotypes shared by the wild and domesticated populations in Group H; similar trends are observed for haplotype H9 in Group M, and haplotypes H5 and H10 in the Group L. The fact that the haplotype of the domesticated Cannabis cultivars are not limited to one of the three haplogroups indicates that there are probably multiregional domestication origins for this crop from the three subspecies of Cannabis. Otherwise, the same genotype (haplogroup) should have been detected in different cultivars from high-middle-low latitude regions if the cultivars were domesticated from one single region. AMOVA analyses results also demonstrate that there is no significant difference (Fst = 0.023) between the wild population group and domesticated cultivar group based on cpDNA data. This molecular evidence is in accordance with the multiregional origin of human use of the cannabis plant proposed based on archaeological investigation (Long et al., 2016) and Fossil pollen studies (Mcpartland et al., 2018). Actually, contemporaneous cannabis achenes (5,000–10,200 years ago) have been found in more than ten different archaeological sites located in the two distal parts (both Europe and East Asia) of the continent (Long et al., 2016). Thus the domestication of C. sativa could have occurred in more than three areas in Eurasia.
Author Contributions
QZ designed and performed the research, analyzed the data, and wrote the manuscript. QZ and XC contributed equally as first author. LT and ES carried out pre-experiment research and revised the manuscript in detail. HG, RG, MG, and YX conducted field work. XC and MY provided the technical assistance. MY organized this work. All authors contributed to and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31360350) and the project of China Agriculture Research System (CAR-16-E07).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are very grateful to Dr. Zai-Wei Ge and Bang Feng from Kunming Institute of Botany, Chinese Academy of Sciences, for helping in dating analyses.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.01876/full#supplementary-material
References
Amaducci, S., Colauzzi, M., Bellocchi, G., Cosentino, S. L., Pahkala, K., Stomph, T.J., et al. (2012). Evaluation of a phenological model for strategic decisions for hemp (Cannabis sativa L.) biomass production across European sites. Ind. Crops Prod. 37, 100–112. doi: 10.1016/j.indcrop.2011.11.012
Amaducci, S., Scordia, D., Liu, F. H., Zhang, Q., Guo, H., Testa, G., et al. (2015). Key cultivation techniques for hemp in Europe and China. Ind. Crops Prod. 68, 2–16. doi: 10.1016/j.indcrop.2014.06.041
Avise, J. C. (2000). Phylogeography: The History and Formation of Species. Cambridge: Harvard University Press, 248.
Avise, J. C. (2004). Molecular Markers Natural History, and Evolution. Sunderland: Sinauer and Associates Press, 78–79.
Avise, J. C. (2009). Phylogeography: retrospect and prospect. J. Biogeogr. 36, 3–15. doi: 10.1111/j.1365-2699.2008.02032.x
Bandelt, H. J., Forster, P., and Rohl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Beutler, J. A., and Marderosian, A. H. (1978). Chemotaxonomy of Cannabis I. crossbreeding between Cannabis sativa and C. ruderalis, with analysis of cannabinoid content. Econ. Bot. 32, 387–394. doi: 10.1007/BF02907934
Chen, X., Guo, R., Wan, R. X., Xu, Y. P., Zhang, Q. Y., Guo, M.B., et al. (2015). Genetic structure of five dioecious industrial hemp varieties in Yunnan. Mol. Plant Breed. 13, 2069–2075. doi: 10.13271/j.mpb.013.002069 (in Chinese)
Clarke, R. C., and Merlin, M. D. (2016). Cannabis domestication, breeding history, present-day genetic diversity, and future prospects. Crit. Rev. Plant Sci. 35, 293–327. doi: 10.1080/07352689.2016.1267498
de Candolle, A. (ed.). (1885). “Hemp – Cannabis sativa L.” in Origin of Cultivated Plants (New York, NY: D. Appleton), 148–149. doi: 10.5962/bhl.title.29067
Doyle, J. (1991). “DNA protocols for plants-CTAB total DNA isolation.” in Molecular Techniques in Taxonomy, eds G. M. Hewitt, A. W. B. Johnston, and J. P. W. Young (Berlin: Springer), 283–293. doi: 10.1007/978-3-642-83962-7_18
Drummond, A. J., and Rambaut, A. (2007). BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Ervín, K., Hans, D. M, and František, H. (1986). Monographie der Frúchte und Samen in der Kreide von Mitteleuropa. Prague: Vydal ústrední ústav geologický v Academii, nakladatelství Ceskoslovenské akademie ved, 219.
Excoffier, L., and Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Gao, C., Xin, P., Cheng, C., Tang, Q., Chen, P., Wang, C., et al. (2014). Diversity analysis in Cannabis sativa based on large-scale development of expressed sequence tag-derived simple sequence repeat markers. PLoS ONE 9:e110638. doi: 10.1371/journal.pone.0110638
Gilmore, S., Peakall, R., and Robertson, J. (2003). Short tandem repeat (STR) DNA are hypervariable and informative in Cannabis sativa L.: implications for forensic investigations. Forensic Sci. Intl. 9, 65–74. doi: 10.1016/S0379-0738(02)00397-3
Gilmore, S., Peakall, R., and Robertson, J. (2007). Organelle DNA haplotypes reflect crop-use characteristics and geographic origins of Cannabis sativa. Forensic Sci. Intl. 172, 179–190. doi: 10.1016/j.forsciint.2006.10.025
Hillig, K. W. (2005). Genetic evidence for speciation in Cannabis (Cannabaceae). Genet. Resour. Crop Evol. 53, 161–180. doi: 10.1007/s10722-003-4452-y
Hillig, K. W., and Mahlberg, P. G. (2004). A chemotaxonomic analysis of cannabinoid variation in Cannabis (Cannabaceae). Am. J. Bot. 91, 966–975. doi: 10.3732/ajb.91.6.966
Johnson, R. (2013). Hemp as an Agricultural Commodity. CRS Report for Congress. Congressional Research Service. Available online at: https://www.fas.org/sgp/crs/misc/RL32725.pdf (Accessed August 15, 2018).
Keppen, T. (1886). Догадка о происхожденiи большинства индоевропейскихъ названiй конопли. Журнал Министерства народнаго просвˇeщенiя. 245, 73–86.
Li, H. L. (1974). An archaeological and historical account of Cannabis in China. Econ. Bot. 28, 437–448. doi: 10.1007/BF02862859
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Long, T., Wagner, M., Demske, D., Leipe, C., and Tarasov, P. E. (2016). Cannabis in eurasia: origin of human use and bronze age trans-continental connections. Veget. Hist. Archaeobot. 26, 245–258. doi: 10.1007/s00334-016-0579-6
Mabberley, D.J. (2008). Mabberley's Plant-Book: A Portable Dictionary of Plants, their Classification and Uses. New York, NY: Cambridge University Press.
McPartland, J. M. (2018). Cannabis systematics at the levels of family, genus, and species. Cannabis Cannabinoid Res. 3, 203–212. doi: 10.1089/can.2018.0039
McPartland, J. M., and Guy, G. W. (2004). “The evolution of Cannabis and coevolution with the cannabinoid receptor - a hypothesis,” in The Medicinal Uses of Cannabis and Cannabinoids, eds G. W. Guy, B. A. Whittle, and P. J. Robson (London: Pharmaceutical Press), 71–101.
McPartland, J. M., and Guy, G. W. (2014). “A question of rank: Using DNA barcodes to classify Cannabis sativa and Cannabis indica,” in Proceedings of the 24th annual symposium on the Cannabinoids. International Cannabinoid Research Society (Research Triangle Park, NC).
Mcpartland, J. M., and Guy, G. W. (2017). Models of cannabis, taxonomy, cultural bias, and conflicts between scientific and vernacular names. Bot. Rev. 83, 1–55. doi: 10.1007/s12229-017-9187-0
Mcpartland, J. M., Guy, G. W., and Hegman, W. (2018). Cannabis, is indigenous to europe and cultivation began during the copper or bronze age: a probabilistic synthesis of fossil pollen studies. Veget. Hist. Archaeobot. 27, 635–638. doi: 10.1007/s00334-018-0678-7
Mcpartland, J. M., and Hegman, W. (2018). Cannabis, utilization, and diffusion patterns in prehistoric europe: a critical analysis of archaeological evidence. Veget. Hist. Archaeobot. 27, 627–634. doi: 10.1007/s00334-017-0646-7
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from small number of individuals. Genetics 89, 583–590.
Nylander, J. A. A. (2004). MrModeltest v2. Program Distributed by the Author. Evolutionary Biology Centre, Uppsala University.
Oh, H., Seo, B., Lee, S., Ahn, D.H., Jo, E., Park, J.K., et al. (2016). Two complete chloroplast genome sequences of Cannabis sativa varieties. Mitochondrial DNA Part A 27, 2835–2837. doi: 10.3109/19401736.2015.1053117
Pahkala, K., Pahkala, E., and Syrjäläl, H. (2008). Northern limits to fibre hemp production in Europe. J. Ind. Hemp. 13, 104–116. doi: 10.1080/15377880802391084
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Piluzza, G., Delogu, G., Cabras, A., Marceddu, S., and Bullitta, S. (2013). Differentiation between fiber and drug types of hemp (Cannabis sativa) from a collection of wild and domesticated accessions. Genet. Resour. Crop Evol. 60, 2331–2342. doi: 10.1007/s10722-013-0001-5
Piomelli, D., and Russo, E. B. (2016). The cannabis sativa versus cannabis indica debate: an interview with Ethan Russo, MD. Cannabis Cannabinoid Res. 1, 44–46. doi: 10.1089/can.2015.29003.ebr
R Core Team (2015). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.r-project.org
Rambaut, A., Suchard, M., Xie, D., and Drummond, A. (2014). Tracer v1. 6. Computer Program and Documentation Distributed by the Author. Available online at: http://beast.bio.ed.ac.uk/Tracer
Rendle, A.B. (1925). The Classification of Flowering Plants, Vol. 2, London: Cambridge University Press.
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Salentijn, E. M. J., Zhang, Q. Y., Amaducci, S., Yang, M., and Trindade, L. M. (2015). New developments in fiber hemp (Cannabis sativa L.) breeding. Ind. Crops Prod. 68, 32–41 doi: 10.1016/j.indcrop.2014.08.011
Sawler, J., Stout, J. M., Gardner, K. M., Hudson, D., Vidmar, J., Butler, L., et al. (2015). The genetic structure of marijuana and hemp. PLoS ONE 10:e0133292. doi: 10.1371/journal.pone.0133292
Schaal, B. A., Hayworth, D. A., Olsen, K. M., Rauscher, J. T., and Smith, W. A. (1998). Phylogeographic studies in plants: problems and prospects. Mol. Ecol. 7, 465–474. doi: 10.1046/j.1365-294x.1998.00318.x
Schultes, R., Klein, W., Plowman, T., and Lockwood, T. (1974). Cannabis: an example of taxonomic neglect. Bot. Museum Leaflets Harvard Univ. 23, 337–364.
Small, E. (2015). Evolution and classification of Cannabis sativa, (marijuana, hemp) in relation to human utilization. Bot. Rev. 81, 189–294. doi: 10.1007/s12229-015-9157-3
Small, E., and Cronquist, A. (1976). A practical and natural taxonomy for Cannabis. Taxon 25, 405–435. doi: 10.2307/1220524
Soler, S., Gramazio, P., Figàs, M. R., Vilanova, S., Rosa, E., Llosa, E. R., et al. (2017). Genetic structure of Cannabis sativa var. indica cultivars based on genomic ssr (gssr) markers: implications for breeding and germplasm management. Ind. Crops Prod. 104, 171–178. doi: 10.1016/j.indcrop.2017.04.043
Spitters, C. J. T., Toussaint, H. A. J. M., and Goudriaan, J. (1986). Separating the diffuse and direct component of global radiation and its implications for modeling canopy photosynthesis. Part I. Components of incoming radiation. Agric. Forest Meteorol. 38, 217–229. doi: 10.1016/0168-1923(86)90060-2
Sytsma, K. J., Morawetz, J., Pires, J. C., Nepokroeff, M., Conti, E., Zjhra, M., et al. (2002). Urticalean rosids: circumscription, rosid ancestry, and phylogenetics based on rbcL, trnL-trnF, and ndhF sequences. Am. J. Bot. 89, 1531–1546. doi: 10.3732/ajb.89.9.1531
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Thiébaut de Berneaud, A. (1835). “Chanvre,” in Dictionnaire Pittorosque d'histoire Naturelle et des Phénomènes de la Nature, Tome 2, ed F. E. Guérin-Méneville (Paris: De Cosson), 87–89.
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Tiffney, B.H. (1986). Fruit and seed dispersal and the evolution of the Hamamelidae. Ann. Mo Bot. Gard. 73, 394–416. doi: 10.2307/2399119
Vavilov, N. I. (1926). “The origin of the cultivation of ‘primary' crops, in particular cultivated hemp,” in Studies on the Origin of Cultivated Plants, Leningrad, USSR: Institute of Applied Botany and Plant Breeding, 221–233.
Vergara, D., White, K. H., Keepers, K. G., and Kane, N. C. (2016). The complete chloroplast genomes of Cannabis sativa and Humulus lupulus. Mitochondrial DNA Part A 27, 3793–3794. doi: 10.3109/19401736.2015.1079905
Welling, M. T., Shapter, T., Rose, T. J., Lei, L., Stanger, R., and King, G. J. (2016). A belated green revolution for cannabis: virtual genetic resources to fast-track cultivar development. Front. Plant Sci. 7:1113. doi: 10.3389/fpls.2016.01113
Yang, M. Q., Velzen, R. V., Bakker, F. T., Sattarian, A., Li, D. Z., and Yi, T. S. (2013). Molecular phylogenetics and character evolution of Cannabaceae. Taxon 62, 473–485. doi: 10.12705/623.9
Yang, X. Y. (1991). History of cultivation on hemp, sesame and flax. Agric. Archaeol. 3, 267–274. (in Chinese)
Yuan, Z. X., Xin, F., and Du, C. X. (2014). Analysis of the variation of photoperiod with latitude and season. Sustainable Energy 4, 41–50. (in Chinese) doi: 10.12677/SE.2014.44007
Zhang, Q. Y., Chen, X., Guo, M. B., Guo, R., Xu, Y. P., Yang, M., et al. (2017). Screening and development of chloroplast polymorphic molecular markers on wild hemp (Cannabis sativa L.), Mol. Plant Breed. 5, 979–985. doi: 10.13271/j.mpb.015.000827 (in Chinese).
Keywords: Cannabaceae, industrial hemp, genetic diversity, phylogeography, cpDNA
Citation: Zhang Q, Chen X, Guo H, Trindade LM, Salentijn EMJ, Guo R, Guo M, Xu Y and Yang M (2018) Latitudinal Adaptation and Genetic Insights Into the Origins of Cannabis sativa L.. Front. Plant Sci. 9:1876. doi: 10.3389/fpls.2018.01876
Received: 19 August 2018; Accepted: 05 December 2018;
Published: 21 December 2018.
Edited by:
Donald Lawrence Smith, McGill University, CanadaReviewed by:
Yoshiko Shimono, Kyoto University, JapanUmesh K. Reddy, West Virginia State University, United States
Copyright © 2018 Zhang, Chen, Guo, Trindade, Salentijn, Guo, Guo, Xu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingying Zhang, aGVtcHpoYW5ncWluZ3lpbmdAMTI2LmNvbQ==;
Ming Yang, eW1oZW1wQDE2My5jb20=
†These authors have contributed equally to this work