 Silvia Crasto
Silvia Crasto Ilaria My
Ilaria My Elisa Di Pasquale
Elisa Di Pasquale- 1Humanitas Clinical and Research Center – IRCCS, Rozzano, Italy
- 2Institute of Genetic and Biomedical Research (IRGB) – UOS of Milan, National Research Council (CNR), Milan, Italy
Mutations of Lamin A/C gene (LMNA) cause laminopathies, a group of disorders associated with a wide spectrum of clinically distinct phenotypes, affecting different tissues and organs. Heart involvement is frequent and leads to cardiolaminopathy LMNA-dependent cardiomyopathy (LMNA-CMP), a form of dilated cardiomyopathy (DCM) typically associated with conduction disorders and arrhythmias, that can manifest either as an isolated event or as part of a multisystem phenotype. Despite the recent clinical and molecular developments in the field, there is still lack of knowledge linking specific LMNA gene mutations to the distinct clinical manifestations. Indeed, the severity and progression of the disease have marked interindividual variability, even amongst members of the same family. Studies conducted so far have described Lamin A/C proteins involved in diverse biological processes, that span from a structural role in the nucleus to the regulation of response to mechanical stress and gene expression, proposing various mechanistic hypotheses. However, none of those is per se able to fully justify functional and clinical phenotypes of LMNA-CMP; therefore, the role of Lamin A/C in cardiac pathophysiology still represents an open question. In this review we provide an update on the state-of-the-art studies on cardiolaminopathy, in the attempt to draw a line connecting molecular mechanisms to clinical manifestations. While investigators in this field still wonder about a clear genotype/phenotype correlation in LMNA-CMP, our intent here is to recapitulate common mechanistic hypotheses that link different mutations to similar clinical presentations.
Introduction
LMNA Gene and Its Products
LMNA gene maps to chromosome 1q21.1-21.2 and is composed of 12 exons spanning around 25 kb. It encodes A-type nuclear lamins via alternative splicing (Lin and Worman, 1993; Wydner et al., 1996): Lamin A and C (Lamin A/C) represents the two main isoforms, while Lamin C2 and AD10 are described in germ cells and cancer cells, respectively (Machiels et al., 1996; Alsheimer et al., 2000). A-type lamin proteins have a tripartite domain organization with a central rod domain, a short N-terminal head domain and a tail C-terminal domain. Unlike Lamin C, Lamin A is translated as Prelamin A, containing a carboxyl-terminal CaaX motif, which is then modified by carboxymethylation and farnesylation and undergoes sequential post-translational modifications to form mature Lamin A (Herrmann and Aebi, 2004; Young et al., 2005). Nuclear lamins are classified as type V intermediate filament (IF) proteins and represent the major elements that constitute the nuclear lamina (NL). In addition to their well-known structural role in the nucleus, Lamin A/C are increasingly considered as key players in regulating gene transcription through both direct and indirect modulation of chromatin organization, DNA replication and signal transduction pathways (Gruenbaum et al., 2005; Dechat et al., 2008; Dittmer and Misteli, 2011).
Unlike B-type lamins, the expression of A-type lamins is developmentally regulated and mostly occurs in differentiated cells (Worman and Bonne, 2007). During mouse embryogenesis, Lamin A/C start to be expressed around day 8–9 in extra-embryonic tissues and few days later (day 10–12) in the embryo (Dittmer and Misteli, 2011). The cell-specific and temporally regulated expression of these proteins supports a fundamental role of Lamin A/C in cell differentiation, lineage specification and tissue development (Worman and Bonne, 2007; Butin-Israeli et al., 2012).
Laminopathies
Mutations in the LMNA gene cause laminopathies, a group of disorders characterized by phenotypically heterogeneous manifestations. Up to now a total of 498 LMNA mutations have been described1 associated to more than 15 different phenotypes. Indeed, laminopathies can either specifically affect distinct tissues, including striated muscles, the peripheral nerves, or the adipose tissue, or present as a systemic disease affecting concomitantly several organs similar to premature aging syndromes. However, there is growing evidence of overlapping phenotypes, suggesting the presence of a real continuum within the disease.
Among the different phenotypes, cardiac involvement is one of the most prevalent and severe manifestations, being a hallmark of several laminopathies, such as Emery-Dreifuss muscular dystrophy, Limb-girdle muscular dystrophy 1B and Hutchinson-Gilford Progeria Syndrome (HGPS) (Fatkin et al., 1999; Arbustini et al., 2002; van Tintelen et al., 2007a). Despite the recent developments in the field, the understanding of the pathogenetic role of Lamin A/C in cardiac disease is still incomplete.
In this review we aim to provide an update on the state-of-the-art investigations in this area of research, drawing a definite line from the molecular mechanisms toward the clinical manifestations. Although investigators are still working on defining genotype/phenotype correlations, our goal is to highlight common molecular features that could link different LMNA variants to a similar clinical presentation.
From Lmna Variants to Cardiac Phenotypes
LMNA Variants
The first reports that associate LMNA mutations to cardiac diseases date back to 1999 (Bonne et al., 1999; Fatkin et al., 1999): these reports represented a revolution in the field, identifying for the first time LMNA mutations as responsible for dilated cardiomyopathy (DCM) associated with conduction system disease. Jakobs et al. (2001) described several novel LMNA mutations, one missense (p.E203K, nucleotide c.G607A) in 14 individuals and one nonsense (p.R225X, nucleotide c.C673T) in 10 subjects: these patients were diagnosed with DCM and variable conduction system disease, and were free from any skeletal muscle phenotype. Few years later, other mutations in LMNA gene were described (van Tintelen et al., 2007a,van Tintelen et al., 2007b; Saga et al., 2009): one nonsense mutation (815_818delinsCCAGAC) and some missense variants (p.N195K; p.Y259H; p.R166P). Overtime, LMNA gene emerged as the second most commonly mutated gene associated to familial cardiomyopathy (CMP), accounting for ∼6–8% of the cases (Hershberger and Siegfried, 2011) of idiopathic DCM, with this number raising up to 33% for cases presenting with both DCM and conduction defects (McNally and Mestroni, 2017).
Cardiolaminopathy
Cardiomyopathy caused by mutations in LMNA gene is referred to as cardiolaminopathy (LMNA-CMP). The disease is generally characterized by variable extent of ventricular dilation (Bonne et al., 1999; Fatkin et al., 1999) or, less frequently, by left ventricular (LV) non-compaction (Sedaghat-Hamedani et al., 2017).
LMNA-CMP has been linked to 165 unique mutations, distributed along the entire gene (Tesson et al., 2014). However, the majority of the mutations occur in the head and in the rod domains and rarely in the tail domain (Fatkin et al., 1999; Jakobs et al., 2001). Pathogenic variants are mainly missense and nonsense mutations, while fewer small deletions/insertions have been identified (Dittmer et al., 2014; Zahr and Jaalouk, 2018). Haploinsufficiency has been suggested as mechanism of disease in patients carrying truncating variants, while missense mutations have been proposed to act mainly through a dominant negative pathway. Subjects with truncating mutations have been associated to an earlier onset of cardiac conduction defects and atrial arrhythmias and a lower LV ejection fraction (EF), than those with missense mutations (Nishiuchi et al., 2017). However, so far there is still lack of knowledge explaining the link between specific LMNA mutations and a defined phenotype, and the severity and the progression of the disease have marked interindividual variability, not only among unrelated probands, but also within members of the same family. This supports the concept that the final phenotype results not only from the single LMNA mutation, but is also influenced by modifying genes or environmental cues, similarly to what has been reported in some families with titin and desmin mutations (Muntoni et al., 2008; Granger et al., 2011; Roncarati et al., 2013). Whole-exome and whole-genome sequencing studies will unequivocally facilitate the investigation of the genetics behind the disease and allow a comprehensive understanding of the mechanisms underlying the final clinical presentation.
Cardiac Conduction System Disease and Arrhythmias
The clinical course of LMNA-CMP is characterized by a high rate of major cardiac events such as sudden cardiac death (SCD), malignant ventricular tachycardia (VT), extreme bradycardia due to a high degree of atrioventricular block (AVB) and end-stage heart failure. What is intriguing is the common finding that laminopathies often manifest as primary arrhythmia. Bradyarrhythmias and supraventricular tachyarrhythmias as atrial fibrillation and flutter often anticipate by decades the development of DCM. For this reason, genetic screening should be considered in young patients presenting with new AVB or atypical atrial arrhythmias, even in the absence of LV dysfunction. In the study by Kumar et al. (2016), only one-half of patients have an LVEF < 50% at the initial medical consultation.
The pathophysiological mechanisms underlying the arrhythmic phenotype are still not well elucidated. Systolic dysfunction, male sex, non-missense mutations and non-sustained VT are considered predictors of malignant ventricular arrhythmias in LMNA-CMP (Van Rijsingen et al., 2012; Hasselberg et al., 2014; Kumar et al., 2016; Nishiuchi et al., 2017). However, such risk factors are debated in some studies (Nishiuchi et al., 2017; Peretto et al., 2019) and it was recently shown in a large cohort of DCM patients, that carriers of LMNA variants experience the highest rates of SCD/VT/ventricular fibrillation (VF), which was independent of the LV EF (Gigli et al., 2019). Further studies need to be conducted in order to clarify to which extent phenotypic differences among the different cohorts of patients are dependent on genetic background rather than on specific LMNA variants.
Clinical Presentation
The presentation of DCM does not show specific characteristics and the expression of the DCM pattern was found to be age-dependent, with development of the phenotype between 20 and 39 years of age in two thirds of the cases and complete penetrance by 60 years (Arbustini et al., 2002). The “red flags” predicting higher chance of LMNA mutations are the concomitant presence of conduction defects and skeletal muscle involvement, even if creatine phosphokinase (CPK – serum marker of muscular damage) is elevated only in one third of the cases (Fatkin et al., 1999; Rapezzi et al., 2013). Arbustini et al. (2002) showed that approximately 33% of patients with AVB and cardiomyopathy carries a LMNA mutation. For this reason, screening for LMNA mutations in young patients with idiopathic DCM, especially when it is associated with atrial arrhythmias and/or AVB, is important for prognosis and genetic counseling.
Patients who have both cardiac and neuromuscular manifestations more commonly experience bradyarrhythmias and atrial fibrillation compared to patients with solely cardiac phenotypes, despite having no differences in structural heart disease. In this group of patients cardiac abnormalities do not strictly correlate with the severity of the neuromuscular involvement, which may suggest distinct pathogenetic mechanisms (Benedetti et al., 2007; Hasselberg et al., 2018; Peretto et al., 2019). However, a recent study on a small cohort of patients showed that the neuromuscular presentation was associated with earlier cardiac involvement, characterized by a linear and progressive evolution from rhythm disorders to cardiomyopathy (Ditaranto et al., 2019).
Prognosis and Risk Stratification
Cardiolaminopathy often presents an aggressive and rapid evolution, with a worse natural history compared to other forms of non-ischemic dilated cardiomyopathies and have higher prevalence of malignant arrhythmias and cardiac transplantation (Pasotti et al., 2008; Kayvanpour et al., 2017).
Positive genetic testing for LMNA mutations has crucial clinical and prognostic implications. Mortality in patients with LMNA-CMP is estimated to be 40% at 5 years (Pasotti et al., 2008), whereas 45% suffered SCD or aborted SCD. Currently, LMNA mutations represent the only genetic background in DCM that influences international guidelines-based timing of ICD therapy in primary prevention, regardless of LV EF values (Priori et al., 2013; Priori and Blomström-Lundqvist, 2015).
Recently, a new 5-year prediction model for life threatening ventricular tachyarrhythmia (LTVTA) has been proposed (Wahbi et al., 2019) to assist us in deciding whether or not a candidate is eligible for the placement of an ICD implantation (online calculator available at http://lmna-risk-vta.fr/). Predictors of LTVTA in the analyzed sample were male sex, non-missense LMNA mutation, 1st degree and higher AV block, non-sustained VT and LV EF. The risk threshold used enabled reclassification of 28.8% of patients compared with the guidelines-based approach.
New Molecular Targets Under Clinical Investigation
Currently, three new molecular targets, emerged from biochemical studies, are under investigation as possible pharmacological strategies to treat LMNA-CMP. The first approach involves a selective oral inhibitor of the p38 MAPK pathway is currently under a phase III clinical trial2. The second approach regards the application of mTOR pathway inhibitors that have shown promising improvements in terms of LV size and function in animal models (Choi et al., 2012), and the third approach focuses on PDGFR blockers and their ability to ameliorate the arrhythmic phenotype in in vitro models (Lee et al., 2019).
Molecular Hypotheses Behind the “Clinical Scenario”
As mentioned above, 498 different mutations in LMNA gene have been reported, of which 165 associated with LMNA-CMP. Since the discovery of the LMNA gene as causative of laminopathies, more than 1000 research and clinical studies have been published, aiming to establish a causal correlation between morphological and functional defects of laminopathic cells and the heterogeneous clinical phenotypes of this group of disorders. Although several hypotheses on the potential mechanisms have emerged from these studies, a unique view on the role of Lamin A/C defects in laminopathies is still missing. The same scenario also applies to CMPs due to LMNA mutations and the functional role of A-type lamins in the mammalian heart. Studies on animal models either lacking Lmna gene or transgenic for specific human LMNA variants, together with evidence from human cardiac models generated through iPSC (induced Pluripotent Stem Cell) technology have significantly contributed to increase our knowledge in the field of cardiolaminopathy. Independent investigations have proposed several hypotheses to explain molecular mechanisms underlying Lamin A/C action in the heart and their link to disease traits.
Nikolova et al. (2004) proposed that the softer nuclei’s structure associated to LMNA mutations and the resulting morphological abnormalities may be the molecular events at the basis of the Lamin-related cardiac phenotypes. This has been specifically referred to as the “structural hypothesis,” attributing the lamin-associated phenotypes to structural defects. Furthermore, being an integral part of the LINC complex (Linker of Nucleoskeleton and Cytoskeleton), A-type lamins may also contribute in regulating the structural architecture of the contractile tissue, conferring resistance and protection against any mechanical stress (Swift et al., 2013); this view was later referred to as the “mechano-transduction hypothesis.”
Alongside these “structural hypotheses,” a “gene transcription hypothesis” also emerged, starting from previous evidence suggesting a role of the inner nuclear membrane in regulating chromatin organization (and gene expression). A study from Ye and Worman (1996) provided the first piece of evidence of a link between NL and chromatin regulation: the authors finely demonstrated a specific binding between the human chromodomain proteins HP1 and the integral protein of the inner nuclear membrane LBR (Lamin B Receptor) in Drosophila, portraying for the first time, the association between heterochromatin and the inner nuclear membrane in eukaryotic cells. These findings added a new function to Lamin A/C proteins, demonstrating that their biological role is not just restricted to the control of nuclear shape and mechano-transduction, but extends to the active regulation of gene transcription. Following this trail, many other studies subsequently showed that several binding factors are able to simultaneously interact with the chromatin and the NL (Cohen et al., 2001; Wilson et al., 2001; Wilson and Foisner, 2010; Cesarini et al., 2015). This gene transcription hypothesis relies on a two-step model, in which the NL regulates transcription factors, either by sequestering them to the nuclear periphery – usually transcriptionally inactive – or by a mechanism that involves activation of specific signaling pathways, such as ERK1/2 (Arimura et al., 2005; Muchir et al., 2007; Chatzifrangkeskou et al., 2018). In addition to this, Lamin A/C is also able to directly bind chromatin, modulating its spatial organization. The interaction between chromatin and A-type lamins occurs at definite genomic regions, defined as lamin-associated domains (LADs) (Pickersgill et al., 2006). Interestingly, there is evidence showing a remodeling of LADs, in particular those tissue-specific (facultative LADs), in presence of Lamin A/C mutations. These rearrangements do not exclusively occur at the nuclear periphery, but also affect expression of genes located at the nuclear interior (Briand and Collas, 2018). For example, repositioning of T/BRACHYURY gene from the nuclear periphery toward the nuclear interior has been reported in fibroblasts of patients affected by familial partial lipodystrophy of Dunnigan type 2 (FPLD2) (Briand et al., 2018). Muck et al. (2012) also showed that knockdown of Lamin A/C in human HeLa cells, cause relocation of CFTR gene in the nuclear interior, without affecting the position of the neighboring GASZ and CORTBP2 genes. On the contrary, we recently demonstrated a preferential localization of SCN5A gene by the nuclear periphery in human iPSC-CMs carrying the p.K219T LMNA mutation, resulting in the inhibition of the expression of the gene and thus leading to reduction of sodium currents and impaired cell excitability (Salvarani et al., 2019).
In addition to this, lamin-chromatin interactions may also occur through several binding factors, identified as interacting-mediators between Lamin A/C and chromatin compartments (Kubben et al., 2010); interestingly some of these have been described as essential elements to regulate transcription of key tissue-specific genes, also in the heart (Briand et al., 2018; Salvarani et al., 2019). From this perspective, this mechanistic model of Lamin A/C action can be more accurately defined as “chromatin hypothesis”; importantly, Lamin A/C-driven chromatin regulation has been recently started to be addressed also in the field of cardiolaminopathy by studies from different research groups including ours (Bertero et al., 2019; Mozzetta and Tedesco, 2019; Salvarani et al., 2019). The general view emerging from chromosome conformation capture studies (HiC), a method to study three-dimensional architecture of genomes enabling to distinguish between transcriptionally active (A) and inactive (B) compartments, is that Lamin A/C haploinsufficiency is associated to an increase of intra-chromosomal interactions (meaning interactions between two active compartments – A-A), without interfering with inter-chromosome interactions (with different transcriptional status – A-B) or inducing massive changes in gene transcription (Bertero et al., 2019). According to that, four dimensional genome conformation (referred as spatial assemblies of heterochromatic topological associated domains – TADs, or “TAD cliques”) showed silencing of developmental genes in human adipose-derived stromal cells (ASCs), without reporting any A to B switch (Paulsen et al., 2019). On the other hand, the study from Lee et al. (2019) associated Lamin A/C haploinsufficiency, caused by a different frameshift LMNA mutation, with relevant LAD alterations, that are responsible for substantial changes in gene expression profile leading to altered activation of PDGF pathway. Although the “chromatin hypothesis” has been associated so far to few specific variants, it is likely that similar regulatory mechanisms are driven also by other Lamin A/C mutations, reinforcing the link between changes in transcriptional regulation and chromatin organization occurring in Lamin A/C mutant cells and the respective clinical phenotype.
Consistently, chromatin remodeling and the associated transcriptional changes of disease-relevant genes, is also the prevalent model proposed for the HGPS, the most severe LMNA-dependent disease (Goldman et al., 2004; Gordon et al., 2014; Aguado et al., 2019). Interestingly, a very recent study by Ikegami et al. (2020) further contributed to the field describing a new regulatory mechanism by Lamin A/C. Specifically, the authors describe a new regulatory function of the nucleoplasmic Ser22-phosphorylated (pS22) Lamin A/C, that they found specifically bound to a subset of active enhancers, and show the acquisition of new pS22-Lamin A/C binding site in HGPS cells, leading to upregulation of clinically relevant genes and potentially underlying the mechanism behind the disease phenotype.
Overall, data obtained so far from different studies in the field support a model by which all the proposed hypotheses can be causally connected, rather than mutually exclusive. As suggested by Osmanagic-Myers and Foisner (2019) in their Perspective, it is likely that a robust link between the structural abnormalities of laminopathic nuclei (Nikolova et al., 2004; Galiová et al., 2008) and either chromatin modifications or reorganization exists, thus contributing to a multifaceted mechanistic model underlying the pathogenesis of the disease.
Mouse Models
In the past two decades, several mouse models have been generated, either reproducing patients’ mutations (LMNA transgenic mice) or studying the consequences of Lmna deficiency (Lmna knockout mice). Lmna–/– mouse developed muscular dystrophy, DCM, neuropathy and displayed retarded growth rate, reduced stores of white fat and cardiac arrhythmia (Sullivan et al., 1999). These mice generally die by 8 weeks of age.
The heterozygous (Lmna±), with 50% of Lamin A/C expression, manifest conduction system diseases, ventricular dilatation later in adult life and generally die by 8 months of age (Chandar et al., 2010). At the molecular level, both the Lmna null and the Lmna haploinsufficiency mice showed altered desmin pathway and defective force transmission, leaning toward the mechano-transduction hypothesis. Furthermore, those models also showed an increase of pro-adipogenic factors (PPAPγ and CEBP/α), whereas Wnt-10/β-catenin levels were decreased (Tong et al., 2011). Another two Lmna knock-out models, LmnaGT–/– and LmnadelK32/delK32, have also contributed significantly to establish the role of Lamin A/C in postnatal maturation, showing developmental impairment and growth retardation, that mostly affect the adipose tissue, the skeletal muscle and the heart (Kubben et al., 2011; Bertrand et al., 2012).
Recently, the generation of LmnaH222P and LmnaN195K transgenic mice (Arimura et al., 2005; Mounkes et al., 2005) successfully recapitulated a faithful model of skeletal muscle and DCM, with no phenotype at neonatal stage and displaying disease traits similar to patients. The pathogenesis of LmnaH222P mice was linked to elevated MAP kinase 1/2 (ERK1/2) and AKT/mTor signaling pathways (Muchir et al., 2007; Chatzifrangkeskou et al., 2016). Antoku et al. (2019) recently further characterized this model, describing a novel relationship between the elevated levels of ERK1/2 and nuclear positioning in cardiomyocytes (CMs) isolated from LmnaH222P/H222P. The authors showed that a single phosphorylation site in the formin homology domain-containing proteins (FHOD) can be targeted by ERK1/2, changing cell polarity and negatively regulating cell migration. Nuclear positioning is strictly linked to the formation of the sarcomeres during skeletal muscle maturation (Sewry et al., 2001). Consistently, the authors showed that H222P mutation in the LMNA gene is associated to altered nuclear positioning and formation and function of sarcomeres, contributing to the pathogenesis of cardiomyopathy. On the other hand, LmnaN195K mice displayed an abnormal expression and/or localization of connexin 40 and connexin 43 and sarcomeres disorganization associated to the main DCM phenotype and conduction system defects (Mounkes et al., 2005). A complete list of the described mouse models and their relevant cardiac phenotypes and molecular characteristics are provided in the Table 1.
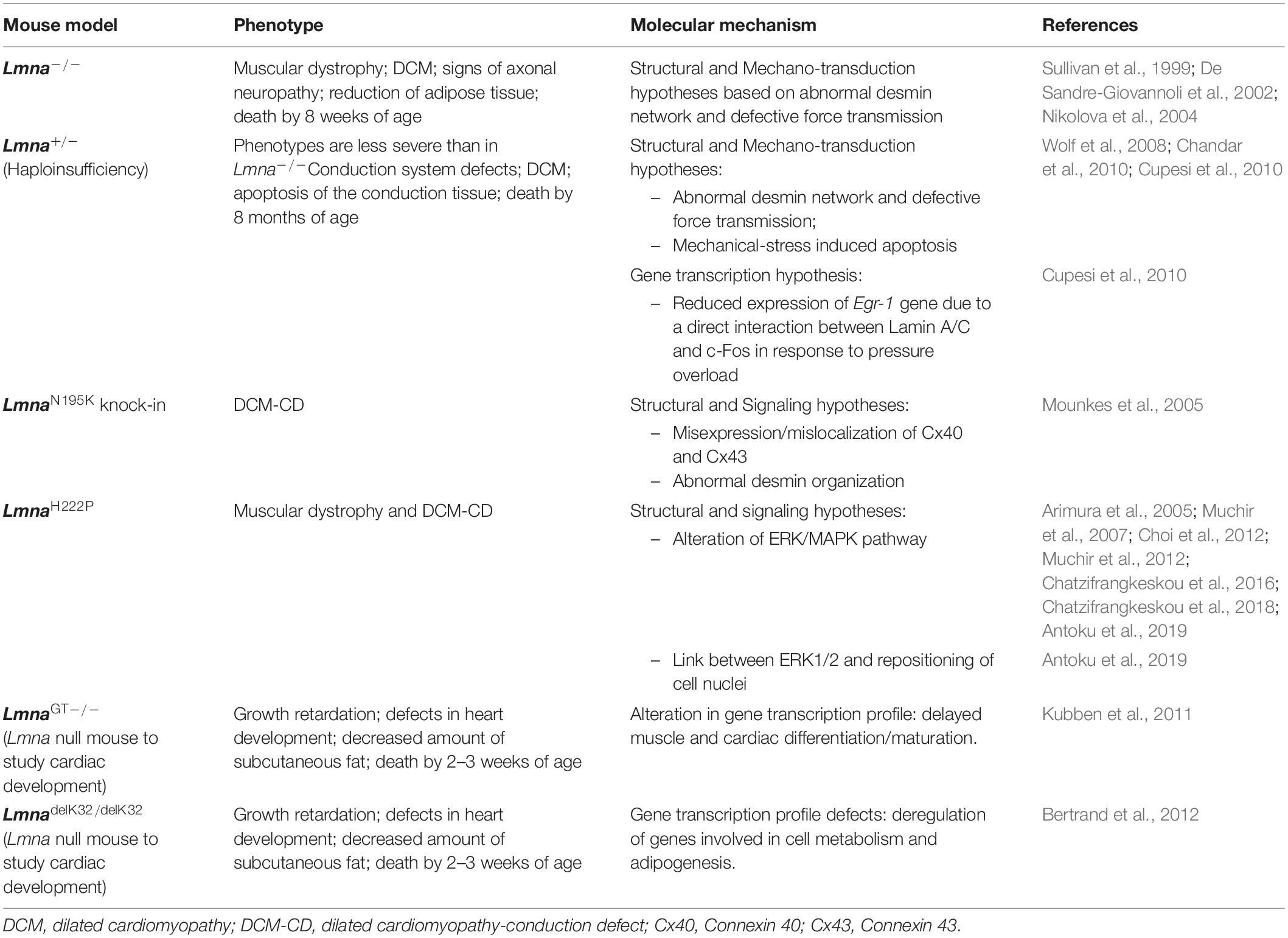
Table 1. Mouse models of cardiolaminopathy.
To conclude, despite likely species-specific limitations, studies conducted so far using mouse models have been fundamental to identify signaling pathways relevant to development of new therapeutics, such as the previously mentioned p38 inhibitor ARRY-371797 – already in Phase III clinical trial (NCT02057341) – and the N-acetyltransferare 10 (NAT10) inhibitor, Remodelin (Balmus et al., 2018). However, although these are important achievements, there is still an important lack of knowledge on the pathogenetic mechanisms underlying this complex disease and therefore further research in the field is mandatory to comprehensively tackle its molecular basis and develop more specific and effective therapies (Stewart et al., 2007; Zhang et al., 2013).
Cardiomyocytes From iPscs: a Powerful Platform to Model Human Cardiolaminopathy
The development of iPSC technology has been a groundbreaking revolution in all areas of research, allowing us to generate in vitro any cell type of interest, including human CMs, in which pathogenic mechanisms of diseases may be fully investigated (Xu et al., 2002; Moretti et al., 2010; Lodola et al., 2016; Lee J.H. et al., 2017). Research in the cardiolaminopathy field has also benefit of this technology, since use of iPSC-based models allowed us to overcome the major limitations linked to investigation on animal models and non-cardiac primary cells. Indeed, recreating human phenotypes in mice may not be feasible, due to species-specific differences between mice and humans, especially in relation to the cardiovascular system (i.e., electrophysiological properties of cardiac cells) (Davis et al., 2011). On the other hand, obtaining cells from the human heart may be difficult, due to the limited access the organ and the poor survival of the heart cells grown ex vivo. For these reasons studies on cardiolaminopathies so far have been mainly conducted on other somatic cell types, such as fibroblasts, skeletal muscle cells and adipocytes, which are easy to access and to maintain in vitro (Ho et al., 2011).
Thus far seven independent studies have investigated human LMNA-CMP using iPSCs either derived from patients carrying different LMNA mutations or generated through genome editing (Supplementary Table S1). The first set of studies – those published from 2011 to late 2018 – were mainly descriptive, reporting morphological and functional characteristics of cardio-laminopathic cells: these studies thus confirmed the abnormal morphology of cell nuclei and investigated the susceptibility to electrical and mechanical stresses and the alteration of excitation-contraction coupling in contractile cardiac cells (Ho et al., 2011; Siu et al., 2012; Lee Y.K. et al., 2017). The reported phenotypes were generally ascribed to a structural and/or mechano-transduction hypotheses, in which a generated physical force is responsible for the sarcomeres’ disorganization and contribute to alter the NL structure. In a few studies, defects due to LMNA mutations were associated to alteration of ERK1/2 signaling pathway, that itself influences the correct assembly of the sarcomeres (Siu et al., 2012; Chatzifrangkeskou et al., 2018).
More recently, similarly to what reported in other cellular models of laminopathy, the pathogenesis of the LMNA-CMP has been linked to the “gene expression/chromatin organization hypothesis,” showing a causal association between LMNA mutations, transcription and specific cell phenotypes (Bertero et al., 2019; Lee et al., 2019; Salvarani et al., 2019). More specifically, these studies utilized models of cardiolaminopathy carrying different mutations (i.e., K219T, R225X, and R190W) and found an altered expression of genes of key pathways for CMs functionality. Whether these effects on gene expression are preferentially mediated by remodeling of LADs or the results of a rearrangement at a different level of chromatin organization/regulation (i.e., TADs; change of affinity of mutant Lamin A/C for chromatin binding proteins) still need to be clearly determined.
What is important to highlight, from the functional point of view, is that the robust association between the disease phenotypes and LMNA defects was supported by the experiments conducted in isogenic iPSC paired lines, in which the LMNA mutations were corrected or inserted through CRISPR/Cas9-based (Bertero et al., 2019; Salvarani et al., 2019) or TALEN (Lee et al., 2019) technologies. Also, these studies provided the identification of new potential therapeutic targets (SCN5A gene and PDGF pathway) to treat cardiolaminopathy.
These are important achievements in the field but, with the constant amelioration of the protocols for CM differentiation, we expect to improve even further and to gain more insights into the pathogenetic mechanisms of the disease. Indeed, data available so far come from studies on human CMs obtained from iPSCs mostly through 2D-based differentiation protocols, known to generate CMs with an immature fetal-like phenotype, meaning that they lack some of the structural (i.e., T-tubules, sarcomeric alignment), functional (i.e., calcium handling and contractile properties differences) and metabolic (i.e., glycolysis vs. fatty acid oxidation) properties typical of an adult CM. These properties are highly desirable for drug screening and for modeling specific, adult-onset traits of disease (Karbassi et al., 2020; Mazzola and Di Pasquale, 2020). Given the developmentally regulated expression of Lamin A/C and the complexity of disease phenotype, cell immaturity is likely to impact also on LMNA-CMP modeling, and some important traits of the disease may have been missed so far. The iPSC field is now moving toward the application of 3D-based differentiation protocols, shown to improve maturation of the differentiated CMs, enhancing the potential of iPSC-based cardiac platforms for both disease modeling and drug screening purposes, and from which the cardiolaminopathy field will also benefit in the near future.
Conclusion
Consensus has yet to be reached on the pathogenetic mechanism of cardiolaminopathy and researchers in the field are still trying to finely dissect the molecular role of A-type lamins in the heart and to establish clear genotype/phenotype correlations. Indeed, cardiolaminopathy is a quite complex diseases, with patients exhibiting extremely variable phenotypes, as regards of either specific clinical manifestations or the severity and progression of the disease.
Decades of studies in the field clearly sustain a view in which epigenetic regulations and environmental stimuli are likely to act alongside a single LMNA mutation in determining the final cardiolaminopathy phenotype. More recent studies have started to unravel those mechanisms showing (a) the involvement of other “actors” cooperating with a defective Lamin A/C (Salvarani et al., 2019) and (b) the dual impairment of mechano-signaling and gene expression in laminopathies (Osmanagic-Myers and Foisner, 2019), supporting a holistic model in which the different mechanistic hypotheses cooperate with different extents to the final clinical phenotypes observed in patients (Figure 1).
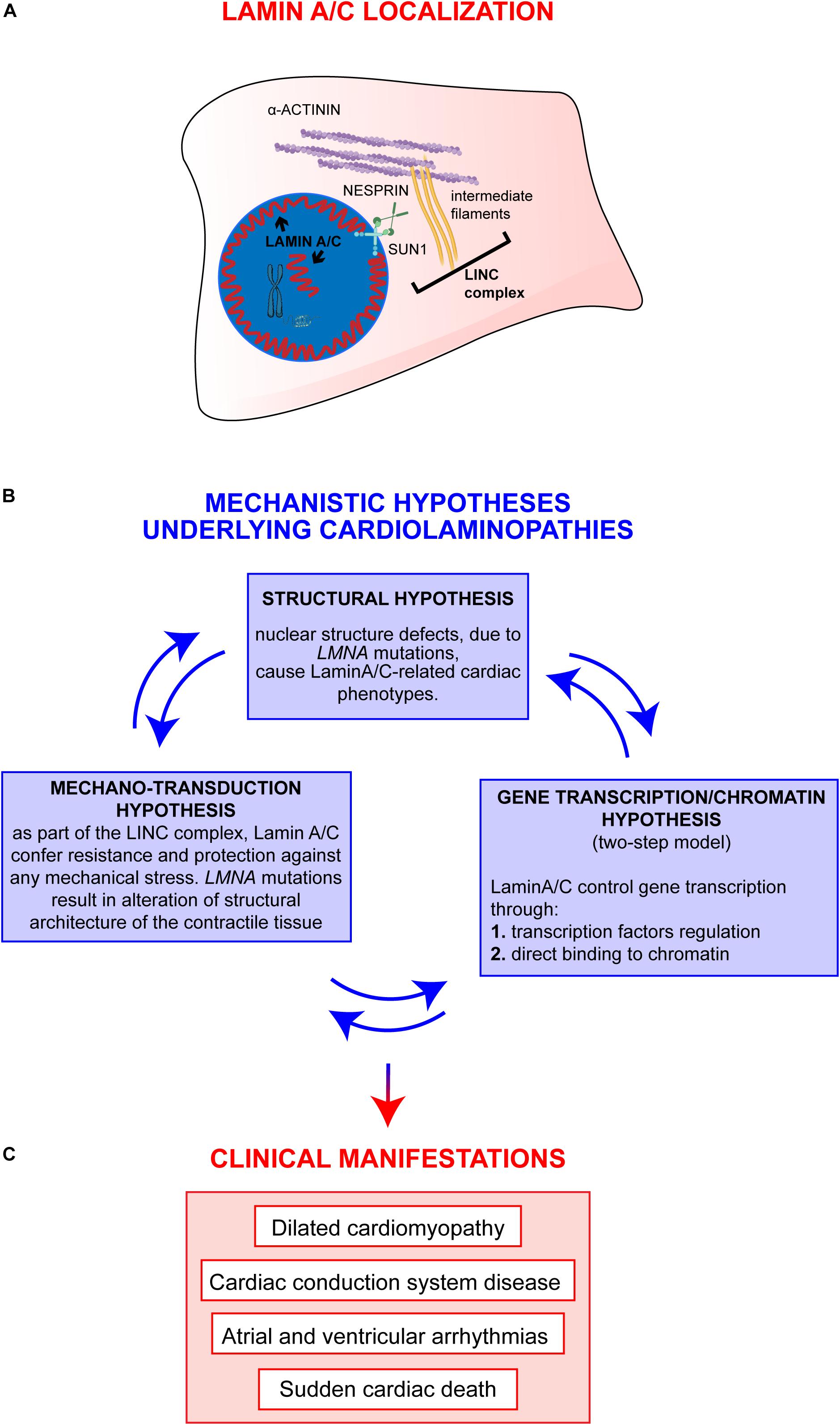
Figure 1. Overview on Lamin A/C proteins localization and on major mechanistic hypotheses underlying cardiac phenotypes. (A) Graphic representation of Lamin A/C proteins localization within the cell: Lamin A/C can be found both, at the periphery and in the nuclear interior. In the nucleus, Lamin A/C have a key structural function and are also involved in chromatin organization and regulation of genes transcription. This latter function can be mediated by both peripheral and nucleoplasmic Lamin A/C forms. Besides their role inside the nucleus, Lamin A/C also impact on cellular processes taking place at the outer part of the nuclear envelope: Lamin A/C indeed interact with the nucleo-cytoskeletal proteins, here indicated as LINC complex (i.e., SUN1, NESPRIN, intermediate filaments). The LINC proteins, in turn, interact with other cytoskeletal proteins (i.e., alpha-actinin), contributing to the maintenance of nuclear and cytoskeletal structure and effectors of specific signaling pathways. (B) The diagram shows the main mechanistic hypotheses underlying clinical manifestations of cardiolaminopathies. The arrows connecting the blue boxes indicate that these hypotheses are not mutually exclusive, but, instead, are potentially interconnected and all contribute to the final phenotype. (C) Clinical manifestations typically associated to LMNA-CMP.
We strongly believe that future studies will further support this global model, unveiling novel disease-relevant pathways and specific targets to treat LMNA-CMP.
Author Contributions
SC and IM performed the bibliographic search and wrote the manuscript. ED wrote and revised the manuscript, provided funding. All authors contributed to the article and approved the submitted version.
Funding
SC was supported by the Italian Ministry of Education, University and Research (2015583WMX), and European Union (ERC-CoG-2017 under the grant agreement no. 772168).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Andrew Kadies for language editing and proofreading.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.00761/full#supplementary-material
Footnotes
- ^ http://www.umd.be/LMNA/
- ^ www.clinicalTrials.gov identifier NCT03439514
References
Aguado, J., Sola-Carvajal, A., Cancila, V., Rev̂echon, G., Ong, P. F., Jones-Weinert, C. W., et al. (2019). Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson–Gilford Progeria Syndrome. Nat. Commun. 10, 1–11.
Alsheimer, M., von Glasenapp, E., Schnölzer, M., Heid, H., and Benavente, R. (2000). Meiotic lamin C2: the unique amino-terminal hexapeptide GNAEGR is essential for nuclear envelope association. Proc. Natl. Acad. Sci. U.S.A. 97, 13120–13125. doi: 10.1073/pnas.240466597
Antoku, S., Wu, W., Joseph, L. C., Morrow, J. P., Worman, H. J., and Gundersen, G. G. (2019). ERK1/2 phosphorylation of FHOD connects signaling and nuclear positioning alternations in cardiac laminopathy. Dev. Cell. 51, 602.e12–616.e12.
Arbustini, E., Pilotto, A., Repetto, A., Grasso, M., Negri, A., Diegoli, M., et al. (2002). Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J. Am. Coll. Cardiol. 39, 981–990. doi: 10.1016/s0735-1097(02)01724-2
Arimura, T., Helbling-Leclerc, A., Massart, C., Varnous, S., Niel, F., Lacène, E., et al. (2005). Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 14, 155–169. doi: 10.1093/hmg/ddi017
Balmus, G., Larrieu, D., Barros, A. C., Collins, C., Abrudan, M., Demir, M., et al. (2018). Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun. 9:1700.
Benedetti, S., Menditto, I., Degano, M., Rodolico, C., Merlini, L., D’Amico, A., et al. (2007). Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology 69, 1285–1292. doi: 10.1212/01.wnl.0000261254.87181.80
Bertero, A., Fields, P. A., Smith, A. S., Leonard, A., Beussman, K., Sniadecki, N. J., et al. (2019). Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J. Cell Biol. 218, 2919–2944. doi: 10.1083/jcb.201902117
Bertrand, A. T., Renou, L., Papadopoulos, A., Beuvin, M., Lacène, E., Massart, C., et al. (2012). DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 21, 1037–1048. doi: 10.1093/hmg/ddr534
Bonne, G., Di Barletta, M. R., Varnous, S., Bécane, H.-M., Hammouda, E.-H., Merlini, L., et al. (1999). Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 21, 285–288. doi: 10.1038/6799
Briand, N., and Collas, P. (2018). Laminopathy-causing lamin A mutations reconfigure lamina-associated domains and local spatial chromatin conformation. Nucleus 9, 216–226. doi: 10.1080/19491034.2018.1449498
Briand, N., Guénantin, A.-C., Jeziorowska, D., Shah, A., Mantecon, M., Capel, E., et al. (2018). The lipodystrophic hotspot lamin A p. R482W mutation deregulates the mesodermal inducer T/Brachyury and early vascular differentiation gene networks. Hum. Mol. Genet. 27, 1447–1459. doi: 10.1093/hmg/ddy055
Butin-Israeli, V., Adam, S. A., Goldman, A. E., and Goldman, R. D. (2012). Nuclear lamin functions and disease. Trends Genet. 28, 464–471. doi: 10.1016/j.tig.2012.06.001
Cesarini, E., Mozzetta, C., Marullo, F., Gregoretti, F., Gargiulo, A., Columbaro, M., et al. (2015). Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 211, 533–551. doi: 10.1083/jcb.201504035
Chandar, S., Yeo, L. S., Leimena, C., Tan, J.-C., Xiao, X.-H., Nikolova-Krstevski, V., et al. (2010). Effects of mechanical stress and carvedilol in lamin A/C-deficient dilated cardiomyopathy. Circ. Res. 106:573. doi: 10.1161/circresaha.109.204388
Chatzifrangkeskou, M., Le Dour, C., Wu, W., Morrow, J. P., Joseph, L. C., Beuvin, M., et al. (2016). ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 25, 2220–2233. doi: 10.1093/hmg/ddw090
Chatzifrangkeskou, M., Yadin, D., Marais, T., Chardonnet, S., Cohen-Tannoudji, M., Mougenot, N., et al. (2018). Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 27, 3060–3078. doi: 10.1093/hmg/ddy215
Choi, J. C., Muchir, A., Wu, W., Iwata, S., Homma, S., Morrow, J. P., et al. (2012). Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 4:144ra102. doi: 10.1126/scitranslmed.3003875
Cohen, M., Gruenbaum, Y., Lee, K. K., and Wilson, K. L. (2001). Transcriptional repression, apoptosis, human disease and the functional evolution of the nuclear lamina. Trends Biochem. Sci. 26, 41–47. doi: 10.1016/s0968-0004(00)01727-8
Cupesi, M., Yoshioka, J., Gannon, J., Kudinova, A., Stewart, C. L., and Lammerding, J. (2010). Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J. Mol. Cell Cardiol. 48, 1290–1297. doi: 10.1016/j.yjmcc.2009.10.024
Davis, R. P., van den Berg, C. W., Casini, S., Braam, S. R., and Mummery, C. L. (2011). Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol. Med. 17, 475–484. doi: 10.1016/j.molmed.2011.05.001
De Sandre-Giovannoli, A., Chaouch, M., Kozlov, S., Vallat, J.-M., Tazir, M., Kassouri, N., et al. (2002). Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. J. Peripher. Nerv. Syst. 7, 205–205. doi: 10.1046/j.1529-8027.2002.02026_2.x
Dechat, T., Pfleghaar, K., Sengupta, K., Shimi, T., Shumaker, D. K., Solimando, L., et al. (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832–853. doi: 10.1101/gad.1652708
Ditaranto, R., Boriani, G., Biffi, M., Lorenzini, M., Graziosi, M., Ziacchi, M., et al. (2019). Differences in cardiac phenotype and natural history of laminopathies with and without neuromuscular onset. Orphanet J. Rare Dis. 14:263.
Dittmer, T. A., and Misteli, T. (2011). The lamin protein family. Genome Biol. 12:222. doi: 10.1186/gb-2011-12-5-222
Dittmer, T. A., Sahni, N., Kubben, N., Hill, D. E., Vidal, M., Burgess, R. C., et al. (2014). Systematic identification of pathological lamin A interactors. Mol. Biol. Cell 25, 1493–1510. doi: 10.1091/mbc.e14-02-0733
Fatkin, D., MacRae, C., Sasaki, T., Wolff, M. R., Porcu, M., Frenneaux, M., et al. (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. New Engl. J. Med. 341, 1715–1724. doi: 10.1056/nejm199912023412302
Galiová, G., Bártová, E., Raška, I., Krejèí, J., and Kozubek, S. (2008). Chromatin changes induced by lamin A/C deficiency and the histone deacetylase inhibitor trichostatin A. Eur. J. Cell Biol. 87, 291–303. doi: 10.1016/j.ejcb.2008.01.013
Gigli, M., Merlo, M., Graw, S. L., Barbati, G., Rowland, T. J., Slavov, D. B., et al. (2019). Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 74, 1480–1490. doi: 10.1016/j.jacc.2019.06.072
Goldman, R. D., Shumaker, D. K., Erdos, M. R., Eriksson, M., Goldman, A. E., Gordon, L. B., et al. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. U.S.A. 101, 8963–8968. doi: 10.1073/pnas.0402943101
Gordon, L. B., Rothman, F. G., López-Otín, C., and Misteli, T. (2014). Progeria: a paradigm for translational medicine. Cell 156, 400–407. doi: 10.1016/j.cell.2013.12.028
Granger, B., Gueneau, L., Drouin-Garraud, V., Pedergnana, V., Gagnon, F., Yaou, R. B., et al. (2011). Modifier locus of the skeletal muscle involvement in Emery–Dreifuss muscular dystrophy. Hum. Genet. 129, 149–159. doi: 10.1007/s00439-010-0909-1
Gruenbaum, Y., Margalit, A., Goldman, R. D., Shumaker, D. K., and Wilson, K. L. (2005). The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 6, 21–31. doi: 10.1038/nrm1550
Hasselberg, N. E., Edvardsen, T., Petri, H., Berge, K. E., Leren, T. P., Bundgaard, H., et al. (2014). Risk prediction of ventricular arrhythmias and myocardial function in Lamin A/C mutation positive subjects. Europace 16, 563–571. doi: 10.1093/europace/eut291
Hasselberg, N. E., Haland, T. F., Saberniak, J., Brekke, P. H., Berge, K. E., Leren, T. P., et al. (2018). Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 39, 853–860. doi: 10.1093/eurheartj/ehx596
Herrmann, H., and Aebi, U. (2004). Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular scaffolds. Annu. Rev. Biochem. 73, 749–789. doi: 10.1146/annurev.biochem.73.011303.073823
Hershberger, R. E., and Siegfried, J. D. (2011). Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 57, 1641–1649. doi: 10.1016/j.jacc.2011.01.015
Ho, J. C., Zhou, T., Lai, W.-H., Huang, Y., Chan, Y.-C., Li, X., et al. (2011). Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging 3:380. doi: 10.18632/aging.100277
Ikegami, K., Secchia, S., Almakki, O., Lieb, J. D., and Moskowitz, I. P. (2020). Phosphorylated Lamin A/C in the nuclear interior binds active enhancers associated with abnormal transcription in progeria. Dev. Cell 52, 699.e11–713.e11.
Jakobs, P. M., Hanson, E. L., Crispell, K. A., Toy, W., Keegan, H., Schilling, K., et al. (2001). Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J. Card. Fail. 7, 249–256. doi: 10.1054/jcaf.2001.26339
Karbassi, E., Fenix, A., Marchiano, S., Muraoka, N., Nakamura, K., Yang, X., et al. (2020). Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 17, 341–359. doi: 10.1038/s41569-019-0331-x
Kayvanpour, E., Sedaghat-Hamedani, F., Amr, A., Lai, A., Haas, J., Holzer, D. B., et al. (2017). Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 106, 127–139. doi: 10.1007/s00392-016-1033-6
Kubben, N., Voncken, J. W., Konings, G., van Weeghel, M., van den Hoogenhof, M. M., Gijbels, M., et al. (2011). Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus 2, 195–207. doi: 10.4161/nucl.2.3.15731
Kubben, N., Voncken, J. W., and Misteli, T. (2010). Mapping of protein-and chromatin-interactions at the nuclear lamina. Nucleus 1, 460–471. doi: 10.4161/nucl.1.6.13513
Kumar, S., Baldinger, S. H., Gandjbakhch, E., Maury, P., Sellal, J.-M., Androulakis, A. F., et al. (2016). Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J. Am. Coll. Cardiol. 68, 2299–2307. doi: 10.1016/j.jacc.2016.08.058
Lee, J., Termglinchan, V., Diecke, S., Itzhaki, I., Lam, C. K., Garg, P., et al. (2019). Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340. doi: 10.1038/s41586-019-1406-x
Lee, J. H., Protze, S. I., Laksman, Z., Backx, P. H., and Keller, G. M. (2017). Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell 21, 179.e4–194.e4.
Lee, Y. K., Lau, Y. M., Cai, Z. J., Lai, W. H., Wong, L. Y., Tse, H. F., et al. (2017). Modeling treatment response for lamin A/C related dilated cardiomyopathy in human induced pluripotent stem cells. J. Am. Heart Assoc. 6:e005677.
Lin, F., and Worman, H. J. (1993). Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 268, 16321–16326.
Lodola, F., Morone, D., Denegri, M., Bongianino, R., Nakahama, H., Rutigliano, L., et al. (2016). Adeno-associated virus-mediated CASQ2 delivery rescues phenotypic alterations in a patient-specific model of recessive catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 7:e2393. doi: 10.1038/cddis.2016.304
Machiels, B. M., Zorenc, A. H., Endert, J. M., Kuijpers, H. J., van Eys, G. J., Ramaekers, F. C., et al. (1996). An alternative splicing product of the lamin A/C gene lacks exon 10. J. Biol. Chem. 271, 9249–9253. doi: 10.1074/jbc.271.16.9249
Mazzola, M., and Di Pasquale, E. (2020). Toward cardiac regeneration: combination of pluripotent stem cell-based therapies and bioengineering strategies. Front. Bioeng. Biotechnol. 8:455. doi: 10.3389/fbioe.2020.00455
McNally, E. M., and Mestroni, L. (2017). Dilated cardiomyopathy: genetic determinants and mechanisms. Circ. Res. 121, 731–748. doi: 10.1161/circresaha.116.309396
Moretti, A., Bellin, M., Welling, A., Jung, C. B., Lam, J. T., Bott-Flügel, L., et al. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. New Engl. J. Med. 363, 1397–1409.
Mounkes, L. C., Kozlov, S. V., Rottman, J. N., and Stewart, C. L. (2005). Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum. Mol. Genet. 14, 2167–2180. doi: 10.1093/hmg/ddi221
Mozzetta, C., and Tedesco, F. S. (2019). Challenging the “chromatin hypothesis” of cardiac laminopathies with LMNA mutant iPS cells. J. Cell Biol. 218, 2826–2828. doi: 10.1083/jcb.201907166
Muchir, A., Pavlidis, P., Bonne, G., Hayashi, Y. K., and Worman, H. J. (2007). Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery–Dreifuss muscular dystrophy. Hum. Mol. Genet. 16, 1884–1895. doi: 10.1093/hmg/ddm137
Muchir, A., Wu, W., Choi, J. C., Iwata, S., Morrow, J., Homma, S., et al. (2012). Abnormal p38α mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 21, 4325–4333. doi: 10.1093/hmg/dds265
Muck, J. S., Kandasamy, K., Englmann, A., Günther, M., and Zink, D. (2012). Perinuclear positioning of the inactive human cystic fibrosis gene depends on CTCF, A-type lamins and an active histone deacetylase. J. Cell. Biochem. 113, 2607–2621. doi: 10.1002/jcb.24136
Muntoni, F., Torelli, S., and Brockington, M. (2008). Muscular dystrophies due to glycosylation defects. Neurotherapeutics 5, 627–632. doi: 10.1016/j.nurt.2008.08.005
Nikolova, V., Leimena, C., McMahon, A. C., Tan, J. C., Chandar, S., Jogia, D., et al. (2004). Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C–deficient mice. J. Clin. Invest. 113, 357–369. doi: 10.1172/jci200419448
Nishiuchi, S., Makiyama, T., Aiba, T., Nakajima, K., Hirose, S., Kohjitani, H., et al. (2017). Gene-based risk stratification for cardiac disorders in LMNA mutation carriers. Circ. Cardiovasc. Genet. 10:e001603.
Osmanagic-Myers, S., and Foisner, R. (2019). The structural and gene expression hypotheses in laminopathic diseases—not so different after all. Mol. Biol. Cell 30, 1786–1790. doi: 10.1091/mbc.e18-10-0672
Pasotti, M., Klersy, C., Pilotto, A., Marziliano, N., Rapezzi, C., Serio, A., et al. (2008). Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 52, 1250–1260. doi: 10.1016/j.jacc.2008.06.044
Paulsen, J., Ali, T. M. L., Nekrasov, M., Delbarre, E., Baudement, M.-O., Kurscheid, S., et al. (2019). Long-range interactions between topologically associating domains shape the four-dimensional genome during differentiation. Nat. Genet. 51, 835–843. doi: 10.1038/s41588-019-0392-0
Peretto, G., Di Resta, C., Perversi, J., Forleo, C., Maggi, L., Politano, L., et al. (2019). Cardiac and neuromuscular features of patients with LMNA-related cardiomyopathy. Ann. Intern. Med. 171, 458–463.
Pickersgill, H., Kalverda, B., De Wit, E., Talhout, W., Fornerod, M., and van Steensel, B. (2006). Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet. 38, 1005–1014. doi: 10.1038/ng1852
Priori, S. G., and Blomström-Lundqvist, C. (2015). 2015 European Society of Cardiology Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co-chairs. Eur. Heart J. 36, 2757–2759.
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., et al. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 10, 1932–1963. doi: 10.1016/j.hrthm.2013.05.014
Rapezzi, C., Arbustini, E., Caforio, A. L., Charron, P., Gimeno-Blanes, J., Heliö, T., et al. (2013). Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 34, 1448–1458. doi: 10.1093/eurheartj/ehs397
Roncarati, R., Anselmi, C. V., Krawitz, P., Lattanzi, G., Von Kodolitsch, Y., Perrot, A., et al. (2013). Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur. J. Hum. Genet. 21, 1105–1111. doi: 10.1038/ejhg.2013.16
Saga, A., Karibe, A., Otomo, J., Iwabuchi, K., Takahashi, T., Kanno, H., et al. (2009). Lamin A/C gene mutations in familial cardiomyopathy with advanced atrioventricular block and arrhythmia. Tohoku J. Exp. Med. 218, 309–316. doi: 10.1620/tjem.218.309
Salvarani, N., Crasto, S., Miragoli, M., Bertero, A., Paulis, M., Kunderfranco, P., et al. (2019). The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 10, 1–16.
Sedaghat-Hamedani, F., Haas, J., Zhu, F., Geier, C., Kayvanpour, E., Liss, M., et al. (2017). Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 38, 3449–3460.
Sewry, C., Brown, S., Mercuri, E., Bonne, G., Feng, L., Camici, G., et al. (2001). Skeletal muscle pathology in autosomal dominant Emery-Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol. Appl. Neurobiol. 27, 281–290. doi: 10.1046/j.0305-1846.2001.00323.x
Siu, C.-W., Lee, Y.-K., Ho, J. C.-Y., Lai, W.-H., Chan, Y.-C., Ng, K.-M., et al. (2012). Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 4:803. doi: 10.18632/aging.100503
Stewart, C. L., Kozlov, S., Fong, L. G., and Young, S. G. (2007). Mouse models of the laminopathies. Exp. Cell Res. 313, 2144–2156. doi: 10.1016/j.yexcr.2007.03.026
Sullivan, T., Escalante-Alcalde, D., Bhatt, H., Anver, M., Bhat, N., Nagashima, K., et al. (1999). Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147, 913–920. doi: 10.1083/jcb.147.5.913
Swift, J., Ivanovska, I. L., Buxboim, A., Harada, T., Dingal, P. D. P., Pinter, J., et al. (2013). Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341:1240104. doi: 10.1126/science.1240104
Tesson, F., Saj, M., Uvaize, M. M., Nicolas, H., Płoski, R., and Biliñska, Z. (2014). Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 21, 331–342. doi: 10.5603/cj.a2014.0037
Tong, J., Li, W., Vidal, C., Yeo, L. S., Fatkin, D., and Duque, G. (2011). Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech. Ageing Dev. 132, 552–559. doi: 10.1016/j.mad.2011.09.004
Van Rijsingen, I. A., Arbustini, E., Elliott, P. M., Mogensen, J., Hermans-van Ast, J. F., Van Der Kooi, A. J., et al. (2012). Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: a European cohort study. J. Am. Coll. Cardiol. 59, 493–500.
van Tintelen, J. P., Hofstra, R. M., Katerberg, H., Rossenbacker, T., Wiesfeld, A. C., and du Marchie, et al. (2007a). High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am. Heart. J. 154, 1130–1139. doi: 10.1016/j.ahj.2007.07.038
van Tintelen, J. P., Tio, R. A., Kerstjens-Frederikse, W. S., van Berlo, J. H., Boven, L. G., Suurmeijer, A. J., et al. (2007b). Severe myocardial fibrosis caused by a deletion of the 5’end of the lamin A/C gene. J. Am. Coll. Cardiol. 49, 2430–2439. doi: 10.1016/j.jacc.2007.02.063
Wahbi, K., Ben Yaou, R., Gandjbakhch, E., Anselme, F., Gossios, T., Lakdawala, N. K., et al. (2019). Development and validation of a new risk prediction score for life-threatening ventricular tachyarrhythmias in laminopathies. Circulation 140, 293–302.
Wilson, K. L., and Foisner, R. (2010). Lamin-binding proteins. Cold Spring Harb. Perspect. Biol. 2:a000554. doi: 10.1101/cshperspect.a000554
Wilson, K. L., Zastrow, M. S., and Lee, K. K. (2001). Lamins and disease: insights into nuclear infrastructure. Cell 104, 647–650. doi: 10.1016/s0092-8674(02)01452-6
Wolf, C. M., Wang, L., Alcalai, R., Pizard, A., Burgon, P. G., Ahmad, F., et al. (2008). Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell Cardiol. 44, 293–303. doi: 10.1016/j.yjmcc.2007.11.008
Worman, H. J., and Bonne, G. (2007). “Laminopathies”: a wide spectrum of human diseases. Exp. Cell Res. 313, 2121–2133. doi: 10.1016/j.yexcr.2007.03.028
Wydner, K. L., McNeil, J. A., Lin, F., Worman, H. J., and Lawrence, J. B. (1996). Chromosomal assignment of human nuclear envelope protein genes LMNA, LMNB1, and LBR by fluorescencein situhybridization. Genomics 32, 474–478. doi: 10.1006/geno.1996.0146
Xu, C., Police, S., Rao, N., and Carpenter, M. K. (2002). Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ. Res. 91, 501–508. doi: 10.1161/01.res.0000035254.80718.91
Ye, Q., and Worman, H. J. (1996). Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J. Biol. Chem. 271, 14653–14656. doi: 10.1074/jbc.271.25.14653
Young, S. G., Fong, L. G., and Michaelis, S. (2005). Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res. 46, 2531–2558. doi: 10.1194/jlr.r500011-jlr200
Zahr, H. C., and Jaalouk, D. E. (2018). Exploring the crosstalk between LMNA and splicing machinery gene mutations in dilated cardiomyopathy. Front. Genet. 9:231. doi: 10.3389/fgene.2018.00231
Keywords: LMNA gene, cardiolaminopathy, Lamin A/C, clinical phenotype, molecular mechanisms
Citation: Crasto S, My I and Di Pasquale E (2020) The Broad Spectrum of LMNA Cardiac Diseases: From Molecular Mechanisms to Clinical Phenotype. Front. Physiol. 11:761. doi: 10.3389/fphys.2020.00761
Received: 26 February 2020; Accepted: 11 June 2020;
Published: 03 July 2020.
Edited by:
Martina Calore, Maastricht University, NetherlandsReviewed by:
J. Carter Ralphe, University of Wisconsin-Madison, United StatesMichelle S. Parvatiyar, Florida State University, United States
Copyright © 2020 Crasto, My and Di Pasquale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa Di Pasquale, elisa.dipasquale@humanitasresearch.it; elisadipa@gmail.com
†These authors have contributed equally to this work