 Fabio A. Vigil
Fabio A. Vigil Chase M. Carver
Chase M. Carver Mark S. Shapiro
Mark S. Shapiro- Department of Cellular and Integrative Physiology, University of Texas Health San Antonio, San Antonio, TX, United States
Kv7 (“M-type,” KCNQ) K+ currents, play dominant roles in controlling neuronal excitability. They act as a “brake” against hyperexcitable states in the central and peripheral nervous systems. Pharmacological augmentation of M current has been developed for controlling epileptic seizures, although current pharmacological tools are uneven in practical usefulness. Lately, however, M-current “opener” compounds have been suggested to be efficacious in preventing brain damage after multiple types of insults/diseases, such as stroke, traumatic brain injury, drug addiction and mood disorders. In this review, we will discuss what is known to date on these efforts and identify gaps in our knowledge regarding the link between M current and therapeutic potential for these disorders. We will outline the preclinical experiments that are yet to be performed to demonstrate the likelihood of success of this approach in human trials. Finally, we also address multiple pharmacological tools available to manipulate different Kv7 subunits and the relevant evidence for translational application in the clinical use for disorders of the central nervous system and multiple types of brain insults. We feel there to be great potential for manipulation of Kv7 channels as a novel therapeutic mode of intervention in the clinic, and that the paucity of existing therapies obligates us to perform further research, so that patients can soon benefit from such therapeutic approaches.
Introduction
Kv7 channels, also known as M-type, or KCNQ channels, are low-threshold voltage gated K+ channels first described almost 40 years ago as underlying the cholinergic slow excitatory post-synaptic potential in sympathetic neurons (Brown and Adams, 1980; Constanti and Brown, 1981). Kv7 channels can be composed of homo- or heterotetrameric assembly of Kv7.1-Kv7.5 subunits; however, only Kv7.2-5 are expressed in the nervous system (Jentsch, 2000). In a wide variety of central and peripheral neurons, M-channels play a significant role in controlling active and passive discharge properties, including action potential threshold, resting membrane potential, spike afterhyperpolarization (AHPs), and shunting conductance (Jones et al., 1995; Yue and Yaari, 2004; Peters et al., 2005; Shah et al., 2008). Consistent with that role, channels composed of Kv7.2 and 7.3 in varying composition are mainly localized in brain to the axon initial segment (Cooper et al., 2001; Pan et al., 2006; Rasmussen et al., 2007), adjacent to the NaV channels that generate action potentials. Since M channels deactivate slowly, they contribute to AHP currents (Tzingounis and Nicoll, 2008). Excessive Kv7-channel suppression or channel dysfunction often leads to seizures or other epileptic syndromes (Biervert et al., 1998; Singh et al., 1998; Ambrosino et al., 2015; Miceli et al., 2015; Greene and Hoshi, 2017), leading to the idea that proper M-channel function acts as a “brake” to prevent excess hyperexcitability (Maljevic et al., 2008; Soldovieri et al., 2011), by accumulated M-current activation increasing the threshold for firing (Peters et al., 2005; Tzingounis and Nicoll, 2008) and increasing the interspike interval (Lawrence et al., 2006).
M current is so named for its discovery as a K+ conductance suppressed by stimulation of muscarinic acetylcholine receptors in sympathetic ganglia neurons (Brown et al., 1995). In those cells, the action is via Gq/11-mediated activation of phospholipase C, which hydrolyzes phosphatidylinositol-4,5-bisphospate (PIP2), reducing its abundance in the membrane (Haley et al., 1998; Suh and Hille, 2005), and by activation of protein kinase C (Hoshi et al., 2003). Since PIP2 binding is required for M-channel opening (Zhang et al., 2003; Li et al., 2005; Suh et al., 2006; Sun and MacKinnon, 2020), its depletion reduces M-current amplitudes in a voltage-independent manner (Shapiro et al., 2000; Nakajo and Kubo, 2005; Choveau et al., 2018). However, in those same neurons, other Gq/11-coupled receptors suppress M current via release of Ca2+ from IP3-gated stores, loading of Ca2+ into calmodulin (Gamper and Shapiro, 2003; Winks et al., 2005; Zaika et al., 2007) and changes in configuration of CaM molecules bound to the proximal carboxy terminus of Kv7.1-7.5 subunits (Yus-Najera et al., 2002; Haitin and Attali, 2008) in varying configurations (Haitin and Attali, 2008; Kosenko and Hoshi, 2013; Strulovich et al., 2016; Sun and MacKinnon, 2017; Chang et al., 2018; Archer et al., 2019).
Pharmacological manipulation of M current has been studied extensively as a therapeutic option for epilepsy (Kapetanovic et al., 1995; Rostock et al., 1996; Armand et al., 1999; Miceli et al., 2008; Amabile and Vasudevan, 2013; Splinter, 2013) and for analgesia (Blackburn-Munro and Jensen, 2003; Munro and Dalby-Brown, 2007; Szelenyi, 2013; Hayashi et al., 2014; Abd-Elsayed et al., 2015; Zheng et al., 2015; Busserolles et al., 2016; Wang and Li, 2016; Du et al., 2018; Li et al., 2019). Retigabine (RTG) was developed some 20 years ago as an anti-epileptic drug that acts by augmenting M current (Main et al., 2000; Rundfeldt and Netzer, 2000; Tatulian and Brown, 2003; Wuttke et al., 2005) and is widely used in research labs. Retigabine induces a hyperpolarizing shift of Kv7.2-5 channel activation (but not Kv7.1), resulting in current enhancement at potentials positive to −80 mV (Tatulian et al., 2001). However, although FDA-approved, its long-term side-effects (e.g., dilation of smooth muscle, blue tinting to skin over time) has led to its withdrawal from the market. Recently, however, a plethora of more selective “next-generation” M channel-targeting compounds have been developed. That may make manipulation of M current a modality used for myriad of brain disorders and insults, besides as anti-convulsants. In this review we will explore some of the possible new therapeutic uses of pharmacological M-current manipulation in treating brain dysfunction.
M Current and Neurovascular Injuries
Two research groups first explored the role of M current during metabolic stress induced by oxygen and glucose deprivation (OGD) using cell culture models. They observed RTG-induced enhancement of M current to significantly reduce neuronal death in organotypic cultures of hippocampal slices subjected to 30 min of OGD, whereas M-current block with XE991 (Zaczek et al., 1998) resulted in increased neuronal death (Boscia et al., 2006; Gamper et al., 2006). Similar observations were reported by Barrese et al. (2015), using rat caudate brain slices. All these groups observed that OGD-induced damage was reduced by pharmacological M-current augmentation. Therefore, it seemed possible that pharmacological M-current enhancement could reduce brain damage after a stroke. Indeed, our group showed M-current augmentation to be neuroprotective after occlusive stroke. M-current augmentation strongly reduced stroke-induced neuronal death, the maladaptive immune response, and locomotor deficits (Bierbower et al., 2015). In a rat model, RTG treatment impaired stroke-induced increases in blood brain barrier (BBB) permeability, opening of tight junctions from microvascular endothelial cells, and cerebral infarct area (Zhao et al., 2018). An evident connection between stroke and the previous OGD models is that both involve cellular metabolic stress.
More recently, Vigil et al. (2020) also showed pharmacological M-current augmentation to prevent brain damage after traumatic brain injury (TBI). With only one i.p. injection of RTG 30 min post-injury, we observed significant reductions in post-traumatic seizures and seizure susceptibility, cellular energetic demand, the maladaptive inflammatory/immune response, breakdown of the BBB, and cell death (Vigil et al., 2020). Thus, we believe that prevention of initial TBI-induced hyperexcitability, even before a post-traumatic seizure can occur, severely hampered the damaging TBI-induced cascade of events. Interestingly, an increase in Kv7.2 expression in TBI-subjected animals treated with RTG was observed in cortical and dentate gyrus hippocampal cells up to 6 days after TBI, although this transcriptional up-regulation is likely not to last much longer than 10 days (Carver et al., 2020; Vigil et al., 2020). As RTG has a half-life of 2 h in animals (Valeant Pharmaceuticals), it is reasonable to assume that M-current augmentation facilitated a later increase in Kv7.2 transcription in neurons that survive the insult. This elevated expression of the kcnq2 gene could represent a second longer-term therapeutic window, as one could take advantage of the increased expression of Kv7.2 channels to maximize the effects of therapeutic treatment. Recently, acute RTG administration was also reported to improve pain and motor neuron recovery after spinal-cord injury (SCI). Wu et al. (2020) observed RTG treatment to be effective up to 3 days after SCI if local delivery of RTG was performed by a pump implant.
Based on the above, a reasonable hypothesis is that pharmacological M-current augmentation reduces neuronal firing after TBI, SCI and, stroke, and as in the OGD model, reduces cellular energy demand. Therefore, reducing Na+/K+ ATPase activity, osmotic unbalance, and cell lysis. This hypothesis is summarized in Figure 1. However, we are only beginning to use in vivo models to confirm the cause of cell death after post-traumatic seizures. Moreover, the various elements of the injury-induced cascade of events are likely to further cross-activate each other resulting in the secondary injury often observed in TBI (Beez et al., 2017; Simon et al., 2017), SCI (Ahuja et al., 2017), and stroke (Hemphill et al., 2015; Beez et al., 2017). By initially preventing this cascade of events at the start, M-current augmentation should have long-lasting beneficial effects in these neurovascular injury events (Figure 1).
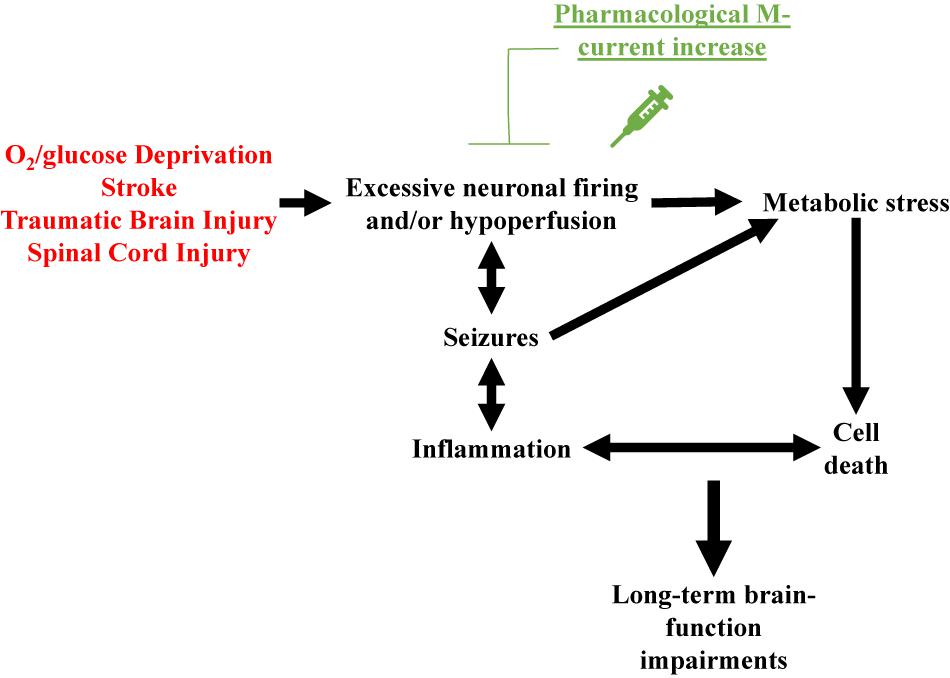
Figure 1. Schematic representation of the over-arching hypothesis proposed to explain the therapeutic effects of pharmacological M-current augmentation after O2/glucose deprivation, stroke, traumatic brain injury and spinal cord injury. These neurovascular insults all share brain hypoperfusion and/or excessive neuronal firing, leading to cellular metabolic stress, cell lysis. Cell death hyper-activates the immune system of the brain, increasing insult-related inflammation. If uncontrolled, this hyper-inflammation may result in further cell death, creating a deleterious cycle of positive feedback between these events. Insult-induced excessive neuronal firing also promotes seizures, which further increase cellular metabolic stress and enhanced inflammatory responses, facilitating the occurrence of more seizures. Acute pharmacological M-current increase post-insult stops this cascade of events by reducing the initial increased cellular energy demand.
The secondary effects of TBI can be observed in epileptogenesis in which the injury converts a healthy brain into a brain in which synchronous neuronal activity and seizures are more likely to occur. Traumatic brain injury is responsible for 20% of symptomatic epilepsies and 5–6% of all epilepsy (Garga and Lowenstein, 2006). Higher risk of post-traumatic epilepsy may persist for up to a decade after an initial TBI, but an indeterminate latent period can last between months and years without any presentation of overt seizures (Frey, 2003; Christensen et al., 2009; Lowenstein, 2009). Hence, prior to seizure presentation, TBI must induce pathophysiological changes in the brain that increase seizure susceptibility and epileptogenesis. Post-traumatic epileptogenesis entails a wide scope of regulatory plasticity from many different ion channels, including GABAA receptors, HCN channels, and Kv7 channels, which often provide inhibitory opposition in response to neuronal hyperexcitability. However, both excitatory and inhibitory circuit reorganizations after TBI lead to maladaptive synaptic connectivity, contributing to epileptogenesis (Hunt et al., 2013). Due to the capacity of M-channel openers to act as an inhibitory force to the brain during susceptible periods of the post-traumatic cascade, they could provide control, and possibly prevention, of TBI-induced epilepsy.
Another role played by Kv7 channels, specifically Kv7.4 and Kv7.5, are as regulators of excitability in blood vessels smooth muscle (Yeung et al., 2008; Joshi et al., 2009). Thus, it is possible that part of the beneficial effect of M-current augmentation might be ascribed to an acute increase in blood flow that would lead to a greater supply of glucose and O2 to support metabolic demands. However, the dilation of bladder smooth muscle, leading to urinary incontinence, has been suggested to be due to RTG actions on afferent nerve activity, rather than direct regulation of bladder myocyte contraction (Tykocki et al., 2019). In TBI and stroke models, increases in BBB permeability and infarct area were reduced by RTG treatment (Zhao et al., 2018; Vigil et al., 2020). Zhao et al. (2018) suggest that RTG may reduce BBB permeability by inhibition of injury-induced increase in expression of protein kinase C delta (PKCδ) and of the extracellular matrix proteinases, MMP-2 and MMP-9. Phosphorylation by PKCδ activates MMP-2/9, which degrades tight junction-associated proteins of cerebral vascular endothelial cells, resulting in increased BBB permeability. How RTG treatment reduces injury-induced expression of MMP-2/9 and PKCδ is unknown. Additionally, more experiments measuring brain blood flow and BBB permeability at different time points after injury in animals treated with RTG are still necessary to further investigate this matter.
An additional confound to consider is that RTG seems to affect other ion channels besides Kv7 channels. Retigabine at 10 μM concentration reduces Kv2.1 current by ∼20% and at 100 μM, RTG inhibits ∼80% of Kv2.1 current (Stas et al., 2016). Retigabine at 10 μM also reduces current throughout L-type voltage-gated Ca2+ channels by >50% (Mani et al., 2013). Experimental evidence shows that RTG also acts on GABAA receptors at concentrations above 10 μM (Treven et al., 2015). Inhibition of these channels may also play a role in the aforementioned therapeutic effects. For treatment of epilepsy in patients, the mean free average plasma concentrations of RTG is approximately 0.83 μM and maximum mean free plasma concentrations (Cmax) is approximately 1 μM (Gunthorpe et al., 2012). Hence, if the same doses are used for treatment of other diseases/injuries, off-target effects are likely to be avoided.
M Current and Drug Addiction
Alcohol Addiction
One of the first demonstrations of the relationship between M current and drugs of abuse centers on alcohol addiction. Moore et al. (1990) showed that M current from hippocampal CA1 pyramidal neurons was inhibited by ethanol. In that same year, M current was recorded for the first time in ventral tegmental area (VTA) neurons (Lacey et al., 1990), a region that plays a key role in reward, mood and drug addiction (Di Chiara and Imperato, 1988; Lammel et al., 2012; Morales and Pickel, 2012), including to alcohol (Gessa et al., 1985; Rodd-Henricks et al., 2000). Koyama et al. (2007) showed that ethanol increases spontaneous firing frequency and suppresses M current in VTA dopaminergic neurons, with a correlation between the two actions. Ethanol seems to inhibit Kv7.2/7.3 heteromers by a PIP2-related mechanism (Kim et al., 2019). Ethanol has also been shown to reduce Kv7.2 trafficking to the membrane in neurons of the nucleus accumbens (NAc; McGuier et al., 2016), a region that is heavily innervated by the VTA and fundamental for reward and drug addiction (Weiss et al., 1993; Robinson and Berridge, 2003; Morales and Pickel, 2012). Retigabine injection, either systemically or into the NAc, significantly reduced voluntary ethanol consumption in rats, without any significant effect in sucrose or water consumption, whereas injection of XE991 increased it (Knapp et al., 2014; McGuier et al., 2016, 2018). Finally, systemic treatment with the Kv7.2 and Kv7.4 opener, ML213 (Yu et al., 2010), also reduced ethanol intake in rats (McGuier et al., 2018). Taken together, this evidence suggests regulation of M current to be linked to alcoholism, and that M-current augmentation may represent a mode of therapeutic intervention to treat alcoholism disease (Figure 2).
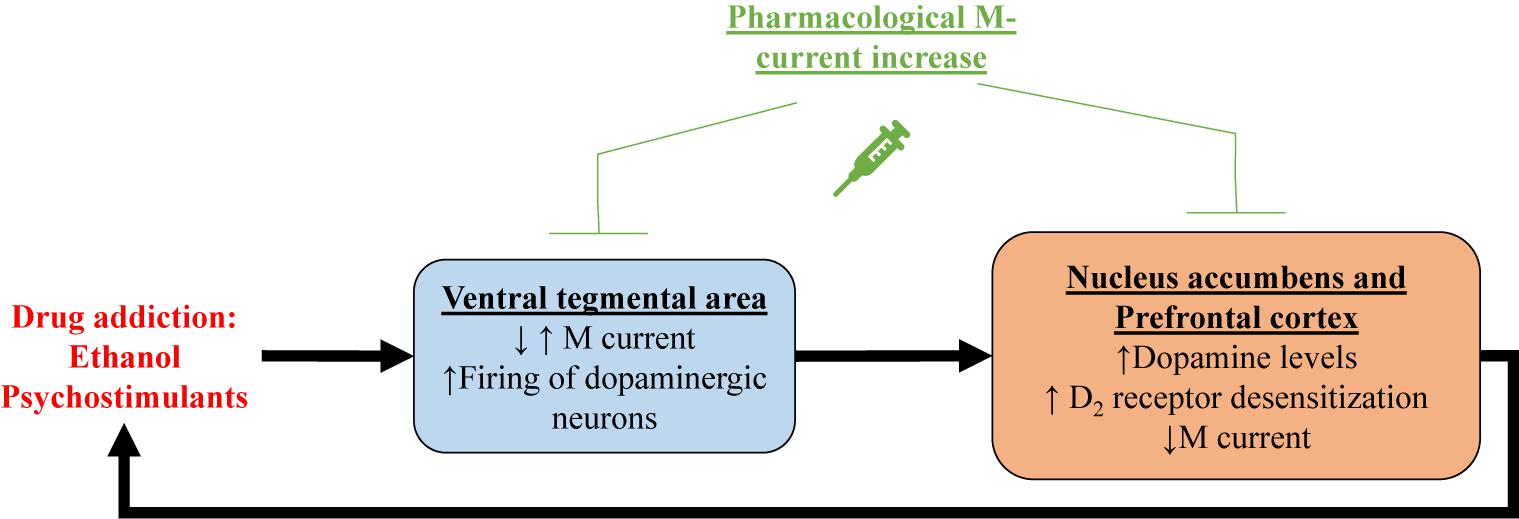
Figure 2. Shown is a proposed mechanism by which drug addiction may involve M current. Psychostimulant-induced increase in dopamine levels within the ventral tegmental area (VTA)could correlate with decreased M current in dopaminergic neurons, and/or increased M current in GABAergic neurons. In either case, increase firing of dopaminergic neurons in the ventral tegmental area releases more dopamine in the nucleus accumbens and the prefrontal cortex, inducing reward. Prolonged increases in dopamine levels result in desensitization of dopamine receptors. Concomitantly, M current is reduced in nucleus accumbens and prefrontal cortex, possibly caused by dopamine receptor desensitization. All of these addiction-induced alterations lead to increased drug intake, creating a deleterious cycle, which can be broken by pharmacological M-current increases in all of these brain regions.
Psychostimulant Addiction
Pharmacological M-current augmentation has also been tested as a treatment for addiction to the psychostimulants, cocaine, methylphenidate (Ritalin) and phencyclidine (PCP) in rat models (Hansen et al., 2007). Retigabine injection was shown to significantly reduce cocaine, methylphenidate and PCP-induced locomotor activity and c-Fos expression in the NAc and the primary motor cortex. Retigabine treatment also impaired methylphenidate-induced overflow of dopamine in the striatum (Hansen et al., 2007). More recently, Parrilla-Carrero et al. (2018) showed that training to self-administer cocaine reduced spike frequency adaptation (SFA) and AHPs in a subpopulation of prelimbic prefrontal cortex (PL-PFC) neurons. This increase in excitability was resistant to extinction training and enhanced by a cued reinstatement test. These neurons also show decreased M current amplitudes and reduced sensitivity to dopamine. Ex vivo treatment with RTG restored the SFA and AHPs of these PL-PFC neurons. Moreover, RTG injection directly into the PL-PFC was shown to reduce reinstatement-induced drug-seeking behavior (Parrilla-Carrero et al., 2018), a model of relapse in rodents. Psychostimulants are known to increase dopamine levels in the brain, resulting in desensitization of D2 receptors (D2Rs; Volkow et al., 2010; Juarez and Han, 2016). Experiments with heterologously expressed dopamine D2R and Kv7.1-7.4 channels revealed D2R stimulation to increase M current in a mechanism involving G proteins of the Go/i subtype (Ljungstrom et al., 2003). Hence, it is possible that the decrease of M-current amplitudes observed with psychostimulants is related to desensitization of D2Rs, involving diminished D2R facilitation of Kv7 channel opening (Figure 2). This hypothesis could explain the hyperexcitability observed in VTA and PL-PFC. Reduction in membrane levels of Kv7 channels could also play a role in psychostimulant-induced reduction of M current amplitudes.
In brief, addiction to both ethanol and psychostimulants seem to result in reduced M-current expression and/or amplitudes, and this reduction may be related to enhanced drug-seeking behavior (Figure 2). Indeed, pharmacological M-current augmentation has shown beneficial effects in a number of addiction models. We believe the VTA to be an ideal target for the use of pharmacological M-current augmentation as a novel treatment for addiction, although we do not yet know if the mechanism of action would be due to changes in the excitability of VTA dopaminergic neurons, GABAergic neurons, or both. Ventral tegmental area projections are the main source of dopamine in all the brain regions mentioned above (Ikemoto, 2007; Ferreira et al., 2008; Hosp et al., 2011; Morales and Pickel, 2012; Han et al., 2017). Even though RTG injection into the VTA does reduce ethanol consumption (McGuier et al., 2018), for example, intra-VTA injections are not of course feasible clinically. Nonetheless, the unique composition of Kv7 channels in the VTA could represent a therapeutic opportunity. Ventral tegmental area expresses high levels of neuronal Kv7.4 subunits (Li et al., 2017), compared to neurons from other regions in the brain, in which Kv7.4 has little to no expression (Kharkovets et al., 2000; Saganich et al., 2001; Greene and Hoshi, 2017). This could allow for the use of drugs that specifically target Kv7.4, such as fasudil (Li et al., 2017), as a treatment for addiction. However, continuous pharmacological M-current augmentation through Kv7.4 channels is likely to induce hearing and blood pressure problems (Kharkovets et al., 2000; Kharkovets et al., 2006; Yeung et al., 2008; Joshi et al., 2009). Thus, more preclinical studies are necessary for pharmacological M-current augmentation to be used as a treatment for drug addiction.
M Current and Mood Disorders
Depression
The VTA also plays a major role in mood disorders such as depression (Nestler and Carlezon, 2006; Figure 3), a widespread chronic illness characterized by low mood, lack of energy, sadness, and anhedonia (Cui, 2015). Pharmacological M-current augmentation, either systemically or in the VTA, reduces depression-like behavior in the social defeat depression mouse model, measured by different depression paradigms (Friedman et al., 2016). Additionally, intra-VTA viral vector transfection of Kv7.3 channels also reduced depression like behavior, as did anterograde expression of Kv7.3 in the NAc via intra-VTA injections of viral vector (Friedman et al., 2016). Li et al. (2017) observed that systemic pharmacological augmentation of Kv7.4 channels with fasudil also reduced depression-like behavior and excitability of VTA neurons in the social defeat depression mice model.
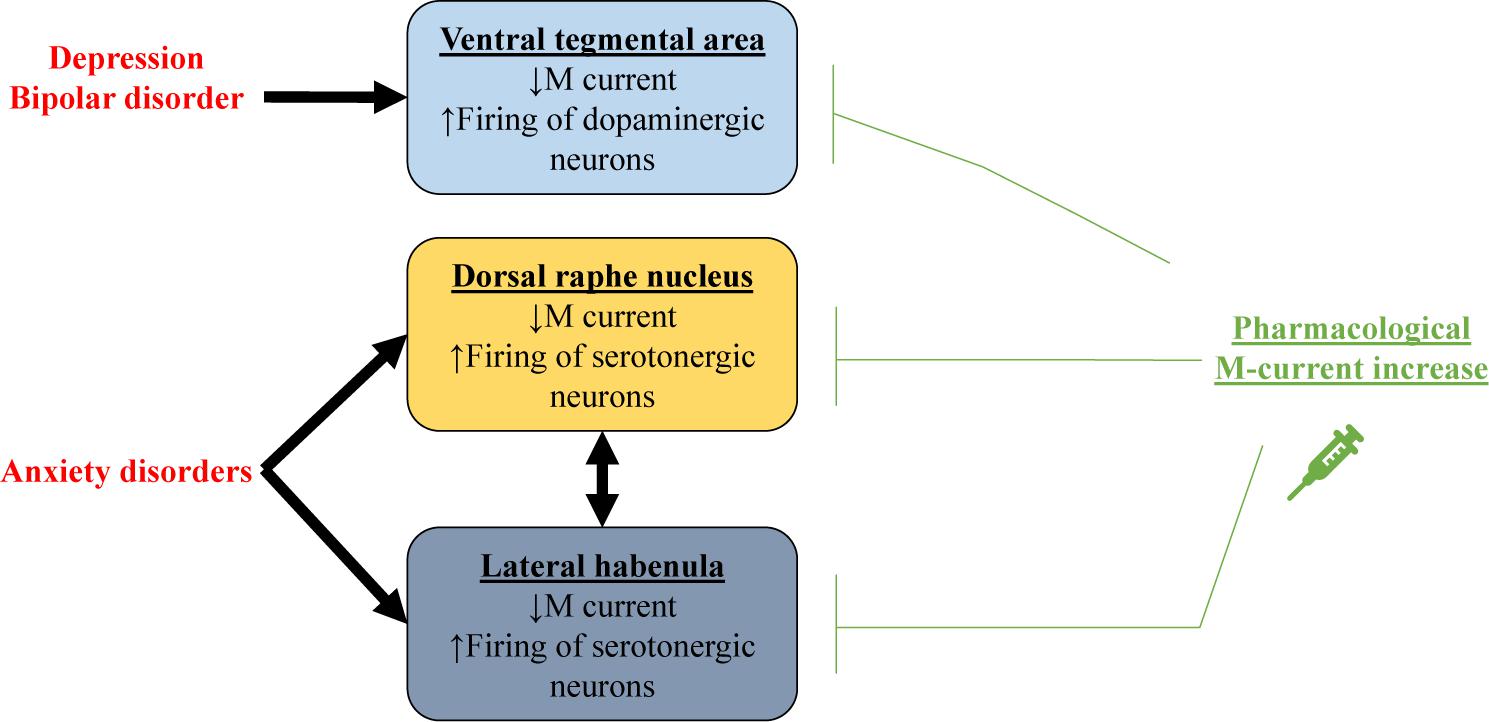
Figure 3. Schematic representation showing how decreased M current could be related to mood disorders. In depression and bipolar disorder, impaired M current in dopaminergic neurons of the ventral tegmental area (VTA) results in excessive firing. For anxiety disorders, M current is decreased in serotonergic neurons of the dorsal raphe nucleus and the lateral habenula. Pharmacological M-current increase in these regions could thus be used as a therapeutic tool for treating these mood disorders.
Bipolar Disorder
Bipolar disorder is a severe chronic mood dysfunction that is characterized by oscillation between episodes of depression and of mania (American Psychiatric Association, 2013). During manic episodes, patients experience euphoria, hyperactivity, and high levels of risk-taking behavior (American Psychiatric Association, 2013). In rodents, manic episodes can be modeled by injection of amphetamine (AMPH), combined with the benzodiazepine, chlordiazepoxide (CDP). The combination of these drugs induces hyperactivity and this phenotype can be examined with drugs used for treating bipolar disorder in the clinic, such as lithium (Dencker et al., 2008; Redrobe and Nielsen, 2009). M-current augmentation has also displayed beneficial effects in rodent models of mania (Figure 3). Studies have found that administration of RTG 30 min before testing impaired AMPH + CDP-induced hyperactivity with no effects on basal locomotor activity (Dencker et al., 2008). Redrobe and Nielsen (2009) observed that enhancement of M current with ICA-27243, which selectively augments currents from Kv7.2/7.3 channels, also reduced AMPH + CDP-induced hyperactivity. On the other hand, enhancement of Kv7.4-7.5 currents by BMS-204352 did not have significant anti-manic effects. Further studies using the AMPH + CDP model showed RTG to reduce the AMPH + CDP-induced increase in cellular metabolic demand in the thalamus, striatum and retrosplenial cortex (Kristensen et al., 2012). Furthermore, intravenous injection of RTG was shown to reduce AMPH-induced locomotor activity, neuronal firing in the VTA, and dopamine release in the NAc, whereas XE991 had the opposite effect (Sotty et al., 2009). Retigabine was also shown to impair sensitization after multiple AMPH injections in rodents (Dencker and Husum, 2010).
A new genetic dimension involves glycogen synthase kinase 3 beta (GSK3β), which is best known for intra-nuclear re-phosphorylation of the transcription factor, nuclear factor of activated T-lymphocytes (NFAT), which despite its name, is ubiquitous in brain and critical to synaptic plasticity (Graef et al., 1999). Retigabine was found to increase phosphorylation of GSK3β in hippocampus and in the pre-frontal cortex (Kristensen et al., 2012). Such phosphorylation of GSK3β at serine 9 reduces kinase activity, similar to the effect of standard anti-manic agents, such as lithium (Stambolic et al., 1996; De Sarno et al., 2002). Using in vitro studies, GSK3β was seen to phosphorylate Kv7.2 subunits, suppressing M current and lithium impairs this phosphorylation, rescuing M current (Borsotto et al., 2007). This relationship could offer a plausible novel target for the treatment of bipolar disorder.
Valproic acid (VPA) is an anti-epileptic agent commonly used in the clinic as a mood stabilizer for treatment of patients with bipolar disorder (Chiu et al., 2013; Cipriani et al., 2013). Using in vitro and in vivo mice models, part of the antiepileptic effect of VPA was shown to be due to inhibition of muscarinic-induced suppression of M current and to be dependent of Kv7.2 phosphorylation at S558 (Kay et al., 2015; Greene et al., 2018). It is possible that the beneficial effect of VPA as a mood stabilizer may also be related to drug-induced M-current increases. Nonetheless, VPA has various mechanism of action that are not related to M current (Tomson et al., 2016; Collins-Yoder and Lowell, 2017). Therefore, a direct link between VPA-induced facilitation of M current and its effect as a mood stabilizer remains to be proven.
In humans, associations between bipolar disorder and single nucleotide polymorphisms (SNPs) in the kcnq2 gene have been found (Borsotto et al., 2007; Judy et al., 2013). These SNPs disturb the interaction of Kv7.2 with ankyrin G (Borsotto et al., 2007) and protein phosphatase 2A (Judy et al., 2013), which could impair channel assembly and dephosphorylation, respectively. Finally, bipolar patients have decrease methylation of exon 11 in the kcnq3 gene, resulting in lower expression of Kv7.3 (Kaminsky et al., 2015).
Anxiety
Both BMS-204352 and RTG have anxiolytic-like effects in the zero-maze and marble-burying rodent paradigms. Those effects were blocked by XE991 without any observed motor alterations, supporting pharmacological M-current enhancement as an anxiolytic treatment (Korsgaard et al., 2005). Anxiolytic-like dose-dependent effects of RTG were also observed in the conditioned emotional-response paradigm (Munro et al., 2007). As proposed by Hansen et al. (2008), the anxiolytic-like effects of BMS-204352, observed by Korsgaard et al. (2005), suggest Kv7.4 and 7.5 to play an important role in anxiety. Immunostaining experiments show Kv7.4 channels to be highly expressed in serotonergic neurons of the dorsal raphe nucleus (DRN; Hansen et al., 2008; Zhao et al., 2017), a region of the brain known to play a central role in anxiety regulation (Graeff et al., 1997; Lowry et al., 2008). For example, increased activity of serotonergic neurons from the DRN are observed in rodent models of induced anxiety (Maier and Watkins, 2005). The excitability of serotonergic neurons from the DRN can be manipulated by pharmacological and genetic manipulation of Kv7.4 (Zhao et al., 2017). Hence, pharmacological M-current augmentation may have anxiolytic effects due to an increase in M current composed of Kv7.4 homomers in serotonergic DRN neurons, resulting in reduced firing (Figure 3). Corroborating with this hypothesis, pharmacological M-current augmentation has been shown to reduce preoperative anxiety of human patients (Yadav et al., 2017). M current in the lateral habenula (LHb) also seems to play a role in anxiety disorders. Hyperexcitability in LHb was observed in a mouse model of ethanol withdrawal, concomitant with reduced M current, specifically from Kv7.2 and 7.3. Additionally, infusion of RTG in the LHb impaired ethanol withdrawal-induced anxiety behavior (Kang et al., 2017). Curiously, intra-LHb injection of SB242084, an antagonist of the serotonin receptor 5-HT2C, also reduced ethanol withdrawal-induced anxiety behavior and increased Kv7.2 and 7.3 membrane protein levels (Fu et al., 2020). Serotonin 5-HT2C receptors are coupled to Gq/11 and therefore activate phospholipase C (Martin et al., 2014), which could modulate M current. But how these events could affect Kv7 channel membrane levels remains to be understood.
The evidence presented in this section highlight the promise of pharmacological M-current augmentation to be an effective treatment for multiple mood disorders, with different specificities of brain regions and channel subunits among the disorders. M-current regulation of dopaminergic VTA neurons may play a major role in depression and bipolar disorders (Figure 3). The VTA is an interesting therapeutic target due to its peculiarly high expression of Kv7.4 compared to other brain regions (Kharkovets et al., 2000; Saganich et al., 2001; Greene and Hoshi, 2017). Nevertheless, brain-specific drug delivery would presently be necessary to avoid peripherical effects (Kharkovets et al., 2000; Kharkovets et al., 2006; Yeung et al., 2008; Joshi et al., 2009). High expression of Kv7.4 channels can also be found in the DRN, where it is a potential therapeutic target for anxiety disorders, although for such disorders, augmentation of Kv7.2 and 7.3 in the LHb may also be necessary/beneficial. It is important to remember that the LHb and the raphe nuclei have reciprocal innervations between each other (Vertes et al., 1999; Yang et al., 2008; Metzger et al., 2017; Zhang et al., 2018). Moreover, M current might even be important in the etiology of bipolar and anxiety disorders (Borsotto et al., 2007; Judy et al., 2013; Kaminsky et al., 2015; Kang et al., 2017; Fu et al., 2020). It is likely that in both disorders, regulation of M-current by serotonergic and dopaminergic receptors (Ljungstrom et al., 2003; Fu et al., 2020) is disturbed by disease-induced alterations in these neurotransmitters and their receptors.
Conclusion
Currently pharmacological M-current manipulation is only approved by the FDA for treatment of epilepsy, although as mentioned above, RTG is off the market and its predecessor, flupertine (Szelenyi, 2013), has unacceptable liver toxicity (Puls et al., 2011; Michel et al., 2012). However, the plethora of “next-generation” M-channel openers (Miceli et al., 2018), of which many were highlighted at the recent International Kv7 symposium in Naples, Italy in 2019, show great translational promise. Animal research indicates M current to be a therapeutic target for multiple brain disorders, including those with no current treatments, such as TBI and psychostimulant addiction. But which compound should be tested first for these? RTG has a clinical history as an adjunctive treatment for epilepsy (Brodie et al., 2010; Tompson et al., 2013). Thus, repurposing RTG could represent the fastest way to the clinic, as it is already FDA-approved (in oral, but not injectable, form) and is the compound most incorporated into preclinical research involving M-channel augmentation. However, the prolonged use of RTG entails adverse side effects, such as reversible skin discoloration, retinal pigmentation abnormalities, cognitive changes, and urinary incontinence (Beacher et al., 2015; Zaugg et al., 2017). The discoloration effects are largely due to the properties of the dimerized compound that may be mitigated in more recent derivatives of RTG that have yet to undergo clinical trials. It has been suggested that other side effects may also be reduced by identifying RTG-derivatives that are more potent and selective for Kv7.2/7.3 channels (Grunnet et al., 2014; Kumar et al., 2016). Another benefit of selectivity for Kv7.2/7.3 has been suggested in the treatment of tinnitus involving M-current dependent plasticity in the dorsal cochlear nucleus (Li et al., 2013; Kalappa et al., 2015). As previously mentioned, RTG also has dose-dependent side effects on Kv2.1 (Stas et al., 2016), L-type voltage-gated Ca2+ channels (Mani et al., 2013), and GABAA receptors (Treven et al., 2015). In addition, newer insights suggest that GABA interacts with, and activates, certain Kv7 subtypes due to a conserved binding pocket (Manville and Abbott, 2018, 2020). Thus, the use of more potent and specific M-channel compounds, as well as alternative strategies to regulate M-channel transcription, need to be further explored.
Much is still unknown about how M current is involved in the variety of neurological diseases mentioned in this review. Even basic preclinical tests remain to be performed for most of them. For example, for the new RTG-derivatives the minimum and maximum effective doses and possible side effects of long-term use are unknown. In neurovascular injuries, the necessity of one or only a few doses of treatment after the injury should eliminate the risks associated with long-term use of a drug. Since mood disorders are chronic diseases, the clinical use of pharmacological M-current augmentation necessitates prolonged use, presenting an extra challenge. It is also necessary to start investigating brain-specific delivery of M-current modulators. Addiction is a case that remains a bigger challenge. In rodent models, M current has been described to reduce both drug intake and relapse-like drug seeking behavior. Translating this to the clinic, M-current augmentation might be useful for treatment of an active drug user and/or to reduce relapse of a patient that is no longer using the drug. Both possibilities can only be truly tested in clinical trials. Pharmacological M-current augmentation is likely to be a therapeutic tool for a spectrum of pathological situations, as discussed here. However, there is still a long road ahead until clinical trials establish the true value of this mode of therapeutic intervention.
Author Contributions
All authors wrote the manuscript.
Funding
This work was supported by the U.S. Department of Defense CDMRP grant W81XWH-15-1-0284 (MS), National Institutes of Health grant R01 159040 (MS), American Heart Association post-doctoral fellowship 20POST35180050 (FV), and the Morrison Trust of San Antonio (MS).
Conflict of Interest
The authors have submitted a pending US patent for the use of Kv7 channel openers as a novel therapy for traumatic brain injury, held in the name of Advanced Neuroresearch Therapeutics, LLC.
References
Abd-Elsayed, A. A., Ikeda, R., Jia, Z., Ling, J., Zuo, X., Li, M., et al. (2015). KCNQ channels in nociceptive cold-sensing trigeminal ganglion neurons as therapeutic targets for treating orofacial cold hyperalgesia. Mol. Pain 11:45.
Ahuja, C. S., Wilson, J. R., Nori, S., Kotter, M. R. N., Druschel, C., Curt, A., et al. (2017). Traumatic spinal cord injury. Nat. Rev. Dis. Primers 3:17018.
Amabile, C. M., and Vasudevan, A. (2013). Ezogabine: a novel antiepileptic for adjunctive treatment of partial-onset seizures. Pharmacotherapy 33, 187–194. doi: 10.1002/phar.1185
Ambrosino, P., Alaimo, A., Bartollino, S., Manocchio, L., De Maria, M., Mosca, I., et al. (2015). Epilepsy-causing mutations in Kv7.2 C-terminus affect binding and functional modulation by calmodulin. Biochim. Biophys. Acta 1852, 1856–1866. doi: 10.1016/j.bbadis.2015.06.012
American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th Edn. Washington, DC: American Psychiatric Association.
Archer, C. R., Enslow, B. T., Taylor, A. B., De la Rosa, V., Bhattacharya, A., and Shapiro, M. S. (2019). A mutually induced conformational fit underlies Ca2+-directed interactions between calmodulin and the proximal C terminus of KCNQ4 K+ channels. J. Biol. Chem. 294, 6094–6112. doi: 10.1074/jbc.ra118.006857
Armand, V., Rundfeldt, C., and Heinemann, U. (1999). Effects of retigabine (D-23129) on different patterns of epileptiform activity induced by 4-aminopyridine in rat entorhinal cortex hippocampal slices. Naunyn Schmiedebergs Arch. Pharmacol. 359, 33–39. doi: 10.1007/pl00005320
Barrese, V., Taglialatela, M., Greenwood, I. A., and Davidson, C. (2015). Protective role of Kv7 channels in oxygen and glucose deprivation-induced damage in rat caudate brain slices. J. Cereb. Blood Flow Metab. 35, 1593–1600. doi: 10.1038/jcbfm.2015.83
Beacher, N. G., Brodie, M. J., and Goodall, C. (2015). A case report: retigabine induced oral mucosal dyspigmentation of the hard palate. BMC Oral Health 15:122. doi: 10.1186/s12903-015-0102-y
Beez, T., Steiger, H. J., and Etminan, N. (2017). Pharmacological targeting of secondary brain damage following ischemic or hemorrhagic stroke, traumatic brain injury, and bacterial meningitis - a systematic review and meta-analysis. BMC Neurol. 17:209. doi: 10.1186/s12883-017-0994-z
Bierbower, S. M., Choveau, F. S., Lechleiter, J. D., and Shapiro, M. S. (2015). Augmentation of M-type (KCNQ) potassium channels as a novel strategy to reduce stroke-induced brain injury. J. Neurosci. 35, 2101–2111. doi: 10.1523/jneurosci.3805-14.2015
Biervert, C., Schroeder, B. C., Kubisch, C., Berkovic, S. F., Propping, P., Jentsch, T. J., et al. (1998). A potassium channel mutation in neonatal human epilepsy. Science 279, 403–406. doi: 10.1126/science.279.5349.403
Blackburn-Munro, G., and Jensen, B. S. (2003). The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur. J. Pharmacol 460, 109–116. doi: 10.1016/s0014-2999(02)02924-2
Borsotto, M., Cavarec, L., Bouillot, M., Romey, G., Macciardi, F., Delaye, A., et al. (2007). PP2A-Bgamma subunit and KCNQ2 K+ channels in bipolar disorder. Pharmacogenomics J. 7, 123–132. doi: 10.1038/sj.tpj.6500400
Boscia, F., Annunziato, L., and Taglialatela, M. (2006). Retigabine and flupirtine exert neuroprotective actions in organotypic hippocampal cultures. Neuropharmacology 51, 283–294. doi: 10.1016/j.neuropharm.2006.03.024
Brodie, M. J., Lerche, H., Gil-Nagel, A., Elger, C., Hall, S., Shin, P., et al. (2010). Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology 75, 1817–1824. doi: 10.1212/wnl.0b013e3181fd6170
Brown, D. A., and Adams, P. R. (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283, 673–676. doi: 10.1038/283673a0
Brown, D. A., Buckley, N. J., Caulfield, M. P., Duffy, S. M., Jones, S., Lamas, J. A., et al. (1995). “Coupling of muscarinic acetylcholine receptors to neural ion channels: closure of K+ channels,” in Molecular Mechanisms of Muscarinic Acetylcholine Receptor Function, ed. J. Wess (Austin, TX: RG Lanes Co), 165–182.
Busserolles, J., Tsantoulas, C., Eschalier, A., and Lopez Garcia, J. A. (2016). Potassium channels in neuropathic pain: advances, challenges, and emerging ideas. Pain 157(Suppl. 1), S7–S14.
Carver, C. M., Hastings, S. D., Cook, M. E., and Shapiro, M. S. (2020). Functional responses of the hippocampus to hyperexcitability depend on directed, neuron-specific KCNQ2 K+ channel plasticity. Hippocampus 30, 435–455. doi: 10.1002/hipo.23163
Chang, A., Abderemane-Ali, F., Hura, G. L., Rossen, N. D., Gate, R. E., and Minor, D. L. Jr. (2018). A calmodulin C-lobe Ca2+-dependent switch governs Kv7 channel function. Neuron 97, 836.e6–852.e6.
Chiu, C. T., Wang, Z., Hunsberger, J. G., and Chuang, D. M. (2013). Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol. Rev. 65, 105–142. doi: 10.1124/pr.111.005512
Choveau, F., de la Rosa, V., Bierbower, S. M., Hernandez, C. C., and Shapiro, M. S. (2018). Phosphatidylinositol 4,5-bisphosphate (PIP2) regulates KCNQ3 K+ channels through multiple sites of action. J. Biol. Chem. 293, 19411–19428. doi: 10.1074/jbc.ra118.005401
Christensen, J., Pedersen, M. G., Pedersen, C. B., Sidenius, P., Olsen, J., and Vestergaard, M. (2009). Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 373, 1105–1110. doi: 10.1016/s0140-6736(09)60214-2
Cipriani, A., Reid, K., Young, A. H., Macritchie, K., and Geddes, J. (2013). Valproic acid, valproate and divalproex in the maintenance treatment of bipolar disorder. Cochrane Database Syst. Rev. 2013:CD003196.
Collins-Yoder, A., and Lowell, J. (2017). Valproic acid: special considerations and targeted monitoring. J. Neurosci. Nurs. 49, 56–61. doi: 10.1097/jnn.0000000000000259
Constanti, A., and Brown, D. A. (1981). M-Currents in voltage-clamped mammalian sympathetic neurones. Neurosci. Lett. 24, 289–294. doi: 10.1016/0304-3940(81)90173-7
Cooper, E. C., Harrington, E., Jan, Y. N., and Jan, L. Y. (2001). M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 21, 9529–9540. doi: 10.1523/jneurosci.21-24-09529.2001
Cui, R. (2015). Editorial: a systematic review of depression. Curr. Neuropharmacol. 13:480. doi: 10.2174/1570159x1304150831123535
De Sarno, P., Li, X., and Jope, R. S. (2002). Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 43, 1158–1164. doi: 10.1016/s0028-3908(02)00215-0
Dencker, D., Dias, R., Pedersen, M. L., and Husum, H. (2008). Effect of the new antiepileptic drug retigabine in a rodent model of mania. Epilepsy Behav. 12, 49–53. doi: 10.1016/j.yebeh.2007.09.023
Dencker, D., and Husum, H. (2010). Antimanic efficacy of retigabine in a proposed mouse model of bipolar disorder. Behav. Brain Res. 207, 78–83. doi: 10.1016/j.bbr.2009.09.040
Di Chiara, G., and Imperato, A. (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. U.S.A. 85, 5274–5278. doi: 10.1073/pnas.85.14.5274
Du, X., Gao, H., Jaffe, D., Zhang, H., and Gamper, N. (2018). M-type K+ channels in peripheral nociceptive pathways. Br. J. Pharmacol. 175, 2158–2172. doi: 10.1111/bph.13978
Ferreira, J. G., Del-Fava, F., Hasue, R. H., and Shammah-Lagnado, S. J. (2008). Organization of ventral tegmental area projections to the ventral tegmental area-nigral complex in the rat. Neuroscience 153, 196–213. doi: 10.1016/j.neuroscience.2008.02.003
Frey, L. C. (2003). Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia 44, 11–17. doi: 10.1046/j.1528-1157.44.s10.4.x
Friedman, A. K., Juarez, B., Ku, S. M., Zhang, H., Calizo, R. C., Walsh, J. J., et al. (2016). KCNQ channel openers reverse depressive symptoms via an active resilience mechanism. Nat. Commun. 7:11671.
Fu, R., Mei, Q., Shiwalkar, N., Zuo, W., Zhang, H., Gregor, D., et al. (2020). Anxiety during alcohol withdrawal involves 5-HT2C receptors and M-channels in the lateral habenula. Neuropharmacology 163:107863. doi: 10.1016/j.neuropharm.2019.107863
Gamper, N., and Shapiro, M. S. (2003). Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J. Gen. Physiol. 122, 17–31. doi: 10.1085/jgp.200208783
Gamper, N., Zaika, O., Li, Y., Martin, P., Hernandez, C. C., Perez, M. R., et al. (2006). Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 25, 4996–5004. doi: 10.1038/sj.emboj.7601374
Garga, N., and Lowenstein, D. H. (2006). Posttraumatic epilepsy: a major problem in desperate need of major advances. Epilepsy Curr. 6, 1–5. doi: 10.1111/j.1535-7511.2005.00083.x
Gessa, G. L., Muntoni, F., Collu, M., Vargiu, L., and Mereu, G. (1985). Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain Res. 348, 201–203. doi: 10.1016/0006-8993(85)90381-6
Graef, I. A., Mermelstein, P. G., Stankunas, K., Neilson, J. R., Deisseroth, K., Tsien, R. W., et al. (1999). L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401, 703–708. doi: 10.1038/44378
Graeff, F. G., Viana, M. B., and Mora, P. O. (1997). Dual role of 5-HT in defense and anxiety. Neurosci. Biobehav. Rev. 21, 791–799. doi: 10.1016/s0149-7634(96)00059-0
Greene, D. L., and Hoshi, N. (2017). Modulation of Kv7 channels and excitability in the brain. Cell Mol. Life. Sci. 74, 495–508. doi: 10.1007/s00018-016-2359-y
Greene, D. L., Kosenko, A., and Hoshi, N. (2018). Attenuating M-current suppression in vivo by a mutant Kcnq2 gene knock-in reduces seizure burden and prevents status epilepticus-induced neuronal death and epileptogenesis. Epilepsia 59, 1908–1918. doi: 10.1111/epi.14541
Grunnet, M., Strobaek, D., Hougaard, C., and Christophersen, P. (2014). Kv7 channels as targets for anti-epileptic and psychiatric drug-development. Eur. J. Pharmacol. 726, 133–137. doi: 10.1016/j.ejphar.2014.01.017
Gunthorpe, M. J., Large, C. H., and Sankar, R. (2012). The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 53, 412–424. doi: 10.1111/j.1528-1167.2011.03365.x
Haitin, Y., and Attali, B. (2008). The C-terminus of Kv7 channels: a multifunctional module. J. Physiol. 586, 1803–1810. doi: 10.1113/jphysiol.2007.149187
Haley, J. E., Abogadie, F. C., Delmas, P., Dayrell, M., Vallis, Y., Milligan, G., et al. (1998). The alpha subunit of Gq contributes to muscarinic inhibition of the M- type potassium current in sympathetic neurons. J. Neurosci. 18, 4521–4531. doi: 10.1523/jneurosci.18-12-04521.1998
Han, X., Jing, M. Y., Zhao, T. Y., Wu, N., Song, R., and Li, J. (2017). Role of dopamine projections from ventral tegmental area to nucleus accumbens and medial prefrontal cortex in reinforcement behaviors assessed using optogenetic manipulation. Metab. Brain Dis. 32, 1491–1502. doi: 10.1007/s11011-017-0023-3
Hansen, H. H., Andreasen, J. T., Weikop, P., Mirza, N., Scheel-Kruger, J., and Mikkelsen, J. D. (2007). The neuronal KCNQ channel opener retigabine inhibits locomotor activity and reduces forebrain excitatory responses to the psychostimulants cocaine, methylphenidate and phencyclidine. Eur. J. Pharmacol. 570, 77–88. doi: 10.1016/j.ejphar.2007.05.029
Hansen, H. H., Waroux, O., Seutin, V., Jentsch, T. J., Aznar, S., and Mikkelsen, J. D. (2008). Kv7 channels: interaction with dopaminergic and serotonergic neurotransmission in the CNS. J. Physiol. 586, 1823–1832. doi: 10.1113/jphysiol.2007.149450
Hayashi, H., Iwata, M., Tsuchimori, N., and Matsumoto, T. (2014). Activation of peripheral KCNQ channels attenuates inflammatory pain. Mol. Pain 10:15.
Hemphill, J. C. III, Greenberg, S. M., Anderson, C. S., Becker, K., Bendok, B. R., Cushman, M., et al. (2015). Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the american heart association/american stroke association. Stroke 46, 2032–2060. doi: 10.1161/str.0000000000000069
Hoshi, N., Zhang, J. S., Omaki, M., Takeuchi, T., Yokoyama, S., Wanaverbecq, N., et al. (2003). AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat. Neurosci. 6, 564–571. doi: 10.1038/nn1062
Hosp, J. A., Pekanovic, A., Rioult-Pedotti, M. S., and Luft, A. R. (2011). Dopaminergic projections from midbrain to primary motor cortex mediate motor skill learning. J. Neurosci. 31, 2481–2487. doi: 10.1523/jneurosci.5411-10.2011
Hunt, R. F., Boychuk, J. A., and Smith, B. N. (2013). Neural circuit mechanisms of post-traumatic epilepsy. Front. Cell Neurosci. 7:89. doi: 10.3389/fncel.2013.00089
Ikemoto, S. (2007). Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res. Rev. 56, 27–78. doi: 10.1016/j.brainresrev.2007.05.004
Jentsch, T. J. (2000). Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 1, 21–30. doi: 10.1038/35036198
Jones, S., Brown, D. A., Milligan, G., Willer, E., Buckley, N. J., and Caulfield, M. P. (1995). Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and Gαq/11. Neuron 14, 399–405. doi: 10.1016/0896-6273(95)90295-3
Joshi, S., Sedivy, V., Hodyc, D., Herget, J., and Gurney, A. M. (2009). KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J. Pharmacol. Exp. Ther. 329, 368–376. doi: 10.1124/jpet.108.147785
Juarez, B., and Han, M. H. (2016). Diversity of dopaminergic neural circuits in response to drug exposure. Neuropsychopharmacology 41, 2424–2446. doi: 10.1038/npp.2016.32
Judy, J. T., Seifuddin, F., Pirooznia, M., Mahon, P. B., Jancic, D., Goes, F. S., et al. (2013). Converging evidence for epistasis between ANK3 and potassium channel gene KCNQ2 in bipolar disorder. Front. Genet. 4:87. doi: 10.3389/fgene.2013.00087
Kalappa, B. I., Soh, H., Duignan, K. M., Furuya, T., Edwards, S., Tzingounis, A. V., et al. (2015). Potent KCNQ2/3-specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J. Neurosci. 35, 8829–8842. doi: 10.1523/jneurosci.5176-14.2015
Kaminsky, Z., Jones, I., Verma, R., Saleh, L., Trivedi, H., Guintivano, J., et al. (2015). DNA methylation and expression of KCNQ3 in bipolar disorder. Bipolar Disord. 17, 150–159. doi: 10.1111/bdi.12230
Kang, S., Li, J., Zuo, W., Fu, R., Gregor, D., Krnjevic, K., et al. (2017). Ethanol withdrawal drives anxiety-related behaviors by reducing M-type potassium channel activity in the lateral habenula. Neuropsychopharmacology 42, 1813–1824. doi: 10.1038/npp.2017.68
Kapetanovic, I. M., Yonekawa, W. D., and Kupferberg, H. J. (1995). The effects of D-23129, a new experimental anticonvulsant drug, on neurotransmitter amino acids in the rat hippocampus in vitro. Epilepsy Res. 22, 167–173. doi: 10.1016/0920-1211(95)00050-x
Kay, H. Y., Greene, D. L., Kang, S., Kosenko, A., and Hoshi, N. (2015). M-current preservation contributes to anticonvulsant effects of valproic acid. J. Clin. Invest. 125, 3904–3914. doi: 10.1172/jci79727
Kharkovets, T., Dedek, K., Maier, H., Schweizer, M., Khimich, D., Nouvian, R., et al. (2006). Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 25, 642–652. doi: 10.1038/sj.emboj.7600951
Kharkovets, T., Hardelin, J. P., Safieddine, S., Schweizer, M., El-Amraoui, A., Petit, C., et al. (2000). KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc. Natl. Acad. Sci. U.S.A. 97, 4333–4338. doi: 10.1073/pnas.97.8.4333
Kim, K. W., Kim, K., Lee, H., and Suh, B. C. (2019). Ethanol elevates excitability of superior cervical ganglion neurons by inhibiting Kv7 channels in a cell type-specific and PI(4,5)P2-dependent manner. Int. J. Mol. Sci. 20:4419. doi: 10.3390/ijms20184419
Knapp, C. M., O’Malley, M., Datta, S., and Ciraulo, D. A. (2014). The Kv7 potassium channel activator retigabine decreases alcohol consumption in rats. Am. J. Drug Alcohol Abuse 40, 244–250. doi: 10.3109/00952990.2014.892951
Korsgaard, M. P., Hartz, B. P., Brown, W. D., Ahring, P. K., Strobaek, D., and Mirza, N. R. (2005). Anxiolytic effects of Maxipost (BMS-204352) and retigabine via activation of neuronal Kv7 channels. J. Pharmacol. Exp. Ther. 314, 282–292. doi: 10.1124/jpet.105.083923
Kosenko, A., and Hoshi, N. (2013). A change in configuration of the calmodulin-KCNQ channel complex underlies Ca2+-dependent modulation of KCNQ channel activity. PLoS One 8:e82290. doi: 10.1371/journal.pone.0082290
Koyama, S., Brodie, M. S., and Appel, S. B. (2007). Ethanol inhibition of m-current and ethanol-induced direct excitation of ventral tegmental area dopamine neurons. J. Neurophysiol. 97, 1977–1985. doi: 10.1152/jn.00270.2006
Kristensen, L. V., Sandager-Nielsen, K., and Hansen, H. H. (2012). K(v) 7 (KCNQ) channel openers normalize central 2-deoxyglucose uptake in a mouse model of mania and increase prefrontal cortical and hippocampal serine-9 phosphorylation levels of GSK3beta. J. Neurochem. 121, 373–382. doi: 10.1111/j.1471-4159.2012.07704.x
Kumar, M., Reed, N., Liu, R., Aizenman, E., Wipf, P., and Tzounopoulos, T. (2016). Synthesis and evaluation of potent KCNQ2/3-specific channel activators. Mol. Pharmacol. 89, 667–677. doi: 10.1124/mol.115.103200
Lacey, M. G., Calabresi, P., and North, R. A. (1990). Muscarine depolarizes rat substantia nigra zona compacta and ventral tegmental neurons in vitro through M1-like receptors. J. Pharmacol. Exp. Ther. 253, 395–400.
Lammel, S., Lim, B. K., Ran, C., Huang, K. W., Betley, M. J., Tye, K. M., et al. (2012). Input-specific control of reward and aversion in the ventral tegmental area. Nature 491, 212–217. doi: 10.1038/nature11527
Lawrence, J. J., Saraga, F., Churchill, J. F., Statland, J. M., Travis, K. E., Skinner, F. K., et al. (2006). Somatodendritic Kv7/KCNQ/M channels control interspike interval in hippocampal interneurons. J. Neurosci. 26, 12325–12338. doi: 10.1523/jneurosci.3521-06.2006
Li, L., Li, J., Zuo, Y., Dang, D., Frost, J. A., and Yang, Q. (2019). Activation of KCNQ channels prevents paclitaxel-induced peripheral neuropathy and associated neuropathic pain. J. Pain 20, 528–539. doi: 10.1016/j.jpain.2018.11.001
Li, L., Sun, H., Ding, J., Niu, C., Su, M., Zhang, L., et al. (2017). Selective targeting of M-type potassium Kv 7.4 channels demonstrates their key role in the regulation of dopaminergic neuronal excitability and depression-like behaviour. Br. J. Pharmacol. 174, 4277–4294. doi: 10.1111/bph.14026
Li, S., Choi, V., and Tzounopoulos, T. (2013). Pathogenic plasticity of Kv7.2/3 channel activity is essential for the induction of tinnitus. Proc. Natl. Acad. Sci. U.S.A. 110, 9980–9985. doi: 10.1073/pnas.1302770110
Li, Y., Gamper, N., Hilgemann, D. W., and Shapiro, M. S. (2005). Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol (4,5)-bisphosphate. J. Neurosci. 25, 9825–9835. doi: 10.1523/jneurosci.2597-05.2005
Ljungstrom, T., Grunnet, M., Jensen, B. S., and Olesen, S. P. (2003). Functional coupling between heterologously expressed dopamine D(2) receptors and KCNQ channels. Pflugers Arch. 446, 684–694. doi: 10.1007/s00424-003-1111-2
Lowenstein, D. H. (2009). Epilepsy after head injury: an overview. Epilepsia 50(Suppl. 2), 4–9. doi: 10.1111/j.1528-1167.2008.02004.x
Lowry, C. A., Hale, M. W., Evans, A. K., Heerkens, J., Staub, D. R., Gasser, P. J., et al. (2008). Serotonergic systems, anxiety, and affective disorder: focus on the dorsomedial part of the dorsal raphe nucleus. Ann. N. Y. Acad. Sci. 1148, 86–94.
Maier, S. F., and Watkins, L. R. (2005). Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neurosci. Biobehav. Rev. 29, 829–841. doi: 10.1016/j.neubiorev.2005.03.021
Main, M. J., Cryan, J. E., Dupere, J. R., Cox, B., Clare, J. J., and Burbidge, S. A. (2000). Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 58, 253–262. doi: 10.1124/mol.58.2.253
Maljevic, S., Wuttke, T. V., and Lerche, H. (2008). Nervous system Kv7 disorders: breakdown of a subthreshold brake. J. Physiol. 586, 1791–1801. doi: 10.1113/jphysiol.2008.150656
Mani, B. K., O’Dowd, J., Kumar, L., Brueggemann, L. I., Ross, M., and Byron, K. L. (2013). Vascular KCNQ (Kv7) potassium channels as common signaling intermediates and therapeutic targets in cerebral vasospasm. J. Cardiovasc. Pharmacol. 61, 51–62. doi: 10.1097/fjc.0b013e3182771708
Manville, R. W., and Abbott, G. W. (2018). Gabapentin is a potent activator of KCNQ3 and KCNQ5 potassium channels. Mol. Pharmacol. 94, 1155–1163. doi: 10.1124/mol.118.112953
Manville, R. W., and Abbott, G. W. (2020). Potassium channels act as chemosensors for solute transporters. Commun. Biol. 3:90.
Martin, C. B., Hamon, M., Lanfumey, L., and Mongeau, R. (2014). Controversies on the role of 5-HT(2C) receptors in the mechanisms of action of antidepressant drugs. Neurosci. Biobehav. Rev. 42, 208–223. doi: 10.1016/j.neubiorev.2014.03.001
McGuier, N. S., Griffin, W. C. III, Gass, J. T., Padula, A. E., Chesler, E. J., and Mulholland, P. J. (2016). Kv7 channels in the nucleus accumbens are altered by chronic drinking and are targets for reducing alcohol consumption. Addict. Biol. 21, 1097–1112. doi: 10.1111/adb.12279
McGuier, N. S., Rinker, J. A., Cannady, R., Fulmer, D. B., Jones, S. R., Hoffman, M., et al. (2018). Identification and validation of midbrain Kcnq4 regulation of heavy alcohol consumption in rodents. Neuropharmacology 138, 10–19. doi: 10.1016/j.neuropharm.2018.05.020
Metzger, M., Bueno, D., and Lima, L. B. (2017). The lateral habenula and the serotonergic system. Pharmacol. Biochem. Behav. 162, 22–28. doi: 10.1016/j.pbb.2017.05.007
Miceli, F., Soldovieri, M. V., Ambrosino, P., Manocchio, L., Mosca, I., and Taglialatela, M. (2018). Pharmacological targeting of neuronal Kv7.2/3 channels: a focus on chemotypes and receptor sites. Curr. Med. Chem. 25, 2637–2660. doi: 10.2174/0929867324666171012122852
Miceli, F., Soldovieri, M. V., Martire, M., and Taglialatela, M. (2008). Molecular pharmacology and therapeutic potential of neuronal Kv7-modulating drugs. Curr. Opin. Pharmacol. 8, 65–74. doi: 10.1016/j.coph.2007.10.003
Miceli, F., Striano, P., Soldovieri, M. V., Fontana, A., Nardello, R., Robbiano, A., et al. (2015). A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia 56, e15–e20. doi: 10.1111/epi.12887
Michel, M. C., Radziszewski, P., Falconer, C., Marschall-Kehrel, D., and Blot, K. (2012). Unexpected frequent hepatotoxicity of a prescription drug, flupirtine, marketed for about 30 years. Br. J. Clin. Pharmacol. 73, 821–825. doi: 10.1111/j.1365-2125.2011.04138.x
Moore, S. D., Madamba, S. G., and Siggins, G. R. (1990). Ethanol diminishes a voltage-dependent K+ current, the M-current, in CA1 hippocampal pyramidal neurons in vitro. Brain Res. 516, 222–228. doi: 10.1016/0006-8993(90)90922-x
Morales, M., and Pickel, V. M. (2012). Insights to drug addiction derived from ultrastructural views of the mesocorticolimbic system. Ann. N. Y. Acad. Sci. 1248, 71–88. doi: 10.1111/j.1749-6632.2011.06299.x
Munro, G., and Dalby-Brown, W. (2007). Kv7 (KCNQ) channel modulators and neuropathic pain. J. Med. Chem. 50, 2576–2582. doi: 10.1021/jm060989l
Munro, G., Erichsen, H. K., and Mirza, N. R. (2007). Pharmacological comparison of anticonvulsant drugs in animal models of persistent pain and anxiety. Neuropharmacology 53, 609–618. doi: 10.1016/j.neuropharm.2007.07.002
Nakajo, K., and Kubo, Y. (2005). Protein kinase C shifts the voltage dependence of KCNQ/M channels expressed in Xenopus oocytes. J. Physiol. 569, 59–74. doi: 10.1113/jphysiol.2005.094995
Nestler, E. J., and Carlezon, W. A. Jr. (2006). The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 59, 1151–1159. doi: 10.1016/j.biopsych.2005.09.018
Pan, Z., Kao, T., Horvath, Z., Lemos, J., Sul, J. Y., Cranstoun, S. D., et al. (2006). A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26, 2599–2613. doi: 10.1523/jneurosci.4314-05.2006
Parrilla-Carrero, J., Buchta, W. C., Goswamee, P., Culver, O., McKendrick, G., Harlan, B., et al. (2018). Restoration of Kv7 channel-mediated inhibition reduces cued-reinstatement of cocaine seeking. J. Neurosci. 38, 4212–4229. doi: 10.1523/jneurosci.2767-17.2018
Peters, H. C., Hu, H., Pongs, O., Storm, J. F., and Isbrandt, D. (2005). Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat. Neurosci. 8, 51–60. doi: 10.1038/nn1375
Puls, F., Agne, C., Klein, F., Koch, M., Rifai, K., Manns, M. P., et al. (2011). Pathology of flupirtine-induced liver injury: a histological and clinical study of six cases. Virchows Arch. 458, 709–716. doi: 10.1007/s00428-011-1087-9
Rasmussen, H. B., Frokjaer-Jensen, C., Jensen, C. S., Jensen, H. S., Jorgensen, N. K., Misonou, H., et al. (2007). Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J. Cell Sci. 120, 953–963. doi: 10.1242/jcs.03396
Redrobe, J. P., and Nielsen, A. N. (2009). Effects of neuronal Kv7 potassium channel activators on hyperactivity in a rodent model of mania. Behav. Brain Res. 198, 481–485. doi: 10.1016/j.bbr.2008.12.027
Rodd-Henricks, Z. A., McKinzie, D. L., Crile, R. S., Murphy, J. M., and McBride, W. J. (2000). Regional heterogeneity for the intracranial self-administration of ethanol within the ventral tegmental area of female Wistar rats. Psychopharmacology 149, 217–224. doi: 10.1007/s002139900347
Rostock, A., Tober, C., Rundfeldt, C., Bartsch, R., Engel, J., Polymeropoulos, E. E., et al. (1996). D-23129: a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 23, 211–223. doi: 10.1016/0920-1211(95)00101-8
Rundfeldt, C., and Netzer, R. (2000). The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells tranfected with human KCNQ2/3 subunits. Neurosci. Lett. 282, 73–76. doi: 10.1016/s0304-3940(00)00866-1
Saganich, M. J., Machado, E., and Rudy, B. (2001). Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J. Neurosci. 21, 4609–4624. doi: 10.1523/jneurosci.21-13-04609.2001
Shah, M. M., Migliore, M., Valencia, I., Cooper, E. C., and Brown, D. A. (2008). Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 7869–7874. doi: 10.1073/pnas.0802805105
Shapiro, M. S., Roche, J. P., Kaftan, E. J., Cruzblanca, H., Mackie, K., and Hille, B. (2000). Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J. Neurosci. 20, 1710–1721. doi: 10.1523/jneurosci.20-05-01710.2000
Simon, D. W., McGeachy, M. J., Bayir, H., Clark, R. S., Loane, D. J., and Kochanek, P. M. (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 13, 171–191. doi: 10.1038/nrneurol.2017.13
Singh, N. A., Charlier, C., Stauffer, D., DuPont, B. R., Leach, R. J., Melis, R., et al. (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 18, 25–29. doi: 10.1038/ng0198-25
Soldovieri, M. V., Miceli, F., and Taglialatela, M. (2011). Driving with no brakes: molecular pathophysiology of Kv7 potassium channels. Physiology 26, 365–376. doi: 10.1152/physiol.00009.2011
Sotty, F., Damgaard, T., Montezinho, L. P., Mork, A., Olsen, C. K., Bundgaard, C., et al. (2009). Antipsychotic-like effect of retigabine [N-(2-Amino-4-(fluorobenzylamino)-phenyl)carbamic acid ester], a KCNQ potassium channel opener, via modulation of mesolimbic dopaminergic neurotransmission. J. Pharmacol. Exp. Ther. 328, 951–962. doi: 10.1124/jpet.108.146944
Splinter, M. Y. (2013). Efficacy of retigabine in adjunctive treatment of partial onset seizures in adults. J. Cent. Nerv. Syst. Dis. 5, 31–41.
Stambolic, V., Ruel, L., and Woodgett, J. R. (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664–1668.
Stas, J. I., Bocksteins, E., Jensen, C. S., Schmitt, N., and Snyders, D. J. (2016). The anticonvulsant retigabine suppresses neuronal Kv2-mediated currents. Sci. Rep. 6:35080.
Strulovich, R., Tobelaim, W. S., Attali, B., and Hirsch, J. A. (2016). Structural insights into the M-channel proximal C-terminus/calmodulin complex. Biochemistry 55, 5353–5365. doi: 10.1021/acs.biochem.6b00477
Suh, B.-C., and Hille, B. (2005). Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 15, 370–378. doi: 10.1016/j.conb.2005.05.005
Suh, B. C., Inoue, T., Meyer, T., and Hille, B. (2006). Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314, 1454–1457. doi: 10.1126/science.1131163
Sun, J., and MacKinnon, R. (2017). Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long-QT syndrome. Cell 169, 1042.e9–1050.e9.
Sun, J., and MacKinnon, R. (2020). Structural basis of human KCNQ1 modulation and gating. Cell 180, 340.e9–347.e9.
Szelenyi, I. (2013). Flupirtine, a re-discovered drug, revisited. Inflamm. Res. 62, 251–258. doi: 10.1007/s00011-013-0592-5
Tatulian, L., and Brown, D. A. (2003). Effect of the KCNQ potassium channel opener retigabine on single KCNQ2/3 channels expressed in CHO cells. J. Physiol. 549, 57–63. doi: 10.1113/jphysiol.2003.039842
Tatulian, L., Delmas, P., Abogadie, F. C., and Brown, D. A. (2001). Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J. Neurosci. 21, 5535–5545. doi: 10.1523/jneurosci.21-15-05535.2001
Tompson, D. J., Crean, C. S., Reeve, R., and Berry, N. S. (2013). Efficacy and tolerability exposure-response relationship of retigabine (ezogabine) immediate-release tablets in patients with partial-onset seizures. Clin. Ther. 35, 1174.e4–1185.e4.
Tomson, T., Battino, D., and Perucca, E. (2016). Valproic acid after five decades of use in epilepsy: time to reconsider the indications of a time-honoured drug. Lancet Neurol. 15, 210–218. doi: 10.1016/s1474-4422(15)00314-2
Treven, M., Koenig, X., Assadpour, E., Gantumur, E., Meyer, C., Hilber, K., et al. (2015). The anticonvulsant retigabine is a subtype selective modulator of GABAA receptors. Epilepsia 56, 647–657. doi: 10.1111/epi.12950
Tykocki, N. R., Heppner, T. J., Dalsgaard, T., Bonev, A. D., and Nelson, M. T. (2019). The Kv 7 channel activator retigabine suppresses mouse urinary bladder afferent nerve activity without affecting detrusor smooth muscle K(+) channel currents. J. Physiol. 597, 935–950. doi: 10.1113/jp277021
Tzingounis, A. V., and Nicoll, R. A. (2008). Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc. Natl. Acad. Sci. U.S.A. 105, 19974–19979. doi: 10.1073/pnas.0810535105
Vertes, R. P., Fortin, W. J., and Crane, A. M. (1999). Projections of the median raphe nucleus in the rat. J. Comp. Neurol. 407, 555–582. doi: 10.1002/(sici)1096-9861(19990517)407:4<555::aid-cne7>3.0.co;2-e
Vigil, F. A., Bozdemir, E., Bugay, V., Chun, S. H., Hobbs, M., Sanchez, I., et al. (2020). Prevention of brain damage after traumatic brain injury by pharmacological enhancement of KCNQ (Kv7, “M-type”) K(+) currents in neurons. J. Cereb. Blood Flow Metab. 40, 1256–1273. doi: 10.1177/0271678X19857818
Volkow, N. D., Wang, G. J., Fowler, J. S., Tomasi, D., Telang, F., and Baler, R. (2010). Addiction: decreased reward sensitivity and increased expectation sensitivity conspire to overwhelm the brain’s control circuit. Bioessays 32, 748–755. doi: 10.1002/bies.201000042
Wang, J. J., and Li, Y. (2016). KCNQ potassium channels in sensory system and neural circuits. Acta Pharmacol. Sin. 37, 25–33. doi: 10.1038/aps.2015.131
Weiss, F., Lorang, M. T., Bloom, F. E., and Koob, G. F. (1993). Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J. Pharmacol. Exp. Ther. 267, 250–258.
Winks, J. S., Hughes, S., Filippov, A. K., Tatulian, L., Abogadie, F. C., Brown, D. A., et al. (2005). Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J. Neurosci. 25, 3400–3413. doi: 10.1523/jneurosci.3231-04.2005
Wu, Z., Li, L., Xie, F., Xu, G., Dang, D., and Yang, Q. (2020). Enhancing KCNQ channel activity improves neurobehavioral recovery after spinal cord injury. J. Pharmacol. Exp. Ther. 373, 72–80. doi: 10.1124/jpet.119.264010
Wuttke, T. V., Seebohm, G., Bail, S., Maljevic, S., and Lerche, H. (2005). The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol. Pharmacol. 67, 1009–1017. doi: 10.1124/mol.104.010793
Yadav, G., Jain, G., and Singh, M. (2017). Role of flupirtine in reducing preoperative anxiety of patients undergoing craniotomy procedure. Saudi J. Anaesth. 11, 158–162.
Yang, L. M., Hu, B., Xia, Y. H., Zhang, B. L., and Zhao, H. (2008). Lateral habenula lesions improve the behavioral response in depressed rats via increasing the serotonin level in dorsal raphe nucleus. Behav. Brain Res. 188, 84–90. doi: 10.1016/j.bbr.2007.10.022
Yeung, S., Schwake, M., Pucovsky, V., and Greenwood, I. (2008). Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br. J. Pharmacol. 155, 62–72. doi: 10.1038/bjp.2008.231
Yu, H., Wu, M., Hopkins, C., Engers, J., Townsend, S., Lindsley, C., et al. (2010). “A small molecule activator of KCNQ2 and KCNQ4 channels,” in Proceedings of the Probe Reports from the NIH Molecular Libraries Program, (Bethesda, MD: National Center for Biotechnology Information).
Yue, C., and Yaari, Y. (2004). KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J. Neurosci. 24, 4614–4624. doi: 10.1523/jneurosci.0765-04.2004
Yus-Najera, E., Santana-Castro, I., and Villarroel, A. (2002). The identification and characterization of a non-continuous calmodulin binding site in non-inactivating voltage-dependent KCNQ potassium channels. J. Biol. Chem. 24:24.
Zaczek, R., Chorvat, R. J., Saye, J. A., Pierdomenico, M. E., Maciag, C. M., Logue, A. R., et al. (1998). Two new potent neurotransmitter release enhancers, 10,10-bis(4- pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4- pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J. Pharmacol. Exp. Ther. 285, 724–730.
Zaika, O., Tolstykh, G. P., Jaffe, D. B., and Shapiro, M. S. (2007). Inositol triphosphate-mediated Ca2+ signals direct purinergic P2Y-receptor regulation of neuronal ion channels. J. Neurosci. 27, 8914–8926. doi: 10.1523/jneurosci.1739-07.2007
Zaugg, B. E., Bell, J. E., Taylor, K. Y., and Bernstein, P. S. (2017). Ezogabine (Potiga) maculopathy. Retin. Cases Brief. Rep. 11, 38–43. doi: 10.1097/icb.0000000000000283
Zhang, H., Craciun, L. C., Mirshahi, T., Rohacs, T., Lopes, C. M., Jin, T., et al. (2003). PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975. doi: 10.1016/s0896-6273(03)00125-9
Zhang, H., Li, K., Chen, H. S., Gao, S. Q., Xia, Z. X., Zhang, J. T., et al. (2018). Dorsal raphe projection inhibits the excitatory inputs on lateral habenula and alleviates depressive behaviors in rats. Brain Struct. Funct. 223, 2243–2258. doi: 10.1007/s00429-018-1623-3
Zhao, C., Su, M., Wang, Y., Li, X., Zhang, Y., Du, X., et al. (2017). Selective modulation of K+ Channel Kv7.4 significantly affects the excitability of DRN 5-HT neurons. Front. Cell Neurosci. 11:405. doi: 10.3389/fncel.2017.00405
Zhao, Y. J., Nai, Y., Li, S. Y., and Zheng, Y. H. (2018). Retigabine protects the blood-brain barrier by regulating tight junctions between cerebral vascular endothelial cells in cerebral ischemia-reperfusion rats. Eur. Rev. Med. Pharmacol. Sci. 22, 8509–8518.
Keywords: Kv7, potassium channels, stroke, traumatic brain injury, drug addiction, anxiety, bipolar disorder
Citation: Vigil FA, Carver CM and Shapiro MS (2020) Pharmacological Manipulation of Kv7 Channels as a New Therapeutic Tool for Multiple Brain Disorders. Front. Physiol. 11:688. doi: 10.3389/fphys.2020.00688
Received: 21 April 2020; Accepted: 27 May 2020;
Published: 19 June 2020.
Edited by:
Francesco Miceli, University of Naples Federico II, ItalyReviewed by:
Edward C. Cooper, Baylor College of Medicine, United StatesNaoto Hoshi, University of California, Irvine, United States
Copyright © 2020 Vigil, Carver and Shapiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark S. Shapiro, c2hhcGlyb21AdXRoc2NzYS5lZHU=