 J. Snyder
J. Snyder R. Zhai
R. Zhai A. I. Lackey
A. I. Lackey P. Y. Sato
P. Y. Sato- Department of Pharmacology and Physiology, Drexel University College of Medicine, Philadelphia, PA, United States
Heart disease is widely recognized as a major cause of death worldwide and is the leading cause of mortality in the United States. Centuries of research have focused on defining mechanistic alterations that drive cardiac pathogenesis, yet sudden cardiac death (SCD) remains a common unpredictable event that claims lives in every age group. The heart supplies blood to all tissues while maintaining a constant electrical and hormonal feedback communication with other parts of the body. As such, recent research has focused on understanding how myocardial electrical and structural properties are altered by cardiac metabolism and the various signaling pathways associated with it. The importance of cardiac metabolism in maintaining myocardial function, or lack thereof, is exemplified by shifts in cardiac substrate preference during normal development and various pathological conditions. For instance, a shift from fatty acid (FA) oxidation to oxygen-sparing glycolytic energy production has been reported in many types of cardiac pathologies. Compounded by an uncoupling of glycolysis and glucose oxidation this leads to accumulation of undesirable levels of intermediate metabolites. The resulting accumulation of intermediary metabolites impacts cardiac mitochondrial function and dysregulates metabolic pathways through several mechanisms, which will be reviewed here. Importantly, reversal of metabolic maladaptation has been shown to elicit positive therapeutic effects, limiting cardiac remodeling and at least partially restoring contractile efficiency. Therein, the underlying metabolic adaptations in an array of pathological conditions as well as recently discovered downstream effects of various substrate utilization provide guidance for future therapeutic targeting. Here, we will review recent data on alterations in substrate utilization in the healthy and diseased heart, metabolic pathways governing cardiac pathogenesis, mitochondrial function in the diseased myocardium, and potential metabolism-based therapeutic interventions in disease.
Introduction
Cardiovascular disease is the number one cause of death in the United States and worldwide, accounting for about 17.9 million deaths globally in 2015 (Benjamin et al., 2018). As the incidence of cardiac disease increases exponentially, estimates show that by 2035, expenditures related to chronic heart disease will exceed one trillion dollars (Benjamin et al., 2018). A common terminal event for cardiovascular disease is sudden cardiac arrest (SCA) leading to sudden cardiac death (SCD). SCA often results from ventricular tachyarrhythmias or ventricular fibrillation initiated by dysfunctional excitation contraction coupling. The genesis of the heartbeat is an intrinsic electrical phenomena of the heart, with electrical impulses driven by pacemaker cells at the sino-atrial node that propagate through the His-Purkinje bundle system, ultimately leading to synchronous ventricular contraction (Jalife et al., 2009). Ventricular contraction is a highly energetic process, as the heart is the most metabolically demanding organ in our body, contracting approximately 3 billion times during an average lifespan of 75 years. In spite of this extremely high energetic requirement, myocardial ATP stores are relatively low with a complete turnover of myocardial ATP pool every 10s (Neely and Morgan, 1974). To accomplish this highly bioenergetic coupling to mechanical contraction, the myocardium utilizes a gamut of circulating energetic substrates such as FAs, glucose, lactate, branched chain amino acids (BCAAs), and ketone bodies (Figure 1). These cytosolic metabolites are enzymatically processed, ultimately converging in the generation of acetyl-CoA, and/or reduced nucleotide electron carriers (FADH2 and NADH), that through the electron transport chain (ETC) are used to develop the proton motive force driving ATP production.
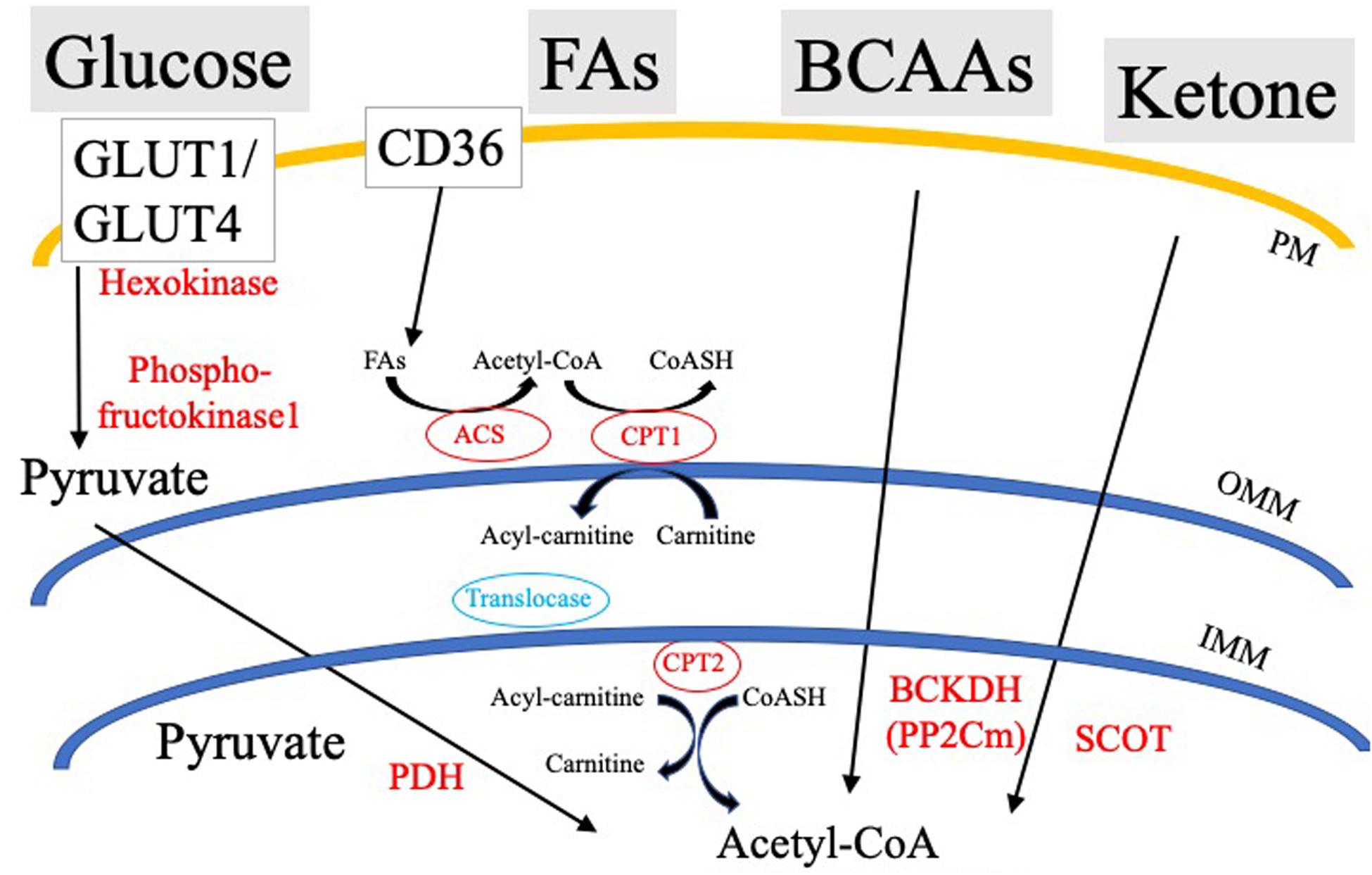
Figure 1. Simplified schematic of cardiac substrate utilization and substrate-specific rate-limiting/key processes. PDH, Pyruvate dehydrogenase; SCOT, CoA Transferase; BCKDH, Branched-chain α-keto acid dehydrogenase; ACS, Acyl-CoA Synthetase; OMM, Outer mitochondrial membrane; IMM, Inner mitochondrial membrane; PM, Plasma membrane; and CPT, Carnitine palmitoyltransferase.
While metabolic flexibility is an essential cardiac attribute, the use of each substrate is not only regulated but preference altered during development and in various cardiac pathologies. Under normal physiological conditions, energetic demand is primarily met by carbohydrates and FAs, with the latter being the major fuel source in the adult heart, where β-oxidation contributes to 60–80% of ATP production (Opie, 1968, 1969; Neely and Morgan, 1974). Nevertheless, in several cardiomyopathies, such as left ventricular hypertrophy, the adult heart reverts to a more fetal-like cardiometabolic state, switching from FA oxidation as the primary energy source to oxygen sparing glycolytic energy production (Taegtmeyer and Overturf, 1988; Depre et al., 1998). This review will focus on alterations in metabolism that occur as the heart progresses from healthy to diseased states (Figure 2). We will discuss substrate transport, the role of substrate metabolism, metabolic pathways, mitochondrial function, and ion channel modifications that precede and contribute to SCA/SCD. Lastly, we will briefly discuss potential interventions that target myocardial energy metabolism, as an approach to treat and/or prevent heart failure (HF) and SCD.
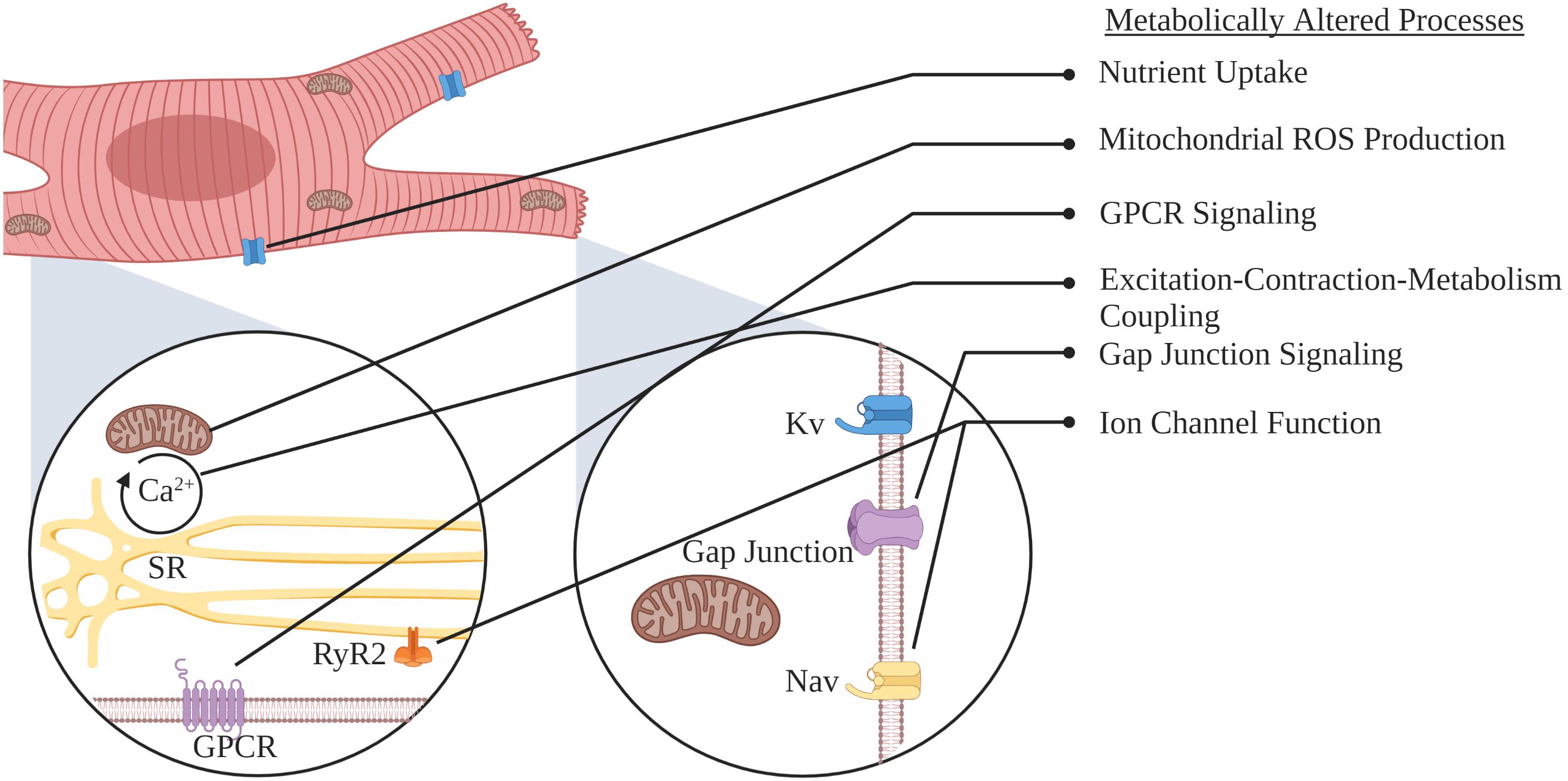
Figure 2. Schematic representation of myocardial adaptations preceding SCA or SCD.
Regulation of Glucose and Fatty Acid Entry in Cardiomyocytes
In order to meet its own bioenergetic demands, the heart expresses specialized proteins that regulate substrate entry from the blood into the cytosol of cardiomyocytes. While glucose and FAs are the major cardiac metabolic substrates, the heart can also use ketone bodies and BCAAs to meet its energetic demands (Figure 1). Transport of substrates is specific and dependent on concentration gradients and hormonal signaling. In this section, we will discuss current knowledge of various modes for glucose and FA entry into cardiomyocytes.
Transporters of Glucose
In the adult heart, about 25 to 30% of energy used for contractile function is generated by carbohydrates (Shao and Tian, 2015). There are three known modes of glucose transport across cellular plasma membranes: (i) facilitative glucose transporters (GLUTs), which are the major form of cardiac glucose entry; (ii) sodium-glucose cotransporters (SGLTs), which use the energy of a favorable sodium gradient to transport glucose against its concentration gradient; and (iii) recently identified sugars will eventually be exported transporters (SWEETs), which promote cellular efflux of glucose (Banerjee et al., 2009; Wright, 2013). SWEETS are novel transporters of glucose that are mostly found in plants, and although understudied in humans, the human SWEET1 gene has been identified in mammary glands where it is thought to provide glucose for lactose synthesis (Chen et al., 2010). Thus far, no other major physiological function of SWEETs is known in humans.
The major path of glucose entry into the heart is via GLUTs, a family of proteins consisting of 14 members, where the main cardiac isoforms are GLUT1 and GLUT4. The expression of GLUT1 and GLUT4 is dysregulated in various cardiac diseases. In response to chronic hypoxia and ischemia, GLUT1 expression increases, leading to cardioprotection via increased glucose influx independent of insulin signaling (Brosius et al., 1997). GLUT1 is localized on the plasma membrane and plays an important role in regulating basal metabolism of cardiomyocytes. GLUT1 can be regulated by fasting and is mainly expressed in embryonic or neonatal stages of cardiac development (Kraegen et al., 1993). Genetically modified animal models have revealed an important role for GLUT1 in regulating glucose utilization for cardiac function. Murine cardiac-specific overexpression of GLUT1 enhanced cardiac glucose utilization, attenuated the development of contractile dysfunction, and improved long-term survival after transverse aortic constriction (TAC; Liao et al., 2002; Luptak et al., 2007). Moreover, acute increases in glucose uptake mediated by inducible overexpression of GLUT1 improved mitochondrial function and reduced cardiac structural remodeling but did not prevent ventricular dysfunction (Pereira et al., 2013).
Following birth, the expression of GLUT4 increases and becomes the predominant glucose transporter in adult hearts. GLUT4 is located in intracellular vesicles that translocate to the plasma membrane upon insulin stimulation or muscular contraction (Rattigan et al., 1991; Slot et al., 1991; Kraegen et al., 1993). During the development of cardiac hypertrophy, and in response to myocardial ischemia (MI), GLUT4 translocates from intracellular vesicles to the plasma membrane, increasing glucose intracellular levels (Slot et al., 1991; Young et al., 1997; Shi et al., 2016). However, GLUT4 expression is decreased in human HF with a concurrent decrease in glucose-mediated energy production (Razeghi et al., 2001; Doehner et al., 2010). Similarly, decreases in GLUT4 expression are also observed in hypertensive models showing insulin-resistance (Paternostro et al., 1995). Mouse models of GLUT4 have revealed that this transporter plays a prominent role in cardiac function. Cardiac overexpression of GLUT4 increased glycolysis but not glucose oxidation (Belke et al., 2001), while cardiac-specific deletion of GLUT4 led to cardiac hypertrophy with preserved contractile function (Abel et al., 1999). Notably, following downregulation of GLUT4, SGLT1 may mediate glucose utilization, as it is increased 2- to 3-fold in ischemic and type 2 diabetic mellitus (T2DM) hearts (Banerjee et al., 2009) and other studies have reported that SGLT1 colocalizes with GLUT1 at the sarcolemma (Turk et al., 1991; Zhou et al., 2003). Yet, the precise functional role of SGLT1 and any compensatory role in the heart remains poorly understood.
Noteworthy is the ability of incretins to promote GLUT1 expression in cardiomyocytes. In fact, incretins like glucagon-like peptide 1 (GLP-1; produced by enteroendocrine L-cells), and glucose-dependent insulinotropic polypeptide (GIP; produced by enteroendocrine K-cells; Baggio and Drucker, 2007) can regulate appetite, glucose-mediated insulin secretion from pancreatic β-cells (Varndell et al., 1985), and increase GLUT1 expression and translocation to the sarcolemma in cardiomyopathy models (Bhashyam et al., 2010). Interestingly, the impact of incretins is not limited to diabetes-related cardiac dysfunction, as GLP-1 agonists restored cardiac function in dogs with advanced dilated cardiomyopathy (DCM; Nikolaidis et al., 2004). GIP also reduces angiotensin II-induced cardiac hypertrophy and fibrosis in mice (Hiromura et al., 2016). Nonetheless, the importance of incretin signaling in metabolism and chronic disease in humans has been underscored by the association of genetic variations in GLP-1 and GIP receptors with body weight, pancreatic islet function, and risk for T2DM (Sathananthan et al., 2010; Finan et al., 2016).
Transporters of Fatty Acids
As the healthy heart greatly depends on the oxidation of FAs for energy production, FA transport through the plasma membrane is key to myocardial metabolism (Nagoshi et al., 2011). Although FAs can passively diffuse into the plasma membrane of cells (Schaffer and Lodish, 1995), several proteins facilitate cardiomyocyte FA uptake and transport, including cluster of differentiation 36 (CD36 or FAT; Abumrad et al., 1993), plasma membrane-associated FA-binding protein or plasmalemmal FA-binding protein (FABPpm; Stremmel et al., 1985), and FA transport proteins (FATPs; Schaffer and Lodish, 1994; Gimeno et al., 2003). In addition to these membrane bound lipid transport proteins, cardiac tissue also expresses heart-FABP (HFABP or FABP3), a cytosolic lipid binding protein that binds and transports hydrophobic lipid species through the cytoplasm (Binas et al., 1999; Storch and Thumser, 2010). In the heart, CD36 is the main transporter responsible for FA uptake.
CD36 was first identified as a cellular transporter of FAs in 1993 (Abumrad et al., 1993). Several ex vivo and in vivo studies support the hypothesis that CD36 is necessary for efficient myocardial FA uptake and accumulation (Coburn et al., 2000; Bharadwaj et al., 2010). Additionally, CD36 can transduce signals that influence how cardiac FAs are utilized. For example, CD36 induces a signaling cascade that favors FA oxidation by activation of 5’ adenosine monophosphate-activated protein kinase (AMPK). CD36 forms as complex with LKB1 (AMPK Kinase) and Fyn (src kinase; Samovski et al., 2015). Binding of FA to CD36 disassociates the complex, allowing for LKB1 to phosphorylate and activate AMPK (Samovski et al., 2015). Although CD36 is thought to be the main regulator of cardiac FA transport through the plasma membrane, there does not appear to be a consensus on the role of CD36 in pathological cardiac hypertrophy (Nakamura et al., 1999). For example, in mice that were subjected to TAC, cardiomyocyte-specific ablation of CD36 resulted in a more rapid progression from compensated hypertrophy to HF (Sung et al., 2017). Conversely, in diet induced obese (DIO) mice, cardiomyocyte-specific knock down of CD36 was protective and reduced signs of pathological cardiac remodeling when compared to control mice (Zhang et al., 2015). In response to hypertrophy, the expression of FATPs are significantly decreased (Vork et al., 1992), but the physiological significance of this finding remains unclear. In order for FAs to enter the mitochondria for energy production, FAs must be activated by forming fatty-acyl-CoA. The mitochondrial enzyme that mediates FA entry, carnitine palmitoyltransferase 1 (CPT1), is located on the outer mitochondrial membrane, and modulation of its activity or expression results in alterations in FA substrate utilization in the myocardium (Opie and Knuuti, 2009). CPT1 catalyzes the transesterifcation of acetyl-CoA into acylcarnitine. Subsequently, carnitine acylcarnitine translocase (CACT) transports acylcarnitine across the inner mitochondrial membrane in exchange for a free carnitine molecule. The acylcarnitine is then reconverted into acetyl-CoA via CPT2, which is located on the inner mitochondrial membrane (Houten et al., 2016). CPT1 is the rate limiting enzyme of FA entry into the mitochondria, and its regulation is integral to the control of FA oxidation. An important post-transcriptional step in the regulation of FA oxidation involves the inhibition of CPT1 by malonyl CoA, which is formed from acetyl-CoA via acetyl-CoA carboxylase (ACC; Foster, 2012). Thus, the negative feedback inhibition of CPT1 via malonyl-CoA can avert FA-driven ATP generation, and lead to the incorporation of FA into lipid droplets in the cytosol. Excess intracellular palmitate can also lead to ceramide production and protein palmitoylation of ionic channels and mediators of cardiac conduction (Chien et al., 1996; Pei et al., 2016). Relevant to HF and SCD are the studies suggesting that reversible protein S-pamitoylation plays an important role for sodium and potassium channel biosynthesis (Schmidt and Catterall, 1987). Particularly pertinent to cardiac arrhythmias is the recent study showing that palmitoylation of the major cardiac sodium channel (Nav1.5) leads to increased channel availability and late sodium current activity, promoting the generation of arrhythmogenic events (Pei et al., 2016). Although it is currently not known if palmitoylation impacts the inward rectifier potassium channel (Ik1), there is a known reciprocity between the sodium and potassium channels at the plasma membrane that impacts excitability and arrhythmia generation (Milstein et al., 2012). Thus at the very least, the inward rectifier current could be indirectly impacted by palmitoylation of Nav1.5, this could be significant as it could also alter resting membrane potential. Palmitoylation is also known to modify the L-type calcium beta2a subunit impacting channel function (Chien et al., 1996). Thus, while it is evident that a link between cardiac metabolism and SCD exists, specific mechanisms dictating this spatiotemporal relationship remain poorly understood.
Metabolic Pathways Governing Cardiac Function and Pathogenesis
Metabolic Flexibility
Metabolic flexibility is a key functional aspect of a healthy heart and it relates to the ability of a cell to adapt to its environment as determined by the utilization of nutrients, oxygen, and hormonal input. It is specifically important in the heart due to the energetic demands of continuous repetitive cardiac muscle contractions and the detrimental feedback loops initiated in its absence, to be reviewed in this section. Loss of this flexibility is thought to be one of the earliest indications of cardiac dysfunction leading to HF (Harris and Das, 1991). Indeed, the failing heart has reduced bioenergetic capacity and phosphocreatine/ATP ratio (Neubauer, 2007). Glucose and FA oxidation are the two main cardiac energy sources, but chronic overreliance or overabundance of one substrate class without the corresponding bioenergetic coupling may have deleterious consequences for myocardial function (Sharma et al., 2004; Chess and Stanley, 2008). Exemplifying this metabolic-contractile link is the potential role for glucose and FA metabolism in regulating the sarcoplasmic-reticulum calcium pump (SERCA) 2a expression via SP1 (a glucose regulated transcription factor) and peroxisome proliferation-activated receptors (PPAR; a FA regulated transcription factor), respectively (Rupp and Zarain-Herzberg, 2002). Table 1 shows current models of cardiac pathology associated with SCD and their observed metabolic impact.
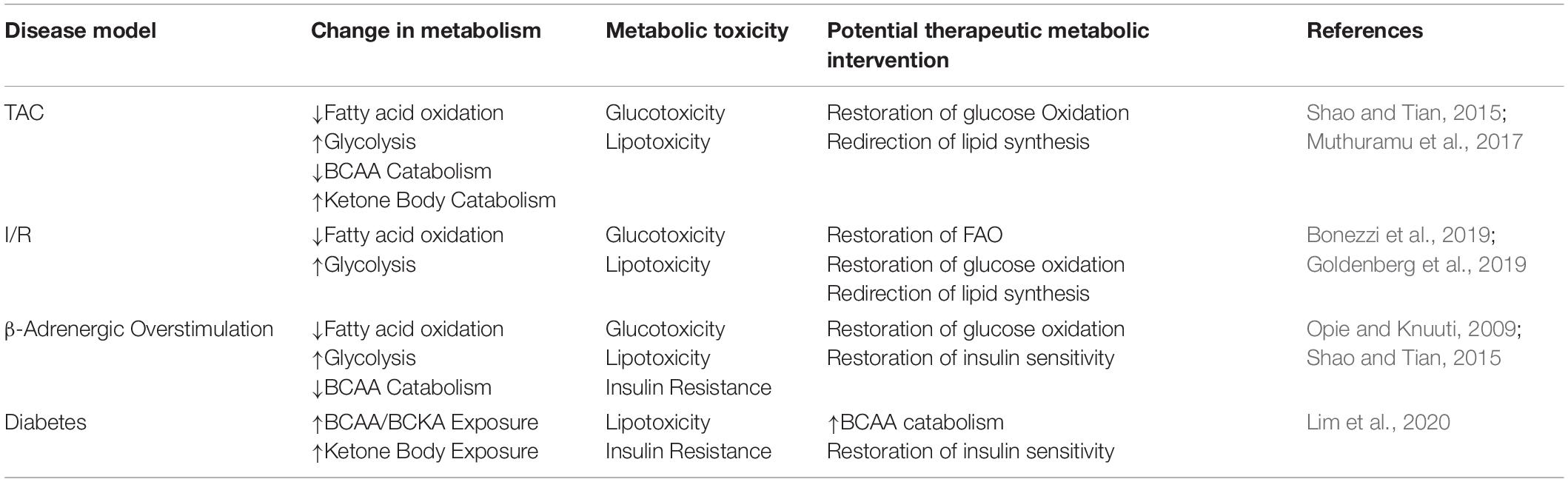
Table 1. SCD-disease models with associated metabolic profiles and resultant toxicity.
Cardiac substrate utilization is intertwined, crossing over and impacting parallel metabolic pathways and signaling integrations. For instance, cardiac-specific deletion of ACC prevented TAC-induced decreases in FA oxidation and increased reliance on glucose (Kolwicz et al., 2012). Conversely, cardiac-specific inducible pyruvate dehydrogenase α (PDHα) deletion led to decreased glucose oxidation, decreased AMPK phosphorylation, and ischemic-induced cardiac injury (Sun W. et al., 2016). These mechanistic studies are particularly relevant to pathological conditions that involve altered substrate preferences. For example, in pathological hypertrophy and ischemic heart disease, the heart heavily relies on glucose to meet its bioenergetic needs (Eberli et al., 1991). In contrast, in T2DM cardiac glucose uptake is reduced with concurrent increase in FA oxidation enzymes leading to exacerbated cardiac hypertrophy (Paternostro et al., 1999; Domenighetti et al., 2010).
Under normal physiological conditions, more than 90% of myocardial ATP production is generated via mitochondrial oxidative phosphorylation, with the remainder being produced by anaerobic glycolysis and GTP from the tricarboxylic acid (TCA) cycle (Harris and Das, 1991). Oxidative phosphorylation in the fetal heart mainly relies on glucose, while in adult stages, there is a shift in preference toward FA metabolism. Notably, in the heart, the majority of ATP produced is quickly consumed by myofilaments during contraction. Additionally, about 25% is used to fuel cardiac sarcolemmal and sarcoplasmic reticulum (SR) ion channels and transporters (Schramm et al., 1994). The link between cardiac rhythm, myocardial work, and metabolism was first explored in the early 1900s, when notable physiologists including E. H Starling developed in situ models of studying these relationships (Knowlton and Starling, 1912; Patterson et al., 1914). They were able to establish relationships between preload and afterload with energetic rates, oxygen consumption, and changing blood parameters (Evans, 1939). Notably, metabolic dysfunction is ubiquitously observed in chronic heart diseases that are associated with increased risk for SCD.
Lipotoxicity
In 1963, Philip Randle and colleagues hypothesized that the heart must utilize both glucose and FA to meet its high energy demands (Randle et al., 1963). However, successful oxidation of FA produces several factors that inhibit glycolysis, including NADH, ATP, citrate, and acetyl CoA. Thus, an environment where high glycolytic flux and FA oxidation simultaneously occur is rare. Flux through these pathways is influenced by feed/fast cycles and by exercise paradigms such that at different points, glucose and FA are used in different proportions and neither are over-accumulated (Randle et al., 1963). Overabundance of glucose or FA availability or dysregulation of the usage thereof may contribute to cytotoxic effects classified as lipotoxicity and glucotoxicity (Lundsgaard et al., 2019).
Lipotoxicity is induced by the abnormal accumulation of intra-myocellular fatty acids and lipid metabolites in non-adipose tissues such as the heart. Lipid storage in non-adipose sites contributes to altered cellular physiology and perhaps most notably insulin resistance. The summation of these detrimental effects of lipid accumulation is termed lipotoxicity (Sharma et al., 2004). Lipotoxicity can be mimicked via high fat diets that model western feeding habits and/or increased lipid transport into the myocardium (Chiu et al., 2001, 2005). However, the roles of various classes of lipid molecules in myocardial pathogenesis have only been recently explored. Increased levels of triacylglycerol (TG), the main dietary form of lipid, are associated with diseases such as obesity and T2DM. Yet, the role for TGs in inducing cellular lipotoxicity remains controversial (Drosatos and Schulze, 2013). Studies suggest that TG levels act as a marker of general lipid load, while an array of other lipid metabolites play a larger role in increasing or decreasing lipotoxicity. For example, while the role of TG in directly inducing lipotoxicity remains inconclusive, there is evidence that diacylglycerol (DG), a TG metabolite, is linked to lipotoxicity and induction of insulin resistance in muscle and liver (Inoguchi et al., 1992; Erion and Shulman, 2010). More recently, Law and colleagues have demonstrated that very long ceramide species (≥24 carbons) diminish mitochondrial function and increase apoptosis, autophagy, and mitophagy in cardiomyocytes (Law et al., 2018). Conversely, in a model where long-chain FA synthesis was re-directed toward long-chain ceramide synthesis (20–22 carbons) a cardioprotective phenotype was demonstrated in a TAC model of left ventricular hypertrophy, demonstrating the importance of lipid saturation and chain length. This latter study used a mouse model overexpressing cardiac acyl-coenzyme A synthetase 1, and thereby affected multiple lipid metabolism parameters (Goldenberg et al., 2019). However, the lipid profile following the TAC intervention specifically deviated in ceramide species accumulation (Goldenberg et al., 2019). Additionally, saturated FAs, such as palmitic acid, have been associated with increased ceramide synthesis and cardiomyocyte apoptosis when compared to unsaturated FAs, such as oleic acid (Okere et al., 2006). In fact, restoring FA oxidation in the heart by overexpression of Long-chain-FA-CoA ligase 1 (ACSL1) in a TAC model preserved cardiac function (Goldenberg et al., 2019). Together, this emphasizes the importance of lipid diversity in the heart and how they may differently impact the development of lipotoxicity.
The main source of FAs that fuel mitochondrial oxidative metabolism are circulating free FAs (FFAs) bound to albumin and/or released from TGs that are present in very-low-density lipoproteins (VLDL) or chylomicrons (Niu et al., 2004; Bharadwaj et al., 2010). As mentioned above, extracellular FAs can enter cardiomyocytes via passive diffusion, FATPs, or CD36 (Schaffer and Lodish, 1994; Gimeno et al., 2003; Bharadwaj et al., 2010). Once FFAs are in the cytosol, they are esterified to CoA via fatty acyl-CoA synthase enzymes, forming long-chain fatty acyl-CoAs (Chiu et al., 2001, 2005). These fatty acyl-CoAs can then shuttle into mitochondria via CPT1 and CPT2. CPT1 converts the long chain fatty acyl-CoA to long chain acylcarnitine in the outer mitochondrial membrane, which is then subsequently converted back to long chain fatty acyl CoA by CPT2 in the mitochondrial inner membrane (Murthy and Pande, 1984; McGarry and Brown, 1997). As mentioned previously, a major point of regulation for fatty acyl-CoA transport into the mitochondria involves CPT1, which is robustly inhibited by the presence of malonyl CoA (Foster, 2012). The levels of malonyl CoA are determined by the balance of acetyl CoA synthesis via ACC, and degradation of malonyl CoA by malonyl CoA decarboxylase (Dyck and Lopaschuk, 2002; Ussher and Lopaschuk, 2008). For example, citrate produced from the TCA cycle can move to the cytosol and activate ACC (Lane et al., 1970; Beaty and Lane, 1983), which leads to greater production of malonyl CoA and a feedback response that reduces mitochondrial FA oxidation.
During HF, fatty acyl carnitines in the cytosol and sarcolemma are accumulated up to 10 times their normal levels, particularly in cardiac regions exposed to ischemia. The relationship between lipid accumulation and electrical abnormalities has been hypothesized in the 1980s (Corr et al., 1989). Since then multiple pathways have been established by which fatty acyl carnitines affect ion channel function and therefore arrhythmogenic events. Reactive oxygen species (ROS) generated from fatty acyl carnitines interact with redox sensitive channels, such as the ryanodine receptor, thus modulating calcium release from the SR during contraction. Palmitoyl carnitine, a fatty acyl carnitine, increases the oxidation of the ryanodine receptor 2 resulting in prolonged calcium leak following opening (Roussel et al., 2015). Direct infusion of palmitoyl carnitine at levels observed in diabetic cardiomyopathy is independently arrhythmogenic in healthy animals (Roussel et al., 2015). Palmitoyl carnitine exposure to cardiomyocytes induces the slow-inactivating sodium current, impacting current inactivation thereby augmenting intracellular sodium concentration. Subsequently, palmitoyl carnitine inhibited the sodium potassium exchanger by approximately 13%, inducing an elevation in intracellular calcium (Wu and Corr, 1995). Moreover, long-chain acyl carnitines are known to reduce gap junctional conductance by 68%, a phenomenon which has been linked to the preferential accumulation of endogenous long-chain acyl carnitines at the sarcolemma during hypoxia (Wu et al., 1993). These effects alter the incidence of early after depolarizations, delayed after depolarizations, and action potential propagation promoting the development of arrhythmias.
In addition to certain lipids that may directly induce lipotoxicity, such as ceramides and DGs, ROS is generated as a byproduct of the ETC which can induce cellular stress. ROS are generated when electrons leak from the mitochondrial ETC, allowing free electrons to reduce molecular oxygen (Turrens, 2003). Certain pathological conditions, such as MI and HF, can increase ROS production via mitochondrial membrane leakage. Elevation of mitochondrial ROS can damage ETC complexes, further impairing ATP production in a failing heart (Murray et al., 2003). This leads to a vicious circle in which the electron leak increases ROS formation, and ROS formation alters the ETC to favor additional production of ROS. ROS can damage ETC complex proteins and can react with polyunsaturated FAs, which enhances the formation of peroxidized lipids that can dysregulate phospholipid membranes (Mylonas and Kouretas, 1999). In humans with diabetic cardiomyopathy and obesity-related cardiomyopathy, insulin resistance diminishes glucose uptake and utilization enhancing cardiac lipid accumulation (Boudina and Abel, 2007; Montaigne et al., 2014). As such, impairments in cardiac lipid metabolism that reduce intracellular TG-derived FA mobilization and oxidation promote an accumulation of intracellular TG (McGavock et al., 2007; O’Donnell et al., 2008). Interestingly, similar to citrate, insulin also inhibits FA oxidation through activation of ACC (Witters and Kemp, 1992). In the failing heart, insulin-induced inhibition of FA oxidation is impaired, but the effect of chronic dysregulation of genes associated with FA metabolism dominates such that TG and DG are still accumulated (Inoguchi et al., 1992; Zhang et al., 2013; Fukushima and Lopaschuk, 2016).
Glucotoxicity
Glucotoxicity is characterized by an intracellular accumulation of glucose metabolites and subsequent impairment in glucose-mediated oxidative phosphorylation. In multiple HF etiologies, such as left ventricular hypertrophy, the heart increases its reliance on oxygen sparing glycolytic energy production. Although glucose uptake is increased, oxidative phosphorylation is diminished, or unchanged (Mori et al., 2013; Li et al., 2017a). In HF patients, augmented glycolysis is uncoupled from oxidative phosphorylation, or mitochondrial energy production, leading to the accumulation of glycolytic byproducts such as lactate and protons (Diakos et al., 2016). These glycolytic byproducts can lead to acidosis, inducing aberrations in ATPases that regulate cytosolic Na+ and Ca2+, ultimately reducing cardiac contractility (Fiolet and Baartscheer, 2000; Jaswal et al., 2011).
High glucose alone can promote pathological cardiac development by accelerating hypertrophy, promoting ER stress, increasing ROS production, and ultimately leading to apoptosis (Ng et al., 2018; Zhang X. et al., 2018; Shi et al., 2019). When uncoupled from oxidative phosphorylation, glycolysis produces glycolytic intermediates that can accelerate flux through non-anapleurotic pathways, such as the pentose phosphate pathway (PPP), hexosamine biosynthetic pathway, or lactate shuttling (Gupte et al., 2006; Gibb et al., 2017). Specifically, increased flux through the PPP occurs in both hypertrophic and chronic HF (Meerson et al., 1967; Zimmer and Peffer, 1986). Li and colleagues recently demonstrated that restoring glucose oxidation by inhibiting pyruvate dehydrogenase kinase (PDK) 4, a rate controlling enzyme of glucose oxidation, improved cardiac function, and glucose uptake post-ischemia reperfusion injury (Li et al., 2017a). Increased glucose exposure over long periods reduces BCAA catabolism, leading to an accumulation of BCAA and BCAA metabolites (Zhang X. et al., 2018). It is well established that leucine, a BCAA, is a potent activator of the mammalian target of rapamycin (mTOR) signaling pathway (Xu and Brink, 2016). Increased mTOR signaling, a known regulator of cell growth and metabolism, promotes the progression of cardiac hypertrophy and cell death (Xu and Brink, 2016). Moreover, increased glucose exposure has been shown to alter miRNAs that control protein expression and thereby regulate gross cell metabolism. For instance, elevation of miRNA-195 upregulates multiple genes associated with hypertrophic development. Exogenous administration of this oligonucleotide can promote hypertrophy and alter cardiac mitochondrial function, while the complementary oligonucleotide provides a protective effect (Shi et al., 2019; Wang et al., 2019). Lastly, increased use of glucose has been shown to induce cell growth (Kolwicz et al., 2013; Shao et al., 2018) and alter epigenetic regulation in the nucleus (Shao et al., 2018; Lombardi et al., 2019). Even though elevation in glucose appears to be an important signaling mechanism for cell growth and differentiation, specific mechanisms detailing how chronic exposure to high levels of glucose leads to glucotoxicity remains to be further characterized.
Glucotoxicity promotes protein glycation and glycosylation, which are, respectively, non-enzymatic and enzymatic protein modifications mediated by carbohydrates. These changes impact many pathways relevant to myocardial structure-functional relationships. Increased glycation drives the formation of advanced glycation end-products, which accumulate in tissues and contribute to inflammation and subsequent structural alterations, namely fibrosis (Dziubak et al., 2018). The deposition of advanced glycation end products contributes to electrical abnormalities through alterations of the myocyte cytoskeleton, altered ion channel activity, and damage to the local autonomic nervous system that contribute to regulating cardiac rhythm (Balcıoğlu and Müderrisoğlu, 2015). Advanced glycation end products specifically inhibit voltage gated K+ channels, Kv channels, that play a primary role in cardiac microcirculation and a secondary role in atrial repolarization, contributing to metabolic remodeling and arrhythmogenic susceptibility (Bai et al., 2015; Su et al., 2015). Relevant to atrial repolarization and His-Purkinje conduction is increased N-glycosylation of the K2P potassium channel, enhancing its function and transport to the cell membrane. Interestingly, while acute or chronic high glucose exposure increase N-glycosylation of the K2P channel, chronic exposure results in downregulation of its expression (Wiedmann et al., 2019). Acute hyperglycemia promotes the covalent modification of CaMKII by O-linked N-acetylglucosamine (O-GlcNAc) at Ser279, which activates CaMKII autonomously, leading to molecular memory as calcium concentration declines (Erickson et al., 2013). Increased O-GlcNAc-modified CaMKII, as observed in human diabetic hearts, enhances CaMKII-dependent activation of spontaneous SR calcium release, leading to augmented premature ventricular complexes that contribute to arrhythmogenicity (Erickson et al., 2013). Moreover, voltage gated sodium channels, the major ionic current responsible for ventricular membrane depolarization, are also heavily modulated by glucose-based and other posttranslational modifications that can be dysregulated by metabolism. For instance, regulation of Nav1.5 glycosylation and sialyltransferase activity impacts the sodium voltage-gated activity and propensity for arrhythmias (Ufret-Vincenty et al., 2001; Ednie et al., 2013). Moreover, exposure of high glucose in CHO cells transiently overexpressing the human Nav1.5 channel led to a rightward shift in voltage dependence of conductance and steady-state fast inactivation, which was attributed to the observed increase in ROS (Fouda et al., 2020). Whether this mechanism is also pertinent to cardiac cells remains to be determined.
Ketone Body Metabolism
In HF, cardiac substrate utilization of glucose and FAs decrease with a concurrent increased reliance on ketone bodies and BCAAs (Kolwicz et al., 2013; Aubert et al., 2016; Bedi et al., 2016). In the hypertrophied and failing heart, ketone body metabolism supplements energy production following decreased FA oxidation (Aubert et al., 2016). Ketones are primarily produced within hepatocytes and form acetyl-CoA via 3-oxoacid-CoA transferase (SCOT) in extrahepatic tissue to generate energy (Laffel, 1999). The major determinant of cardiac ketone oxidation rates is circulating ketone levels (Laffel, 1999; Ikegami et al., 2017). In human HF, ketone utilization plays an important role in maintaining cardiac metabolism (Aubert et al., 2016; Bedi et al., 2016). Recently, Schugar and colleagues reported that mice subjected to TAC with a cardiomyocyte-specific ablation of SCOT (the rate limiting enzyme in ketone oxidation) displayed increased mitochondrial dysfunction, and accelerated pathological cardiac remodeling (Schugar et al., 2014). Additionally, overexpression of cardiac D-β-hydroxybutyrate dehydrogenase 1 (BDH1) decreased ROS and apoptosis in mice subjected to TAC (Uchihashi et al., 2017). Studies in isolated rat cardiomyocytes exposed to simulated hypoxia showed that β-hydroxybutyrate increases contraction and calcium in a dose-dependent manner (Klos et al., 2019). Experiments in isolated working hearts subjected to 4-weeks of TAC showed that enhancing ketone body oxidation increased energy production although it did not show significantly improvement in cardiac efficiency (Ho et al., 2019). Thus, increased myocardial ketone oxidation may be an adaptive mechanism for the failing heart. These results from animal models are supported by recent findings in humans with chronic HF that show decreased expression of lipid utilization proteins with increased expression of ketone catabolic genes (Aubert et al., 2016; Bedi et al., 2016).
Recently, the “ketogenic diet” has become popular, yet it remains unknown whether enhancing long-term ketone oxidation is adaptive or maladaptive to the human heart. There is concern about the common use of this diet, especially in obese and diabetic individuals with an increased risk for SCA/SCD. Currently, it appears that the benefit of weight loss associated with this diet may outweigh potential adverse cardiac metabolic changes (Harvey et al., 2019). In wild-type mice, ketogenic diets increase ketone oxidation genes, decrease gene profiles associated with glucose utilization, and do not alter expression of FA utilization genes (Shimizu et al., 2018). Nevertheless, long-term effects of increasing ketone bioenergetics remain to be fully detailed.
BCAA Metabolism
Branched chain amino acids can be metabolized in the cardiac muscle via the branched-chain α-ketoacid dehydrogenase (BCKDH) complex (Neinast et al., 2019). Alterations in cardiac BCAA metabolism are associated with cardiac pathologies. For example, defective BCAA catabolism disrupts glucose signaling and sensitizes the heart to ischemic-reperfusion injury (Li et al., 2017b). The expression of BCAA catabolic enzymes is decreased in HF patients, in conjunction with increased levels of BCAA and branched chain keto acids (Sun H. et al., 2016). In fact, studies in TAC-induced mice suggest that increasing BCAA metabolism preserves cardiac structure and function (Chen et al., 2019). BCAA supplementation and more generally high protein diets are common practices in order to obtain desired physiological outcomes such as in resistance training. In a large observational study, it appears that both low (<1 g/kg body weight) and high protein (>1.38 g/kg body weight) intake associate with increased risk for cardiovascular events as well as all-cause mortality even when accounting for risk factors and renal function (Halbesma et al., 2009). Specific conclusions regarding the BCAA element of these dietary classifications, however, require further studies. BCAAs only represent a small part of total protein intake, and high protein diets in observational studies will most likely differ in macronutrient and micronutrient makeup. BCAA supplementation in addition to resistance exercise training in late-stage HF patients does not improve physical and functional capacities (Pineda-Juarez et al., 2016). Nevertheless, multiple clinical trials continue to investigate a potential interaction between BCAA metabolism, BCAA supplementation, and exercise in different types of HF (Halbesma et al., 2009; Pineda-Juarez et al., 2016).
Pathological Mitochondrial Function
Mitochondrial Calcium Handling
Deterioration in contractile mechanics of a heart that is hypertrophied, dilated, or fibrotic correlates with altered excitation-contraction coupling and failing bioenergetics (Franz et al., 1992; Nishimura et al., 2006). Contractile force generation requires calcium signaling; while intracellular SR calcium is reviewed in detail elsewhere (Eisner et al., 2020), in this section, we will specifically provide a brief overview of mitochondrial calcium handling. Mitochondrial calcium dynamics are determined by ATP/ADP ratio, transporter expression levels, and other regulatory signals (Zhou and Tian, 2018). Calcium entry in the mitochondrial inner membrane can be facilitated by the mitochondrial calcium uniporter (MCU). Systemic MCU knockout studies did not reveal any major baseline phenotype although mitochondrial calcium handling was diminished (Pan et al., 2013). Cardiac-specific MCU deletion led to improved cardiac function post-IR (Luongo et al., 2015), supporting the notion that MCU does not impact baseline physiology but instead modulates energetic upregulation by increasing contractility or sympathetic stress signaling. Moreover, MICU1, a regulator of the MCU (Mallilankaraman et al., 2012), was recently shown to possess an additional function where it controls cristae junction and anchoring of the MCU complex (Gottschalk et al., 2019). Mitochondrial calcium extrusion, however, is dependent on mitochondrial Na/Ca exchanger (NCLX). Murine models of inducible cardiac-specific NCLX knockout increased arrhythmogenicity and SCD, while overexpression of NCLX was cardioprotective post-IR (Luongo et al., 2017). Calcium extrusion can also be elicited via the mitochondrial permeability transition pore (MPTP), which promotes apoptotic signaling. Although the molecular identity of the MPTP is debated and unknown, recent studies have suggested that dimerization of the F1F0 ATP synthase is responsible for the pore forming MPTP (Urbani et al., 2019), with a prominent role for the c-subunit of the ATPase in MPTP formation (Neginskaya et al., 2019). Most importantly, recent studies have linked the relevance of mitochondrial calcium dynamics to human cardiac pathology. In diabetic rats, cardiac mitochondrial MCU expression is decreased, diminishing mitochondrial calcium and subsequently blunting pyruvate dehydrogenase activity, which it is the rate-limiting process in glucose oxidation (Suarez et al., 2018). In agreement are studies showing decreased levels of MCU and MICU1 in cardiac tissue from ischemic HF patients (Luongo et al., 2015). The feasibility and specificity of pharmacologically targeting these mitochondrial channels and signaling pathways remain to be determined.
Mitochondrial ROS Generation
ROS are generated when electrons leak from the mitochondrial ETC, allowing free electrons to reduce molecular oxygen (Turrens, 2003). Certain pathological conditions, such as MI and HF, increase ROS production via mitochondrial membrane leakage. Elevation of mitochondrial ROS damage ETC complexes further impairing ATP production in the failing heart (Murray et al., 2003; Redout et al., 2007; Dubouchaud et al., 2018). ROS generation is in concert with previously discussed alterations in mitochondrial calcium dynamics and reduced oxidative phosphorylation (Ide et al., 1999; Zhang H. et al., 2018). Dysfunction of complex 1, 2, and 3 of the ETC have been associated with ROS production (Redout et al., 2007; Dubouchaud et al., 2018). ROS can also trigger the reversible, transient, and minimally concerted opening of the MPTP, which is thought to have a housekeeping physiological function of releasing ROS to the cytosol. Prolonged ROS generation triggers MPTP opening, which leads to ROS-induced-ROS release promoting the destruction of the mitochondrion that may be propagated inter-mitochondrially, culminating in cell death (Zorov et al., 2000). Interestingly, deletion of cyclophilin-D, a regulator of the MPTP, led to resistance to MPTP opening and improved cell survival post-IR (Baines et al., 2005). Additionally, loss of cyclophilin-D led to greater hypertrophy and HF (Elrod et al., 2010). Mitochondrial oxygen levels can be sensed by the mitochondrially localized NADPH-oxidase 4 (Nox4). Unlike other Nox family members, Nox4 is constitutively active where 90% of the electron flux through isolated Nox4 produces H2O2 and 10% forms superoxide (Nisimoto et al., 2014). Cardiac-specific Nox4 overexpression is detrimental to cardiac function post-ischemia-reperfusion injury (Ago et al., 2010), re-enforcing the crucial role that Nox4 plays in regulating myocyte function. An extensive review of mitochondrial ROS is found elsewhere (Cadenas, 2018).
There is a strong link between ROS and cardiac ion channel dysfunction directly impacting cardiac electrophysiology, contractile deficiencies, and SCD. For instance, the ryanodine receptor, can be oxidized by ROS resulting in altered calcium release (Roussel et al., 2015). The carotid body is a group of neurosensory cells that sense blood oxygenation and inform ventilation and adrenergic stimulation to increase blood flow in response to ischemia. Voltage-gated potassium channels (Kv) channels are significantly impacted by redox-based reactions in the coronary vasculature and the carotid body. As a result of altered metabolism there is an increase in ROS in the coronary vasculature and carotid body, resulting in greater oxygenation and perfusion rates in an attempt to reduce hypoxic exposure (Rogers et al., 2006; Moreno-Dominguez et al., 2020). Therein ROS, plays an important direct role in the hyperemic response to increased cardiac output. Yet, long-term activation of this mechanism, either due to metabolic or structural remodeling, has adverse consequences as ROS alters a multitude of vital cardiac pathways that promote SCD. Although Kv channels are also found elsewhere in the heart, including the sino-atrial node and His-Purkinje system, how ROS alters action potential propagation in these specialized cardiac cells are currently unknown. Importantly, Kv channels have varying subunit composition in various tissues as well as modulation by other cellular redox agents such as NADH and NADPH which increase the activity of Kv channels (Dwenger et al., 2018). Kv channel activation via ROS in an acutely adaptive mechanism, such as increasing oxygenation and perfusion, contrasts Kv channel inhibition by glycation and products discussed in the section “Glucotoxicity” (Su et al., 2015; Moreno-Dominguez et al., 2020). While it is reasonable to conclude that metabolic toxicity would dysregulate this family of ion channels, further studies are required to delineate how ROS modification of various ionic channels are integrated in the resultant arrhythmic incidence of the heart.
Past and On Going Therapeutic Interventions to Improve Glucose and FA Metabolism
Clinical Approaches to Modulate Glucose Metabolism
The interaction between insulin resistance and diabetic cardiomyopathy has been a major driver for unraveling therapies that enhance glucose metabolism. Enhancing glucose uptake has been attempted by insulin infusion called glucose–insulin–potassium (GIK) therapy, which involved exposure to high concentrations of insulin during acute ischemia (Sodi-Pallares et al., 1962). Some studies have suggested that GIK infusion during ischemia reduced infarct size and improved post-ischemic cardiac dysfunction, but clinical trials were inconsistent and a meticulous glucose control was essential for those patients (Van den Berghe et al., 2003; Malmberg et al., 2005). GLP1 can increase glucose uptake by increasing GLUT2 expression in pancreatic islets (Villanueva-Penacarrillo et al., 2001). Exenatide is a human GLP1 receptor agonist that is currently being investigated as a potential adjunct therapy for HF. A study conducted in 2007 showed that patients treated with exenatide had decreased mortality from cardiovascular causes when compared to patients treated with placebo or standard of care (Erdmann et al., 2007). Exenatide can also contribute to vasorelaxation due to the effect on opening KATP channels and therefore preventing vessel dysfunction (Selley et al., 2014). Conversely, recent studies showed no significant benefit on cardiovascular events in T2DM patients (Mentz et al., 2018). Liraglutide is an FDA-approved drug that is currently used to treat T2DM patients. Liraglutide also activates the GLP1 receptor, and is currently being investigated as a potential treatment for HF. T2DM patients treated with liraglutide have improved mortality rates compared to placebo treated patients (Cohen and Beckey, 2016). Circulating GLP1 is quickly metabolized by dipeptidyl peptidase (DPP)-IV, and thus, a DPP-IV inhibitors can be used as adjuvant with GLP1 agonists to improve GLP1 efficacy (Deacon, 2004). One study demonstrated that an inhibitor of DPP-IV, sitagliptin, improves left ventricular function in patients with coronary artery disease (Read et al., 2010). It has been suggested that the mechanism for the cardioprotective effect of sitagliptin is via upregulation of the transient receptor potential channel (TRP), therefore increasing Ca2+ influx (Al-Awar et al., 2018). In contrast, a recent study revealed no benefit on reducing HF risk in patients with diabetes treated with sitagliptin (Nauck et al., 2019).
Pyruvate is a key glycolytic metabolite which has positive inotropic actions in both healthy and dysfunctional hearts, improving left ventricular function (Mentzer et al., 1989). Pyruvate can potentiate the inotropic response of β-adrenergic receptor compounds and result in increased intracellular Ca2+ transients via improvement of SERCA in failing human myocardium (Hermann et al., 2002). The pyruvate dehydrogenase complex (PDC) metabolizes pyruvate to acetyl-CoA, and PDK inactivates this complex via phosphorylation, leading to a buildup of glucose metabolites. Inhibiting PDK with dichloroacetate (DCA) is one approach that increases the utilization of glucose in the heart (Stacpoole et al., 1998). Clinical evidence has shown that DCA can weakly improve cardiac function through the regulation of voltage-gated K+ channels, but unfortunately, this compound has a major toxicity risk for the liver, kidney, and nervous system (Michelakis et al., 2002). Other alternative compounds currently under development for PDK inhibition have also shown substantial side effects in multiple organs (Morrell et al., 2003; Roche and Hiromasa, 2007). Currently, another effective modulator of PDK, PS10, is being evaluated as potential drug target (Wu et al., 2018). Moreover, SGLT2s are expressed in the early proximal tubule of the kidney (Vallon et al., 2011), and while SGLT2 inhibitors have been developed with the intent to improve glucose control in diabetic patients via its impact on renal sodium-glucose re-absorptive function, it has sparked enthusiasm for the treatment of HF. The EMPA-trial revealed that T2DM patients treated with empagliflozin possessed a 38% relative risk reduction in death from cardiovascular causes (Zinman et al., 2015). Empagliflozin also significantly regulates calcium and sodium exchange, which can be a potential mechanism for the cardioprotective effect (Lee et al., 2019). While there is a consensus that improving glucose metabolism is an attractive pharmacological intervention, specific, safe, and effective compounds that provide cardiovascular benefits still need to be developed.
Therapeutic Strategies Targeting Fatty Acid Utilization
As mitochondrial FA utilization is dependent on CPT-1, this transporter is an attractive pharmacological target for HF, such that inhibiting FA oxidation can result in parallel increase of glucose utilization (Xu et al., 1995). Perhexiline maleate was developed as an antianginal drug in the 1970s (Ashrafian et al., 2007). It potentially inhibits the uptake of fatty-acyl-CoA via mitochondrial CPT-1 (Murray et al., 2005), and this inhibition of FA metabolism improves left ventricular ejection fraction (LVEF) in patients with chronic HF (Lee et al., 2005). Perhexiline also potentially protects the heart from cardiac dysfunction by inhibiting Human ether-a-go-go-related gene (HERG) channels (Walker et al., 1999). Etomoxir is an irreversible inhibitor of CPT-1 (Lopaschuk et al., 1988) that also increased SERCA2a expression, improving SR Ca2+ handling and cardiac function (Vetter and Rupp, 1994). It was first developed and tested in clinical trials in 1984, passing phase I and II, and becoming a promising drug for HF patients (Schmidt-Schweda and Holubarsch, 2000). Unfortunately, etomoxir treatment resulted in abnormally high liver transaminase levels and now is only used as an experimental tool for inhibiting FA oxidation (Holubarsch et al., 2007).
Trimetazidine is an inhibitor of 3-ketoacyl CoA thiolase, the terminal enzyme of FA β-oxidation (Kantor et al., 2000; Lopaschuk et al., 2003). Trimetazidine treatment reduced FA oxidation and induced a compensatory increase in glucose oxidation. Trimetazidine also reduced the accumulation of lactic acid caused by the buildup of fatty acids. Further, this drug modified calcium dynamics by inhibiting SERCA activity and modifying the density of Ca2+ channels in cardiomyocytes impacting left ventricular systolic and diastolic function (Kiyosue et al., 1986; Banach et al., 2006; Meng et al., 2006). In clinical trials trimetazidine significantly increased LVEF (Vitale et al., 2004), myocardial perfusion, oxidative metabolism, and work efficiency (Fragasso et al., 2006), and is being investigated as a potential therapeutic strategy for HF patients (Dezsi, 2016).
Ranolazine was FDA-approved in 2006 for the treatment of stable angina pectoris (Reddy et al., 2010). Ranolazine was first thought to inhibit FA metabolism similarly to trimetazidine, but the drug concentration required to inhibit FA β-oxidation was much higher than the recommended therapeutic dose, indicating an alternative mechanism of action (McCormack et al., 1996). Studies suggest that the primary mechanism of action for ranolazine is via inhibition of the late sodium current and diastolic Ca2+overload, which benefit patients with diastolic dysfunction (Antzelevitch et al., 2004; Belardinelli et al., 2006; Fraser et al., 2006). Ranolazine has been shown in clinical studies to have therapeutic efficacy for many kinds of cardiomyopathies (Sossalla et al., 2008; Maier et al., 2013).
Lastly, PPARs are key transcriptional factors that regulate FA uptake and metabolism, and modulation of various PPAR family members may have a beneficial impact for HF. The expression of PPARα is decreased during TAC-induced HF (Kaimoto et al., 2017). However, ischemic failing rats hearts treated with the PPARα agonist fenofibrate did not show improvements in left ventricular function (Morgan et al., 2006). Additionally, fenofibrate treatment may lead to left ventricular dysfunction in mice with hypertension (Ogata et al., 2004; Brigadeau et al., 2007). Post-trial follow up from the ACCORD study confirmed the original overall neutral results of the study but revealed a beneficial impact of fenofibrate therapy in reducing cardiovascular diseases in a specific group of participants with diabetes, hypertriglyceridemia, and low high-density lipoprotein cholesterol (Elam et al., 2017). PPARγ agonists are currently used in the treatment of T2DM with suggested indirect benefits to cardiovascular disease (Elam et al., 2011). Table 2 summarizes previous and ongoing therapeutic interventions for glucose and FA metabolism.
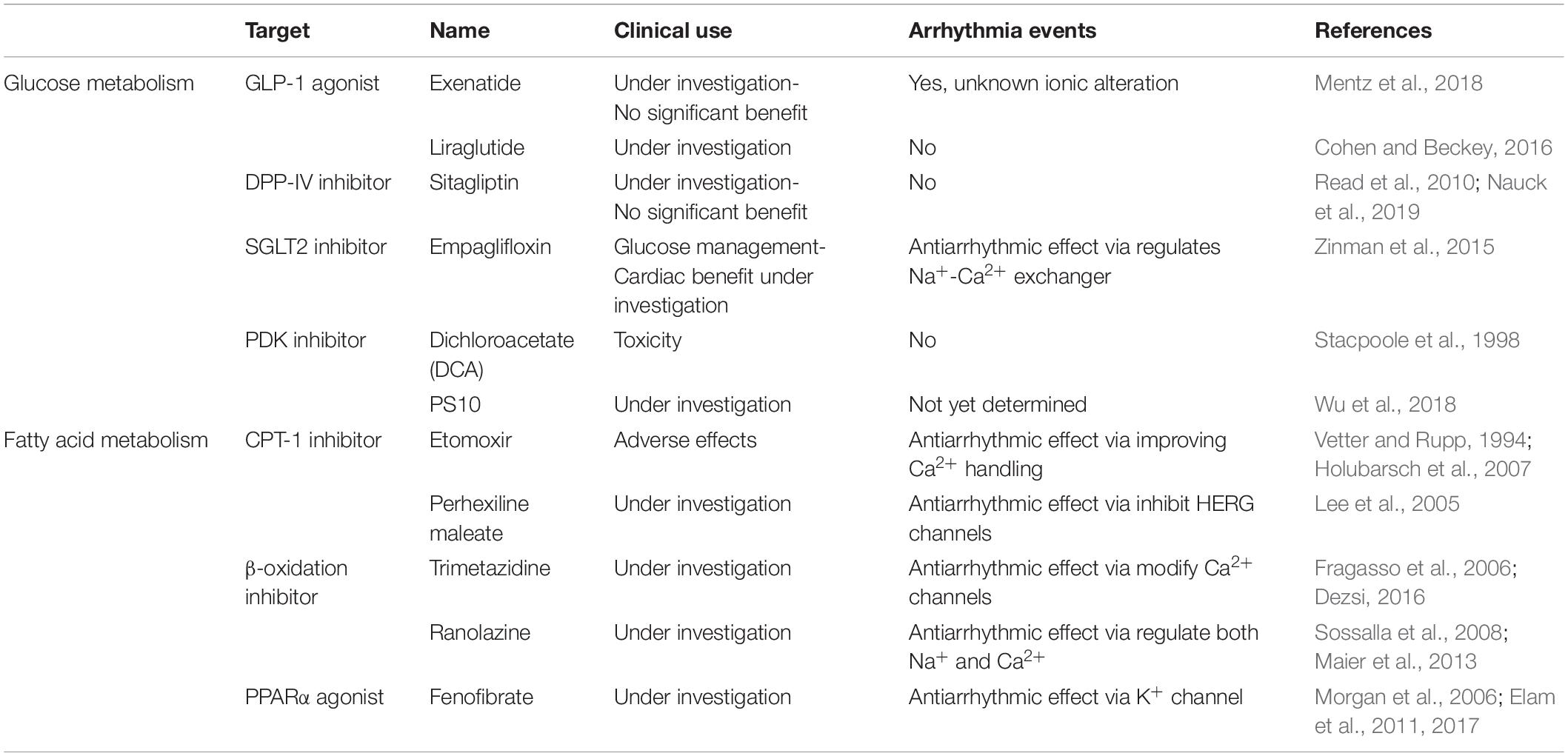
Table 2. Therapeutic interventions that may regulate glucose and FA metabolism.
Conclusion
Atrial fibrillation, tachyarrhythmias, and dysfunction in mechanical junctions preceding cardiac structural remodeling and fibrosis may all contribute to SCD (Jalife et al., 2009; Sato et al., 2009, 2011, 2018). More recently, cardiac metabolism has been studied as a potential early causal mode that promotes and supports SCD (Wu et al., 1993; Roussel et al., 2015). Metabolic flexibility and shifts in metabolic utilization are major determinants of adaptive and maladaptive cardiac signaling. This is particularly evident in T2DM patients, where aberrant glucose control is thought to contribute to cardiac dysfunction in more than 50% of patients (Dunlay et al., 2019). While metabolic pathways are somewhat plastic, detangling specific substrate utilization pathways has been particularly challenging because substrate preference alterations include horizontal crosstalk of various metabolic pathways. Undoubtedly, intervening at early maladaptive stages is key to delay or impede the development of HF and SCD. Detailing which enzymatic reactions are critical and pharmacologically targetable is essential to developing specific compounds that intervene accurately and efficaciously in the fight against SCD events. Future studies will be paramount in unraveling key enzymatic events for the development of novel compounds and determining which population groups are more prone to be responsive to these interventions.
Author Contributions
JS and RZ contributed equally to this work. JS, RZ, AL, and PS reviewed the literature, drafted the manuscript, and critically revised the manuscript.
Funding
This work was supported by the American Heart Association (17SDG33660407) to PS. We also acknowledge the NIH Funded Diabetes Research Center NIH DK 19525.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abel, E. D., Kaulbach, H. C., Tian, R., Hopkins, J. C., Duffy, J., Doetschman, T., et al. (1999). Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J. Clin. Invest. 104, 1703–1714. doi: 10.1172/jci7605
Abumrad, N. A., El-Maghrabi, M. R., Amri, E. Z., Lopez, E., and Grimaldi, P. A. (1993). Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J. Biol. Chem. 268, 17665–17668.
Ago, T., Kuroda, J., Pain, J., Fu, C., Li, H., and Sadoshima, J. (2010). Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 106, 1253–1264. doi: 10.1161/circresaha.109.213116
Al-Awar, A., Almasi, N., Szabo, R., Takacs, I., Murlasits, Z., Szucs, G., et al. (2018). Novel Potentials of the DPP-4 inhibitor sitagliptin against Ischemia-Reperfusion (I/R) injury in Rat Ex-Vivo heart model. Int. J. Mol. Sci. 19:3226. doi: 10.3390/ijms19103226
Antzelevitch, C., Belardinelli, L., Zygmunt, A. C., Burashnikov, A., Di Diego, J. M., Fish, J. M., et al. (2004). Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 110, 904–910. doi: 10.1161/01.cir.0000139333.83620.5d
Ashrafian, H., Horowitz, J. D., and Frenneaux, M. P. (2007). Perhexiline. Cardiovasc. Drug Rev. 25, 76–97. doi: 10.1111/j.1527-3466.2007.00006.x
Aubert, G., Martin, O. J., Horton, J. L., Lai, L., Vega, R. B., Leone, T. C., et al. (2016). The failing heart relies on Ketone bodies as a fuel. Circulation 133, 698–705. doi: 10.1161/circulationaha.115.017355
Baggio, L. L., and Drucker, D. J. (2007). Biology of incretins: GLP-1 and GIP. Gastroenterology 132, 2131–2157. doi: 10.1053/j.gastro.2007.03.054
Bai, J. Y., Ding, W. G., Kojima, A., Seto, T., and Matsuura, H. (2015). Putative binding sites for arachidonic acid on the human cardiac Kv 1.5 channel. Br. J. Pharmacol. 172, 5281–5292. doi: 10.1111/bph.13314
Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. doi: 10.1038/nature03434
Balcıoğlu, A. S., and Müderrisoğlu, H. (2015). Diabetes and cardiac autonomic neuropathy: clinical manifestations, cardiovascular consequences, diagnosis and treatment. World J. Diabetes 6, 80–91.
Banach, M., Okonski, P., Rysz, J., Drozdz, J., and Zaslonka, J. (2006). The role of Trimetazidine in the Treatment of Heart Diseases. Poznan: Termedia Publishing House.
Banerjee, S. K., Mcgaffin, K. R., Pastor-Soler, N. M., and Ahmad, F. (2009). SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 84, 111–118. doi: 10.1093/cvr/cvp190
Beaty, N. B., and Lane, M. D. (1983). Kinetics of activation of acetyl-CoA carboxylase by citrate. Relationship to the rate of polymerization of the enzyme. J. Biol. Chem. 258, 13043–13050.
Bedi, K. C. Jr., Snyder, N. W., Brandimarto, J., Aziz, M., Mesaros, C., Worth, A. J., et al. (2016). Evidence for Intramyocardial disruption of lipid metabolism and increased Myocardial Ketone utilization in advanced human heart failure. Circulation 133, 706–716. doi: 10.1161/circulationaha.115.017545
Belardinelli, L., Shryock, J. C., and Fraser, H. (2006). Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92(Suppl. 4), iv6–iv14. doi: 10.1136/hrt.2005.078790
Belke, D. D., Larsen, T. S., Gibbs, E. M., and Severson, D. L. (2001). Glucose metabolism in perfused mouse hearts overexpressing human GLUT-4 glucose transporter. Am. J. Physiol. Endocrinol. Metab. 280, E420–E427.
Benjamin, E. J., Virani, S. S., Callaway, C. W., Chamberlain, A. M., Chang, A. R., Cheng, S., et al. (2018). Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation 137, e67–e492.
Bharadwaj, K. G., Hiyama, Y., Hu, Y., Huggins, L. A., Ramakrishnan, R., Abumrad, N. A., et al. (2010). Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J. Biol. Chem. 285, 37976–37986. doi: 10.1074/jbc.m110.174458
Bhashyam, S., Fields, A. V., Patterson, B., Testani, J. M., Chen, L., Shen, Y. T., et al. (2010). Glucagon-like peptide-1 increases myocardial glucose uptake via p38alpha MAP kinase-mediated, nitric oxide-dependent mechanisms in conscious dogs with dilated cardiomyopathy. Circ. Heart Fail 3, 512–521. doi: 10.1161/circheartfailure.109.900282
Binas, B., Danneberg, H., Mcwhir, J., Mullins, L., and Clark, A. J. (1999). Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. 13, 805–812. doi: 10.1096/fasebj.13.8.805
Bonezzi, F., Piccoli, M., Dei Cas, M., Paroni, R., Mingione, A., Monasky, M. M., et al. (2019). Sphingolipid Synthesis inhibition by Myriocin administration enhances lipid consumption and Ameliorates lipid response to Myocardial Ischemia reperfusion injury. Front. Physiol. 10:986. doi: 10.3389/fphys.2019.00986
Boudina, S., and Abel, E. D. (2007). Diabetic cardiomyopathy revisited. Circulation 115, 3213–3223. doi: 10.1161/circulationaha.106.679597
Brigadeau, F., Gele, P., Wibaux, M., Marquie, C., Martin-Nizard, F., Torpier, G., et al. (2007). The PPARalpha activator fenofibrate slows down the progression of the left ventricular dysfunction in porcine tachycardia-induced cardiomyopathy. J. Cardiovasc. Pharmacol. 49, 408–415. doi: 10.1097/fjc.0b013e3180544540
Brosius, F. C. III, Liu, Y., Nguyen, N., Sun, D., Bartlett, J., and Schwaiger, M. (1997). Persistent myocardial ischemia increases GLUT1 glucose transporter expression in both ischemic and non-ischemic heart regions. J. Mol. Cell Cardiol. 29, 1675–1685. doi: 10.1006/jmcc.1997.0405
Cadenas, S. (2018). Mitochondrial uncoupling. ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 1859, 940–950. doi: 10.1016/j.bbabio.2018.05.019
Chen, L. Q., Hou, B. H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X. Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532.
Chen, M., Gao, C., Yu, J., Ren, S., Wang, M., Wynn, R. M., et al. (2019). Therapeutic effect of targeting branched-chain Amino acid catabolic flux in pressure-overload induced heart failure. J. Am. Heart Assoc. 8:e011625.
Chess, D. J., and Stanley, W. C. (2008). Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc. Res. 79, 269–278. doi: 10.1093/cvr/cvn074
Chien, A. J., Carr, K. M., Shirokov, R. E., Rios, E., and Hosey, M. M. (1996). Identification of palmitoylation sites within the L-type calcium channel beta2a subunit and effects on channel function. J. Biol. Chem. 271, 26465–26468. doi: 10.1074/jbc.271.43.26465
Chiu, H. C., Kovacs, A., Blanton, R. M., Han, X., Courtois, M., Weinheimer, C. J., et al. (2005). Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ. Res. 96, 225–233. doi: 10.1161/01.res.0000154079.20681.b9
Chiu, H. C., Kovacs, A., Ford, D. A., Hsu, F. F., Garcia, R., Herrero, P., et al. (2001). A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Invest. 107, 813–822. doi: 10.1172/jci10947
Coburn, C. T., Knapp, F. F. Jr., Febbraio, M., Beets, A. L., Silverstein, R. L., and Abumrad, N. A. (2000). Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 275, 32523–32529. doi: 10.1074/jbc.m003826200
Cohen, S., and Beckey, C. (2016). Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 375:1797. doi: 10.1056/nejmc1611289
Corr, P. B., Creer, M. H., Yamada, K. A., Saffitz, J. E., and Sobel, B. E. (1989). Prophylaxis of early ventricular fibrillation by inhibition of acylcarnitine accumulation. J. Clin. Invest. 83, 927–936. doi: 10.1172/jci113978
Deacon, C. F. (2004). Circulation and degradation of GIP and GLP-1. Horm. Metab. Res. 36, 761–765. doi: 10.1055/s-2004-826160
Depre, C., Shipley, G. L., Chen, W., Han, Q., Doenst, T., Moore, M. L., et al. (1998). Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat. Med. 4, 1269–1275. doi: 10.1038/3253
Dezsi, C. A. (2016). Trimetazidine in practice: review of the clinical and experimental evidence. Am. J. Ther. 23, e871–e879. doi: 10.1097/mjt.0000000000000180
Diakos, N. A., Navankasattusas, S., Abel, E. D., Rutter, J., Mccreath, L., Ferrin, P., et al. (2016). Evidence of Glycolysis up-regulation and Pyruvate Mitochondrial oxidation mismatch during mechanical unloading of the failing human heart: implications for cardiac reloading and conditioning. JACC Basic Transl. Sci. 1, 432–444. doi: 10.1016/j.jacbts.2016.06.009
Doehner, W., Gathercole, D., Cicoira, M., Krack, A., Coats, A. J., Camici, P. G., et al. (2010). Reduced glucose transporter GLUT4 in skeletal muscle predicts insulin resistance in non-diabetic chronic heart failure patients independently of body composition. Int. J. Cardiol. 138, 19–24. doi: 10.1016/j.ijcard.2008.07.004
Domenighetti, A. A., Danes, V. R., Curl, C. L., Favaloro, J. M., Proietto, J., and Delbridge, L. M. (2010). Targeted GLUT-4 deficiency in the heart induces cardiomyocyte hypertrophy and impaired contractility linked with Ca(2+) and proton flux dysregulation. J. Mol. Cell Cardiol. 48, 663–672. doi: 10.1016/j.yjmcc.2009.11.017
Drosatos, K., and Schulze, P. C. (2013). Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr. Heart Fail Rep. 10, 109–121. doi: 10.1007/s11897-013-0133-0
Dubouchaud, H., Walter, L., Rigoulet, M., and Batandier, C. (2018). Mitochondrial NADH redox potential impacts the reactive oxygen species production of reverse Electron transfer through complex I. J. Bioenerg. Biomembr. 50, 367–377. doi: 10.1007/s10863-018-9767-7
Dunlay, S. M., Givertz, M. M., Aguilar, D., Allen, L. A., Chan, M., Desai, A. S., et al. (2019). Type 2 Diabetes Mellitus and Heart Failure: a Scientific Statement From the American Heart Association and the Heart Failure Society of America: this statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline update. Circulation 140, e294–e324.
Dwenger, M. M., Ohanyan, V., Navedo, M. F., and Nystoriak, M. A. (2018). Coronary microvascular Kv1 channels as regulatory sensors of intracellular pyridine nucleotide redox potential. Microcirculation 25:e12426. doi: 10.1111/micc.12426
Dyck, J. R., and Lopaschuk, G. D. (2002). Malonyl CoA control of fatty acid oxidation in the ischemic heart. J. Mol. Cell Cardiol. 34, 1099–1109. doi: 10.1006/jmcc.2002.2060
Dziubak, A., Wójcicka, G., Wojtak, A., and Bełtowski, J. (2018). Metabolic effects of metformin in the failing heart. Int. J. Mol. Sci. 19:2869. doi: 10.3390/ijms19102869
Eberli, F. R., Weinberg, E. O., Grice, W. N., Horowitz, G. L., and Apstein, C. S. (1991). Protective effect of increased glycolytic substrate against systolic and diastolic dysfunction and increased coronary resistance from prolonged global underperfusion and reperfusion in isolated rabbit hearts perfused with erythrocyte suspensions. Circ. Res. 68, 466–481. doi: 10.1161/01.res.68.2.466
Ednie, A. R., Horton, K. K., Wu, J., and Bennett, E. S. (2013). Expression of the sialyltransferase, ST3Gal4, impacts cardiac voltage-gated sodium channel activity, refractory period and ventricular conduction. J. Mol. Cell Cardiol. 59, 117–127. doi: 10.1016/j.yjmcc.2013.02.013
Eisner, D. A., Caldwell, J. L., Trafford, A. W., and Hutchings, D. C. (2020). The control of diastolic calcium in the heart: basic mechanisms and functional implications. Circ. Res. 126, 395–412. doi: 10.1161/circresaha.119.315891
Elam, M., Lovato, L. C., and Ginsberg, H. (2011). Role of fibrates in cardiovascular disease prevention, the ACCORD-Lipid perspective. Curr. Opin. Lipidol. 22, 55–61. doi: 10.1097/mol.0b013e328341a5a8
Elam, M. B., Ginsberg, H. N., Lovato, L. C., Corson, M., Largay, J., Leiter, L. A., et al. (2017). Association of Fenofibrate therapy with long-term cardiovascular risk in Statin-treated patients with type 2 diabetes. JAMA Cardiol. 2, 370–380.
Elrod, J. W., Wong, R., Mishra, S., Vagnozzi, R. J., Sakthievel, B., Goonasekera, S. A., et al. (2010). Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687. doi: 10.1172/jci43171
Erdmann, E., Charbonnel, B., Wilcox, R. G., Skene, A. M., Massi-Benedetti, M., Yates, J., et al. (2007). Pioglitazone use and heart failure in patients with type 2 diabetes and preexisting cardiovascular disease: data from the PROactive study (PROactive 08). Diabetes Care 30, 2773–2778. doi: 10.2337/dc07-0717
Erickson, J. R., Pereira, L., Wang, L., Han, G., Ferguson, A., Dao, K., et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376. doi: 10.1038/nature12537
Erion, D. M., and Shulman, G. I. (2010). Diacylglycerol-mediated insulin resistance. Nat. Med. 16, 400–402. doi: 10.1038/nm0410-400
Finan, B., Muller, T. D., Clemmensen, C., Perez-Tilve, D., Dimarchi, R. D., and Tschop, M. H. (2016). Reappraisal of GIP Pharmacology for Metabolic Diseases. Trends Mol. Med. 22, 359–376. doi: 10.1016/j.molmed.2016.03.005
Fiolet, J. W., and Baartscheer, A. (2000). Cellular calcium homeostasis during ischemia; a thermodynamic approach. Cardiovasc. Res. 45, 100–106. doi: 10.1016/s0008-6363(99)00294-1
Foster, D. W. (2012). Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J. Clin. Invest. 122, 1958–1959. doi: 10.1172/jci63967
Fouda, M. A., Ghovanloo, M. R., and Ruben, P. C. (2020). Cannabidiol protects against high glucose-induced oxidative stress and cytotoxicity in cardiac voltage-gated sodium channels. Br. J. Pharmacol. doi: 10.1111/bph.15020 [Online ahead of print]
Fragasso, G., Palloshi, A., Puccetti, P., Silipigni, C., Rossodivita, A., Pala, M., et al. (2006). A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J. Am. Coll. Cardiol. 48, 992–998. doi: 10.1016/j.jacc.2006.03.060
Franz, M. R., Cima, R., Wang, D., Profitt, D., and Kurz, R. (1992). Electrophysiological effects of myocardial stretch and mechanical determinants of stretch-activated arrhythmias. Circulation 86, 968–978. doi: 10.1161/01.cir.86.3.968
Fraser, H., Belardinelli, L., Wang, L., Light, P. E., Mcveigh, J. J., and Clanachan, A. S. (2006). Ranolazine decreases diastolic calcium accumulation caused by ATX-II or ischemia in rat hearts. J. Mol. Cell Cardiol. 41, 1031–1038. doi: 10.1016/j.yjmcc.2006.08.012
Fukushima, A., and Lopaschuk, G. D. (2016). Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta 1861, 1525–1534. doi: 10.1016/j.bbalip.2016.03.020
Gibb, A. A., Lorkiewicz, P. K., Zheng, Y.-T., Zhang, X., Bhatnagar, A., Jones, S. P., et al. (2017). Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem. J. 474, 2785–2801. doi: 10.1042/bcj20170474
Gimeno, R. E., Ortegon, A. M., Patel, S., Punreddy, S., Ge, P., Sun, Y., et al. (2003). Characterization of a heart-specific fatty acid transport protein. J. Biol. Chem. 278, 16039–16044. doi: 10.1074/jbc.m211412200
Goldenberg, J. R., Carley, A. N., Ji, R., Zhang, X., Fasano, M., Schulze, P. C., et al. (2019). Preservation of Acyl Coenzyme a attenuates pathological and metabolic cardiac remodeling through selective lipid trafficking. Circulation 139, 2765–2777. doi: 10.1161/circulationaha.119.039610
Gottschalk, B., Klec, C., Leitinger, G., Bernhart, E., Rost, R., Bischof, H., et al. (2019). MICU1 controls cristae junction and spatially anchors mitochondrial Ca(2+) uniporter complex. Nat. Commun. 10:3732.
Gupte, S. A., Levine, R. J., Gupte, R. S., Young, M. E., Lionetti, V., Labinskyy, V., et al. (2006). Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J. Mol. Cell Cardiol. 41, 340–349. doi: 10.1016/j.yjmcc.2006.05.003
Halbesma, N., Bakker, S. J. L., Jansen, D. F., Stolk, R. P., De Zeeuw, D., De Jong, P. E., et al. (2009). High protein intake associates with cardiovascular events but not with loss of renal function. J. Am. Soc. Nephrology 20, 1797–1804. doi: 10.1681/asn.2008060649
Harris, D. A., and Das, A. M. (1991). Control of mitochondrial ATP synthesis in the heart. Biochem. J. 280(Pt 3), 561–573. doi: 10.1042/bj2800561
Harvey, K. L., Holcomb, L. E., and Kolwicz, S. C. Jr. (2019). Ketogenic diets and exercise performance. Nutrients 11:2296. doi: 10.3390/nu11102296
Hermann, H. P., Zeitz, O., Lehnart, S. E., Keweloh, B., Datz, N., Hasenfuss, G., et al. (2002). Potentiation of beta-adrenergic inotropic response by pyruvate in failing human myocardium. Cardiovasc. Res. 53, 116–123. doi: 10.1016/s0008-6363(01)00437-0
Hiromura, M., Mori, Y., Kohashi, K., Terasaki, M., Shinmura, K., Negoro, T., et al. (2016). Suppressive effects of Glucose-Dependent insulinotropic polypeptide on cardiac hypertrophy and fibrosis in Angiotensin II-Infused mouse models. Circ. J. 80, 1988–1997. doi: 10.1253/circj.cj-16-0152
Ho, K. L., Zhang, L., Wagg, C., Al Batran, R., Gopal, K., Levasseur, J., et al. (2019). Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 115, 1606–1616. doi: 10.1093/cvr/cvz045
Holubarsch, C. J., Rohrbach, M., Karrasch, M., Boehm, E., Polonski, L., Ponikowski, P., et al. (2007). A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. 113, 205–212. doi: 10.1042/cs20060307
Houten, S. M., Violante, S., Ventura, F. V., and Wanders, R. J. (2016). The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 78, 23–44. doi: 10.1146/annurev-physiol-021115-105045
Ide, T., Tsutsui, H., Kinugawa, S., Utsumi, H., Kang, D., Hattori, N., et al. (1999). Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 85, 357–363. doi: 10.1161/01.res.85.4.357
Ikegami, R., Shimizu, I., Yoshida, Y., and Minamino, T. (2017). Metabolomic Analysis in Heart Failure. Circ. J. 82, 10–16. doi: 10.1253/circj.cj-17-1184
Inoguchi, T., Battan, R., Handler, E., Sportsman, J. R., Heath, W., and King, G. L. (1992). Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc. Natl. Acad. Sci. U.S.A. 89, 11059–11063. doi: 10.1073/pnas.89.22.11059
Jalife, J. D. M., Anumonwo, J., Berenfeld, O., and Kalifa, J. (2009). Basic Cardiac Electrophysiology for the Clinician. Hoboken, NJ: Wiley-Blackwell.
Jaswal, J. S., Keung, W., Wang, W., Ussher, J. R., and Lopaschuk, G. D. (2011). Targeting fatty acid and carbohydrate oxidation–a novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta 1813, 1333–1350. doi: 10.1016/j.bbamcr.2011.01.015
Kaimoto, S., Hoshino, A., Ariyoshi, M., Okawa, Y., Tateishi, S., Ono, K., et al. (2017). Activation of PPAR-alpha in the early stage of heart failure maintained myocardial function and energetics in pressure-overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 312, H305–H313.
Kantor, P. F., Lucien, A., Kozak, R., and Lopaschuk, G. D. (2000). The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ. Res. 86, 580–588. doi: 10.1161/01.res.86.5.580
Kiyosue, T., Nakamura, S., and Arita, M. (1986). Effects of trimetazidine on action potentials and membrane currents of guinea-pig ventricular myocytes. J. Mol. Cell Cardiol. 18, 1301–1311. doi: 10.1016/s0022-2828(86)80433-3
Klos, M., Morgenstern, S., Hicks, K., Suresh, S., and Devaney, E. J. (2019). The effects of the ketone body beta-hydroxybutyrate on isolated rat ventricular myocyte excitation-contraction coupling. Arch. Biochem. Biophys. 662, 143–150. doi: 10.1016/j.abb.2018.11.027
Knowlton, F. P., and Starling, E. H. (1912). Experiments on the consumption of sugar in the normal and the diabetic heart. J. Physiol. 45, 146–163. doi: 10.1113/jphysiol.1912.sp001542
Kolwicz, S. C. Jr., Olson, D. P., Marney, L. C., Garcia-Menendez, L., Synovec, R. E., and Tian, R. (2012). Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 111, 728–738. doi: 10.1161/circresaha.112.268128
Kolwicz, S. C. Jr., Purohit, S., and Tian, R. (2013). Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 113, 603–616. doi: 10.1161/circresaha.113.302095
Kraegen, E. W., Sowden, J. A., Halstead, M. B., Clark, P. W., Rodnick, K. J., Chisholm, D. J., et al. (1993). Glucose transporters and in vivo glucose uptake in skeletal and cardiac muscle: fasting, insulin stimulation and immunoisolation studies of GLUT1 and GLUT4. Biochem. J. 295(Pt 1), 287–293. doi: 10.1042/bj2950287
Laffel, L. (1999). Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 15, 412–426. doi: 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8
Lane, M. D., Moss, J., Ryder, E., and Stoll, E. (1970). The activation of acetyl CoA carboxylase by tricarboxylic acids. Adv. Enzyme Regul. 9, 237–251. doi: 10.1016/s0065-2571(71)80047-x
Law, B. A., Liao, X., Moore, K. S., Southard, A., Roddy, P., Ji, R., et al. (2018). Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J. 32, 1403–1416. doi: 10.1096/fj.201700300r
Lee, L., Campbell, R., Scheuermann-Freestone, M., Taylor, R., Gunaruwan, P., Williams, L., et al. (2005). Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation 112, 3280–3288. doi: 10.1161/circulationaha.105.551457
Lee, T. I., Chen, Y. C., Lin, Y. K., Chung, C. C., Lu, Y. Y., Kao, Y. H., et al. (2019). Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and reverses cardiac remodeling in streptozotocin-induced diabetic rats. Int. J. Mol. Sci. 20:1680. doi: 10.3390/ijms20071680
Li, T., Xu, J., Qin, X., Hou, Z., Guo, Y., Liu, Z., et al. (2017a). Glucose oxidation positively regulates glucose uptake and improves cardiac function recovery after myocardial reperfusion. Am. J. Physiol. Endocrinol. Metab. 313, E577–E585.
Li, T., Zhang, Z., Kolwicz, S. C. Jr., Abell, L., Roe, N. D., Kim, M., et al. (2017b). Defective Branched-Chain Amino Acid catabolism disrupts glucose metabolism and sensitizes the Heart to Ischemia-reperfusion injury. Cell Metab. 25, 374–385. doi: 10.1016/j.cmet.2016.11.005
Liao, R., Jain, M., Cui, L., D’agostino, J., Aiello, F., Luptak, I., et al. (2002). Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 106, 2125–2131. doi: 10.1161/01.cir.0000034049.61181.f3
Lim, L. L., Lau, E. S. H., Fung, E., Lee, H. M., Ma, R. C. W., Tam, C. H. T., et al. (2020). Circulating branched-chain amino acids and incident heart failure in type 2 diabetes: the Hong Kong Diabetes Register. Diabetes Metab. Res. Rev. 36:e3253.
Lombardi, A. A., Gibb, A. A., Arif, E., Kolmetzky, D. W., Tomar, D., Luongo, T. S., et al. (2019). Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat. Commun. 10:4509.
Lopaschuk, G. D., Barr, R., Thomas, P. D., and Dyck, J. R. (2003). Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ. Res. 93, e33–e37.
Lopaschuk, G. D., Wall, S. R., Olley, P. M., and Davies, N. J. (1988). Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ. Res. 63, 1036–1043. doi: 10.1161/01.res.63.6.1036
Lundsgaard, A. M., Fritzen, A. M., Nicolaisen, T. S., Carl, C. S., Sjoberg, K. A., Raun, S. H., et al. (2019). Glucometabolic consequences of acute and prolonged inhibition of fatty acid oxidation. J. Lipid Res. 61, 10–19. doi: 10.1194/jlr.ra119000177
Luongo, T. S., Lambert, J. P., Gross, P., Nwokedi, M., Lombardi, A. A., Shanmughapriya, S., et al. (2017). The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 545, 93–97.
Luongo, T. S., Lambert, J. P., Yuan, A., Zhang, X., Gross, P., Song, J., et al. (2015). The Mitochondrial Calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 12, 23–34. doi: 10.1016/j.celrep.2015.06.017
Luptak, I., Yan, J., Cui, L., Jain, M., Liao, R., and Tian, R. (2007). Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation 116, 901–909. doi: 10.1161/circulationaha.107.691253
Maier, L. S., Layug, B., Karwatowska-Prokopczuk, E., Belardinelli, L., Lee, S., Sander, J., et al. (2013). RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction: the RALI-DHF proof-of-concept study. JACC Heart Fail 1, 115–122. doi: 10.1016/j.jchf.2012.12.002
Mallilankaraman, K., Doonan, P., Cardenas, C., Chandramoorthy, H. C., Muller, M., Miller, R., et al. (2012). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644. doi: 10.1016/j.cell.2012.10.011
Malmberg, K., Ryden, L., Wedel, H., Birkeland, K., Bootsma, A., Dickstein, K., et al. (2005). Intense metabolic control by means of insulin in patients with diabetes mellitus and acute myocardial infarction (DIGAMI 2): effects on mortality and morbidity. Eur. Heart J. 26, 650–661. doi: 10.1093/eurheartj/ehi199
McCormack, J. G., Barr, R. L., Wolff, A. A., and Lopaschuk, G. D. (1996). Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 93, 135–142. doi: 10.1161/01.cir.93.1.135
McGarry, J. D., and Brown, N. F. (1997). The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 244, 1–14. doi: 10.1111/j.1432-1033.1997.00001.x
McGavock, J. M., Lingvay, I., Zib, I., Tillery, T., Salas, N., Unger, R., et al. (2007). Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation 116, 1170–1175. doi: 10.1161/circulationaha.106.645614
Meerson, F. Z., Spiritchev, V. B., Pshennikova, M. G., and Djachkova, L. V. (1967). The role of the pentose-phosphate pathway in adjustment of the heart to a high load and the development of myocardial hypertrophy. Experientia 23, 530–532. doi: 10.1007/bf02137950
Meng, D., Feng, L., Chen, X. J., Yang, D., and Zhang, J. N. (2006). Trimetazidine improved Ca2+ handling in isoprenaline-mediated myocardial injury of rats. Exp. Physiol. 91, 591–601. doi: 10.1113/expphysiol.2005.032615
Mentz, R. J., Thompson, V. P., Aguilar, D., Choi, J., Gustavson, S. M., Iqbal, N., et al. (2018). Effects of Once-Weekly exenatide on clinical outcomes in patients with preexisting cardiovascular disease. Circulation 138, 2576–2578. doi: 10.1161/circulationaha.118.036811
Mentzer, R. M. Jr., Van Wylen, D. G., Sodhi, J., Weiss, R. J., Lasley, R. D., Willis, J., et al. (1989). Effect of pyruvate on regional ventricular function in normal and stunned myocardium. Ann. Surg. 209, 629–633.
Michelakis, E. D., Mcmurtry, M. S., Wu, X. C., Dyck, J. R., Moudgil, R., Hopkins, T. A., et al. (2002). Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 105, 244–250. doi: 10.1161/hc0202.101974
Milstein, M. L., Musa, H., Balbuena, D. P., Anumonwo, J. M., Auerbach, D. S., Furspan, P. B., et al. (2012). Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 109, E2134–E2143.
Montaigne, D., Marechal, X., Coisne, A., Debry, N., Modine, T., Fayad, G., et al. (2014). Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130, 554–564. doi: 10.1161/circulationaha.113.008476
Moreno-Dominguez, A., Ortega-Saenz, P., Gao, L., Colinas, O., Garcia-Flores, P., Bonilla-Henao, V., et al. (2020). Acute O2 sensing through HIF2alpha-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci. Signal. 13:eaay9452. doi: 10.1126/scisignal.aay9452
Morgan, E. E., Rennison, J. H., Young, M. E., Mcelfresh, T. A., Kung, T. A., Tserng, K. Y., et al. (2006). Effects of chronic activation of peroxisome proliferator-activated receptor-alpha or high-fat feeding in a rat infarct model of heart failure. Am. J. Physiol. Heart Circ. Physiol. 290, H1899–H1904.
Mori, J., Alrob, O. A., Wagg, C. S., Harris, R. A., Lopaschuk, G. D., and Oudit, G. Y. (2013). ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. Am. J. Physiol. Heart Circ. Physiol. 304, H1103–H1113.
Morrell, J. A., Orme, J., Butlin, R. J., Roche, T. E., Mayers, R. M., and Kilgour, E. (2003). AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem. Soc. Trans. 31, 1168–1170. doi: 10.1042/bst0311168
Murray, A. J., Panagia, M., Hauton, D., Gibbons, G. F., and Clarke, K. (2005). Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes 54, 3496–3502. doi: 10.2337/diabetes.54.12.3496
Murray, J., Taylor, S. W., Zhang, B., Ghosh, S. S., and Capaldi, R. A. (2003). Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J. Biol. Chem. 278, 37223–37230. doi: 10.1074/jbc.m305694200
Murthy, M. S., and Pande, S. V. (1984). Mechanism of carnitine acylcarnitine translocase-catalyzed import of acylcarnitines into mitochondria. J. Biol. Chem. 259, 9082–9089.
Muthuramu, I., Amin, R., Postnov, A., Mishra, M., Jacobs, F., Gheysens, O., et al. (2017). Coconut Oil aggravates pressure overload-induced cardiomyopathy without inducing obesity, systemic insulin resistance, or Cardiac Steatosis. Int. J. Mol. Sci. 18:1565. doi: 10.3390/ijms18071565
Nagoshi, T., Yoshimura, M., Rosano, G. M., Lopaschuk, G. D., and Mochizuki, S. (2011). Optimization of cardiac metabolism in heart failure. Curr. Pharm. Des. 17, 3846–3853.
Nakamura, T., Sugihara, H., Inaba, T., Kinoshita, N., Adachi, Y., Hirasaki, S., et al. (1999). CD36 deficiency has little influence on the pathophysiology of hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 31, 1253–1259. doi: 10.1006/jmcc.1999.0957
Nauck, M. A., Mcguire, D. K., Pieper, K. S., Lokhnygina, Y., Strandberg, T. E., Riefflin, A., et al. (2019). Sitagliptin does not reduce the risk of cardiovascular death or hospitalization for heart failure following myocardial infarction in patients with diabetes: observations from TECOS. Cardiovasc. Diabetol. 18:116.
Neely, J. R., and Morgan, H. E. (1974). Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 36, 413–459. doi: 10.1146/annurev.ph.36.030174.002213
Neginskaya, M. A., Solesio, M. E., Berezhnaya, E. V., Amodeo, G. F., Mnatsakanyan, N., Jonas, E. A., et al. (2019). ATP Synthase C-Subunit-Deficient Mitochondria have a small Cyclosporine a-sensitive channel, but lack the permeability transition pore. Cell Rep. 26:e12.
Neinast, M. D., Jang, C., Hui, S., Murashige, D. S., Chu, Q., Morscher, R. J., et al. (2019). Quantitative analysis of the whole-body metabolic fate of Branched-Chain Amino Acids. Cell Metab. 29:417-429.e4.
Neubauer, S. (2007). The failing heart–an engine out of fuel. N. Engl. J. Med. 356, 1140–1151. doi: 10.1056/nejmra063052
Ng, K.-M., Lau, Y.-M., Dhandhania, V., Cai, Z.-J., Lee, Y.-K., Lai, W.-H., et al. (2018). Empagliflozin Ammeliorates high glucose induced-Cardiac Dysfuntion in human iPSC-Derived Cardiomyocytes. Sci. Rep. 8, 14872–14872.
Nikolaidis, L. A., Elahi, D., Hentosz, T., Doverspike, A., Huerbin, R., Zourelias, L., et al. (2004). Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation 110, 955–961. doi: 10.1161/01.cir.0000139339.85840.dd
Nishimura, S., Kawai, Y., Nakajima, T., Hosoya, Y., Fujita, H., Katoh, M., et al. (2006). Membrane potential of rat ventricular myocytes responds to axial stretch in phase, amplitude and speed-dependent manners. Cardiovasc. Res. 72, 403–411. doi: 10.1016/j.cardiores.2006.08.011
Nisimoto, Y., Diebold, B. A., Cosentino-Gomes, D., and Lambeth, J. D. (2014). Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53, 5111–5120. doi: 10.1021/bi500331y
Niu, Y. G., Hauton, D., and Evans, R. D. (2004). Utilization of triacylglycerol-rich lipoproteins by the working rat heart: routes of uptake and metabolic fates. J. Physiol. 558, 225–237. doi: 10.1113/jphysiol.2004.061473
O’Donnell, J. M., Fields, A. D., Sorokina, N., and Lewandowski, E. D. (2008). The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J. Mol. Cell Cardiol. 44, 315–322. doi: 10.1016/j.yjmcc.2007.11.006
Ogata, T., Miyauchi, T., Sakai, S., Takanashi, M., Irukayama-Tomobe, Y., and Yamaguchi, I. (2004). Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. J. Am. Coll. Cardiol. 43, 1481–1488. doi: 10.1016/j.jacc.2003.11.043
Okere, I. C., Chandler, M. P., Mcelfresh, T. A., Rennison, J. H., Sharov, V., Sabbah, H. N., et al. (2006). Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution, and serum leptin. Am. J. Physiol. Heart Circ. Physiol. 291, H38–H44.
Opie, L. H. (1968). Metabolism of the heart in health and disease. I. Am. Heart J. 76, 685–698. doi: 10.1016/0002-8703(68)90168-3
Opie, L. H. (1969). Metabolism of the heart in health and disease. II. Am. Heart J. 77, 100–122. doi: 10.1016/0002-8703(69)90135-5
Opie, L. H., and Knuuti, J. (2009). The adrenergic-fatty acid load in heart failure. J. Am. Coll. Cardiol. 54, 1637–1646. doi: 10.1016/j.jacc.2009.07.024
Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. doi: 10.1038/ncb2868
Paternostro, G., Clarke, K., Heath, J., Seymour, A. M., and Radda, G. K. (1995). Decreased GLUT-4 mRNA content and insulin-sensitive deoxyglucose uptake show insulin resistance in the hypertensive rat heart. Cardiovasc. Res. 30, 205–211. doi: 10.1016/0008-6363(95)00019-4
Paternostro, G., Pagano, D., Gnecchi-Ruscone, T., Bonser, R. S., and Camici, P. G. (1999). Insulin resistance in patients with cardiac hypertrophy. Cardiovasc. Res. 42, 246–253. doi: 10.1016/s0008-6363(98)00233-8
Patterson, S. W., Piper, H., and Starling, E. H. (1914). The regulation of the heart beat. J. Physiol. 48, 465–513. doi: 10.1113/jphysiol.1914.sp001676
Pei, Z., Xiao, Y., Meng, J., Hudmon, A., and Cummins, T. R. (2016). Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation. Nat. Commun. 7:12035.
Pereira, R. O., Wende, A. R., Olsen, C., Soto, J., Rawlings, T., Zhu, Y., et al. (2013). Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J. Am. Heart Assoc. 2:e000301.
Pineda-Juarez, J. A., Sanchez-Ortiz, N. A., Castillo-Martinez, L., Orea-Tejeda, A., Cervantes-Gaytan, R., Keirns-Davis, C., et al. (2016). Changes in body composition in heart failure patients after a resistance exercise program and branched chain amino acid supplementation. Clin. Nutr. 35, 41–47. doi: 10.1016/j.clnu.2015.02.004
Randle, P. J., Garland, P. B., Hales, C. N., and Newsholme, E. A. (1963). The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789. doi: 10.1016/s0140-6736(63)91500-9
Rattigan, S., Appleby, G. J., and Clark, M. G. (1991). Insulin-like action of catecholamines and Ca2+ to stimulate glucose transport and GLUT4 translocation in perfused rat heart. Biochim. Biophys. Acta 1094, 217–223. doi: 10.1016/0167-4889(91)90012-m
Razeghi, P., Young, M. E., Alcorn, J. L., Moravec, C. S., Frazier, O. H., and Taegtmeyer, H. (2001). Metabolic gene expression in fetal and failing human heart. Circulation 104, 2923–2931. doi: 10.1161/hc4901.100526
Read, P. A., Khan, F. Z., Heck, P. M., Hoole, S. P., and Dutka, D. P. (2010). DPP-4 inhibition by sitagliptin improves the myocardial response to dobutamine stress and mitigates stunning in a pilot study of patients with coronary artery disease. Circ. Cardiovasc. Imaging 3, 195–201. doi: 10.1161/circimaging.109.899377
Reddy, B. M., Weintraub, H. S., and Schwartzbard, A. Z. (2010). Ranolazine: a new approach to treating an old problem. Tex. Heart Inst. J. 37, 641–647.
Redout, E. M., Wagner, M. J., Zuidwijk, M. J., Boer, C., Musters, R. J., Van Hardeveld, C., et al. (2007). Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res. 75, 770–781. doi: 10.1016/j.cardiores.2007.05.012
Roche, T. E., and Hiromasa, Y. (2007). Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol. Life Sci. 64, 830–849. doi: 10.1007/s00018-007-6380-z
Rogers, P. A., Dick, G. M., Knudson, J. D., Focardi, M., Bratz, I. N., and Swafford, A. N. Jr., et al. (2006). H2O2-induced redox-sensitive coronary vasodilation is mediated by 4-aminopyridine-sensitive K+ channels. Am. J. Physiol. Heart Circ. Physiol. 291, H2473–H2482.
Roussel, J., Thireau, J., Brenner, C., Saint, N., Scheuermann, V., Lacampagne, A., et al. (2015). Palmitoyl-carnitine increases RyR2 oxidation and sarcoplasmic reticulum Ca2+ leak in cardiomyocytes: role of adenine nucleotide translocase. Biochim. Biophys. Acta 1852, 749–758. doi: 10.1016/j.bbadis.2015.01.011
Rupp, H., and Zarain-Herzberg, A. (2002). Therapeutic potential of CPT I inhibitors: cardiac gene transcription as a target. Expert Opin. Invest. Drugs 11, 345–356. doi: 10.1517/13543784.11.3.345
Samovski, D., Sun, J., Pietka, T., Gross, R. W., Eckel, R. H., Su, X., et al. (2015). Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes 64, 353–359. doi: 10.2337/db14-0582
Sathananthan, A., Man, C. D., Micheletto, F., Zinsmeister, A. R., Camilleri, M., Giesler, P. D., et al. (2010). Common genetic variation in GLP1R and insulin secretion in response to exogenous GLP-1 in nondiabetic subjects: a pilot study. Diabetes Care 33, 2074–2076. doi: 10.2337/dc10-0200
Sato, P. Y., Chuprun, J. K., Grisanti, L. A., Woodall, M. C., Brown, B. R., Roy, R., et al. (2018). Restricting mitochondrial GRK2 post-ischemia confers cardioprotection by reducing myocyte death and maintaining glucose oxidation. Sci. Signal. 11:eaau0144. doi: 10.1126/scisignal.aau0144
Sato, P. Y., Coombs, W., Lin, X., Nekrasova, O., Green, K. J., Isom, L. L., et al. (2011). Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ. Res. 109, 193–201. doi: 10.1161/circresaha.111.247023
Sato, P. Y., Musa, H., Coombs, W., Guerrero-Serna, G., Patino, G. A., Taffet, S. M., et al. (2009). Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 105, 523–526. doi: 10.1161/circresaha.109.201418
Schaffer, J. E., and Lodish, H. F. (1994). Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 79, 427–436. doi: 10.1016/0092-8674(94)90252-6
Schaffer, J. E., and Lodish, H. F. (1995). Molecular mechanism of long-chain fatty acid uptake. Trends Cardiovasc. Med. 5, 218–224. doi: 10.1016/1050-1738(95)00102-6
Schmidt, J. W., and Catterall, W. A. (1987). Palmitylation, sulfation, and glycosylation of the alpha subunit of the sodium channel. Role of post-translational modifications in channel assembly. J. Biol. Chem. 262, 13713–13723.
Schmidt-Schweda, S., and Holubarsch, C. (2000). First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin. Sci. 99, 27–35. doi: 10.1042/cs0990027
Schramm, M., Klieber, H. G., and Daut, J. (1994). The energy expenditure of actomyosin-ATPase, Ca(2+)-ATPase and Na+,K(+)-ATPase in guinea-pig cardiac ventricular muscle. J. Physiol. 481(Pt 3), 647–662. doi: 10.1113/jphysiol.1994.sp020471
Schugar, R. C., Moll, A. R., André D’avignon, D., Weinheimer, C. J., Kovacs, A., and Crawford, P. A. (2014). Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol. Metab. 3, 754–769. doi: 10.1016/j.molmet.2014.07.010
Selley, E., Kun, S., Szijarto, I. A., Laczy, B., Kovacs, T., Fulop, F., et al. (2014). Exenatide induces aortic vasodilation increasing hydrogen sulphide, carbon monoxide and nitric oxide production. Cardiovasc. Diabetol. 13:69. doi: 10.1186/1475-2840-13-69
Shao, D., and Tian, R. (2015). Glucose transporters in cardiac metabolism and hypertrophy. Compr. Physiol. 6, 331–351. doi: 10.1002/cphy.c150016
Shao, D., Villet, O., Zhang, Z., Choi, S. W., Yan, J., Ritterhoff, J., et al. (2018). Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nat. Commun. 9:2935.
Sharma, S., Adrogue, J. V., Golfman, L., Uray, I., Lemm, J., Youker, K., et al. (2004). Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 18, 1692–1700. doi: 10.1096/fj.04-2263com
Shi, T., Papay, R. S., and Perez, D. M. (2016). alpha1A-Adrenergic receptor prevents cardiac ischemic damage through PKCdelta/GLUT1/4-mediated glucose uptake. J. Recept. Signal. Transduct. Res. 36, 261–270. doi: 10.3109/10799893.2015.1091475
Shi, Y., Yan, C., Li, Y., Zhang, Y., Zhang, G., Li, M., et al. (2019). Expression signature of miRNAs and the potential role of miR-195-5p in high-glucose-treated rat cardiomyocytes. J. Biochem. Mol. Toxicol. 34:e22423.
Shimizu, K., Saito, H., Sumi, K., Sakamoto, Y., Tachi, Y., and Iida, K. (2018). Short-term and long-term ketogenic diet therapy and the addition of exercise have differential impacts on metabolic gene expression in the mouse energy-consuming organs heart and skeletal muscle. Nutr. Res. 60, 77–86. doi: 10.1016/j.nutres.2018.09.004
Slot, J. W., Geuze, H. J., Gigengack, S., James, D. E., and Lienhard, G. E. (1991). Translocation of the glucose transporter GLUT4 in cardiac myocytes of the rat. Proc. Natl. Acad. Sci. U.S.A. 88, 7815–7819. doi: 10.1073/pnas.88.17.7815
Sodi-Pallares, D., Testelli, M. R., Fishleder, B. L., Bisteni, A., Medrano, G. A., Friedland, C., et al. (1962). Effects of an intravenous infusion of a potassium-glucose-insulin solution on the electrocardiographic signs of myocardial infarction. A preliminary clinical report. Am. J. Cardiol. 9, 166–181. doi: 10.1016/0002-9149(62)90035-8
Sossalla, S., Wagner, S., Rasenack, E. C., Ruff, H., Weber, S. L., Schondube, F. A., et al. (2008). Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts–role of late sodium current and intracellular ion accumulation. J. Mol. Cell Cardiol. 45, 32–43. doi: 10.1016/j.yjmcc.2008.03.006
Stacpoole, P. W., Henderson, G. N., Yan, Z., and James, M. O. (1998). Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 106(Suppl. 4), 989–994. doi: 10.1289/ehp.98106s4989
Storch, J., and Thumser, A. E. (2010). Tissue-specific functions in the fatty acid-binding protein family. J. Biol. Chem. 285, 32679–32683. doi: 10.1074/jbc.r110.135210
Stremmel, W., Strohmeyer, G., Borchard, F., Kochwa, S., and Berk, P. D. (1985). Isolation and partial characterization of a fatty acid binding protein in rat liver plasma membranes. Proc. Natl. Acad. Sci. U.S.A. 82, 4–8. doi: 10.1073/pnas.82.1.4
Su, W., Li, W., Chen, H., Liu, H., Huang, H., and Li, H. (2015). Advanced Glycation end products impair Voltage-Gated K+ Channels-Mediated Coronary vasodilation in diabetic rats. PLoS One 10:e0142865. doi: 10.1371/journal.pone.0142865
Suarez, J., Cividini, F., Scott, B. T., Lehmann, K., Diaz-Juarez, J., Diemer, T., et al. (2018). Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 293, 8182–8195. doi: 10.1074/jbc.ra118.002066
Sun, H., Olson, K. C., Gao, C., Prosdocimo, D. A., Zhou, M., Wang, Z., et al. (2016). Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 133, 2038–2049.
Sun, W., Quan, N., Wang, L., Yang, H., Chu, D., Liu, Q., et al. (2016). Cardiac-Specific Deletion of the Pdha1 gene sensitizes heart to toxicological actions of ischemic stress. Toxicol. Sci. 151, 193–203. doi: 10.1093/toxsci/kfw035
Sung, M. M., Byrne, N. J., Kim, T. T., Levasseur, J., Masson, G., Boisvenue, J. J., et al. (2017). Cardiomyocyte-specific ablation of CD36 accelerates the progression from compensated cardiac hypertrophy to heart failure. Am. J. Physiol. Heart Circ. Physiol. 312, H552–H560.
Taegtmeyer, H., and Overturf, M. L. (1988). Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension 11, 416–426. doi: 10.1161/01.hyp.11.5.416
Turk, E., Zabel, B., Mundlos, S., Dyer, J., and Wright, E. M. (1991). Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature 350, 354–356. doi: 10.1038/350354a0
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. doi: 10.1113/jphysiol.2003.049478
Uchihashi, M., Hoshino, A., Okawa, Y., Ariyoshi, M., Kaimoto, S., Tateishi, S., et al. (2017). Cardiac-Specific Bdh1 Overexpression Ameliorates oxidative stress and cardiac remodeling in pressure overload-induced heart failure. Circ. Heart Fail. 10:e004417.
Ufret-Vincenty, C. A., Baro, D. J., Lederer, W. J., Rockman, H. A., Quinones, L. E., and Santana, L. F. (2001). Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J. Biol. Chem. 276, 28197–28203. doi: 10.1074/jbc.m102548200
Urbani, A., Giorgio, V., Carrer, A., Franchin, C., Arrigoni, G., Jiko, C., et al. (2019). Purified F-ATP synthase forms a Ca(2+)-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 10:4341.
Ussher, J. R., and Lopaschuk, G. D. (2008). The malonyl CoA axis as a potential target for treating ischaemic heart disease. Cardiovasc. Res. 79, 259–268. doi: 10.1093/cvr/cvn130
Vallon, V., Platt, K. A., Cunard, R., Schroth, J., Whaley, J., Thomson, S. C., et al. (2011). SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 22, 104–112. doi: 10.1681/asn.2010030246
Van den Berghe, G., Wouters, P. J., Bouillon, R., Weekers, F., Verwaest, C., Schetz, M., et al. (2003). Outcome benefit of intensive insulin therapy in the critically ill: insulin dose versus glycemic control. Crit. Care Med. 31, 359–366. doi: 10.1097/01.ccm.0000045568.12881.10
Varndell, I. M., Bishop, A. E., Sikri, K. L., Uttenthal, L. O., Bloom, S. R., and Polak, J. M. (1985). Localization of glucagon-like peptide (GLP) immunoreactants in human gut and pancreas using light and electron microscopic immunocytochemistry. J. Histochem. Cytochem. 33, 1080–1086. doi: 10.1177/33.10.3900195
Vetter, R., and Rupp, H. (1994). CPT-1 inhibition by etomoxir has a chamber-related action on cardiac sarcoplasmic reticulum and isomyosins. Am. J. Physiol. 267, H2091–H2099.
Villanueva-Penacarrillo, M. L., Puente, J., Redondo, A., Clemente, F., and Valverde, I. (2001). Effect of GLP-1 treatment on GLUT2 and GLUT4 expression in type 1 and type 2 rat diabetic models. Endocrine 15, 241–248. doi: 10.1385/endo:15:2:241
Vitale, C., Wajngaten, M., Sposato, B., Gebara, O., Rossini, P., Fini, M., et al. (2004). Trimetazidine improves left ventricular function and quality of life in elderly patients with coronary artery disease. Eur. Heart J. 25, 1814–1821. doi: 10.1016/j.ehj.2004.06.034
Vork, M. M., Trigault, N., Snoeckx, L. H., Glatz, J. F., and Van Der Vusse, G. J. (1992). Heterogeneous distribution of fatty acid-binding protein in the hearts of Wistar Kyoto and spontaneously hypertensive rats. J. Mol. Cell Cardiol. 24, 317–321. doi: 10.1016/0022-2828(92)93168-j
Walker, B. D., Valenzuela, S. M., Singleton, C. B., Tie, H., Bursill, J. A., Wyse, K. R., et al. (1999). Inhibition of HERG channels stably expressed in a mammalian cell line by the antianginal agent perhexiline maleate. Br. J. Pharmacol. 127, 243–251. doi: 10.1038/sj.bjp.0702502
Wang, L., Qin, D., Shi, H., Zhang, Y., Li, H., and Han, Q. (2019). MiR-195-5p Promotes Cardiomyocyte Hypertrophy by Targeting MFN2 and FBXW7. Biomed. Res. Int. 2019:1580982.
Wiedmann, F., Schlund, D., Voigt, N., Ratte, A., Kraft, M., Katus, H. A., et al. (2019). N-glycosylation-dependent regulation of hK(2P)17.1 currents. Mol. Biol. Cell 30, 1425–1436. doi: 10.1091/mbc.e18-10-0687
Witters, L. A., and Kemp, B. E. (1992). Insulin activation of acetyl-CoA carboxylase accompanied by inhibition of the 5’-AMP-activated protein kinase. J. Biol. Chem. 267, 2864–2867.
Wright, E. M. (2013). Glucose transport families SLC5 and SLC50. Mol. Aspects Med. 34, 183–196. doi: 10.1016/j.mam.2012.11.002
Wu, C. Y., Satapati, S., Gui, W., Wynn, R. M., Sharma, G., Lou, M., et al. (2018). A novel inhibitor of pyruvate dehydrogenase kinase stimulates myocardial carbohydrate oxidation in diet-induced obesity. J. Biol. Chem. 293, 9604–9613. doi: 10.1074/jbc.ra118.002838
Wu, J., and Corr, P. B. (1995). Palmitoylcarnitine increases [Na+]i and initiates transient inward current in adult ventricular myocytes. Am. J. Physiol. 268, H2405–H2417.
Wu, J., Mchowat, J., Saffitz, J. E., Yamada, K. A., and Corr, P. B. (1993). Inhibition of gap junctional conductance by long-chain acylcarnitines and their preferential accumulation in junctional sarcolemma during hypoxia. Circ. Res. 72, 879–889. doi: 10.1161/01.res.72.4.879
Xu, K. Y., Zweier, J. L., and Becker, L. C. (1995). Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ. Res. 77, 88–97. doi: 10.1161/01.res.77.1.88
Xu, L., and Brink, M. (2016). mTOR, cardiomyocytes and inflammation in cardiac hypertrophy. Biochim. Biophys. Acta 1863, 1894–1903. doi: 10.1016/j.bbamcr.2016.01.003
Young, L. H., Renfu, Y., Russell, R., Hu, X., Caplan, M., Ren, J., et al. (1997). Low-flow ischemia leads to translocation of canine heart GLUT-4 and GLUT-1 glucose transporters to the sarcolemma in vivo. Circulation 95, 415–422. doi: 10.1161/01.cir.95.2.415
Zhang, H., Gong, G., Wang, P., Zhang, Z., Kolwicz, S. C., Rabinovitch, P. S., et al. (2018). Heart specific knockout of Ndufs4 ameliorates ischemia reperfusion injury. J. Mol. Cell Cardiol. 123, 38–45. doi: 10.1016/j.yjmcc.2018.08.022
Zhang, X., Lin, Q., Chen, J., Wei, T., Li, C., Zhao, L., et al. (2018). High glucose-induced cardiomyocyte death may be linked to unbalanced branched-chain amino acids and energy metabolism. Molecules 23:807. doi: 10.3390/molecules23040807
Zhang, L., Jaswal, J. S., Ussher, J. R., Sankaralingam, S., Wagg, C., Zaugg, M., et al. (2013). Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ. Heart Fail 6, 1039–1048. doi: 10.1161/circheartfailure.112.000228
Zhang, Y., Bao, M., Dai, M., Wang, X., He, W., Tan, T., et al. (2015). Cardiospecific CD36 suppression by lentivirus-mediated RNA interference prevents cardiac hypertrophy and systolic dysfunction in high-fat-diet induced obese mice. Cardiovasc. Diabetol. 14:69.
Zhou, B., and Tian, R. (2018). Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Invest. 128, 3716–3726. doi: 10.1172/jci120849
Zhou, L., Cryan, E. V., D’andrea, M. R., Belkowski, S., Conway, B. R., and Demarest, K. T. (2003). Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J. Cell Biochem. 90, 339–346. doi: 10.1002/jcb.10631
Zimmer, H. G., and Peffer, H. (1986). Metabolic aspects of the development of experimental cardiac hypertrophy. Basic Res. Cardiol. 81(Suppl. 1), 127–137. doi: 10.1007/978-3-662-11374-5_13
Zinman, B., Wanner, C., Lachin, J. M., Fitchett, D., Bluhmki, E., Hantel, S., et al. (2015). Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 373, 2117–2128. doi: 10.1056/NEJMoa1504720
Keywords: mitochondria, heart failure, sudden cardiac death, substrate utilization, lipotoxicity, glucotoxicity
Citation: Snyder J, Zhai R, Lackey AI and Sato PY (2020) Changes in Myocardial Metabolism Preceding Sudden Cardiac Death. Front. Physiol. 11:640. doi: 10.3389/fphys.2020.00640
Received: 26 March 2020; Accepted: 20 May 2020;
Published: 16 June 2020.
Edited by:
Brian P. Delisle, University of Kentucky, United StatesReviewed by:
Martin Young, The University of Alabama at Birmingham, United StatesMatthew W. Kay, The George Washington University, United States
Douglas Andres, University of Kentucky, United States
Copyright © 2020 Snyder, Zhai, Lackey and Sato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: P. Y. Sato, cHlzMjZAZHJleGVsLmVkdQ==
†These authors have contributed equally to this work